Autoimmune Thyroid Disease
The Syndromes of Thyroid Autoimmunity
The concept of thyroid autoimmunity, the original exemplar of an organ-specific autoimmune disorder, arose from the seminal observations of Rose and Witebsky, who showed that thyroid autoantibodies and thyroiditis developed in rabbits that had been immunized with thyroid extract,1 and from the observations of Roitt and colleagues, who first described thyroglobulin (TG) antibodies in the serum of patients with Hashimoto’s thyroiditis.2 Graves’ disease became defined as an autoimmune disorder after the discovery of a long-acting thyroid stimulator in the serum of such patients3 that subsequently was shown to be a specific immunoglobulin G (IgG) directed against the thyroid-stimulating hormone receptor (TSHR). Over the past 40 years, a huge volume of work founded on these discoveries has continued to improve our understanding of the origin and pathogenesis of these disorders. It is now appreciated that autoimmune hypothyroidism and Graves’ disease share many features, with some patients progressing from one to the other, and that lesser degrees of thyroid autoimmunity (subclinical hypothyroidism, focal thyroiditis, postpartum thyroiditis) are remarkably common in the general population.
The main types of thyroid autoimmunity are summarized in Table 8-1. The prevalence of subclinical thyroid autoimmunity is difficult to define because the prevalence depends in part on the sensitivity of assays for thyroid autoantibodies, with sensitive assays detecting TG and thyroid peroxidase (TPO) antibodies in up to 20% of women. However, focal thyroiditis, which is closely associated with circulating thyroid autoantibodies, is present in up to 40% of white women and 20% of men at autopsy,4 thus indicating that these figures do reflect the real prevalence of subclinical thyroid autoimmunity. Careful community studies have shown that such subclinical disease progresses only slowly and infrequently to overt thyroid dysfunction; the weighted prevalence of autoimmune hypothyroidism in the United States is 0.8 per 100 population, 95% of whom are women.5

Subclinical autoimmune thyroiditis may become clinically apparent in the postpartum period as the result of an exacerbation of the autoimmune process for reasons that are not yet clear. Typically, such women have TPO autoantibodies antepartum and experience transient thyroid dysfunction (thyrotoxicosis followed by hypothyroidism, or either phase alone) accompanied by a small painless goiter, with full recovery within a year after delivery.6 It is now known that postpartum thyroiditis is a risk factor for future permanent hypothyroidism, which ensues in 20% to 30% of cases in the subsequent 5 years. Around 5% of pregnant white women have one or more episodes of postpartum thyroiditis.
Overt hypothyroidism caused by autoimmunity has two main forms: Hashimoto’s (or goitrous) thyroiditis and primary myxedema, also known as atrophic thyroiditis. The former is characterized by a variably sized, firm goiter, often with an irregular surface; the goiter is typically painless, although a rare, painful variant occurs that, in contrast to subacute thyroiditis, might respond poorly to steroid treatment.7 Such patients may have a goiter and normal thyroid function or subclinical or overt hypothyroidism, depending on the extent of the autoimmune destructive process. However, in primary myxedema, the typical manifestation is clinically evident hypothyroidism, obviously without a goiter. Many attempts have been made to ascribe distinct genetic or pathogenic factors to these two disorders, but in most cases, a unique pathogenesis is not evident, and it seems most likely that they are simply at opposite ends of a spectrum of clinical features, with gradual diminution in the size of the goiter noted as disease progresses.
Similarly, Graves’ disease shares many immunologic features with autoimmune hypothyroidism, and even the hallmark of Graves’ disease, TSHR autoantibodies, can be found in some patients with autoimmune hypothyroidism, although their effects are masked by a more vigorous destructive autoimmune process (see below). Graves’ disease is the most common autoimmune disorder in the United States, with an estimated weighted prevalence of 1.2 per 100, 88% of whom are women.5 Many of these individuals progress to hypothyroidism, either spontaneously after successful treatment with antithyroid drugs or iatrogenically after radioiodine therapy or surgery.
Pathology
The typical appearance of Hashimoto’s thyroiditis is termed struma lymphomatosa to indicate the extensive infiltration of the thyroid by lymphocytes, plasma cells, and macrophages.7 As well as a diffuse infiltrate, germinal center formation may be particularly prominent, and giant (Langerhans) cells can occur. The thyroid follicular cells are destroyed to a variable extent, depending on the chronicity of the disease, and during this process, the remaining cells become hyperplastic and undergo oxyphil metaplasia, which gives rise to the so-called Askanazy or Hürthle cells. Variable fibrosis and, in rare cases, concurrent changes typical of Graves’ disease appear—so-called hashitoxicosis.
Primary myxedema is characterized by extensive fibrosis, loss of normal lobular architecture, and gland atrophy; lymphocytic infiltration varies from minor to moderate. It remains to be established how frequently Hashimoto’s thyroiditis progresses to primary myxedema. The extent of fibrosis in autoimmune hypothyroidism is directly correlated with the age of the patient,8 compatible with the occurrence of such a progression, although little change in the pathologic appearance has been found in sequential biopsies over 10 to 20 years in those with Hashimoto’s thyroiditis.9 The histologic appearances of postpartum thyroiditis and silent thyroiditis resemble Hashimoto’s thyroiditis, although oxyphil metaplasia and germinal centers are less frequent, whereas in focal thyroiditis, mild Hashimoto-like changes are seen in localized areas of the thyroid, but most follicles are preserved.
The pathologic features of Graves’ disease usually are obscured by prior treatment with antithyroid drugs, whose effects on the autoimmune process are considered later. Hypertrophy and hyperplasia of the thyroid follicles may be noted, the epithelium is columnar, and the colloid shrinks.7 In addition, a variable degree of lymphocytic infiltration is present, sometimes with germinal center formation. Lymphoid hyperplasia also can be found in the thymus, lymph nodes, and spleen. Follicular involution and reversal of the lymphocytic infiltrate and hyperplasia occur with antithyroid drug treatment.
Thus, all forms of thyroid autoimmunity are associated with a lymphocytic infiltrate in the thyroid, and these lymphocytes are largely responsible for generating both T and B cell–mediated autoreactivity, although other sites such as thyroid-draining lymph nodes and bone marrow contain thyroid-autoreactive lymphocytes in autoimmune thyroid disease.10 Many investigations have used peripheral blood lymphocytes from patients, but although these cells are readily accessible, they reflect the behavior of only a small proportion of autoreactive lymphocytes migrating in the blood compartment in what is essentially a localized disease.
The Basis of Autoimmune Disease
This section provides only an attenuated overview of the basic mechanisms underlying autoimmunity; the reader is referred elsewhere for a more comprehensive review.11 For the purposes of this chapter, however, a brief summary of the normal immune response is followed by a description of how autoreactivity can arise.
The Normal Immune Response
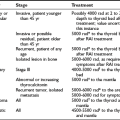
The essence of the immune response is shown in Fig. 8-1. The initial step involves presentation of an antigen by an antigen-presenting cell (APC), such as a dendritic cell or macrophage, to a helper T cell, which recognizes the antigen by means of a specific T cell receptor (TCR).12 Such T cells can be identified phenotypically by expression of the molecule CD4 (CD stands for cluster of differentiation and is used to define an array of surface molecules on lymphocytes and other cells). The antigen is first taken up by simple phagocytosis or by involvement of specific surface receptors, and it is processed by the APC so that fragments of 12 to 18 amino acids are generated. These fragments are the “epitopes” of the antigen, which binds to major histocompatibility complex (MHC) class II molecules expressed constitutively by the APC. The MHC (termed HLA in humans and H-2 in mice) is a huge complex of genes that is of fundamental immunologic importance because class I (described below) and class II genes encode polymorphic molecules capable of binding a wide range of antigenic peptides.13 The ability of an individual to mount an immune response to any given antigen depends in part on the inheritance of class I and II genes, which determine (1) whether appropriate T cells develop in the thymus, because MHC molecules can positively or negatively select T cells with particular antigenic specificities, as determined by their TCR and stage of development; and (2) whether an antigenic epitope can bind to an appropriate MHC molecule and therefore be presented to a mature T cell in adult life.
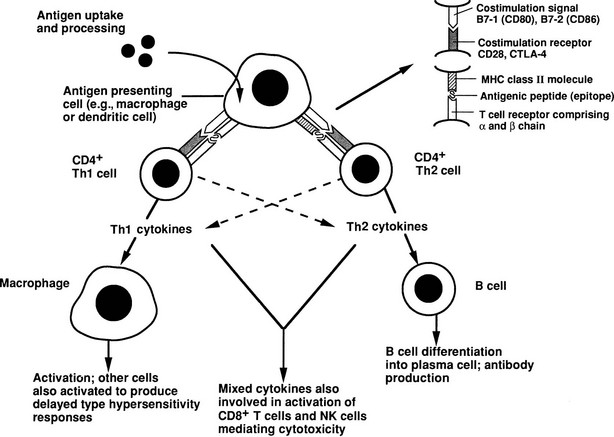
FIGURE 8-1 Simplified diagram of the key elements in a normal immune response, starting with antigen presentation. The type of immune response that is induced depends on the cytokine profile of the T helper cell that is stimulated. (From Weetman AP: Recent progress in thyroid autoimmunity: an overview for the clinician. Thyroid Today 19[2]:1–9, 1996.)
Formation of the trimolecular complex between the MHC class II molecule, antigenic epitope, and TCR is followed by activation of the CD4+ T cell, a process that involves expression of the interleukin-2 (IL-2) receptor and autocrine stimulation by IL-2 release and leads to T cell proliferation, secretion of other cytokines, and thus the development of effector function. However, many other molecules are involved in this interaction and act as stabilizers, signal transducers, or providers of so-called costimulatory or second signals (Fig. 8-2). In brief, interaction of a number of adhesion molecule ligands and receptors, such as intercellular adhesion molecule-1 (ICAM-1, CD54)/lymphocyte function-associated antigen-1 (LFA-1, CD11a/CD18) and LFA-3 (CD58)/CD2, allows initial binding of the T cell to an APC. CD4 interacts with a nonpolymorphic region on the MHC molecule to stabilize the trimolecular complex; this activity explains why helper T cells recognize antigen that is presented by class II rather than class I molecules. CD3 is composed of five peptides that are expressed uniquely by all T cells and serves to initiate the intercellular events after TCR ligation. Finally, a host of costimulatory signals can determine whether antigen recognition proceeds or is terminated because of the absence of an essential costimulator or the presence of an inhibitory signal.
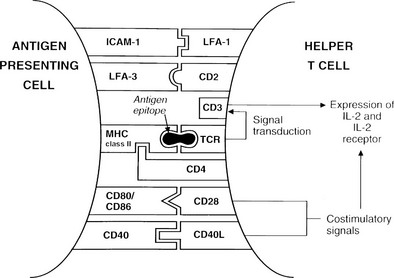
FIGURE 8-2 Diagram of the key molecular interactions in CD4+ T cell activation by an antigen-presenting cell (APC).
The best defined costimulatory pathway involves the interaction of B7-1 (CD80) and B7-2 (CD86) on the APC with CD28, which transduces a stimulatory second signal, or cytotoxic T lymphocyte antigen type 4 (CTLA-4, now named CD152), which transduces an inhibitory second signal on the surface of the T cell.14 Interaction of the TCR on naïve T cells with an MHC class II molecule plus epitope, in the absence of CD28 ligation, induces anergy (rather than stimulation) in the T cell; that is, the T cell is paralyzed and is unable to respond (Fig. 8-3). Similarly, engagement of CTLA-4 results in T-cell anergy. As is discussed below, induction of anergy is an important mechanism in preventing autoimmune responses. Other important membrane-bound costimulatory signals exist, for instance, ICOS (inducible costimulator) ligand, which seems to be particularly important for eliciting responses from previously activated (or memory) T cells.15 Thus, strikingly different outcomes are possible after antigen presentation, depending on which costimulatory signals are delivered, and the signal that is delivered in turn depends on the maturity of the T cell and the type of APC involved.
FIGURE 8-3 Mechanisms for anergy induction. The normal pathway for antigen presentation is shown in the upper panel; failure to provide an appropriate costimulatory signal (lower panels) results in anergy. Classic antigen-presenting cells (APCs) express costimulatory molecules, but class II–positive nonclassic APCs (e.g., thyroid cells) do not.
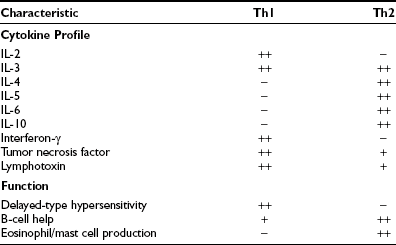
Activated CD4+ T cells can follow two broad pathways of function depending on their pattern of cytokine secretion (Table 8-2). Type 1 helper T (Th1) cells mediate delayed-type hypersensitivity responses, essentially the kind of destructive process that is seen in organ-specific endocrinopathies, whereas Th2 cells promote antibody production.13 A number of factors, including TCR affinity and ligand density, the nature of the APC, and the presence of non–T-cell–derived IL-4 and IL-12, determine which pathway is followed.16,17 Although this paradigm is undoubtedly useful, many helper T cells, particularly in humans, cannot be neatly classified according to the Th1/Th2 dichotomy. For example, a third subset, Th0 cells, produces a mix of Th1 and Th2 cytokines and is thought to be a precursor population; evidence suggests linear development of Th1 into Th2 cells via Th0 in humans.18 Another recently identified subset, Th17, produces the cytokine IL-17 and is highly proinflammatory, with a potent role in causing autoimmunity.17 Expanded clones of T cells arising from naïve T cells after ligation of the T cell receptor give rise to memory T cells, which help antibody production, promote inflammation, or regulate immune responses.19
Table 8-2
Characteristics of Murine CD4+ T Helper (Th) Cell Subsets; Similar but Not Identical Profiles Have Been Identified in Humans
CD8+ T cells are typically cytotoxic but have in the past been assigned suppressor functions. Th1 and Th2 responses are mutually inhibitory; therefore, many suppressor phenomena may be due to a population of cells that are capable of secreting appropriate cytokines that can switch off an ongoing immune response (termed immune deviation). The best defined regulatory T cell subset is CD25+ CD4+, Foxp3+ and is generated in the thymus; the emergence of autoimmune disease in animals when such thymic development is interfered with provides compelling evidence for the importance of these cells.20,21 The cytotoxic function of activated CD8+ T cells is directed against antigenic epitopes that are synthesized within the target cell and presented by MHC class I molecules; the two classic groups of antigens that are recognized by these cells are the products of viral infection or malignant transformation.13 Destruction of target cells is mediated by two mechanisms: (1) the granule exocytosis pathway, which utilizes perforin and granzyme A and B; and (2) apoptosis, or programmed cell death. In the latter, engagement of Fas on the surface of the target cell with Fas ligand (FasL) on the T cell leads to activation of a chain of intracellular events that results in target cell apoptosis (Fig. 8-4). Fas expression is generalized, whereas FasL is restricted to cells of the immune system and to sites of immune privilege, as is described in the next section.
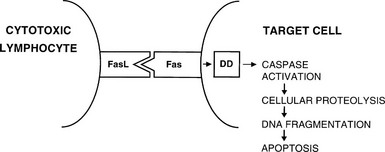
FIGURE 8-4 Interaction between Fas ligand (FasL) on cytotoxic lymphocytes and Fas on a target cell leads to signaling via the death domain (DD) proteins and apoptosis.
Mention should also be made of a further subdivision of T cells. Most T cells have a TCR that is a heterodimer composed of an α and a β chain. The genes for these chains are rearranged to ensure adequate diversity for antigen recognition. However, a small proportion of T cells have a TCR composed of a γ and a δ chain, and these receptors have a more restricted diversity.22 The role of γδ T cells is unclear, but they may be important in mucosal immunity and protection against particular microorganisms.
Two other cell types are essential components of the immune response. B cells produce antibodies after differentiation into plasma cells, a process that is regulated by Th2 cytokines and CD40/CD40 ligand interaction with a helper T cell. Up to 107 different antibody specificities can be generated in humans by recombination and somatic mutation of the immunoglobulin genes.23 During an immune response, these processes ensure selection of the most appropriate antibodies so that IgG molecules with the highest affinity for antigen are produced. Antibodies generally recognize specific determinants on an antigen that is intact rather than processed or fragmented, and these determinants are termed epitopes; however, unlike T cell epitopes, most B cell epitopes are conformational and are formed from discontinuous regions of the molecule.24 As well as producing antibodies, B cells are important APCs in that they are able to take up antigen via this surface-bound immunoglobulin and enter into cognate recognition of appropriate T cells. This type of antigen presentation may be important in diversifying the T cell response because B cells present multiple epitopes of the antigen to T cells.
Killer and natural killer cells are CD3 negative and spontaneously destroy target cells with altered surface antigens (particularly reduced MHC class I), such as tumor cells.13 Because killer and natural killer cells express receptors (CD16) for the constant (Fc) region of immunoglobulins, they also can bind to and kill antibody-coated targets, a process called antibody-dependent cell-mediated cytotoxicity (ADCC). This function also can be mediated by macrophages. Thus, although natural killer cells have little antigen specificity, they can be focused on a target via a specific antibody (Fig. 8-5).
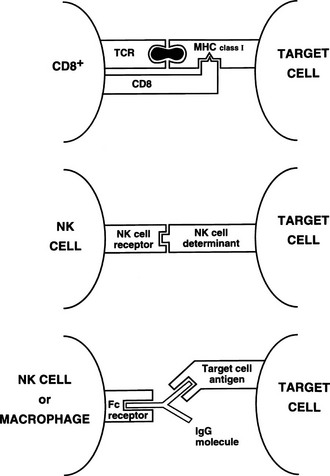
Self-Tolerance and Autoimmunity
The immune system exists to eliminate foreign antigens but must remain tolerant of (i.e., must not respond to) autoantigens. We now know, particularly through experiments with transgenic mice, that individual mechanisms for ensuring self-tolerance (Table 8-3) are practically never completely successful; a few autoreactive T cells are present in all healthy individuals, although in various states of nonreactivity. Failure of the control mechanisms for ensuring nonreactivity allows clonal expansion of these cells, with subsequent autoimmune disease if the response is sufficiently vigorous.

During development, the thymus is mainly responsible for eliminating autoreactive T cells (clonal deletion) and for positively selecting appropriate T cells to constitute the immune repertoire.25,26 These processes depend on the interaction of endogenous peptides, presented by thymic APCs, with the naive TCR repertoire. Inevitably, a few T cells escape tolerance, which is particularly likely if specific endogenous antigens are unavailable for presentation in the thymus because of low abundance outside a developing organ, for example. For these antigens, peripheral tolerance, including both deletion and anergy (see Fig. 8-3), may be very important for regulation of self-reactivity.
As well as deletion and anergy, clonal ignorance provides effective protection against autoreactivity. Simply put, autoantigen-specific lymphocytes are harmless unless they become activated by autoantigen; therefore, autoimmune disease will not result from such cells if they do not come into contact with autoantigen, or if the necessary costimulatory signals are not provided. Autoreactive CD8+ T cells and B cells remain harmless or “ignorant” of self-antigens unless activated by helper T cells; therefore, provided that the latter are controlled, autoimmune disease will not result. However, it should be noted that CD8+ T cells and B cells are subject to central tolerance mechanisms in the fetal thymus or bone marrow and liver, respectively.27 Another mechanism for conferring immunologic privilege at an anatomic site, whereby it becomes protected from recognition, is localized expression of FasL, which induces apoptosis in potentially autoaggressive lymphocytes entering the site; examples of such FasL expression are seen in Sertoli cells, corneal cells, and the placental trophoblast.
Subsidiary control over autoreactive T cells is provided by a number of suppressor mechanisms that often are now referred to as regulatory pathways and remain to be fully characterized. Inhibitory cytokines, such as those that produce reciprocal inhibition of Th1 and Th2 subset responses, provide one means of suppression of harmful autoimmune responses,18 but antigen-specific suppressor phenomena have been defined in many situations, especially in animal cell transfer experiments.20,28,29
Tolerance to a self-antigen can be broken at one or more of the levels at which it operates (see Table 8-3) and may involve both genetic and environmental factors:
1. Autoreactive T cells might not be deleted or rendered anergic in the thymus (e.g., through inheritance of particular MHC genes or, in animals, after neonatal thymectomy).
2. T cells that escape thymic tolerance might fail to be deleted or rendered anergic in the periphery (e.g., because of abnormal provision of costimulatory signals; see Fig. 8-3).
3. Failure of immunologic tolerance might occur (e.g., because of altered FasL expression).
4. Cross-reactive exogenous antigens might induce a response against a normally “silent” autoantigen (e.g., myocarditis after streptococcal infection).
5. Suppressor mechanisms might fail to occur (e.g., provision of high concentrations of endogenous or exogenous cytokines).
Experimental Autoimmune Thyroiditis
The development of animal models of experimental autoimmune thyroiditis (EAT) (Table 8-4) has allowed profound insight into the development of thyroid autoimmunity, for example, Fig. 8-6 shows how autoimmune thyroid disease can arise in mice through modulation at the different stages of T cell tolerance, some of the principles of which were described in the preceding section. Several different types of EAT have been described,30 and these types more or less resemble Hashimoto’s thyroiditis, with lymphocytic infiltration of the thyroid, TG antibodies, and variable degrees of hypothyroidism.30 Most recently, attempts to produce animal models of Graves’ disease31,32 have had some success, although they remain incompletely characterized. Key lessons from these animal models can be summarized as follows.
Table 8-4
Summary of the Main Animal Models of Experimental Autoimmune Thyroiditis
| Model | Antigen | Comments |
| Immunization usually with adjuvant (mouse, rat, rabbit, guinea pig) | TG, TPO, TSHR | Strain dependent, transient, and transferable via T cells; TSHR does not induce a Graves’ disease–like model because the TSHR antibodies are without stimulatory action |
| Thymectomy induced (mouse, rat) | TG | Depends on time of thymectomy and strain, may require sublethal irradiation; transferable with T cells |
| T cell manipulations (mouse) | TG | Cyclosporine A and transfer of specific T cells to T cell–depleted animals induce thyroiditis |
| Spontaneous (chicken, dog, rat) | TG + other autoantigens | Thyroiditis occurs in OS chickens, beagles, NOD mice, and BB and Buffalo strain rats (NOD and BB animals also have autoimmune diabetes) |
| Virus induced (mouse) | TG + polyendocrine autoantigens | Reovirus infection of certain strains of mice |
| SCID mouse | TG, TPO, TSHR | SCID mice allow long-term study of transplanted thyroid tissue from patients with Graves’ and Hashimoto’s disease; disease does not develop in the mice themselves |
| cDNA Immunization (mouse) | TSHR | Allows production of monoclonal TSHR antibodies |
| Immunization with fibroblasts transfected with TSHR and MHC class II (mouse) | TSHR | Hyperthyroidism and histologic features of Graves’ disease develop, but without lymphocytic infiltration |
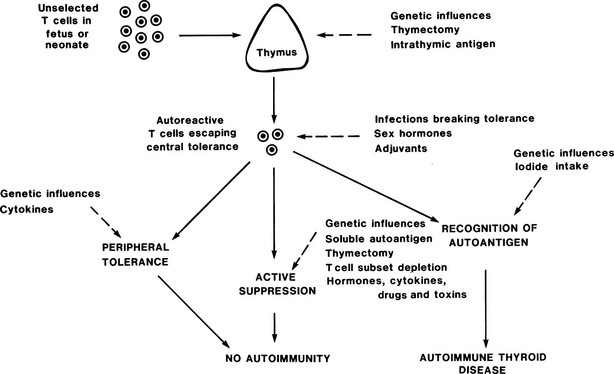
FIGURE 8-6 Control of autoreactive T cells in experimental autoimmune thyroiditis in mice. The sites at which genetic and other factors act are shown as dashed lines; the development of autoimmune thyroid disease depends on the balance between these multiple influences. (From Weetman AP, McGregor AM: Autoimmune thyroid disease: further developments in our understanding. Endocr Rev 15:788–830. © 1994, The Endocrine Society.)
A strong genetic tendency is apparent in all models such that manipulations that readily induce EAT in one strain may induce no disease or a different autoimmune disease (such as oophoritis or gastritis) in a different strain. In both spontaneous and immunization-induced models, the MHC makes a contribution to genetic susceptibility, but it is clear that other non-MHC loci are also involved; in the OS chicken, for example, these loci control T cell responsiveness, glucocorticoid tonus, and intrinsic properties of the thyroid, which makes the OS chicken more susceptible to autoimmunity.33 Perhaps the most elegant demonstration of susceptibility is the creation of HLA-DRB1*0301 (DR3) transgenic mice; HLA-DR3 is an MHC specificity that is known to confer susceptibility to autoimmune disease in humans (see below). Thyroiditis develops in HLA-DR3 but not HLA-DR2 transgenic mice after TG immunization, thereby confirming that this HLA-DRB1 polymorphism determines, at least in part, susceptibility to autoimmune thyroiditis.34
In addition, a number of environmental and endogenous factors contribute to susceptibility, and more must await discovery. Excess iodine exacerbates spontaneous thyroiditis, possibly through the generation of toxic metabolites that are formed with oxygen in the thyroid, but it is also known that a major T cell epitope on TG requires iodination for recognition by autoreactive T cells.35 Antithyroid drugs suppress EAT with no reduction in thyroid hormone levels, which confirms the direct immunomodulatory effects of these drugs.36
The potential adverse effects of infection are illustrated by the absence of EAT in suitably thymectomized rats that are reared under germ-free conditions; transfer of normal gut microflora will induce disease.37 It is possible that this result is due to the nonspecific effects of gut microflora, such as polyclonal lymphocytic activation or release of cytokines, or to some thyroid cross-reactive antigen. Environmental toxins, such as 3-methylcholanthrene, can induce EAT in genetically susceptible strains of rats.30 Similar to humans, female animals generally have more severe EAT than males do; this difference is dependent on sex hormones; estrogen administration worsens thyroiditis, whereas testosterone has the opposite effect.38
In all models, the role of T cells is paramount, as is illustrated by the effects of T cell manipulation on disease induction (see Table 8-4), and disease is transferred only poorly, if at all, by thyroid antibodies.39 In general, transfer of both CD4+ and CD8+ autoreactive T cells from a donor with EAT is needed to induce EAT in a recipient. A key role for T regulatory cells in preventing EAT has been obvious for decades based on cell transfer experiments.30 It is now established that naturally occurring CD4+, CD25+ T cells have a partial role in this regard, and additional contributions from antigen-activated T regulatory cells may occur.40 Additional layers of regulation, including cytokine profiles, are likely to be involved, and it is clear from the multiple models of Graves’ disease that a paradigm of TH1/Th2 balance is too simplistic to explain the type of autoimmune response involved.41
No correlation has been found between the level of TG antibodies and the severity of EAT,30 further demonstrating the uncertain pathologic role for these autoantibodies in EAT. Indeed, in transgenic mice, it has been shown that tolerance is not induced in B cells by membrane-bound antigen expressed on thyroid cells, presumably because the preimmune B cells are sequestered from the antigen by basement membrane and endothelium.42 Because tolerance is induced in T cells in this model, it appears that somehow thyroid surface antigens can affect the development of these cells, either by transport to the thymus or by some type of peripheral tolerance. This observation is important and implies that thyroid autoreactive B cells are frequent but remain ignorant; the frequent appearance of thyroid autoantibodies in otherwise healthy populations could be explained by the activation of these B cells if T cell tolerance breaks down or is bypassed.
Thyroid Autoantigens
TG is a 660 kDa glycoprotein that is composed of two identical subunits and is secreted by thyroid follicular cells into the follicular lumen, where it is stored as colloid. Each TG molecule consists of around 100 tyrosine residues, a quarter of which are iodinated; at four to eight so-called hormonogenic sites, these residues couple to form the thyroid hormones triiodothyronine (T3) and thyroxine (T4). The sequence of human TG has been determined.43 Despite considerable work, the exact location of T and B cell epitopes within TG is still uncertain, even in EAT, in which it is possible to immunize animals with defined peptides.44 A key T cell epitope in the spontaneous thyroiditis of OS chickens contains iodine, and poorly iodinated TG is only weakly immunogenic.35
It has long been recognized that each 330 kDa TG subunit has two major B cell epitopes and a minor epitope.45 However, as the titer of TG antibodies rises in Hashimoto’s thyroiditis over time, other regions of the molecule become targets for autoantibodies—a process that is reflected in mice that are immunized with human TG, in which an array of epitopes subsequently develop.46 The two major B cell epitopes on TG are conformational,47 although linear epitopes have been identified that react with a small proportion of Hashimoto sera.48
Thyroid Peroxidase
TPO, an apical 100 to 105 kDa protein that is responsible for tyrosine iodination and coupling, is the key enzyme in thyroid hormone synthesis. Many studies have confirmed the identity of the previously defined thyroid microsomal antigen as TPO.49 Multiple T cell epitopes exist within the molecule, and individual patients respond to different sets of epitopes with no obvious clinical correlations.50,51
It is also apparent that multiple B cell epitopes are present, some conformational and some linear, because TPO antibodies can recognize native, denatured, or denatured and reduced antigen.52 As has been reviewed extensively elsewhere,53 studies using human and murine monoclonal TPO antibodies have defined two neighboring major domains—A and B—that constitute the antibody reactivity of more than 80% of Hashimoto sera (Fig. 8-7). Some sera bind to epitopes overlapping the two domains. It is striking that the antibody response to TPO is restricted at the level of the germline heavy and light chain variable (V) regions.53,54 Cocrystallization of TPO with monoclonal TPO antibody fragments is required for further elucidation of the epitopes within the main domains of this autoantigen.
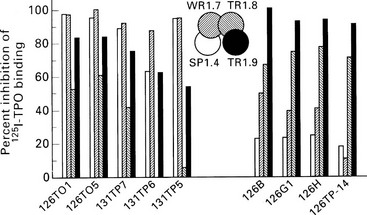
FIGURE 8-7 Epitopic domains on thyroid peroxidase (TPO). Domains A1, A2, B1, and B2 have been defined by the recombinant monoclonal TPO antibodies SP1.4, WR1.7, TR1.8, and TR1.9, respectively (center of diagram), and the bars represent the inhibition of binding of these four monoclonal reagents (shading of bars corresponds to the shading of the domains) by a separate panel of TPO monoclonal antibodies. Those that bind predominantly to domain A are shown on the left, and those that bind predominantly to domain B are shown on the right. (From Guo J, McIntosh RS, Czarnocka B, et al: Relationship between autoantibody epitopic recognition and immunoglobulin gene usage. Clin Exp Immunol 111:408–414, 1998.)
Thyroid-Stimulating Hormone Receptor
TSHR is a member of the G protein–coupled receptor family; activation of TSHR by TSH or that subset of TSHR antibodies with stimulatory activity leads to intracellular signaling by the cyclic adenosine monophosphate (cAMP) pathway, although other signaling pathways operate at high ligand concentrations. TSHR has an extracellular domain, the A subunit, which is linked via a C peptide region to the B subunit, which comprises the transmembrane domain (organized in seven loops) and an intracellular domain.55 Variants of TSHR have been described, in particular a form that does not include the transmembrane region and in which shed A subunits may induce or amplify the immune response to the receptor in Graves’ disease.56 The extrathyroidal expression of TSHR, particularly in the orbit, where it could serve as a cross-reactive autoantigen, may be crucial to the complications of Graves’ disease.57
As with TPO, multiple T cell epitopes, including TSHR sequences that are recognized by 10% to 20% of healthy individuals, have been defined.58,59 B cell epitopes are generally conformational, and it is clear that the response is heterogeneous both within and between patients because some antibodies can stimulate via the receptor in several ways, and other TSHR antibodies bind to the receptor without stimulating it, with a proportion of these antibodies interfering with TSH-mediated stimulation (Table 8-5).60 Further characterization of these B cell epitopes has required the development of human TSHR monoclonal antibodies—a daunting task given the low frequency of TSHR antibody–secreting B cells and the difficulty of expressing the receptor in vitro in its native form.61 Such reagents have shown far greater similarity of binding sites for blocking and stimulatory antibodies than had previously been suspected, and it is likely that these antibodies, together with TSH, bind to similar regions in the leucine-rich horseshoe portion of the TSHR ectodomain.41,60
Table 8-5
Classification of Main Thyroid-Stimulating Hormone Receptor Antibodies
| Antibody | Assay |
| TSAbs | Bioassay; usually measurement of cAMP production by primary cultures of thyroid cells, thyroid cell lines, (e.g., FRTL-5), or cells transfected with TSHR |
| Thyroid-blocking antibodies | Bioassay; measurement of inhibition of cAMP production after TSH-mediated stimulation of the TSHR; may operate at the level of TSH binding or receptor signaling |
| TSH binding-inhibiting immunoglobulins | Measurement of inhibition of radiolabeled TSH binding to TSHR by antibodies; unable to define functional activity |
| Long-acting thyroid stimulator | Original description of TSAb; used bioassay in whole mouse to assess effects of stimulator antibodies on radioiodine release |
Other Autoantigens
Cloning and sequencing of the Na+/I− symporter (NIS) have allowed confirmation of NIS as a fourth major thyroid autoantigen, first demonstrated with the use of cultured dog thyroid cells.62 Up to a third of Graves’ disease sera and 15% of Hashimoto sera contain antibodies that inhibit NIS-mediated iodide uptake in vitro.63 Antibodies to thyroid hormones can be found in 10% to 25% of patients with autoimmune thyroid disease,64 and nonspecific autoantibodies against DNA, tubulin, and other cytoskeletal proteins can be detected in a small proportion of patients.
Genetic Factors
The role of heredity in autoimmune thyroid disease has been illustrated by numerous studies showing a higher frequency of autoimmune thyroid disease or thyroid antibodies in family members of patients with autoimmune hypothyroidism and Graves’ disease.30 That both types of thyroid disease cluster together in families provides additional support for the notion that these conditions share causative and pathogenic features. A number of patterns of inheritance and candidate genes have been suggested, but ascertainment artifacts and the drawbacks of genetic association studies have produced many inconsistencies in results. Meticulous twin studies have shown a concordance rate of only 22% for Graves’ disease—much lower than was previously thought.65
The most important susceptibility factor that has thus far been recognized is the association with particular HLA-DR alleles; the role that these MHC class II genes play in the immune response makes them excellent candidates.66 HLA-DR3 is associated with Graves’ disease and Hashimoto’s thyroiditis in whites and gives a relative risk of between 2 and 6, whereas HLA-DR4 and HLA-DR5 have been associated with goitrous but not atrophic thyroiditis in some white populations.67 Postpartum thyroiditis has only a weak association with HLA-DR5. It should be noted that nonwhite populations have very different HLA associations.68
Detailed family studies have found only weak evidence of linkage between the HLA region and autoimmune thyroid disease.69,70 These results imply the existence of other susceptibility loci. Of the candidates that have been tested, which include genes that encode immunoglobulins, TCR, and various cytokines, the most robust association has been seen with two linked polymorphisms of the CTLA-4 gene, which exists in both Graves’ disease and autoimmune hypothyroidism and confers a relative risk of around 2.67,68 The same polymorphism confers susceptibility for type 1 diabetes mellitus and therefore presumably reflects some generalized effect of a common functional variant of the CTLA-4 gene on autoreactive T cell regulation. A number of studies have employed genome-wide scans to identify new susceptibility genes, but their results indicate that no genes exert a major effect and, conversely, no upper limit is placed on the number of genes that have a small effect.68a The candidate gene approach has established that polymorphisms in PTPN22 and the IL-2 receptor (CD25), both integrally involved in T cell regulation, contribute to susceptibility, and a contribution to the Fc receptor–like-3 locus is likely.69–72 The TSHR has been confirmed as a susceptiblity locus for Graves’ disease but not Hashimoto’s thyroiditis.73
Environmental Factors
Work in EAT has identified several environmental influences on thyroiditis (see earlier), and epidemiologic evidence supports a role for some of these environmental factors in humans; only these factors can explain the rapid changes in the prevalence of Hashimoto’s thyroiditis recently described in Italy.74 An excess of thyroid autoantibodies and thyroiditis occurs after iodination programs75; as in EAT, iodine might increase the immunogenicity of thyroid autoantigens or may have a role in the generation of toxic metabolites. Radioiodine given therapeutically can rarely induce autoimmune responses in patients with nodular goiters.76 The mechanism is obscure but could relate to the release of thyroid autoantigens (possibly altered by radiation damage and therefore more immunogenic) or to the effect of 131I on radiation-sensitive regulatory T cell subsets. Occupational exposure to ionizing radiation also may be a risk factor.77
No convincing evidence has indicated a role for infection in autoimmune hypothyroidism, except for the high frequency of this condition in patients with congenital rubella syndrome; however, an association has been proposed between Yersinia infection and Graves’ disease, and Yersinia contains proteins that mimic TSHR immunologically.78 The relative importance of this association has not been established but seems low, given the relative frequencies of Graves’ disease and Yersinia infection. Autoimmune thyroid disease occurs only rarely, if at all, after subacute thyroiditis.79 Epidemiologic studies suggest that inferior levels of prosperity and hygiene may actually protect against the development of thyroid autoimmunity.80
A number of retrospective surveys have identified stress during the year before the initial evaluation as an important risk factor for Graves’ disease, but these surveys suffer from potential flaws in their dependence on recall and other sources of bias.81 Any such effect presumably results from the neuroendocrine responses to stress that alter the regulation of autoreactive lymphocytes. Attacks of allergic rhinitis are associated with increased risk for relapse in those with Graves’ disease, most likely because of the Th2-dominant effects of cytokines produced during the attack.82 Exogenous cytokines given therapeutically, particularly interferon-alpha (IFN-α), exacerbate preexisting thyroid and other types of autoimmunity and lead to the development of autoimmune hypothyroidism in predisposed individuals.83 Other immunologically active agents can cause Graves’ disease, particularly during the recovery phase after administration of lymphocyte-depleting monoclonal antibodies given to patients with multiple sclerosis,84 possibly by deviating the immune response to a Th2-predominant phase. The roles of toxins and pollutants remain underexplored, although the adverse effects of smoking on Graves’ disease and ophthalmopathy provide a clear example that these factors could be important.85
T Cell–Mediated Responses
Many studies have been performed to characterize the circulating T cell population in autoimmune thyroid disease, but because the functional consequences of alterations in T cell phenotype are still not clear, particularly within this lymphocyte compartment, the meaning of any changes is open to debate. Although complete consensus has not been achieved, it seems that CD8+ T cell numbers are decreased in active Hashimoto’s thyroiditis and in Graves’ disease, with an increase in activated T cells expressing markers such as HLA-DR.86 Both CD4+ and CD8+ T cells occur in the thyroid lymphocytic infiltrate, with a preponderance of activated CD4+ cells.87,88
Attention has focused on possible clonal restriction of intrathyroidal T cells, as shown by limited usage of the V gene families encoding the α chain of the TCR.89 Although this concept seems plausible in the earliest stages of an autoimmune response, by the time that disease is recognizable clinically, multiple antigens and epitopes are responsible for T cell autoreactivity (so-called spreading of the immune response), and such restriction therefore would not be expected; little evidence of TCR restriction is apparent when IL-2 receptor–positive (and hence recently activated) intrathyroidal T cells in Graves’ disease are analyzed.90 However, restricted V gene usage by CD8+ T cells seen in Hashimoto’s thyroiditis could reflect clonal expansion of cytotoxic populations.91 Certainly, cytotoxic T cells have been cloned from this population.92 These cells have the αβ TCR, but a second cytotoxic population with γδ TCRs has also been identified in Graves’ disease; the nature of the thyroid surface autoantigen and the mode of its presentation to these T cells are unknown.93
A wide array of cytokines, including IL-2, IFN-γ, tumor necrosis factor-alpha (TNF-α), IL-4, IL-6, IL-10, IL-12, IL-13, and IL-15, are produced by the lymphocytic infiltrate in autoimmune thyroid disease, with some variations noted between patients and yet no predominance of a Th1 or Th2 response.94 Once again, this situation could reflect the late stage of disease and the mixed cell populations that have been analyzed in such studies. Ideally, one would wish to examine only thyroid autoantigen–specific CD4+ T cells to determine the pattern of helper T cell cytokine production, but establishing such clones has been difficult, and the very process of expanding these cells in vitro might well distort their behavior. Weak in vitro responses to TG, TPO, and TSHR, used as whole antigen or as putative peptide epitopes, have been detected in circulating and intrathyroidal T cell populations in assays of proliferation, secretion of migration inhibition factor, or B cell helper activity as readouts.30,50,51,58,59 Attempts to define thyroid antigen–specific suppressor T cells with such systems have also been made, but the suppressor cell defects that such assays have suggested in autoimmune thyroid disease95 have been disputed on grounds of specificity and the nonphysiologic nature of the assay systems.96
B Cell Responses
TG and TPO antibodies occur, often in very high concentrations, in patients with Hashimoto’s thyroiditis and primary myxedema (Fig. 8-8); in patients without circulating antibodies, they can be detected by culture of intrathyroidal lymphocytes.97 These antibodies are less common but still frequent in Graves’ disease, whereas TPO rather than TG antibodies are frequent in postpartum thyroiditis.6 Both antibodies show partial restriction to the IgG1 and IgG4 subclass and κ light chain restriction of TPO antibodies.53,98 The pathogenic role of TG antibodies is unclear because the epitopes on the antigen are too widely spaced to allow bound autoantigen to cross-link and thus fix complement, but these antibodies can mediate ADCC, at least in vitro.98 In contrast, TPO antibodies do fix complement, and immunohistochemical evidence has established the formation of terminal complement complexes within the thyroid in autoimmune thyroid disease.99 Because TPO is located at the apical surface and within the thyroid cell cytoplasm, it seems that under normal circumstances, TPO antibodies do not gain access to their autoantigen, which accounts for the euthyroidism seen in healthy individuals with TPO antibodies and in neonates born to mothers with high levels of TPO antibodies. Cell-mediated injury may be necessary for TPO antibodies to gain access to their antigen and become pathogenic. For instance, cytokines such as IL-1α could induce dissociation of the junctional complexes between thyroid cells within a follicle (see Fig. 8-8).100
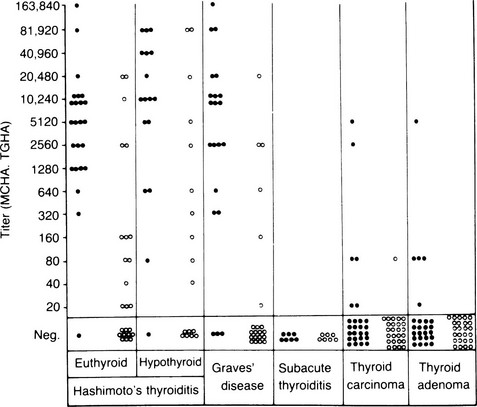
FIGURE 8-8 Titers of microsomal/thyroid peroxidase (TPO) hemagglutination antibodies (MCHAs), shown as solid circles, and thyroglobulin hemagglutination antibodies (TGHAs) in various thyroid diseases. (From Amino N, Hagan SR, Yamada N, Refetoff S: Measurement of circulating thyroid microsomal antibodies by the tanned red cell haemagglutination technique: its usefulness in the diagnosis of autoimmune thyroid disease. Clin Endocrinol 5:115–126, 1976.)
Thyroid-stimulating antibodies (TSAbs) directed against the TSHR are the hallmark of Graves’ disease, and the best antibody assays currently have a sensitivity of up to 99%. These antibodies are often κ chain restricted and of the IgG1 subclass,101,102 which suggests origin from a small number of B cell clones. It is now established that TSAbs also occur in a small proportion of patients with autoimmune hypothyroidism, but their effects are obscured by TSHR-blocking antibodies and destructive processes.103 In some patients, fluctuation in the relative proportion of the two types of TSHR antibody may produce a confusing clinical picture of alternating hyperthyroidism and hypothyroidism, and such fluctuation also may occur after pregnancy.104 Blocking antibodies have been found in 10% to 20% of patients with autoimmune hypothyroidism, and in Asian populations, they seem to be most closely associated with atrophic rather than goitrous thyroiditis.105 In whites, however, they appear in Hashimoto’s thyroiditis as well, and in such patients, the goiter is most likely to be the result of lymphocytic infiltration.106 The existence of separate populations of growth-stimulating and growth-inhibiting immunoglobulins, operating independently of the TSHR, remains disputed.107,108 In Graves’ disease at least, it is TSAbs that mediate goitrogenesis.
Pathogenic Mechanisms
Graves’ disease clearly results from the action of TSAbs, primarily via the cAMP pathway, although other signaling pathways may be used by TSHR antibodies in some patients, which suggests a subdivision based on the effector function of these antibodies.109 TSAbs also increase the vascularity of the Graves’ thyroid by enhancing local expression of vascular endothelial growth factor and its receptor.110 In around 15% of patients with Graves’ disease who are treated with antithyroid drugs, hypothyroidism supervenes years later, thus indicating that similar destructive mechanisms operate in Graves’ disease and autoimmune hypothyroidism. These shared pathogenic mechanisms are detailed after consideration of the role of thyroid follicular cells as APCs.
The discovery that thyroid cells express MHC class II molecules in autoimmune thyroid disease, but not under normal circumstances, led to the suggestion that such expression could permit thyroid autoantigen presentation, which in turn could initiate or exacerbate disease.111 It is now clear that thyroid cells generally express class II molecules only after stimulation with IFN-γ, which implies that a T cell infiltrate must precede such expression, so class II expression is a secondary event and transgenic expression of class II molecules on thyroid cells in mice alone does not induce EAT.112 Furthermore, thyroid cells do not express B7-1 or B7-2 costimulatory molecules and therefore can act as APCs only for T cells that no longer require such costimulation, in general those that have been activated previously. In vitro experiments confirm that thyroid cells can act as APCs under such circumstances, but they also are able to induce anergy in naïve T cells that require costimulation113 (see Fig. 8-3). Teleologically, MHC class II expression is likely to be an important means of ensuring peripheral T cell tolerance under normal circumstances, but such expression is damaging under conditions of thyroid autoimmunity (Fig. 8-9). Class II expression is more readily induced by IFN-γ in Graves’ thyroid cells than in those from multinodular goiter, thus suggesting a genetically regulated component to this response.114
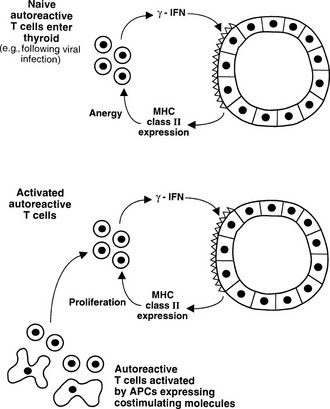
FIGURE 8-9 Alternative outcomes after major histocompatibility complex (MHC) class II expression by thyroid cells. Naïve T cells require costimulation for activation, and anergy can be induced by interaction with the MHC class II molecule/antigenic epitope alone (upper panel, peripheral tolerance). If T cells receive costimulation from classic antigen-presenting cells (APCs), class II expression by thyroid cells can enhance the T cell response (lower panel).
Cytokines have a large number of other effects on thyroid cells that might be of pathogenic relevance. As well as adversely affecting thyroid growth and function, a number of immunologically important molecules are expressed by thyroid cells in response to cytokines that are known to be produced locally by the infiltrating leukocytes in Graves’ disease and autoimmune hypothyroidism94 (Fig. 8-10). Expression of ICAM-1, LFA-3, CD40, and MHC class I molecules is enhanced by IL-1, TNF, and IFN-γ, and this response increases the ability of cytotoxic T cells to mediate lysis.94 A complex series of interactions, including secretion of chemokines, is important in allowing lymphocytes to enter the thyroid and in some cases to develop tertiary lymphoid structures within the gland.115 Thyroid cell destruction is mediated both by perforin-containing T cells, which accumulate in the thyroid,116 and by Fas-dependent mechanisms.117 A unique type of suicide has been suggested by reports that IL-1β–stimulated thyroid cells in Hashimoto’s thyroiditis express FasL, which could lead to self-ligation with Fas and thus cell death,117 but these findings have not been reproduced consistently, and the final outcome of Fas ligation depends on a complex regulatory pathway that might also involve the death ligand TRAIL.118 Cytokines and other toxic molecules such as nitric oxide and reactive oxygen metabolites probably also contribute directly to cell-mediated tissue injury.
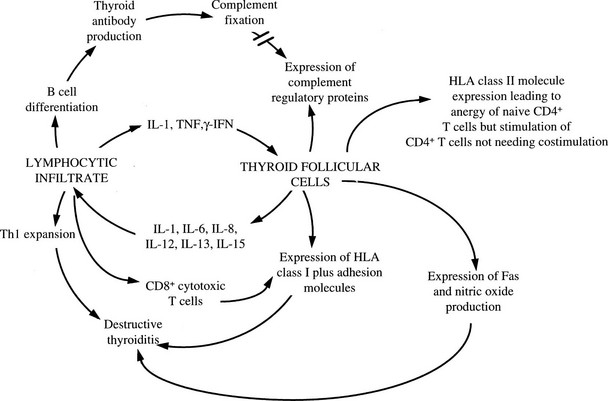
FIGURE 8-10 Interaction between thyroid cells and the immune system via cytokines. Expression of complement regulatory proteins in response to interleukin-1 (IL-1), tumor necrosis factor (TNF), and interferon-γ (IFN-γ) will protect against complement-mediated injury, and class II expression may induce T cell anergy under appropriate conditions; the other cytokine-induced events will amplify the autoimmune process. (From Weetman AP, Ajjan RA, Watson PF: Cytokines and Graves’ disease. Baillieres Clin Endocrinol Metab 11:481–497, 1997.)
Humoral immunity most likely exacerbates cell-mediated damage in a secondary fashion, both by direct complement fixation (for TPO antibodies) and by ADCC.98,119 These effects occur in addition to the inhibitory effects of TSHR-blocking antibodies on thyroid cell function. Thyroid cells increase their expression of a number of regulatory proteins (CD46, CD55, CD59) in response to cytokines, and these proteins prevent cell death in the face of widespread complement damage in autoimmune thyroid disease.87,120 Nonetheless, a sublethal complement attack, initiated via the classic or alternative pathway, impairs the metabolic function of thyroid cells and induces them to secrete IL-1, IL-6, reactive oxygen metabolites, and prostaglandins, all of which could enhance the autoimmune response.121 As well as T and B cells, dendritic cells and monocytes/macrophages accumulate in the thyroid, where they presumably play a major role as APCs that are capable of providing costimulatory signals. Besides acting as APCs, these cells are important sources of cytokines, as shown by the inhibition of thyroid cell growth by IL-1 and IL-6 derived from dendritic cells.122 An additional group of inflammatory cells, namely, NK-like T cells, has been identified in the thyroid infiltrate, in turn suggesting a role for lipid-containing molecules in thyroid autoimmunity.123
Natural History and Response to Treatment
The natural history of subclinical hypothyroidism, present in many cases of focal thyroiditis, is now well established from epidemiologic surveys: The presence of thyroid autoantibodies in addition to this biochemical picture confers a considerable future risk for permanent hypothyroidism—much greater than the presence of autoantibodies alone.124 Around a quarter of patients with postpartum thyroiditis progress to permanent hypothyroidism,6 but as in the case of subclinical hypothyroidism, it is unclear which factors predispose to this outcome. One speculation is that maternal microchimerism, caused by the transfer of fetal cells bearing paternal antigens at delivery, leads to the enhancement of any ongoing autoimmune response, and the most severe disease therefore would accompany the greatest cell transfer. Certainly, evidence for intrathyroidal fetal microchimerism in autoimmune thyroid disease is growing.125 Hormonal disturbances during and after pregnancy also may be involved in these changes, as postpartum thyroiditis has been associated with low cortisol levels in the last trimester.126
Spontaneous resolution of Graves’ disease and autoimmune hypothyroidism does occur but seems unusual, and thus far, no prospective study has fully established whether any remission is permanent rather than temporary. However, patients with autoimmune hypothyroidism and TSHR-blocking antibodies seem particularly likely to enter remission after T4 treatment, although no consensus has been reached on whether this remission is associated with decreases in antibody levels.105,127 Intermittent exposure to a crucial environmental agent could explain some remissions, and it is noteworthy that many animal models of EAT spontaneously remit. One potential factor that has recently been tried therapeutically is selenium: supplementation with this trace element appears to lower TPO antibody levels.128
Treatment with antithyroid drugs (carbimazole, methimazole, and propylthiouracil) for Graves’ disease leads to a decline in TSAbs and other thyroid antibodies and a decline in the severity of thyroiditis, as well as other immunologic changes (Table 8-6). This may occur because antithyroid drugs inhibit the release of proinflammatory molecules by thyroid cells, which would explain both the altered immune response during treatment and its thyroid specificity.121
Table 8-6
Immunologic Effects of Antithyroid Drugs
In Vivo
Reduction in levels of TSHR, TG, and TPO antibodies but not nonthyroid autoantibodies
Reduction in thyroid lymphocytic infiltration in Graves’ disease and experimental/spontaneous autoimmune thyroiditis in animals
Reversal of thymic hyperplasia
Restoration of elevated circulating levels of activated and CD69+ T cells and increased CD4/CD8 ratio
Reduction in circulating levels of soluble markers of immune response (terminal complement complexes, CD8, ICAM-1)
In Vitro
Radioiodine therapy is followed after 3 to 6 months by a striking rise in thyroid autoantibodies, and the possibility of differential effects on TSAbs and TSHR-blocking antibodies could explain transient thyroid dysfunction at this time. Thyroid-associated ophthalmopathy may worsen transiently after radioiodine treatment in some patients.129 Both events may be related to the release of thyroid autoantigens or radiation effects on T cell subpopulations, inasmuch as activated T cells increase in the circulation weeks after 131I administration.130
Undoubtedly, improved understanding of the immunologic basis of Graves’ disease ultimately will lead to immunologically based treatment aimed at reinducing tolerance to TSHR. Pilot studies have been performed to assess the potential for oral tolerance with the use of TG,131 and it is possible that any such treatment might have benefit for ophthalmopathy as well. On the other hand, T4 is such a simple treatment for autoimmune hypothyroidism that, at present, novel treatments are most unlikely.
Relation to Other Diseases
Thyroiditis and thyroid antibodies are found in a quarter to a third of patients with thyroid cancer, and such patients have an improved prognosis.132 Preexisting Hashimoto’s thyroiditis is the major risk factor for the development of primary thyroid lymphoma.133 Other novel associations have been suggested by studies that show an increased frequency of autoimmune thyroiditis in women with breast cancer,134 infertility and persistent miscarriage,135 and depression.136 In all these examples, the reasons for the links with thyroid autoimmunity remain unclear. Finally, autoimmune thyroid disease is associated with many other autoimmune diseases and is a well-recognized component of autoimmune polyglandular syndrome type 2, as well as a minor component of the type 1 syndrome.137–139
References
1. Rose, NR, Witebsky, E. Changes in the thyroid glands of rabbits following active immunization with rabbit thyroid extracts. J Immunol. 1956;76:417–427.
2. Roitt, IM, Doniach, D, Campbell, PN, et al. Autoantibodies in Hashimoto’s disease (lymphadenoid goitre). Lancet. 1956;2:820–821.
3. Adams, DD. The presence of an abnormal thyroid-stimulating hormone in the serum of some thyrotoxic patients. J Clin Endocrinol Metab. 1958;18:699–712.
4. Okayasu, I, Hara, Y, Nakamura, K, et al. Racial and age-related differences in incidence and severity of focal autoimmune thyroiditis. Anat Pathol. 1993;101:698–702.
5. Jacobson, DL, Gange, SJ, Rose, NR, et al. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243.
6. Stagnaro-Green, A. Postpartum thyroiditis. J Clin Endocrinol Metab. 2002;87:4042–4047.
7. LiVolsi, VA. Pathology of thyroid disease. In: Falk SA, ed. Thyroid Disease: Endocrinology, Surgery, Nuclear Medicine and Radiotherapy. New York: Raven Press; 1990:127–175.
8. Mizukami, Y, Michigishi, T, Kawato, M, et al. Thyroid function and histologic correlations in 601 cases. Hum Pathol. 1991;23:980–988.
9. Hayashi, Y, Tamai, H, Fukata, S, et al. A long term clinical, immunological, and histological follow-up study of patients with goitrous chronic lymphocytic thyroiditis. J Clin Endocrinol Metab. 1985;61:1172–1177.
10. Weetman, AP, McGregor, AM, Wheeler, MH, et al. Extrathyroidal sites of autoantibody synthesis in Graves’ disease. Clin Exp Immunol. 1984;56:330–336.
11. Goodnow, CC, Sprent, J, Fazekas de St Groth, B, et al. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597.
12. Garcia, KC, Teyton, L, Wilson, IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397.
13. Delves, PJ, Roitt, IM. The immune system. N Engl J Med. 2000;343:37–49.
14. Sansom, DM, Manzotti, CN, Zheng, Y. What’s the difference between CD80 and CD86? Trends Immunol. 2003;24:314–319.
15. Frauwirth, KA, Thompson, CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest. 2002;109:295–299.
16. Jiang, H, Chess, L. Regulation of immune responses by T cells. N Engl J Med. 2006;354:1166–1176.
17. Bettelli, E, Oukka, M, Kuchroo, V. TH-17 cells in the circle of immunity and autoimmunity. Nature Immunol. 2007;8:345–350.
18. Perussia, B, Loza, MJ. Linear “2-0-1” lymphocyte development: hypotheses on cellular bases for immunity. Trends Immunol. 2003;24:235–241.
19. Beverley, PCL. Primer: making sense of T-cell memory. Nature. 2008;4:43–49.
20. D’Ambrosio, D, Sinigaglia, F, Adorini, L. Special attractions for suppressor T cells. Trends Immunol. 2003;24:122–126.
21. Sakaguchi, S, Yamaguchi, T, Nomura, T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787.
22. Hirsh, MI, Junger, WG. Roles of heat shock proteins and gamma-delta T cells in inflammation. Am J Respir Cell Mol Biol. 2008;39:50–513.
23. Matsuda, F, Honjo, T. Organization of the human immunoglobulin heavy-chain locus. Adv Immunol. 1996;62:1–29.
24. Laver, WG, Air, GM, Webster, RG, et al. Epitopes on protein antigens: misconceptions and realities. Cell. 1990;61:553–556.
25. MacKay, IR, Rosen, FS. Tolerance and autoimmunity. N Engl J Med. 2001;344:655–664.
26. Cheng, MH, Shum, AK, Anderson, MS. What’s new in the Aire. Trends Immunol. 2007;28:321–327.
27. Goodnow, CC. Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci U S A. 1996;93:2264–2271.
28. Cone, RE, Malley, A. Soluble, antigen-specific T-cell proteins: T-cell-based humoral immunity? Immunol Today. 1996;17:318–322.
29. Goldschneider, I, Cone, RE. A central role for peripheral dendritic cells in the induction of acquired thymic tolerance. Trends Immunol. 2003;24:77–81.
30. Weetman, AP, McGregor, AM. Autoimmune thyroid disease: further developments in our understanding. Endocr Rev. 1994;15:788–830.
31. Ando, T, Imaizumi, M, Graves, P, et al. Induction of thyroid-stimulating hormone receptor autoimmunity in hamsters. Endocrinology. 2003;144:671–680.
32. Schwarz-Lauer, L, Pichurin, PN, Chen, C-R. The cysteine-rich amino terminus of the thyrotropin receptor is the immunodominant linear antibody epitope in mice immunized using naked deoxyribonucleic acid or adenovirus vectors. Endocrinology. 2003;144:1718–1725.
33. Wick, G, Cole, R, Dietrich, H, et al. The obese strain of chickens with spontaneous thyroiditis as a model for Hashimoto disease. In: Cohen IR, Miller A, eds. Autoimmune Disease Models. San Diego, CA: Academic Press; 1994:107–122.
34. Kong, Y-C, Lomo, LC, Mott, RW, et al. HLA-DRB1 polymorphism determines susceptibility to autoimmune thyroiditis in transgenic mice: definitive association with HLA-DRB1*0301 (DR3) gene. J Exp Med. 1996;184:1167–1172.
35. Barin, JG, Talor, MV, Sharma, RB, et al. Iodination of murine thyroglobulin enhances autoimmune reactivity in the NOD.H2h4 mouse. Clin Exp Immunol. 2005;142:251–259.
36. Rennie, DP, McGregor, AM, Keast, D, et al. The influence of methimazole on thyroglobulin-induced autoimmune thyroiditis in the rat. Endocrinology. 1983;112:326–330.
37. Penhale, WJ, Young, PR. The influence of the normal microbial flora on the susceptibility of rats to experimental autoimmune thyroiditis. Clin Exp Immunol. 1988;72:288–292.
38. Okayasu, I, Kong, YM, Rose, NR. Effect of castration and sex hormones on experimental autoimmune thyroiditis. Clin Immunol Immunopathol. 1981;20:240–245.
39. Quaratino, S, Badami, E, Pang, YY, et al. Degenerate self-reactive human T-cell receptor causes spontaneous autoimmune disease in mice. Nature Med. 2004;10:920–926.
40. Yu, S, Maiti, PK, Dyson, M, et al. B cell-deficient NOD.H-2h4 mice have CD4+ CD25+ T regulatory cells that inhibit the development of spontaneous autoimmune thyroiditis. J Exp Med. 2006;203:349–358.
41. McLachlan, S, Nagayama, Y, Rapoport, B. Insight into Graves’ hyperthyroidism from animal models. Endocrine Reviews. 2005;26:800–832.
42. Akkaraju, S, Canaan, K, Goodnow, CC. Self-reactive B cells are not eliminated or inactivated by autoantigen expressed on thyroid epithelial cells. J Exp Med. 1997;186:2005–2012.
43. Malthiery, Y, Lissitzky, S. Primary structure of human thyroglobulin deduced from the sequence of its 8848-base complementary DNA. Eur J Biochem. 1987;105:491–498.
44. Carayanniotis, G, Rao, VP. Searching for pathogenic epitopes in thyroglobulin: parameters and caveats. Immunol Today. 1997;18:83–88.
45. Nye, L, Pontes de Carvalho, L, Roitt, I. Restriction in the response to autologous thyroglobulin in the human. Clin Exp Immunol. 1980;41:252–263.
46. Bresler, HS, Burek, CL, Hoffman, WH, et al. Autoantigenic determinants on human thyroglobulin: II. Determinants recognized by autoantibodies from patients with chronic autoimmune thyroiditis compared to autoantibodies from healthy subjects. Clin Immunol Immunopathol. 1990;54:76–86.
47. Prentice, L, Kiso, Y, Fukuma, N, et al. Monoclonal thyroglobulin autoantibodies: variable region analysis and epitope recognition. J Clin Endocrinol Metab. 1995;80:977–986.
48. Tomer, Y. Anti-thyroglobulin autoantibodies in autoimmune thyroid diseases: cross-reactive or pathogenic? Clin Immunol Immunopathol. 1997;82:3–11.
49. McLachlan, SM, Rapoport, B. The molecular biology of thyroid peroxidase: cloning, expression, and role as autoantigen in autoimmune thyroid disease. Endocr Rev. 1992;13:192–206.
50. Tandon, N, Freeman, M, Weetman, AP. T cell responses to synthetic thyroid peroxidase peptides in autoimmune thyroid disease. Clin Exp Immunol. 1991;86:56–60.
51. Fisfalen, M-E, Soliman, M, Okamoto, Y, et al. Proliferative responses of T cells to thyroid antigens and synthetic thyroid peroxidase peptides in autoimmune thyroid disease. J Clin Endocrinol Metab. 1995;80:1597–1604.
52. Hamada, N, Jaeduck, N, Portman, L, et al. Antibodies against denatured and reduced thyroid microsomal antigen in autoimmune thyroid disease. J Clin Endocrinol Metab. 1987;64:230–238.
53. McIntosh, R, Watson, P, Weetman, A. Somatic hypermutation in autoimmune thyroid disease. Immunol Rev. 1998;162:219–231.
54. Chazenbalk, GD, Portolano, S, Russo, D, et al. Human organ-specific autoimmune disease. Molecular cloning and expression of an autoantibody gene repertoire for a major autoantigen reveals an antigenic immunodominant region and restricted immunoglobulin gene usage in the target organ. J Clin Invest. 1993;92:62–74.
55. Vassart, G, Dumont, JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr Rev. 1992;13:596–611.
56. Chen, C-R, Pichurin, P, Nagayama, Y, et al. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J Clin Invest. 2003;111:1897–1904.
57. Bahn, RS, Dutton, CM, Natt, N, et al. Thyrotropin receptor expression in Graves’ orbital adipose/connective tissues: potential autoantigen in Graves’ ophthalmopathy. J Clin Endocrinol Metab. 1998;83:998–1002.
58. Tandon, N, Freeman, MA, Weetman, AP. T cell responses to synthetic TSH receptor peptides in Graves’ disease. Clin Exp Immunol. 1992;89:468–473.
59. Martin, A, Nakashima, M, Zhou, A, et al. Detection of major T cell epitopes on human thyroid stimulating hormone receptor by overriding immune heterogeneity in patients with Graves’ disease. J Clin Endocrinol Metab. 1997;82:3361–3366.
60. Schott, M, Scherbaum, WA, Morgenthaler, NG. Thyrotropin receptor autoantibodies in Graves’ disease. Trends Endocrinol Metab. 2005;16:243–248.
61. Sanders, J, Evans, M, Premawardhana, LDKE, et al. Human monoclonal thyroid stimulating autoantibody. Lancet. 2003;362:126–128.
62. Raspe, E, Costagliola, S, Ruf, J, et al. Identification of the thyroid Na+/I− symporter in the sera of patients with autoimmune thyroid disease. Biochem Biophys Res Commun. 1996;224:399–405.
63. Ajjan, RA, Findlay, C, Metcalfe, RA, et al. The modulation of the human sodium iodide symporter activity by Graves’ disease sera. J Clin Endocrinol Metab. 1998;83:1217–1221.
64. Benvenga, S, Trimarchi, F, Robbins, J. Circulating thyroid hormone autoantibodies. J Endocrinol Invest. 1987;10:605–610.
65. Brix, TH, Kyvik, KO, Hegedüs, L. What is the evidence of genetic factors in the etiology of Graves’ disease? A brief review. Thyroid. 1998;8:627–635.
66. Zeitlin, AA, Simmonds, MJ, Gough, SCL. Genetic developments in autoimmune thyroid disease: an evolutionary process. Clin Endocrinol. 2008;68:671–682.
67. Yanagawa, T, Hidaka, Y, Guimaraes, V, et al. CTLA-4 gene polymorphism associated with Graves’ disease in Caucasian population. J Clin Endocrinol Metab. 1995;80:41–45.
68. Kavvoura, FK, Akamizu, T, Awata, T, et al. Cytotoxic T-lymphocyte associated antigen 4 gene polymorphisms and autoimmune thyroid disease: a meta-analysis. J Clin Endocrinol Metab. 2007;92:3162–3170.
68a. Taylor, JC, Gough, SC, Hunt, PJ, et al. A genome-wide screen in 1119 relative pairs with autoimmune thyroid disease. J Clin Endocrinol Metab. 2006;91:646–653.
69. Velaga, MR, Wilson, V, Jennings, CE, et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab. 2004;89:5862–5865.
70. Brand, OJ, Lowe, CE, Franklyn, JA, et al. Association of the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region with Graves’ disease using a multilocus test and tag SNPs. Clin Endocrinol. 2007;66:508–512.
71. Kochi, Y, Yamada, R, Suzuki, A, et al. A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nature Genetics. 2005;37:478–485.
72. Simmonds, MJ, Heward, JM, Carr-Smith, J, et al. Contribution of single nucleotide polymorphisms within FRCL3 and MAP3K7IP2 to the pathogenesis of Graves’ disease. J Clin Endocrinol Metab. 2005;91:1056–1061.
73. Dechairo, BM, Zabaneh, D, Collins, J, et al. Association of the TSHR gene with Graves’ disease: the first disease specific locus. Eur J Human Genetics. 2005;13:1223–1230.
74. Benvenga, S, Trimarchi, F. Changed presentation of Hashimoto’s thyroiditis in north-eastern Sicily and Calabria (Southern Italy) based on a 31 year experience. Thyroid. 2008;18:429–441.
75. Papanastiou, L, Alebizaki, M, Piperingos, G, et al. The effect of iodine administration on the development of thyroid autoimmunity in patients with non-toxic goiter. Thyroid. 2000;10:493–496.
76. Huysmans, DAKC, Hermus, ADRMM, Edelbroek, MAL, et al. Autoimmune hyperthyroidism occurring late after radioiodine treatment for volume reduction of large multinodular goiters. Thyroid. 1997;7:535–538.
77. Völzke, H, Werner, A, Wallaschofski, H, et al. Occupational exposure to ionizing radiation is associated with autoimmune thyroid disease. J Clin Endocrinol Metab. 2005;90:4587–4592.
78. Tomer, Y, Davies, TF. Infection, thyroid disease, and autoimmunity. Endocr Rev. 1993;14:107–120.
79. Bartalena, L, Bogazzi, F, Percori, F, et al. Graves’ disease occurring after subacute thyroiditis: report of a case and review of the literature. Thyroid. 1996;6:345–348.
80. Kondrashova, A, Viskari, H, Haapla, A-M, et al. Serological evidence of thyroid autoimmunity among schoolchildren in two different socioeconomic environments. J Clin Endocrinol Metab. 2008;93:729–734.
81. Chiovato, L, Pinchera, A. Stressful life events and Graves’ disease. Eur J Endocrinol. 1996;134:680–682.
82. Sato, A, Takemura, Y, Yamada, T, et al. A possible role of immunoglobulin E in patients with hyperthyroid Graves’ disease. J Clin Endocrinol Metab. 1999;84:3602–3605.
83. Carella, C, Mazziotti, G, Amato, G, et al. Interferon-α related thyroid disease: pathophysiological, epidemiological and clinical aspects. J Clin Endocrinol Metab. 2004;88:3656–3661.
84. Coles, A, Wing, M, Smith, S, et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet. 1999;354:1691–1695.
85. Krassas, GE, Wiersinga, W. Smoking and autoimmune thyroid disease: the plot thickens. Eur J Endocrinol. 2006;154:777–780.
86. Iwatani, Y, Amino, N, Hidaka, Y, et al. Decreases in αβ T cell receptor negative T cells and CD8+ cells, and an increase in CD4+, CD8+ cells in active Hashimoto’s disease and subacute thyroiditis. Clin Exp Immunol. 1992;87:444–449.
87. Aichinger, G, Fill, H, Wick, G. In situ immune complexes, lymphocyte subpopulations, and HLA-DR positive epithelial cells in Hashimoto’s thyroiditis. Lab Invest. 1985;52:132–140.
88. Ueki, YM, Eguchi, K, Otsubo, T, et al. Phenotypic analysis of concanavalin-A-induced suppressor cell dysfunction of thyroidal lymphocytes from patients with Graves’ disease. J Clin Endocrinol Metab. 1988;67:1018–1024.
89. Davies, TF, Martin, A, Concepcion, ES, et al. Evidence for selective accumulation of intrathyroidal T lymphocytes in human autoimmune thyroid disease based on T cell receptor V gene usage. J Clin Invest. 1992;89:157–162.
90. McIntosh, RS, Tandon, N, Pickerill, AP, et al. IL-2 receptor positive intrathyroidal lymphocytes in Graves’ disease: analysis of Vα transcript microheterogeneity. J Immunol. 1993;91:3884–3893.
91. McIntosh, RS, Watson, PF, Weetman, AP. Analysis of T cell receptor Vα repertoire in Hashimoto’s thyroiditis: evidence for the restricted accumulation of CD8+ T cells in the absence of CD4+ T cell restriction. J Clin Endocrinol Metab. 1997;82:1140–1146.
92. MacKenzie, WA, Davies, TF. An intrathyroidal T-cell clone specifically cytotoxic for human thyroid cells. Immunology. 1987;61:101–103.
93. Catalfamo, M, Roura-Mir, C, Sospedra, M, et al. Self-reactive cytotoxic γδ T lymphocytes in Graves’ disease specifically recognize thyroid epithelial cells. J Immunol. 1996;156:804–811.
94. Ajjan, RA, Watson, PF, Weetman, AP. Cytokines in Graves’ disease. In: Rapoport B, McLachlan SM, eds. Graves’ Disease: Pathogenesis and Treatment. Norwell, MA: Kluwer Academic Publishers; 2000:79.
95. Volpe, R. Immunoregulation in autoimmune thyroid disease. Thyroid. 1994;4:373–377.
96. Martin, A, Davies, TF. T cells in human autoimmune thyroid disease: emerging data shows lack of need to invoke suppressor T cell problems. Thyroid. 1992;2:247–261.
97. Baker, JR, Saunders, NB, Tseng, YC, et al. Seronegative Hashimoto thyroiditis with thyroid autoantibody production localized to the thyroid. Ann Intern Med. 1988;108:26–30.
98. Weetman, AP, Black, CM, Cohen, SB, et al. Affinity purification of IgG subclasses and the distribution of thyroid autoantibody reactivity in Hashimoto’s thyroiditis. Scand J Immunol. 1989;30:73–82.
99. Weetman, AP, Cohen, SB, Oleesky, DA, et al. Terminal complement complexes and C1/C1 inhibitor complexes in autoimmune thyroid disease. Clin Exp Immunol. 1989;77:25–30.
100. Rebufaat, SA, Nguyen, B, Robert, B, et al. Antithyroperoxidase antibody-dependent cytotoxicity in autoimmune thyroid disease. J Clin Endocrinol Metab. 2008;93:929–932.
101. Williams, RC, Marshall, NJ, Kilpatrick, K, et al. Kappa/lambda immunoglobulin distribution of Graves’ thyroid stimulating antibodies: simultaneous analysis of Cλ gene polymorphisms. J Clin Invest. 1988;82:1306–1312.
102. Weetman, AP, Yateman, ME, Ealey, PA, et al. Thyroid-stimulating antibody activity between different immunoglobulin G subclasses. J Clin Invest. 1990;86:723–727.
103. Kohn, LD, Suzuki, K, Hoffman, WH, et al. Characterization of monoclonal thyroid-stimulating and thyrotropin binding-inhibiting autoantibodies from a Hashimoto’s patient whose children had intrauterine and neonatal thyroid disease. J Clin Endocrinol Metab. 1997;82:3998–4009.
104. Kung, AWC, Jones, BM. A change from stimulatory to blocking antibody activity in Graves’ disease during pregnancy. J Clin Endocrinol Metab. 1998;83:514–518.
105. Cho, BY, Kim, WB, Chung, JH, et al. High prevalence and little change in TSH receptor blocking antibody titres with thyroxine and antithyroid drug therapy in patients with non-goitrous autoimmune thyroiditis. Clin Endocrinol. 1995;43:465–471.
106. Kraiem, Z, Lahat, N, Glaser, B, et al. Thyrotrophin receptor blocking antibodies: incidence, characterization and in vivo synthesis. Clin Endocrinol. 1987;27:409–421.
107. Drexhage, HA. Autoimmunity and thyroid growth: where do we stand? Eur J Immunol. 1996;135:39–45.
108. Vitti, P, Chiovato, L, Tonacchera, M, et al. Failure to detect thyroid growth-promoting activity in immunoglobulin G of patients with endemic goiter. J Clin Endocrinol Metab. 1994;78:1020–1025.
109. Di Cerbo, A, Di Paola, R, Bonati, M, et al. Subgroups of Graves’ patients identified on the basis of the biochemical activities of their immunoglobulins. J Clin Endocrinol Metab. 1995;80:2785–2790.
110. Sato, K, Yamazaki, K, Shizume, K, et al. Stimulation by thyroid-stimulating hormone and Graves’ immunoglobulin G of vascular endothelial growth factor mRNA expression in human thyroid follicles in vitro and flt mRNA expression in the rat thyroid in vivo. J Clin Invest. 1995;96:1295–1302.
111. Bottazzo, GF, Pujol-Borrell, R, Hanafusa, T, et al. Role of aberrant HLA-DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet. 1983;2:1115–1119.
112. Kimura, H, Kimura, M, Tzou, S-C, et al. Expression of class II major histocompatiblity complex molecules on thyrocytes does not cause spontaneous thyroiditis but mildly increases its severity after immunization. Endocrinol. 2005;146:1154–1162.
113. Marelli-Berg, F, Weetman, AP, Frasca, L, et al. Antigen presentation by epithelial cells induces anergic immunoregulatory CD45R0+ T cells and deletion of CD45RA+T cells. J Immunol. 1997;159:5853–5861.
114. Sospedra, M, Obiols, G, Babi, LFS, et al. Hyperinducibility of HLA class II expression of thyroid follicular cells from Graves’ disease. J Immunol. 1995;154:4213–4222.
115. Marinkovic, T, Garin, A, Yokota, Y, et al. Interaction of mature CD3+CD4+ T cells with dendritic cells triggers the development of tertiary lymphoid structures in the thyroid. J Clin Invest. 2006;116:2622–2632.
116. Wu, Z, Podack, ER, McKenzie, JM, et al. Perforin expression by thyroid-infiltrating T cells in autoimmune thyroid disease. Clin Exp Immunol. 1994;98:470–477.
117. Giordano, C, Stassi, G, De Maria, R, et al. Potential involvement of Fas and its ligand on the pathogenesis of Hashimoto’s thyroiditis. Science. 1997;275:960–963.
118. Bretz, JD, Baker, JR, Jr. Apoptosis and autoimmune thyroid disease: following a TRAIL to destruction? Clin Endocrinol. 2001;55:1–11.
119. Chiovato, L, Bassi, P, Santini, F, et al. Antibodies producing complement-mediated thyroid cytotoxicity in patients with atrophic or goitrous autoimmune thyroiditis. J Clin Endocrinol Metab. 1993;77:1700–1705.
120. Tandon, N, Yan, SL, Morgan, BP, et al. Expression and function of multiple regulators of complement activation in autoimmune thyroid disease. Immunology. 1994;84:643–647.
121. Weetman, AP, Tandon, N, Morgan, BP. Antithyroid drugs and release of inflammatory mediators by complement-attacked thyroid cells. Lancet. 1992;340:633–636.
122. Simons, PJ, Delemarre, FGA, Drexhage, HA. Antigen-presenting dendritic cells as regulators of the growth of thyrocytes: a role of interleukin-1β and interleukin-6. Endocrinology. 1998;139:3148–3156.
123. Roura-Mir, C, Catálfamo, M, Cheng, T-Y, et al. CD1a and CD1c activate intrathyroidal T cells during Graves’ disease and Hashimoto’s thyroiditis. J Immunol. 2005;174:3773–3780.
124. Vanderpump, MPJ, Tunbridge, WMG, French, JM, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham survey. Clin Endocrinol. 1995;43:55–68.
125. Ando, T, Davies, TF. Postpartum autoimmune thyroid disease: the potential role of fetal michochimerism. J Clin Endocrinol Metab. 2003;88:2965–2971.
126. Kokandi, AA, Parkes, AB, Premawardhana, LDKE, et al. Association of postpartum thyroid dysfunction with antepartum hormonal and immunological changes. J Clin Endocrinol Metab. 2003;88:1126–1132.
127. Takasu, N, Yamada, Y, Takasu, M, et al. Disappearance of thyrotropin-blocking antibodies and spontaneous recovery from hypothyroidism in autoimmune thyroiditis. N Engl J Med. 1992;326:513–518.
128. Turker, O, Kumanlioglu, K, Karapolat, I, et al. Selenium treatment in autoimmune thyroiditis: 9-month follow-up with variable doses. J Endocrinol. 2006;190:151–156.
129. Bartalena, L, Marcocci, C, Bogazzi, F, et al. Relation between therapy for hyperthyroidism and the course of Graves’ ophthalmopathy. N Engl J Med. 1998;338:73–78.
130. Teng, WP, Stark, R, Munro, AJ, et al. Peripheral blood T cell activation after radioiodine treatment for Graves’ disease. Acta Endocrinol. 1990;122:233–240.
131. Lee, S, Scherberg, N, DeGroot, LJ. Induction of oral tolerance in human autoimmune thyroid disease. Thyroid. 1998;8:229–234.
132. Baker, JR, Jr., Fosso, CK. Immunological aspects of cancers arising from thyroid follicular cells. Endocr Rev. 1993;13:729–746.
133. Thieblemont, C, Mayer, A, Dumontet, C, et al. Primary thyroid lymphoma is a heterogeneous disease. J Clin Endocrinol Metab. 2002;87:105–111.
134. Giani, C, Fierabracci, P, Bonacci, R, et al. Relationship between breast cancer and thyroid disease: relevance of autoimmune thyroid disorders in breast malignancy. J Clin Endocrinol Metab. 1996;81:990–994.
135. Prummel, MF, Wiersinga, W. Thyroid autoimmunity and miscarriage. Eur J Endocrinol. 2004;150:751–755.
136. Pop, VJ, Maartens, LH, Leusink, G, et al. Are autoimmune thyroid dysfunction and depression related? J Clin Endocrinol Metab. 1998;83:3194–3197.
137. Jenkins, RC, Weetman, AP. Disease associations with autoimmune thyroid disease. Thyroid. 2002;12:975–986.
138. Cetani, F, Barbesino, G, Borsari, S, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab. 2001;86:4747–4752.
139. Dittmar, M, Kahaly, GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J Clin Endocrinol Metab. 2003;88:2983–2992.