Chapter 15 Atrial Fibrillation
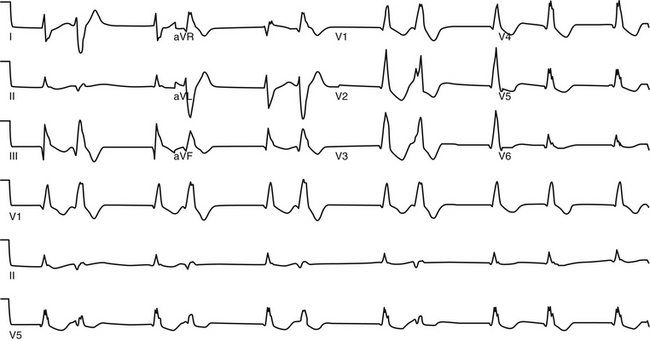
ELECTROCARDIOGRAPHIC FEATURES,
CATHETER ABLATION OF ATRIAL FIBRILLATION,
FOCAL ABLATION OF PULMONARY VEIN TRIGGERS,
SEGMENTAL OSTIAL PULMONARY VEIN ISOLATION,
CIRCUMFERENTIAL ANTRAL PULMONARY VEIN ISOLATION,
CIRCUMFERENTIAL LEFT ATRIAL ABLATION,
ABLATION OF COMPLEX FRACTIONATED ATRIAL ELECTROGRAMS,
ABLATION OF NON–PULMONARY VEIN TRIGGERS,
OUTCOME OF CATHETER ABLATION OF ATRIAL FIBRILLATION,
RECOMMENDATIONS AND CONTROVERSIES,
ATRIOVENTRICULAR JUNCTION ABLATION,
Pathophysiology
Classification of Atrial Fibrillation
Atrial fibrillation (AF) has been described in various ways, such as paroxysmal or persistent, lone, idiopathic, nonvalvular, valvular, or self-terminating. Each of these classifications has implications regarding mechanisms, as well as response to therapy. At the initial detection of AF, it may be difficult to be certain of the subsequent pattern of duration and frequency of recurrences. Thus, a designation of first-detected episode of AF is made on the initial diagnosis, irrespective of the duration of the arrhythmia. When the patient has experienced two or more episodes, AF is classified as recurrent. After the termination of an episode of AF, the rhythm can be classified as paroxysmal or persistent. Paroxysmal AF is characterized by self-terminating episodes that generally last less than 7 days. Persistent AF generally lasts longer than 7 days and often requires electrical or pharmacological cardioversion. Permanent AF refers to AF in which cardioversion has failed or AF that has been sustained for more than 1 year, or when further attempts to terminate the arrhythmia are deemed futile. With the advent of catheter ablation interventions for AF, patients with persistent AF for longer than 1 year who are considered for ablation have been referred to as having longstanding persistent AF, to distinguish them from patients with permanent AF in whom attempts to restore normal sinus rhythm (NSR) were unsuccessful or have been abandoned.1,2
Although useful, this arbitrary classification does not account for all presentations of AF. Paroxysmal AF often progresses to longer, non–self-terminating episodes. Additionally, the pattern of AF can change in response to treatment. AF that has been persistent can become paroxysmal with antiarrhythmic drug therapy, and AF that had been permanent may be cured or made paroxysmal by surgical or catheter-based ablation. Furthermore, the distinction between persistent and permanent AF is not only a function of the underlying arrhythmia but also a reflection of the clinical pragmatism of the patient and physician. The severity of symptoms associated with AF, anticoagulation status, and patient preference all affect the decision of whether and when cardioversion will be attempted. This decision would then affect the duration of sustained AF and could lead to a diagnosis of persistent or permanent AF.3
Mechanism of Atrial Fibrillation
Advanced mapping technologies, along with studies in animal models, have suggested the potential for complex pathophysiological substrates and modifiers responsible for AF, including the following: (1) continuous aging or degeneration of atrial tissue and the cardiac conduction system; (2) progression of structural heart disease (e.g., valvular heart disease and cardiomyopathy); (3) myocardial ischemia, local hypoxia, electrolyte derangement, and metabolic disorders (e.g., atherosclerotic heart disease, chronic lung disease, hypokalemia, and hyperthyroidism); (4) inflammation related to pericarditis or myocarditis, with or without cardiac surgery; (5) genetic predisposition; (6) drugs; and (7) autonomic effects.2
Mechanism of Initiation of Atrial Fibrillation
The factors responsible for the onset of AF include triggers that induce the arrhythmia and the substrate that sustains it. The triggers are diverse yet may not cause AF in the absence of other contributors. There are two different types of arrhythmias that can potentially play a role in generating AF: premature atrial complexes (PACs) that initiate AF (focal triggers) and focal tachycardia that either induces fibrillation in the atria or mimics AF by creating a pattern of rapid and irregular depolarization wavefronts in the atria for as long as the focus continues to discharge.2,4
The mechanism of initiation of AF is not certain in most cases and likely is multifactorial. Triggers propagating into the atrial myocardium can initiate multiple reentering wavelets and AF. In some patients with paroxysmal AF, impulses initiated by ectopic focal activity propagate into the left atrium (LA) and encounter heterogeneously recovered tissue. If reentry were assumed to be the mechanism of AF, initiation would require an area of conduction block and a wavelength of activation short enough to allow the reentrant circuits to persist in the myocardium.2,4
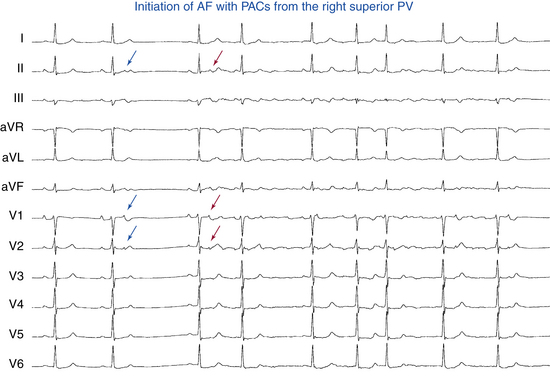
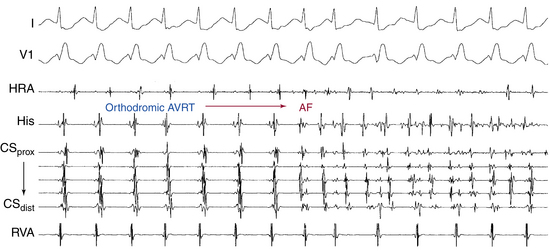
AF triggering factors include sympathetic or parasympathetic stimulation, bradycardia, PACs (which are the most common cause; Fig. 15-1), atrial flutter (AFL), supraventricular tachycardias (SVTs; especially those mediated by atrioventricular [AV] bypass tracts [BTs]; Fig. 15-2), and acute atrial stretch. Identification of these triggers has clinical importance because treatment approaches directed at elimination of the triggers (e.g., radiofrequency [RF] ablation of the initiating PACs or SVT) can be curative in selected patients.
Pulmonary Vein Triggers
Triggering foci of rapidly firing cells within the sleeves of atrial myocytes extending into the pulmonary veins (PVs) have been clearly shown to be the underlying mechanism in most cases of paroxysmal AF.4 Supporting this idea are clinical studies of impulses generated by single foci propagating from individual PVs or other atrial regions to the remainder of the atria as fibrillatory waves and abolition of AF by RF ablation to eliminate or isolate the venous foci.
Based on several features, the thoracic veins are highly arrhythmogenic. The PV-LA junction has discontinuous myocardial fibers separated by fibrotic tissues and hence is highly anisotropic. Insulated muscle fibers can promote reentrant excitation, automaticity, and triggered activity. These regions likely resemble the juxtaposed islets of atrial myocardium and vascular smooth muscle in the coronary sinus (CS) and AV valves that, under normal circumstances, manifest synchronous electrical activity but develop delayed afterdepolarizations and triggered activity in response to catecholamine stimulation, rapid atrial pacing, or acute stretch.5
Furthermore, the PVs of patients with paroxysmal AF demonstrate abnormal properties of conduction so that there can be markedly reduced refractoriness within the PVs, progressive conduction delay within the PV in response to rapid pacing or programmed stimulation, and often conduction block between the PV and the LA. Such findings are much more common in patients with paroxysmal AF than in normal subjects.2,6 Rapidly firing foci can often be recorded within the PVs with conduction block to the LA. Administration of catecholamines such as isoproterenol can lead to shortening of the LA refractory period, thereby allowing these foci to propagate to the LA with the induction of AF.6 These discontinuous properties of conduction within the PV can also provide a substrate for reentry within the PV itself, although this remains to be proven.5
Non–Pulmonary Vein Triggers
Although more than 90% of triggering foci that are mapped during electrophysiological (EP) studies in patients with paroxysmal AF occur in the PVs, foci within the superior vena cava (SVC), small muscle bundles in the ligament of Marshall, and the musculature of the CS have been identified. Although these latter locations of triggering foci are uncommon in patients with paroxysmal AF, the common factor is that the site of origin is often within a venous structure that connects to the atrium. Other sites of initiating foci can be recorded in the LA wall or along the crista terminalis in the right atrium (RA).5
Mechanism of Maintenance of Atrial Fibrillation
Having been initiated, AF can be brief; however, various factors can act as perpetuators, thus ensuring the maintenance of AF.2 One factor is the persistence of the triggers and initiators that induced the AF, which then act as an engine driving the continuation of AF. In this setting, maintenance of AF is dependent on the continued firing of the focus (the so-called focal driver). However, AF can persist even in the absence of the focal drivers. Without focal drivers, persistence of AF results from a combination of electrical and structural remodeling processes characterized by atrial dilation and shortening of atrial refractoriness (see later). These factors can be present at baseline or, alternatively, induced by the AF itself.
Multiple Wavelet Theory
For many years, the most widely held theory on the maintenance of AF was the multiple wavelet hypothesis, which was a key development in our understanding of the mechanism of AF. Moe and associates noted that, “The grossly irregular wavefront becomes fractionated as it divides about islets or strands of refractory tissue, and each of the daughter wavelets may now be considered as independent offspring. Such a wavelet may accelerate or decelerate as it encounters tissue in a more or less advanced state of recovery.”7 Moe and associates subsequently hypothesized that AF is sustained by multiple randomly wandering wavelets that collided with each other and were extinguished, or divided into daughter wavelets that continually reexcited the atria.7 Those functional reentrant circuits are therefore unstable; some disappear, whereas others reform. These circuits have variable, but short, cycle lengths (CLs), resulting in multiple circuits to which atrial tissue cannot respond in a 1:1 fashion. As a result, functional block, slow conduction, and multiple wavefronts develop. It has been suggested that at least four to six independent wavelets are required to maintain AF. These wavelets rarely reenter themselves but can reexcite portions of the myocardium recently activated by another wavefront, a process called random reentry. As a result, there are multiple wavefronts of activation that can collide with each other and extinguish themselves or create new wavelets and wavefronts, thereby perpetuating the arrhythmia.
The persistence of multiple-circuit reentry depends on the ability of a tissue to maintain enough simultaneously reentering wavefronts so that electrical activity is unlikely to extinguish simultaneously in all parts of the atria. Therefore, the more wavelets are present, the more likely it is that the arrhythmia will be sustained. The number of wavelets on the heart at any moment depends on the atrial mass, refractory period, conduction velocity, and anatomical obstacles in different portions of the atria. In essence, a large atrial mass with short refractory periods and conduction delay would yield increased wavelets and would present the most favorable situation for AF to be sustained.2,8
Single Circuit Reentry Theory
Studies in isolated human atrial preparations questioned the randomness of atrial activity and suggested the presence of a single source of stable reentrant activity (“mother rotor”) that serves as a periodic background focus, with break-up of emanating waves in atrial tissue of variable electrical properties and anatomical obstacles into multiple wavelets spreading in various directions. Although well represented in animal studies, rotors have not been observed in whole human atria studied at the time of thoracic surgery. More recent data using different techniques have shown that functional reentry (or anatomical reentry with a functional component), in the form of spiral waves rotating around microreentrant circuits approximately 1 cm in diameter, was suggested to be the most likely cause of AF. Other studies have shown that these dominant rotors that drive AF invariably originate and anchor in the LA, with the RA activated passively.8,9
Focal Drivers with Fibrillatory Conduction
Although multiple wandering wavelets probably account for most cases of AF, occasionally a single, rapidly firing focus can be identified with EP mapping. Impulses initiated by ectopic focal activity propagate into the atria to encounter heterogeneously recovered tissue. When cardiac impulses are continuously generated at a rapid rate from any source or any mechanism, they activate the tissue of that cardiac chamber in a 1:1 manner, up to a critical rate. However, when this critical rate is exceeded, so that not all the tissue of that cardiac chamber can respond in a 1:1 fashion (e.g., because the CL of the driver is shorter than the refractory periods of those tissues), fibrillatory conduction develops. Fibrillatory conduction can be caused by spatially varying refractory periods or by the structural properties of atrial tissue, with source-sink mismatches providing spatial gradients in the response.2 Thus, fibrillatory conduction is characterized by activation of tissues at variable CLs, all longer than the CL of the driver, because of variable conduction block. In that manner, activation is fragmented.2 This is the mechanism of AF in several animal models in which the driver consists of a stable, abnormally automatic focus of a very short CL, a stable reentrant circuit with a very short CL, or an unstable reentrant circuit with a very short CL. It also appears to be the mechanism of AF in patients in whom activation of the atria at very short CLs originates in one or more PVs. The impulses from the PVs seem to precipitate and maintain AF. Autonomic influences (parasympathetic or sympathetic) can cause some of these rapid discharges. Of note, it has also been suggested that fibrillatory conduction caused by a reentrant driver can potentially be the cause of ventricular fibrillation (VF).
Substrate for Atrial Fibrillation
AF results from the interplay between a trigger for initiation and a vulnerable EP substrate for maintenance. The fact that most potential triggers do not initiate AF suggests some role for functional and structural substrates in most patients. However, the relative contribution of triggers versus substrate can vary with the clinical context, and the exact nature of the interaction between triggers and substrate remains to be elucidated.8
AF commonly occurs in the context of other cardiac or noncardiac pathological conditions, such as valvular disease, hypertension, ischemic heart disease, heart failure, or hyperthyroidism. Depending on the type, extent, and duration of such external stressors, a cascade of time-dependent adaptive, as well as maladaptive, atrial responses develops in order to maintain homeostasis (so-called atrial remodeling), including changes at the ionic channel level, cellular level, or extracellular matrix level, or a combination of these, thus resulting in structural, functional, and electrical consequences. A hallmark of atrial structural remodeling is atrial dilation, often accompanied by a progressive increase in interstitial fibrosis. Atrial arrhythmias, especially AF, are the most common manifestations of electrical remodeling.10
Increased dispersion in atrial refractoriness and inhomogeneous dispersion of conduction abnormalities, including block, slow conduction, and dissociation of neighboring atrial muscle bundles, are key elements in the development of the substrate of AF. Importantly, different pathological conditions can be associated with a different set of remodeling responses in the atria.11
Even in the setting of lone AF, whereby no structural heart disease is apparent, there is accumulating evidence that occult abnormalities (e.g., patchy fibrosis, inflammatory infiltrates, loss of myocardial voltage, conduction slowing, altered sinus node function, and vascular dysfunction) can be observed and likely represent an early stage of atrial remodeling contributing to the substrate of AF.12
Atrial Fibrosis
Atrial fibrosis plays an important role in the pathophysiology of AF. Atrial fibrosis results from various cardiac insults that share common fibroproliferative signaling pathways. Fibrotic myocardium exhibits slow and inhomogeneous conduction, likely secondary to reduced intercellular coupling, discontinuous branching architecture, and zigzagging circuits. When combined with inhomogeneous dispersion of refractoriness within the atria, conduction block provides an ideal substrate for reentry. The greater the slowing of conduction velocity is in scarred myocardium, the shorter the anatomical circuit will need to be to sustain a reentrant wavelet. In fact, reentrant circuits need be only a few millimeters in length in discontinuously conducting tissue. Thus, atrial regions with advanced fibrosis can be local sources for AF. Such a hypothesis would not preclude the remainder of the atria from showing fibrillatory conduction or intact, functional reentrant waves, or both.10,13
The normal aging process results in anatomical changes likely to yield inhomogeneous conduction that can potentially create the milieu necessary for the development of reentry. These changes are probably magnified by the presence of certain disease processes, such as hypertension, coronary artery disease, and heart failure. The strong association of sinus node dysfunction and AF (the bradycardia-tachycardia syndrome) also suggests that replacement of atrial myocytes by interstitial fibrosis likely plays an important part in the pathogenesis of AF in older adults, although in some instances the bradycardia component is a functional response to the tachycardia. Furthermore, AF itself seems to produce various alterations of atrial architecture that further contribute to atrial remodeling, mechanical dysfunction, and perpetuation of fibrillation. Longstanding AF results in loss of myofibrils, accumulation of glycogen granules, disruption in cell-to-cell coupling at gap junctions, and organelle aggregates.10
Changes in AF characteristics during evolving fibrosis also have a direct impact on why electrical or drug treatment, or both, ultimately fails to achieve conversion to NSR. In the markedly fibrotic and discontinuous atrial tissue, characterized by discontinuous anisotropy, a marked degree of gap junctional uncoupling, and fiber branching, the safety factor for propagation is higher than in normal tissue. As a consequence, blocking of the Na+ current to the same degree as is necessary for the termination of functional reentry may not terminate reentry caused by slow and fractionated conduction in fibrotic scars of remodeled atria. Conduction in discontinuous tissue is mostly structurally determined and leads to excitable gaps behind the wavefronts. If a gap is of critical size, the effectiveness of drugs that prolong atrial refractoriness will be limited. Furthermore, scar tissue is likely to exhibit multiple entry and exit points and multiple sites at which unidirectional block occurs. This can potentially lead to activity whose appearance in local extracellular electrograms changes from beat to beat, as well as beat-to-beat CL variability. Although such regions can be expected to respond to defibrillation, AF may resume after extrasystoles or normal sinus beats immediately after conversion, with unidirectional block recurring as a result of the presence of scar.
Atrial Stretch
Atrial stretch and dilation can play a role in the development and persistence of AF. Clinically, AF episodes occur more frequently in association with conditions known to cause elevated LA pressure and atrial stretch, such as acutely decompensated systolic or diastolic heart failure. Additionally, the echocardiographic LA volume index and restrictive transmitral Doppler flow patterns are strong predictors of the development of AF.8,14
The structure of the dilated atria can potentially have important EP effects related to stretch of the atrial myocardium (so-called electromechanical feedback). Acute atrial stretch reduces the atrial refractory period and action potential duration and depresses atrial conduction velocity, potentially through a reduction of cellular excitability by the opening of stretch-activated channels or changes in cable properties (membrane resistance, capacitance, core resistance), or both. Regional stretch for less than 30 minutes turns on the immediate early gene program, thus initiating hypertrophy and altering action potential duration in affected areas. Moreover, acutely altered stress and strain patterns augment the synthesis of angiotensin II, which induces myocyte hypertrophy. By regionally increasing L-type calcium (Ca2+) current (ICaL) and decreasing the transient outward potassium (K+) current (Ito), angiotensin II can contribute to arrhythmogenic electrical dispersion. Altered stretch of atrial myocytes also results in opening of stretch-activated channels, increasing G protein–coupled pathways. This leads to increased protein kinase A and C activity, and enhanced ICaL through the cell membrane, and increased release of Ca2+ from the sarcoplasmic reticulum, thus promoting afterdepolarizations and triggered activity.10 Furthermore, acute stretch can promote an increase in dispersion of refractoriness and spatial heterogeneity by causing conduction block and potentially contributing to the development of AF.8,14,15
Inflammation and Atrial Fibrillation
There is increasing evidence that implicates inflammation in the pathogenesis of AF. Clinically, AF occurs frequently in the setting of inflammatory states such as cardiac surgery and acute pericarditis. Additionally, the levels of inflammatory biomarkers (C-reactive protein [CRP] and interleukin-6 [IL-6]) are significantly increased in patients with AF, findings suggesting the presence of systemic inflammation in these patients. Elevation of the levels of CRP and IL-6 has been shown to predict future development, recurrence, and burden of AF.16–18 There is also evidence suggesting that inflammation is involved in electrical and structural atrial remodeling. Furthermore, inflammation appears to increase the inhomogeneity of atrial conduction directly, potentially via disruption of expression of connexin proteins, leading to impaired intercellular coupling.16
It is also likely that inflammation can be a consequence of AF. CRP levels decrease following restoration of sinus rhythm. Rapid atrial activation in AF results in Ca+2 overload in atrial myocytes that can potentially result in cell death, which induces a low-grade inflammatory response. The inflammation, in turn, can induce healing and repair that likely enhance remodeling and promote perpetuation of the arrhythmia.16
Currently, the exact role of inflammation in AF is poorly defined, and it remains unclear whether inflammation is actually involved in the mechanisms underlying AF or whether it is simply an epiphenomenon. Therapies directed at attenuating the inflammatory burden (e.g., glucocorticoids, statins, and angiotensin II inhibitors) appear promising, although early clinical trials do not support a significant benefit.19
Atrial Remodeling in Atrial Fibrillation
It is well known from clinical practice that AF is a progressive arrhythmia. Eventually, in 14% to 24% of patients with paroxysmal AF, persistent AF will develop, even in the absence of progressive underlying heart disease. Furthermore, conversion of AF to NSR, electrically or pharmacologically, becomes more difficult when the arrhythmia has been present for a longer period. In fact, the arrhythmia itself results in a cascade of electrical and structural changes in the atria that are themselves conducive to the perpetuation of the arrhythmia (“AF begets AF”), a process known as remodeling.20 Recurrent AF can potentially lead to irreversible atrial remodeling and eventually permanent structural changes that account for the progression of paroxysmal to persistent and finally to permanent AF, characterized by the failure of electrical cardioversion or pharmacological therapy, or both, to restore and maintain NSR. Even after cessation of AF, these abnormalities persist for periods that vary in proportion to the duration of the arrhythmia.17,21
Changes in atrial EP features that are induced by AF can occur through alterations in ion channel activities that cause partial depolarization and abbreviation of atrial refractoriness. These changes promote the initiation and perpetuation of AF (electrical remodeling) and the modification of cellular Ca2+ handling, which causes contractile dysfunction (contractile remodeling), as well as atrial dilation with associated structural changes (structural remodeling). Electrical remodeling can potentially begin within a few hours after the onset of AF, whereas the structural changes are slower, likely starting after several weeks.17
Electrical remodeling results from the high rate of electrical activation. The EP changes typical of atrial myocytes during AF are shortening of the atrial refractory period and action potential duration, reduction in the amplitude of the action potential plateau, and loss of response of action potential duration to changes in rate (abnormal restitution). Whereas the normal atrial action potential duration shortens in response to pacing at shorter CLs, AF results in loss of this rate dependence of atrial action potential duration, and the atrial refractory period fails to lengthen appropriately at slow rates (e.g., with return to NSR). These changes can explain the increased duration of AF because, according to the multiple wavelet theory, a short wavelength results in smaller wavelets, which increase the maximum number of wavelets, given a certain atrial mass.2,20 Tachycardia-induced changes in refractoriness are spatially heterogeneous, and there is increased variability both within and among various atrial regions that may promote atrial vulnerability and AF maintenance and provide a substrate for reentry.
The mechanism for electrical remodeling and shortening of the atrial refractory period is not entirely clear. Several potential explanations exist, including ion channel remodeling, angiotensin II, and atrial ischemia. Shortening of the atrial action potential can be caused by a net decrease of inward ionic currents (Na+ or Ca2+), a net increase of outward currents (K+), or a combination of both. The decrease of ICaL seems to be responsible for shortening of the atrial action potential, whereas the decrease of Ito is considered to result in loss of physiological rate adaptation of the action potential. The reduction in ICaL can be explained by a decreased expression of the L-type Ca2+ channel α1C subunit, likely as a compensatory mechanism to minimize the potential for cytosolic Ca2+ overload secondary to increased Ca2+ influx during the rapidly repetitive action potentials during AF. Verapamil, an L-type Ca2+ channel blocker, was shown to prevent electrical remodeling and hasten complete recovery without affecting inducibility of AF, whereas intracellular Ca2+ overload, induced by hypercalcemia or digoxin, enhances electrical remodeling. Electrical remodeling can be attenuated by the sarcoplasmic reticulum’s release of the Ca2+ antagonist ryanodine, a finding suggesting the importance of increased intracellular Ca2+ to the maladaptation of the atrial myocardium during AF. Angiotensin II may also be involved in electrical and atrial myocardial remodeling, and angiotensin II inhibitors may prevent atrial electrical remodeling. Angiotensin-converting enzyme inhibitors reduce the incidence of AF in patients with left ventricular (LV) dysfunction after myocardial infarction and in patients with chronic ischemic cardiomyopathy. Atrial ischemia is another possible contributor to electrical remodeling and shortening of the atrial refractory period via activation of the Na+-H+ exchanger.10,11
Furthermore, persistent AF can result in other changes within the atria, including gap junctional remodeling, cellular remodeling, and sinus node remodeling. Gap junctional remodeling is manifest as an increase in the expression and distribution of connexin 43 and heterogeneity in the distribution of connexin 40, both of which are intercellular gap junction proteins.20 Cellular remodeling is caused by the apoptotic death of myocytes with myolysis, which may not be entirely reversible. AF results in marked changes in atrial cellular substructures, including loss of myofibrils, accumulation of glycogen, changes in mitochondrial shape and size, fragmentation of sarcoplasmic reticulum, and dispersion of nuclear chromatin.20
Sustained AF has also been associated with structural changes, such as myocyte hypertrophy, myocyte death, impaired atrial contractility, and atrial stretch and dilation, which act to reduce conduction velocity.20 Atrial dilation increases electrical instability by shortening the effective refractory period and slowing atrial conduction.10 These structural changes, many of which probably are irreversible, appear to occur more slowly, over periods of weeks to months.
In addition to remodeling of the atria, the sinus node can undergo remodeling, resulting in sinus node dysfunction and bradyarrhythmias caused by reduced sinus node automaticity or prolonged sinoatrial conduction. The phenomenon of sinus node remodeling may contribute to the episodes of bradycardia seen in the tachycardia-bradycardia syndrome and may reduce sinus rhythm stability and increase the stability of AF.20 As mentioned earlier, elements of the sinus bradycardia appear to be functionally reversible if the tachycardia is prevented.
Studies suggest that the PVs are more susceptible to electrical alterations resulting from AF than the atria. Although the PVs display significantly longer refractory periods at baseline than the atria, they exhibit more prominent shortening of refractoriness after a brief episode of pacing-induced AF. Moreover, the short-term presence of AF does influence PV EP features by slowing the conduction velocity without affecting the conduction times of the atria. Structural changes in the atria after remodeling, such as stretch, can also result in increased PV activity. Atrial stretch can lead to increased intraatrial pressure, causing a rise in the rate and spatiotemporal organization of electrical waves originating in the PVs. These changes imply that electrical and structural remodeling increases the likelihood of ectopic PV automaticity and AF maintenance. Therefore, rather than AF begets AF, one can vary that theme: “PV-induced paroxysmal AF begets PV-induced chronic AF.”22
Atrial tachycardia (AT)–induced remodeling can potentially underlie various clinically important phenomena, such as the tendency of patients with other forms of supraventricular arrhythmias to develop AF, the tendency of AF to recur early after electrical cardioversion, the resistance of longer duration AF to antiarrhythmic medications, and the tendency of paroxysmal AF to become persistent.2
Role of Autonomic Nervous System in Atrial Fibrillation
Cardiac function is modulated by both the extrinsic and the intrinsic cardiac autonomic nervous systems. The extrinsic (central) system is composed of the vagosympathetic system from the brain and spinal cord to the heart. The intrinsic system is composed of a large network of autonomic cardiac ganglia buried throughout the epicardial fat within the pericardial space. Groups of several cardiac ganglia comprise plexuses that coalesce in specific locations, and different groups of ganglia have different sites of innervation throughout the heart. Atrial ganglia contain afferent neurons from the atrial myocardium and from the central autonomic nervous system, and efferent cholinergic and adrenergic neurons, with heavy innervation of the PV myocardium and the atrial myocardium surrounding the ganglionic plexuses. Additionally, an extensive array of interconnecting neurons creates a communication network among the different ganglionic plexuses, as well as between the ganglionic plexuses and the atrium and PV myocardium. The intrinsic system receives input from the extrinsic system but acts independently to modulate numerous cardiac functions, including automaticity, contractility, and conduction.23,24
Several studies have suggested that both divisions of the autonomic nervous system are involved in the initiation, maintenance, and termination of AF, with a predominant role of the parasympathetic system. Electrical stimulation of autonomic nerves on the heart itself can facilitate the induction of AF. Increased vagal tone is frequently involved in the onset of AF in patients with structurally normal hearts. Parasympathetic stimulation shortens the atrial refractory period, increases the dispersion of refractoriness, and decreases the wavelength of reentrant circuits that facilitate initiation and perpetuation of AF. Long-term vagal denervation of the atria renders AF less easily inducible in animal experiments, presumably because of increased EP homogeneity. On the other hand, vagal stimulation results in maintenance of AF, and catheter ablation of the parasympathetic autonomic nerves entering the RA from the SVC prevents vagally induced AF in animal models. Sympathetic stimuli also shorten the atrial refractory period and increase the vulnerability to AF.23,25
Experimental evidence suggests that the electrical properties of the PVs are also modulated by changes in autonomic tone.6 Anatomical studies revealed that the LA and PVs are innervated by adrenergic and cholinergic nerve fibers. A collection of ganglia is localized on the posterior wall of the LA between the superior PVs. Subsequent studies found that ganglionated plexuses clustered at the PV entrances (within fat pads) could be stimulated without atrial excitation. For patients with PV foci, a primary increase in adrenergic tone followed by a marked vagal predominance was reported just prior to the onset of paroxysmal AF. A similar pattern of autonomic tone was reported in an unselected group of patients with paroxysmal AF and various cardiac conditions.25 Activation of the ganglionic plexuses at the PV-LA junction can potentially result in conversion of PV ectopy to AF. Furthermore, ablation of the ganglionated plexuses located at the atrial entrances or antra of the PVs can potentially abolish or reduce AF inducibility.
Role of the Pulmonary Veins in Atrial Fibrillation
There is little controversy now that the PVs play a major role in triggering and maintaining AF, as established by animal and human models, especially in the setting of paroxysmal AF. First, fibrillatory conduction is likely initiated by rapid discharges from one or several focal sources within the atria; in most patients with AF (94%), the focus is in one of the PVs (Fig. 15-1). Extra-PV sites can also trigger AF, but this occurs in a few cases, likely no more than 6% to 10% of patients. AF is also perpetuated by microreentrant circuits, or rotors, that exhibit high-frequency periodic activity from which spiral wavefronts of activation radiate into surrounding atrial tissue. Conduction becomes slower and less organized with increasing distance from the rotors, likely because of atrial structural remodeling, resulting in fibrillatory conduction. Interestingly, the dominant rotors in AF are localized primarily in the junction between the LA and PVs. One study also demonstrated that the PV-LA region has heterogeneous EP properties capable of sustaining reentry (microreentry or macroreentry). Finally, vagal inputs may be important in triggering and maintaining AF, and many of these inputs are clustered close to the PV-LA junction. Thus, the PVs play a critical role in triggering and maintaining AF.
Pulmonary Vein Anatomy
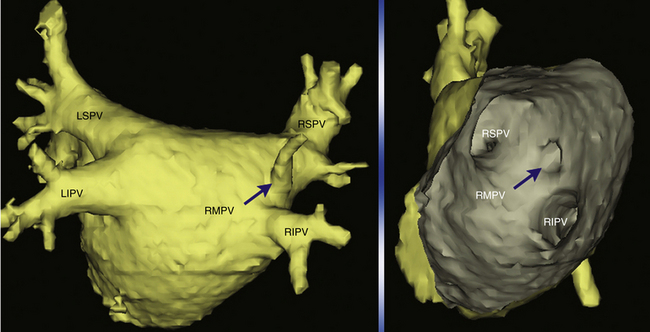
PVs can have variable anatomy. Most hearts examined are found to have four PVs with discrete ostia, but the remainder (approximately 25%) has a common ostium, either on the left or on the right (Fig. 15-3).4 The PV ostia are ellipsoid with a longer superoinferior dimension, and funnel-shaped ostia are frequently noted in patients with AF. The right superior PV is located close to the SVC or RA, and the right inferior PV projects horizontally. The left superior PV is close to the vicinity of the LA appendage, and the left inferior PV courses near the descending aorta. PVs are larger in patients with AF than in normal subjects, in men than in women, and in persistent versus paroxysmal AF. Significant variability of PV morphologies exists, however, including supernumerary right PVs (in 8% to 29% of patients; Fig. 15-4), multiple ramification and early branching (especially of the right inferior PV), and common ostium of left-sided or, less frequently, right-sided PVs.
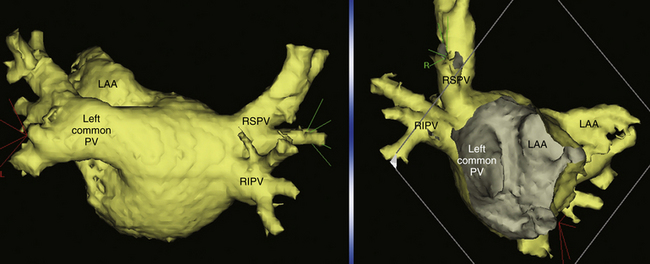
The PVs are covered by myocardial sleeves formed by one or more layers of myocardial fibers oriented in a circular, longitudinal, oblique, or spiral direction. These sleeves, continuing from the LA into the PV, vary from 2 to 25 mm in length, with a mean extent of 13 mm. The length of the myocardial sleeves usually has a distinctive distribution; superior PVs have longer and better developed myocardial sleeves than inferior PVs, which may explain why arrhythmogenic foci are found more often in the superior PVs than in the inferior PVs.4,5 It should be noted that all PVs in all individuals have such myocardial sleeves, regardless of the presence or absence of AF.
The walls of the PVs are composed of a thin endothelium, a media of smooth muscle, and a thick outer fibrous adventitia. The transition from atrial to venous walls is gradual because the myocardial sleeves from the LA overlap with the smooth muscle of the venous wall. The myocardial sleeves are thickest at the venoatrial junction (mean, 1.1 mm) and then gradually taper distally. Furthermore, the thickness of the sleeves is not uniform, with the inferior walls of the superior veins and the superior walls of the inferior veins having the thicker sleeves. Throughout the PV, and even at the venoatrial junction, there are gaps in the myocardial sleeves mainly composed of fibrous tissue. The arrangement of the myocyte bundles within the sleeves is rather complex. There appears to be a mesh-like arrangement of muscle fascicles made up of circularly oriented bundles (spiraling around the long axis of the vein) that interconnect with bundles that run in a longitudinal orientation (along the long axis of the vein). Such an arrangement, together with the patchy areas of fibrosis seen, may be relevant to the role of the PVs in the initiation of AF.4
Electrophysiology of Pulmonary Vein Musculature
As noted, the PVs play a crucial role in the initiation and maintenance of AF. However, it is not clear what makes this region so susceptible to the arrhythmia.4,6 There are, at present, limited data available on the ionic mechanisms that may underlie the arrhythmogenicity of PVs. Detailed mapping studies have suggested that reentry within the PVs is most likely responsible for their arrhythmogenicity, although focal or triggered activity cannot be excluded.
The EP features of the PV, with its distinct area of slow conduction, decremental conduction, nonuniform anisotropy, and heterogeneous repolarization, are potential substrates for reentry. The heterogeneous fiber orientation in the transition from the LA to the PV sleeve results in unique conduction properties in this area.4,6 It is possible that the complex arrangement of muscle fibers within the myocardial sleeves and the uneven distribution of interspersed connective and adipose tissue account for the greater degree of decremental conduction observed in the myocardial sleeves than in the LA and for the heterogeneity in conduction properties and refractory periods among the fascicles in the myocardial sleeves. Therefore, the fractionation of PV potentials commonly observed during premature stimulation (which usually indicates local slowing of conduction) is consistent with anisotropic properties that can be attributable to the complex arrangement of muscle fascicles within the myocardial sleeves.5
Several studies suggested that abnormal automaticity or triggered activity, either alone or in combination with the reentrant mechanisms described previously, can play a role in the initiation of AF. These studies suggested that the propensity of PVs to exhibit focal or triggered activity is enhanced by pathological conditions.4 Further work also implicated the posterior LA in the genesis of AF. Studies suggested that the PVs, together with the posterior LA, have an important role in the persistent form of AF. However, the nature of the relationship between this arrhythmogenic region and the pathological conditions that provide a substrate for AF has not been elucidated. Whether the critical region is the posterior LA, the PVs, or both, has been the source of ongoing debate.6
Clinical Considerations
Epidemiology
AF is the most common sustained arrhythmia encountered in clinical practice. It accounts for approximately one-third of hospitalizations for cardiac rhythm disturbance. AF affects 1% to 2% of the general population, and it has been estimated that 2.3 million people in the United States and 4.5 million in the European Union have paroxysmal or persistent AF. However, the true prevalence of AF is probably higher given the common occurrence of asymptomatic (silent) AF.26
The lifetime risk of the development of AF at age 40 years has been estimated to be approximately 25%. The age-adjusted prevalence is higher in men. Blacks have less than half the age-adjusted risk of developing AF than whites.27 Given the increasing prevalence of AF with age, coupled with our aging population, the number of affected U.S. residents is expected to increase to nearly 16 million by 2050.
AF is a progressive disease. Approximately half of individuals who experience an initial episode of AF will eventually develop recurrent AF, typically within the first 2 years of follow-up. AF progresses from paroxysmal to persistent despite antiarrhythmic therapy in approximately 10% at 1 year, in 25% to 30% at 5 years, and in more than 50% beyond 10 years. Furthermore, progression from paroxysmal and persistent AF to permanent AF occurs in up to 34% of patients at 4 years after initial diagnosis. Maintenance of sinus rhythm is progressively more difficult as the duration of persistent AF increases. Only 40% to 60% of patients with persistent AF of less than 1 year’s duration remain in sinus rhythm 1 year after initiation of antiarrhythmic drug therapy, despite multiple cardioversions, whereas patients with persistent AF of more than 3 year’s duration have only a 15% likelihood of long-term sinus rhythm. Increased age, diabetes, and heart failure are among potential predictors of progression to permanent AF. Lone AF is less likely to progress.28
AF is associated with an increased long-term risk of stroke, heart failure, hospitalization, and all-cause mortality, especially in women, even after controlling for CHADS2 score and other covariables. The mortality rate of patients with AF is approximately double that of patients in NSR and is linked to the severity of underlying heart disease.27,29
Clinical Risk Factors Predisposing to Atrial Fibrillation
The most frequent causes of acute AF are myocardial infarction (5% to 10% of patients with infarct) and cardiothoracic surgery (up to 50% of patients). The most common clinical settings for permanent AF are hypertension and ischemic heart disease, with the subset of patients having congestive heart failure most likely to experience the arrhythmia. In the developing world, hypertension and rheumatic valvular (usually mitral) and congenital heart diseases are the most commonly related conditions.30
Heart failure is present in 34% of patients with AF, and AF develops in up to 42% of patients with heart failure. AF and heart failure have a grim prognosis, with a 1-year mortality of 9.5% and worsening of heart failure in almost 25%.31 In patients with heart failure who are undergoing defibrillator implantation, a history of AF at time of procedure identifies additional risk of heart failure and death, and the new detection of AF afterward is associated with even higher rates of death.32
There is accumulating evidence demonstrating an independent association between sleep apnea and AF, likely related to the effects of sleep apnea and nocturnal hypoxemia on LA electrical remodeling, fibrosis, and chamber enlargement. AF occurs in 5% of persons with severe sleep apnea and only 1% of those without sleep apnea. The prevalence of at least moderate sleep apnea was reported in up to 32% of patients with lone AF. Furthermore, cross-sectional studies demonstrated that patients with AF had a significantly higher risk of obstructive sleep apnea than matched controls (49% versus 33%), and multivariate analysis demonstrated a strong independent association between sleep apnea and AF (odds ratio, 2.2). Additionally, several prospective studies showed that obstructive sleep apnea predicts the occurrence of future AF. Untreated obstructive sleep apnea was associated with a remarkably high rate of AF recurrence at 1 year compared with patients with unknown sleep apnea status (83% versus 53%). The presence of sleep apnea can also predict a higher rate of early recurrence of PV conduction and AF following catheter ablation of AF.33 Several investigations identified obesity (found in approximately 25% of patients with AF) as a risk factor for the development of AF, and there exists a linear association between the body mass index and AF risk. Obesity is associated with increased LA size and impaired LV diastolic function, which can potentially explain its predisposition to AF.34
Clinical Presentation
Symptoms associated with AF vary, depending on the ventricular rate, the underlying functional status, the duration of AF, the presence and degree of structural heart disease, and the individual patient’s perception. The hemodynamic consequences of AF are related to loss of coordinated atrial contraction, irregularity of ventricular response, and fast heart rate, as well as long-term consequences such as atrial and ventricular cardiomyopathy. Loss of effective atrial contraction can potentially reduce cardiac output by 15% to 20%. These consequences are magnified in the presence of impaired diastolic ventricular filling, hypertension, mitral stenosis, LV hypertrophy, and restrictive cardiomyopathy. Irregularity of the cardiac cycle, especially when accompanied by short coupling intervals, and rapid heart rates in AF can lead to reduction in diastolic filling, stroke volume, and cardiac output.27
Most patients with AF complain of palpitations, chest discomfort, dyspnea, generalized fatigue, or dizziness. Although palpitation, or awareness of the irregularity of the heartbeat, is prominent in more than half of patients with AF, its correlation with documented arrhythmia is unimpressive.35 Furthermore, AF with a chronically rapid heart rate (more than 120 to 130 beats/min) can lead to tachycardia-mediated cardiomyopathy and heart failure. Syncope is an uncommon complication of AF that can occur on conversion in patients with sinus node dysfunction or because of rapid ventricular rates in patients with hypertrophic cardiomyopathy, in patients with valvular aortic stenosis, or in patients with ventricular preexcitation over an accessory pathway. The first presentation of asymptomatic AF can be catastrophic—an embolic complication or acute decompensation of heart failure.27,35
In some patients, paroxysmal AF can be classified as either vagal or adrenergic, depending on the types of triggers and the temporal distribution of the arrhythmic episodes. Vagal AF typically occurs in young male patients without structural heart disease and characteristically develops during sleep or postprandial. In contrast, patients with adrenergic AF are usually older, often with evidence of underlying heart disease, and the episodes of AF usually occur during the day and are associated with physical or emotional stress. In patients with paroxysmal AF, the prevalence of vagal AF probably ranges between 6% and 25%, whereas that of adrenergic AF ranges between 7% and 16%. Approximately 12% of patients with paroxysmal AF exhibit features of mixed vagal and adrenergic patterns.25
Many patients with persistent or permanent AF have one or more comorbid conditions that can considerably contribute to specific complaints and to overall quality of life. Therefore, it is important to establish a correlation between any symptoms and AF, as well as ventricular response rates. The effect of regulation of ventricular rate during persistent AF or conversion to NSR on a patient’s symptoms and quality of life can help assess the relative contribution of AF to the patient’s complaints. The Canadian Cardiovascular Society Severity in Atrial Fibrillation (CCS-SAF) scale is a simple, five-point semiquantitative scale (Table 15-1) that has been proposed for use at the bedside to assess the functional consequences of symptoms during AF and can provide objective assessment of the patient’s subjective state (analogous to the New York Heart Association [NYHA] congestive heart failure functional class and the CCS angina severity class).35
TABLE 15-1 Canadian Cardiovascular Society Severity of Atrial Fibrillation Scale
| Step 1: Symptoms | |
| Identify the presence of the following symptoms: |
AF = atrial fibrillation; CCS = Canadian Cardiovascular Society; QOL = quality of life; SAF = severity of atrial fibrillation.
From Dorian P, Guerra PG, Kerr CR, et al: Validation of a new simple scale to measure symptoms in atrial fibrillation: the Canadian Cardiovascular Society Severity in Atrial Fibrillation (CCS-SAF) scale, Circ Arrhythm Electrophysiol 2:268-275, 2009.
Risk of Thromboembolism
AF is a major risk factor for thromboembolism, causing approximately 15% of the ischemic strokes in the United States and up to 25% of those in patients older than 80 years. In the Framingham Heart Study, patients with rheumatic heart disease and AF had a 17-fold increased risk of stroke compared with age-matched controls, and the attributable risk was 5 times greater than in those with nonrheumatic AF. For nonvalvular AF, the risk of stroke is estimated to be 2 to 7 times that of subjects without arrhythmia, thus resulting in an average incidence of stroke of 5% per year. This rate may increase to 7% per year when silent cerebral ischemic events and transient ischemic attacks are taken into account.27 Patients with paroxysmal and persistent AF appear to have a stroke risk similar to that in patients with permanent AF.
Although patients with AF in the setting of rheumatic valvular disease are accepted to be at high risk of stroke, stroke risk in nonvalvular AF is not homogeneous across the various subgroups of patients. The risk ranges from less than 1.5% per year in patients with lone AF who are less than 59 years old to more than 10% per year in older patients, especially when AF is associated with specific conditions or comorbidities. Prior history of stroke, transient ischemic attack, or thromboembolism, age, gender, hypertension, diabetes, coronary artery disease, peripheral artery disease, cardiomyopathy, and heart failure are important risk factors. Also, smoking was identified on multivariate analysis in one study as a significant predictor of thromboembolism.36–38 The presence of moderate to severe LV systolic dysfunction on transthoracic echocardiography is the only independent echocardiographic risk factor for stroke on multivariable analysis. On transesophageal echocardiography (TEE), the presence of an LA thrombus (relative risk, 2.5), complex aortic plaques (relative risk, 2.1), spontaneous echo contrast (relative risk, 3.7), and low LA appendage velocities (up to 20 cm/sec; relative risk, 1.7) are independent predictors of stroke and thromboembolism.36 Limited data suggest that large LA appendage dimensions on magnetic resonance imaging (MRI) may predict a higher risk of thromboembolism.39
Several prominent risk stratification schemes have been developed to help distinguish those patients with AF who are at high risk of ischemic stroke and other systemic thromboembolism from those with a risk sufficiently low that anticoagulation may not be beneficial when considering the associated bleeding risks. Two schemes were developed from multivariable analyses of pooled data from randomized trial participants: the Atrial Fibrillation Investigators (AFI) and the Stroke Prevention in Atrial Fibrillation (SPAF) risk schemes. The CHADS2 index, named for a combination of clinical risk factors (Congestive heart failure, Hypertension, Age 75 years or more, Diabetes mellitus, and prior Stroke or transient ischemic attack), was subsequently developed using data from the AFI and SPAF studies and was validated in a retrospective cohort of hospitalized patients with AF. The CHADS2 score assigns two points to a history of prior cerebral ischemia and one point for the presence of congestive heart failure, hypertension, age 75 years or more, or diabetes mellitus. The CHADS2 score has been associated with higher rates of thromboembolic stroke in a linear fashion for patients, both those taking anticoagulants and those not receiving anticoagulation. The stroke rate per 100 patient-years without antithrombotic therapy increases by a factor of approximately 1.5 for each one-point increase in the CHADS2 score: from 1.9% for a score of 0 to 18.2% for a score of 6.40
The CHADS2 score was compared with four other risk stratification schemes to predict thromboembolism in persons with nonvalvular AF. No risk scheme was superior, and all of them had relatively poor ability to predict thromboembolism. Nonetheless, the CHADS2 scheme remains the most widely used because of its simplicity and ease, and it is useful for assessment of when a patient has sufficiently high risk (CHADS2 score of 2 or higher) to warrant anticoagulation. However, the CHADS2 scheme does not incorporate other stroke risk factors and can lead to inadequate discrimination of risk. In fact, many patients (more than 60%) are classified as having intermediate risk, for whom the ideal thromboprophylaxis strategy is uncertain.40
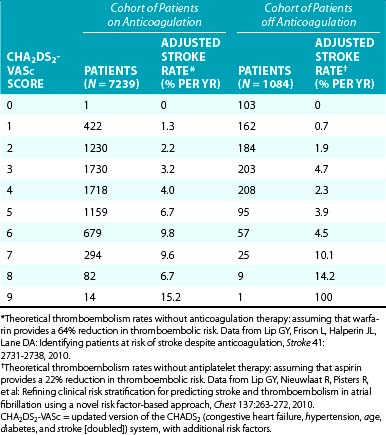
A newer scheme (the CHA2DS2-VASc score or Birmingham 2009 schema) incorporates additional risk factors, including vascular disease, female gender, and age 65 to 74 years (Table 15-2). This scheme showed a modest improvement of thromboembolic risk stratification over the CHADS2 score and classified a much smaller proportion of patients (15.1%) into the intermediate risk category than the original CHADS2 score (61.9%), hence allowing less uncertainty regarding treatment decisions. Also, those in the low-risk category in the CHA2DS2-VASc system were truly at low risk, with a 0% rate of thromboembolic events at 1 year, in contrast to the subjects classified as low risk using the CHADS2 schema (i.e., a score of 0) who still had an annual event rate of 1.4%.38
TABLE 15-2 2009 Birmingham (CHA2DS2-VASc) Scoring System for Predicting Stroke and Thromboembolism in Atrial Fibrillation
| RISK FACTOR | SCORE |
|---|---|
| Congestive heart failure or left ventricular dysfunction | 1 |
| Hypertension | 1 |
| Age ≥75 yr | 2 |
| Diabetes mellitus | 1 |
| Stroke, transient ischemic attack, or thromboembolism | 2 |
| Vascular disease (prior myocardial infarction, peripheral artery disease, or aortic plaque) | 1 |
| Age 65-74 yr | 1 |
| Sex category (i.e., female gender) | 1 |
CHA2DS2-VASc = updated version of the CHADS2 (congestive heart failure, hypertension, age, diabetes, and stroke [doubled]) system, with additional risk factors.
From Lip GY, Nieuwlaat R, Pisters R, et al: Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach, Chest 137:263-272, 2010.
The CHA2DS2-VASc score was validated in a cohort of more than 1000 patients with nonvalvular AF followed during 1 year without anticoagulation, as well as in a large anticoagulated clinical trial cohort (Table 15-3).37,38 The risk of thromboembolism for patients with a CHA2DS2-VASc score of 1 who were not receiving anticoagulation was 0.6% in the first year, but in those with a score of 0, no thromboembolic events were observed. Nonetheless, it is important to recognize that all the available clinical prediction tools have only modest predictive ability.36
In patients with AF who are undergoing implantation of a dual-chamber pacemaker, risk stratification for thromboembolic events can be improved by combining the CHADS2 score with data on the presence and duration of AF episodes detected by the pacemaker. Patients with a CHADS2 score of 2 or less who have no or only very short (less than 5 minutes’ duration) episodes of AF, those with a CHADS2 score of 1 or less who have AF episodes lasting between 5 minutes and 24 hours, and those with a CHADS2 score of 0 regardless of the duration of AF episodes all appear to exhibit a low risk of stroke (0.8% per year).41
Importantly, the assessment of stroke risk is independent of the type of AF (i.e., paroxysmal, persistent or permanent). Although current guidelines consider patients with paroxysmal AF as having a stroke risk similar to those with persistent or permanent AF, in the presence of risk factors, the threshold burden of paroxysmal AF required to justify chronic anticoagulant therapy has not been clearly defined. In patients with AF who have implanted pacemakers or defibrillators, the risk for thromboembolism appears to be quantitatively linked to AF burden as quantified by the implanted devices. An AF burden for more than 5.5 hours on any day in the most recent 30 days seems to confer a doubling of the risk of thromboembolism compared with an AF burden of less than 5.5 hours, even after adjusting for baseline stroke risk factors and antithrombotic therapy. Further research is necessary to identify precisely the amount of AF burden associated with a significantly increased risk of stroke.36,42
It is still unclear how to assess the risk of thromboembolism and guide antithrombotic therapy in patients with implanted pacemakers or defibrillators (but no prior clinical history of AF) in whom atrial high-rate episodes consistent with AT or AF have been detected by the implanted devices. Limited data suggest that any strategy of anticoagulation therapy should take into consideration not only the duration of the atrial arrhythmias but also the individual’s risk factor profile according to the previously mentioned risk stratification schemes.26 Importantly, episodes detected by the device and designated as indicative of AF must be validated because some detection algorithms are overly sensitive and diagnose AF episodes when apparent rapid atrial recordings are, in fact, caused by atrial noncapture during NSR, erroneous counting of far-field R waves, or other phenomena.
Initial Evaluation
The initial evaluation of a patient with suspected or documented AF includes characterizing the pattern of the arrhythmia (e.g., paroxysmal or persistent), determining underlying causes (e.g., heart failure, pulmonary problems, hypertension, or hyperthyroidism), defining associated cardiac and extracardiac conditions, and identifying potential complications of AF. Additionally, a thorough history should be obtained to estimate of the risk of stroke (using the CHADS2 or CHA2DS2-VASc schemes) and quantify AF-related symptoms (e.g., CCS-SAF score). A careful history results in a well-planned focused work-up that serves as an effective guide to therapy.
Exercise testing is often used to assess the adequacy of rate control with exercise in permanent AF, to reproduce exercise-induced AF, and to evaluate for associated ischemic heart disease. Although ischemic heart disease is not a common cause of AF, identifying underlying coronary artery disease is particularly important if the use of a class IC antiarrhythmic drug is being considered. Ambulatory cardiac monitoring can also be required for documentation of AF, its relation to symptoms, and evaluation of the adequacy of heart rate control.27
Principles of Management
Management of AF should be aimed at identifying and treating underlying causes of the arrhythmia, as well as reducing symptoms, improving quality of life, and preventing cardiovascular morbidity and mortality associated with AF. There are two main issues that must be addressed in the treatment of AF: (1) prevention of systemic embolization, and (2) control of symptoms related to AF, typically involving rate or rhythm control. The choice of therapy is influenced by patient preference, associated structural heart disease, severity of symptoms, and whether the AF is recurrent paroxysmal, recurrent persistent, or permanent. In addition, patient education is critical, given the potential morbidity associated with AF and its treatment. For control of symptoms, a safety-driven approach is of paramount importance because most treatments (drug, surgery, ablation) have the capacity to produce significant morbidity and even mortality.27
Prevention of Systemic Embolization
Vitamin K Antagonists
Oral anticoagulation therapy with vitamin K antagonists (warfarin) reduces stroke risk by approximately 64%, corresponding to an absolute annual risk reduction in all strokes of 2.7% as compared with placebo. Warfarin is superior to aspirin, with relative risk reduction of 39% for stroke and 29% for cardiovascular events. However, warfarin increases the risk of major bleeding by approximately 70% compared with aspirin. Although the risk of intracranial hemorrhage is doubled with adjusted-dose warfarin compared with aspirin, the absolute risk increase appears to be small (0.2% per year). Furthermore, randomized clinical trials have shown that warfarin is superior to the combination of aspirin plus clopidogrel for prevention of vascular events in patients with AF at high risk of stroke (relative risk reduction of 40%), with similar risks for major bleeding events. Combinations of warfarin (international normalized ratio [INR], 2.0 to 3.0) with antiplatelet therapy offer no incremental benefit in stroke risk reduction while they increase the risk of bleeding.36,43,44
The reduction in ischemic stroke with warfarin in patients with paroxysmal AF is probably similar to that in patients with persistent or permanent AF. The benefit of warfarin is greatest for patients at higher risk of stroke, and there appears to be little benefit for those with no risk factors. The true efficacy of warfarin is likely to be even higher than suggested by trial results because many of the strokes in the warfarin-treated groups occurred in patients who were noncompliant at the time of the stroke.27 It has been found that if a patient’s INR is not maintained in the therapeutic range at least 65% of the time, the advantage of taking warfarin over aspirin is nullified.
Furthermore, it is estimated that up to 44% of patients with AF have one or more absolute or relative contraindications for long-term oral anticoagulation therapy, most commonly related to increased risk of bleeding. The estimated annual incidence of bleeding associated with oral anticoagulation is 0.6% for fatal bleeding, 3.0% for major bleeding, and 9.6% for major or minor bleeding. The risk of bleeding appears to be especially high during the first year of treatment. Notably, the risk of major bleeding in older patients (at least 80 years of age) receiving warfarin therapy, although higher than younger patients, is acceptably low (2.5% per year), and these patients can still benefit from warfarin prophylaxis when a good quality of anticoagulation is obtained.42 The risk of falling and intracranial bleeding should be considered but not overstated, because a patient may need to fall almost 300 times per year for the risk of intracranial hemorrhage to outweigh the benefit of warfarin therapy in stroke prevention.
Direct Thrombin Inhibitors
Disadvantages of dabigatran include lack of long-term safety data, twice-daily dosing, tolerability issues secondary to dyspepsia, lack of an antidote, and when compared with warfarin, a trend toward increased incidence of myocardial infarction and higher cost. Bleeding with dabigatran remains a hazard and increases over time, out to 2.5 years of follow-up, albeit at a lower rate than with warfarin.
Dabigatran was approved by the U.S. Food and Drug Administration (FDA) for stroke prevention in nonvalvular AF and is likely to improve compliance by obviating the need for dietary restrictions and frequent blood monitoring required for warfarin therapy.45
Factor Xa Inhibitors
Several oral factor Xa inhibitors are being considered. Rivaroxaban was shown to be noninferior to warfarin in stroke prevention in patients with AF and two or more stroke risk factors, and it seems to be associated with a lower risk of intracranial and fatal bleeding.46 In patients for whom vitamin K–antagonist therapy was considered unsuitable, apixaban was found more than 50% superior to aspirin for the prevention of stroke or systemic embolism, with a similar risk of bleeding.47 Also, when compared with warfarin for stroke prevention in patients with AF with one or more additional stroke risk factors, apixaban was found superior to warfarin for stroke and systemic embolism in patients with AF with one or more additional stroke risk factors, thus reducing the risk, with less risk of major bleeding and lower mortality.48
Nonpharmacological Interventions
Given that the LA appendage is the most common site of thrombi in patients with nonvalvular AF, several approaches have targeted exclusion of the LA appendage from the systemic circulation to help prevent systemic thromboembolism. These approaches can potentially become important clinical options in many patients, given the fact that up to 44% of patients with AF have one or more absolute or relative contraindications for chronic oral anticoagulation therapy, most commonly related to increased risk of bleeding.49,50
Open surgical LA appendage amputation, suture ligation, or stapling is commonly performed in patients with AF who are undergoing valvular or coronary artery bypass surgery or as an adjunct to the maze procedure. LA excision appears more efficacious than ligation or stapling, largely because achieving complete LA appendage closure with suture ligation or stapling is quite challenging and operator dependent.50
Additionally, various minimally invasive thoracoscopic LA appendage exclusion techniques (LA appendage excision, stapling, and clipping) have emerged as isolated surgical procedures or in conjunction with minimally invasive maze procedures for ablation of AF. However, data are still limited regarding its efficacy and safety.51
Percutaneous catheter-based closure of the LA appendage using various closure devices (e.g., the Watchman device [Atritech, Plymouth, Minn.], PLAATO implant [ev3 Endovascular, Inc., North Plymouth, Minn.], or Amplatzer cardiac plug [AGA Medical Corporation, Golden Valley, Minn.]) is an emerging approach. The feasibility, safety, and efficacy of these interventions have been demonstrated in experimental and clinical studies; however, further evaluation is required to demonstrate long-term efficacy.49,50
Recommendations for Long-Term Stroke Prevention
The choice of therapy (oral anticoagulation versus aspirin) varies with the estimated thromboembolic risk. Patients with valvular AF (those with mitral stenosis or valvular prosthesis) should be managed with oral anticoagulation. For nonvalvular AF, the CHADS2 (Table 15-4) and CHA2DS2-VASc (see Tables 15-2 and 15-3) scoring systems are currently the best validated and most clinically useful for risk stratification. Patients with a CHADS2 score of 2 or higher are at high risk and should be managed with oral anticoagulation therapy, unless contraindicated. Most patients with AF, however, have a CHADS2 score of less than 2, and a more comprehensive risk factor–based approach, such as the CHA2DS2-VASc scheme, can be used for better risk assessment. Patients with a CHA2DS2-VASc score higher than 1 should be considered for oral anticoagulation, whereas patients with a score of 0 have very low risk and can be managed with aspirin therapy (75 to 325 mg daily). Patients with a score of 1 probably represent an intermediate-risk category and can be managed with either aspirin or oral anticoagulation. When possible, patients at intermediate risk should be considered for oral anticoagulation rather than for aspirin because undertreatment is more harmful than overtreatment. Importantly, those recommendations apply to all patients with AF irrespective of the type of AF.27,38
TABLE 15-4 Stroke Risk in Patients with Nonvalvular Atrial Fibrillation According to CHADS2 Index*
| CHADS2 RISK CRITERIA | POINT(S) | |
|---|---|---|
| Prior stroke or transient ischemic attack | 2 | |
| Age >75 yr | 1 | |
| Hypertension | 1 | |
| Diabetes mellitus | 1 | |
| Heart failure | 1 | |
| PATIENTS (N= 1,733) | ADJUSTED STROKE RATE† (5%/YR) (95% CI) | CHADS2 SCORE |
| 120 | 1.9 (1.2-3.0) | 0 |
| 463 | 2.8 (2.0-3.8) | 1 |
| 523 | 4.0 (3.1-5.1) | 2 |
| 337 | 5.9 (4.6-7.3) | 3 |
| 220 | 8.5 (6.3-11.1) | 4 |
| 65 | 12.5 (8.2-17.5) | 5 |
| 5 | 18.2 (10.5-27.4) | 6 |
CHADS2 = congestive heart failure, hypertension, age, diabetes, and stroke (doubled); CI = confidence interval.
* Not treated with anticoagulation.
† The adjusted stroke rate was derived from multivariate analysis assuming no aspirin usage.
From Gage BF, Waterman AD, Shannon W, et al: Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation, JAMA 285:2864-2870, 2010.
An INR between 2.0 and 3.0 is recommended for most patients with AF who receive warfarin therapy. The risk of stroke doubles when the INR falls to 1.7, and values up to 3.5 do not convey an increased risk of bleeding complications.27 A higher goal (INR between 2.5 and 3.5) is reasonable for patients at particularly high risk for embolization (e.g., prior thromboembolism, rheumatic heart disease, prosthetic heart valves). Similarly, in patients who sustain ischemic stroke or systemic embolism during treatment with therapeutic doses of warfarin (INR, 2.0 to 3.0), raising the intensity of anticoagulation to a higher INR range of 3.0 to 3.5 should be considered. This approach is probably preferable to adding an antiplatelet agent because an appreciable risk in major bleeding is seen with warfarin only when the INR is greater than 3.5 and is likely to be less than that associated with combination therapy.
Oral anticoagulation is associated with increased risk of bleeding; therefore, an assessment of bleeding risk should be part of the patient assessment before starting anticoagulation. At therapeutic INR levels, the rates of intracerebral hemorrhage are typically between 0.1% and 0.6%, with no increment in bleeding risk with INR values between 2.0 and 3.0 compared with lower INR levels. However, an appreciable increase in bleeding risk is observed with higher INR values (more than 3.5 to 4.0). The risk of bleeding should be weighed against the potential benefit of stroke prevention in individual patients considered for anticoagulation therapy. Various bleeding risk scores have been validated for bleeding risk in anticoagulated patients. Among those is the HAS-BLED score, which was found to correlate well with the risk of major bleeding, hospitalization, or death (Table 15-5). Patients are categorized as low, intermediate, and high bleeding risk according to HAS-BLED scores 0 to 1, 2, and 3 or higher, respectively. A score higher than 2 suggests a risk of major bleeding of 1.9% per year, whereas a score of 5 is associated with a risk of major bleeding of up to 12.5% per year.52 One report also proposed a simple 5-variable risk score for quantifying the risk of warfarin-associated hemorrhage, including anemia (3 points), severe renal disease (3 points), age 75 years or more (2 points), prior bleeding (1 point), and hypertension (1 point). Major bleeding annual rates were 0.8% for the low-risk group (0 to 3 points), 2.6% for the intermediate-risk group (4 points), and 5.8% for the high-risk group (5 to 10 points).53
TABLE 15-5 Clinical Characteristics Comprising the HAS-BLED Bleeding Risk Score
| LETTER | CLINICAL CHARACTERISTIC* | POINTS AWARDED |
|---|---|---|
| H | Hypertension | 1 |
| A | Abnormal renal and liver function (1 point each) | 1 or 2 |
| S | Stroke | 1 |
| B | Bleeding | 1 |
| L | Labile INR | 1 |
| E | Older patients (e.g., age >65 years) | 1 |
| D | Drugs or alcohol use (1 point each) | 1 or 2 |
| Maximum 9 points |
INR = international normalized ratio.
* Hypertension is defined as systolic blood pressure higher than160 mmHg. Abnormal kidney function is defined as the presence of long-term dialysis or renal transplantation or serum creatinine concentration of at least 200 µmol/liter. Abnormal liver function is defined as chronic hepatic disease (e.g., cirrhosis) or biochemical evidence of significant hepatic derangement (e.g., bilirubin more than twice the upper limit of normal, in association with aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase more than three times the upper limit of normal). Bleeding refers to previous bleeding history or predisposition to bleeding, or both (e.g., bleeding diathesis, anemia). Labile INRs refers to unstable or high INRs or poor time in therapeutic range (e.g., less than 60%). Drugs or alcohol use refers to concomitant use of drugs (e.g., antiplatelet agents, nonsteroidal antiinflammatory drugs, or alcohol abuse).
From Pisters R, Lane DA, Nieuwlaat R, et al: A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey, Chest 138:1093-1100, 2010.
In high-risk patients who cannot be treated with oral anticoagulation because of poor tolerance or noncompliance issues or because of strong patient preference, dual antiplatelet therapy (aspirin plus clopidogrel) can be used. However, dual antiplatelet therapy is not an alternative to oral anticoagulation in patients at high bleeding risk because the risk of major bleeding associated with dual antiplatelet therapy is broadly similar to that with oral anticoagulation. In the latter group, aspirin monotherapy is associated with lesser bleeding risk, although at the expense of less protection from systemic thromboembolism.36,43,44 Percutaneous LA appendage exclusion procedures may become an important therapeutic alternative to long-term antithrombotic therapy in these patients.36
Anticoagulation in Patients with Coronary Artery Disease
The combined use of antiplatelet and anticoagulant drugs can be required in patients with AF and coronary artery disease. In these patients, it is a common practice to add low-dose aspirin therapy in conjunction with warfarin. Although this strategy appears to be appropriate in patients with acute coronary syndromes, it is important to recognize that in patients with AF with stable coronary, carotid artery disease, or peripheral artery disease, adding aspirin to warfarin does not reduce the risk of stroke, systemic embolism, or myocardial infarction, but it substantially increases the risk of major bleeding (3.9% per year versus 2.3% per year).54
In the absence of randomized trials, antithrombotic therapy should be individualized, taking into account the risks of bleeding, thromboembolism, and stent thrombosis. It appears reasonable to resume anticoagulation as soon as feasible after percutaneous coronary interventions and to use triple therapy (aspirin [75 to 100 mg] plus clopidogrel [75 mg] plus warfarin) in patients with AF with moderate or high stroke risk in the initial period following an acute coronary syndrome (3 to 6 months) and following placement of coronary stents (4 weeks for a bare-metal stent, 6 to 12 months for a drug-eluting stent), especially in patients with low risk of serious bleeding. Afterward, warfarin is continued in addition to a single antiplatelet agent (either aspirin or clopidogrel). During triple therapy, it is advisable to target a lower INR (approximately 2.0), limit the duration of triple therapy to the period required for stent endothelialization, consider prophylactic proton pump inhibition, and follow patients closely to help minimize the high risk of bleeding. Additionally, it is advisable to avoid drug-eluting stents (which are associated with delayed endothelialization and an apparently higher incidence of late stent thrombosis) in patients requiring anticoagulation to reduce the need for prolonged triple therapy.36,54
Anticoagulation in the Pericardioversion Period
Patients without a contraindication to oral anticoagulation who have been in AF for more than 48 hours should receive 3 to 4 weeks of warfarin prior to and after cardioversion. This approach is also recommended for patients with AF who have valvular disease, evidence of LV dysfunction, recent thromboembolism, or AF of unknown duration.27
The rationale for anticoagulation prior to cardioversion is based on observational studies showing that more than 85% of LA thrombi resolve after 4 weeks of warfarin therapy. Thromboembolic events have been reported in 1% to 7% of patients who did not receive anticoagulation before cardioversion. The recommended target INR is 2.0 to 3.0. It has been suggested that it may be prudent to aim for an INR greater than 2.5 before cardioversion to provide the greatest protection against embolic events. Probably more important is to document that the INR has consistently been higher than 2.0 in the weeks before cardioversion.27
After cardioversion, it is recommended to continue warfarin therapy for at least 4 weeks, with a target INR of 2.5 (range, 2.0 to 3.0). This recommendation deals only with protection from embolic events related to the cardioversion period. Subsequently, the long-term recommendations for patients who have been cardioverted to NSR but are at high risk for thromboembolism are similar to those for patients with chronic AF, even though the patients are in NSR.27
A different approach with respect to anticoagulation can be used in low-risk patients (no mitral valve disease, severe LV dysfunction, or history of recent thromboembolism) in whom there is reasonable certainty that AF has been present for less than 48 hours. Such patients have a low risk of clinical thromboembolism if converted early (0.8% in one study), even without screening TEE. The American College of Cardiology/American Heart Association guidelines do not recommend long-term anticoagulation prior to cardioversion in such patients, but they do recommend heparin use at presentation and during the pericardioversion period. The optimal therapy after cardioversion in this group is uncertain. A common practice is to administer aspirin for a first episode of AF that converts spontaneously and warfarin for at least 4 weeks to all other patients. Aspirin should not be considered for patients with AF of less than 48 hours’ duration if there is associated rheumatic mitral valve disease, severe LV dysfunction, or recent thromboembolism. Such patients should be treated the same as patients with AF of longer duration: 3 to 4 weeks of oral anticoagulation with warfarin or shorter term anticoagulation with screening TEE prior to elective electrical or pharmacological cardioversion, followed by prolonged warfarin therapy after cardioversion.27
Rate Control
Adequacy of rate control should be assessed at rest and with exertion. However, parameters for optimal rate control in AF remain controversial. It appears reasonable to target a resting heart rate of 60 to 80 beats/min and 90 to 115 beats/min during moderate exercise. Additionally, ambulatory monitoring can help assess adequacy of rate control; goals of therapy include a 24-hour average heart rate lower than 100 beats/min and no heart rate higher than 100% of the maximum age-adjusted predicted exercise heart rate. Also, a maximum heart rate of 110 beats/min during a 6-minute walk test is a commonly used target.27,47
Nevertheless, one study found that a more lenient rate control (resting heart rate less than 110 beats/min) is not inferior to strict rate control (heart rate less than 80 beats/min at rest and less than 110 beats/min during moderate exercise). Such an approach can be more convenient in clinical practice and may be considered especially in asymptomatic patients with permanent AF and no significant structural heart disease, but periodic monitoring of LV function is necessary to evaluate for the potential risk of tachycardia-mediated cardiomyopathy.47,55 Of note, lenient rate control does not seem to increase the risk of adverse atrial or ventricular remodeling.
In some patients with tachycardia-bradycardia syndrome, pacemaker implantation can be required to protect from severe bradycardia while allowing the use of AV nodal (AVN) blockers for adequate control of fast ventricular rates during AF (see Fig. 8-4).
AVN ablation and permanent pacemaker implantation are highly effective means of improving symptoms in patients with AF who experience symptoms related to a rapid ventricular rate during AF that cannot be adequately controlled with antiarrhythmic or AVN blockers. AV junction ablation is especially useful when excessive ventricular rates induce a tachycardia-mediated decline in LV systolic function, despite appropriate medical therapy. Additionally, in patients with AF and advanced heart failure requiring cardiac resynchronization therapy, AVN ablation can potentially improve long-term survival, NYHA class, and LV systolic function, even in patients with mean ventricular rates lower than 100 beats/min during AF. In these patients, AVN ablation ensures a high percentage of resynchronized QRS complexes and attenuates cardiac output impairment resulting from R-R interval variability.56
Rhythm Control
Restoration and maintenance of sinus rhythm in patients with AF can have several potential benefits, including relief of symptoms, improved functional status and quality of life, prevention of systemic thromboembolism, and avoidance of cardiomyopathy. Reduction in atrial remodeling and retardation of progression of AF, and improvement in LV function also have been described. The impact of rhythm control on mortality, however, remains to be determined.21
Unfortunately, complete restoration of sinus rhythm (100% freedom from AF recurrence) often is unachievable with current drug therapies and remains an impractical treatment goal. It has been estimated that the average 1-year recurrence rate associated with amiodarone approximates 35%, and the recurrence rate for other currently available antiarrhythmic drug therapies is even higher (approximately 50%). However, it is likely that individuals with AF can derive benefit from even partial restoration of sinus rhythm, including improvements in symptoms and attenuation of electrical and structural atrial remodeling associated with AF and hence retardation of progression of AF.21
Reversion to Normal Sinus Rhythm
When rhythm control is chosen, both electrical and pharmacological cardioversion methods are appropriate options. The timing of attempted cardioversion is influenced by the duration of AF. As noted, in patients with AF of a duration longer than 48 hours or unknown duration, cardioversion should be delayed until the patient has been anticoagulated at appropriate levels for 3 to 4 weeks or TEE has excluded atrial thrombi.27
Electrical cardioversion is indicated for patients with rapid ventricular rates not responding promptly to medical therapy and ongoing myocardial ischemia, hypotension, acute heart failure, or ventricular preexcitation. In stable patients in whom spontaneous reversion caused by correction of an underlying condition is not likely, electrical or pharmacological cardioversion can be performed. Electrical cardioversion is usually preferred because of greater efficacy and a low risk of proarrhythmia; however, it requires conscious sedation or anesthesia. Importantly, electrical cardioversion is contraindicated in patients with ongoing toxic reactions from digitalis or patients with hypokalemia.57
The overall success rate of electrical cardioversion (at any level of energy) for AF is 75% to 93% and is related inversely to the duration of AF and LA size. Occasionally, electrical cardioversion can fail to terminate AF, or AF recurs shortly after transient restoration of sinus rhythm. When AF fails to terminate, using higher energy levels, applying external pressure on the cardioversion patches, and performing cardioversion during exhalation can improve effectiveness. When AF recurs early after a successful cardioversion, pretreatment with amiodarone, dofetilide, flecainide, ibutilide, propafenone, or sotalol can facilitate successful electrical cardioversion.57,58 It is important to distinguish failure to terminate AF with a certain shock energy (the solution for which is higher energy shock) from successful termination of AF with nearly immediate recurrence (for which repeated shocks at any energy are unlikely to have greater benefit). Sinus pauses or transient sinus arrest after successful cardioversion are common; rarely, several-second pauses result from sinus node dysfunction and the effect of sedation. Almost all external defibrillators have the capability of back-up bradycardia pacing through the defibrillation patches, which can be used transiently if needed. The need for more prolonged pacing after cardioversion is rare.
Certain antiarrhythmic drugs have been found to be more effective than placebo for chemical cardioversion of AF, by converting 30% to 60% of patients. Evidence of efficacy from randomized trials has been best established for dofetilide, ibutilide, flecainide, propafenone, amiodarone, and quinidine. The benefit from specific antiarrhythmic drugs varies with the duration of AF: dofetilide, flecainide, ibutilide, propafenone or, to a lesser degree, amiodarone if less than 7 days’ duration; and dofetilide or, to a lesser degree, amiodarone or ibutilide if AF is more prolonged. Flecainide and propafenone should be avoided in patients with abnormal LV function and ischemia.57
Rate control with an AVN blocker (beta blockers, diltiazem, verapamil, digoxin) should be attained before instituting a class IA or IC drug because of possible conversion to AFL and a very rapid ventricular rate.27 Importantly, continuous cardiac monitoring is required (to detect sinus node dysfunction, AV block, ventricular arrhythmias, and conversion into AFL) for an interval that is dependent on the agent used (usually approximately half the drug elimination half-life). Once the safety of pharmacological conversion with propafenone or flecainide has been established, repeat patient-administered cardioversion using oral propafenone (450 to 600 mg) or flecainide (200 to 300 mg) may be appropriate on an outpatient basis (“pill-in-the-pocket” approach).36,57
Maintenance of Normal Sinus Rhythm
Only 20% to 30% of patients who are successfully cardioverted maintain NSR for more than 1 year without chronic antiarrhythmic therapy. This is more likely to occur in patients with AF for less than 1 year, no enlargement of the LA (less than 4.0 cm), and a reversible cause of AF, such as hyperthyroidism, pericarditis, pulmonary embolism, or cardiac surgery. It has been thought that the drugs most likely to maintain NSR suppress triggering ectopic beats and arrhythmias and affect atrial EP properties to diminish the likelihood of AF. There is therefore a strong rationale for prophylactic antiarrhythmic drug therapy in patients who have a moderate to high risk of recurrence, provided that the therapy is effective and that toxic and proarrhythmic effects are low. Prophylactic drug treatment is seldom indicated in patients with a first-detected episode of AF and can also be avoided in patients with infrequent and well-tolerated paroxysmal AF.27
Evidence of efficacy from randomized trials is best for amiodarone, propafenone, disopyramide, sotalol, flecainide, dofetilide, dronedarone, and quinidine. Amiodarone is the most effective agent but is associated with substantial noncardiac toxic effects. The relative efficacy of the other antiarrhythmic drugs for the management of AF remains unknown. Drug selection is largely driven by the safety profile. The presence and extent of concomitant cardiovascular disease have to be carefully considered. A safer, although possibly less efficacious, drug is usually recommended before resorting to more effective but less safe therapies (Fig. 15-5).36
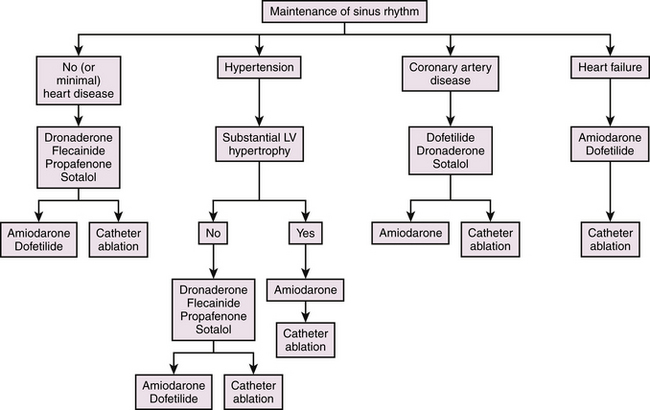
In patients with AF with and minimal or no heart disease (lone AF), flecainide, propafenone, sotalol, and dronedarone are preferred. In patients with adrenergically mediated AF, beta blockers represent first-line treatment, followed by sotalol. The anticholinergic activity of long-acting disopyramide makes it a relatively attractive choice for patients with vagally mediated AF. In contrast, propafenone is not recommended in vagally mediated AF because its (weak) intrinsic beta-blocking activity may aggravate this type of paroxysmal AF. In patients with lone AF, amiodarone should be chosen later in the sequence of drug therapy because of its potential toxicity.36
In patients with substantial LV hypertrophy (LV wall thickness greater than 1.4 cm), sotalol is thought to be associated with an increased incidence of proarrhythmia. It is also reasonable to avoid flecainide and propafenone because of concern of increased proarrhythmic risk. Dronedarone, although not specifically tested in this population, is likely to be safe. Amiodarone is usually considered when symptomatic AF recurrences continue to affect the quality of life of these patients.27,36
Flecainide and propafenone are contraindicated in patients with coronary artery disease. Sotalol, dofetilide, or dronedarone are recommended as first-line therapy. Amiodarone is considered the drug of last resort in this population because of its potential toxicity.36
Dronedarone, dofetilide, and amiodarone are the only agents available for patients with AF with concomitant heart failure; other antiarrhythmic agents can be associated with substantial toxicity and proarrhythmia. Dronedarone is contraindicated in patients with NYHA Class III to IV or recently (within the previous month) decompensated heart failure, especially when the LV ejection fraction is 35% or lower. In such patients, dofetilide and amiodarone are preferred. Importantly, concerning results emerged from a study (the Permanent Atrial fibriLLAtion Outcome Study Using Dronedarone on Top of Standard Therapy [PALLAS]) conducted to assess the potential clinical benefit of dronedarone in patients more than 65 years of age with permanent AF in the reduction of major cardiovascular events (stroke, systemic arterial embolism, myocardial infarction, or cardiovascular death), or unplanned cardiovascular hospitalization or death from any cause. The study was stopped early because of a twofold increase in death, as well as twofold increases in stroke and hospitalization for heart failure in patients receiving dronedarone compared with patients taking a placebo. These data currently are being reviewed by the FDA to determine whether those results apply to patients who use dronedarone for the approved indications in nonpermanent (paroxysmal or persistent) AF.
Although amiodarone is associated with organ toxicity, it appears to have a lower risk of proarrhythmia than other agents. Class IC agents can be associated with substantial toxicity in patients with heart failure because of their proarrhythmic and negative inotropic effects.36
When treatment with a single drug fails, combinations of antiarrhythmic drugs can be tried. Useful combinations include a beta blocker, sotalol, or amiodarone, in addition to a class IC agent.27 When AF recurrences are infrequent and tolerated, patients experiencing breakthrough arrhythmias may not require a change in antiarrhythmic drug therapy.
Upstream Therapy
Given the limited effectiveness and marked risks of serious complications associated with currently available antiarrhythmic drug therapy, the role of therapies targeting the atrial substrate underlying AF (“upstream therapy”) is of particular clinical interest. The goal of this approach is attenuation and reversal of atrial structural remodeling. Given the role of fibrosis and inflammation in the pathogenesis of AF, drugs that suppress fibrosis, as well as antiinflammatory and antioxidative drugs, are being investigated both alone and in combination with traditional antiarrhythmic drugs. Among these drugs are several angiotensin-converting enzyme inhibitors, angiotensin II type 1 receptor blockers, antialdosterone agents, statins (3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors), and omega-3 poly-unsaturated fatty acids. These agents seem to reduce atrial fibrosis and were found potentially to reduce structural remodeling and AF susceptibility in various AF models. Nonetheless, current evidence does not support the routine use of these agents for the sole purpose of preventing or treating AF.13,16,59,60
Rhythm Control Versus Rate Control
In the past, many physicians preferred rhythm control to rate control. Reversion of AF and maintenance of NSR restores normal hemodynamics and had been thought to reduce the frequency of embolism. However, two major clinical trials—AFFIRM (Atrial Fibrillation Follow-Up Investigation of Rhythm Management) and RACE (RAte Control versus Electrical Cardioversion for Persistent Atrial Fibrillation)— compared rhythm and rate control and found that embolic events occurred with equal frequency, regardless of whether a rate control or rhythm control strategy was pursued, and this occurred most often after warfarin had been stopped or when the INR was subtherapeutic. Both studies also showed an almost significant trend toward a lower incidence of the primary endpoint with rate control. There was no difference in the functional status or quality of life. Thus, both rhythm control and rate control are acceptable approaches, and both require long-term anticoagulation for stroke prevention. The trials provided supportive evidence for the rate control option in most patients. The AF-Congestive Heart Failure (AF-CHF) study demonstrated similar results among patients with AF with concomitant heart failure.61
However, it would be incorrect to extrapolate that NSR offers no benefit over AF and that effective treatments to maintain NSR need not be pursued. First, these trials were not comparisons of NSR and AF. They compared a rate control strategy to a rhythm control strategy that attempted to maintain NSR but fell short. The failure of AFFIRM and RACE in showing any difference between rate and rhythm control is not so much a positive statement for rate control but rather a testimony to the ineffectiveness of the rhythm control methods used. Failure of rhythm control to demonstrate superiority over rate control is caused by failure to achieve maintenance of NSR over the long term. Several trials demonstrated that the success of antiarrhythmic therapy in maintaining NSR is borderline, at best, with increasing failure rates over time. When the data from these trials were analyzed according to the patient’s actual rhythm (as opposed to his or her treatment strategy), the benefit of NSR over AF became apparent: the presence of NSR was found to be one of the most powerful independent predictors of survival, along with the use of warfarin, even after adjustment for all other relevant clinical variables. Patients in NSR are almost half as likely to die compared with those with AF. This benefit, however, is offset by the use of antiarrhythmic drug therapy, which increases the risk of death.21,61,62
Achieving and maintaining sinus rhythm remain viable and important treatment goals. However, because currently available antiarrhythmic agents commonly fail to suppress AF completely and have safety profiles that are less than ideal, it is reasonable to reserve it to the populations of patients likely to derive the greatest benefit from rhythm control. The selection of appropriate rhythm control or rate control strategies should be individualized and take into consideration the nature, intensity, and frequency of symptoms, patient preferences, comorbid conditions, and the risk of recurrent AF. According to analyses of available data, these groups may include young patients and those with newly diagnosed AF, significant symptoms, poorly controlled ventricular response, or tachycardia-mediated cardiomyopathy. On the other hand, asymptomatic or mildly symptomatic patients, especially those older than 65 years, and women with persistent AF who have hypertension or other underlying heart diseases may be better suited for rate control therapy.21,61,62
Nonpharmacological Approaches for Rhythm Control
Catheter Ablation of Atrial Fibrillation
Several studies have found catheter ablation superior to medical therapy for the control of AF. In a meta-analysis of 6 randomized, controlled trials with a total of 693 patients with AF that compared catheter ablation (PV isolation) and medical therapy, the efficacy of PV isolation for the maintenance of NSR was more than twofold greater than that of antiarrhythmic drug therapy (77% versus 29% at 12 months of follow-up). The benefit of PV isolation was even greater in patients with paroxysmal AF. Moreover, catheter ablation was associated with a two-thirds reduction in hospitalization for cardiovascular causes, with a relatively small (2.6%) risk of major procedural complications.25 Nonetheless, in the absence of large, well-designed, multicenter randomized trials of catheter ablation as a first-line therapy for AF in symptomatic or asymptomatic patients, a history of symptoms, despite therapeutic doses of antiarrhythmic medication (type IA, IC, or III), is widely accepted as the minimum qualifying criterion for catheter ablation. With improved efficacy of ablation techniques, however, the threshold for ablation will continue to fall.
Catheter ablation was also found to be associated with better quality of life, higher rates of freedom from both AF and antiarrhythmic medications, and lower rates of AF progression when compared with AVN ablation and biventricular pacing in symptomatic patients with AF and cardiomyopathy (LV ejection fraction 40% or less).63
Surgical Ablation of Atrial Fibrillation
The classic Cox-maze procedure involves creating a series of incisions in both the left and right atria that were originally designed to direct the propagation of the sinus impulse through both atria while interrupting the multiple macroreentrant circuits thought to be responsible for AF. This procedure is the most effective means of curing this arrhythmia, eliminating AF in 75% to 95% up to 15 years after surgery. Improvements and simplifications culminated in the Cox-maze III procedure, which became the gold standard for the surgical treatment of AF. Nonetheless, because of its complexity, technical difficulty, and risk of mortality and other complications, the maze procedure did not gain widespread acceptance.64
To simplify the procedure, the standard cut-and-sew surgical technique has been replaced with linear epicardial ablation using unipolar or bipolar RF ablation, cryoablation, laser, high-frequency ultrasound, and microwave energy. Most surgical epicardial ablation procedures have been performed in conjunction with mitral valve surgery; the combination of mitral valve repair and cure of AF can enable selected patients to avoid life-long anticoagulation.64,65
Current surgical instrumentation now enables minimally invasive approaches to be performed epicardially on the beating heart through mini-thoracotomies with video assistance. However, despite elimination of the need for median sternotomy and cardiopulmonary bypass, these procedures are still relatively invasive. To minimize the invasiveness of the procedure further, a totally thoracoscopic approach has been developed. Bipolar RF is the predominant energy source used, and bilateral PV isolation is the most common lesion set, with some approaches adding ganglionic plexus ablation, as well as exclusion of the LA appendage. Although multiple series described high success rates for paroxysmal AF (89% at 12 months of follow-up), success has been limited in patients with persistent and longstanding persistent AF (25% to 87%).64,65 Limited follow-up has left uncertainty about overall results.
More extensive ablation lines, in addition to PV antral isolation and ganglionic plexus ablation, as well as documentation of complete PV isolation by demonstration of EP entrance or exit block, conduction block across ablation lines, and the detection and confirmation of ablation of the parasympathetic component of the ganglionic plexuses, are being evaluated to improve outcome. Some series reported a single procedure success rate of 86% at 1 year without the use of antiarrhythmic drugs.66,67
Currently, stand-alone surgical epicardial AF ablation may be considered for symptomatic patients with AF who were refractory to one or more attempts at catheter ablation or who are not candidates for catheter ablation (e.g., patients not candidates for long-term anticoagulation and those with an LA thrombus). There have been no randomized studies comparing surgical and catheter ablation of AF. Hence, the decision to recommend surgical AF ablation before considering catheter ablation for patients with symptomatic AF refractory to drug therapy and no other indication for cardiac surgery remains controversial, and it should be based on each institution’s experience with both techniques, the relative outcomes and risks of each in the individual patient, and patient preference.64,65 Given the degree of patient discomfort, longer hospitalizations and recovery times, and the risk of bleeding following LA appendage excision, most patients prefer catheter to surgical ablation.68
Electrocardiographic Features
Atrial Activity
AF is characterized by rapid and irregular atrial fibrillatory waves (f waves) and a lack of clearly defined P waves, with an undulating baseline that may alternate between recognizable atrial activity and a nearly flat line (Fig. 15-6). Atrial fibrillatory activity is generally best seen in lead V1 and in the inferior leads. Less often, the f waves are most prominent in leads I and aVL.
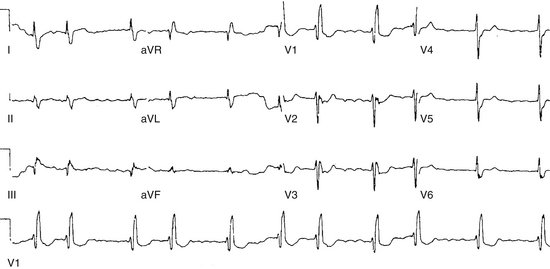
The rate of the fibrillatory waves is generally between 350 and 600 beats/min. With up to 600 pulses generated every minute, syncytial contraction of the atria is replaced by irregular atrial twitches. Therefore, the fibrillating atria look like a bag of worms in that the contractions are very rapid and irregular. The f waves vary in amplitude, morphology, and intervals, thus reflecting the multiple potential types of atrial activation that may be present at the same time at different locations throughout the atria. The f waves can be fine (amplitude less than 0.5 mm on ECG) or coarse (amplitude more than 0.5 mm). On occasion, the f waves can be inapparent on the standard and precordial leads, which is most likely to occur in permanent AF. It was initially thought that the amplitude of the f waves correlated with increasing atrial size; however, echocardiographic studies have failed to show a correlation among the amplitude of the f waves, the size of the atria, and the type of heart disease. The amplitude, however, may correlate with the duration of AF.
AF should be distinguished from other rhythms in which the R-R intervals are irregularly irregular. These include multifocal AT (see Fig. 11-1), wandering atrial pacemaker, multifocal PACs, and AT or AFL with varying AV block (see Fig. 12-4). In general, distinct (although often abnormal and possibly variable) P (or flutter) waves are present during these arrhythmias, in contrast to AF. Patients with rheumatic mitral stenosis often demonstrate large-amplitude fibrillatory waves in the anterior precordial leads (V1 and V2), which can be confused with AFL. However, careful examination of the fibrillatory waves reveals them to have a varying CL and morphology. The distinction between AF and AFL can also be confusing in patients who demonstrate a transition between these arrhythmias. Thus, AF may organize to AFL or AFL may degenerate to AF (Fig. 15-7). Occasionally, extracardiac artifacts (e.g., 60 cycle/min muscle tremors, as in parkinsonism) can mimic f waves.
Atrioventricular Conduction during Atrial Fibrillation
The ventricular response in AF is typically irregularly irregular, and the ventricular rate depends on multiple factors, including the EP properties of the AVN, the rate and organization of atrial inputs to the AVN, the level of autonomic tone, the effects of medications that act on the AV conduction ystem, and the presence of preexcitation over an AV BT.
The ventricular rate in untreated patients usually ranges from 90 up to 170 beats/min. Ventricular rates that are clearly outside this range suggest some concurrent problem. Ventricular rates slower than 60 beats/min are seen with AVN disease and can be associated with the sick sinus syndrome, drugs that affect conduction, and high vagal tone, as can occur in a well-conditioned athlete. The ventricular rate in AF can become rapid (more than 200 beats/min) during exercise, with catecholamine excess (Fig. 15-8), parasympathetic withdrawal, thyrotoxicosis, or preexcitation (Fig. 15-9). The ventricular rate can be very rapid (more than 300 beats/min) in patients with the Wolff-Parkinson-White syndrome, with conduction over AV BTs having short anterograde refractory periods.
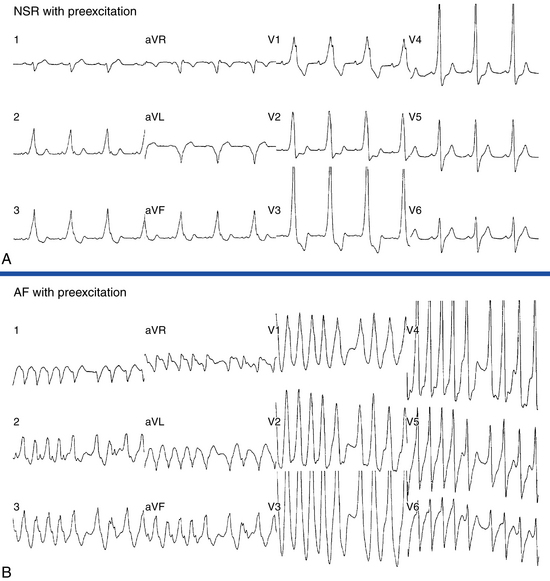
Preexcitation During Atrial Fibrillation
The presence of a grossly irregular, very rapid ventricular response (more than 250 beats/min) with QRS longer than 120-millisecond duration during AF rarely results from conduction over the AVN and strongly implies conduction over an AV BT, with rare exceptions (see Fig. 15-9). At very fast heart rates, there is usually a tendency toward regularization of the R-R intervals; therefore, distinguishing the preexcited AF from ventricular tachycardia (VT) or preexcited SVT can be difficult. However, careful measurement always discloses definite irregularities. Moreover, very rapid and irregular VTs are usually unstable and quickly degenerate into VF. Thus, when a rapid, irregular wide QRS complex tachycardia is noted in a patient who has a reasonably stable hemodynamic state, preexcited AF is the most likely diagnosis.
Regular Ventricular Rate During Atrial Fibrillation
Regular ventricular rate during AF indicates associated abnormalities. A regular, slow ventricular rhythm during AF suggests a junctional, subjunctional, or ventricular rhythm, either as an escape mechanism with complete AV block or as an accelerated pacemaker activity with AV dissociation (see Fig. 9-27). Rarely, the R-R interval can be regularly irregular and show group beating with the combination of complete heart block and a lower nodal pacemaker with a Wenckebach type of exit block. Patients with severe underlying heart disease may develop the combination of AF and VT, leading to a rapid, regular, wide QRS complex tachycardia.
Effect of Digitalis Toxicity on the Ventricular Response
With increasing degrees of digitalis toxicity, high-grade but not complete AV block during AF initially leads to single junctional, subjunctional, or ventricular escape beats. Higher degrees of AV block result in so few atrial impulses being conducted that the lower pacemaker takes over, thus leading to an escape junctional, subjunctional, or ventricular rhythm with a regular R-R interval for two or more cycles. On occasion, the junctional rate can increase, possibly because of digitalis-induced triggered activity, and it is called nonparoxysmal junctional tachycardia. Increasing digitalis toxicity can result in a Wenckebach exit block and give the appearance of an irregular ventricular rhythm with restoration of AF conduction, but the rhythm shows repetitive group beating as a result of the exit block. Complete AV block is marked by a regular escape rhythm with no conducted beats, a finding that may lead to the erroneous assumption that the patient has converted to NSR. Infrequently, impulses from the lower pacemaker travel alternately down the right and left bundle branches or alternate fascicles of the left bundle branch, resulting in a bidirectional tachycardia. This arrhythmia, which is also frequently a reflection of marked digitalis toxicity, may appear to be ventricular bigeminy. In true bigeminy, however, the ventricular beat in the bigeminal pattern is premature. In comparison, the R-R interval is regular with a bidirectional tachycardia, because all the beats arise from a single pacemaker.
QRS Morphology
The QRS complexes during AF are narrow and normal unless AV conduction is abnormal because of functional (rate-related) aberration, preexisting bundle branch block (BBB; see Fig. 15-6), or preexcitation over an AV BT (see Fig. 15-9).
The aberrancy caused by the Ashman phenomenon can be present for one beat and have a morphology that resembles a premature ventricular complex (PVC), or it can involve several sequential complexes, suggesting VT. The persistence of aberrancy may reflect a time-dependent adjustment of refractoriness of the bundle branch to an abrupt change in CL, or it may be the result of concealed transseptal activation.
The proper diagnosis of aberrant conduction is a continuing challenge, but it can usually be accomplished by careful analysis of the rhythm strip and application of certain criteria. An aberrantly conducted beat caused by BBB generally has the pattern of a classic bundle branch or fascicular block. A PVC is usually followed by a longer R-R cycle, indicating the occurrence of a compensatory pause, the result of retrograde conduction into the AVN and anterograde block of the impulse originating in the atrium. The presence of long R-R cycles after the wide QRS complex that have identical CLs also suggests a ventricular origin (see Fig. 10-3). Furthermore, the absence of a long-short cycle sequence associated with the wide or aberrant QRS complex suggests that it is of ventricular origin. Aberrancy is not present if, with inspection of a long ECG rhythm strip, there are R-R interval combinations that are longer and shorter than those associated with the wide QRS complex. Also, a ventricular origin is likely if there is a fixed coupling interval between the normal and wide QRS complexes (Fig. 15-10).