Asthma
Chapter 4 discussed the normal structure of airways and considered several aspects of airway function. The most common disorders disrupting the normal structure and function of the airways—asthma and chronic obstructive pulmonary disease—are discussed here and in Chapter 6, respectively. Several other miscellaneous diseases affecting airways are covered in Chapter 7.
Etiology and Pathogenesis
Airway Inflammation and Bronchial Hyperresponsiveness
No single factor or cell appears to be responsible for asthma; rather, a complex and interrelated series of events, including cellular infiltration, cytokine release, and airway remodeling, likely culminates in airway hyperresponsiveness and episodes of airflow obstruction (Fig. 5-1). A variety of mediators released from inflammatory cells can alter the extracellular milieu of bronchial smooth muscle, increasing its responsiveness to bronchoconstrictive stimuli. Mediators that have been proposed to play such a role include prostaglandin and leukotriene products of arachidonic acid metabolism. Some cytokine mediators released from inflammatory cells have various effects on other inflammatory cells, thus perpetuating the inflammatory response. For example, lymphocytes of the TH2 phenotype, which are thought to be a prominent component of the inflammatory response in asthma, release interleukin (IL)-5, which has a chemoattractant effect for eosinophils. IL-5 also stimulates growth, activation, and degranulation of eosinophils. IL-4, another cytokine released from TH2 lymphocytes, exerts a different type of proinflammatory effect by activating B lymphocytes, enhancing synthesis of IgE and promoting differentiation of TH2 cells.
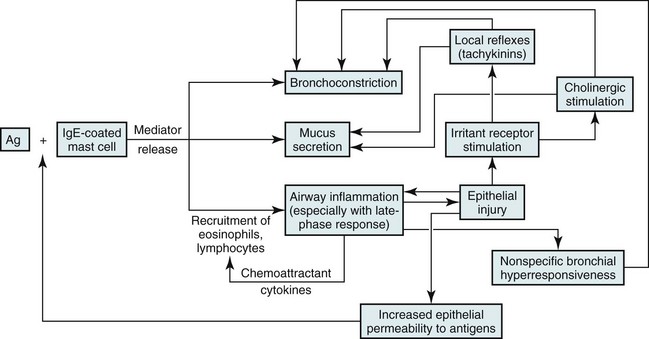
Mediators released from inflammatory cells may produce tissue damage that contributes to asthma pathogenesis. For example, when eosinophils degranulate, they release several toxic proteins from their granules, such as major basic protein and eosinophil cationic protein. These and other eosinophil products may contribute to the epithelial damage found in the asthmatic airway. Once the epithelium is injured or denuded, its barrier function is disrupted, allowing access of inhaled material to deeper layers of the mucosa. Additionally, the epithelial cells themselves may become actively involved in amplifying the inflammatory process (through production of cytokine and chemokine mediators) and in perpetuating airway edema (through vasodilation mediated by release of nitric oxide, leukotrienes, and prostaglandins). Finally, sensory nerve endings in the airway epithelial layer may become exposed, triggering a reflex arc and release of tachykinin mediators (e.g., substance P, neurokinin A), as shown in pathway 4 of Figure 4-3. These peptide mediators, released at bronchial smooth muscle, submucosal glands, and blood vessels, are capable of causing bronchoconstriction and airway edema.
Common Provocative Stimuli
Allergen Exposure
The pathogenetic mechanisms leading to bronchoconstriction are best defined for allergen-induced asthma. Allergens to which an asthmatic person may be sensitized are widespread throughout nature. Although patients and clinicians often first consider seasonal outdoor allergens such as pollen, many indoor allergens may play a more critical role. These allergens include antigens from house dust mites (Dermatophagoides and others), domestic animals, and cockroaches. Inhaled antigens are initially identified and processed by antigen-presenting cells called dendritic cells, which in turn present the antigenic material to T lymphocytes. Chemicals released by TH2 cells, especially IL-4 and IL-13, signal B lymphocytes to produce antigen-specific IgE antibodies. When an asthmatic person has IgE antibody against a particular antigen, the antibody binds to high-affinity IgE receptors on the surface of tissue mast cells and circulating basophils (see Fig. 5-1). If that particular antigen is inhaled, it binds to and cross-links IgE antibody (against the antigen) bound to the surface of mast cells in the bronchial lumen. The mast cell then is activated, leading to release of both preformed and newly synthesized mediators. Mediators released from the mast cell induce bronchoconstriction and increase airway epithelial permeability, allowing the antigen access to the much larger population of specific IgE-containing mast cells deeper within the epithelium. Binding of antigen to antibody on this larger population of mast cells again initiates a sequence of events leading to release of chemical mediators capable of inducing bronchoconstriction and inflammation. Several mediators have been recognized (Table 5-1), but the discussion here is limited to the few that have been primarily implicated in the pathogenesis of allergic asthma; major mediators include histamine and leukotrienes.
Table 5-1
Histamine: This relatively small (molecular weight 111) compound is found preformed within the mast cell and is released on exposure to the appropriate antigen. Histamine has several effects that may be important in asthma, including contraction of bronchial smooth muscle, augmentation of vascular permeability with formation of airway edema, and stimulation of irritant receptors (that can trigger a reflex neurogenic pathway via the vagus nerve, causing secondary bronchoconstriction). Despite these varied effects, the fact that the clinical manifestations of asthma do not respond to antihistamines suggests that histamine is not the most important chemical mediator involved.
Leukotrienes: The leukotrienes include a series of compounds (LTC4, LTD4, and LTE4) that formerly were called slow-reacting substance of anaphylaxis (SRS-A). Unlike histamine, leukotrienes are not preformed in the mast cell but synthesized after antigen exposure and then released. To some extent, their actions are similar to those of histamine; they also have a direct bronchoconstrictor action on smooth muscle, increase vascular permeability, and stimulate excess production of airway mucus. Leukotrienes are synthesized from arachidonic acid (also the precursor for prostaglandins) but along a different pathway involving a lipoxygenase enzyme, as opposed to the cyclooxygenase enzyme used for prostaglandin synthesis (Fig. 5-2). LTC4 and LTD4 in particular are extraordinarily potent bronchoconstrictors and may have an important role in the pathogenesis of bronchial asthma. An interesting sidelight is provided by knowledge that some persons with asthma experience exacerbations of their disease after taking aspirin or other nonsteroidal antiinflammatory drugs (NSAIDs). These drugs are known inhibitors of the cyclooxygenase enzyme and may result in preferential shifting of the pathway shown in Figure 5-2 toward production of the bronchoconstrictor leukotrienes.
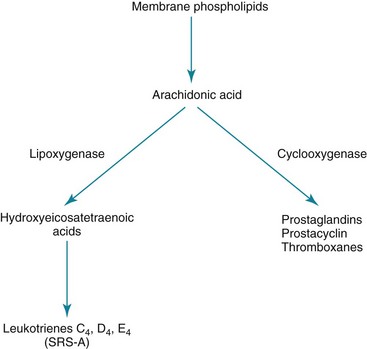
The role of other mediators listed in Table 5-1 in asthma pathogenesis is less clear. Platelet-activating factor has been proposed to play a role in recruiting eosinophils to the lung, and platelet-activating factor activates eosinophils, stimulating them to release proteins toxic to airway epithelial cells.
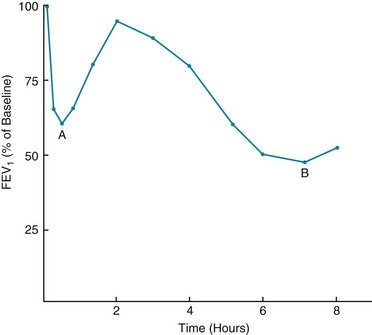
Late-Phase Asthmatic Response: The airway response to antigen challenge, as measured by changes in forced expiratory volume in 1 second (FEV1), appears to be more complicated and involves more than just the rapid mediator-induced bronchoconstriction seen within the first half hour following exposure. In many patients, the return of FEV1 to normal is followed by a secondary delayed fall in FEV1 occurring hours after antigen exposure (Fig. 5-3). This delayed fall in FEV1 is accompanied histologically by inflammatory changes in the airway wall. At the same time, increased bronchial hyperresponsiveness to nonspecific stimuli, such as histamine or methacholine, can be demonstrated and can last for days.
Pathology
1. Edema and cellular infiltrates within the bronchial wall, especially with eosinophils and lymphocytes
2. Epithelial damage, with a “fragile” appearance of the epithelium and detachment of surface epithelial cells from basal cells
3. Hypertrophy and hyperplasia of the smooth muscle layer
4. Increased deposition of collagen in a layer beneath the epithelium (referred to in the past as basement membrane thickening)
5. Enlargement of the mucus-secreting apparatus, with hypertrophy of mucous glands and an increased number of goblet cells
Pathophysiology
FRC, the resting point of the lungs after a normal expiration, may be increased for at least two reasons. First, because more time is required for expiration when airways are obstructed, patients may not have sufficient time before the next breath to fully exhale the volume from the previous breath. This phenomenon, sometimes called dynamic hyperinflation, is a particular problem when the asthmatic person is breathing at a rapid respiratory rate. Another reason for increase in FRC is related to persistent activity of the inspiratory muscles during expiration, maintaining lung volume at a level higher than expected throughout expiration. A physiologic advantage to breathing at a higher-than-normal FRC is having airways held open at a greater diameter. A disadvantage is increased work of breathing due to reduced compliance of the respiratory system at higher lung volumes (see Fig. 1-3, C) and a mechanical disadvantage for diaphragmatic function when the diaphragm is lower and flatter (see Mechanisms of Abnormal Gas Exchange in Chapter 6).
The increased resistance to airflow in asthma exerts a toll on gas exchange, which is generally disturbed during acute attacks. The most common pattern of arterial blood gases consists of a low PO2 accompanied by a low PCO2 (respiratory alkalosis). The mechanism for the hypoxemia is ventilation-perfusion mismatch. The increased airway resistance in asthma is not evenly distributed, such that some airways are affected more than others. Therefore, inspired air is not distributed evenly but tends to go to less diseased areas. However, blood flow remains relatively preserved in the regions that are ventilating poorly. The regions of low ventilation-perfusion (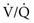 ) ratio contribute blood with a low PO2 that cannot be compensated for by increases in the
) ratio contribute blood with a low PO2 that cannot be compensated for by increases in the 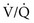 ratio from other regions of the lung (see Chapter 1).
ratio from other regions of the lung (see Chapter 1).
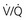 mismatch) and low P
mismatch) and low PClinical Features
The overall severity of an individual’s asthma can be characterized on the basis of the frequency of exacerbations, nocturnal symptoms, and magnitude of abnormality and variability in pulmonary function. The features used to define four categories of severity (intermittent asthma, mild persistent asthma, moderate persistent asthma, and severe persistent asthma) are listed in Table 5-2.
Table 5-2
CLASSIFICATION OF ASTHMA BY SEVERITY: CLINICAL ASPECTS AND TREATMENT
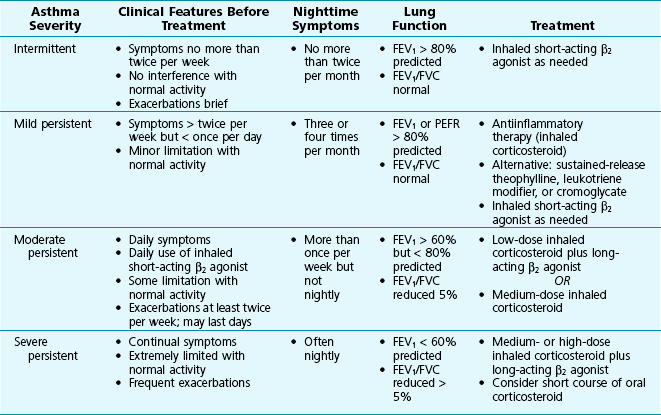
FEV1 = Forced expiratory volume in 1 second; PEFR = peak expiratory flow rate.
Adapted from National Asthma Education and Prevention Program: Guidelines for the diagnosis and management of asthma: Expert panel report 3, Bethesda, MD, 2007, National Institutes of Health, NIH publication 08-4051.
Treatment
The major categories of drugs used to treat asthma are those that dilate smooth muscle of the bronchial wall and those that have an antiinflammatory action. Agents targeted at blocking production or activity of specific mediators are also used, and a monoclonal antibody against IgE has become available. The main categories of drugs used to treat asthma are listed in Table 5-3. Several of the drugs are also used for treatment of other types of pulmonary disease, particularly chronic obstructive pulmonary disease, and are mentioned in other chapters.
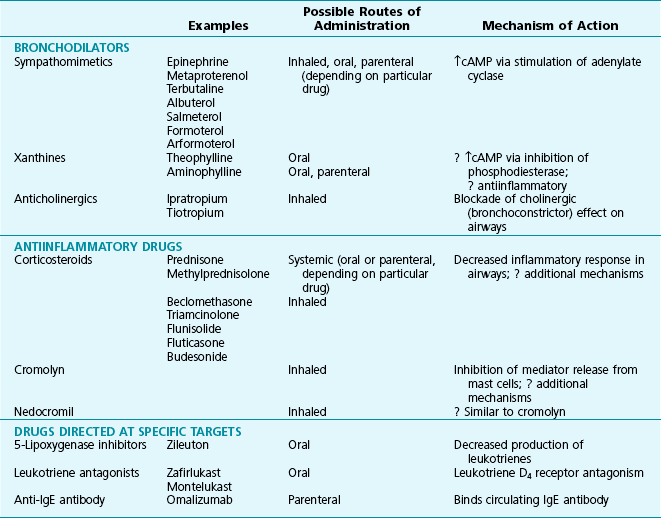
Bronchodilators
The most common bronchodilator agents used for treatment of asthma are the sympathomimetic agents, which act on β2 receptors to activate adenylate cyclase and increase intracellular cyclic adenosine monophosphate (cAMP). Increased levels of cAMP in bronchial smooth muscle, resulting specifically from stimulation of β2 receptors, activate protein kinase A, which phosphorylates several regulatory proteins that mediate bronchodilation. Additionally, β stimulation increases intracellular cAMP in mast cells, inhibiting release of chemical mediators that secondarily cause bronchoconstriction. Specific examples of available sympathomimetic drugs are listed in Table 5-3. To avoid some of the adverse cardiac effects induced by stimulation of β1 receptors, the preferred agents have action limited primarily to stimulation of β2 receptors. The β2-specific agents most commonly used are albuterol (short-acting β2 agonist) and salmeterol or formoterol (long-acting β2 agonists), and the typical route of administration is inhalation. Although some sympathomimetic agents can be given orally or parenterally, the inhaled route is preferred because it has fewer systemic side effects and provides direct delivery to the site of action in the airways.
The third class of bronchodilator agents, also used less frequently than β2 agonists in asthma patients, consists of drugs that have an anticholinergic action. Anticholinergic agents dilate bronchial smooth muscle by decreasing bronchoconstrictor cholinergic tone to airways. Ipratropium, available as an aerosol for inhalation, is the primary short-acting example of this class of agents. It is formally approved in the United States only for use in chronic obstructive lung disease (COPD; see Chapter 6), not in asthma. The major use of ipratropium for asthma has been as adjunctive therapy to inhaled β2 agonists in patients during an acute asthma attack. Tiotropium, a long-acting anticholinergic agent, is approved for and frequently used in patients with COPD, but it is neither approved for nor commonly used in patients with asthma.
Management Strategy
At present, the overall strategy for management of asthma commonly proceeds in the following manner. A patient with relatively infrequent attacks, with symptom-free periods and normal pulmonary function between attacks, is managed with inhaled short-acting sympathomimetics (β2 agonists). These drugs are used on an as-needed basis, both for management of bronchospasm once it occurs and before exposure to stimuli often known to precipitate attacks (e.g., exercise or allergen exposure). These general guidelines are summarized in Table 5-2 according to the categories of clinical severity of disease.
Henneberger, PK, Redlich, CA, Callahan, DB, et al. An official American Thoracic Society statement: work-exacerbated asthma. ATS Ad Hoc Committee on Work-Exacerbated Asthma. Am J Respir Crit Care Med. 2011;184:368–378.
Holtzman, MJ. Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest. 2012;122:2741–2748.
National Asthma Education and Prevention Program. Guidelines for the diagnosis and management of asthma. Expert Panel Report 3. Bethesda, MD: National Institutes of Health, NIH publication 08-4051; 2007.
Barnes, PJ. Neurogenic inflammation in the airways. Respir Physiol. 2001;125:145–154.
Busse, WW, Lemanske, RF, Jr., Gern, JE. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet. 2010;376:826–834.
Chung, KF. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. 2011;139:1470–1479.
Fixman, ED, Stewart, A, Martin, JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J. 2007;29:379–389.
Holgate, ST. Pathophysiology of asthma: what has our current understanding taught us about new therapeutic approaches? J Allergy Clin Immunol. 2011;128:495–505.
Holgate, ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673–683.
Holgate, ST, Yang, Y, Haitchi, HM, et al. The genetics of asthma. ADAM33 as an example of a susceptibility gene. Proc Am Thorac Soc. 2006;3:440–443.
Holt, PG, Sly, PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med. 2012;18:726–735.
Holt, PG, Sly, PD. Interaction between adaptive and innate immune pathways in the pathogenesis of atopic asthma: operation of a lung/bone marrow axis. Chest. 2011;139:1165–1171.
Jacoby, DB. Virus-induced asthma attacks. JAMA. 2002;287:755–761.
Kaur, D, Brightling, C. OX40/OX40 ligand interactions in T-cell regulation and asthma. Chest. 2012;141:494–499.
Koppelman, GH, Sayers, I. Evidence of a genetic contribution to lung function decline in asthma. J Allergy Clin Immunol. 2011;128:479–484.
Martinez, FD. Genes, environments, development and asthma: a reappraisal. Eur Respir J. 2007;29:179–184.
Martinez, FD. New insights into the natural history of asthma: primary prevention on the horizon. J Allergy Clin Immunol. 2011;128:939–945.
Murphy, DM, O’Byrne, PM. Recent advances in the pathophysiology of asthma. Chest. 2010;137:1417–1426.
Paul, G, Brehm, JM, Alcorn, JF, et al. Vitamin D and asthma. Am J Respir Crit Care Med. 2012;185:124–132.
Postma, DS, Kerkhof, M, Boezen, HM, et al. Asthma and chronic obstructive pulmonary disease: common genes, common environments? Am J Respir Crit Care Med. 2011;183:1588–1594.
Robinson, DS. The role of the T cell in asthma. J Allergy Clin Immunol. 2010;126:1081–1091.
Thomson, NC, Chaudhuri, R, Livingston, E. Asthma and cigarette smoking. Eur Respir J. 2004;24:822–833.
Clinical Features and Diagnostic Approach
Anderson, SD. Indirect challenge tests: Airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest. 2010;138:25S–30S.
Beach, J, Russell, K, Blitz, S, et al. A systematic review of the diagnosis of occupational asthma. Chest. 2007;131:569–578.
Corrao, WM, Braman, SS, Irwin, RS. Chronic cough as the sole presenting manifestation of bronchial asthma. N Engl J Med. 1979;300:633–637.
Jackson, DJ, Sykes, A, Mallia, P, et al. Asthma exacerbations: origin, effect, and prevention. J Allergy Clin Immunol. 2011;128:1165–1174.
Johnston, NW, Sears, MR. Asthma exacerbations. 1: Epidemiology. Thorax. 2006;61:722–728.
Katial, RK, Covar, RA. Bronchoprovocation testing in asthma. Immunol Allergy Clin North Am. 2012;32:413–431.
Martin, RJ. Nocturnal asthma: circadian rhythms and therapeutic interventions. Am Rev Respir Dis. 1993;147:S25–S28.
McFadden, ER, Jr., Gilbert, IA. Exercise-induced asthma. N Engl J Med. 1994;330:1362–1367.
McFadden, ER, Jr., Kiser, R, DeGroot, WJ. Acute bronchial asthma: relations between clinical and physiologic manifestations. N Engl J Med. 1973;288:221–225.
Naureckas, ET, Solway, J. Mild asthma. N Engl J Med. 2001;345:1257–1262.
Pratter, MR, Irwin, RS. The clinical value of pharmacologic bronchoprovocation challenge. Chest. 1984;85:260–265.
Singh, AM, Busse, WW. Asthma exacerbations. 2: Etiology. Thorax. 2006;61:809–816.
van den Berge, M, ten Hacken, NH, Cohen, J, et al. Small airway disease in asthma and COPD: clinical implications. Chest. 2011;139:412–423.
Wenzel, SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–725.
Barnes, PJ. Severe asthma: advances in current management and future therapy. J Allergy Clin Immunol. 2012;129:48–59.
Barnes, PJ. Airway pharmacology. In: Mason RJ, Murray JF, Broaddus VC, et al, eds. Textbook of respiratory medicine. ed 4. Philadelphia: WB Saunders; 2005:235–279.
Barnes, PJ. Inhaled glucocorticoids for asthma. N Engl J Med. 1995;332:868–875.
Busse, WW. Long- and short-acting β2-adrenergic agonists. Arch Intern Med. 1996;156:1514–1520.
Chan, WW, Chiou, E, Obstein, KL, et al. The efficacy of proton pump inhibitors for the treatment of asthma in adults: a meta-analysis. Arch Intern Med. 2011;171:620–629.
Durrani, SR, Viswanathan, RK, Busse, WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol. 2011;128:439–448.
Fanta, CH. Drug therapy: asthma. N Engl J Med. 2009;360:1002–1014.
Glassroth, J. The role of long-acting β-agonists in the management of asthma: analysis, meta-analysis, and more analysis. Ann Intern Med. 2006;144:936–937.
Kazani, S, Wechsler, ME, Israel, E. The role of pharmacogenomics in improving the management of asthma. J Allergy Clin Immunol. 2010;125:295–302.
Lazarus, SC. Clinical practice. Emergency treatment of asthma. N Engl J Med. 2010;363:755–764.
O’ Byrne, PM, Parameswaran, K. Pharmacological management of mild or moderate asthma. Lancet. 2006;368:794–803.
Peters-Golden, M, Henderson, WR. Leukotrienes. N Engl J Med. 2007;357:1841–1854.
Price, D, Musgrave, SD, Shepstone, L, et al. Leukotriene antagonists as first-line or add-on asthma-controller therapy. N Engl J Med. 2011;364:1695–1707.
Strunk, RC, Bloomberg, GR. Omalizumab for asthma. N Engl J Med. 2006;354:2689–2695.
van den Berge, M, Hiemstra, PS, Postma, DS. Genetics of glucocorticoids in asthma. N Engl J Med. 2011;365:2434–2435.
Wahidi, MM, Kraft, M. Bronchial thermoplasty for severe asthma. Am J Respir Crit Care Med. 2012;185:709–714.
Weiss, ST. New approaches to personalized medicine for asthma: where are we? J Allergy Clin Immunol. 2012;129:327–334.