Anesthesia for myotonic dystrophy
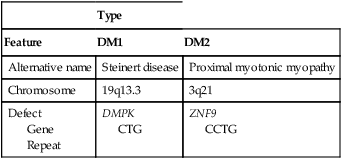
Myotonic dystrophies are autosomal dominantly transmitted disorders and classified as DM1 (Steinert disease) or DM2 (proximal myotonic myopathy), with DM1 further subdivided by age of onset. Prominent features of classic DM1, the most common myotonic dystrophy, include slowly progressive muscle weakness (dystrophy), cataracts, endocrine disturbances, and functional abnormalities of the cardiorespiratory system and gastrointestinal tract (Table 209-1).
Table 209-1
Genetics of Myotonic Dystrophy
| Type | ||
| Feature | DM1 | DM2 |
| Alternative name | Steinert disease | Proximal myotonic myopathy |
| Chromosome | 19q13.3 | 3q21 |
| Defect Gene Repeat |
DMPK CTG |
ZNF9 CCTG |