The electrical discharge of neurons associated with seizure activity stimulates a marked rise in cerebral metabolic activity. Estimates from animal experiments indicate that energy utilization during seizures increases by more than 200%, while tissue adenosine triphosphate (ATP) levels remain at more than 95% of control, even during prolonged status epilepticus (1,2). Cerebral metabolic activity can be measured using 2-deoxy-2-[18F]-fluoro-D-glucose positron emission tomography (FDG PET) (3), and magnetic resonance spectroscopy (MRS) can be used to measure the turnover of glutamate, the major excitatory neurotransmitter (4). Comparison of FDG PET and MRS in patients with seizures indicates that glucose metabolism and the turnover of glutamate are tightly coupled (3). This is because the transporters that remove glutamate as well as gamma-aminobutyric acid (GABA), the major inhibitory neurotransmitter, from the synaptic cleft require substantial energy (5). These transporters are predominantly located on astroglia that surround synapses, and they remove glutamate and GABA soon after their release from presynaptic nerve terminals to terminate their activity. These transporters are also located on excitatory neuron terminals and are responsible for synaptosomal glutamate uptake (6). The transporters are linked to a Na+/K+-ATPase, which in turn is powered by anaerobic metabolism of glucose (4). Additionally, chloride cotransporters that maintain the transmembrane Cl– gradient are also energy dependent (7). The brain generally withstands the metabolic challenge of seizures quite well because enhanced cerebral blood flow delivers additional oxygen and glucose. Mild to moderate degrees of hypoxemia that commonly accompany seizures are usually harmless. In addition, the immature brain is less vulnerable to injury from seizures than the adult brain (8,9). However, severe seizures (ie, lasting >5 min) and status epilepticus (>30 min) can sometimes produce an imbalance between metabolic demands and cerebral perfusion, especially if severe hypotension or hypoglycemia is present. Injury can also occur through a direct effect on synapses known as excitotoxicity (10,11).
Excitotoxicity refers to a process of injury triggered by excessive activation of neuronal receptors for the excitatory amino acid (EAA) neurotransmitters glutamate and aspartate. Under normal circumstances, activation of EAA receptors mediates more than half of the neuronal communication in the brain (12). Seizures initiate release of EAA neurotransmitters from nerve terminals into the synaptic cleft, where they bind to specific neuronal receptors and cause postsynaptic excitation. In the developing brain, EAA receptors perform an additional role as regulators of neuronal development (13). During a prolonged seizure, if the EAA neurotransmitter release continues unabated to raise the synaptic concentration of glutamate, excessive EAA receptor stimulation may damage and eventually kill neurons.
An important component of the excitotoxicity theory holds that EAAs cause intracellular calcium overload, which intoxicates neurons (14–16). Calcium and sodium are allowed to enter neurons through pores or ionophores controlled by EAA receptors. The concentration of calcium within the neuron is normally 10,000 times lower than in the extracellular fluid, and calcium entry is tightly controlled. The neuron has several mechanisms for protecting itself from excessive calcium influx, including sequestration of calcium within mitochondria and endoplasmic reticulum, and energy-dependent pumps that move calcium outward across the cell membrane. Continued bombardment of the neuron by EAA neurotransmitters can overwhelm these protective mechanisms, allowing calcium to poison the metabolic machinery of the neuron (17). The EAA theory proposes that excitatory neurotransmitters are a major direct link between excessive seizure activity and neuronal injury; however, metabolic derangements such as hypoxia may play a contributing destructive role by reducing the efficiency of energy-dependent protective mechanisms and hastening EAA-stimulated metabolic exhaustion (18).
PHARMACOLOGY OF EXCITATORY AMINO ACID RECEPTORS
Glutamate is released from presynaptic nerve terminals, but the synaptic concentration is quickly reduced by an active reuptake process mediated by its specific, energy-dependent reuptake pump. Impairment in the activity of the EAA reuptake pump may play an important role in EAA neurotoxicity in disease states. In experimental hypoxia–ischemia, synaptosomal uptake of glutamate is transiently reduced, contributing to marked elevations in extracellular glutamate concentrations, which may reach the micromolar range (19). Metabolic disorders may contribute to EAA neurotoxicity by this mechanism.
The excitatory responses to glutamate are mediated by several receptor subtypes that may be classified into two broad categories as either N-methyl-D-aspartate (NMDA)-type or non-NMDA-type EAA receptors (20,21) (Figures 4.1 and 4.2). Glutamate is a relatively flexible amino acid molecule that can assume several different conformations, and NMDA is a rigid glutamate analog that represents one of them (22). NMDA maximally activates a glutamate receptor that is linked to an ion channel. Together, the receptor and its channel are referred to as the NMDA receptor/channel complex (see Figure 4.1). Molecular cloning of NMDA receptors showed that their diverse characteristics are related to variations in specific combinations of subunits (23). The NMDA channel is permeable to cations such as sodium and calcium, and at normal membrane potential the channel is blocked by magnesium ions (24). Partial depolarization of the membrane potential is necessary for the magnesium block to be removed. As a result, the NMDA receptor/channel complex is not generally involved in ordinary rapid impulse flow.
Several antagonists of the NMDA receptor/channel complex have been developed, some of which block the NMDA receptor (competitive NMDA antagonists) whereas others block the associated cation channel (noncompetitive antagonists). Drugs such as 3-(2-carboxylpiperazin-4-yl)-propyl-1-phosphonic acid (CPP), CGS-19755, and 2-amino-5-phosphonovaleric acid (AP5) are competitive blockers of the NMDA receptor (25). The channel-binding site is sometimes referred to as the phencyclidine receptor. Two potent preclinical noncompetitive channel-blocking drugs are MK-801 and phencyclidine (26,27). The clinically approved anesthetic ketamine (28) and the antitussive dextromethorphan (29), as well as the anticonvulsant felbamate (30), are also NMDA channel blockers. Memantine, an “un-competitive” NMDA channel blocker, has recently been approved by the Food and Drug Administration for use in Alzheimer disease. It differs from noncompetitive NMDA blockers in that it binds more avidly to open channels, rather than indiscriminately blocking all channel conformations (31). This mechanism may be associated with fewer psychotomimetic side effects than are associated with more complete channel blockade.
The NMDA receptor/channel complex also includes several regulatory sites. One receptor is specific for the simple amino acid glycine (32,33). Unlike the inhibitory glycine site in the spinal cord, this site is not sensitive to strychnine, and activation facilitates opening of the NMDA channel by glutamate. Recognition sites for zinc and polyamines also appear to regulate NMDA receptor/channel complex activity (13). Drugs that block the glycine site on the NMDA receptor are being developed to attempt to avoid the psychotomimetic side effects of noncompetitive NMDA antagonists (34).
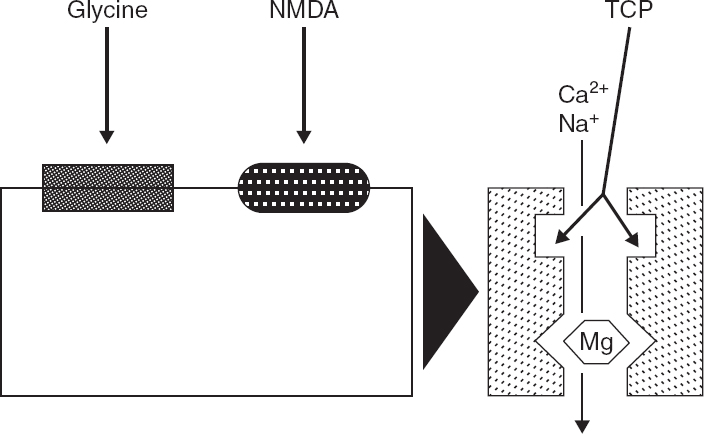
FIGURE 4.1 Current understanding of the components of the NMDA receptor/channel complex, based on biochemical and electro-physiologic evidence, is outlined in this schematic diagram. The NMDA recognition site is coupled to a cationic channel that is permeable to Ca2+ and Na+. NMDA receptor activation is modulated by several regulatory sites. Activation of a closely associated glycine recognition site is required for channel activation and markedly enhances responses to NMDA. Physiologic concentrations of Mg2+ block the NMDA-associated ionophore, and membrane depolarization relieves this blockade. Thus, NMDA receptor/channel activation requires activation of both NMDA and glycine receptors and concurrent membrane depolarization. NMDA-receptor channel activation also can be modulated by Zn2+ and polyamines (not illustrated). Noncompetitive NMDA receptor antagonists, such as phencyclidine and its thienyl derivative TCP, block NMDA responses by binding within the NMDA-operated ionophore. Cerebral events that impair the mechanisms that regulate Ca2+ homeostasis, can produce a prolonged elevation of intracellular Ca2+ concentration and cytotoxicity.
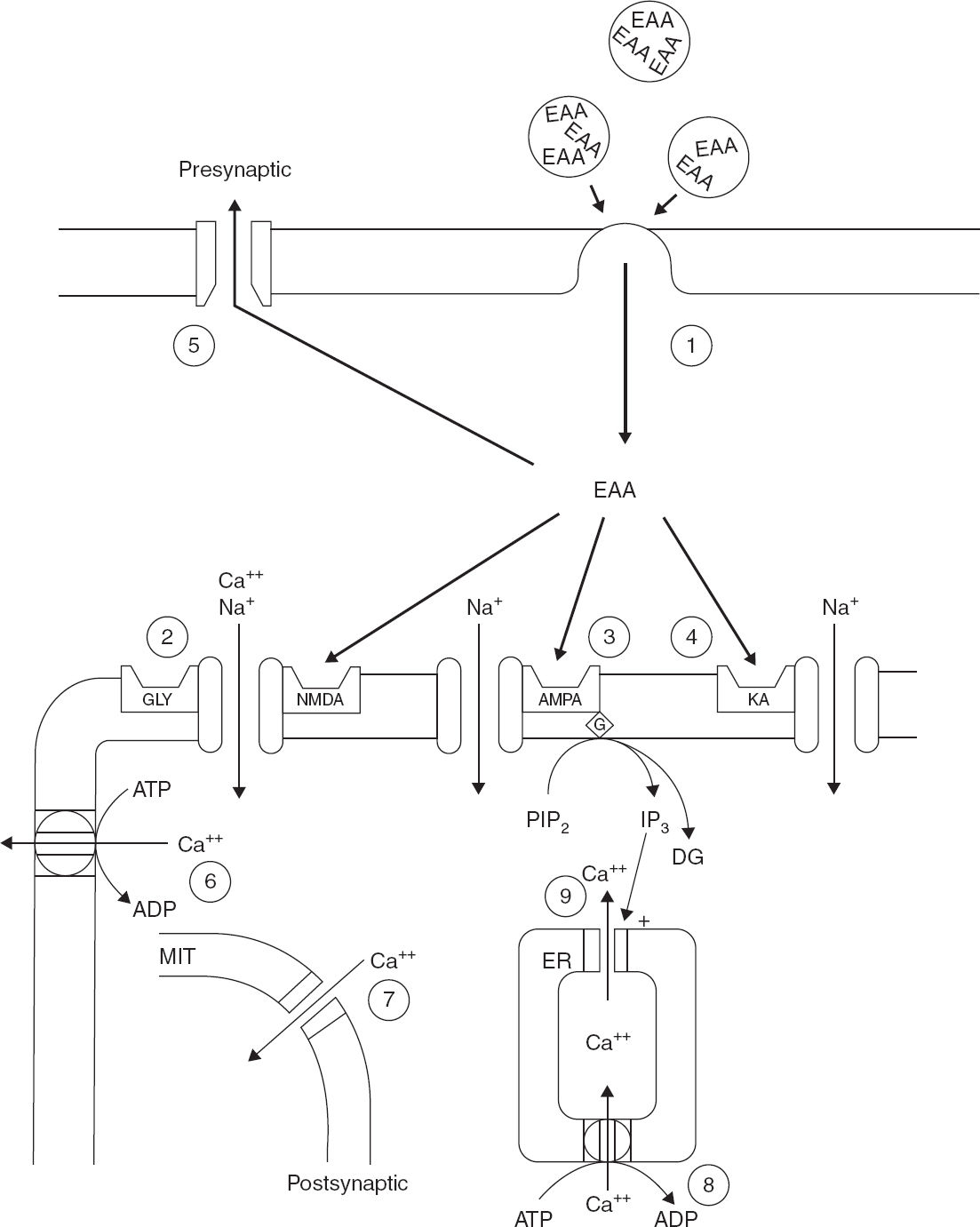
FIGURE 4.2 This schematic diagram outlines the synaptic components that contribute to EAA-mediated synaptic transmission, second-messenger generation, and calcium homeostasis. (1) EAAs such as glutamate are released from presynaptic terminals in a calcium-dependent process by presynaptic depolarization. (2–4) Glutamate in the synaptic cleft depolarizes the postsynaptic membrane by binding to at least three subsets of EAA receptors. Activation of (2) the NMDA receptor/channel complex, (3) AMPA receptors, and (4) kainate receptors produced calcium (Ca2+) and sodium (Na+) entry through receptor-associated ionophores. Furthermore, activation of a subset of metabotropic receptors, which are linked to phospholipase C, produces phosphoinositol hydrolysis and generation of the second messengers inositol triphosphate and diacylglycerol. (5) The excitatory action of synaptically released EAA is terminated by a presynaptic, high-affinity, energy-dependent transport process. (6–9) Mechanisms that regulate post-synaptic Ca2+ homeostasis.
Research investigating differences in function between synaptic and extrasynaptic NMDA receptors have found significant differences (35). Activity-dependent neuronal firing patterns, neuromodulators, and cerebral metabolism modulate the synaptic release of glutamate and therefore extrasynaptic glutamate concentrations. Extrasynaptic glutamate, in turn, can affect neural function and can be neurotoxic on its own. Synaptic and extrasynaptic NMDA receptors differ in their subunit composition, downstream transducing cascades, and effects on cell survival (36). Extrasynaptic NMDAR-dependent neuronal death cascades are mediated by several transduction pathways; many of them antagonize those triggered by synaptic NMDARs (Figure 4.3). Advances in the understanding of selective pharmacology and function of NMDAR subunits will advance the field of new therapeutic strategies given their differential effects on synaptic and extrasynaptic receptors and, therefore, on cell survival (37).
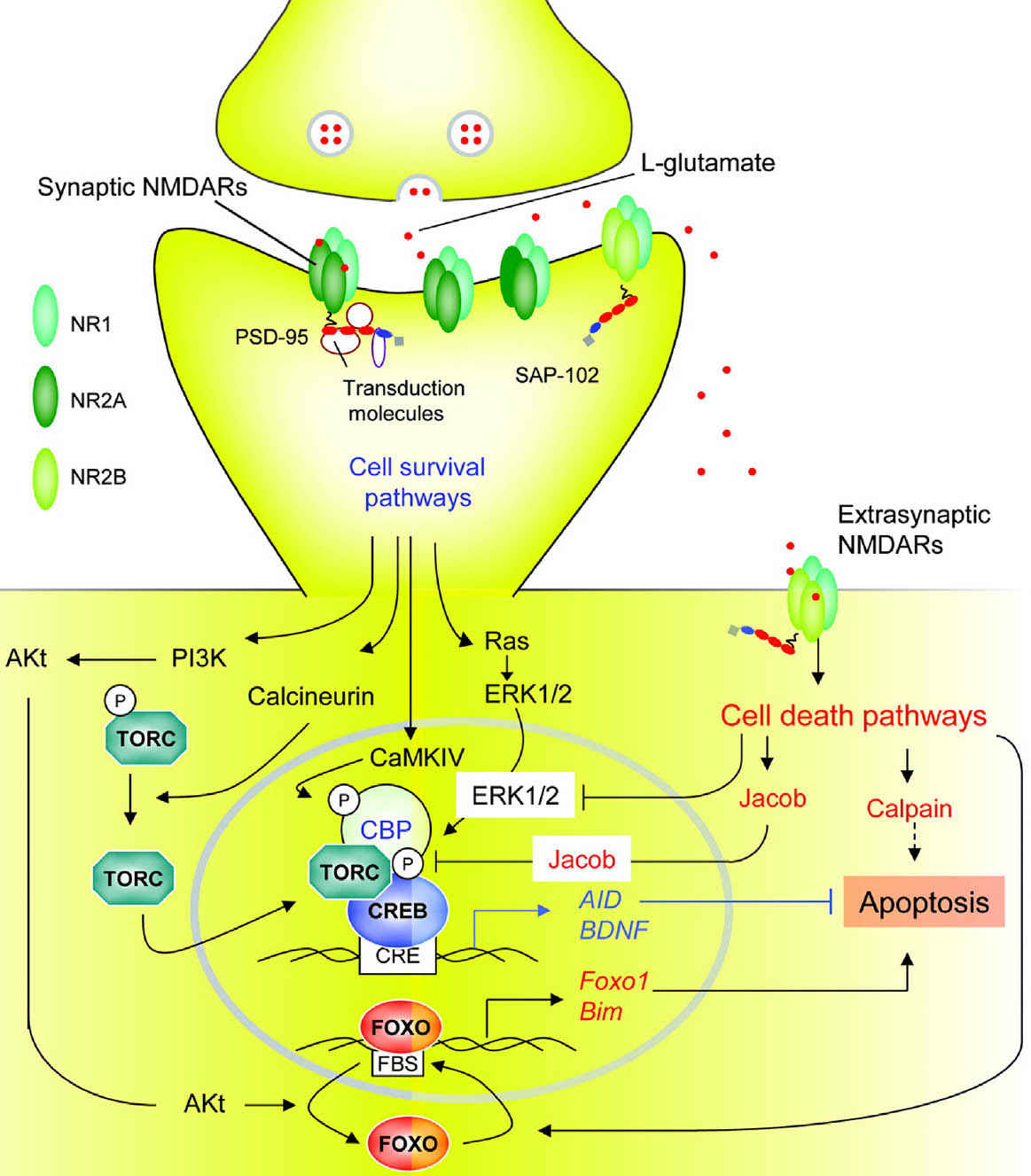
FIGURE 4.3 Transduction pathways involved in cell survival or cell death triggered by synaptic or extrasynaptic NMDA receptors (NMDARs). According to a recent model, synaptic NMDARs, composed primarily of NR1/NR2A subunits, trigger neuroprotective transduction pathways, whereas extrasynaptic NMDARs, composed predominantly by NR1/NR2B subunits, initiate cell death pathways. Extrasynaptic NMDARs are present at perisynaptic sites distributed on dendrites and in some cases adjacent to glia-like processes or axons. Synaptic NMDAR-dependent neuroprotection involves transduction cascades that elicit induction of survival genes, suppression of death genes, or protection against oxidative stress. Calcium influx via synaptic NMDARs triggers Ca2+/calmodulin (CaM) kinase IV and extracellular receptor kinase (ERK)1/2 mediated phosphorylation and activation of CREB (cyclic-AMP response element binding protein) and its coactivator CREB binding protein (CBP); calcineurin elicits dephosphorylation and nuclear translocation of the transducer of regulated CREB-activity (TORC), a key step in CREB activation. CREB promotes transcription of activity-regulated inhibitors of death (AID) and brain-derived neurotrophic factor (BDNF) genes, which provide broad-spectrum neuroprotective effects. Synaptic NMDAR activity also triggers transcriptional suppression of core components of the intrinsic apoptosis cascade, in part via suppressing activity forehead box protein O (FOXO)–dependent expression of proapoptotic genes. This antiapoptotic effect results from Akt-mediated phosphorylation and nuclear export of FOXO. Synaptic NMDAR activation also enhances thioredoxin activity, which elicits antioxidant effects (not shown). In contrast, extrasynaptic NMDARs trigger cell death via several transduction pathways; many of them antagonize those triggered by synaptic NMDARs. These include CREB dephosphorylation by Jacob (juxtasynaptic attractor of caldendrin on dendritic spines) protein, inhibition of the ERK1/2 pathway, promotion of nuclear import of FOXO, and activation of calpain.
Bim, bcl2-interacting mediator of cell death; PI3K, phosphatidyl inositol 3’kinase; Txnp, thioredoxin interacting protein.
Non-NMDA receptors were initially identified because they were activated preferentially by the glutamate analogs quisqualic acid and kainic acid (19). Kainic acid is a very potent neurotoxine and convulsant agent in the adult brain (38,39). Currently, two major types of non-NMDA receptors are recognized: one that is linked to an ion channel that fluxes sodium and, to a lesser extent, calcium and another that is linked to stimulation of phosphoinositide (PPI) turnover or downregulation of cyclic-AMP production (20). The so-called ionotropic non-NMDA receptors are now divided into kainate receptors and AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate) receptors. Molecular studies indicate that these two receptors have similar molecular characteristics, with AMPA receptors made up from combinations of GluR1 to GluR4 receptor sub-units, and kainate receptors made up of GluR5 to GluR7 subunits. AMPA receptors mediate most of the fast excitatory activity in the brain, and they include most of the receptors previously referred to as quisqualate receptors (20). Perampanel is the selective noncompetitive AMPA-antagonist (40). The anticonvulsant topiramate is known to have several cellular targets for its therapeutic actions that, in addition to AMPA receptors, also include the GluR5 subunit of the kainate-receptor (41), voltage-gated sodium channels (42), and specific GABA-A receptor isoforms (43). Kainate receptors stimulate presynaptic release of glutamate as well as activating postsynaptic receptors. The glutamate receptors linked to second messengers are called metabotropic receptors. A marked increase in glutamate release, which occurs during a prolonged seizure, is likely to result in the activation of all types of glutamate receptors.
DEVELOPMENT OF EXCITATORY AND INHIBITORY SYNAPTIC MARKERS
Synaptic markers for EAA neurotransmitters undergo marked changes during postnatal development (44–47). Studies of the ontogeny of NMDA and AMPA-sensitive receptor subtypes during postnatal development indicate that NMDA-type and non-NMDA-type glutamate receptors have a unique ontogenetic profile in the hippocampal formation (47). When receptors for NMDA-sensitive 3H-glutamate binding, strychnine-insensitive glycine binding, and 3H-N-(1[2-thienyl]cyclohexyl)3, 4-piperidine (TCP) binding to the channel were examined independently, all were found to be overexpressed in the neonatal period relative to the adult. NMDA-sensitive binding exceeded adult levels by 50% to 120% in all regions of the hippocampus examined, with peak densities occurring between postnatal days 10 and 28. The ontogenetic profiles of the glycine modulatory site and the phencyclidine receptor were similar to each other but were delayed with respect to the timing of the NMDA recognition site. The physiologic relevance of overexpression of NMDA recognition sites relative to other components of the NMDA receptor/channel complex is unclear. However, the temporal expression of NMDA recognition sites correlates with the molecular changes in receptor subunits and synaptogenesis (48–50). Studies of NMDA receptors in human cerebral cortex in early life have also demonstrated enhanced levels and different characteristics compared to adults (48,51).
The ontogeny of NMDA receptor binding also appears to correlate with physiologic changes. Long-term potentiation, a synaptic correlate of memory, is enhanced during the neonatal period in rats, at the same time that NMDA-mediated neurotransmission predominates over AMPA-mediated activity (52). Postsynaptic excitation appears to predominate over inhibition during the first few postnatal weeks of hippocampal development, as does enhanced seizure susceptibility (53). In the CA3 region of the hippocampus, superfusion with NMDA elicits recurrent synchronized burst activity, and the epileptogenic effect of NMDA increases from postnatal days 1 to 10 (54). Also, the susceptibility to epileptiform activity increases from postnatal days 4 to 6 to postnatal day 14 (55). This activity is blocked by specific NMDA receptor antagonists (56). The development of NMDA receptors tends to lag behind the development of sensitivity to NMDA neurotoxicity, which transiently peaks on day seven.
AMPA receptors also undergo developmental changes that make them more calcium permeable and more likely to mediate seizures and excitotoxicity in infancy and childhood (57). AMPA receptors that lack GluR2 subunits are calcium permeable, while AMPA receptors that include GluR2 subunits flux only sodium. Jensen et al have studied the development of AMPA receptor subunits in rats and human postmortem tissue and found a correspondence between the expression of GluR2-lacking receptors and regional susceptibility to brain injury and seizures (58–60). GluR2-lacking AMPA receptors appear first in radial glia and oligodendrocytes in the periventricular white matter at 25 to 32 weeks gestation in humans (59). Their expression is delayed to around 40 weeks gestation in pyramidal neurons and nonpyramidal neurons of the cerebral cortex, persisting into early infancy and beyond in selected regions (59). These ontogenetic changes in AMPA receptors are consistent with experimental evidence that AMPA-receptor antagonists provide protection against seizures and damage at these stages in development (61).
The development of markers for GABAergic inhibitory synapses also undergoes major changes in the postnatal period and tends to lag behind development of excitatory synapses (62–64). GABAA receptor antagonists cause prolonged seizure-like discharges in the hippocampus of 2- to 3-week-old rats (53). However, during early life, extending to the end of the first week postnatally in rats, stimulation of GABAA receptors paradoxically produces excitation rather than inhibition as a result of a lag in the development of the chloride KCC2 transporter, which maintains low intracellular Cl– levels (65–68). Without this transporter, high concentrations of Cl– accumulate within the dendrite, resulting in depolarization when the Cl– channel is opened by GABA. This is consistent with the theme that excitatory neurotransmission is enhanced early in development to support activity-dependent neuronal development and plasticity and becomes more balanced with inhibitory neurotransmission later in life (69). These developmental programs contribute to the higher incidence of seizures in infants and children.
DEVELOPMENTAL CHANGES IN GLUTAMATE NEUROTOXICITY
The immature brain is quite sensitive to neurotoxicity from glutamate and certain of its analogs (39,70). Olney discovered a number of years ago that feeding monosodium glutamate to neonatal mice produced characteristic lesions in the hypothalamus (39). More recently, it has become clear that the receptors that mediate glutamate neurotoxicity respond differently in the immature brain compared to the adult brain. These observations have important implications for the EAA neurotransmitter hypothesis of epileptic brain injury in children.
In adults, kainic acid is the most potent neurotoxic analog of glutamate, but in neonatal rodents NMDA is the most toxic analog (70–72). Although kainic acid produces seizures in the immature brain, it produces little cytotoxicity (73). In contrast, NMDA injected into the 7-day-old rat brain produces prolonged seizures and extensive neurotoxicity. At 7 days of age, the relative neurotoxicity of several glutamate analogs is NMDA >>> AMPA > kainic = zero, whereas in the adult kainic acid >> NMDA ≥ AMPA. The severity of brain injury produced by direct injection of NMDA into the 7-day-old rat brain is approximately 60 times greater than in the adult (70). There is a relatively sharp peak of sensitivity to NMDA neurotoxicity at 7 days of age in the rat, with less sensitivity at times before and after, suggesting a potentially important effect of age on sensitivity to EAA-mediated injury. The shift in sensitivity to NMDA neurotoxicity parallels developmental changes in genetic expression of NMDA subunits that make up the receptor channel complex (50,74). These changes appear to be programmed to mediate activity-dependent neuronal plasticity during early development (13). NMDA receptor activity is needed to promote normal development, and excessive blockade of these receptors leads to neuronal apoptosis (75). The NMDA receptor is a potential Achilles heel for the developing brain: both overstimulation and blockade can produce damage under certain circumstances (13).
The susceptibility of the developing brain to NMDA-induced brain injury parallels the susceptibility to hypoxic-ischemic brain injury. Olney’s studies suggest that the acute neurohistologic picture of NMDA-induced injury in the 7-day-old rat brain is virtually identical to that from a hypoxic–ischemic injury at the same age (76). NMDA may induce metabolic changes similar to hypoxia–ischemia. Studies using (34) P MRS in brain slices indicate that NMDA produces a rapid exhaustion of phosphocreatine stores and nucleotide triphosphate levels following its application (77). The noncompetitive antagonist MK-801 protects against hypoxic–ischemic damage in the 7-day-old rat brain (78). These studies suggest that over-activation of NMDA receptors may be a final common pathway for hypoxic–ischemic injury, as well as other types of developmental brain injury.
EVIDENCE THAT EAA NEUROTRANSMITTERS PLAY A ROLE IN SEIZURE-RELATED INJURY
The link between excessive EAA neurotransmitter activity and epileptic brain injury was initially strengthened by Meldrum’s pioneering studies in subhuman primates (11). These studies showed that seizures alone could damage the brain and that the EAA analog kainic acid produced sustained seizures and brain injury with a regional histologic pattern resembling epileptic brain injury. Similar studies of selective injury in the hippocampus from kainic acid injection into the brain have been performed in rodents by Nadler et al (79). Two additional models in adult animals have also provided additional, more direct experimental evidence.
In experiments conducted by Sloviter, electrical stimulation of the perforant pathway that projects into the hippocampus from the entorhinal cortex leads to neuronal degeneration in hippocampal zones innervated by perforant pathway fibers (80). The perforant pathway is the major excitatory glutamatergic afferent pathway into the hippocampus (81). The acute neuropathologic changes produced by the persistent stimulation resemble the histopathology seen when glutamate analogues or other convulsant substances are injected into the hippocampus (82).
The pathology replicates the typical “epileptic” pattern of injury with damage to dentate basket cells, hilar cells, and CA3 and CA1 pyramidal cells of the hippocampus. CA2 neurons are typically spared. Electron microscopy shows acutely swollen dendritic segments distributed in a laminar pattern corresponding to the neuronal-receptive fields of EAA pathways in the perforant projection. Somatostatin-containing neurons are especially vulnerable, whereas GABA neurons are relatively spared (83).
In a second model of epileptic brain injury developed by Collins and Olney, persistent focal seizure activity is induced by focal application of the GABA receptor-blocking drug bicuculline onto the cerebral cortex (10). The seizure activity causes acute neurodegenerative changes in specific thalamic regions innervated by the corresponding corticothalamic pathway. This model also produces acute neuropathologic changes that resemble glutamate-like neuronal degeneration. Using this model, Olney’s group demonstrated that ketamine and MK-801, two powerful blockers of the action of NMDA-type EAA receptors, completely abolish the thalamic damage caused by the focal seizure activity (84). MK-801 reduced but did not prevent the electrical seizure activity itself, suggesting that the neuroprotective action resulted from blockade of NMDA-type EAA receptor activation rather than from a reduction in neurotransmitter release. The results of these experiments, along with the electrical stimulation studies, support the hypothesis that EAA receptor stimulation plays an important role in seizure-induced neuronal injury. They also suggest that the neurotoxic effects of seizures can be dissociated from effects of excessive electrical activity.
In addition to etiology, associated seizure burdens often seem to be the defining factor for the emergence of refractoriness to first line AEDs (85). When it comes to neonatal seizures, the ability to determine the true seizure burden of clinically ambiguous behavioral manifestations remains critical in correctly gauging both seizure frequency and response to treatment. Since electroclinical dissociation is now a well-accepted concept (86,87) where cessation of behavioral manifestations of seizures after drug administration represents a false sense of having effectively managed seizures, it is imperative that to truly understand molecular and pharmacologic consequences of seizures, true seizure burdens are ascertained. It has been shown in rodents (88) that there are regional differences in the ontogeny of Cl(-) transport, wherein most thalamic neurons in perinatal rat pups were found to maintain low [Cl(-)] and were inhibited by GABA as well as phenobarbital. In contrast, most neocortical neurons maintained higher [Cl(-)], and were excited by GABA(A)R activation. This may explain why seizure activity in the cortex is not suppressed by anticonvulsants that block the transmission of seizure activity through subcortical networks.
Consequences of Prolonged Febrile Seizures
The FEBSTAT study prospectively studied the relationship between febrile status epilepticus (FSE) and acute hippocampal damage, as well as the development of mesial temporal sclerosis, epilepsy (particularly temporal lobe epilepsy), and impaired hippocampal function (89). The FEBSTAT study has concluded that the HHV-6B infection is commonly associated with FSE and HHV-7 infection is less frequently associated with FSE. Together, they account for one-third of FSE, a condition associated with an increased risk of both hippocampal injury and subsequent temporal lobe epilepsy (90). Earlier studies have reported that mutations in the HCN2 channel may play an important role in the occurrence of febrile seizures; however, these mutations were only found in 2.4% of subjects with febrile seizures and genetic epilepsy with febrile seizures (91) over the 0.2% incidence in blood bank controls. Additionally, the HCN1 and HCN2 variants that have been identified failed to show statistical differences during functional analyses for channel properties (92). Therefore, if gain of function in the HCN channel does play a role in metabolic and pharmacologic consequences of febrile seizures, that role has not been credibly established to underlie FSE.
Children with FSE are at risk for acute hippocampal injury and a substantial number also have abnormalities in hippocampal development (93). Therefore, the conclusion that the immature brain is not susceptible to cell death drawn from animal models of hyperthermia, induced by external heat, with relatively low seizure burdens (94) may not be replicating the clinical condition. In contrast, modifications in the model by exposing the pups to longer duration of heat have also revealed hippocampal cell death (95). This indicates that the severity of the acute injury in addition to the etiology defines the metabolic consequences of FSE.
Hippocampal sclerosis (HS) is the most common neuropathologic pattern observed in pharmacoresistant epilepsy and represents a critical feature in mesial temporal lobe epilepsy syndrome (96). Since febrile seizures (FS) are a prevalent antecedent of temporal lobe epilepsy (TLE), it is likely that the severity of the initial FSE modulates the severity of HS and, therefore, also pharmacoresistance.
GABAergic Consequences of Seizures and Role in Pathogenesis of Epilepsy
It is well known that prolonged seizures in the young brain can produce injury that is aggravated when the inflammatory component is added to the etiology (97,98). The resulting injury contributes to synaptic reorganization that likely plays a major role in the development of chronic epilepsy. Additionally, specific loss of susceptible subsets of interneurons following SE have been implicated to contribute in the process of epileptogenesis (99). Dominant mutations in SCN1A, which encodes the Nav1.1 VGSC α-subunit, underlie several forms of epilepsy, including Dravet syndrome (DS) and genetic epilepsy with febrile seizures plus (GEFS+). However, mouse model studies have shown that 69% of Nav1.1 immunoreactive neurons were also positive for parvalbumin (100). The inactivation of one SCN1A allele in interneurons of the neocortex and hippocampus in the model was sufficient to reduce thresholds to flurothyl- and hyperthermia-induced seizures, whereas thresholds were unaltered following inactivation in excitatory cells. These findings indicate that the GABAergic system plays a predominant role in the generation and pharmacologic consequences of certain seizure syndromes.
Non-Neuronal Consequences of Seizures: Role of Astrocytes
Investigation of specimens from patients with pharmacoresistant temporal lobe epilepsy and epilepsy models have revealed alterations in expression, localization, and function of astroglial K+ and water channels. In addition, malfunction of glutamate transporters and the astrocytic glutamate-converting enzyme, glutamine synthetase, has been observed in epileptic tissue. Dysfunctional astrocytes in gliotic tissue play a crucial role in epilepsy (101).
Decreased expression of inwardly rectifying potassium (Kir) channels in astrocytes are also known to contribute to impaired K+ buffering following seizures; this phenomenon is tied to the local inflammatory environment (102). Acute inflammatory reaction after seizures has long been suspected due to clinical observation of pleocytosis, without any evidence of infection, in the cerebrospinal fluid (CSF) and peripheral blood of patients with recent generalized convulsions. Steroids or adrenocorticotropic hormone (ACTH), with a suppressive effect on inflammation or immune reactions, have been used to effectively treat children with intractable epilepsy.
Seizures and Inflammation
Brain inflammation may be a common substrate contributing to seizures and following seizures in drug-resistant epilepsies of different etiologies, and recurrent seizures can per se be a major cause of long-term inflammation (103,104). Following this view, new research shows that pharmacologic blockade of specific inflammatory molecules and pathways can significantly reduce seizures in experimental models of seizures and epilepsy (105). From a clinical standpoint, a role of inflammation in the pathophysiology of human epilepsy is still hypothetical, although this possibility is supported by abundant evidence (106). Increased BBB permeability and changes in endothelial transporters function induced by seizures, inflammation, or both have clinical implications. BBB leakage allows the entry of compounds with immunogenic or inflammatory potentials, as predicted—for example, in interferon-induced seizures (107)—but it may also facilitate the entry of compounds with therapeutic potential with limited or no access to the CNS. Activation of the ACTH–GC axis in response to antecedent injury or stress, leading to hyperfunction of CRH–neuronal pathways, has been suggested to play a role in the pathogenesis of West syndrome (108).
Role of Glucose Transport
Hypoxic–ischemic brain injury is associated with transient compensatory changes targeted at protecting glucose delivery to fuel cellular energy metabolism by transient increases in the neuronal brain glucose transporter isoform (GLUT-3), which then may delay the processes of apoptosis and cell necrosis (109,110).
MECHANISMS FOR SEIZURES AND INJURY IN THE IMMATURE BRAIN
[level-membership-for-pediatrics-category]
The immature brain, especially the 7-day-old rat brain, is quite sensitive to seizures produced by NMDA as well as to NMDA-mediated neurotoxicity, but the relationship between the two is not clear. The models of prolonged direct electrical stimulation of the perforant pathway and of bicuculline-induced focal seizures described for adult animals have not been examined in the infant. However, both perforant pathway stimulation and the lithium-pilocarpine model of status epilepticus have been shown to produce neurotoxicity in the hippocampus of 15-day-old rodents (111). In our experiments, anticonvulsants such as phenytoin, diazepam, and pentobarbital markedly reduced NMDA-induced seizure activity but did not reduce the neurotoxic effects of injected NMDA (112). MK-801 and other NMDA antagonists produce significant levels of neuroprotection at doses that do not block behavioral seizures. Seizures induced by injection of NMDA in immature animals have some similarity to electrographic and behavioral changes seen in infantile spasms; this is an active area of investigation (113,114).
Activation of non-NMDA receptors also could contribute to seizure-related injury. A model in which kainic acid is injected into immature rodents has been extensively studied for its effects on brain injury as well as on long-term behavior. This is a useful model for trying to understand subtle effects of seizures themselves, as opposed to direct pathologic effects of neurotoxicity (115). Stimulation of AMPA receptors also produces seizures and brain injury in the immature rat (116). Metabotropic receptor stimulation may also contribute to effects of seizures because seizures stimulate phosphoinositide turnover and release of free fatty acids (117).
EAA mechanisms also could play a role in epileptogenesis and seizure expression (118). In an in vitro model of electrographic seizures in hippocampal slices, NMDA antagonists such as AP4 and MK-801 prevented the progressive development of seizures, but did not block previously induced seizures (119). This suggests that the process of establishing a long-lasting seizure-prone state depends on the NMDA receptor/channel complex, whereas the expression of seizures does not. AMPA receptor–mediated mechanisms also appear to play an important role in seizure susceptibility that follows exposure to hypoxia in rodents (120). This suggests an important distinction between antiepileptogenic and anticonvulsant pharmacologic agents (121).
It is also possible that EAA receptors play a role in other more subtle developmental sequelae of seizures (53). In addition to their role in transmembrane signaling, EAA neurotransmitters participate in a variety of neurodevelopmental events. These include promotion of neuronal survival, growth, and differentiation of neurons; regulation of neuronal circuitry and cytoarchitecture; regulation of activity-dependent synaptic plasticity; and certain forms of learning and memory (122). Excitotoxic amino acids may act as neurotrophic factors promoting neuronal survival growth and differentiation during development. The NMDA receptor/channel complex appears to play a critical role in visually determined plasticity in the visual cortex (123). Drugs that block the NMDA receptor/channel complex may block the physiologically determined ocular dominance shifts that normally occur with monocular deprivation. The NMDA receptor/channel complex also appears to play a critical role in the formation of long-term potentiation, an electrical model of learning and memory (124). Disturbances in these normal developmental mechanisms by excessive EAA neurotransmitters released during repeated seizures could potentially be a mechanism for a variety of neurobehavioral disturbances in patients with seizures. Seizure-induced disturbances in EAA mechanisms involved in memory in the hippocampus might be responsible for disturbances in learning and memory in patients with frequent or prolonged seizures. This encephalopathic disturbance might occur in the absence of permanent injury.
Synaptic inhibitory mechanisms also play an important role in both short- and long–term changes that result from seizures. Short-term changes in GABA receptors appear to reduce the efficacy of anticonvulsant drugs that target these receptors after extended periods of status epilepticus (125–129). Injection of tetanus toxin into the hippocampus of immature rats produces selective impairment of GABA neurotransmission and seizures, and this treatment has been shown to induce long-term behavioral and learning deficits as well as changes in the expression of NMDA receptor subunits (53). These experimental results reflect the plastic responses of developing excitatory and inhibitory synaptic mechanisms to seizures.
METABOLIC AND PHARMACOLOGIC CONSEQUENCES OF SEIZURES
Excitatory Amino Acid Mechanisms in the Pathogenesis of Partial Epilepsy
Whether excitotoxic mechanisms play a role in the pathogenesis of childhood temporal lobe epilepsy associated with hippocampal damage is subject to debate (130,131). Although laboratory evidence discussed earlier suggests this is likely, human epidemiologic evidence suggests that prolonged seizures are usually harmless in children (9). Although a history of FSE is frequently obtained in patients undergoing temporal lobectomy surgery for partial complex seizures, magnetic resonance imaging (MRI) studies suggest that many patients have hippocampal malformations that predated onset of seizures (132). However, recent serial MRI has identified a few children who acquired hippocampal sclerosis following very prolonged febrile seizures (133). This suggests that EAA pathways may have a role in the pathogenesis of certain forms of chronic epilepsy.
Partial complex seizures following hippocampal injury or malformation are sometimes progressive, suggesting that further seizures lead to progressive injury and synaptic reorganization that promotes more seizures (134). EAA mechanisms could contribute to progressive metabolic disturbances and injury that could perpetuate this process. Studies of epileptogenic hippocampus using intracerebral microdialysis in patients undergoing monitoring prior to surgery showed high basal levels of glutamate, a low glutamate to glutamine ratio, and high lactate levels, which suggest poor glucose utilization (135). This is consistent with the observation that FDG PET commonly shows hypometabolism in epileptogenic hippocampi (136,137). Interictal energy deficiency could contribute to impaired glutamate reuptake, persistently elevated glutamate, and EAA neurotoxicity. Abnormal recurrent sprouting of excitatory mossy fibers in the hippocampus that have lost their targets in the CA3 region has also been implicated in this process (138). Autoradiography studies of tissue resected from patients with temporal lobe epilepsy shows reduction in binding to the phencyclidine site of the NMDA receptor/channel complex and a relative increase in associated NMDA receptor sites (136). This resembles the ratio of receptor to channel binding found in the immature rat brain (47). Elevations in non-NMDA receptors have also been reported in human hippocampal tissue from patients with partial complex seizures (139). Studies of human hippocampal AMPA and NMDA messenger RNA and protein levels in brain tissue from temporal lobe epilepsy patients also found elevated levels for AMPA GluR1 and NMDA NR2 subunits (140,141), as well as upregulation of metabotropic mGluR5 receptors (142). These results suggest that alterations in glutamate receptor levels and subunit composition contribute to neuronal hyperexcitability and seizure generation in temporal lobe epilepsy.
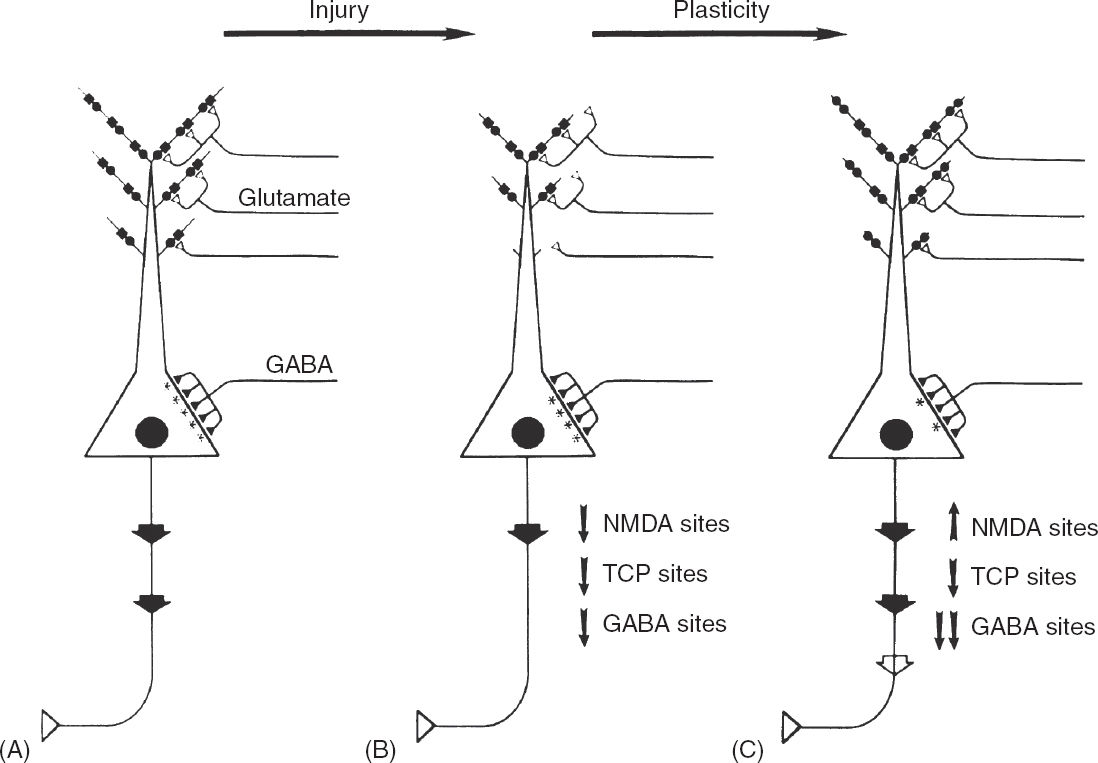
FIGURE 4.4 A speculative proposal of the mechanisms that contribute to the progressive epileptic changes observed in patients with complex partial seizures. (A) Schematic illustration of a typical pyramidal cell from CA1–3 hippocampal subfields. Glutamatergic input (open triangles) from the perforant path originates in the entorhinal cortex and synapses on distal pyramidal cell dendrites and GABAergic interneurons. A severe focal cerebral insult causes overactivation of EAA receptors and produces excitotoxic injury, localized mainly to dendritic regions of the pyramidal cells. (B) The dendritic regions of surviving pyramidal cells are stunted, and the number of functional NMDA recognition sites (solid circles) and corresponding TCP channels (solid squares) is reduced. Moderate loss of functional GABAA sites occurs. As a result of impaired EAA synaptic transmission, the level of pyramidal cell excitation is reduced, as depicted by the large arrow of the emerging axon. In response to reduced pyramidal cell excitation, a series of compensatory synaptic alterations take place. (C) Dendritic zones regenerate, EAA neurotransmission is enhanced by upregulating NMDA recognition sites, and the level of functional inhibition is reduced by downregulating GABAA sites. These changes in turn would compensate for the reduced pyramid cell excitation that results from cerebral injury by increasing excitatory tone. However, these compensatory changes could produce enhanced susceptibility to seizure and seizure-related excitotoxicity by lowering the seizure threshold. The compensatory epileptogenic events lead to a cycle of repeated seizures and injury.
These observations suggest a hypothetical mechanism for progressive epileptic change in certain patients with complex partial seizures (Figure 4.4). In this hypothesis, a severe insult such as prolonged status epilepticus or hypoxia–ischemia leads to EAA release and a glutamate type of injury to neurons in the hippocampus and other susceptible regions. This form of injury causes loss of some pyramidal neurons, but those that remain have stunted dendritic arbors (10,53,143). Perhaps in response to a reduced surface area of dendrites, post-synaptic NMDA receptor/channel complexes adjust by upregulating their NMDA receptors. This would compensate for the reduced dendritic surface area available for excitatory input to the neuron. In addition, a reduction in the number of inhibitory GABA receptors might also serve to increase excitatory tone (136,144).
An increased ratio of NMDA receptors per channel might sensitize the neuron to enhance excitability and further injury from physiologic amounts of synaptic glutamate. Potentially, the seizure threshold might be lower, and further dendritic injury might result from excitatory events that are only mildly superphysiologic. This additional injury in turn leads to a cycle of repeated seizures and injury. An important implication of this model is that pharmacologic attempts could be made to prevent further EAA-mediated injury and thereby halt the progression of the disorder. Based on studies suggesting that there is a distinction between epileptogenesis and epileptic expression, drugs might be developed that would reduce the progressive epileptic process, although they themselves might not be good antiseizure agents (119,120,145).
CONCLUSION
Whether seizures can independently impact neurodevelopmental outcomes has been a topic of active debate for many decades. Recent research and studies designed and conducted to specifically answer this question, indicate that seizures can independently impact neurodevelopmental outcomes (146). However, all pediatric seizures are not equal. Newborns with benign familial neonatal convulsions (BFNC) typically begin having seizures between days 2 and 8 of life and remit by 16 months (147), with very little residual cognitive impairment. This syndrome is due to mutations in KCNQ2 and KCNQ3, which encode the voltage-gated K+ channels, Kv7.2 and Kv7.3. Both KCNQ2 and KCNQ3 are expressed in the brain where the gene products form heteromultimeric channels that mediate the M-current, a slow activating, noninactivating potassium current that serves to inhibit neuronal firing. Therefore, the probability of follow-on metabolic and pharmacologic consequences of seizures heavily depends on the developmental age, underlying etiology, pathology, seizure severity, and efficacy or failure of the acute clinical interventions (148). The early concept that the neonatal brain was less susceptible to injury defined by restricted parameters is now known to be inaccurate both clinically and in animal models. The understanding of the metabolic alterations following neonatal seizures has vastly expanded in recent years from the heavily investigated glutamate/GABA and neuronal centric domains into new and novel domains encompassing non-neuronal cells, glia, inflammation, chloride co-transport, KiR, and infection (149).
REFERENCES
1. Auer RN, Siesjo BK. Biological differences between ischemia, hypoglycemia, and epilepsy. Ann Neurol. 1988;24:699–707.
2. Ingvar M, Siesjo BK. Local blood flow and glucose consumption in the rat brain during sustained bicuculline-induced seizures. Acta Neurol Scand. 1983;68:129–144.
3. Pfund Z, Chugani DC, Juhasz C, et al. Evidence for coupling between glucose metabolism and glutamate cycling using FDG PET and 1H magnetic resonance spectroscopy in patients with epilepsy. J Cereb Blood Flow Metab. 2000;20:871–878.
4. Magistretti PJ, Pellerin L, Rothman DL, et al. Energy on demand. Science. 1999;283:496–497.
5. Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism: Relevance to functional brain imaging and to neurodegenerative disorders. Ann N Y Acad Sci. 1996;777:380–387.
6. Petr GT, Sun Y, Frederick NM, et al. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. 2015;35:5187–5201.
7. Kahle KT, Staley KJ. Neonatal seizures and neuronal transmembrane ion transport. In: Noebels JL, Avoli M, Ragawski MA, et al, eds. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda, MD: National Center for Biotechnology Information (US); 2012:1066–1076.
8. Holmes GL, Khazipov R, Ben-Ari Y. Seizure-induced damage in the developing human: relevance of experimental models. Prog Brain Res. 2002;135:321–334.
9. Shinnar S, Pellock JM, Berg AT, et al. Short-term outcomes of children with febrile status epilepticus. Epilepsia. 2001;42:47–53.
10. Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877.
11. Meldrum BS, Brierly JB. Prolonged epileptic seizures in primates: ischemic cell change and its relationship to ictal physiological events. Arch Neurol. 1973;28:10–17.
12. Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11.
13. McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990;15:41–70.
14. Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155–185.
15. Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379.
16. Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111.
17. Delorenzo RJ, Sun DA. Basic mechanisms in status epilepticus: role of calcium in neuronal injury and the induction of epileptogenesis. Adv Neurol. 2006;97:187–197.
18. Novelli A, Reilly JA, Lysko PG, et al. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212.
19. Silverstein FS, Buchanan K, Johnston MV. Perinatal hypoxia-ischemia disrupts striatal high-affinity [3h]glutamate uptake into synaptosomes. J Neurochem. 1986;47:1614–1619.
20. Watkins JC, Evans RH. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol. 1981;21:165–204.
21. Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402.
22. Watkins JC, Jane DE. The glutamate story. Br J Pharmacol. 2006;147(Suppl 1):S100–S108.
23. Moriyoshi K, Masu M, Ishii T, et al. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37.
24. Ascher P, Nowak L. The role of divalent cations in the N-methyl-D-aspartate responses of mouse central neurons in culture. J Physiol. 1988;399:247–266.
25. Boast CA, Gerhardt SC, Pastor G, et al. The N-methyl-D-aspartate antagonists CGS 19755 and CPP reduce ischemic brain damage in gerbils. Brain Res. 1988;442:345–348.
26. Gill R, Foster AC, Woodruff GN. Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J Neurosci. 1987;7:3343–3349.
27. McDonald JW, Silverstein FS, Johnston MV. Neuroprotective effects of MK-801, TCP, PCP and CPP against N-methyl-D-aspartate induced neurotoxicity in an in vivo perinatal rat model. Brain Res. 1989;490:33–40.
28. Borris DJ, Bertram EH, Kapur J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000;42:117–122.
29. Hamosh A, McDonald JW, Valle D, et al. Dextromethorphan and high-dose benzoate therapy for nonketotic hyperglycinemia in an infant. J Pediatr. 1992;121:131–135.
30. Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and gamma-aminobutyric acid-a receptors. Ann Neurol. 1994;35:229–234.
31. Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170.
32. Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531.
33. Slater P, McConnell SE, D’Souza SW, Barson AJ. Postnatal changes in N-methyl-D-aspartate receptor binding and stimulation by glutamate and glycine of [3H]-MK-801 binding in human temporal cortex. Br J Pharmacol. 1993;108:1143–1149.
34. Wood PL. The NMDA receptor complex: a long and winding road to therapeutics. IDrugs. 2005;8:229–235.
35. Benarroch EE. NMDA receptors: recent insights and clinical correlations. Neurology. 2011;76:1750–1757.
36. Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. The Lancet Neurology. 2008;7:742–755.
37. Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47.
38. Zaczek R, Nelson MF, Coyle JT. Effects of anaesthetics and anticonvulsants on the action of kainic acid in the rat hippocampus. Eur J Pharmacol. 1978;52:323–327.
39. Olney JW, Ho OL, Rhee V. Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp Brain Res. 1971;14:61–76.
40. Hanada T, Hashizume Y, Tokuhara N, et al. Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia. July 2011;52(7):1331–1340.
41. Gryder DS, Rogawski MA. Selective antagonism of glur5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–7074.
42. Curia G, Aracri P, Colombo E, et al. Phosphorylation of sodium channels mediated by protein kinase-c modulates inhibition by topiramate of tetrodotoxin-sensitive transient sodium current. Br J Pharmacol. 2007;150(6):792–797.
43. Simeone TA, Wilcox KS, White HS. Topiramate modulation of β1- and β3-homomeric GABAA receptors. Pharm Res. 2011;64:44–52.
44. Ritter LM, Vazquez DM, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor subunit expression in the rat hippocampus. Brain Res Dev Brain Res. 2002;139:227–236.
45. Brennan EM, Martin LJ, Johnston MV, et al. Ontogeny of non-NMDA glutamate receptors in rat barrel field cortex: ii. alpha-AMPA and kainate receptors. J Comp Neurol. 1997;386:29–45.
46. Blue ME, Johnston MV. The ontogeny of glutamate receptors in rat barrel field cortex. Brain Res Dev Brain Res. 1995;84:11–25.
47. McDonald JW, Johnston MV, Young AB. Differential ontogenic development of three receptors comprising the NMDA receptor/channel complex in the rat hippocampus. Exp Neurol. 1990;110:237–247.
48. Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705.
49. Law AJ, Weickert CS, Webster MJ, et al. Expression of NMDA receptor NR1, NR2A and NR2B subunit MRNAs during development of the human hippocampal formation. Eur J Neurosci. 2003;18:1197–1205.
50. Sheng M, Cummings J, Roldan LA, et al. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147.
51. Slater P, McConnell SE, D’Souza SW, et al. Postnatal changes in N-methyl-D-aspartate receptor binding and stimulation by glutamate and glycine of [3H]-MK-801 binding in human temporal cortex. Br J Pharmacol. 1993;108:1143–1149.
52. Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328.
53. Swann JW. The effects of seizures on the connectivity and circuitry of the developing brain. Ment Retard Dev Disabil Res Rev. 2004;33:96–100.
54. King AE, Cherubini E. Ben-Ari Y. N-methyl-D-aspartate induces recurrent synchronized burst activity in immature hippocampal CA3 neurones in vitro. Brain Res Dev Brain Res. 1989;46:1–8.
55. Swann JW, Brady RJ. Penicillin-induced epileptogenesis in immature rat CA3 hippocampal pyramidal cells. Brain Res. 1984;314:243–254.
56. Brady RJ, Swann JW. Ketamine selectively suppresses synchronized afterdischarges in immature hippocampus. Neurosci Lett. 1986;69:143–149.
57. Pellegrini-Giampietro DE, Gorter JA, Bennett MV, et al. The glur2 (glur-b) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470.
58. Jensen FE. The role of glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci. 2002;20:339–347.
59. Talos DM, Follett PL, Folkerth RD, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. human cerebral white matter and cortex. J Comp Neurol. 2006;497:61–77.
60. Talos DM, Fishman RE, Park H, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. rodent cerebral white matter and cortex. J Comp Neurol. 2006;497:42–60.
61. Koh S, Tibayan FD, Simpson JN, et al. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569–575.
62. Brooks-Kayal AR, Pritchett DB. Developmental changes in human gamma-aminobutyric acida receptor subunit composition. Ann Neurol. 1993;34:687–693.
63. Chandler KE, Princivalle AP, Fabian-Fine R, et al. Plasticity of GABA(B) receptor-mediated heterosynaptic interactions at mossy fibers after status epilepticus. J Neurosci. 2003;23:11382–11391.
64. Johnston MV, Coyle JT. Ontogeny of neurochemical markers for noradrenergic, GABAergic, and cholinergic neurons in neocortex lesioned with methylazoxymethanol acetate. J Neurochem. 1980;34:1429–1441.
65. Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213.
66. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739.
67. Leinekugel X, Khalilov I, McLean H, et al. GABA is the principal fast-acting excitatory transmitter in the neonatal brain. Adv Neurol. 1999;79:189–201.
68. Chudotvorova I, Ivanov A, Rama S, et al. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol. 2005;566:671–679.
69. Ben-Ari Y. Basic developmental rules and their implications for epilepsy in the immature brain. Epileptic Disord. 2006;8:91–102.
70. McDonald JW, Silverstein FS, Johnston MV. Neurotoxicity of N-methyl-D-aspartate is markedly enhanced in developing rat central nervous system. Brain Res. 1988;459:200–203.
71. Campochiaro P, Coyle JT. Ontogenetic development of kainate neurotoxicity: correlates with glutamatergic innervation. Proc Natl Acad Sci U S A. 1978;75:2025–2029.
72. Ikonomidou C, Price MT, Mosinger JL, et al. Hypobaric-ischemic conditions produce glutamate-like cytopathology in infant rat brain. J Neurosci. 1989;9:1693–1700.
73. Holmes GL. Seizure-induced neuronal injury: animal data. Neurology. 2002;59:S3–S6.
74. Burgard EC, Hablitz JJ. Developmental changes in the voltage-dependence of neocortical NMDA responses. Brain Res Dev Brain Res. 1994;80:275–278.
75. Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74.
76. Ikonomidou C, Mosinger JL, Salles KS, et al. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J Neurosci. 1989;9:2809–2818.
77. Jacquin T, Gillet B, Fortin G, et al. Metabolic action of N-methyl-D-aspartate in newborn rat brain ex vivo: 31P magnetic resonance spectroscopy. Brain Res. 1989;497:296–304.
78. McDonald JW, Silverstein FS, Johnston MV. MK-801 protects the neonatal brain from hypoxic-ischemic damage. Eur J Pharmacol. 1987;140:359–361.
79. Nadler JV, Perry BW, Gentry C, et al. Fate of the hippocampal mossy fiber projection after destruction of its postsynaptic targets with intraventricular kainic acid. J Comp Neurol. 1981;196:549–569.
80. Sloviter RS, Damiano BP. Sustained electrical stimulation of the perforant path duplicates kainate-induced electrophysiological effects and hippocampal damage in rats. Neurosci Lett. 1981;24:279–284.
81. Olney JW, Degubareff T, Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path. II. Ultrastructural analysis of acute hippocampal pathology. Brain Res Bull. 1983;10:699–712.
82. Sloviter RS, Dempster DW. “Epileptic” brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Brain Res Bull. 1985;15:39–60.
83. Martin JL, Sloviter RS. Focal inhibitory interneuron loss and principal cell hyperexcitability in the rat hippocampus after microinjection of a neurotoxic conjugate of saporin and a peptidase-resistant analog of substance P. J Comp Neurol. 2001;436:127–152.
84. Clifford DB, Olney JW, Benz AM, et al. Ketamine, phencyclidine, and MK-801 protect against kainic acid-induced seizure-related brain damage. Epilepsia. 1990;31:382–390.
85. Löscher W. High seizure frequency prior to antiepileptic treatment is a predictor of pharmacoresistant epilepsy in a rat model of temporal lobe epilepsy. Epilepsia. 2010;51(1):89–97.
86. Painter MJ. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–489.
87. Weiner SP, Painter MJ, Geva D, et al. Neonatal seizures: electroclinical dissociation. Pediatr Neurol. 1991;7:363–368.
88. Glykys J, Dzhala VI, Kuchibhotla KV, et al. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron. 2009;63:657–672.
89. Hesdorffer DC, Shinnar S, Lewis DV, et al. Design and phenomenology of the FEBSTAT study. Epilepsia. 2012;53(9):1471–1480.
90. Epstein LG, Shinnar S, Hesdorffer DC, et al. Human herpes virus 6 and 7 in febrile status epilepticus: the FEBSTAT study. Epilepsia. 2012;53(9):1481–1488.
91. Dibbens LM, Reid CA, Hodgson B, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. 2010;67:542–546.
92. Tang B, Sander T, Craven KB, et al. Mutation analysis of the hyperpolarization-activated cyclic nucleotide-gated channels HCN1 and HCN2 in idiopathic generalized epilepsy. Neurobiology of Disease. 2008;29:59–70.
93. Shinnar S, Bello JA, Chan S, et al. MRI abnormalities following febrile status epilepticus in children: the FEBSTAT study. Neurology. 2012;79(9):871–877.
94. Dube C, Yu H, Nalcioglu O, et al. Serial MRI after experimental febrile seizures: altered T2 signal without neuronal death. Ann Neurol. 2004;56:709–714.
95. Dube CM, McClelland S, Choy MK, et al. Fever, febrile seizures and epileptogenesis. In: Noebels JL, Avoli M, Ragawski MA, et al, eds. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda, MD: National Center for Biotechnology Information (US); 2012:343–352.
96. Hamelin S, Depaulis A. Revisiting hippocampal sclerosis in mesial temporal lobe epilepsy according to the “two-hit” hypothesis. Rev Neurol (Paris). 2015;171:227–235.
97. Sankar R, Shin DH, Liu H, et al. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393.
98. Sankar R, Auvin S, Mazarati A, et al. Inflammation contributes to seizure-induced hippocampal injury in the neonatal rat brain. Acta Neurol Scand. 2007;115(4 Suppl):16–20.
99. Dudek FE, Shao LR. Loss of GABAergic interneurons in seizure-induced epileptogenesis. Epilepsy Curr. 2003;3:159–161.
100. Dutton SB, Makinson CD, Papale LA, et al. Preferential inactivation of SCN1A in parvalbumin interneurons increases seizure susceptibility. Neurobiology of Disease. 2013;49:211–220.
101. Coulter DA, Steinhäuser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. 2015;5:a022434. doi: 10.1101/cshperspect.a022434.
102. Zurolo E, de GM, Iyer A, et al. Regulation of kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin-1 beta. J Neuroinflammation. 2012;9:280.
103. Vezzani A, Auvin S, Ravizza T, et al. Glia-neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. In: Noebels JL, Avoli M, Ragawski MA, et al, eds. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th ed. Bethesda, MD: National Center for Biotechnology Information (US); 2012:618–633.
104. Vezzani A, Aronica E, Mazarati A, et al. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21.
105. Choi J, Koh S. Role of brain inflammation in epileptogenesis. Yonsei Med J. 2008;49:1–18.
106. Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743.
107. Pavlovsky L, Seiffert E, Heinemann U, et al. Persistent BBB disruption may underlie alpha interferon-induced seizures. J Neurol. 2005;252:42–46.
108. Baram TZ. The brain, seizures and epilepsy throughout life: understanding a moving target. Epilepsy Curr. 2012;12(Suppl 3):7–12.
109. Zovein A, Flowers-Ziegler J, Thamotharan S, et al. Postnatal hypoxic-ischemic brain injury alters mechanisms mediating neuronal glucose transport. Am J Physiol Regul Integr Comp Physiol. 2004;286:R273–R282.
110. Benarroch EE. Brain glucose transporters: implications for neurologic disease. Neurology. 2014;82:1374–1379.
111. Wasterlain CG. Recurrent seizures in the developing brain are harmful. Epilepsia. 1997;38:728–734.
112. McDonald JW, Johnston MV. Excitatory amino acid neurotoxicity in the developing brain. NIDA Res Monogr. 1993;133:185–205.
113. Stafstrom CE, Holmes GL. Infantile spasms: criteria for an animal model. Int Rev Neurobiol. 2002;49:391–411.
114. Rho JM. Basic science behind the catastrophic epilepsies. Epilepsia. 2004;45(Suppl 5):5–11.
115. Stafstrom CE, Chronopoulos A, Thurber S, et al. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–432.
116. McDonald JW, Trescher WH, Johnston MV. Susceptibility of brain to AMPA induced excitotoxicity transiently peaks during early postnatal development. Brain Res. 1992;583:54–70.
117. Iadorola MJ, Nicoletti F, Naranjo JR, et al. Kindling enhances the stimulation of inositol phospholipid hydrolysis elicited by ibotenic acid in rat hippocampal slices. Brain Res. 1986;374:174–178.
118. Hablitz JJ, Lee WL. NMDA receptor involvement in epileptogenesis in the immature neocortex. Epilepsy Res Suppl. 1992;8:139–145.
119. Stasheff SF, Anderson WW, Clark S, et al. NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science. 1989;245:648–651.
120. Jensen FE. The role of glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci. 2002;20:339–347.
121. Suchomelova L, Baldwin RA, Kubova H, et al. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res. 2006;59:237–243.
122. Mattson MP. Neurotransmitters in the regulation of neuronal cytoarchitecture. Brain Res. 1988;472:179–212.
123. Kleinschmidt A, Bear MF, Singer W. Blockade of “NMDA” receptors disrupts experience-dependent plasticity of kitten striate cortex. Science. 1987;238:355–358.
124. Nicoll RA. Expression mechanisms underlying long-term potentiation: a postsynaptic view. Philos Trans R Soc Lond B Biol Sci. 2003;358:721–726.
125. Brooks-Kayal AR. Rearranging receptors. Epilepsia. 2005;46:29–38.
126. Jones-Davis DM, Macdonald RL. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr Opin Pharmacol. 2003;3:12–18.
127. Sankar R, Shin D, Liu H, et al. Epileptogenesis during development: injury, circuit recruitment, and plasticity. Epilepsia. 2002;43(Suppl 5):47–53.
128. Nishimura T, Schwarzer C, Gasser E, et al. Altered expression of GABA(A) and GABA(B) receptor subunit MRNAs in the hippocampus after kindling and electrically induced status epilepticus. Neuroscience. 2005;134:691–704.
129. Naylor DE, Liu H, Wasterlain CG. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733.
130. Lewis DV, Barboriak DP, MacFall JR, et al. Do prolonged febrile seizures produce medial temporal sclerosis? Hypotheses, MRI evidence and unanswered questions. Prog Brain Res. 2002;135:263–278.
131. Lewis DV. Losing neurons: selective vulnerability and mesial temporal sclerosis. Epilepsia. 2005;46(Suppl 7):39–44.
132. Fernandez G, Effenberger O, Vinz B, et al. Hippocampal malformation as a cause of familial febrile convulsions and subsequent hippocampal sclerosis. 1998. Neurology. 2001;57:S13–S21.
133. Vanlandingham KE, Heinz ER, Cavazos JE, et al. Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol. 1998;43:413–426.
134. Sutula TP, Pitkanen A. More evidence for seizure-induced neuron loss: is hippocampal sclerosis both cause and effect of epilepsy? Neurology. 2001;57:169–170.
135. Cavus I, Kasoff WS, Cassaday MP, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–235.
136. McDonald JW, Garofalo EA, Hood T, et al. Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann Neurol. 1991;29:529–541.
137. Chassoux F, Semah F, Bouilleret V, et al. Metabolic changes and electro-clinical patterns in mesio-temporal lobe epilepsy: a correlative study. Brain. 2004;127:164–174.
138. Holmes GL. Epilepsy in the developing brain: lessons from the laboratory and clinic. Epilepsia. 1997;38:12–30.
139. Hosford DA, Crain BJ, Cao Z, et al. Increased AMPA-sensitive quisqualate receptor binding and reduced NMDA receptor binding in epileptic human hippocampus. J Neurosci. 1991;11:428–434.
140. Mathern GW, Pretorius JK, Kornblum HI, et al. Human hippocampal AMPA and NMDA MRNA levels in temporal lobe epilepsy patients. Brain. 1997;120(Pt 11):1937–1959.
141. Mathern GW, Pretorius JK, Mendoza D, et al. Increased hippocampal AMPA and NMDA receptor subunit immunoreactivity in temporal lobe epilepsy patients. J Neuropathol Exp Neurol. 1998;57:615–634.
142. Notenboom RG, Hampson DR, Jansen GH, et al. Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain. 2006;129:96–107.
143. Swann JW, Al-Noori S, Jiang M, et al. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus. 2000;10:617–625.
144. Loup F, Wieser HG, Yonekawa Y, et al. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J Neurosci. 2000;20:5401–5419.
145. McNamara JO, Russell RD, Rigsbee L, et al. Anticonvulsant and antiepileptogenic actions of MK-801 in the kindling and electroshock models. Neuropharmacology. 1988;27:563–568.
146. Holmes GL, Ben-Ari Y. Seizures in the developing brain: perhaps not so benign after all. Neuron. 1998;21:1231–1234.
147. Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–2737.
148. Staley K. Neonatal encephalopathy MRI lesions, and later epilepsy: no harm, no foul? Epilepsy Curr. 2012;12:128–130.
149. Noam Y, Raol YH, Holmes GL. Searching for new targets for treatment of pediatric epilepsy. Epilepsy Behav. 2013;26:253–260.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
[/not-level-membership-for-pediatrics-category]
