Anaphylaxis and Anaphylactic Shock
PATHOGENESIS AND PATHOPHYSIOLOGY
Immunologic Events Leading to Mast Cell and Basophil Activation and Mediator Release
Nonimmunologic Events Leading to Mediator Release
Cellular Characteristics of Anaphylaxis
Biochemical Mediators of Anaphylaxis
Biochemical and Pharmacologic Regulation of Mediator Release
The term anaphylaxis refers to a life-threatening event, allergic in nature, which may result from IgE- or non-IgE-mediated mast cell degranulation. The clinical manifestations of severe anaphylaxis are often explosive in onset and may lead to upper airway obstruction, respiratory failure, and circulatory shock. Milder symptoms can also develop. The term anaphylactoid reaction, which referred to a non-IgE-mediated reaction, is no longer being used. A recent National Institutes of Health (NIH) consensus conference defined anaphylaxis as one of the following three scenarios.1
1. Acute onset of a reaction (minutes to hours) with involvement of the skin, mucosal tissue, or both and at least one of the following: (a) respiratory compromise (b) or reduced blood pressure or symptoms of end-organ dysfunction;
2. Two or more of the following that occur rapidly after exposure to a likely allergen for that patient—involvement of the skin/mucosal tissue, respiratory compromise, reduced blood pressure or associated symptoms, and persistent gastrointestinal symptoms; or
3. Reduced blood pressure after exposure to a known allergen (adapted from Sampson and colleagues1).
The IgE-mediated anaphylactic response is classified as a type I reaction according to the Gell and Coombs classification. The IgE-mediated anaphylactic reaction has clinical features similar to other, milder type I reactions, such as allergic rhinitis, hives, urticaria, and allergic asthma. IgE-mediated anaphylactic reactions are characterized by a well-defined immunologic sequence of events that involves antigen-specific and IgE-specific effector cells. When stimulated, these cells release a variety of inflammatory mediators.2 The effector cells consist of mast cells and basophils, which are based primarily in tissues and in the circulating blood volume. Severe anaphylaxis rapidly progresses to a generalized systemic reaction. Agents that produce well-documented IgE-mediated anaphylactic reactions include medications such as beta lactam antibiotics, biologic agents, nonsteroidal anti-inflammatory drugs (NSAIDs), foods (peanut and other legumes, nuts from trees [walnuts, almonds, etc.], milk, and egg), Hymenoptera venoms (honey bees, yellow jackets, hornets, wasps, and fire ants), natural rubber latex, occupational allergens, and seminal fluid prostate-specific antigen (Table 28.1).
Table 28.1
Agents Frequently Associated with Immune and Nonimmune Types of Anaphylaxis
| Category | Examples |
| Antibiotics | Penicillin and penicillin analogues, β-lactam antibiotics, cephalosporins, tetracyclines, erythromycin |
| Nonsteroidal anti-inflammatory drugs | Salicylates, ibuprofen, indomethacin |
| Narcotic analgesics | Morphine, codeine, meprobamate |
| Local anesthetics | Procaine, lidocaine, cocaine |
| General anesthetics | Thiopental |
| Muscle relaxants | Suxamethonium, tubocurarine, pancuronium |
| Blood products and antisera | Red blood cell, white blood cell, and platelet transfusions; gamma globulin; rabies, tetanus, diphtheria antitoxin; snake and spider antivenom |
| Diagnostic agents | Iodinated radiocontrast agents |
| Foods | Eggs, milk, nuts, legumes (peanuts, soybeans, kidney beans), fish, shellfish |
| Venoms | Bees, wasps, hornets, fire ants, scorpions, snakes |
| Enzymes and other biologic agents | Acetylcysteine, pancreatic enzyme supplements, chymopapain |
| Extracts of potential allergens used in desensitization | Pollen, food, venom extracts |
| Chemotherapeutic agents | Cisplatin, cyclophosphamide, daunorubicin, methotrexate |
| Insulin | Pork, beef, and human insulin |
| Other drugs | Protamine, chlorpropamide, parenteral iron, iodides, thiazide diuretics |
Immune-mediated IgE-dependent or IgE-independent and non-immune-mediated mast cell degranulation-mediated anaphylactic reactions have similar clinical features. Agents capable of producing direct mast cell degranulation include NSAIDs, opiates, ciprofloxacin, and physical factors, (cold, heat, exercise). Patients who are taking beta blockers or ACE inhibitors may develop severe anaphylaxis to an inciting agent and are less likely to respond to first-line agents.3–6
In idiopathic anaphylaxis, the pathophysiology and triggering events are unknown. Idiopathic anaphylaxis is a diagnosis of exclusion. It is mostly seen in adults and adolescents. About half these patients have concomitant atopic disease. They respond to treatment with corticosteroids and antihistamines.7,8 Special testing like serum tryptase, C4 levels may be necessary to exclude conditions such as systemic mastocytosis, hereditary angioedema, and acquired C1 inhibitor deficiency.
History and Incidence
Allergic emergencies have been described in humans since ancient times.9–11 At the turn of the twentieth century, a more detailed description of these events was reported by two French physiologists, Portier and Richet.12 They coined the term anaphylaxis, which originates from the French word anaphylactique, which means “reverse protection.” It was believed that these reactions were in contrast to the attenuated or tachyphylactic reactions that commonly protect subjects from reintroduced antigens such as viruses. More recent research defining the role of IgE; the interactions between IgE, antigen, mast cells, basophils, and eosinophils; and the biochemical mediators from these cells has clarified the events leading to clinical anaphylaxis.2,13–15
The true incidence of the various types of anaphylactic reactions is difficult to determine because these reactions are often spontaneous and unpredictable and are clinically similar to other acute reactions. Lifetime prevalence of anaphylaxis due to all triggers is estimated to be 0.05% to 2%.16 Estimates of the incidence of the most commonly reported episodes are possible, however. In the United States, penicillin alone probably accounts for several hundred fatalities each year.17–19 Anaphylaxis to the cephalosporins also is commonly reported.20 It has been estimated that among patients with an allergic reaction to a penicillin, there is a 3% to 7% rate of allergic reaction to a cephalosporin. Reports of anaphylactic reactions to the newer β-lactam antibiotics are accumulating.21
Insects, especially those of the Hymenoptera order, which includes bees, wasps, hornets, and fire ants, account for numerous immediate hypersensitivity reactions. About 3% of adults and 1% of children are affected and the anaphylaxis can be fatal at the first sting. About 50 people experience fatal reactions to insect stings every year in the United States, and about half of them do not have a prior sting exposure.22–24 Fire ants are aggressive insects from South America that now reside in the southern United States. In some areas, they have been known to sting 58% of the residents yearly and account for serious allergic reactions.25
Snake bites account for probably a dozen or so anaphylactic deaths per year in the United States. Snake bites may be associated with typical anaphylactic symptoms and other problems related to the enzymes, proteins, and peptides in venom. Local tissue necrosis, coagulation problems, hemolysis, and neurologic transmission defects have been described. In the United States, most anaphylactic reactions to snake bites are caused by pit vipers. These snakes include rattlesnakes, water moccasins, and copperheads.26,27
Food-induced anaphylaxis is probably the most common cause of anaphylaxis and accounts for 30% of fatalities. Peanuts (legumes) and typical nuts from trees account for 90% of fatal cases. Additional common food antigens include fish, soybeans, egg whites, and shellfish.28 Biphasic reactions are much more common in food-induced anaphylaxis than in other types of anaphylaxis and have been reported in 25% of fatal cases.29–31
Iodinated contrast agents account for approximately 125 deaths per year32 and lead to clinical symptoms similar to anaphylaxis. Life-threatening reactions are extremely rare, with an incidence of 0.1%.33,34 With the advent of low osmolar iodinated contrast agents, the risk of anaphylaxis to contrast agents has decreased drastically.35
Latex, used in surgical gloves, balloons, condoms, rubber bands, and many other products, may produce anaphylaxis.36,37 The use of universal precautions as a result of the acquired immunodeficiency syndrome epidemic has increased the number of reactions to latex in health care workers. Children with spina bifida and genitourinary tract abnormalities are especially susceptible to latex-induced anaphylaxis because of frequent exposure to latex-containing bladder catheters and other products.
Anaphylactic reactions during anesthesia have been described and typically are associated with hypotension and cardiopulmonary arrest. One review suggests that most cases of intraoperative anaphylaxis are from muscle relaxants (e.g., suxamethonium, tubocurarine, pancuronium).38 Latex, protamine, and blood products also may cause intraoperative anaphylaxis.
Anaphylaxis and other types of IgE-mediated allergic reactions tend to occur in susceptible, genetically predisposed individuals. The reason for the genetic inheritance of sensitivity to the antigens that produce anaphylaxis continues to be speculative. A popular theory is that type I reactions, when confined to an area of parasitic invasion (e.g., intestinal tract), facilitate the killing and removal of parasites and confer a survival advantage to individuals capable of mounting a type I response. Various clinical and laboratory observations support this view.39–41 The sites of IgE synthesis in laboratory subjects correspond to the sites of entry of many parasites. These sites include the lymphoid tissue of the respiratory tract, the gastrointestinal tract, and the skin. Eosinophils, cells that migrate to the site of antigen introduction in anaphylaxis, elaborate mediators that are toxic to the outer parasitic covering.
Pathogenesis and Pathophysiology
Immunologic Mechanisms Leading to Mast Cell and Basophil Activation and Mediator Release
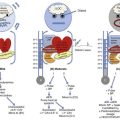
When an antigen to which an individual has previously been sensitized is reintroduced, a sequence of events is initiated that leads to mediator release (Fig. 28.1). At least several weeks are required between the initial exposure to antigen and a subsequent exposure for clinical manifestations of anaphylaxis to occur. The antigen may be introduced through the skin, respiratory tract, or gastrointestinal tract. Antigen also may be introduced intravenously, usually in association with drug administration. Although most venoms are injected subcutaneously, some may access the circulation through an intravascular route.
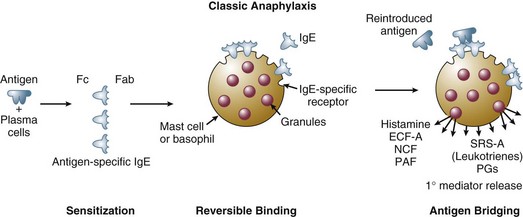
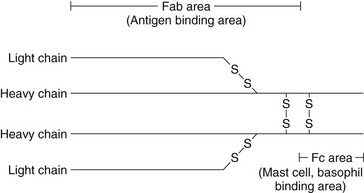
In most cases of anaphylaxis, when antigen is reintroduced into the host, it encounters IgE, previously synthesized by plasma cells in response to a previous introduction of antigen. IgE, similar to other immunoglobulins, is composed of two heavy chains and two light chains linked by disulfide bonds. Two portions of the molecule have well-defined functions. The Fab portion of the molecule recognizes and binds antigen. The Fc portion of the molecule binds reversibly to receptors on the surface of mast cells and basophils (Fig. 28.2).
The combination of reintroduced antigen with antigen-specific IgE sets the stage for a sequence of biochemical and cellular events that produce the clinical syndrome of anaphylaxis. The bivalent antigen cross-bridges two IgE molecules (see Fig. 28.1). Cross-bridging facilitates the approximation of Fc surface receptors on mast cells and basophils, triggering the release of mediators from intracellular granules and membrane-based phospholipids. The systemic release of these mediators leads to the pathophysiologic changes that produce the clinical manifestations of anaphylaxis.
Clinical reactions similar to anaphylaxis mediated by immunoglobulins of the IgG class have been described. IgG molecules may combine with antigens, producing an antigen-antibody complex that activates complement. Activation of complement generates C3a and C5a, also known as anaphylatoxins because they stimulate mediator release from mast cells and basophils. IgG-mediated reactions are considered type III reactions according to the classification of Gell and Coombs or may be referred to as Arthus reactions. These reactions may characterize IgA-deficient individuals who exhibit sudden reactions to blood transfusions. These individuals may develop an antibody of the IgG class to the IgA in the transfused blood product.42,43 This combination of IgG and IgA antibody activates complement, generates C3a and C5a, and produces a sudden reaction typical of anaphylaxis. Because approximately 1 in 700 individuals is IgA deficient, numerous people are susceptible to this type of blood transfusion reaction.43 The anaphylactic responses to protamine may be IgG-, IgE-, or non-immune-mediated.44–46
Nonimmunologic Events Leading to Mediator Release
The mediators released during nonimmune reactions originate from mast cells and basophils and are identical to the mediators of immune-mediated anaphylaxis. The direct activation of surface receptors on mast cells and basophils by antigen may be responsible for mediator release in these reactions. Iodinated contrast agents, opiates, and highly charged polyionic antibiotics seem to activate surface receptors directly. Physical stimuli, including heat, cold, and hyperosmolar stimuli, also are capable of stimulating mast cells and basophils. Exercise-induced anaphylaxis may be associated with the stimulation of mast cells through cooling of the airways. Other possible mechanisms include complement activation without immune complex mediation (old preparations of propafol with cremophor diluent),47 direct mast cell degranulation (merperidine),48 and direct activation of the kinin-kallikrein pathway, resulting in the generation of bradykinin, C3a, and C5a (oversulfated chondroitin sulfate in heparin products).49
Cellular Characteristics of Anaphylaxis
Despite the release of similar mediators during anaphylaxis, mast cells and basophils differ in several ways. Mast cells are more abundant than basophils and generally reside in the connective tissue of subcutaneous and submucosal areas. Basophils characteristically circulate in the blood.50 Despite these differences in location and number, functional differences between the two cell types have not been clearly identified. Both types of cells have receptors for the Fc portion of IgE, and both have granules that bind basic dyes. In addition, the granules of both cells contain histamine and various other mediators that participate in the anaphylactic response.
Eosinophils are commonly identified in the tissues and plasma of patients with both immune- and non-immune-mediated anaphylactic reactions. These cells typically migrate to the site of antigen introduction. They are attracted by a variety of chemotactic factors, including factors derived from mast cells and basophils, antigen-antibody complexes, histamine, and complement. The granules of eosinophils stain with acidophilic dyes and contain a variety of biochemical mediators that are toxic to helminthic parasites. Substances that inactivate leukotrienes and histamines also are elaborated. Eosinophils function as modulators of the inflammatory response triggered by mast cell and basophil activation.51,52
Biochemical Mediators of Anaphylaxis
The biochemical mediators of anaphylaxis are divided into primary and secondary mediators. Mediators directly released from mast cells and basophils are termed primary mediators (Table 28.2). Secondary mediators are released from other cell types in response to primary mediator release (Table 28.3). Primary mediators are subdivided further into preformed and newly synthesized mediators. Preformed mediators are formed and stored in the intracellular granules of mast cells and basophils. Newly synthesized mediators are derived from the metabolism of arachidonic acid, a phospholipid derived from cell membrane.
Table 28.2
Physiologic Effects of Primary Mediators of Anaphylaxis Derived from Mast Cells and Basophils
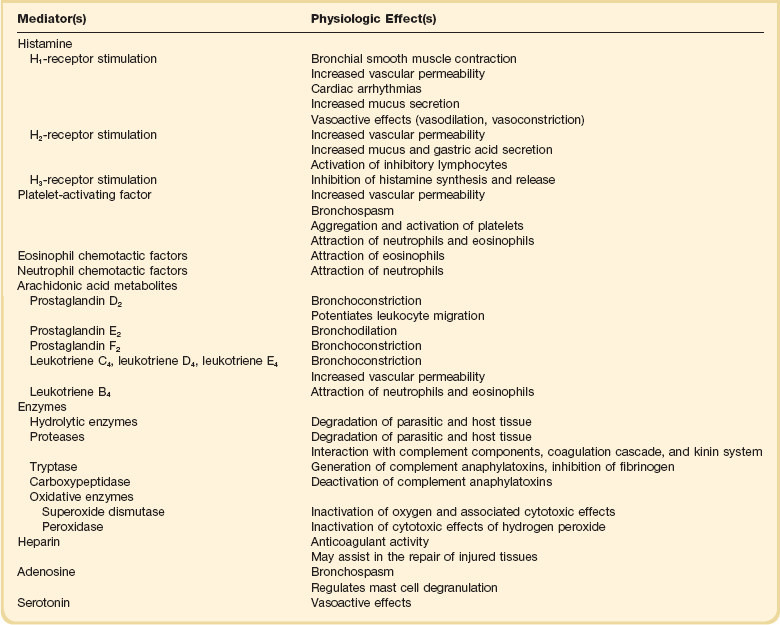
Table 28.3
Secondary Mediators of Anaphylaxis and Physiologic Effects
| Mediator(s) | Physiologic Effect(s) |
| Neutrophil, platelet, and eosinophil-derived mediators | Permeability, coagulation changes, proteolysis |
| Activated complement system | |
| C3a and C5a | Contract bronchial smooth muscle; increase vascular permeability; attract neutrophils, macrophages, and monocytes |
| C6-C9 | Membrane damage |
| Activated coagulation cascade | Intravascular coagulation, permeability changes, tissue injury |
| Activated kinin system (bradykinin) | Increases vascular permeability |
Biochemical and Pharmacologic Regulation of Mediator Release
The biochemical regulation of mediator release provides a rationale for the pharmacologic therapy of anaphylaxis. The release of primary mediators is thought to be modified by intracellular levels of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) and calcium and other bivalent cations. Pharmacologic agents that affect the intracellular levels of these modulators and inhibit mediator release are often used in the treatment of anaphylaxis (Box 28.1).
Mediator release from mast cells and basophils is associated with an influx of calcium. Although calcium blocking agents theoretically may be useful in the treatment of anaphylaxis, clinical experience with use of these agents for this condition is lacking. Conversely, calcium administration may be harmful in anaphylaxis because of its association with enhanced mediator release. Other bivalent cations, such as magnesium and manganese, also enhance mediator release. Multiple tyrosine kinases are activated and regulate by exerting either stimulatory or inhibitory actions on the signal transduction cascade.53–55 Sphingosine-1-phosphate has been identified recently as one of the mediators that serves as a modulator in the mast cell. In addition, it is now labeled as a circulating mediator in anaphylaxis.56
Pathophysiologic Effects of Mediators
The numerous mediators released during anaphylactic crisis have many physiologic effects that have been studied extensively in the laboratory. Although it is difficult to determine the specific actions of each mediator in anaphylaxis, the cumulative effects of mediator release have been described in the clinical setting. These effects include abnormalities secondary to increased vascular permeability; vascular resistance changes, primarily vasodilation; and bronchospasm. Autopsies of fatal cases of anaphylaxis reveal edema of the lungs, upper airway (including the larynx and epiglottis), skin, and viscera. Pulmonary congestion is typical in fatal anaphylaxis, and light microscopy often reveals fluid-filled pulmonary alveoli.57 In another series of fatal cases of anaphylaxis, acute pulmonary emphysema was observed in almost half of cases.58 This condition is characterized by hyperextended alveoli and thinning of the alveolar septum. Because of the association of acute pulmonary emphysema with laryngeal edema, these fatalities were thought to be caused by upper airway obstruction, with alveolar rupture resulting from forced exhalation against the obstruction.
Cardiac abnormalities, including arrhythmias, reduced contractility, and myocardial ischemia, have been described in anaphylaxis but seem to be uncommon.59 These abnormalities may be secondary to the effects of histamine and other mediators on the myocardium. Other contributing factors include circulatory shock, hypotension, increased adrenergic tone, and drugs used to treat anaphylaxis.
Late-Phase or Biphasic Reactions
Both immune-mediated and non-immune-mediated anaphylactic reactions may be followed by late-phase reactions (also termed biphasic reactions). These reactions typically occur 6 to 12 hours after the initial reaction as a result of the migration of mast cells, basophils, and polymorphonuclear leukocytes into areas of antigen introduction. A secondary wave of mediator release and recurrence of symptoms may be observed.60–62
Clinical and Hemodynamic Features
The most life-threatening reactions are usually explosive in nature, often occurring within minutes of exposure to the antigen. Victims of these reactions have been noted to describe a feeling of impending doom before more defined symptoms develop. Generalized cutaneous abnormalities include erythema, urticaria, and flushing. Swelling of the periorbital and perioral areas is characteristic. Upper and lower airway abnormalities are common and especially dangerous. Swelling of the posterior pharynx, uvula, tonsils, and vocal cords may develop rapidly. Auscultation of the chest may reveal generalized wheezing and prolongation of expiration. Auscultatory and radiographic signs of pulmonary edema are characteristic of severe episodes. Signs of circulatory shock include hypotension, oliguria, and lactic acidosis from intravascular volume depletion. In some instances, such as the intravenous injection of venom or a drug, circulatory shock may develop without preceding cutaneous and respiratory abnormalities. The clinical features of anaphylaxis may respond quickly to treatment or, in the most severe cases, may last for several hours to several days. An initial favorable response to treatment may be followed by a late-phase reaction—a recurrence of symptoms resulting from a second wave of mediator release approximately 6 to 12 hours after the initial reaction.60,62
Hemodynamic descriptions of human anaphylaxis are limited to detailed studies of a few cases. The loss of circulating plasma volume is characteristic and is associated with hemoconcentration, hypotension, tachycardia, decreased cardiac filling pressures, and decreased cardiac output.63,64 Vasodilation, associated with a decrease in systemic vascular resistance, may contribute to the reduction in venous return and cardiac output. When oxygen delivery decreases to levels below systemic oxygen demands, anaerobic metabolic pathways are activated, and lactic acidosis emerges.65 Decreases in myocardial contractility seem to be minimal in studies of human anaphylaxis using routine hemodynamic monitoring. This is supported by the observation that most patients with anaphylaxis respond favorably to fluid therapy and do not require inotropic support.63–67 In a few case reports, reduced myocardial contractility has been observed in association with myocardial ischemia and infarction.68–73 Some of these adverse cardiac effects have been associated with epinephrine administration, but in some cases they also have been noted before pharmacologic treatment.68–73
Laboratory studies provide more detailed descriptions of the hemodynamic features of anaphylaxis. After antigenic challenge in primates, a transient increase in cardiac output is observed and is followed by decreases in arterial pressure, right and left ventricular filling pressures, and peripheral vascular resistance.74 The transient increase in cardiac output has been attributed to vasodilation-induced left ventricular unloading or an increase in cardiac contractility or both. Elevated plasma levels of epinephrine, norepinephrine, and histamine have been shown in laboratory animals and humans and may contribute to this increase in contractility.75,76 Cardiac output eventually decreases when hypotension and shock become established. In human and canine models of anaphylaxis, a reduction in venous return has been observed secondary to vasodilation and pooling of blood in the splanchnic circulation.77–79 Loss of plasma volume from increased vascular permeability is probably a contributing factor.
When pulmonary edema fluid is sufficiently copious to be sampled from the airway of patients with anaphylaxis, albumin concentrations and oncotic pressures are nearly identical to plasma values. These findings and the association of pulmonary edema with low pulmonary artery wedge pressures suggest that the pulmonary edema in anaphylaxis is noncardiogenic and secondary to increased microvascular permeability.63 Although transient pulmonary hypertension and increased pulmonary vascular resistance have been observed in primates immediately after antigen challenge,74 it is unknown whether pulmonary hypertension characterizes human anaphylaxis.
Management
Initial Management
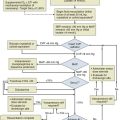
The initial assessment of a patient with suspected anaphylaxis should be brief and specific because immediate therapeutic interventions are required. Because a variety of conditions may appear similar to anaphylaxis (Box 28.2), it is important to rule out these events quickly. Vasovagal episodes are among the most common conditions confused with anaphylaxis. Bradycardia, pale skin, and diaphoresis in an acutely ill patient are suggestive of a vasovagal attack, in contrast to the tachycardic, flushed appearance typical of anaphylaxis.
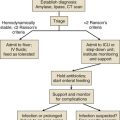
When it is strongly suspected that the patient is experiencing a severe or potentially severe anaphylactic episode, the following steps should proceed rapidly (Fig. 28.3): (1) assurance of a patent airway, (2) removal of toxin at the site of introduction or an attempt to delay the systemic absorption of toxin or both, (3) establishment of intravenous access for fluid therapy, and (4) initiation of pharmacologic support with epinephrine (Box 28.3). A team approach is essential in severe cases of anaphylaxis because assessment and interventions must proceed rapidly and, if possible, simultaneously.
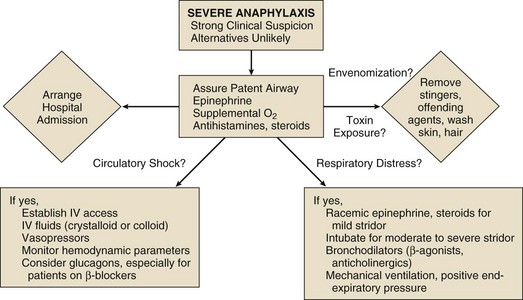
Admission to the hospital is required for all patients experiencing severe anaphylaxis. Hospital personnel should have advanced airway skills and be capable of managing hemodynamic instability. Although symptom recurrence is uncommon in patients who respond favorably to treatment,79 hospital admission and monitoring is recommended for all patients with severe anaphylaxis because of the potential for late-phase reactions, which may be severe and may occur 12 hours after the initial attack. While in the hospital, the patient should be monitored for signs of circulatory shock, respiratory failure, and upper airway obstruction. Blood pressure, urine output, and heart and respiratory rate require frequent evaluation.
If intubated patients are unable to breathe spontaneously or have labored respirations, mechanical ventilation should be initiated. Positive end-expiratory pressure and other advanced ventilatory techniques are often necessary when hypoxemia, pulmonary edema, and decreased pulmonary compliance develop. In nonintubated patients, 6 to 8 L per minute of oxygen through a facemask is recommended.80
If hypotension and other signs of circulatory shock persist after the initial administration of epinephrine and fluids, then intravenous epinephrine is recommended. Bolus doses are no longer recommended because of dosing errors and side effects.81 The initial dose for intravenous epinephrine infusion is 2 to 10 µg per minute, titrated to effect on blood pressure.82 Sometimes patients may need an additional vasopressor along with epinephrine.
Additional Therapeutic Options
Although the role of epinephrine in anaphylaxis is established, the role of antihistamines and corticosteroids continues to be debated.80 Nonetheless, corticosteroids should be administered in severe cases of anaphylaxis because there is evidence that they prevent or attenuate late-phase reactions, increase tissue responsiveness to β-agonists, and inhibit the synthesis of histamine. Methylprednisolone or hydrocortisone may be given for 72 hours and can then be rapidly tapered off because almost all the biphasic reactions occur within 72 hours.83,84
Several other agents have been used in cases of human anaphylaxis or in laboratory models of anaphylaxis with apparent success. Glucagon, a pancreatic hormone that increases intracellular cAMP levels by activating adenylate cyclase, was effective in a case report of a patient with anaphylaxis who was receiving beta-blocker therapy.85,86 Current research focuses on understanding mast cell and basophil biology, targeting tyrosine kinases, and developing antibodies to IgE.87,88
Prophylaxis and Immunotherapy
If a patient must be treated with a drug that has previously produced severe allergic symptoms or anaphylaxis and no alternative exists, premedication should be implemented. Most authorities recommend premedication with H1 and H2 blockers and corticosteroids. Several studies have confirmed that premedication with these agents decreases anaphylactic reactions to radiocontrast media.89–91 In very high risk patients, some authorities believe that epinephrine or isoproterenol should be included as premedication.92 Because fatal anaphylaxis has been described in patients who received premedication, it is preferable, if clinically possible, to avoid all antigens associated with anaphylaxis.
Methods to desensitize individuals immediately before administration of a drug have been described in detail, especially for penicillin,93 aspirin,94 and insulin.95 These techniques, which involve exposure to antigen in 20- to 30-minute increments, may be unsuccessful, however, and occasional fatalities have been reported.96–98
Long-term desensitization may be useful in patients who have experienced anaphylaxis to antigens that are difficult to avoid, especially foods and venoms. This type of immunotherapy involves initial injection of a minute dose of antigen followed by gradual increases in dose at weekly or biweekly intervals according to the patient’s tolerance.99,100 A non-IgE-blocking antibody forms and decreases the reactivity of mast cells and basophils to antigen.
References
1. Sampson, HA, Munoz-Furlong, A, Campbell, RL, et al. Second symposium on the definition and management of anaphylaxis: Summary report Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis network symposium. J Allergy Clin Immunol. 2006; 117:391–397.
2. Serafin, WE, Austin, KF. Mediators of immediate hypersensitivity reactions. N Engl J Med. 1987; 317:30.
3. Simons, FER. Anaphylaxis: Recent advances in assessment and treatment. J Allergy Clin Immunol. 2009; 124:625–636.
4. Lieberman, PL. Anaphylaxis. In: Adkinson NF, Jr., Bochner BS, Busse WW, et al, eds. Middleton’s Allergy: Principles and Practice. 7th ed. St. Louis: Mosby; 2009:1027–1049.
5. Mueller, UR. Cardiovascular disease and anaphylaxis. Curr Opin Allergy Clin Immunol. 2007; 7:337–341.
6. Lang, DM. Do beta-blockers really enhance the risk of anaphylaxis during immunotherapy? Curr Allergy Asthma Rep. 2008; 8:37–44.
7. Ditto, AM, Harris, KE, Krasnick, J, et al. Idiopathic anaphylaxis: A series of 335 cases. Ann Allergy Asthma Immunol. 1996; 77:285–291.
8. Wong, S, Yarnold, PR, Yango, C, et al. Outcome of prophylactic therapy for idiopathic anaphylaxis. Ann Intern Med. 1991; 114:133–136.
9. Waddell, LA. Egyptian Civilization: Its Sumerian Origin and Real Chronology and Sumerian Origin of Egyptian Hieroglyphs. London: Luzac; 1930.
10. Chafee, FH. Insect-sting allergy. J Allergy. 1969; 43:309.
11. Cohen, SG. The pharaoh and the wasp. Allergy Proc. 1989; 10:149.
12. Portier, P, Richet, C. De l’action anaphylactique de certains venins. Comput Rend Soc Biol (Paris). 1902; 54:170.
13. Ishizaka, T. IgE and mechanisms of IgE mediated hypersensitivity. Ann Allergy. 1982; 48:313.
14. Murphy, RC, Hammarstrom, S, Samuelsson, B. Leukotriene C: A slow-reacting substance from murine mastocytoma cells. Proc Natl Acad Sci U S A. 1979; 76:4275.
15. Lewis, RA, Austen, KF, Soberman, RJ. Leukotrienes and other products of the 5-lipoxygenase pathway: Biochemistry and relation to pathobiology in human diseases. N Engl J Med. 1990; 323:645.
16. Lieberman, P, Camargo, CA, Jr., Bohlke, K, et al. Epidemiology of anaphylaxis: Findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol. 2006; 97:596–602.
17. Idsoe, O, Guthe, T, Willcox, RR, et al. Nature and extent of penicillin side-reactions with particular reference to fatalities from anaphylactic shock. Bull World Health Organ. 1968; 38:159.
18. Park, MA, Li, JT. Diagnosis and management of penicillin allergy. Mayo Clin Proc. 2005; 80:405.
19. Weiss, ME, Adkinson, NF. Immediate hypersensitivity reactions to penicillin and related antibiotics. Clin Allergy. 1988; 18:515.
20. Kabins, SA, Eisenstein, B, Cohen, S. Anaphylactoid reaction to an initial dose of sodium cephalothin. JAMA. 1965; 193:165.
21. Saxon, A, Beall, GN, Rohr, AS, et al. Immediate hypersensitivity reactions to beta-lactam antibiotics. Ann Intern Med. 1987; 107:204.
22. Golden, DB. Insect sting allergy and venom immunotherapy: A model and a mystery. J Allergy Clin Immunol. 2005; 115:439–447.
23. Golden, DB, Marsh, DG, Kagey-Sobotka, A, et al. Epidemiology of insect venom sensitivity. JAMA. 1989; 262:240–244.
24. Barnard, JH. Studies of 400 Hymenoptera sting deaths in the United States. J Allergy Clin Immunol. 1973; 52:259–264.
25. DeShazo, RD, Butcher, BT, Banks, WA. Reactions to the stings of the imported fire ant. N Engl J Med. 1990; 323:462.
26. Russell, FE, Carlson, RW, Wainschel, J, et al. Snake venom poisoning in the United States: Experience with 550 cases. JAMA. 1975; 233:341.
27. Kunkel, DB. Bites of venomous reptiles. Emerg Med Clin North Am. 1984; 2:563.
28. Bock, SA, Munoz-Furlong, A, Sampson, HA. Fatalities due to anaphylactic reactions to foods. J Allergy Clin Immunol. 2001; 107:191.
29. Sicherer, SH, Sampson, HA. Food allergy. J Allergy Clin Immunol. 2006; 117:S470–S475.
30. Derby, CJ, Gowland, MH, Hourihane, JO. Sesame allergy in Britain: A questionnaire survey of members of the Anaphylaxis Campaign. Pediatr Allergy Immunol. 2005; 16(2):171–175.
31. Lieberman, P. Biphasic anaphylactic reactions. Ann Allergy Asthma Immunol. 2005; 95:217–226.
32. Katayama, H, Yamaguchi, K, Kozuka, T, et al. Adverse reactions to ionic and nonionic contrast media. A report from the Japanese Committee on the Safety of Contrast Media. Radiology. 1990; 175:621–628.
33. Bush, WH. Treatment of systemic reactions to contrast media. Urology. 1990; 35:145–150.
34. Shehadi, WH, Toniolo, G. Adverse reactions to contrast media: A report from the Committee on Safety of Contrast Media of the International Society of Radiology. Radiology. 1980; 137:299–302.
35. Bettmann, MA. Ionic versus nonionic contrast agents for intravenous use: Are all answers in? Radiology. 1990; 175:616–618.
36. Sussman, GL, Tarlo, S, Dolovich, J. The spectrum of IgE-mediated responses to latex. JAMA. 1991; 265:2844.
37. Slater, JE. Latex allergy. J Allergy Clin Immunol. 1994; 94:139.
38. Galletly, DC, Treuren, BC. Anaphylactoid reactions during anaesthesia. Anaesthesia. 1985; 40:329.
39. Johansson, SGO, Melvin, T, Vahlquist, B. Immunoglobulin levels in Ethiopian preschool children with special reference to high concentrations of immunoglobulin E (IgND). Lancet. 1968; 1:1118.
40. Phils, JA, Harrold, AJ, Whiteman, GV. Pulmonary infiltrates, asthma, and eosinophilia due to Ascaris suum infestation in man. N Engl J Med. 1972; 286:965.
41. Dessein, AJ, Parker, WL, James, SL, et al. IgE antibody and resistance to infection, I: Selective suppression of the IgE antibody response in rats diminishes the resistance and the eosinophil response to Trichinella spiralis infection. J Exp Med. 1981; 153:423.
42. Van Arsdel, PP. Diagnosing drug allergy. JAMA. 1982; 247:2576.
43. Vyas, GN, Holmdahl, L, Perkins, A, et al. Serologic specificity of human anti-IgA and its significance in transfusion. Blood. 1969; 34:573.
44. Best, N, Sinosich, MJ, Teisner, B, et al. Complement activation during cardiopulmonary bypass by heparinprotamine interaction. Br J Anaesth. 1984; 56:339.
45. Sharath, MD, Metzger, WJ, Richerson, HB, et al. Protamine-induced fatal anaphylaxis. J Thorac Cardiovasc Surg. 1985; 90:86.
46. Weiss, ME, Nyhan, D, Zhikang, P, et al. Association of protamine IgE and IgG antibodies with life threatening reactions to intravenous protamine. N Engl J Med. 1989; 320:886.
47. Hüttel, MS, Schou Olesen, A, Stoffersen, E. Complement-mediated reactions to diazepam with Cremophor as solvent (Stesolid MR). Br J Anaesth. 1980; 52:77.
48. Blunk, JA, Schmelz, M, Zeck, S, et al. Opioid-induced mast cell activation and vascular responses is not mediated by mu-opioid receptors: An in vivo microdialysis study in human skin. Anesth Analg. 2004; 98:364.
49. Kishimoto, TK, Viswanathan, K, Ganguly, T, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med. 2008; 358:2457.
50. Galli, SJ, Wedemeyer, J, Tsai, M. Analyzing the role of most cells and basophils in host response and other biological responses. Int J Hematol. 2002; 75:363.
51. Butterworth, AE, David, JR. Eosinophil function. N Engl J Med. 1981; 304:154.
52. Weller, PF. The immunobiology of eosinophils. N Engl J Med. 1991; 324:1110.
53. Peavy, RD, Metcalfe, DD. Understanding the mechanisms of anaphylaxis. Curr Opin Allergy Clin Immunol. 2008; 8:310–315.
54. Metcalfe, DD, Peavy, RD, Gilfillan, AM. Mechanisms of mast cell signaling in anaphylaxis. J Allergy Clin Immunol. 2009; 124:639–646.
55. Bansal, G, Xie, Z, Rao, S, et al. Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008; 9:73–80.
56. Olivera, A, Mizugishi, K, Tikhonova, A, et al. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity. 2007; 26:287–297.
57. Delage, C, Irey, NS. Anaphylactic deaths: A clinicopathologic study of 43 cases. J Forensic Sci. 1972; 17:525.
58. James, LP, Austen, KF. Fatal systemic anaphylaxis in man. N Engl J Med. 1964; 270:597.
59. Fisher, MM. Anaphylaxis. Acute Care. 1988-1989; 14-15:47.
60. Gleich, GJ. The late phase of the immunoglobulin E mediated reaction: A link between anaphylaxis and common allergic disease? J Allergy Clin Immunol. 1982; 70:160.
61. Naclerio, RM, Proud, D, Togias, AG, et al. Inflammatory mediators in late antigen-induced rhinitis. N Engl J Med. 1985; 313:65.
62. Lieberman, P. Biphasic anaphylactic reactions. Ann Allergy Asthma Immunol. 2005; 95:217.
63. Carlson, RW, Schaeffer, RC, Puri, VK, et al. Hypovolemia and permeability pulmonary edema associated with anaphylaxis. Crit Care Med. 1981; 9:883.
64. Silverman, HJ, Van Hook, C, Haponik, EF. Hemodynamic changes in human anaphylaxis. Am J Med. 1984; 77:341.
65. Hanashiro, PK, Weil, MH. Anaphylactic shock in man: Report of two cases with detailed hemodynamic and metabolic studies. Arch Intern Med. 1967; 119:129.
66. Fisher, M. Blood volume replacement in acute anaphylactic cardiovascular collapse related to anaesthesia. Br J Anaesth. 1977; 49:1023.
67. Fisher, MM, Dicks, I. Blood volume replacement in acute anaphylactoid reactions. Anesth Intensive Care. 1979; 7:375.
68. Booth, BH, Patterson, R. Electrocardiographic changes in human anaphylaxis. JAMA. 1970; 211:627.
69. Levine, HD. Acute myocardial infarction following a wasp sting. Am Heart J. 1976; 91:365.
70. Sullivan, TJ. Cardiac disorders in penicillin-induced anaphylaxis. JAMA. 1982; 248:2161.
71. Austin, SM, Barooah, B, Kim, CS. Reversible acute cardiac injury during cefoxitin-induced anaphylaxis in a patient with normal coronary arteries. Am J Med. 1984; 77:729.
72. McLean-Tooke, AP, Bethune, CA, Fay, AC, et al. Adrenaline in the treatment of anaphylaxis: What is the evidence? BMJ. 2003; 327:1332.
73. Raper, RF, Fisher, MM. Profound reversible myocardial depression after anaphylaxis. Lancet. 1988; 1:368.
74. Smedegard, G, Revenas, B, Lundberg, C, et al. Anaphylactic shock in monkeys passively sensitized with human reaginic serum, I: Hemodynamics and cardiac performance. Acta Physiol Scand. 1981; 111:239.
75. Moss, J, Fahmy, NR, Sunder, N, et al. Hormonal and hemodynamic profile of an anaphylactic reaction in man. Circulation. 1981; 63:210.
76. Hamberger, B, Fredholm, BB, Farnebo, LO. Anaphylaxis and plasma catecholamines. Life Sci. 1980; 26:1465.
77. Olinger, GN, Becker, RM, Boncheck, LI. Noncardiogenic pulmonary edema and peripheral vascular collapse following cardiopulmonary bypass: Rare protamine reaction? Ann Thorac Surg. 1980; 29:20.
78. Enjeti, S, Bleeker, ER, Smith, PL, et al. Hemodynamic mechanisms in anaphylaxis. Circ Shock. 1983; 11:297.
79. Brady, WJ, Jr., Luber, S, Carter, CT, et al. Multiphasic anaphylaxis: An uncommon event in the emergency department. Acad Emerg Med. 1997; 4:193.
80. Simons, FE. Anaphylaxis. J Allergy Clin Immunol. 2010; 125(2 Suppl 2):S161–S181.
81. Simons, KJ, Simons, FE. Epinephrine and its use in anaphylaxis: Current issues. Curr Opin Allergy Clin Immunol. 2010; 10:354.
82. Lieberman, P, Nicklas, RA, Oppenheimer, J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol. 2010; 126:477.
83. Tole, JW, Lieberman, P. Biphasic anaphylaxis: Review of incidence, clinical predictors, and observation recommendations. Immunol Allergy Clin North Am. 2007; 27:309–326.
84. Scranton, SE, Gonzalez, EG, Waibel, KH. Incidence and characteristics of biphasic reactions after allergen immunotherapy. J Allergy Clin Immunol. 2009; 123:493–498.
85. Zaloga, GP, Delacey, W, Holmboe, E, et al. Glucagon reversal of hypotension in a case of anaphylactoid shock. Ann Intern Med. 1986; 105:65.
86. Thomas, M, Crawford, I. Best evidence report: Glucagon infusion in refractory anaphylactic shock in patients on beta-blockers. Emerg Med J. 2005; 22:272.
87. Mazuc, E, Villoutreix, BO, Malbec, O, et al. A novel drug-like spleen tyrosine kinase binder prevents anaphylactic shock when administered orally. J Allergy Clin Immunol. 2008; 122:188–194.
88. Chang, TW, Shiung, YY. Anti-IgE as a mast cell-stabilizing therapeutic agent. J Allergy Clin Immunol. 2006; 117:1203–1212.
89. Lasser, EC, Berry, CC, Talner, LB, et al. Pretreatment with corticosteroids to alleviate reactions to intravenous contrast material. N Engl J Med. 1987; 317:845.
90. Miller, WL, Doppman, JL, Kaplan, AP. Renal arteriography following systemic reaction to contrast material. J Allergy Clin Immunol. 1975; 56:291.
91. Tramer, MR, von Elm, E, Loubeyre, P, et al. Pharmacologic prevention of serious anaphylactic reactions due to iodinated contrast media: Systemic review. BMJ. 2006; 333:675.
92. Watkins, J. Adverse anaesthetic reactions. Anaesthesia. 1985; 40:797.
93. Weiss, ME, Adkinson, NF. Immediate hypersensitivity reactions to penicillin and related antibiotics. Clin Allergy. 1988; 18:515.
94. Canonica, GW. Specific immunotherapy: Still young after one century. Allergy Immunol. 2005; 37:301.
95. Mattson, JR, Patterson, R, Roberts, M. Insulin therapy in patients with systemic insulin allergy. Arch Intern Med. 1975; 135:818.
96. Pfaar, O, Klimek, L. Aspirin desensitization in aspirin intolerance: Update on current standards and recent improvements. Curr Opin Allergy Clin Immunol. 2006; 6:161.
97. Grieco, MH, Dubin, MR, Robinson, JL, et al. Penicillin hypersensitivity in patients with bacterial endocarditis. Ann Intern Med. 1964; 60:204.
98. Tuft, L. Fatalities following the reinjection of foreign serum: With report of an unusual case. Am J Med Sci. 1928; 175:325.
99. Graft, DF. Venom immunotherapy for stinging insect allergy. Clin Rev Allergy. 1987; 5:149.
100. Lockey, RF. Immunotherapy for allergy to insect stings. N Engl J Med. 1990; 323:1627.