Adult hypoxic and ischemic lesions
TERMINOLOGY
Hypoxia
 CEREBRAL BLOOD FLOW (CBF)
CEREBRAL BLOOD FLOW (CBF)CBF = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR)
CPP = systemic arterial blood pressure − intracranial pressure (ICP)






exacerbates the damage produced by ischemia. In practice, many causes of stagnant or hypoxemic hypoxia (e.g. cardiac arrest and carbon monoxide poisoning, respectively) also depress cardiac output, resulting in combined hypoxic/global ischemic brain injury.
Global brain ischemia
 CELLULAR MECHANISMS OF ISCHEMIC CELL DEATH
CELLULAR MECHANISMS OF ISCHEMIC CELL DEATH
 Depolarization of neuronal/axonal membranes within 60–180 seconds of global anoxia leads to changes in extracellular and intracellular electrolyte composition and a decrease in ATP (secondary to impaired glycolysis and oxidative phosphorylation), with associated release of lactate and hydrogen ions and acidosis. ATP may decline to <25% of that in normally perfused tissue.
Depolarization of neuronal/axonal membranes within 60–180 seconds of global anoxia leads to changes in extracellular and intracellular electrolyte composition and a decrease in ATP (secondary to impaired glycolysis and oxidative phosphorylation), with associated release of lactate and hydrogen ions and acidosis. ATP may decline to <25% of that in normally perfused tissue.



 Cytoskeletal damage can affect the machinery of protein synthesis, but this may eventually recover.
Cytoskeletal damage can affect the machinery of protein synthesis, but this may eventually recover.
 Lipases, proteases and nucleases are activated, also with deleterious effects.
Lipases, proteases and nucleases are activated, also with deleterious effects.
 Free radicals and NO/peroxynitrite, a mediator of NO toxicity, increase.
Free radicals and NO/peroxynitrite, a mediator of NO toxicity, increase.

Local brain ischemia
 ROLE OF ASTROCYTES
ROLE OF ASTROCYTES




 REPERFUSION AFTER CIRCULATORY ARREST
REPERFUSION AFTER CIRCULATORY ARREST



PATHOPHYSIOLOGIC CONSIDERATIONS
Adult and infant brains react differently to hypoxia and ischemia. In general, infant brains are more resistant than those of adults; hypoxic-ischemic lesions have a different distribution in infants and adults reflecting an age-related differential (selective) vulnerability to such insults (Fig. 8.1). Because of the brain’s immense metabolic demands, after the onset of ischemia levels of brain glycogen, glucose, ATP and phosphocreatine plummet and are often depleted within 10 min of the acute event. After 15 min of cardiac arrest, up to 95% of the brain may be damaged. Primary respiratory arrest (e.g. due to aspiration, anaphylaxis, or airway trauma) may cause transient brain dysfunction, but less severe damage than ischemia. Optimal brain function and respiration are dependent upon the availability of glucose; however, the neuropathology of hypoglycemic brain injury differs from that due to hypoxia-ischemia.
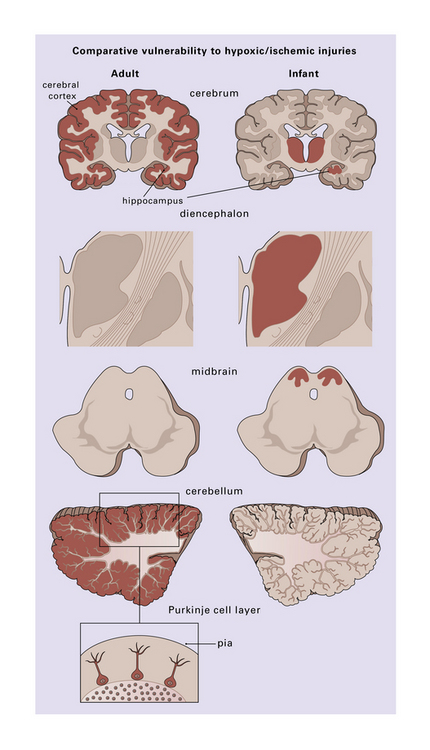
8.1 Regions of selective vulnerability to hypoxic–ischemic damage are different in the adult and infant brain.
Those regions most susceptible to such an insult are colored in the diagram, though individual cases may show much significant variation. Inset (lower left) emphasizes the observation that cerebellar Purkinje cells are especially at risk during hypoxia.
PATHOLOGY
Lesions may be considered as either acute/subacute or chronic.
 HYPOXIC-ISCHEMIC BRAIN INJURY
HYPOXIC-ISCHEMIC BRAIN INJURY Hypoxic-ischemic encephalopathy (HIE) has an extremely variable clinical presentation.
Hypoxic-ischemic encephalopathy (HIE) has an extremely variable clinical presentation.


 The severity and duration of HIE after transient cerebral hypoxia or ischemia depend upon:
The severity and duration of HIE after transient cerebral hypoxia or ischemia depend upon:
• severity and duration of insult
• blood glucose level – high levels are associated with poor outcome
• CNS (core) temperature – a lower temperature is protective.

 In one study of many patients with HIE secondary to cardiac arrest:
In one study of many patients with HIE secondary to cardiac arrest:

 TYPES OF NEURONAL DEATH OF POTENTIAL IMPORTANCE IN HYPOXIC-ISCHEMIC DAMAGE
TYPES OF NEURONAL DEATH OF POTENTIAL IMPORTANCE IN HYPOXIC-ISCHEMIC DAMAGE
MACROSCOPIC APPEARANCES
Acute/subacute lesions include the following:
 Precursors of cystic infarcts – especially in watershed territories.
Precursors of cystic infarcts – especially in watershed territories.


 Bright pink color and edema – after acute CO poisoning.
Bright pink color and edema – after acute CO poisoning.
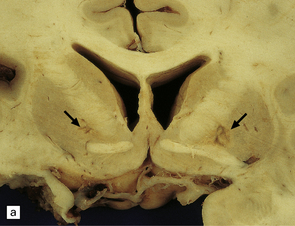
 Generalized dusky discoloration and softening – the appearance of ‘non-perfused’ brain (Fig. 8.2).
Generalized dusky discoloration and softening – the appearance of ‘non-perfused’ brain (Fig. 8.2).
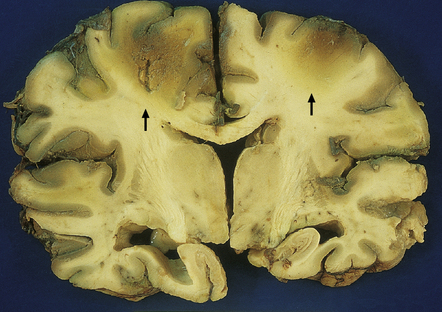
8.2 Coronal section through a nonperfused (‘respirator’) brain.
The brain is edematous with focally accentuated gray-brown discoloration throughout the cortex and extending into the subcortical white matter, most notably in the watershed regions (arrows) between the perfusion territories of the middle and anterior cerebral arteries.
 PRINCIPAL CAUSES OF HYPOXIA
PRINCIPAL CAUSES OF HYPOXIAHypoxemic hypoxia (secondary to low O2 content in blood):
 CO poisoning – mechanisms include displacement of O2 from hemoglobin and direct binding to heme-containing proteins in the CNS.
CO poisoning – mechanisms include displacement of O2 from hemoglobin and direct binding to heme-containing proteins in the CNS.

 Respiratory arrest – drug-induced, status asthmaticus, epiglottitis, airway trauma, aspiration.
Respiratory arrest – drug-induced, status asthmaticus, epiglottitis, airway trauma, aspiration.

Stagnant hypoxia (inadequate supply of oxygenated blood, e.g. ischemia):
 Cardiac arrest with prolonged asystole or hypotension secondary to a myocardial infarct, cardiac tamponade, or major cardiac arrhythmia.
Cardiac arrest with prolonged asystole or hypotension secondary to a myocardial infarct, cardiac tamponade, or major cardiac arrhythmia.

 Severe increases in intracranial pressure (ICP). When ICP reaches mean arterial BP, brain perfusion ceases.
Severe increases in intracranial pressure (ICP). When ICP reaches mean arterial BP, brain perfusion ceases.

Histotoxic hypoxia (inability of brain tissue to utilize available O2):

 NEURONAL SELECTIVE VULNERABILITY AND EXCITOTOXICITY
NEURONAL SELECTIVE VULNERABILITY AND EXCITOTOXICITY
 Hippocampus – especially CA1 segment of pyramidal cell layer.
Hippocampus – especially CA1 segment of pyramidal cell layer.
 Cerebral cortex – especially layers 3 and 5.
Cerebral cortex – especially layers 3 and 5.





In both premature and term infants, the pattern of neuronal susceptibility to hypoxic-ischemic injury differs from that in adults (see Chapter 2).




Chronic lesions that evolve from acute/subacute pathology often become more obvious and include:
 Watershed/borderzone infarcts (Fig. 8.3).
Watershed/borderzone infarcts (Fig. 8.3).
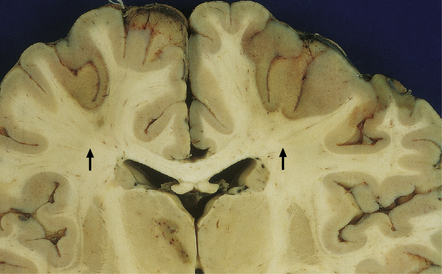
8.3 Relatively recent bihemispheric watershed infarcts in anterior cerebral artery/middle cerebral artery border zones.
Note the dusky gray-brown discoloration of the cortex in these regions (arrows), which is accentuated at the junction between the gray and white matter. There is also a region of necrosis in the left thalamus. The patient was a child aged 2 years with double outlet right ventricle who underwent the Fontan procedure. Intraoperative and postoperative complications led to death 15 days later.
 Cortical laminar necrosis or variable thinning of the cortex (Figs 8.4, 8.5).
Cortical laminar necrosis or variable thinning of the cortex (Figs 8.4, 8.5).
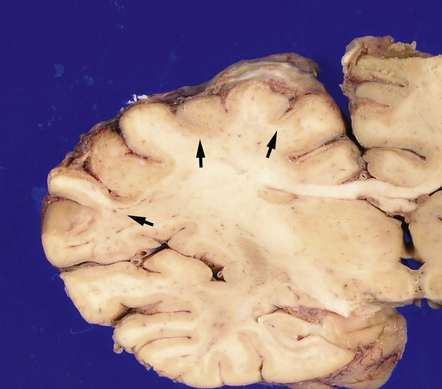
8.4 Coronal brain section from a 73-year-old woman who experienced a cardio-pulmonary arrest with prolonged asystole approximately 1 month prior to death.
Significant neurologic impairment followed the arrest. Coronal sections of the cerebral hemispheres show multiple regions of cortical thinning and discoloration (arrows). Changes are most pronounced in the ACA and MCA vascular territories; by contrast, the inferior aspect of the temporal lobe appears relatively unaffected.
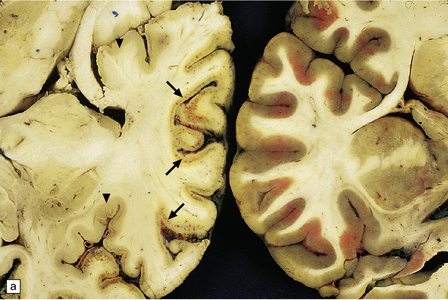
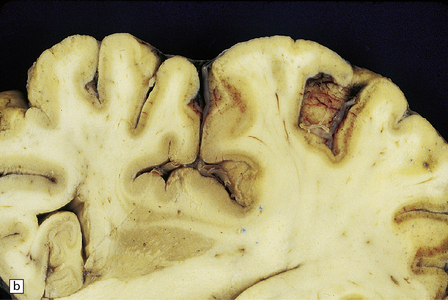
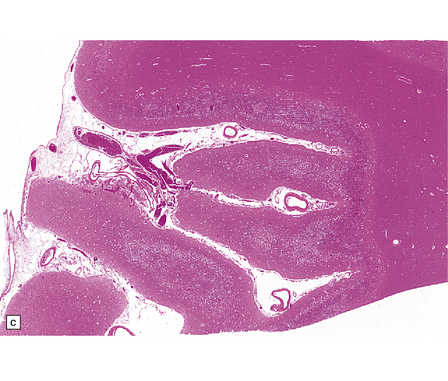
8.5 Cortical laminar necrosis (LN).
(a) Coronal section through the brains of two different patients; a ‘control’ patient, without history of cardiac arrest or neurologic deficit (right), and a patient who survived several days after extended cardiopulmonary arrest (left). Contrasting features between the two include patchy gray discoloration with pronou0nced thinning of the neocortex in a laminar pattern (left). Despite multifocal areas of laminar necrosis (indicated by arrows), many regions are relatively spared (arrowheads). Another slice (b) from brain illustrated in (a, left), highlights the patchy nature of cortical necrosis. (c) Whole mount from a focus of LN showing extensive cortical injury, but with comparative sparing of the superficial cortical layers. Dystrophic calcification had occurred in areas of LN (see Fig. 8.11c). (d) More subtle LN manifests as a thin dark line running through the cortical ribbon (arrowheads).
 Cystic necrosis following CO poisoning. This is usually symmetrical, affecting the globus pallidus but sparing other regions of the basal ganglia. Occasionally, this pattern of cavitation is seen without well-documented CO exposure (Fig. 8.6), occurring as the result of cyanide poisoning, heroin overdose, or other causes of global cerebral hypoxia/ischemia.
Cystic necrosis following CO poisoning. This is usually symmetrical, affecting the globus pallidus but sparing other regions of the basal ganglia. Occasionally, this pattern of cavitation is seen without well-documented CO exposure (Fig. 8.6), occurring as the result of cyanide poisoning, heroin overdose, or other causes of global cerebral hypoxia/ischemia.
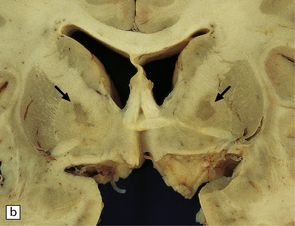
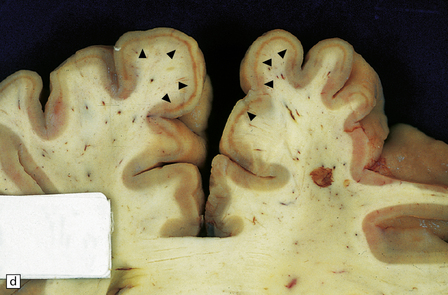
8.6 Bilateral pallidal cystic necrosis.
(a) Pallidal necrosis in a patient who died after surgery for a malignant glioma in the left temporal lobe. The surgical bed is seen at lower left. He had previously attempted suicide by carbon monoxide poisoning several months before his death. Note the bilateral pallidal cystic necrosis (arrows), which is more extensive on the right than on the left. (b) Comparable coronal section from the brain of a 75-year-old woman with severe toxic epidermal necrolysis syndrome and no history of carbon monoxide poisoning. Recent bihemispheric parieto-occipital infarcts (not shown) and bilateral partial necrosis of globus pallidus (arrows) were noted at necropsy.

 Leukoencephalopathy secondary to anoxia or ischemia (see Fig. 8.15).
Leukoencephalopathy secondary to anoxia or ischemia (see Fig. 8.15).
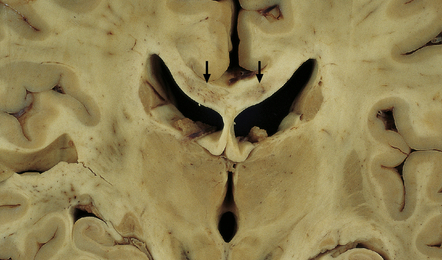
8.15 Hypoxic–ischemic leukoencephalopathy.
Note white matter discoloration and necrosis of central part of the corpus callosum (arrows). This patient aged 69 years underwent complicated aortic surgery and developed ‘mental status changes’ and spinal cord paralysis secondary to a cord infarct 1 month before death. There were also widespread necrotic lesions in the cortex and subcortical white matter. Histologic changes included dense collections of macrophages within affected white matter.
MICROSCOPIC APPEARANCES
Acute/subacute lesions
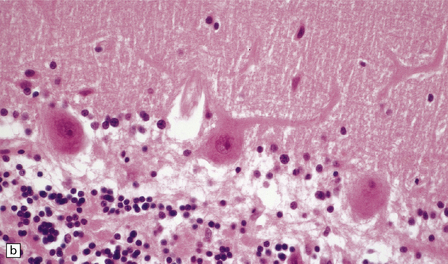
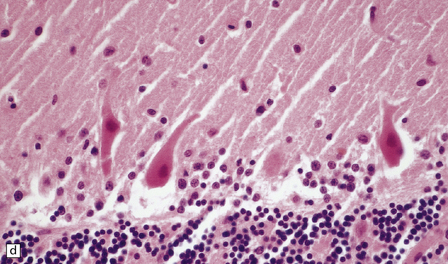
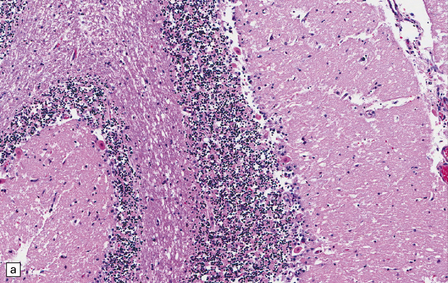
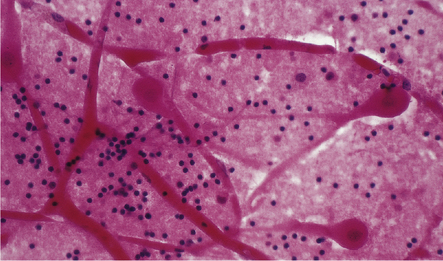
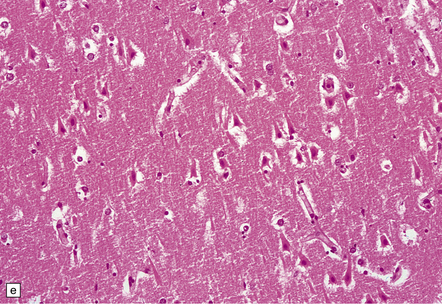
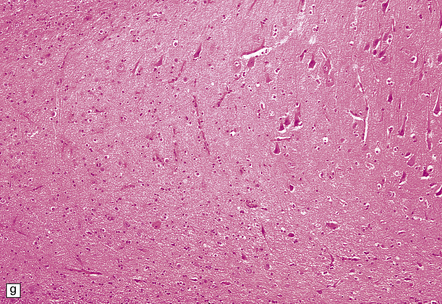
Ischemic (irreversibly damaged) neurons have collapsed pyknotic nuclei and brightly eosinophilic cytoplasm (in H&E-stained sections). At later time points, as chromatin is degraded, nuclei become more eosinophilic and appear to blend with surrounding cytoplasm. These changes are first discernible histologically after survival for several hours (most estimates are 4–6 hours minimum), last for up to 2 weeks, and are particularly likely to be seen in neurons that are sensitive to hypoxia, e.g. hippocampal neurons in the CA1 field, pyramidal neurons in the cerebral cortex, cerebellar Purkinje cells, reticular neurons of the thalamus, and medium-sized neurons of basal ganglia (Figs 8.7–8.9). Neuropil may show slight vacuolization or be normal (in contrast to marked neuropil vacuolization in ischemic infarction). Normal neurons may be seen immediately adjacent to affected nerve cell bodies. Hypoxic-ischemic neuronal change is sometimes demonstrated in smear/squash preparations of affected brain regions (Fig. 8.10).
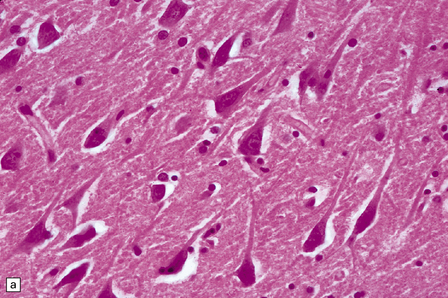
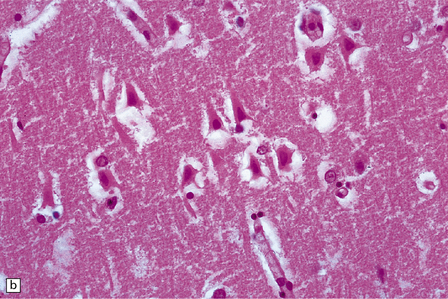
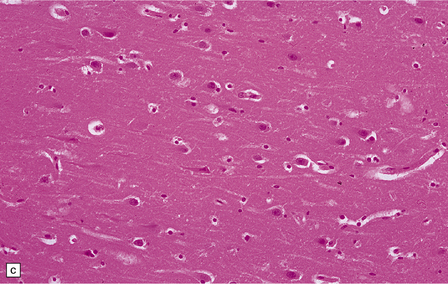
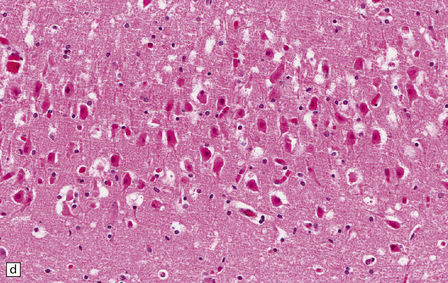
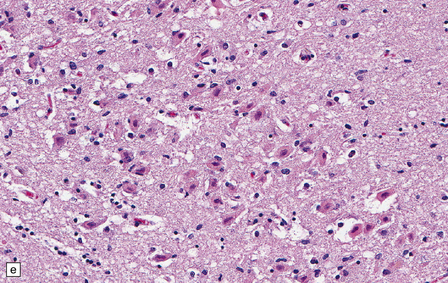
8.7 Acute hypoxic neuronal change.
(a) Normal cortical neurons with basophilic cytoplasm and nuclei with prominent nucleoli. (b) Hypoxic neurons showing pronounced cytoplasmic eosinophilia, collapse of cytoplasm with accentuated (artifactual) pericellular spaces, and pyknotic nuclei with indistinct nucleoli, photographed at magnification comparable to that in panel (b). (c) More subtle and patchy hypoxic–ischemic neuronal change affecting only scattered neurons. (d) Hippocampal pyramidal cell layer (CA1 segment) showing brightly eosinophilic neurons, reflecting severe acute anoxic-ischemic change. (e) Similar region of hippocampus from another patient, with slightly longer interval between anoxia and death. Many eosinophilic neurons are seen, some as ‘ghost’ nerve cell bodies; in addition many reactive cells (probably microglia and some astrocytes) surround dead neurons.
8.8 Hypoxic change in cerebral Purkinje cells.
The cerebellar cortex is especially vulnerable to hypoxic–ischemic lesions. (a) Low-power view of normal cerebellum. (b) High-power view of normal cerebellum. (c) Low-power view of cerebellar cortex from a 74-year-old patient who died shortly after complicated open heart surgery. At low magnification there is little obvious difference between this specimen and that shown in (a). (d) High-power view of (c) showing eosinophilic Purkinje cells with smudged and pyknotic nuclei, which contrast with the normal Purkinje cells that have plump large vesicular nuclei and prominent nucleoli (b).
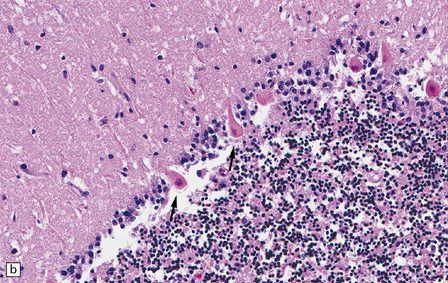
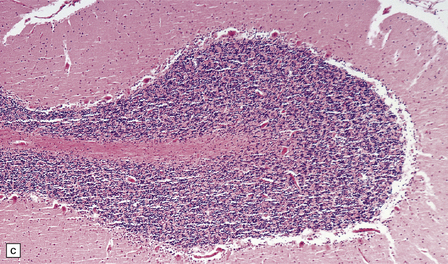
8.9 Sections of cerebellum from a patient who experienced severe hypoxia prior to death.
In contrast to the preserved cerebellar architecture shown in Fig. 8.8, there is microvacuolization of the molecular layer (post-infarction changes) in addition to profound eosinophilia of Purkinje cells (arrows in panel B).
Chronic lesions
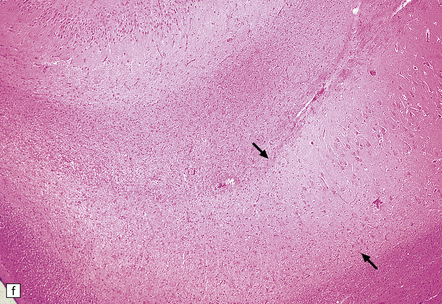
Laminar necrosis (LN) usually affects the middle cortical layers, especially laminae 3 and 5. In extreme instances, there may be full-thickness necrosis (Figs 8.5, 8.11). LN may be suspected antemortem on the basis of high-intensity T1-weighted MRI abnormalities that follow a gyral distribution. Necrosis is usually accentuated in watershed zones.
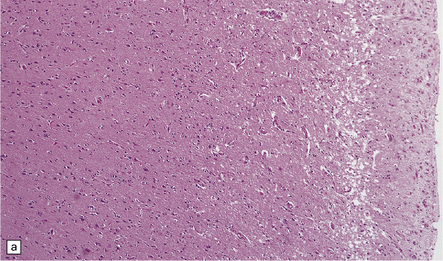
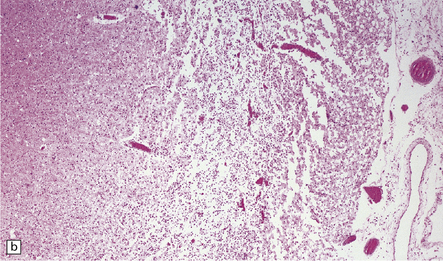
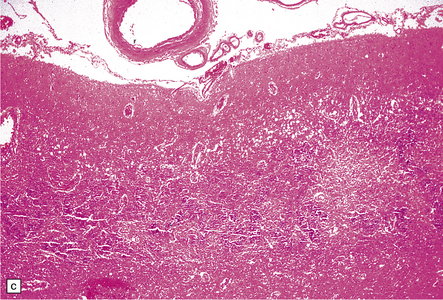
8.11 Hypoxic–ischemic cortical injury.
(a) This patient aged 80 years underwent pituitary surgery 1 month before death. At necropsy there were minimal abnormalities of the brain, but microscopically there was widespread hypoxic neuronal change, vacuolation of the superficial cortex, subtle microvascular proliferation, and gliosis. Hypoxic–ischemic cortical injury. (b) Severe cortical necrosis in a different patient. This is from the brain illustrated in Fig. 8.5b. It shows pancortical necrosis tantamount to infarction. The cellular content of the neuronal layers comprises a mixture of astrocytes and macrophages. (c) High-power view of the cerebral cortex shown in Fig. 8.5c. Note extensive destruction of deep cortical layers, with dystrophic calcification, but relative preservation of superficial cortical layers.
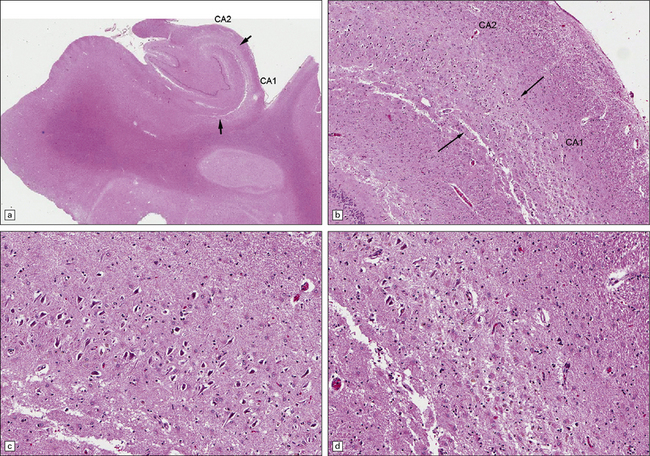
In many brain regions, long-term hypoxic-ischemic damage may appear simply as a focal loss of neurons with variably severe reactive astrocytosis. This is especially dramatic in the pyramidal cell layer of the hippocampus; CA1 is highly vulnerable to hypoxia, whereas CA2 is relatively resistant (Fig. 8.12, 8.13). A distinction is often made between ‘selective neuronal loss and gliosis’, in which neurons disappear with attendant astrocytic gliosis, and ‘infarction’, in which all cellular elements die and macrophages infiltrate to remove cellular debris. In practical terms, the two processes may be difficult to differentiate months or years after the brain injury has occurred, though cystic change and encephalomalacia are certainly more pronounced in an infarct. In infants that experience severe perinatal asphyxia/hypoxia, extensive cystic encephalomalacia (Fig. 8.14) may lead to severe mental retardation and intractable seizures.

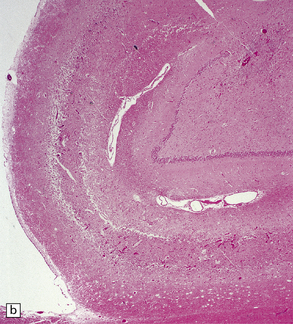
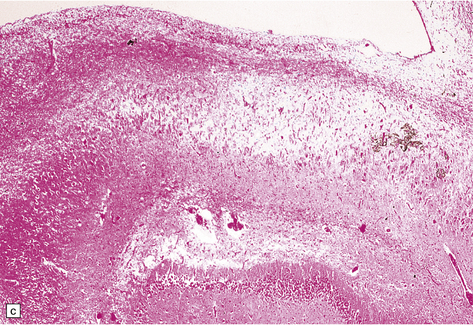
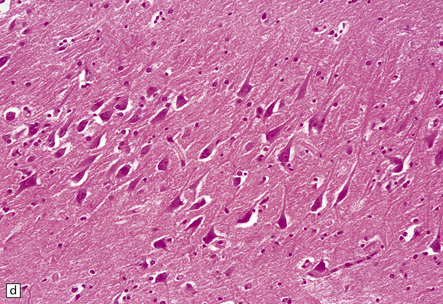
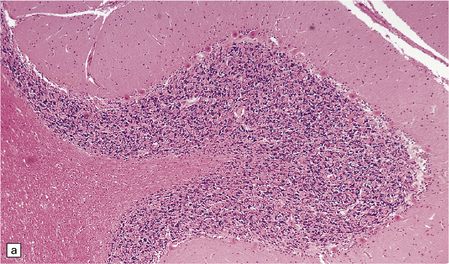
8.12 Selective vulnerability to hypoxic–ischemic change in the hippocampal pyramidal cell layer.
(a) Normal hippocampus at low magnification. The figure includes part of the granule cell layer and part of the pyramidal cell layer as far as the prosubiculum/CA1 junction. (b) Segmental loss of neurons and prominent neuropil vacuolation within the CA1 sector of pyramidal cell layer. (c) There is an infarct involving virtually the entire CA1 field or sector and extending into the prosubiculum. Neuron loss and spongy change are seen in the affected neuropil. Note preservation of the granule cell layer (dentate fascia). (d)Normal CA1 zone of hippocampal pyramidal cell layer, contrasted with (e) a region with severe acute anoxic–ischemic change (neuronal eosinophilia, cytoplasmic and nuclear collapse, etc). (f,g) Severe hippocampal sclerosis in a patient with longstanding temporal lobe epilepsy. (f) Arrows indicate junction between sclerotic CA1 zone (at left) and intact prosubiculum (at right). (g) The junction between the two sectors is highlighted; gliotic tissue and neuron depletion in CA1, intact neurons in prosubiculum.
8.13 Sections of hippocampus from a 76-year-old man with history of therapy for gastric carcinoma complicated by seizures and memory impairment. Autopsy showed many small brain infarcts.
(a) The hippocampus shows atrophy and loss of pyramidal cells in CA1 between arrows. (b) The contrast between grossly abnormal CA1 and relatively normal CA2 segments (arrows – border zone) is demonstrated at higher power in (c) and (d), where there is relative preservation of neurons in CA2 (c) but profound neuronal loss and astrocytic gliosis in CA1 (d).
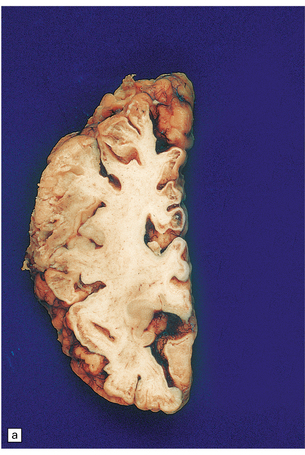
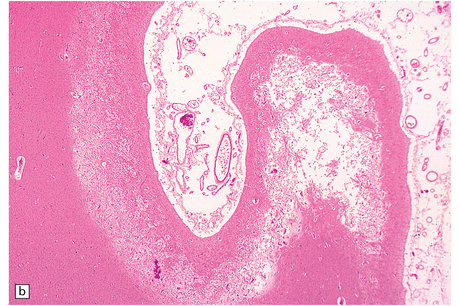
8.14 Multifocal cystic/gliotic encephalopathy. This is a consequence of late intrauterine/perinatal asphyxia in infants.
(a) Representative coronal slice of brain, showing multiple regions of cystic cavitation of the gyral white matter. (b) Low magnification view of a representative gyrus, showing extensive cystic cavitation of its ‘core’, with LN-like change in the adjacent cortical ribbon.
A leukoencephalopathy may coexist with hypoxic-ischemic gray matter damage (Fig. 8.15), and exceptionally may be the predominant finding after prolonged hypoxia in association with hypotension. It has been associated with CO poisoning, but may occur in other settings, such as drug overdose. Histologically, there may be necrosis with abundant macrophages, or loss of myelin in association with relative axonal sparing, reactive astrocytosis, and activation of microglia, as well as overexpression of amyloid precursor protein (APP) within and among adjacent axons. It is important to remember, however, that white matter ‘pallor/degeneration’ in an individual with a past history of severe neocortical HIE may simply represent Wallerian degeneration.
 PERSISTENT VEGETATIVE STATE (PVS) AND ‘BRAIN DEATH’
PERSISTENT VEGETATIVE STATE (PVS) AND ‘BRAIN DEATH’
The vegetative state (VS) is a clinical condition of complete unawareness of self and environment, accompanied by sleep–wake cycles, with either complete or partial preservation of hypothalamic and brainstem autonomic functions.
Diffuse white matter injury (leukoencephalopathy)
The commonest cause is diffuse axonal injury secondary to traumatic brain injury (TBI). PVS due to diffuse white matter damage rarely complicates hypoxic-ischemic injury (see Fig. 8.12), and may also result from leukodystrophies associated with lysosomal or peroxisomal defects and other genetic disorders (e.g. Alexander’s disease).
REFERENCES
Adams, J.H., Brierley, J.B., Connor, R.C.R., et al. The effects of systemic hypotension upon the human brain. Clinical and neuropathological observations in 11 cases. Brain.. 1966;89:235–268.
Adams, J.H., Graham, D.I., Jennett, B. The neuropathology of the vegetative state after an acute brain insult. Brain.. 2000;123:1327–1338.
Armengol, C.G. Acute oxygen deprivation: neuropsychological profiles and implications for rehabilitation. Brain Inj.. 2000;14:237–250.
Armengol, C.G. Acute oxygen deprivation: neuropsychological profiles and implications for rehabilitation. Brain Inj.. 2000;14:237–250.
Auer, R.N. Insulin, blood glucose levels, and ischemic brain damage. Neurology.. 1998;51(suppl 3):S39–S43.
Auer, R.N., Dunn, J.F., Sutherland, G.R. Hypoxia and related conditions. In: Love S., Louis D.N., Ellison D.W., eds. Greenfield’s neuropathology. 8th ed. London: Hodder Arnold; 2008:63–119.
Busl, K.M., Greer, D.M. Hypoxic-ischemic brain injury: Pathophysiology, neuropathology and mechanisms. NeuroRehabilitation.. 2010;26:5–13.
Colbourne, F., Sutherland, G.R., Auer, R.N. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci.. 1999;19:4200–4210.
Dirnagl, U., Iadecola, C., Moskowitz, M.A. Pathobiology of ischaemic stroke: an integrated view. TINS.. 1999;22:391–397.
Donaire, A., Carreno, M., Gomez, B., et al. Cortical laminar necrosis related to prolonged focal status epilepticus. J Neurol Neurosurg Psychiatry.. 2006;77:104–106.
Fogel, F., Krieger, D., Veith, M., et al. Serum neuron-specific enolase as early predictor of outcome after cardiac arrest. Crit Care Med.. 1997;25:1133–1138.
Ginsberg, M.D., Hedley-Whyte, E.T., Richardson, E.P., Jr. Hypoxic-ischemic leukoencephalopathy in man. Arch Neurol.. 1976;33:5–14.
Levy, D.E., Caronna, J.J., Singer, B.H., et al. Predicting outcome from hypoxic-ischemic coma. JAMA.. 1985;253:1420–1426.
Lipton, P. Ischemic cell death in brain neurons. Physiol Rev.. 1999;79:1431–1568.
Maragakis, N.J., Rothstein, J.D. Glutamate transporters in neurologic disease. Arch Neurol.. 2001;58:365–370.
Miyamoto, O., Auer, R.N. Hypoxia, hyperoxia, ischemia, and brain necrosis. Neurology.. 2000;54:362–371.
Newcombe, V.F.J., Williams, G.B., Scoffings, D., et al. Aetiological differences in neuroanatomy of the vegetative state: insights from diffusion tensor imaging and functional implications. J Neurol Neurosurg Psychiatry.. 2010;81:552–561.
Rossi, D.J., Brady, J.D., Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci.. 2007;10:1377–1386.
Shewmon, D.A. Chronic ‘brain death’. Meta-analysis and conceptual consequences. Neurology.. 1998;51:1538–1545.
Shewmon, D.A. Brain death: Can it be resuscitated? Issues Law Med.. 2009;25:3–14.
Smith, M-L., Auer, R.N., Siesjo, B.K. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol (Berl).. 1984;64:319–332.
Sofroniew, M.V., Vinters, H.V. Astrocytes: biology and pathology. Acta Neuropathol (Berl).. 2010;119:7–35.
Takano, T., Oberheim, N., Cotrina, M.L., et al. Astrocytes and ischemic injury. Stroke.. 2009;40(suppl 3):S8–S12.
Weiss, N., Galanaud, D., Carpentier, A., et al. Clinical review: Prognostic value of magnetic resonance imaging in acute brain injury and coma. Crit Care.. 2007;11:230.
Wijdicks, E.F.M. Determining brain death in adults. Neurology.. 1995;45:1003–1011.