Adrenal Insufficiency in the Critically Ill Patient
Adrenal insufficiency (AI) is by far the most common adrenal disorder seen in the ICU and is the focus of this chapter. It occurs more frequently in critically ill patients than in general hospitalized patients and represents a true emergency that requires rapid diagnosis and treatment. If missed, the condition can be fatal. In addition, because critical illness is often the precipitant of overt AI, the intensivist may have the first and only chance to make the diagnosis.1
The term relative adrenal insufficiency has been replaced in recent literature by critical illness–related corticosteroid insufficiency (CIRCI),2,3 which in simple terms is inadequate corticosteroid activity for the severity of the illness of a patient. Similar to type II diabetes (relative insulin deficiency), CIRCI is thought to arise because of corticosteroid tissue resistance and inadequate circulating levels of free cortisol.
Patients at risk of AI vary from young athletes on steroids to persons taking adrenal extracts for “adrenal fatigue syndrome.” Also at risk are those receiving chronic topical glucocorticoids for dermatologic disorders. Patients who are on glucocorticoids and inhibitors (such as itraconazole, diltiazem) of CYP3A4 are at risk as well.4 For all these reasons the intensivist must understand clinical problems associated with the HPA axis and the use of glucocorticoid hormones.
Incidence and Prevalence
The actual incidence of acute AI is unknown. The incidence of HPA axis failure varies depending on the criteria used to make the diagnosis and the patient population studied. The overall incidence of AI in critically ill patients is estimated to be as high as 60% in patients with severe sepsis and septic shock.5 At least 90% of both adrenal glands must be destroyed before clinical and biochemical manifestations of AI occur. Tissue hypoxia, a relatively common disorder in critically ill patients, has little effect on the synthesis of cortisol. Secondary AI may be more common than primary AI. The clinical presentation of secondary AI is relatively nonspecific and often resembles other conditions common in the ICU. Hence it is not uncommon to attribute the clinical features resulting from acute AI to commonly seen medical conditions in the ICU.6
Pathophysiology
The principal mineralocorticoid is aldosterone, which is regulated not only by ACTH but also by serum sodium and potassium levels and by the renin-angiotensin system.7,8 Mineralocorticoids exert their primary effect on distal renal tubule cells, resulting in renal sodium retention at the expense of potassium loss in the urine. A third major class of adrenal steroids is the sex hormones: dehydroepiandrosterone (DHEA), DHEA-sulfate, and androstenedione. Like the glucocorticoids, ACTH primarily regulates these steroid hormones. They function mainly as precursors for the primary circulating androgen, testosterone, and also may undergo separate conversion to estrogen hormones. In critically ill patients, glucocorticoids are the steroid hormones of greatest concern and therefore remain the focus of the remainder of this discussion.
Glucocorticoid Synthesis
Cortisol is normally secreted in a diurnal pattern. The circulating cortisol level is increased in the morning hours, at approximately 8 AM. Serum cortisol concentrations decrease throughout the remainder of the day.9 Similarly, the serum cortisol response to ACTH stimulation also varies in a circadian rhythm. Afternoon responsiveness is much greater because of the decreased circadian level of cortisol at that time. In addition, cortisol is secreted in a series of pulses rather than in a continuous fashion. These factors contribute to make interpretation of a random cortisol level and the ACTH-stimulated value difficult.
“Stress” (exemplified by sepsis, major surgery, or trauma) also affects glucocorticoid synthesis.10–12 The stress response is characterized by continuous ACTH secretion despite a high serum cortisol concentration. Stress overrides all other regulatory mechanisms of cortisol secretion by the adrenal cortex and increases cortisol secretion irrespective of the time of day or the current serum cortisol concentration. The mechanism by which the HPA axis is regulated during stress is not clearly understood. Periventricular neurons in the hypothalamus respond to stress by increasing the levels of CRH messenger ribonucleic acid (mRNA).13,14 It has been shown that production of the cytokines interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor-α (TNF-α) also plays an important role in the regulation of the HPA axis.15–19 The cortisol secretion that occurs because of the activation of the HPA axis causes an inhibitory effect not only on the secretion of CRH and ACTH but also on the liberation of interleukins.20 Thus, there is a functional loop between immune activation and regulation of the HPA axis during stress.
The stress response is biphasic, consisting of an early phase in which both ACTH and cortisol are elevated and a late phase in which the serum cortisol level is elevated but the serum ACTH level is paradoxically low.8 This is explained by the fact that endothelin and atrial natriuretic peptide are both elevated in severe illnesses. Endothelin increases cortisol production by the adrenals, whereas the atrial natriuretic peptide inhibits ACTH production by acting at the hypothalamic-pituitary level. Vasopressin and angiotensin II can increase ACTH secretion during stress conditions, such as sepsis and septic shock
Acute respiratory failure causes a 50% to 100% rise in serum cortisol concentration. A twofold to sixfold rise occurs with septic shock and following surgical procedures and trauma. The rise in serum cortisol correlates positively with severity of illness6 and negatively with survival.7
Glucocorticoid Actions
Cardiovascular Effects
Glucocorticoids help to maintain vascular tone and cardiac contractility. The presence of glucocorticoids is important to the physiologic effects of catecholamines on vascular smooth muscle. Glucocorticoids affect blood pressure by different mechanisms including direct action of glucocorticoids on the vasculature, permissive effects of the glucocorticoids on the vasopressor action of catecholamines, and glucocorticoid-induced decrease in the levels of prostaglandin E2 and kallikrein (vasodilators). Angiotensinogen synthesis is increased by glucocorticoids.21 Glucocorticoids increase the synthesis of β-adrenergic receptors, reverse β2-adrenergic receptor dysfunction, and increase the coupling of the receptor with the second messenger system.22
Two hemodynamic states have been described during acute AI:
1. Low cardiac output, high systemic vascular resistance shock is caused by both decreased myocardial contractility and decreased preload.
2. High cardiac output, low systemic vascular resistance shock mimics septic shock.23 It appears that patients with AI present initially with a combination of cardiogenic shock and hypovolemic shock. Intravascular volume expansion with intravenous fluids results in an increase in cardiac output and a lowering of systemic vascular resistance. The hemodynamic profile that one sees depends on the timing of pulmonary artery catheter placement during the course of treatment in an individual patient. Thus, the hypotension of AI can mimic cardiogenic, hypovolemic, or septic shock (depending on when the hemodynamic assessment was made) and may be poorly responsive or unresponsive to treatment with fluids and vasopressors in the absence of glucocorticoid therapy.
Renal Effects
Glucocorticoids bind to the mineralocorticoid receptors in renal tubules and increase sodium reabsorption and excretion of potassium and hydrogen ions. Glucocorticoids increase the glomerular filtration rate, proximal tubular epithelial sodium transport, and free water clearance.21 Glucocorticoids also increase free water excretion by inhibiting the release of antidiuretic hormone (ADH).
Immunologic Effects
Glucocorticoids suppress immunologic responses, and this is the basis for their use in the treatment of autoimmune and inflammatory disorders. In the peripheral blood, they redistribute lymphocytes from the intravascular compartment to the lymphoid pool in the spleen, lymph nodes, and bone marrow. They therefore decrease lymphocyte counts, but neutrophil counts increase after glucocorticoid administration. Eosinophil counts fall, due to eosinophil apoptosis. The immunologic actions of glucocorticoids are mediated through T and B lymphocytes. Glucocorticoids inhibit immunoglobulin synthesis and cytokine production from lymphocytes. Glucocorticoids also inhibit monocyte differentiation into macrophages and plasminogen activators.21
Calcium Metabolism
Glucocorticoids lower serum calcium levels by several mechanisms. They inhibit calcium absorption from the gut, decrease renal calcium reabsorption (which results in hypercalciuria), and promote shift of calcium from the extracellular compartment to the intracellular compartment. Glucocorticoids inhibit osteoblast function and cause osteoporosis, which occurs in approximately 50% of patients who require long-term glucocorticoids.24 Avascular necrosis is a dreaded complication.21 It mainly affects the femoral head and causes pain and collapse of the bone, often necessitating hip replacement. This effect is not necessarily dose dependent.
Other Effects
Aldosterone secretion is regulated mainly by the renin-angiotensin system. The most potent modulator of this system is renal perfusion. Hyperkalemia inhibits production of renin but increases the synthesis of aldosterone. Aldosterone increases sodium reabsorption in the collecting tubules and at the same time causes potassium and hydrogen ion excretion. This is mediated by the Na+/K+ pump in the presence of the enzyme Na+/K+-ATPase and results in sodium and water retention and an increase in intravascular volume.21
Hyperkalemia, hyponatremia, non–anion gap metabolic acidosis, hemoconcentration, and hypovolemia provide important clinical clues to the diagnosis of primary AI. Because ACTH is not a potent regulator of aldosterone secretion, secondary AI is usually not associated with hyperkalemia. Hyponatremia and hypovolemia may be present in secondary AI but not to the degree found in primary AI. The renin-angiotensin system is activated during AI and serves as a defense mechanism to improve the low intravascular volume and the altered vasomotor tone that results from aldosterone and cortisol deficiency.25,26
Etiology and Pathogenesis
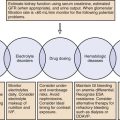
Causes of primary AI have been previously discussed and are shown in Box 59.1.
Autoimmune Disease
Autoimmune disease (Addison’s disease) is currently the most common cause of primary AI and accounts for approximately 80% of cases. For many years tuberculosis was the most common cause. AI may occur as isolated disease or as part of a polyglandular autoimmune syndrome associated with thyroiditis, diabetes mellitus (Schmidt’s syndrome), hypogonadism, vitiligo, and pernicious anemia.27–29 Autoimmune AI is more common in women than men and usually occurs in the third to fifth decades of life. The mean duration of symptoms before diagnosis is approximately 3 years. In this disorder high levels of circulating autoantibodies attack the cytoplasm of adrenal cortical cells and inhibit synthesis of glucocorticoids.27,28
Infectious Disease
Tuberculosis
The second most common cause of primary AI is adrenal gland destruction by Mycobacterium tuberculosis. This infection currently accounts for less than 20% of cases.30 This usually occurs in the presence of tuberculosis elsewhere in the body, especially with involvement of the lungs, genitourinary system, and gastrointestinal system. AI is usually manifest years after the initial presentation of tuberculosis.31 The mean duration of symptoms of AI prior to diagnosis is 6 to 9 months. AI secondary to tuberculosis occurs with equal frequency in men and women. In contrast to autoimmune adrenalitis, tuberculosis-induced adrenal disease is not associated with other endocrine diseases. In addition, with tuberculosis the adrenal glands are enlarged and may be calcified. In contrast, the adrenal glands in autoimmune adrenalitis are usually atrophied and noncalcified.
Fungal Disease
Fungal disease can also cause primary AI. Histoplasma capsulatum is the most common organism. As seen in tuberculosis, fungal infection is usually disseminated and involves organs other than the adrenal glands. Adrenal involvement may be seen during the active phase or may develop years later after the disease has become “inactive.” Sarosi and colleagues32 reported that more than 50% of patients with disseminated histoplasmosis had AI, and AI was the most common cause of death.
Acquired Immunodeficiency Syndrome
Patients with acquired immunodeficiency syndrome (AIDS) are at risk of developing AI by several different mechanisms. Fungal infections are more common in this patient population, and disseminated disease may involve the adrenal glands. Similarly, mycobacterial infection, cytomegalovirus infection, and Kaposi sarcoma may cause involvement of the adrenals in up to 50% of patients.3 In addition, sepsis and spontaneous adrenal hemorrhage are also seen in this group of patients. Because patients with human immunodeficiency virus (HIV) infection now survive longer, an increased incidence of AI is likely.
Neoplasia
Neoplastic metastasis to the adrenal glands has been found on autopsy in 27% to 40% of patients who die of malignancy.33–36 Yet metastatic carcinoma accounts for less than 1% of cases of primary AI.33 This finding is explained by the tremendous functional reserve possessed by the adrenal glands. More than 90% of the adrenal gland must be destroyed before hypofunction occurs. Many patients with metastasis to the adrenals do not develop hormonal deficiency.37 The most common neoplasms to involve the adrenals are lung cancer, breast cancer, melanoma, and lymphoma.35 AI usually occurs in the setting of widespread metastatic disease and is rarely the initial manifestation of malignancy.
Adrenal Hemorrhage
Besides the infectious causes, other conditions may predispose to adrenal hemorrhage, including severe illness (particularly cardiac disease), coagulopathy, anticoagulant therapy, thromboembolism, burns, and trauma.38 Under these circumstances the typical signs and symptoms of AI are often mistaken for those of other common conditions. Typical settings include the patient who is in the first or second postoperative week or a patient who has been started recently on anticoagulation therapy. Common findings include abdominal, back, flank, or chest pain; nausea; vomiting; fever; altered mental status; orthostatic hypotension; and a sudden drop in hematocrit. The hemodynamic crisis associated with adrenal hemorrhage occurs 1 to 3 days after the initial hemorrhage.
Etiology of Secondary Adrenal Insufficiency
Causes of secondary AI have been discussed previously and are shown in Box 59.2. Secondary AI occurs because of a decrease in ACTH caused by either hypothalamic-pituitary disease or suppression of the HPA axis as a result of glucocorticoid therapy. The most common cause today is discontinuation of corticosteroid therapy. Chronic glucocorticoid therapy leads to HPA axis suppression with resulting secondary AI if glucocorticoids are abruptly discontinued. With the exception of AI because of discontinuation of chronic glucocorticoid therapy, secondary adrenocortical insufficiency is much less common than primary AI. Isolated ACTH deficiency is rare, and ACTH is the last pituitary hormone to be impaired by enlarging sellar and suprasellar tumors.
Most patients admitted to the ICU with secondary AI have recently received steroid therapy or have taken steroids within the year prior to admission. No clear evidence indicates that detailing the duration or dose of steroid therapy predisposes patients to adrenal suppression. Doses of 25 mg of prednisone twice a day for 2 days, 12.5 mg per day for 6 months, or 5 mg per day for 5 years have all been shown to cause adrenal suppression. On the other hand, studies have shown that prednisone in doses less than 40 mg per day given every morning for 5 to 7 days did not cause adrenal suppression.39
As a practical guideline, all patients who have taken 40 mg of prednisone per day or its equivalent for a period greater than 2 or 3 weeks should be considered to be adrenal insufficient until proved otherwise. If glucocorticoids have been given to a patient for more than 1 to 2 weeks, they should be tapered off to allow time for the adrenal glands to recover function. AI can occur in response to stress as long as 1 year after steroids are discontinued.40 All of these patients should be evaluated for adrenal function and should be treated with stress doses of steroids during the interim period.
Clinical Features
Common symptoms and signs of AI are shown in Box 59.3. The symptoms, signs, and general laboratory data seen in AI are nonspecific. However, when taken together they form a pattern of findings that should suggest the possibility of AI. Patients with acute AI share many characteristics with patients who have chronic AI, but the symptoms are usually more severe in the acute setting. Virtually all patients complain of weakness, fatigue, and loss of appetite. They also complain of nausea and diarrhea with occasional vomiting and abdominal pain. Infrequently, patients note myalgias, arthralgias, and dizziness caused by orthostatic hypotension. Weight loss can occur. The classic presentation of acute AI is a patient with unexplained hemodynamic instability who is unresponsive to intravascular volume resuscitation and use of vasopressors. Patients with primary AI may have hyperpigmentation of the tongue, buccal mucosa, palmar creases, and scar tissue. This is caused by increased production of ACTH from the pituitary. Hyperpigmentation is notably absent in secondary AI. If the underlying problem is autoimmune adrenalitis, the patient may have vitiligo, pernicious anemia, or one of the other associated endocrinopathies.
Diagnosis
Laboratory Findings
Laboratory evaluation of patients with suspected AI is essential. In a patient with acute worsening of a chronic hypoadrenal state, the common laboratory findings shown in Box 59.4 are more likely to be present and are likely to be more pronounced. Electrolyte abnormalities depend on the type of deficiency: a combined glucocorticoid and mineralocorticoid deficiency (typically seen in primary AI) or an isolated glucocorticoid deficiency (characteristic of secondary AI).
Diagnostic Tests
Serum Cortisol Level
The biochemical diagnosis of AI is controversial. It is based on the demonstration of decreased cortisol production. Most clinical laboratories routinely measure total rather than free cortisol levels. Experts have recently suggested that measurement of free cortisol levels makes more physiologic sense, but studies have not helped establish diagnostic thresholds for free cortisol levels.41 In addition, variability of cortisol assays can confound the diagnosis of AI.42
Given the controversy in diagnosis, the following recommendations are made. A randomly measured serum cortisol level that exceeds 44 µg/dL makes the diagnosis of adrenocortical deficiency unlikely.5 Serum cortisol levels increase significantly in patients with normal adrenal function who are in shock and critically ill. The finding of a random serum cortisol level of less than 10 µg/dL in this setting is highly suggestive of compromised adrenal function and should prompt treatment or a confirmatory ACTH stimulation test.5
One must be careful in the interpretation of random serum cortisol levels in patients treated with several commonly used drugs in the ICU. Propofol produces a temporary reduction in serum cortisol levels. However, it does not seem to inhibit adrenal responsiveness to ACTH. Etomidate, on the other hand, is associated with a reduced serum cortisol concentration despite ACTH stimulation.43
Rapid ACTH Stimulation Test
The ACTH stimulation test measures the response of the adrenal gland to stimulation by exogenous ACTH. This test can be performed at any time of the day because the normal diurnal variation of cortisol is lost in the setting of critical illness. A blood sample is drawn, and a baseline serum cortisol level is measured. Cosyntropin (synthetic ACTH) 250 µg is then administered intravenously. Repeat samples are drawn at 60 minutes. Although controversial, some studies have shown that a low dose of ACTH (1 to 5 µg) produces a similar response as the 250 µg dose, especially in patients with AI that is recent or new onset.44,45
An increase in serum cortisol of less than 9 µg/dL following 250 µg of cosyntropin is highly suggestive of AI regardless of the baseline cortisol level. An increase in serum cortisol of greater than 17 µg/dL or total cortisol level of 44 µg/dL or greater suggests adrenal competence. When the baseline cortisol level is between 10 and 44 µg/dL, and the cortisol increment after cosyntropin stimulation is between 9 and 17 µg/dL, metyrapone testing is needed to assess adrenal function.5
The rapid ACTH stimulation test is a relatively simple test for evaluating AI.46 It does not, however, differentiate between primary and secondary AI. To differentiate between primary and secondary AI, a basal plasma ACTH determination is made. A serum cortisol measurement is then made following a continuous 48-hour infusion of ACTH. An increased basal ACTH (>250 pg/mL) or a serum cortisol level (<20 µg/dL) after 48 hours of ACTH stimulation is compatible with primary AI. On the other hand, a decreased basal ACTH and a high cortisol level after ACTH administration suggest secondary AI.
Critical Illness–Related Corticosteroid Insufficiency
Despite no conclusive evidence of benefit, in the 1950s, 1960s, and into the 1970s cortisol at low doses over days was often used in patients with severe manifestations of sepsis to counter the AI that was assumed to be present. This was based on autopsy studies that revealed adrenal necrosis in patients dying with severe infection. The subsequent recognition of the systemic effects of inflammation in sepsis and the discovery that the majority of patients in septic shock had normal or increased cortisol levels led to a paradigm shift in treating septic shock with massive doses of steroids given for a short period of time. This practice was based on animal studies showing that large doses of steroids given prior to boluses of endotoxin or gram-negative bacteria prevented death.42 Clinical trials testing the utility of several large doses of steroids in patients with septic shock failed to show benefit.47,48
One study in patients with septic shock demonstrated that regardless of baseline cortisol level, the inability to raise the cortisol level following ACTH stimulation by at least 10 µg/dL signified poor prognosis.49 Of this poor prognostic group, the higher the baseline cortisol level with failure to produce a 10 µg/dL increase, the worse the prognosis.
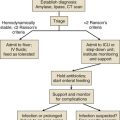
Management
Once the diagnosis of AI is made, a search for the cause should be initiated as the patient is being stabilized. To rule out tuberculosis, a purified protein derivative (PPD) skin test must be placed and a chest radiograph performed. An abnormal prothrombin time, partial thromboplastin time, or platelet count may point to an unsuspected coagulopathy suggesting the possibility of adrenal hemorrhage. Antiadrenal antibodies are found in about 70% of patients with autoimmune adrenal disease and in less than 0.1% of normal subjects.50 Computed tomography scanning of the abdomen is useful in determining the size and presence of calcification of the adrenal glands. Adrenal calcification can be seen in 53% of cases of tuberculosis.51
Management of AI can be best accomplished by identifying the degree of acuteness and severity of the patient’s illness at the time of presentation.52
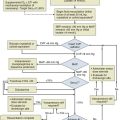
Severe Sepsis and Septic Shock
Six randomized clinical trials suggest that replacement of moderate-dose hydrocortisone (200-300 mg/day) decreases the need for vasopressor support in patients with septic shock.2,53 Among them, the two larger trials were better powered to detect a survival difference but had differing results. These differences were attributed in part to varying demographics and other factors. In the Corticus study, hydrocortisone did not decrease mortality rate in both responders and nonresponders to ACTH, but the patients who received hydrocortisone had more rapid resolution of shock, which has been seen in other studies as well. These results were different from the Annane study, in which the nonresponders to ACTH had both reduction in mortality rate and reversal of shock. The differences were attributed in part to the fact that in the Annane study, the patients enrolled were sicker, patients had higher SAPS II scores, and time to enrollment differed (8 hours versus 72 hours in the Corticus study). The Corticus study was published some years after the Annane study; it is possible that variations in the supportive care of the critically ill with advances in the care of the septic patient in the last few years could have made it difficult to show a mortality rate difference in Corticus. More patients in the Corticus study had a surgical source of sepsis, and thus source control may have played a bigger role in improving outcomes. In addition, fludrocortisones was not administered in the Corticus study. The duration of steroid therapy was different in the two studies, and this could have caused the differing results. In addition, Corticus had a higher rate of superinfection. Compounding all these issues is the difficulty surrounding the accurate diagnosis of AI.53,54 However, in a recent study, the addition of enteral fludrocortisone did not result in a significant improvement in hospital mortality rates.55
In the trials mentioned here, there was resolution of shock in both responders and nonresponders to ACTH stimulation. The decision to treat patients with septic shock with hydrocortisone should, therefore, probably not be based on the results of a random total cortisol level or the response to cosyntropin. If a clinician decides to perform an ACTH test, until the test is performed, treatment with dexamethasone in patients with septic shock should not be done because of the possibility that a single dose of a long-acting corticosteroid may cause prolonged suppression of the hypothalamic-pituitary axis.2
To date, no studies document an improved outcome with corticosteroid use in the absence of septic shock (Fig. 59.1).
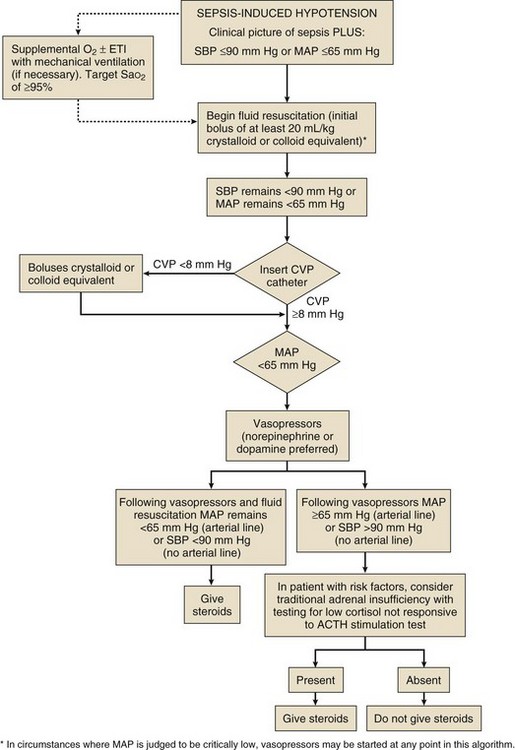
Additional Indications for Corticosteroids
Observational studies suggest that stress doses of corticosteroids may have a role in the management of critically ill patients with liver failure and cardiac surgery. Additional large randomized studies are needed in these areas.3
References
1. Cooper, MS, Stewart, PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003; 348:727–734.
2. Marik, PE, Pastores, SM, Annane, D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008; 36:1937–1949.
3. Marik, PE. Critical illness-related corticosteroid insufficiency. Chest. 2009; 135(1):181–193.
4. Bornstein, SR. Predisposing factors for adrenal insufficiency. N Engl J Med. 2009; 360:2328–2339.
5. Annane, D, Maxime, V, Ibrahim, F, et al. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am J Respir Crit Care Med. 2006; 174:1319–1326.
6. Span, LFR, Hermus, ARMM, Bartelink, AKM, et al. Adrenocortical function: An indicator of severity of disease and survival in chronically ill patients. Intensive Care Med. 1992; 18:93.
7. Jurney, TH, Cockrell, JL, Lindberg, JS, et al. Spectrum of serum cortisol response to ACTH in patients: Correlation with degree of illness and mortality. Chest. 1987; 92:292.
8. Vermes, I, Beishuizen, A, Hampsink, RM, et al. Dissociation of plasma ACTH and cortisol levels in critically ill patients: Possible role of endothelin and atrial natriuretic hormone. J Clin Endocrinol Metab. 1995; 80:1238.
9. Horrocks, PM, Jones, AF, Ratcliffe, WA, et al. Pattern of ACTH and cortisol pulsatility over 24 hours in normal males and females. Clin Endocrinol. 1990; 32:127.
10. Chernow, B, Alexander, HR, Smallridge, RC, et al. Hormonal responses to graded surgical stress. Arch Intern Med. 1987; 147:1273.
11. Nto, T, Fukata, J, Tam, S, et al. Biphasic changes in hypothalamo-pituitary-adrenal function during the early recovery period after major abdominal surgery. J Clin Endocrinol Metab. 1991; 73:111.
12. Ellis, MJ, Schmidli, RS, Livesey, JH, et al. Plasma corticotropin releasing factor and vasopressin responses to hypoglycemia in normal man. Clin Endocrinol. 1990; 32:93.
13. Wittert, GA, Stewart, DE, Graves, MP, et al. Plasma corticotropin releasing factor and vasopressin responses to exercise in normal man. Clin Endocrinol. 1991; 35:311.
14. Bartanusz, V, Jezova, D, Bertini, LC, et al. Stress induced increase in vasopressin and corticotropin releasing factor expression in hypophysiotropic paraventricular neurons. Endocrinology. 1993; 132:895.
15. Gllard, WO, Turnhill, D, Sappino, P, Muller, AF. Tumor necrosis factor alpha inhibits the hormonal response of the pituitary gland to hypothalamic releasing factors. Endocrinology. 1990; 127:101.
16. Darling, G, Goldstein, DS, Stull, R, et al. Tumor necrosis factor: Immune endocrine interaction. Surgery. 1989; 106:1155.
17. Jaattela, M, Ilvesmaki, V, Voutilnen, R, et al. Tumor necrosis factor as a potent inhibitor of adrenocorticotropin induced cortisol production and steroidogenic P450 enzyme gene expression in cultured human fetal adrenal cells. Endocrinology. 1991; 128:623.
18. Sharp, BM, Matta, SG, Peterson, PK, et al. Tumor necrosis factor alpha is a potent ACTH secretagogue: Comparison to interleukin 1 beta. Endocrinology. 1989; 124:3131.
19. Bateman, A, Singh, A, Kral, T, Solomon, S. The immune hypothalamo-pituitary axis. Endocrinol Rev. 1989; 10:92.
20. Besedovsky, H, Delrey, A, Sorkin, E, Dinarello, CA. Immunoregulatory feedback between interleukin 1 and glucocorticoid hormone. Science. 1986; 233:652.
21. Stewart, PM. The adrenal cortex and endocrine hypertension. In: Kronenberg HM, Melmed S, Polonsky KS, et al, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia: WB Saunders; 2011:479–534.
22. Svedmyr, N. Action of corticosteroids on beta-adrenergic receptors. Am Rev Respir Dis. 1990; 141:S31.
23. Bouachour, G, Tirot, P, Varache, N, et al. Hemodynamic changes in acute AI. Intensive Care Med. 1994; 20:138.
24. Luckert, BP, Rsz, LG. Glucocorticoid induced osteoporosis: Pathogenesis and management. Ann Intern Med. 1990; 112:352.
25. Munck, A, Náray-Fejes-Toth, A. Glucocorticoid physiology. In: DeGroot L, Jameson JL, eds. Endocrinology. 5th ed. Philadelphia: WB Saunders; 2005:2287–2309.
26. Schwartz, J, Keil, LC, Masseli, J, Reid, I. Role of vasopressin in regulating blood pressure in AI. Endocrinology. 1983; 112:234.
27. Bright, GM, Singh, I. Adrenal autoantibodies bind to adrenal subcellular fractions enriched in cytochrome c reductase and 5′-nucleotidase. J Clin Endocrinol Metab. 1990; 70:95.
28. De Bellis, A, Bizzarro, A, Rossi, R, et al. Remission of subclinical adrenocortical failure in subjects with adrenal autoantibodies. J Clin Endocrinol Metab. 1993; 76:1002.
29. Loriaux, DL. The polyendocrine deficiency syndromes. N Engl J Med. 1985; 312:1568.
30. Irwine, WJ, Barnes, EW. AI. Clin Endocrinol Metab. 1972; 1:549.
31. Guttma, PH. Addison’s disease. Arch Pathol. 1930; 10:742.
32. Sarosi, GA, Voth, DW, Dahl, BA, et al. Disseminated histoplasmosis: Results of long term follow up. A CDC cooperative mycoses study. Ann Intern Med. 1971; 75:511.
33. Knowlton, AI. Adrenal insufficiency in the intensive care setting. J Intensive Care Med. 1989; 4:35.
34. Dluhy, RG. The growing spectrum of HIV-related endocrine abnormalities. J Clin Endocrinol. 1990; 70:563.
35. Redman, BG, Pazdur, R, Zingas, AP, Loredo, R. Prospective evaluation of AI in patients with adrenal metastasis. Cancer. 1987; 60:103.
36. Irwine, W. Autoimmunity in endocrine disease. Proc R Soc Med. 1974; 67:548.
37. Kung, AWC, Pun, KK, Lam, K, et al. Addisonian crisis as presenting feature in malignancies. Cancer. 1990; 65:177.
38. Rao, RH, Vagnucci, AH, Amico, JA. Bilateral massive adrenal hemorrhage: Early recognition and treatment. Ann Intern Med. 1989; 110:227.
39. Christy, NP, Wallace, EZ, Jler, JW. Comparative effects of prednisone and cortisone in suppressing the response of the adrenal cortex to exogenous adrenocorticotropin. J Clin Endocrinol Metab. 1956; 16:1059.
40. Graber, AL, Ney, RL, Nicholson, WE, et al. Natural history of pituitary-adrenal recovery following long term suppression with corticosteroids. J Clin Endocrinol. 1956; 25:11.
41. Hamrahain, AH, Oseni, TS, Awafah, BM, et al. Measurement of serum free cortisol in critically ill patients. N Engl J Med. 2004; 350:1629–1639.
42. Cohen, J, Ward, G, Prins, J, et al. Variability of cortisol assays can confound the diagnosis of adrenal insufficiency in the critically ill population. Intensive Care Med. 2006; 32:1901–1911.
43. Jackson, WL. Should we use etomidate as an induction agent for endotracheal intubation in patients with septic shock? A critical appraisal. Chest. 2005; 127:1031–1038.
44. Dickstein, G, Shechner, C, Nicholson, WE, et al. Adrenocorticotropin stimulation test: Effects of basal cortisol level, time of day, and suggested new sensitive low dose test. J Clin Endocrinol Metab. 1991; 72:773.
45. Crowley, S, Hindmarsh, C, Honour, JW, Brook, CGD. Reproducibility of the cortisol response to stimulation with dose of ACTH: The effect of basal cortisol levels and comparison of low dose with high dose secretory dynamics. Endocrinology. 1993; 136:167.
46. Motsay, GJ, Alho, A, Jaeger, T, et al. Effects of corticosteroids on the circulation in shock: Experimental and clinical results. Fed Proc. 1970; 29:1861–1873.
47. Sprung, CL, Caralis, PV, Marcial, EH, et al. The effects of high-dose corticosteroids in patients with septic shock. N Engl J Med. 1984; 311:1137–1143.
48. Bone, RC, Fisher, CJ, Clemmer, TP, et al. and the Methylprednisolone Severe Sepsis Study Group: A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987; 317:653–658.
49. Annane, D, Sébille, V, Troche, G, et al. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000; 283:1038–1045.
50. Nerup, J. Addison’s disease—A review of some clinical, pathological and immunological features. Dan Med Bull. 1974; 21:201.
51. Doppman, JL, Gill, JR, Nienhuis, AW, et al. CT findings in Addison’s disease. J Comput Assist Tomogr. 1982; 6:757.
52. Lamberts, SWJ, Bruining, HA, Dejong, FH. Drug therapy: Corticosteroid therapy in severe illness. N Engl J Med. 1997; 337:1285–1292.
53. Annane, D, Sebille, V, Charpentier, C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002; 288:862–871.
54. Sprung, CL, Annane, D, Keh, D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008; 358:111–124.
55. Annane, D, Cariou, A, Maxime, V, et al. COIITSS Study Investigators: Corticosteroid treatment and intensive insulin therapy for septic shock in adults: A randomized controlled trial. JAMA. 2010; 303(4):341–348.