Adrenal Causes of Hypercortisolism
Clinical Features of Hypercortisolism
Diagnosis of Cushing’s Syndrome
Diagnosis of Adrenal Cushing’s Syndrome
Differential Diagnosis of Adrenal Causes of Cushing’s Syndrome
Pathology of Cortisol-Secreting Adrenal Lesions
Pathogenesis of Adrenal Tumors
ACTH-Independent Adrenal Hyperplasia
Primary Pigmented Nodular Adrenal Dysplasia
Adrenal causes of Cushing’s syndrome are diverse, including both tumoral and genetic disorders. Tumors arising from the zona fasciculata of the adrenal cortex may be benign or malignant and are thus classified as adenomas and carcinomas, respectively, a distinction that is by no means always straightforward (see also Chapter 11). ACTH-independent adrenal hyperplasia, both macro- and micronodular, is characterized by nodular hyperplasia of the zona fasciculata, possibly a consequence of inappropriate or excessive stimulation by factors other than ACTH. Lastly, familial causes of adrenal Cushing’s syndrome include primary pigmented nodular adrenal dysplasia (PPNAD), usually in the context of Carney’s complex, and the McCune-Albright syndrome (MAS).
Historical Notes
The first description of Cushing’s syndrome dates back to 1899 when William Osler presented a patient with “an acute mixoedematous condition,” which he ascribed to thyroid gland dysfunction, but in hindsight was actually due to excess glucocorticoid secretion.1 Some 10 years later, Harvey Cushing summarized the symptoms featured in his namesake syndrome and established the causative link with “pituitary basophilism.” The second case in his landmark monograph, however, was ascribed to an adrenal disorder, and he himself recognized that the syndrome of “diabetes in bearded women” was often associated with hyperplasia or tumors of the suprarenal glands.2 In 1934, Walters et al. demonstrated that removal of the adrenals was curative in these patients,3 thus validating the importance of the adrenal gland as a target of pituitary hyperproduction or primary cause of glucocorticoid excess. This distinction was by no means straightforward; pituitary Cushing’s disease was also being called “ACTH-dependent adrenal hyperplasia,” necessitating bilateral adrenalectomy, and “nodular adrenal hyperplasia” was considered partially ACTH dependent.
Epidemiology
Cushing’s syndrome is a rare disease. Its incidence has been estimated at around 2 to 10 patients per million population per year, with less than half due to adrenal causes.4,5 Of interest, adrenal adenomas appear to affect the Japanese population with greater frequency, reportedly presenting over 20 new patients per million per year.6 The ratio between benign and malignant adrenal tumors varies from roughly equal in some series5,7 to 10:1 in Japanese studies.6 Oncologic surveys report an annual incidence of adrenal neoplasms of around 2 per million8,9 and 0.6 to 1 per million for adrenocortical cancer.8 Approximately one third of these patients will present with Cushing’s syndrome,10,11 resulting in 0.2 to 0.3 cortisol-secreting adrenal cancers per million per year, which tallies nicely with estimates garnered from endocrinologic studies.5
Age and gender distribution vary somewhat between the different adrenal etiologies of Cushing’s syndrome, with adrenal cancer presenting a bimodal distribution (peak in childhood/adolescence then again after 45 years of age), and adrenal adenoma occurring mostly in young adults (25 to 45 years of age) (Fig. 7-1). PPNAD gives rise to hypercortisolism during infancy and adolescence in over half of cases,12 whereas ACTH-independent macronodular adrenocortical hyperplasia (AIMAH) has been reported mostly in adults older than 50.13,14 Adrenal Cushing’s syndrome in the majority of adult patients is due to benign adenomas (50% to 80%, according to different series). Adrenal-cortex carcinoma accounts for another 20%, and a scattering of cases are due to ACTH-independent adrenal hyperplasia and PPNAD.15,16 Conversely, in childhood, malignant forms make up the major share, the remainder due to adenomas or even genetic causes such as PPNAD or MAS.12,17
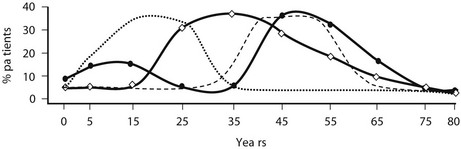
FIGURE 7-1 Age distribution in adrenal Cushing’s syndrome according to etiology, depicted as percentage of patients at different ages: adenoma (-◊-), carcinoma (-•-), PPNAD (…….), and AIMAH (- – – ).
Adrenal adenoma occurs mostly in females, as does—to a lesser extent—cortisol-secreting adrenal cancer.5,18,19 No clear-cut sex-related pattern has been observed for Cushing’s associated with AIMAH and PPNAD.
Mortality from untreated Cushing’s syndrome is high, with 50% survival 5 years after the diagnosis,20 mostly due to cerebrovascular events and infections. Upon removal of a benign adenoma, taking perisurgical mortality into consideration, 5-year survival averaged 77%21 in the 1980s, but is nowadays comparable to mortality in the general population.5,22 As regards mortality from malignant adrenocortical cancer, outcome is dismal, with high mortality for stage III and IV disease and slightly better survival for patients diagnosed with localized disease.23–25 In Cushing’s syndrome due to PPNAD within Carney’s complex, a familial multiple endocrine neoplasia syndrome, extraendocrine manifestations such as cardiac myxomas or malignant Schwannomas contribute to excess mortality.26
Clinical Features of Hypercortisolism
Cushing’s syndrome features all the consequences of tissue exposure to excessive cortisol concentrations, including aspecific alterations such as obesity, hypertension, and mood changes, and more unique signs such as proximal muscle wasting, purple striae, and easy bruising. The severity of clinical manifestations varies according to the extent and duration of cortisol hypersecretion, with adrenal cancer characterized by rapid attainment of very high cortisol levels and severe clinical signs; adenomas present milder glucocorticoid hypersecretion and a more indolent course. In fact, the clinical presentation of adrenal adenomas closely resembles that of pituitary-driven Cushing’s disease, whereas adrenal cancer mimics ectopic ACTH secretion due to poorly differentiated neuroendocrine tumors. On average, over 2 years are necessary for a cortisol-secreting adrenal adenoma to be diagnosed after the appearance of the first symptoms, but the remarkable clinical presentation of adrenal carcinoma leads to the diagnosis in half that time.7,27 Indeed, the time to diagnosis is inversely related to mean cortisol levels (Fig. 7-2).
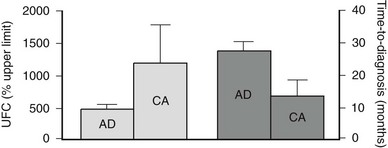
FIGURE 7-2 Urinary free cortisol (UFC) and time to diagnosis in patients with Cushing’s syndrome due to adrenal adenoma (AD) or adrenal carcinoma (CA). UFC is expressed as percent of the upper limit of the normal range.
Specifically, patients with hypercortisolism usually present with central obesity, a round and erythematous face, supraclavicular fat pads, cervical fat accumulation (the so-called “buffalo hump”), thinning of the skin, purple striae, and proximal muscle weakness. Hirsutism, a common feature of ACTH-dependent Cushing’s syndrome, is usually absent in benign tumors that secrete only cortisol but, conversely, accompanies malignant transformation.7 Hypertension as well as osteopenia and pathologic fractures (ribs, feet, and vertebrae are frequently affected sites) are common, and osteopenia in young patients with Cushing’s syndrome should raise the suspicion for PPNAD (see later). Derangements in blood chemistry include increased white blood cell count with granulocytosis, lymphopenia, and eosinopenia; a slightly elevated red blood cell count; increased VLDL, LDL, and HDL cholesterol and triglycerides; and frequently, impaired glucose tolerance or overt type 2 diabetes mellitus. Electrolyte imbalances such as hypokalemia are more common in severe hypercortisolism. Case history will reveal menstrual irregularities or even amenorrhea, reduced libido or impotence, fatigue, easy bruising, insomnia, and psychiatric alterations. These latter range from changes in mood, irritability with rage fits, anxiety, impaired memory, and concentration to psychosis with frank depression and manic episodes. Curiously, in children and adolescents, hypercortisolism has been associated with overachievement both in school and in extracurricular activities. Less frequent signs are cutaneous fungal infections, wound dehiscence, exophthalmos, nephrolithiasis, glaucoma, and headache. As mentioned before, severity of the clinical presentation often reflects the underlying etiology and features such as extreme muscle atrophy, hypokalemia, hypercalcemia, and the abrupt appearance of diabetes should increase the suspicion of adrenocortical cancer (Fig. 7-3). Except for signs of virilization, however, no single sign or symptom is significantly overrepresented in either etiology.7,16 The hormonal secretory pattern of adrenal carcinoma may change over time. Curiously, Cushing’s syndrome developed in two previously asymptomatic patients after recurrence of adrenocortical carcinoma, but conversely, no sign of hypercortisolism accompanied relapse of a previously symptomatic adrenocortical carcinoma.18
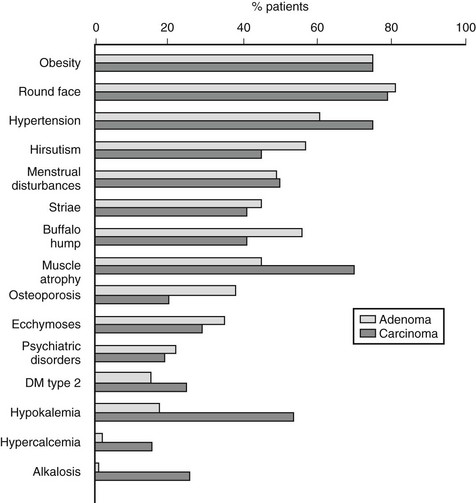
FIGURE 7-3 Clinical presentation in patients with Cushing’s syndrome due to adrenal adenoma or adrenal carcinoma.
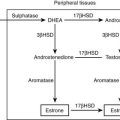
In addition to cortisol hypersecretion, adrenal androgens may also be produced in excess, most notably in adrenocortical cancer and more rarely in adenomas. Androgen secretion reflects inefficient steroidogenesis, since enzymes involved in the later steps of cortisol synthesis (e.g., 11β-hydroxylase or 21β-hydroxylase) are unable to transform their substrate overload. Precursors, therefore, accumulate and are shunted to alternative biosynthetic pathways, leading to accumulation of DHEA, DHEAS, androstenedione, and estrogen (see Chapter 1). This alteration is the biochemical correlate of progressive dedifferentiation of adrenal tumors, with steroidogenesis proceeding to its final product, cortisol, in benign and hyperplastic lesions, whereas poorly differentiated carcinomas are unable to efficiently carry steroidogenesis to term.28 From a clinical viewpoint, androgen hypersecretion gives rise to significant hirsutism, acne, and temporal balding in adult patients and contributes to menstrual alterations in women. In children, hyperandrogenism may arrest linear growth in boys or lead to sexual precocity and virilization in girls.29 Excess estrogen secretion may lead to gynecomastia in men.30 Mixed cortisol- and aldosterone-producing adenomas have also been reported,31 as have mixed cortisol-, androgen-, and estrogen-producing adenomas.32
Adrenal cortex carcinomas may present with mass-related signs, symptoms of metastatic disease, or nonspecific features of malignancy such as weight loss, fever, and anorexia. Palpable and painful abdominal masses, an acute abdomen, and vascular obstruction by tumor thrombi of hepatic veins (Budd-Chiari syndrome) or the inferior vena cava may first raise suspicion of adrenal cancer. Metastases occur most frequently to liver, lungs, retroperitoneal lymph nodes, and bone and may give rise to back pain and osteoporotic fractures, or hypoglycemia if the liver is extensively infiltrated18,24 (see Chapter 11).
Diagnosis
Diagnosis of Cushing’s Syndrome
Urinary free cortisol (UFC) levels are usually very high in patients with adrenocortical cancer (up to 100 times greater than normal), whereas such striking elevations are less frequent in patients with cortisol-secreting adenoma (Fig. 7-4). Indeed, UFC concentrations may occasionally fall within the normal range in these latter patients,27 and this underlines the need for multiple UFC measurements.33,34 In patients with AIMAH, UFC concentrations are rarely very high and may fluctuate according to stimulation by the causative aberrant receptor. For example, patients in whom hypercortisolism was dependent upon adrenal cortex stimulation by LH, who exhibit markedly elevated UFC concentrations and develop Cushingoid features only during pregnancy and menopause.35,36 In young patients with PPNAD, again, UFC rarely reaches very high levels and may present slow, periodic progression.12,37,38 Other tests usually involve assessment of cortisol daily variations or its suppressibility by dexamethasone. In patients with full blown hypercortisolism, two clearly pathologic results or even one are sufficient to establish the diagnosis of Cushing’s syndrome and proceed with the diagnostic workup. However, in patients with mildly elevated or fluctuating cortisol values (as often occurs with AIMAH or even PPNAD), repeat evaluations over time are necessary. A secure diagnosis of Cushing’s syndrome is essential before performing tests for the etiologic diagnosis of hypercortisolism, because the results obtained with these procedures may overlap with normal physiology.
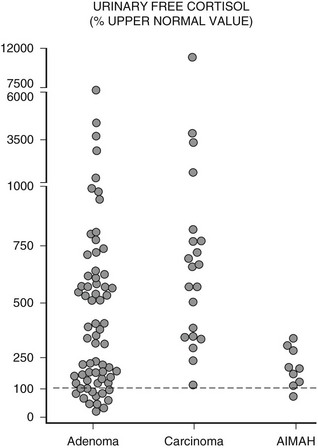
FIGURE 7-4 Distribution of urinary free cortisol (UFC) levels in patients with adrenal Cushing’s syndrome. Values are expressed as percentage of the upper limit of normal range (dashed line = 100%). (Adapted from Invitti C, Pecori Giraldi F, De Martin M, Cavagnini F: Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study, J Clin Endocrinol Metab 84:442, 1999. © Endocrine Society.)
Diagnosis of Adrenal Cushing’s Syndrome
In the vast majority of patients with adrenal Cushing’s syndrome, plasma ACTH concentrations are suppressed, thus providing clear distinction from ACTH-dependent Cushing’s syndrome. Initially formulated guidelines indicated that morning ACTH concentrations lower than 10 pg/mL (2 pmol/L) or evening ACTH levels lower than 5 pg/mL (1 pmol/L) were indicative of ACTH-independent Cushing’s syndrome.39 In RIA assays with 5 to 10 pg/mL sensitivity, this corresponds to values below the detection limit. With the newly developed IRMA or chemiluminescent assays capable of measuring plasma ACTH concentrations as low as 1 pg/mL (0.2 pmol/L), ACTH values may be detectable in patients with adrenal Cushing’s syndrome but usually are below the lower limit of the normal range.16,27,40,41 However, in an Italian study on over 50 patients with adrenal Cushing’s syndrome, up to one fourth presented ACTH concentrations within the normal range27 (Fig. 7-5), but whether this is because of issues in assay methodology, as seems more likely, or to incomplete suppression of pituitary ACTH secretion remains to be established. Occasionally, ACTH concentrations are frankly non-suppressed40,42,43; in one patient bearing an adrenal carcinoma, ACTH concentrations were persistently high because the tumor itself produced ACTH.44 In addition to patients with adrenal Cushing’s syndrome presenting measurable or normal ACTH concentrations, some 10% of patients with Cushing’s disease display ACTH values below 10 pg/mL (2 pmol/L),27 thus blurring the boundaries between the two etiologies. To correctly diagnose patients falling into this grey area, stimulation with CRH proves of use; patients with pituitary-dependent hypercortisolism will mount an ACTH response to the stimulus, whereas patients with ACTH-independent hypercortisolism will not27,45,46 (Fig. 7-6).
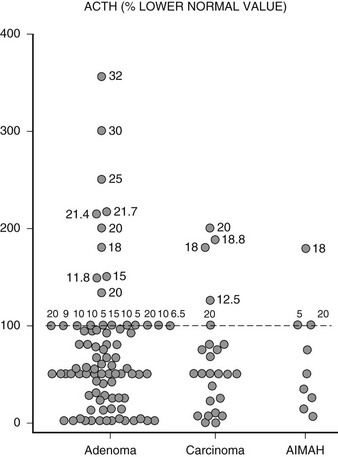
FIGURE 7-5 Distribution of plasma ACTH levels in patients with adrenal Cushing’s syndrome. Values are expressed as percentage of the lower limit of the normal range. For values within the normal range, numbers close to individual points indicate absolute ACTH concentrations in pg/mL measured using different assays. (Adapted from Invitti C, Pecori Giraldi F, De Martin M, Cavagnini F: Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study, J Clin Endocrinol Metab 84:442, 1999. © Endocrine Society.)
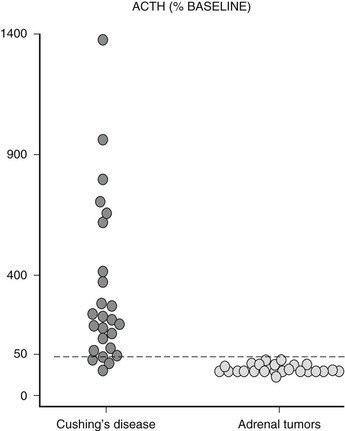
FIGURE 7-6 ACTH response (% baseline) to stimulation with CRH in patients with Cushing’s syndrome presenting with adrenal nodules.
In the past, tests such as stimulation with metyrapone or suppression with high-dose dexamethasone have been employed to distinguish between adrenal and pituitary Cushing’s syndrome,47 but their suboptimal specificity has superseded their routine use for this purpose. However, the high-dose dexamethasone suppression test has been re-proposed for the etiologic diagnosis of adrenal Cushing’s syndrome (see later).
Differential Diagnosis of Adrenal Causes of Cushing’s Syndrome
The etiology of adrenal Cushing’s syndrome is usually established by imaging of the adrenals (see also Chapter 10), since biochemical and clinical features overlap considerably between adenoma, carcinoma, and adrenal hyperplasia.7,14,16 Hormonal measurements provide useful adjunctive information but rarely establish the diagnosis with certainty. Genetic causes, such as PPNAD or MAS, may be recognized by features typical to the syndrome (e.g., lentigines or myxomas and fibrous osseous dysplasia, respectively; see later).
Steroid Markers
Dehydroepiandrosterone Sulfate: Measurement of serum dehydroepiandrosterone sulfate (DHEAS) may aid in the distinction between benign and malignant adrenal tumors. Indeed, although no cutoff value ensures absolute diagnostic accuracy, low age-and-gender-adjusted DHEAS levels are usually found in Cushing’s syndrome due to adrenal adenoma, whereas DHEAS levels are markedly increased in adrenal carcinoma (Fig. 7-7), especially in children.16,29,48,49 Two mechanisms underlie this diversity: on the one hand, DHEAS is an ACTH-dependent androgen, so inhibition of the normal adrenal gland in patients with cortisol-secreting adrenal adenoma will lead to reduced DHEAS secretion. On the other hand, inefficient steroidogenesis in malignant neoplasms leads to the release of several steroid precursors, including delta-5 steroids such as DHEAS. Of interest, DHEAS levels often remain suppressed long after removal of the hypersecreting adrenal gland, occasionally up to 8 years after surgery.48,50 This is in contrast with the recovery of ACTH and cortisol secretion, which usually occurs 12 to 18 months after surgery and indicates a dissociation of adrenal DHEAS and cortisol regulation.48,50 Enhanced sensitivity of the reticular zone to ACTH deficiency and preferential inhibition of 17,20-lyase in the atrophic adrenal gland have been hypothesized as causal factors.
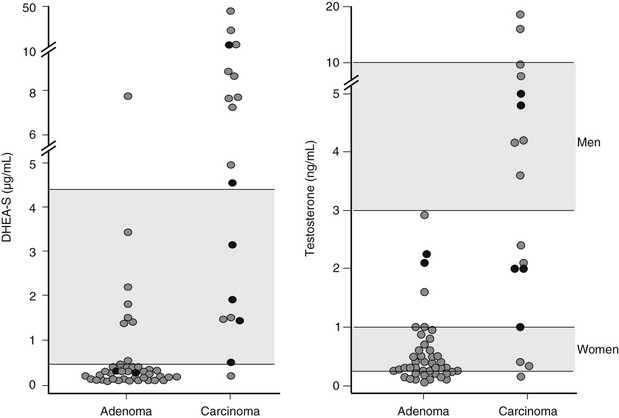
Testosterone and Androstenedione: Plasma testosterone levels are within the lower centiles of the normal range in men with cortisol-secreting adrenal adenoma,51 reflecting testosterone suppression (see Fig. 7-7). This is in line with the 30% to 50% fall in testosterone levels reported after administration of high doses of cortisol or dexamethasone52 and its rebound increase following removal of the adenoma.51 Normal testosterone levels have been reported in women with cortisol-secreting adrenal adenomas7 (see Fig. 7-7). Androstenedione levels are mostly low-normal in patients of both sexes with cortisol-secreting adrenal adenomas.29,53 Conversely, in adrenal carcinoma, androstenedione and testosterone levels may be high in both men and women (see Fig. 7-7) when impairment of downstream steroidogenic enzymes in the tumor, in particular 21-hydroxylase and 11β-hydroxylase, leads to accumulation of progesterone or pregnenolone and shunting of these precursors towards 17,20-lyase53 (see Figure 1-4 in Chapter 1). Accumulated DHEA can be transformed into androstenedione and subsequently into testosterone. Indeed, the detection of elevated testosterone or androstenedione in a patient with an adrenal tumor is virtually diagnostic for adrenal carcinoma.29,53 In children with adrenal carcinoma, Cushing’s syndrome is often accompanied by virilization, and testosterone, androstenedione, and DHEAS levels may reach extremely high levels.24,29
17-Hydroxyprogesterone and 11-Deoxycortisol: These two cortisol precursors are usually normal in patients with benign cortisol-secreting adrenal lesions and elevated in malignant adrenocortical tumors.24,29,53 Normal plasma hormone levels, however, do not allow adrenocortical cancer to be ruled out.
Urinary Steroid Profile: The hallmark of hypercortisolism is excess urinary excretion of cortisol and its main metabolites, such as tetrahydrocortisol, tetrahydro-11-deoxycortisol, tetrahydrocortisone and derivate cortols and cortolones (see Chapter 1). In addition, minor cortisol metabolic pathways, such as 6β-hydroxylation, are recruited to metabolize excess cortisol. In cortisol-producing adenomas, steroid synthesis is efficient, and only minor amounts of early products of the steroid cascade are excreted in urine.54,55 Conversely, in adrenocortical carcinoma, steroid precursors accumulate, and 3β-hydroxysteroids, desoxycorticosterone, and pregnanetriol—a 17-hydroxyprogesterone metabolite—become abundant in urine.54,56 Urinary 3β-hydroxy DHEA metabolites, such as 16alpha DHEA, androstenediol, and androstenetriol, as well as androstenedione-derived products such as etiocholanolone, androsterone, and androstanediols (Fig. 7-8), are usually high in adrenocortical carcinomas.55,57 In addition to these metabolites of early steroid synthesis products, tetrahydro-11-deoxycortisol, the metabolite of the cortisol substrate, is also typically increased in adrenocortical carcinoma, a consequence of relative 11β-hydroxylase deficiency.55,56 Investigation of the urinary steroid profile by gas chromatography mass spectrometry may reveal unique steroid excretory patterns, but the wide variability of excreted steroids and its changes over time hamper its routine use in clinical setting.
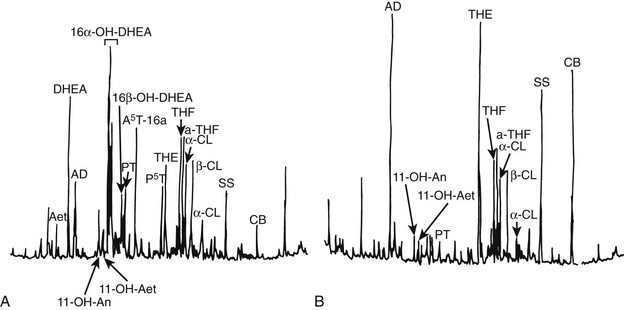
FIGURE 7-8 Chromatographic profile of urinary steroids in (A) a 6-year-old girl bearing an adrenal carcinoma and (B) an age- and sex-matched normal subject. Androsterone (An), etiocholanolone (Aet), androsten-3β, 16α, 17β-triol (A5T), pregnanetriol (PT), pregnentriol-5-ene-3βhydroxy (P5T), tetrahydrocortisone (THE), tetrahydrocortisol (THF), allo-tetrahydrocortisol (a-THF), cortols (C), cortolones (CL); internal standards (AD, SS, CB). (From Homoki J, Holl R, Teller WM: Urinary steroid profiles in Cushing’s syndrome and tumors of the adrenal cortex, Klin Wochensch 65:721, Fig. 1, 1987; © Springer).
High-Dose Dexamethasone Testing
Adrenal tumors, both benign and malignant, fail to suppress UFC secretion after 2 days of 2 mg of oral dexamethasone every 6 hours; cutoffs for suppression are a 50% decrease from baseline of UFC levels. Patients with PPNAD often exhibit a marked paradoxical rise in UFC levels during high-dose dexamethasone testing.15,37,41 This test falls short of providing diagnostic certainty in the differential diagnosis of adrenal Cushing’s syndrome, since some patients with adrenal adenoma also exhibit an increase in UFC levels after 8 mg dexamethasone for 2 days,15,27,37 but a doubling of UFC levels is highly suggestive of PPNAD. Parallel evaluation of creatinine clearance should be performed to exclude gross changes in glomerular filtration rate.58 However, patients with malignancy may also exhibit a paradoxical increase in cortisol levels after high-dose dexamethasone testing,43,59 so this procedure should be performed only after imaging has narrowed down the choice to benign adrenal lesions.
Subclinical Cushing’s Syndrome
This condition, characterized by subtle alterations of HPA-axis parameters in patients who do not present stigmata of Cushing’s syndrome, is being increasingly recognized in subjects with an adrenal incidentaloma. Indeed, a variable percentage of adrenal incidentalomas will reveal themselves as cortisol-secreting adenomas, 5% to 20% according to various series,60,61 possibly higher (up to 47%) in the Japanese population.62 This variability can be ascribed in part to the different criteria adopted for the biochemical diagnosis of subclinical Cushing’s and in part to the variable degree of cortisol excess; this condition is by no means homogeneous. Usually, two or more of the following are considered indicative of subclinical Cushing’s syndrome: (1) incomplete cortisol suppression after low-dose dexamethasone; (2) low baseline ACTH levels; (3) absent cortisol circadian rhythm; (4) blunted ACTH response to CRH; (5) elevated UFC; (6) decreased DHEAS.63 Iodocholesterol scintigraphy may also be useful because exclusive uptake by the adrenal mass is indicative of excessive autonomous cortisol secretion with functional inhibition of the contralateral gland.64 On the other hand, the degree of uptake does not differentiate between overt and subclinical Cushing’s syndrome. Patients diagnosed with subclinical Cushing’s are somewhat older (mostly older than 50) than those with Cushing’s syndrome due to benign cortisol-secreting tumors, and the risk of developing overt Cushing’s syndrome in the short term appears low.46,65 However, even the mild excess in cortisol secretion observed in these patients is deleterious for metabolic, cardiovascular, and bone physiology, and removal of the “pretoxic” adrenal gland is followed in a number of cases by amelioration of diabetes, hypertension, insulin resistance, obesity, and parameters of bone turnover.66 A note of caution is necessary upon removal of the lesion: Suppression of the contralateral gland may lead to postsurgical adrenal insufficiency.65
Imaging
Radiologic procedures for visualization of adrenal lesions comprise abdominal x-ray, sonography, computed tomography (CT), and magnetic resonance imaging (MRI), while iodocholesterol scintigraphy and the recently developed adrenal-specific positron emission tomography (PET) provide functional assessment of adrenal masses. Additional procedures such as arteriography and CT-guided biopsy are only rarely used in patients with adrenal Cushing’s syndrome (for detailed discussion see Chapter 10).
The aim of adrenal radiology in patients with adrenal Cushing’s syndrome is to visualize the lesion, define its morphologic features and eventual extension into neighboring structures and provide insights into the nature and function of the tissue. In addition, unrelated adrenal lesions such as silent pheochromocytoma, metastases, collision tumors, and even accessory spleens must be recognized.67,68 As with adrenal pathology, the results of individual imaging studies may be unequivocal and allow a straightforward diagnosis, whereas in other cases, CT, MRI, and scintigraphy may all be necessary. Unenhanced CT is usually the first-line procedure and may provide sufficient information to diagnose a benign lesion. Should malignancy be suspected, chemical-shift MRI may be performed to confirm the diagnosis and allow the staging of the lesion.
Computed Tomography: This technique provided a breakthrough in adrenal radiology, enabling easy visualization of the glands, given their location, morphology, and composition. Indeed, retroperitoneal fat provides an ideal background for recognition of the Y-shaped hyperlucent adrenal gland. The adrenal rim is usually straight or slightly concave, and any convexity should increase suspicion for underlying lesions. Adrenal size measurements are of little use because they largely depend on gland orientation.69
Adrenocortical adenomas, carcinomas, and macronodular hyperplasia are easily identified at unenhanced, thin-section (3 to 5 mm) CT scanning. As to their differentiation, adenomas usually present as a small (<3 cm), homogeneous, round mass with smooth margins and thin peripheral capsule (Fig. 7-9, left panel). Given the high steroid content of clear and compact cells, density of the mass is typically low, near that of water, and unenhanced attenuation values below 10 HU are commonly considered indicative of adenoma.19,70 Black adenomas present somewhat higher density than clear-cell yellow adenomas.71 Injection of a contrast medium, when performed, leads to homogeneous enhancement of the entire lesion, followed by rapid washout; attenuation values after enhancement do not allow clear distinction from nonadenomatous masses, whereas washout of contrast medium provides useful information for the differentiation from malignant lesions such as metastases.70 The unaffected gland is usually normal sized or occasionally smaller.15
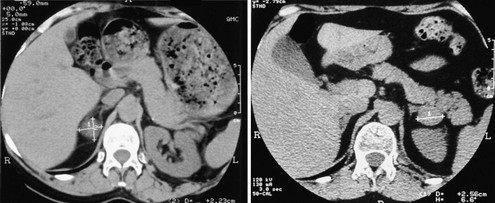
FIGURE 7-9 CT scan of cortisol-secreting adrenal adenoma (left panel) and adrenal nodular hyperplasia (right panel). This latter patient presented with a left-sided macronodule and bilateral enlarged glands.
Adrenocortical carcinomas commonly appear as large (>6 cm), irregularly shaped, inhomogeneous lesions with soft tissue density (>20 HU) and patchy enhancement. The borders of the lesion are often “smudged” and only rarely sharp. The heterogeneity of the mass is due to intratumoral necrosis, cystic degeneration, or calcification.19,69 These latter features, while extremely frequent in adrenal cancer, may be encountered also in degenerated adenomas72 and are not absolute criteria. Conversely, findings diagnostic for carcinoma are local spread (e.g., extension into the inferior vena cava, lymph node enlargement) or metastases. The size of the lesion, initially considered a useful criterion for the distinction of benign from malignant lesions, has increasingly been questioned; both large adenomas (>6 cm) and small carcinomas (<3 cm) are not infrequent.24 In addition, both CT and MRI may underestimate the size of the lesion by as much as 20%.73
In patients with AIMAH, CT scan most commonly reveals prominent glands with or without multiple large nodules. Massive macronodular hyperplasia is usually easily recognized because the glands appear markedly hypertrophic and multiple macronodules (>1 cm) can be seen in both adrenals14 (see Fig. 7-9, right panel). In other cases, only one gland appears to be affected.74 Conversely, in micronodular adrenal hyperplasia, the glands appear only slightly enlarged or even indistinguishable from normal. Indeed, a normal adrenal scan in a patient with ACTH-independent Cushing’s syndrome may provide indirect evidence of AIMAH.75
Adrenal radiology in patients with PPNAD varies from a normal appearance, slight enlargement of one or both adrenal glands, to unilateral or bilateral micronodules.12,76,77 Macronodules are more rarely encountered and mostly in patients older than 25 years of age.77 One feature typical to PPNAD is the “knobbly” and irregular outline of the glands, most likely due to internodular atrophy, resulting in a “beads-on-string” appearance.19,77
Magnetic Resonance Imaging: MRI easily visualizes both normal adrenal glands and adrenal masses by virtue of the abundant hydrogen ions in lipid tissues. In addition to two-dimensional imaging, which may reveal features typical of benign or malignant adrenocortical tumors (see previous paragraph), MRI allows the assessment of its lipid content and soft-tissue characterization via study of T2 signal intensity and relaxation times, dynamic perfusion studies, or chemical-shift opposed-phase images. Adrenocortical adenomas are isointense and slightly hyperintense relative to the liver on T1- and T2-weighted sequences, respectively,78,79 whereas carcinomas may display very high signal intensity on T2-weighted images.77,79 After gadolinium, carcinomas show marked enhancement and slow washout, in contrast with the rapid washout of benign lesions.79 Chemical-shift imaging reveals loss of signal in out-of-phase images in lipid-rich tissues and is used to differentiate adenomas from metastases80 but does not reliably distinguish between adenoma and carcinoma, since both usually display a loss of signal intensity.78 On balance, although some features may be highly indicative, the accuracy of MRI in differentiating adenoma from carcinoma is not absolute,16,67 and routine MRI is not recommended for patients with adrenal Cushing’s syndrome.19,79 In patients with adrenal carcinoma, however, MRI enables visualization of the mass along sagittal and coronal planes, which allows the definition of anatomic boundaries, compression of surrounding organs, and extent of caval invasion.24 MRI is the procedure of choice for Cushing’s syndrome during pregnancy81 and in childhood, because of safety concerns and because it allows visualization of the glands even in absence of retroperitoneal fat. The drawback of MRI in pediatric patients is the requirement for absolute standstill, which may be difficult to achieve.
Adrenal Scintigraphy: The advent of CT has shifted adrenal scintigraphy from the center stage of adrenal imaging to an adjunctive, complementary procedure. In the past, evaluation of adrenal uptake was an integral part of the diagnostic workup, providing information also for the differential diagnosis between ACTH-dependent and ACTH-independent Cushing’s syndrome. Nowadays it is mostly reserved for differentiation of adenoma from hyperplasia and for those rare cases of ectopic adrenal tissue or adrenal remnants after adrenalectomy. Adrenal scintigraphy relies on the ability of the adrenal cortex to capture and accumulate radiolabeled cholesterol. Tracers are 131I-19 iodocholesterol or its derivative 131I-6β-iodomethyl-19 norcholesterol (NP59), the latter offering sharper adrenal images owing to a fivefold greater adrenal concentration,82 and 75Se-6β-selenomethyl-19 norcholesterol—used mostly in Europe. For scintigraphy with 131I, adequate preparation with Lugol iodine drops or potassium iodide and laxatives reduces thyroid uptake and intestinal background. Scans can be obtained as early as 48 hours after tracer injection, but usable images are registered up to 7 to 10 days after administration.
Adrenal scintigraphy provides quantitative and qualitative data on adrenal function. In patients with benign adrenal hypercortisolism, radiolabeled cholesterol uptake may be increased compared to normal subjects, usually 0.7% of the administered dose compared to 0.3% in normal subjects83,84 (Fig. 7-10). This percentage, although indicative of adrenal hyperfunction, does not provide absolute certainty of excessive corticosteroid production and, indeed, does not appear to be directly correlated with indices of adrenal function in these patients.83 Conversely, qualitative data is of great help for localizing steroid-secreting lesions. Scintigraphy may yield several uptake patterns, as proposed by Gross83 (Table 7-1).
Table 7-1
Uptake Patterns at Adrenal Scintigraphy
| Exclusive | Uptake on the side of the lesion, with nonvisualization of the contralateral gland |
| Prevalent | Asymmetric uptake by the tumor, with visualization of the contralateral gland |
| Symmetric | Comparable uptake by both glands |
| Discordant | Reduced or absent uptake on the side of the mass |
| Bilateral nonvisualization | No uptake on either side |
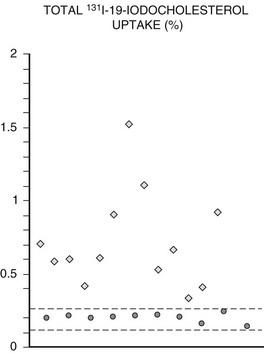
FIGURE 7-10 131I-19-iodocholesterol total adrenal uptake in normal subjects (•) and patients with Cushing’s syndrome ( ). Dotted lines depict mean ±2 SD. (Adapted from Ortega D, Foz M, Doménech-Torné FM, Tresánchez JM: The diagnosis of Cushing’s syndrome using 131I-19-iodocholesterol uptake and adrenal imaging, Clin Endocrinol 13:148, 1980.)
). Dotted lines depict mean ±2 SD. (Adapted from Ortega D, Foz M, Doménech-Torné FM, Tresánchez JM: The diagnosis of Cushing’s syndrome using 131I-19-iodocholesterol uptake and adrenal imaging, Clin Endocrinol 13:148, 1980.)
Exclusive or prevalent uptake on the side of the mass are considered concordant with the mass and confirm its functional activity as a steroid hormone–producing tissue and are taken to indicate a benign adrenocortical adenoma74,85 (Fig. 7-11A). Inhibition of ACTH secretion by autonomous cortisol production prevents the imaging of the contralateral gland. False-negative results have occasionally been reported with black adenomas, possibly a consequence of the prevalence of lipid-poor compact cells.86 In adrenocortical carcinoma, bilateral nonvisualization is the rule because the carcinoma itself does not accumulate tracer sufficiently, possibly a consequence of deranged steroidogenesis, and the contralateral gland is inhibited.85,87 However, some well-differentiated adrenocortical carcinomas and, rarely, also high-grade malignant carcinomas may accumulate the tracer, yielding exclusive or prevalent images.87 Malignant collision tumors may also display concordant uptake because of residual compressed cortical parenchyma within the nonadrenal neoplasm.59,88 It follows, therefore, that while bilateral nonvisualization is highly indicative of a malignant mass, concordant uptake does not uniformly represent benign disease. On the other hand, in patients with carcinoma concentrating the tracer, scintigraphy may enable the visualization of unsuspected metastases, thus providing invaluable preoperative information.89
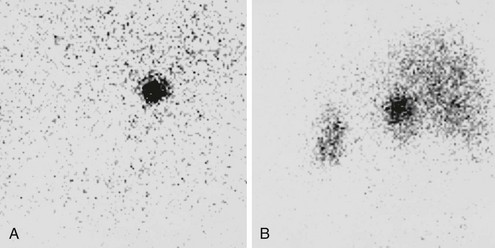
FIGURE 7-11 Scintigraphy in two patients with ACTH-independent Cushing’s syndrome due to primary adrenal disease. Images are posterior views obtained 1 week after 131I-19-norcholesterol injection. A, Exclusive uptake by a right-sided adrenocortical adenoma. B, Asymmetric uptake in a patient with macronodular adrenocortical hyperplasia. (Courtesy Dr. Eugenio Reschini, Ospedale Maggiore, Milan, Italy.)
In patients with ACTH-independent Cushing’s syndrome and apparently normal adrenal radiology, scintigraphy becomes paramount in order to localize the lesion and establish the etiologic diagnosis.85,90 In those rare cases of adrenal adenoma not identified by CT or MRI, exclusive uptake will reveal the side of the lesion and direct the surgeon.90 In addition, scintigraphy may identify ectopic adenomas or lesions not recognized as adrenals at standard imaging.91 Bilateral symmetric or slightly asymmetric uptake characterizes adrenal hyperplasia (see Fig. 7-11B) and PPNAD,12,74,85,92 and adrenal scintigraphy may be the sole procedure to correctly diagnose these clinical entities. Both AIMAH and PPNAD may in fact present with normal or slightly enlarged glands or unilateral or bilateral nodules and need to be differentiated from adrenal adenoma.74,90,93 However, scintigraphy itself is not foolproof. Bilateral nonvisualization has been reported in patients with hyperplasia or dysplasia,15,93 as has unilateral uptake in a patient with PPNAD.12,94
Lastly, scintigraphy may be employed in recurrence of adrenal Cushing’s syndrome or Cushing’s disease after adrenalectomy to localize hyperfunctioning adrenal remnants.82,93
Adrenal Venous Sampling: Adrenal venous sampling, performed only in experienced centers, is profitably used to lateralize the source of aldosterone hypersecretion but has been little used in adrenal Cushing’s syndrome. It might prove an interesting alternative to adrenal scintigraphy for those patients with bilateral adrenal masses in whom unilateral involvement is suspected95; large-scale studies are needed to validate its usefulness in adrenal Cushing’s syndrome.
Positron Emission Tomography: Whole-body positron emission tomography (PET) has been used in patients with adrenocortical carcinoma as an adjunctive procedure to detect and monitor metastases. Tracers employed so far are 18fluorine-fluorodeoxyglucose (FDG), the classical marker of enhanced glycolysis in malignant tumors, and 11carbon etomidate or metomidate, two inhibitors of 11β-hydroxylase. FDG-PET proved useful for the identification of neoplastic spread both prior to surgery and during follow-up, with markedly increased uptake detected in the site of the neoplasm and in metastases.96,97 Its use for the distinction between carcinoma and adenoma has been confirmed in larger series,98 although adrenocortical adenomas and hyperplasias may occasionally yield positive scans.98 PET using adrenal-specific tracers such as carbon-labeled etomidate or metomidate has yielded promising results for the detection of adrenocortical tumors and their metastases because uptake is clearly increased in adrenal cancer and derived lesions. The aim of this procedure, in contrast to 18F-FDG PET, is to establish the adrenocortical origin of the adrenal lesion; pheochromocytomas, metastases, and nonadrenal masses yield negative scans.99 Currently, only 11carbon-labeled metomidate is available, but 18fluorine tracers are being developed, and these may allow a more widespread use of adrenal-specific tracers in the future.
Interpretation of Adrenal Radiology: Adrenal radiology in patients with adrenal Cushing’s syndrome may reveal the presence of normal glands, unilateral or bilateral masses, or uniformly enlarged adrenals. A normal CT scan virtually excludes the presence of adenomatous or malignant lesions and can be taken to indicate micronodular hyperplasia or nodular dysplasia, the latter being more probable in young patients. Unilateral masses need to be categorized as malignant or benign, and this distinction remains most fraught in cases with intermediate radiologic features, since no procedure is decisive. Benign unilateral masses are most likely adrenocortical adenomas, although dominant nodules in AIMAH and even nodular dysplasia cannot be entirely excluded, and adrenal scintigraphy may be needed to discriminate between unilateral or bilateral disease. On the other hand, hypersecreting and nonhypersecreting adrenocortical adenomas present roughly the same CT/MRI features and cannot be distinguished by radiology.19,74 Bilateral masses with benign appearance are in all likelihood due to adrenal macronodular hyperplasia and, to a lesser extent, PPNAD. However, the possibility of bilateral carcinoma or adenoma, although exceptional, should not be forgotten. Lastly, bilaterally enlarged glands can be encountered both in adrenal hyperplasia and adrenal dysplasia.
Pathology of Cortisol-Secreting Adrenal Lesions
Four main adrenal lesions may be detected in patients with adrenal Cushing’s syndrome: a nodule (either isolated or within hyperplastic adrenals), adenoma, carcinoma, and nodular dysplasia. Distinction of hyperplastic nodules from adenoma, as well as adenoma from carcinoma, requires careful pathologic examination not only of the nodule itself but also of the surrounding adrenal tissue (Table 7-2). Indeed, different pathologic systems are used for the diagnosis of these lesions; each offers different advantages and is burdened by different drawbacks (see Chapter 11).
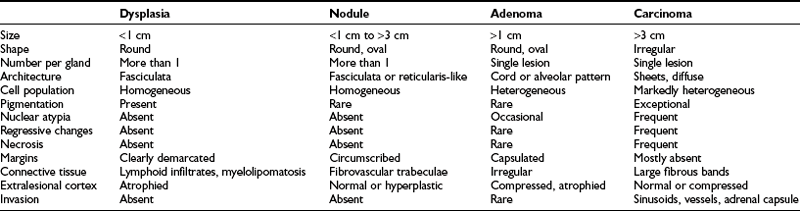
Hyperplastic nodules are usually distinguished from adenomas by the following features: less than 1 cm in diameter, oval or round shape, homogeneous clear-cell population organized in zona fasciculata– or zona reticularis–like architecture, rare smooth cell nests or cordlike structures, with well developed fibrovascular trabeculae.100–102 Nodules are usually more than one per gland, circumscribed but not encapsulated, and adjacent tissue is normal or hyperplastic. Conversely, adenomas are round or oval lesions of more than 1 cm in diameter, with heterogeneous cell population comprising both clear and compact cells in irregular cord or alveolar patterns. The adenoma is usually a single, encapsulated lesion surrounded by atrophic and compressed cortex. Both hyperplastic nodules and adenomatous lesions are surrounded by a rich network of sinusoidal vessels and dilated intercellular spaces and present well-developed, variably sized and shaped mitochondria and prominent smooth endoplasmic reticulum.103 Hyperplastic nodules and adenomas present as yellow, lipid-rich lesions, but occasionally lesions are composed of lipofuscin-containing compact cells with abundant eosinophilic cytoplasm, resembling zona reticularis cells and giving rise to black nodules or the so-called black adenoma.71,102 In patients with AIMAH, adrenal pathology depends on the progression and the extent of the lesions. Most patients present with huge adrenals (up to 500 g) in which the normal architecture is subverted by the presence of big, multiple, yellow or black nodules, some as large as 7 cm, surrounded by internodular hyperplasia (macronodular hyperplasia). Occasionally, the adrenals may be only slightly enlarged, with multiple small nodules (micronodular hyperplasia) or only one gland might be affected, presenting one or multiple nodules (unilateral hyperplasia). Intriguingly, hyperplastic nodules and adenomas have been reported in the same adrenal gland of patients with Cushing’s syndrome,102 as have bilateral adenomas or multiple adenomas in the same gland.104,105 In these cases, nodules or adenomas may present different cell composition (e.g., clear or compact cells) and secretory status (hypersecreting or nonhypersecreting) and affect the two glands at different time points. Indeed, bilateral adenomas as well as hyperplastic nodules have been removed several years apart.106,107
A unique variant of bilateral nodular involvement is PPNAD, a condition in which adrenals are small to normal-sized and occupied by multiple small, dark nodules (from 1 to 2 mm to 3 cm in diameter) containing large, heavily pigmented, granular eosinophilic cells (rarely clear cells). Pigmented granules contain lipofuscin and neuromelanin.26,108 Nodules are sharply demarcated, although not encapsulated, and surrounded by atrophic cortical tissue—this provides the major distinction from bilateral hyperplasia. The lesions are usually positioned deep within the adrenal cortex, nearly straddling the corticomedullary junction, whereas the remaining cortex may lose the normal zonation pattern12 (Fig. 7-12). Some nodules may show lymphocytic infiltration (mostly T-helper cells) within and without the nodule, as well as myelolipomatosis.109,110 As occurs for cortical tumors, positive immunostaining for neuroendocrine markers, such as synaptophysin and neuron-specific enolase, may be detected.111 Occasionally, nodularity may be asymmetric and present as a unilateral mass, or more rarely, nodules are markedly prominent, and adrenals appear enlarged,77 mimicking adrenal macronodular hyperplasia.
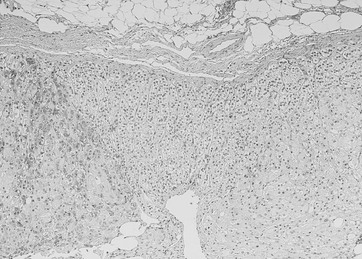
FIGURE 7-12 Pathologic findings in PPNAD. The two nodules are separated by internodular atrophic cortex. Note absence of capsule around nodules and foci of myelolipomatosis. (Courtesy Dr. Aidan Carney, Mayo Clinic, Rochester, MN.)
The diagnosis of adrenal carcinoma is straightforward if the neoplasm presents typical malignant features (“anaplastic carcinoma”) such as necrosis, hemorrhage, abundant mitoses, and nuclear atypia, together with local or hematogenous spread. In most cases, however, Cushing’s syndrome is associated with “well-differentiated carcinoma,” because steroidogenesis proceeds inefficiently in anaplastic tumors, and these precursors are devoid of significant biological activity.112 The distinction of well-differentiated carcinoma from adrenocortical adenoma relies on tallies from different pathologic scoring systems, the most used being those of Weiss,113 Hough,114 and van Slooten.115 These diagnostic algorithms are based upon histologic features such as necrosis, architectural changes, nuclear atypia, hyperchromasia, mitotic activity and atypical mitosis, nucleolar appearance, cytoplasmic vacuolization, cellular pleomorphism, fibrosis, and capsular and vascular invasion. The Hough scoring system also takes tumor weight and clinical/endocrine features into account (e.g., 17-ketosteroid excretion and response to ACTH, Cushing’s syndrome with or without virilization, body weight loss).114 While widely accepted, none of these systems offers complete diagnostic certainty,116 and the definite diagnosis of malignancy remains local invasion or metastasis. Borderline pathologic grading for benign lesions should lead to close follow-up because development of malignant tumors on the same site of a previously removed adenoma117,118 questions the initial diagnosis of “benign adrenocortical adenoma.” Ultrastructural analysis usually reveals lipid-depleted cells with altered mitochondrial and smooth endoplasmic reticulum architecture.103 Immunohistochemical studies evaluating p53 expression, MiB-1 or Ki-67 labeling, AgNOR counting, MHC class II antigen expression, and c-myc intracellular distribution revealed a differential pattern in benign and malignant lesions, but heterogeneity of neoplastic phenotypes and the resulting overlap between the two neoplasms precludes the identification of a secure profile of malignancy.119 Likewise, DNA ploidy analysis, comparative genomic hybridization, candidate gene mutations, or differential expression (most notably IGF-II; see Pathogenesis of Adrenal Tumors heading) revealed distinct differences between adenoma and carcinoma but, again, none appeared to ensure absolute diagnostic accuracy.119 Thus the current state of the art calls for prolonged follow-up also for apparently benign lesions for timely detection of signs of malignancy.
Several variants of adrenocortical tumors have been associated with Cushing’s syndrome, such as myxoid adrenocortical carcinoma, composed of polygonal basophilic cells arranged in arborizing cords surrounding myxoma-like large acellular spaces,120 or oncocytic adrenal tumors, usually benign nonfunctioning lesions composed of mitochondria-rich eosinophilic cells.121 Combined adrenal adenoma-myelolipoma characterized by intermingled cortical, bone marrow, and lipid cells has also been reported in conjunction with adrenal Cushing’s syndrome.122 Even more complex are mixed corticomedullary tumors, in which tumoral cortical cells are intimately admixed with pheochromocytes,123 or cortical tumors with neuroendocrine differentiation.124,125 In the latter case, malignant or benign adrenocortical cells present neuroendocrine features, such as dense core granules and positivity for synaptophysin and neurospecific enolase, and the tumor may give rise to excess secretion of various peptides, even ACTH itself.44 Pigmented adrenal lesions are usually benign (i.e., “black adenoma” or PPNAD), although a malignant pigmented lesion has also been reported.126 Finally, cortisol-secreting adenoma and carcinoma have been reported in the two adrenals of the same subject,127 as have bilateral carcinomas.128 On balance, tumors of the adrenal cortex usually display obvious benign or malignant features but occasionally may present with mixed forms or bizarre combinations that challenge the pathologist.
Pathogenesis of Adrenal Tumors
Mechanisms leading to adrenocortical tumorigenesis will be discussed in detail in Chapter 11; this issue will be reviewed briefly here, focusing on features of cortisol-secreting tumors.
Studies performed in the past decade suggest a common multistep process leading from polyclonal hyperplasia, to polyclonal or monoclonal adenomatous lesions, then to single-clone malignant cell expansion. This process, which is in keeping with Knudson’s two-hit hypothesis, is apparent in adrenal lesions at different stages,129,130 possibly even within the same adrenal.131 Further, genetic heterogeneity in terms of clonality, chromosomal alterations, and proliferation kinetics characterizes all three adrenal lesions, and no single feature allows the distinction between these forms. The common origin of adenoma and carcinoma is also supported by the fact that the same chromosomal regions appear to be affected in benign and malignant lesions.132 Loci of putative oncogenes (sites of chromosomal gain) and tumor-suppressor genes (sites of chromosomal loss) have been identified on chromosomes 17, 4, 5, and 9 and chromosomes 1, 2, and 11, respectively.133 Loss of heterozygosity is frequently detected on 11p15.5 (site of IGF2/H19/CDKN1C), 17p13 (site of TP53), 11q13 (site of MEN1), 13q (site of the retinoblastoma gene RB1), 9p (site of CDKN2A), and less frequently on 1p (site of neuroblastoma candidate gene NBL1) and 3p (site of the von Hippel-Lindau gene VHL).133,134 These changes, most notably the first two, are common in malignant adrenal tumors and are believed to represent pivotal events for adrenocortical tumorigenesis (see Chapter 11). Further, alterations in IGF-2 and p53 can be encountered in Beckwith-Wiedemann and Li-Fraumeni syndromes, respectively, two hereditary conditions presenting, among others, adrenocortical tumors. Germinal TP53 mutations are also linked to adrenal cancer predisposition in southern Brazil, a region with inexplicably high incidence of pediatric adrenal carcinoma.135 Mutation analysis of factors involved in tumorigenesis in other endocrine tissues (e.g., G proteins, ras) failed to identify significant alterations,119 nor did the analysis of cortex-specific genes, such as the 21-hydroxylase gene CYP21A2.136 The ACTH receptor appears unchanged in patients with adrenal disease,137 although a mutation in MC2R has been reported in one patient with adrenal hyperplasia138 (see heading ACTH-Independent Adrenal Hyperplasia). Of interest, somatic mutations of PRKAR1A, the gene involved in Carney’s complex, and loss of heterozygosity at its locus, 17q22-24, have been detected in adrenal tumors and adrenal hyperplasia,139,140 as well as alterations in protein kinase A subunits,141 suggesting an extensive involvement of the cAMP pathway in adrenocortical disease. Lastly, recent microarray studies opened several new avenues of research for adrenocortical lesions, most notably into the Wnt/β-catenin signaling pathway142 and the inhibin/activin complex.143
Inherited Familial Syndromes
Multiple Endocrine Neoplasia Type I
A link between MEN1 (Wermer’s syndrome) and adrenal tumors is suggested by the frequent loss of heterozygosity at 11q13, site of the menin gene, in adrenal cancers.133 However, no mutation in the MEN1 gene itself has been reported in cortisol-secreting sporadic adrenocortical tumors.144 Conversely, up to 70% of patients with MEN1 present adrenal lesions, mostly nodular and diffuse hyperplasia, adenomas, or rarely, carcinomas.145 Cushing’s syndrome in the context of MEN1, however, is mostly associated with pituitary ACTH-secreting adenoma, since adrenal lesions are only rarely accompanied by hypercortisolism.146
McCune-Albright Syndrome
Since the first description of bony lesions, skin pigmentation, and precocious puberty by Albright in 1937, the spectrum of endocrine abnormalities associated with this syndrome has progressively increased to include thyroid, parathyroid, pituitary, and adrenal disorders. Adrenal Cushing’s syndrome develops in less than 10% of patients with MAS, usually in the first year after birth, possibly even before sexual precocity and fibrous dysplasia.17 Adrenal histology usually reveals bilateral nodular hyperplasia or, rarely, adenoma; interestingly, these lesions have also been detected at autopsy in patients who did not present Cushingoid signs during their lifetime. MAS is known to be due to early postzygotic mutations of the stimulatory guanine nucleotide-binding protein (Gsα; GNAS1), which can be detected in multiple tissues, including the adrenal gland,147 and also in patients with MAS and adrenal Cushing’s syndrome described so far.17,148 Conversely, GNAS1 mutations are exceptional in patients with sporadic cortisol-secreting adrenal adenomas.149 GNAS1 mutations have been reported in adult patients with Cushing’s syndrome due to bilateral adrenal hyperplasia without other features of MAS,150,151 suggesting that mutational Gsα activation could be involved in adrenal hyperplasia.
Familial Adenomatous Polyposis
Patients affected by this autosomal dominant inherited disorder develop extensive adenomatous polyps of the colon and a variety of extracolonic manifestations comprising desmoid tumors, osteomas, pigmented retinal lesions, and adrenocortical neoplasms. Adrenocortical lesions consist in adenomas, bilateral nodular hyperplasia, or rarely, carcinomas and are mostly nonhypersecreting.152 Cushing’s syndrome is a rare occurrence both in classic familial adenomatous polyposis152 and its variant, Gardner’s syndrome.153 Familial adenomatous polyposis has been causally linked to germline mutations of the adenomatous polyposis coli (APC) gene located on chromosome 5, usually inactivating mutations accompanied by loss of the normal allele. It is worth recalling that inactivation of the APC gene leads to degradation of β-catenin, a recent putative player in adrenal tumorigenesis.142 Somatic mutations of APC have been detected in a wide variety of other human carcinomas but none so far in sporadic adrenocortical tumors.
Carney’s Complex
Carney’s complex represents the only etiology of Cushing’s syndrome in which a genetic defect has been identified. Indeed, intensive work by two groups of researchers led to the short-listing of two probable loci, on the short arm of chromosome 2 and on 17q24, followed by the identification of inactivating mutations in the protein kinase A regulatory subunit type 1α.154,155 Carney’s complex is discussed in greater detail later (see heading Primary Pigmented Nodular Adrenal Dysplasia).
Steroidogenic Enzymes in Adrenal Lesions
Several studies have evaluated steroidogenesis in adrenal Cushing’s syndrome. Cortisol concentration in tumoral specimens is 2- to 50-fold greater in adenomas than in normal glands,156 attesting to the greater secretory output. Steroidogenesis usually proceeds without hindrance to its principal end product, cortisol, but individual steroidogenic enzymes may become rate-limiting in hypersecreting adrenals,157 the extent and involvement of individual enzymes being quite variable. Analysis of steroidogenic enzymes yielded somewhat conflicting results, with increased 21-hydroxylase and 17α-hydroxylase synthesis and activity in cortisol-secreting adenomas as the most constant finding.112,156,158 Increased 17,20-lyase activity reportedly occurs in adenomas producing androgens in addition to cortisol, possibly a consequence of increased cytochrome b5 expression.159 A relative deficiency of 11β-hydroxylase has been suggested by some authors,160 although not confirmed by others.158 Conversely, 3β-hydroxysteroid dehydrogenase and cholesterol desmolase appear substantially unchanged in cortisol-secreting adenomas.112,158 In adrenal carcinoma, cortisol secretion per gram of tumoral tissue is mostly reduced,160 as are individual steroidogenic enzymes.28,112,161 Of interest, malignant cells may fail to synthesize the full complement of steroidogenic enzymes,28,161 thus possibly explaining ineffective steroidogenesis. Shift from excess aldosterone to excess cortisol production has also been reported in malignant tumors, associated with changes in steroidogenic enzyme synthesis and attesting to the plasticity of tumoral tissues.162 The low efficiency of cortisol secretion is also a feature of macronodular hyperplasia,100 and variable expression of steroidogenic enzymes in individual cortical cells has been observed.163 In contrast, nodules from patients with PPNAD all present intense immunostaining for steroidogenic enzymes.163 Lastly, low levels of expression and activity of 11β-hydroxysteroid dehydrogenase type 2, the enzyme which inactivates cortisol, have been detected in adenomatous cortisol-secreting adrenals and may represent an additional modulator of total cortisol levels.164,165
Regulation of Steroid Synthesis in Adrenal Lesions
In adrenal Cushing’s syndrome, cortisol secretion is by definition ACTH-independent. However, a considerable proportion of adenomas, and to a lesser extent carcinomas, maintain ACTH-responsiveness in vivo and in vitro,7,166 and the relevance of this preserved ACTH sensitivity in tumors which secrete cortisol autonomously remains unclear. The ACTH receptor (also known as the melanocortin 2 receptor, MC2-R) is expressed in the vast majority of cortisol-secreting adenomas, even overexpressed compared with the normal adrenal cortex, according to some authors,166,167 whereas cortisol-secreting carcinomas present low levels of MC2R mRNA, possibly a consequence of dedifferentiation and loss of heterozygosity at this locus.167 ACTH is known to up-regulate its own receptor at low concentrations and exert the opposite effect at higher doses,168 but no direct association between ACTH receptor mRNA and ACTH plasma levels has been detected in various forms of Cushing’s syndrome.169 Stimulation of steroid secretion in adrenocortical tumor cells occurs through activation of protein kinase C, together with the main protein kinase A–cAMP pathway.170 Of interest, factors other than ACTH may also increase cAMP levels in malignant but not in normal adrenal cells, as has been demonstrated for catecholamines and thyrotropin,171 indicating a loss of specificity of the transductional machinery in tumoral tissues (see heading ACTH-Independent Adrenal Hyperplasia). Moreover, ACTH is also an indirect adrenocortical mitogen acting through factors such as basic fibroblast growth factor (bFGF), insulin-like growth factor 2 (IGF-2), epidermal growth factor (EGF), and transforming growth factor (TGF) α or β.172,173 These mediators are responsible for the growth-promoting effect of ACTH in vivo, whereas in vitro, ACTH exerts a direct antimitogenic activity.172 Similarly divergent are the effects of N-terminal pro-opiomelanocortin (POMC), which appears to stimulate mitogenic activity via the secretory serine protease (AsP) pathway in vitro174 but is devoid of mitogenic effects in Pomc-null mice in vivo.175
One brief mention is warranted for the regulation of cortisol synthesis by corticosteroids themselves. Evidence for this ultra-short negative feedback has been garnered by in vivo and in vitro studies, although paradoxically, a stimulatory, permissive effect of cortisol or dexamethasone on ACTH-stimulated cortisol secretion has been reported by some authors.176 The direct action of corticosteroids on steroidogenesis is presumably mediated by glucocorticoid receptors themselves, since both binding and induction of gene synthesis have been demonstrated in the adrenal cortex.167,177 Expression of the glucocorticoid receptor is apparently reduced in cortisol-secreting tumors.167,178
ACTH-Independent Adrenal Hyperplasia
ACTH-independent adrenal hyperplasia was recognized as a cause of Cushing’s syndrome following the description of several case reports which defied classification.179,180 Considerable confusion arose initially from the fact that some features of ACTH-dependent adrenal hyperplasia (i.e., Cushing’s disease) overlapped with ACTH-independent adrenal hyperplasia; indeed, “macronodular Cushing’s syndrome” once comprised both entities. Advances in pathology,100 as well as the identification of factors other than ACTH capable of modulating cortisol hypersecretion, enabled a more precise definition and classification.14,181,182 In view of its most common presentation, we will refer to this entity with the currently used acronym, AIMAH: ACTH-independent macronodular adrenal hyperplasia.
Currently accepted criteria for the diagnosis of AIMAH are the presence of bilaterally enlarged adrenals containing one or more nonpigmented nodules and the presence of suppressed and/or unresponsive ACTH levels.14,100 Internodular tissue was initially described as atrophic100 but may also be normal or even hyperplastic, thus does not represent a distinctive element.14,182 On average, glands weigh five times normal (e.g., 20 to 120 g) and present large, well-circumscribed nodules grossly distorting adrenal cortex architecture (see heading Pathology of Cortisol-Secreting Adrenal Lesions). The size of nodules usually ranges from 0.5 cm to 2 to 3 cm, although exceptionally large lesions, up to 10 cm in diameter, have been described.183 Nodules are composed of lipid-laden clear cells in cord or nest-like structures interspersed by small islands of lipid-poor compact cells,100,184 foci of angiomyelolipomatosis and lymphocytic infiltrates. Cellular and nuclear pleomorphism are usually absent. Unilateral macronodular hyperplasia, as well as diffuse bilateral hyperplasia, have also been described in patients with ACTH-independent hypercortisolism,14,185 possibly representing different points in the continuum of AIMAH.
The large size of adrenals is in marked contrast with the often mild clinical picture, corroborating in vitro evidence of inefficient steroidogenesis by cortical cells.100,163,184 Indeed, cytochrome P450 expression and activity are reduced compared to that of cells derived from adrenal adenoma100,184; thus the adrenal mass may have to attain a considerable volume for hypercortisolism to become manifest. Subclinical Cushing’s syndrome is increasingly reported in patients with ACTH-independent adrenal hyperplasia,186 occasionally evolving with progressive adrenal enlargement and development of frank hypercortisolism.187,188
Salient features of hypercortisolism due to AIMAH are later age at diagnosis (usually around 50 years of age), the even gender distribution, and an unusually long time to diagnosis, on average 4 years from the appearance of the first symptoms, compared to 2 years for cortisol-secreting adenomas.14,182 One patient developed full-blown hypercortisolism 7 years after the incidental detection of bilateral adrenal masses.188 The youngest and oldest reported patients with ACTH-independent adrenal hyperplasia were 17 and 74 years old, respectively. In patients with aberrant receptor-induced hyperplasia (see later), hypercortisolism may follow the course of ligand levels, as exemplified by the course of LH-dependent Cushing’s syndrome or by low morning serum cortisol levels in the fasted patient with food-dependent Cushing’s syndrome.
By definition, ACTH levels are suppressed in patients with ACTH-independent adrenal hyperplasia, but the detection of low-normal ACTH concentrations does not exclude the diagnosis.14,182 As in other causes of adrenal Cushing’s syndrome, CRH testing provides proof of ACTH independency. Inferior petrosal sinus sampling should not be performed, because significant post-CRH center-periphery ACTH gradients have been reported in several patients, presumably reflecting incomplete suppression of pituitary corticotrophs by mild or episodic hypercortisolism.189 Prior to the availability of ACTH assays, adrenal autonomy from ACTH was established by means of absent dexamethasone suppression or metyrapone stimulation of adrenal steroids. These tests, however, do not guarantee complete diagnostic accuracy; indeed, the number of patients with Cushing’s disease and adrenal nodules who fail to suppress after dexamethasone is not negligible,190 and specificity of metyrapone stimulation is known to be suboptimal.47 The distinction between ACTH-independent adrenal hyperplasia and unilateral adrenal lesions or PPNAD relies on imaging, both morphologic and functional, and pathology (see earlier discussion).
Pathogenesis
ACTH-independent adrenal hyperplasia presents a heterogeneous etiology, comprising legitimate and illegitimate adrenal growth factors and gene mutations. In one of the first descriptions of ACTH-independent adrenal hyperplasia, proliferation of small cortical cells with maturational arrest or incomplete maturation was hypothesized to cause hyperplasia.100 Causes identified so far, however, account for only part of reported patients, and other mechanisms are likely to be involved. Indeed, microarray studies in AIMAH tissues identified a host of potential candidate genes,191,192 in part overlapping with genes involved in adrenal tumorigenesis, such as the Wnt signaling pathway, and cytogenetic analysis revealed frequent allelic losses in 2p16 and 17q22, two sites linked to PPNAD,140 in adrenal hyperplastic tissues.
Legitimate Adrenal Growth Factor
Adrenal hyperplasia was initially believed to result from prolonged stimulation by ACTH followed by autonomization of adrenal secretion. Several case reports describing transition from ACTH-dependent to ACTH-independent hypercortisolism supported this hypothesis.193,194 In fact, ACTH induces exaggerated cortisol increases in patients with adrenal hyperplasia, both in vivo and in vitro, attesting to an exquisite adrenal sensitivity to ACTH, in contrast with the barely preserved responsiveness of adrenal adenomas and carcinomas. Indeed, the cortisol response to cosyntropin represents the standard against which responses to other agents are compared (see later). This pathogenetic hypothesis, while theoretically still tenable, has been superseded by subsequent developments.
A role for ACTH cannot be excluded, since locally produced ACTH participates in the intraadrenal CRH/ACTH modulation of corticosteroid release and adrenal proliferation.195 In two patients with adrenal hyperplasia, cortisol secretion was driven by intraadrenal ACTH production. In one patient, ACTH was produced by an adrenocortical adenoma presenting neuroendocrine features,196 whereas in the other patient, steroidogenic cells with positivity for Leydig-cell markers were the site of ACTH synthesis.197 Interestingly, plasma ACTH levels were high in the former but not in the latter patient, and the diagnostic workup comprising adrenal vein sampling was indicative of ectopic ACTH secretion.196 Altered sensitivity to the adrenal ACTH receptor might be an alternative mechanism. Indeed, a homozygous MC2R germline mutation was identified in a patient with Cushing’s syndrome and ACTH-independent adrenal hyperplasia; functional studies revealed constitutive activation of the F278C mutated receptor.138 This case is unique; no mutations in MC2R were detected in other patients with ACTH-independent adrenal hyperplasia.151,198 Quantitation of MC2R expression in adrenals from patients responsive to illicit receptor stimulants yielded contrasting results, with some studies indicative of increased expression,199 others observing reduced expression,192,200 and others again observing expression comparable to normal adrenocortical tissues.201 Variability in clinical and molecular phenotypes might explain these differences.
Illicit Adrenal Receptors
The capacity of ligands other than ACTH to stimulate the adrenal cortex had been demonstrated in both animal and human tumoral adrenal specimens,171,202 but this was initially considered an epiphenomenon to tumoral dedifferentiation. The causative role of aberrant adrenal receptors was later demonstrated by two groups who simultaneously reported that food-induced hypercortisolism was due to stimulation of steroidogenesis by the gastrointestinal-inhibitory peptide (GIP).203,204 Since then, a variety of adrenal cortisol-stimulating receptors have been described (Fig. 7-13), not only in adrenal hyperplasia but also in patients with adenomas149 and unilateral or bilateral incidental adrenal lesions associated with subclinical hypercortisolism.186,205,206 Responses to illicit receptor stimulants appear to occur less frequently in unilateral compared to bilateral lesions.207,208 Different developmental derangements have been postulated to explain the common etiologic pathway of adenoma and hyperplasia. Sensitivity to illicit adrenal stimulants would be acquired through mutations occurring during early embryogenesis in patients with bilateral hyperplasia, whereas somatic mutations in single adrenal cells would result in unilateral adenoma. The importance of aberrant receptor expression in ACTH-independent adrenal hyperplasia has recently been validated by two elegant xenograft studies.209,210 These studies have shown that implantation of bovine adrenal tissue transfected with either the GIP or the LH/human chorionic gonadotropin (hCG) receptor into nude mice was accompanied by adrenal growth, elevated cortisol levels, and development of Cushingoid features.209,210 It appears, therefore, that aberrant expression of the receptor in the adrenal is a primary event, sufficient to initiate adrenal growth and hypersecretion.
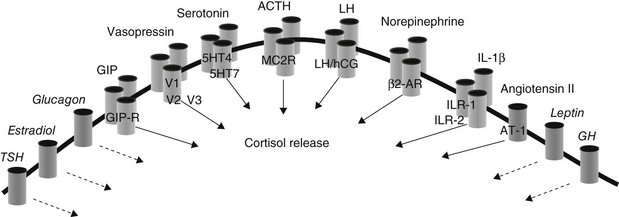
FIGURE 7-13 Ligands and illicit receptors involved in Cushing’s syndrome associated with AIMAH. Single barrelled receptors and dashed arrows indicated putative adrenal-stimulating agents.
The demonstration of ectopic receptors or overexpression of eutopic receptors is essential but not sufficient to diagnose illicit receptor-dependent adrenal Cushing’s syndrome, because ligand-induced cortisol secretion and adrenal growth or, better, remission of hypercortisolism with specific receptor antagonists are necessary to prove its causative role. Expression of the receptor in the adrenal lesion, in fact, is not invariably associated with responsiveness to the ligand,211,212 and further, simultaneous expression of multiple aberrant receptors in the same patient is not unusual.213 Cortisol responsiveness to multiple ligands can often be observed in vivo, and in vitro incubation with potential stimulants may result in significant cortisol increases even if no significant change in cortisol serum levels occurred in vivo.
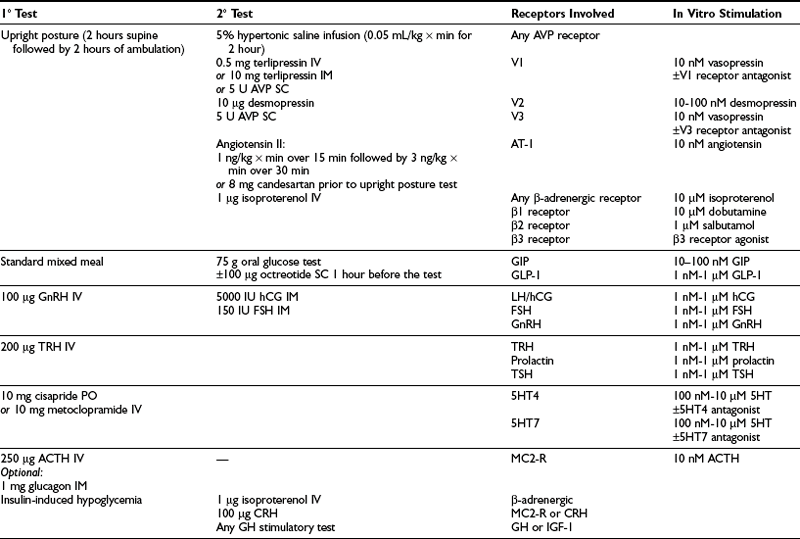
Protocol for adrenal responsiveness testing is illustrated in Table 7-3 and should be applied to all patients with bilateral adrenal lesions and full-blown or subclinical hypercortisolism. Currently, screening of patients with unilateral, incidental adrenal lesions is not recommended until its usefulness is validated in large-scale studies. Testing usually begins with first-tier tests (see Table 7-3), followed by more specific stimulants if a positive cortisol response is observed.
Gastrointestinal Inhibitory Polypeptide: Food-dependent Cushing’s syndrome was the first variant of hypercortisolism due to stimulants other than ACTH to be described. Over 25 cases have been reported since the first description in 1987—mostly in women—so this appears to be the most frequent etiology of ectopic receptor-associated hypercortisolism. A causative role of gastrointestinal hormones was suggested by the increase in cortisol levels after various mixed meals and after oral but not intravenous glucose and by blunting of these responses through octreotide. Candidate hormones were GIP (gastrointestinal or gastric-inhibitory polypeptide or glucose-dependent insulinotropic peptide) and GLP-1 (glucagon-like peptide 1). Evidence in favor of the former accrued with the observation of a close correlation between plasma cortisol and GIP levels in these patients,203 and the demonstration of a direct stimulatory effect of GIP on cortisol release in vivo203,204,208 and on adrenal tissue in vitro.204,213 Conversely, GLP-1 did not stimulate cortisol secretion in either experimental paradigm.107,213 GIP is a 42-amino-acid gastric peptide which increases after orally ingested meals and stimulates insulin secretion. The GIP receptor (GIP-R) is a member of the secretin-vasoactive intestinal polypeptide family of G protein–coupled receptors normally expressed in pancreatic β cells and in the brain. GIP-R is expressed at very low levels in the normal adrenal, where it appears to be nonfunctional, since no changes in cortisol secretion have been observed in normal adrenals incubated with GIP.214,215 In contrast, the GIP-R appears to be abundantly expressed in adenomas, nodules, and internodular hyperplastic tissue of patients with food-dependent Cushing’s syndrome.107,208 GIP-R is observed more frequently in tissues from AIMAH than in adenomas, even in patients without signs of food-dependent hypercortisolism208; indeed, GIP-R mRNA has also been detected in some patients with Cushing’s disease.216 No mutation in the promoter region and coding sequence of GIP-R has been identified so far,214,217 and no specific changes in local transcription factors have been detected in patients with food-dependent Cushing’s syndrome,218 thus the mechanisms responsible for GIP responsiveness remain to be established. In GIP-sensitive adrenal cortex nodules, GIP induces cAMP synthesis, coupled to PKA kinase activity as well as ionic currents,212 thereby activating steroidogenesis. In lesions arising from the zona fasciculata cortisol and to a lesser extent 17OHP will be the main secretory products,107 whereas in the single adenoma of the zona reticularis described so far, DHEA and androstenedione secretion prevailed.219
From a clinical viewpoint, food-dependent Cushing’s syndrome is characterized by typically low morning serum cortisol levels, because cortisol will not be stimulated in the fasted patient, and endogenous ACTH will be suppressed by postprandial cortisol bursts.203,204 Occasionally, ACTH concentrations may fall within the normal range and even respond to CRH stimulation, possibly a consequence of the intermittent nature of food-dependent hypercortisolism.220 Treatment with octreotide (e.g., 100 to 200 µg t.i.d.) or octreotide LAR achieved clinical and biochemical improvement in several patients.203,220
Vasopressin: Arginine vasopressin (AVP) is the second most common “illicit” stimulant in adrenal Cushing’s syndrome. The main action of AVP on the HPA axis is stimulation of ACTH with attendant cortisol release,221 although direct stimulation of adrenocortical cells by AVP has been demonstrated in vitro.222 However, the importance of this action is unclear, since no cortisol response to AVP is observed in vivo in absence of ACTH.223 Direct stimulation of AVP on adrenocortical secretion and cell growth most likely subserves the paracrine action of locally produced AVP.222
Several patients with AIMAH have been reported in whom AVP, LVP, or the prodrug terlipressin increased serum cortisol in the face of suppressed ACTH levels.206,224,225 Endogenous AVP plasma levels are usually suppressed in these patients but may increase after stimuli such as upright posture, hypertonic saline, or insulin-induced hypoglycemia in parallel with cortisol.207,211,226 Aberrant responses to AVP have also been reported in several patients with adrenal hyperplasia and subclinical Cushing’s syndrome,227,228 as well as in a few patients with cortisol-secreting adenoma or carcinoma.206
In vitro studies on human adrenal tissue showed that AVP stimulates cortisol secretion via its phospholipase C–coupled V1 receptor,224,225,229 and that this receptor is abundantly expressed in AVP-responsive patients.225,227,229 The V1 receptor is also expressed in normal cortical cells222 and in patients with adrenal hyperplasia and subclinical Cushing’s syndrome,227 but the mechanisms leading to adrenal hypersensitivity in these patients remain to be established. Administration of an oral V1 receptor antagonist did not significantly affect the acute cortisol response to AVP but slightly reduced UFC concentrations.226 Involvement of other AVP receptor subtypes, namely V2 and V3, has been suspected after the demonstration of these receptors by IHC and RT-PCR in adrenal nodules229,230 but has yet to be convincingly demonstrated. Desmopressin failed to stimulate cortisol release in patients tested so far, both in vitro and in vivo, and no coincubation experiment using AVP together with selective V3 receptor antagonists has been performed to substantiate V2 or V3 receptor functionality. AVP-dependent adrenal Cushing’s syndrome could therefore be due to an aberrant response to the eutopic V1 receptor or ectopic expression of V2 or V3 receptors.
Angiotensin II: Angiotensin induces cortisol secretion by normal and hyperplastic adrenal in vitro, albeit to a consistently greater extent in the latter, especially in patients in whom cortisol increases during the posture test.213 Involvement of the AT-1 receptor can be demonstrated by blockade of the posture-induced cortisol increase by candesartan, and the drug could be used as medical therapy to contain hypercortisolism.231 A full description of angiotensin-mediated hypercortisolism has, however, yet to be provided.
Luteinizing Hormone/Human Chorionic Gonadotropin: The observation that some young women develop clinical and biochemical features of hypercortisolism during pregnancy which resolve spontaneously after delivery has long intrigued endocrinologists (Fig. 7-14). Further, the prevalence of adrenal Cushing’s syndrome is considerably higher in pregnancy.81 This suggested an enhanced adrenal sensitivity to some factor produced by the fetoplacental unit and, indeed, aberrant LH/hCG receptors have been shown to mediate the increased adrenal secretion in these patients.35 The causative role of LH was first demonstrated in a 63-year-old woman who developed Cushingoid features during pregnancy and again after menopause. Cortisol levels increased markedly upon administration of GnRH and chorionic gonadotropin in this patient, whereas FSH was without effect. Treatment with a GnRH agonist over 24 months normalized UFC levels and ameliorated signs of hypercortisolism without, however, affecting adrenal size.35 Subsequent studies demonstrated that LH can stimulate cortisol secretion in vitro, albeit not in all patients responsive to LH in vivo, and that adenomatous lesions express the LH receptor.198,213 Low-level synthesis of LH/hCGR can be detected in the zona fasciculata and reticularis of the normal adrenal cortex,232 but no stimulatory effect of LH/hCG on cortisol secretion by normal adrenals has been observed.198,213 Support for the role of LH as an adrenal stimulant is supported by animal models. LH-transgenic mice, gonadectomized ferrets, and adrenal xenotransplant studies develop bilateral adrenal hyperplasia.210,233,234
FIGURE 7-14 UFC levels during successive pregnancies in a woman with LH-dependent Cushing’s syndrome. The first pregnancy was carried through to the 34th week, the second and third terminated within the first 2 months. Shaded area represents normal range. (Data from Hána V, Dokoupilová M, Marek J, et al: Recurrent ACTH-independent Cushing’s syndrome in multiple pregnancies and its treatment with metyrapone, Clin Endocrinol 54:277–281, 2001.)
Of interest, in the few patients with LH-dependent Cushing’s syndrome studied so far, an aberrant response to other stimulants (e.g., 5HT, GIP, AVP) accompanied LH hypersensitivity.35,186,198,213 In contrast to food-dependent Cushing’s syndrome, sustained increases in LH/hCG levels are necessary to induce clinically relevant adrenal hypersecretion; signs of hypercortisolism appeared only during pregnancies and/or menopause. It appears that the normal pulsatory gonadotropin pattern is insufficient to activate adrenal steroidogenesis. However, not all patients aberrantly expressing the LH receptor in the adrenal cortex will develop hypercortisolism during periods of increased LH secretion, so factors other than LH must contribute to pregnancy-induced Cushing’s syndrome.
Catecholamines: β-Adrenergic receptors were the first aberrant receptors to be demonstrated in cortisol-secreting tumors,235 but recent studies suggest that the β2-adrenergic type 2 receptor (β2-AR) is expressed also in the normal adrenal cortex, albeit without cortisol-secreting ability.201 β-Adrenergic agonists have proven capable of stimulating cortisol secretion in a few patients,207,211,236 and proof of β2-AR involvement was obtained through in vitro experiments with different β-adrenergic receptor agonists.201 These findings have been translated into medical therapy with long-term beneficial effects in patients,201,236 thus proving the existence of β-adrenergic-dependent hypercortisolism.
Serotonin: Serotonin is secreted by intraadrenal mast cells and stimulates cortisol secretion in a paracrine fashion, most likely via its type 4 receptor (5HT4). This action can be replicated by incubation of adrenocortical cells with serotonin but not in vivo, because serotonin agonists do not stimulate cortisol secretion in normal subjects.237 Conversely, significant increases in cortisol levels have been observed in several patients with Cushing’s syndrome due to adrenal hyperplasia or adrenocortical adenomas,149,238 often in patients harboring other types of “aberrant” receptors, attesting to receptor promiscuity in hyperplastic adrenals.13 Initial studies focused on the 5HT4 isoform, but no consistent differences in 5HTR4 expression between normal and hyperplastic adrenal tissues238,239 or mutations in its coding sequence were detected.238 Further, some in vivo cisapride-responsive patients failed to release cortisol with cisapride in vitro, and 5HTR4 antagonists did not block the cortisol increase evoked by serotonin in vitro.212 Evaluation of additional serotonin receptor isoforms identified 5HT7a as a possible illicit mediator of serotonin responsiveness in AIMAH, given that this receptor does not appear to be expressed by the normal adrenal.212 No 5HT receptor antagonist has yet been tested in patients with cortisol-hypersecreting adrenal hyperplasia.
Interleukin 1: A single case of Cushing’s syndrome due to aberrant expression of interleukin-1 type I receptor (IL1R1) has been described so far.240 A role of cytokines in adrenal tumorigenesis was suspected in this patient when pathologic examination of the adrenal adenoma disclosed heavy leukocyte and macrophage infiltration. Immunohistochemistry, as well as in situ hybridization, revealed the presence of IL1R1, and adenomatous cells released substantial amounts of cortisol in vitro upon incubation with interleukin-1β. In normal adrenal glands, the IL1R1 was not detected by immunohistochemistry, and further, interleukin-1β failed to stimulate cortisol secretion from normal and adenomatous adrenals collected from other patients. The proliferative effect of interleukin-1β is well known,241 thus it was hypothesized that high concentrations of interleukin-1β released by local immune cells may have favored clonal expansion of adrenocortical cells aberrantly expressing IL1R1.
Estradiol: Mimicking LH-dependent Cushing’s syndrome, a young woman presented with features of hypercortisolism during two pregnancies, but in contrast to LH-dependent Cushing’s, these features also recurred during contraceptive therapy.242 Pathology of the adrenals revealed enlarged glands with multiple nonpigmented nodules, and in vitro studies showed marked cortisol release in the presence of estradiol. The authors deduced a causative role of estradiol in cortisol hypersecretion.
Other Putative Stimulants: Additional adrenal receptor ligands include FSH, TSH, GH, glucagon, and leptin. Evidence in favor of these substances is currently only circumstantial, since no patient with hormone-driven hypercortisolism has been fully studied. Finally, a proportion of patients with AIMAH does not present identifiable stimulatory ligands, so a careful clinical history together with specific testing would be necessary to define other, at present unknown, aberrant receptors.
Genetic Causes
As discussed above, no mutation in illicit or licit receptor gene sequences in patients with AIMAH has been reported so far. Conversely, somatic mutations of Gsα, the so-called gsp mutations, have been detected in three out of five patients with adrenal nodular hyperplasia,151 which is in keeping with adrenal hyperplasia as the most common cause of hypercortisolism in patients with MAS (see earlier). Other genetic causes of ACTH-independent adrenal hyperplasia cannot be excluded, given the increasing number of familial cases, both with and without overt hypercortisolism.228,230,243
Treatment
Removal of both adrenal glands is obviously the rational approach to curing hypercortisolism in this condition. Laparoscopic adrenalectomy of glands measuring up to 13 cm has been reported, indicating that size need not be a concern in this benign lesion.244 Likewise, development of Nelson’s syndrome has not been reported in patients with ACTH-independent adrenal hyperplasia. Alternative approaches, such as subtotal or unilateral adrenalectomy, may be considered in selected patients. To avoid risks associated with adrenal insufficiency,245 subtotal adrenalectomy has been performed in a patient with multiple malignancies, whereas removal of the largest adrenal achieved long-term remission in several patients with asymmetric involvement.189,246 Transitory hypoadrenalism requiring steroid replacement therapy may occur. Should abnormalities in HPA function persist, as appears to occur in patients with comparable enlargement of both adrenal glands, removal of the contralateral gland may be performed,246 or medical therapy with antagonists to illicit receptors may be used to restrain hypercortisolism.13 Surgery may not be advisable in all patients with AIMAH, given the often advanced age at diagnosis and the attendant, age-related comorbidities. Thus, administration of steroid synthesis inhibitors may be the treatment of choice, and relatively low doses of steroid blocking agents are usually capable of containing hypercortisolism in these patients. In addition to ketoconazole, metyrapone and mitotane have also been administered with success.247,248
Primary Pigmented Nodular Adrenal Dysplasia
PPNAD occurs in a sporadic fashion, without additional clinical conditions, in approximately half of the cases. In the remaining patients, PPNAD is part of Carney’s complex, an inherited association of myxomas, spotty pigmentation, and endocrine overactivity.26 Sporadic and familial PPNAD present similar clinical and pathophysiologic features and, indeed, the same genetic alterations.155,249
PPNAD is associated with overt Cushing’s syndrome in approximately 70% of cases, the remainder presenting subclinical Cushing’s syndrome (i.e., biochemical parameters of adrenal autonomy/hyperfunction without clinical features of hypercortisolism) or latent Cushing’s syndrome (no clinical or laboratory alteration but genetic trait present). Salient features of Cushing’s syndrome due to PPNAD include its occurrence at a young age (i.e., pre- and postpubertal children in the second decade of life, the youngest patient being a 3-month-old infant), slow progression of features of hypercortisolism, and severe osteoporosis. The mildness, sluggishness, and possibly even cyclical development of Cushingoid features might contribute to the delay in diagnosis (approximately 4 years from the appearance of the first symptoms, against less than 2 years in other forms of Cushing’s syndrome). As regards clinical features, osteopenia and stunted growth was the outstanding feature in several young children, whereas hirsutism and menstrual irregularity may be the only sign in young adolescent girls.12,41 Patients with PPNAD are rarely obese, but skin and muscular atrophy are common. In one young woman, Cushing’s syndrome due to PPNAD was diagnosed during pregnancy after vertebral collapse and detection of extensive osteoporosis.250 In patients in whom PPNAD is part of Carney’s complex, other signs of the disease are usually present by the time hypercortisolism develops (Table 7-4), most notably pigmented spots and cardiac myxomas, and thus may aid in the diagnosis. However, given the clinical heterogeneity of the complex, all cases of PPNAD should be assumed familial until accurate examination of first-degree relatives excludes Carney’s complex.
Table 7-4
| Primary Lesions | Affected Patients (%) |
| Myxomas | |
| Cardiac | 60 |
| Cutaneous | 40 |
| Breast | 20 |
| Mucocutaneous spotty pigmentation | 70 |
| Endocrine overactivity | |
| PPNAD (ACTH-independent Cushing’s syndrome) | 50 |
| Testicular tumors (sexual precocity) | 35 |
| Pituitary mammosomatotroph tumors (acromegaly, gigantism) | 10-15 |
Additional Lesions
Diagnosis of Cushing’s syndrome due to PPNAD may follow different strategies, depending on the sporadic or familial nature of the disease. In the former, adrenal Cushing’s syndrome is suspected in a patient without prior knowledge of Carney’s complex and thus requires the differential diagnosis from other forms of adrenal hypercortisolism. Conversely, if the patient or his/her relatives are known to be affected by Carney’s complex, then the diagnosis will be aimed at establishing the existence of overt hypercortisolism. In this context, it should be recalled that subclinical or latent hypercortisolism are most probably present in the vast majority of patients with Carney’s complex, as attested by typical adrenal lesions, as well as Crooke’s hyaline change revealed at autopsy in asymptomatic patients.12 Further, subtle alterations of cortisol circadian rhythmicity and/or suppressibility by dexamethasone may be encountered in older patients with Carney’s complex who did not develop Cushing’s syndrome or in first-degree relatives.12,251
Patients with Cushing’s disease due to PPNAD do not always present suppressed plasma ACTH levels; indeed, low-normal ACTH levels have been reported in several series,37,41,76 possibly a consequence of the mildness of hypercortisolism (see heading Diagnosis). However, no hormonal response to CRH stimulation was observed.12 Administration of 8 mg dexamethasone for 2 days is followed by a paradoxical doubling of UFC levels in approximately half of patients with PPNAD,37 and this may provide useful adjunctive information (see heading Differential Diagnosis of Adrenal Causes of Cushing’s Syndrome). CT scan (and possibly adrenal scintigraphy) should be performed in these patients.
Pathogenesis
The initial pathogenic hypothesis for PPNAD was based upon the finding of adrenal-stimulating antibodies in sera of patients with PPNAD.92,252 These studies revealed that DNA synthesis, adrenal growth, and cortisol release can be stimulated by circulating immunoglobulins present in patients with PPNAD.92,110,252 Stimulating immunoglobulins were present independent of the presence of hypercortisolism, suggesting that these antibodies contribute but are not the sole mediator of increased adrenal activity.252 No additional autoantibodies were detected in sera of affected patients, underlining the specificity of the phenomenon. However, adrenal-stimulating antibodies are not an invariable finding in patients with PPNAD.253 Studies on resected adrenals showed that nodules present intense immunostaining for steroidogenic enzymes and secrete cortisol in normal amounts.163,254 Steroidogenesis appears to be extremely sensitive to inhibition by ketoconazole in vitro254 and increases paradoxically during incubation with dexamethasone.255 This increase, which may occur also in adrenal adenomas (see earlier) and most likely accounts for the paradoxical responsiveness to high-dose dexamethasone in vivo, has been attributed to the intense expression of the glucocorticoid receptor.255
The advent of molecular biology shifted the focus to chromosomal and genetic alterations, leading to the identification of specific chromosomal loci and the first causative genetic alteration. Linkage studies revealed strong LOD scores (≥5.9) for a 6.4 cM region on the short arm of chromosome 2253 and a 17 cM locus on the long arm of chromosome 17.256 In keeping with the genetic heterogeneity of Carney’s complex, some kindreds map to both loci and others do not segregate to either, thus further sites remain to be identified. The region on chromosome 2 is known to harbor genes involved in tumor suppression and cell-cycle progression, and both genetic losses and gains at 2p16 (Carney complex 2 locus, CNC2) have been reported, suggesting the presence of oncogenes which await definition.257 Conversely, loss of heterozygosity at 17q22-24 hinted at a possible tumor-suppressor gene, narrowing the field of candidate genes and leading to the detection of mutations in the protein kinase A regulatory subunit 1 alpha (PRKAR1A) gene, a mediator involved in inhibition of cAMP signaling.154,155 PRKAR1A spans 11 exons and gives rise to a 2890-bp mRNA species, which in turn is translated into a 381-amino-acid protein (Fig. 7-15B). Heterozygous germline mutations in PRKAR1A (Carney complex 1 locus, CNC1) have been detected so far in nearly half of known kindreds with Carney’s complex as well as in patients with “sporadic” PPNAD.155,249 Gene alterations are mostly nonsense point mutations or frameshift mutations with premature stop codons, more rarely large deletions, leading to null alleles due to nonsense-mediated degradation of mutant mRNA or to truncated proteins154,155,258 (see Fig. 7-15B; Table 7-5). Indeed, the amount of PRKAR1A protein is nearly halved in lymphocytes from patients with Carney’s complex compared to normal subjects, reflecting expression of the wild-type allele only.154 More rarely, PRKAR1A mutated genes are transcribed and result in mutant or reduced amounts of protein.259 The PRKAR1A protein is part of the protein kinase A holoenzyme, consisting in two regulatory and two catalytic subunits (see Fig. 7-15A). The tetramer is inactive and requires binding of cAMP to regulatory subunits for catalytic subunits to dissociate and phosphorylate target proteins which in turn will activate downstream events. The two regulatory subunits (R1 and R2) each present two isoforms, alpha and beta, with different tissue-specific patterns of expression. Activity of PKA is dependent upon the availability of free catalytic subunits as well as on the ratio of R1:R2 regulatory subunits, and R1-alpha appears crucial for protection against excessive catalytic activity.260 In patients with Carney’s complex, haploinsufficiency of PRKAR1A due to nonsense-mediated mRNA decay or presence of mutant PRKAR1A proteins, leads to increased basal/stimulated PKA activity in adrenal cells,155,259,261 which in turn affects downstream signaling. The effects exerted by cAMP-stimulated PKA activity vary in different tissues, from growth promotion in pituitary and thyroid adenomas to differentiation of cardiac cells and inhibition of proliferation in Schwann cells. Of interest, PRKAR1A mutations appear to be more frequent in patients with Carney’s complex who develop PPNAD,261 and specific mutations have been shown to recur in patients with sporadic PPNAD.262 However, there is no clear genotype-phenotype correlation, and other disease-modifying loci most likely contribute to the clinical presentation. In this context, deletions and amplifications of CNC2 at chromosome 2p16 have also been observed in patients with PRKAR1A mutations.257
Table 7-5
PRKAR1A Mutations Associated With Carney’s Complex*

*Mutations are listed according to their past and present nomenclature when reported. Genomic location refers to the genomic PRKAR1A sequence (RefSeq NG_007093) and the corresponding amino acid sequence change given if known (codon 1 corresponds to translation initiation codon, RefSeq NP_002725).
FIGURE 7-15 A, Structure of PKA holoenzyme. Four regulatory subunits (R1α, R1β, R2α, and R2β) and three catalytic subunits (Cα, Cβ, and Cγ) have been identified so far. In the resting state, the PKA holoenzyme tetramer consists of four subunits: two homodimers of regulatory and catalytic subunits. Upon interaction of the receptor with its ligand, the Gα subunit dissociates and activates adenylate cyclase (AC). In turn, cAMP binds to regulatory subunits of PKA enabling dissociation of catalytic subunits which can phosphorylate cytoplasmic and nuclear proteins. Interaction with other intracellular pathways and gene transcription ensues. B, Structure of the PRKAR1A gene and protein. Mutations identified so far are shown. Of note, gene structure has been revised and exons renumbered starting with exon 4B. See Table 7-5 for list of mutations.
Interestingly, somatic PRKAR1A mutations have been observed in patients with pigmented adrenal adenoma139 but none in patients with classic macronodular adrenal hyperplasia.140 Among other enzymes involved in the cAMP-signaling cascade, phosphodiesterase 11A has recently been linked to both bilateral adrenocortical hyperplasia and dysplasia, and available evidence suggests germline mutations in PDE11A are low-penetrance predisposing factors for these and other tumors.263 Mutation of another phosphodiesterase, PDE8B, has been detected in a young girl with Cushing’s syndrome due to micronodular adrenal hyperplasia.264
Treament
Bilateral adrenalectomy is the treatment of choice for PPNAD, since both adrenals are affected. The risk of Nelson’s syndrome is nonexistent; plasma ACTH levels may not even rise above the normal range after removal of the adrenal glands.265 Patients with minimal hypercortisolism have occasionally been treated with unilateral adrenalectomy, and this has proven curative in some12,77; in others, however, removal of the remaining adrenal was necessary to contain hypercortisolism.12,266 In patients submitted to removal of a single adrenal gland, subtle alterations of HPA function, such as disruption of cortisol circadian rhythm, may persist26,41,267 and lead to progressive development of Cushingoid features over time.267 Medical adrenalectomy has been obtained in a patient treated with o,p’DDD for some 3 years, followed by remission of hypercortisolism for over 12 years.12 Brief treatments with low doses of adrenal blocking agents such as ketoconazole may prove beneficial prior to surgery. Spontaneous resolution has also been reported, most notably the first case described by Carney.12,76
Carney’s Complex
Carney’s complex is a hereditary disease with autosomal-dominant inheritance and incomplete penetrance (MIM 160980). In 1985, some 40 years after the first description of familial PPNAD, Carney and colleagues assembled a series of patients presenting a constellation of rare tumors (see Table 7-4) unlikely to occur together by chance alone, and the term Carney’s complex was coined. Various acronyms had been used in the past for patients presenting cardiac and cutaneous lesions (e.g., LAMB, for lentigines, atrial myxomas, blue nevi or NAME, for nevi, atrial myxomas, ephelides), but these terms have been abandoned. Given the significant variability in clinical presentation, the diagnosis of Carney’s complex is presumptive if two features are present, definite when three or more occur. Rarely are more than five elements of the complex present. Supplemental criteria for the diagnosis of Carney’s complex are an affected first-degree relative or presence of causative mutations, as of now only established for PRKAR1A.
Endocrine Lesions
PPNAD is the most frequent endocrine disorder in Carney’s complex, diagnosed in nearly 50% of patients affected by the complex.12 In order of frequency, testicular tumors are the next most common, followed by pituitary, thyroid, and ovarian lesions.
Three different testicular tumors arising from Sertoli cells, Leydig cells, and adrenal remnants have been reported in patients with Carney’s complex. The large-cell calcifying Sertoli cell tumor (LCCSCT) is the most common testicular lesion, arising in some 34% of male patients with Carney’s complex.268 LCCSCTs are usually detected at puberty as solid, calcified masses (5 to 10 mm in diameter), not palpable but easily visualized at sonography. These lesions are benign except for rare cases of malignant metastatic spread. LCCSCTs may also induce hormone-related symptoms such as gynecomastia, owing to the abundant expression of aromatase and increased estrogen production within the tumor. More rarely, the testes harbor steroid-type tumors such as the Leydig cell tumor, characterized by unencapsulated masses of large testosterone-secreting cells, or pigmented nodular adrenal rest tumors, arising close to the rete testis.12 Testosterone secretion by the testicular tumor may induce sexual precocity. Orchiectomy is the treatment of choice for all testicular lesions in order to avoid hormone-related symptoms and malignant transformation.
Acromegaly and gigantism are part of the initial description made by Carney26 but rarely occur in more than 10% of patients with the complex, although acromegaly was the leading manifestation of Carney’s complex in one sibship.269 Subclinical alterations of somatotroph function, such as elevated IGF-1 levels and insufficient GH inhibition by glucose, are present in many patients with Carney’s complex270 and have been observed to precede appearance of full-blown acromegaly by several years.271 PPNAD is usually diagnosed prior to acromegaly; indeed, remission of hypercortisolism has been suggested to “unmask” the GH-secreting tumor.272 Conversely, micro- and macroadenomas are found in equal proportion, and pathologic specimens may present adenohypophyseal hyperplasia, well-defined adenomas, or multicentric tumors.269,270 Immunohistochemistry may reveal both GH and prolactin immunoreactivity, thus pointing to mammosomatotroph cells in some patients.270,271 In this context, prolactin levels may also be increased in patients with Carney’s complex, without additional signs of pituitary hyperfunction.270,273
Thyroid lesions in patients with Carney’s complex range from hyperplasia to carcinoma and Hürthle cell tumor. Although published series are small, given the rarity of the complex, and intensive investigation of affected subjects may lead to an over-representation of these lesions, roughly 1 out of 10 patients with Carney’s complex will present thyroid gland abnormalities.274 These lesions are mostly benign nodules, but malignant follicular or papillary thyroid cancer has been described in a few patients, justifying screening with sonography.
Ovarian cysts were present in two patients in the initial series reported by Carney,26 and a later study reported the frequent detection of ovarian cysts in both prospective sonograms and autoptic studies.275 Malignant ovarian lesions, such as endometrioid or mucinous adenocarcinoma, have been described in two patients.275
Nonendocrine Lesions
Myxomas may originate in the heart, the skin, and the breast. These mesenchymal tumors, ubiquitous and recurring, are one of the distinctive features of Carney’s complex. Cardiac myxomas occurring within Carney’s complex present several unique features, most notably the location, multicentricity, and young age of appearance. Myxomas may arise in any of the four cardiac chambers, not just the left atrium, the usual site for sporadic cardiac myxomas, and tend to recur repeatedly. Further, myxomas arise in young adults with Carney’s complex, whereas sporadic tumors are most often diagnosed in middle-aged women.276 Cardiac myxomas, together with metastatic disease, account for the significant excess mortality in patients with Carney’s complex.277 Myxoid fibroadenoma is the most typical breast lesion in patients with Carney’s complex and may occur even in male patients.26 These benign tumors present as bilateral, palpable masses and are usually asymptomatic. In patients presenting with breast myxoid lesions, solid intraductal tumors with features of ductal adenoma have also been detected. Both pathologic examination and mammary gland imaging may indicate malignancy, although none of the ductal adenomas described so far has exhibited malignant behavior.278 Lastly, skin/mucosal myxomas usually occur in the oropharynx, the eyelids and ears, the trunk (especially around the nipples), the axilla, and the genital tract. Appearance of skin myxomas varies from small sessile papules to large pedunculated lesions.26
The so-called “spotty pigmentation” of patients with Carney’s complex is due to the abundance of lentigines (increased melanocytes), ephelides (increased melanin production by normal numbers of melanocytes), and blue nevi (accumulation of spindle-shaped melanocytes in the dermis)279 (Fig. 7-16). Less common are café-au-lait spots and junctional, atypical, or compound nevi. Pigmented lesions may be present at birth or develop shortly thereafter, intensify after puberty, and favor specific sites such as the centrofacial area, the labial border and the lips themselves, the conjunctiva and lachrymal caruncle, the external genitalia, and the anal region.26 Pigmentations tend to fade with age.
FIGURE 7-16 Patient with Carney’s complex. Note multiple lentigines and black spots on the face and neck, in particular on the vermillion border of the lips. (Courtesy Dr. Aidan Carney, Mayo Clinic, Rochester, MN.)
Psammomatous melanotic schwannoma (PMS) is a peripheral nerve sheath tumor characterized by heavy melanin pigmentation and frequent psammoma bodies, features which occur only rarely outside of Carney’s complex. PMS arises most frequently in lumbar nerve roots and in the gastrointestinal tract (esophagus, stomach), rarely in the skin, and may give rise to compressive symptoms. Multiple tumors are not uncommon, likewise local recurrences. Although usually benign, PMS may metastasize, most commonly to lung or liver271 and occasionally represents the cause of death for patients with Carney’s complex.269 Tumors appear dark, circumscribed, with solid or spongy consistency, featuring polygonal epithelium-like cells and spindle cells organized in an organoid pattern with positive S-100 and vimentin staining. The basal lamina is prominent, as are melanosomes.26 Melanotic cell death may lead to liquor coloring.
Osteochondromyxoma is a congenital mixed mesenchymal tumor of the bone. Pathologically, these lesions are circumscribed, erode surrounding bone and soft tissues, but do not appear to metastasize. Tumors are composed of immature and mature tissue, with cells disposed in solid sheets interspersed by abundant intercellular matrix and mesenchymal, cartilaginous, osseous, and hyaline fibrous tissue.280 Exeresis of the lesion, when possible, is curative.
• Sequencing of affected PRKAR1A and, if no mutation is detected, large gene rearrangements should be excluded
• Cardiac, testes, and thyroid ultrasound
• If schwannomas are suspected, thoracic, abdominal, and pelvic CT scan
• Cortisol circadian rhythm and overnight suppression test (The paradoxical cortisol response to high-dose dexamethasone, while highly suggestive of PPNAD, does not provide absolute diagnostic certainty, and the test should be reserved for later stages in the diagnostic workup.)
• Baseline GH, IGF-1, and prolactin levels; if necessary, assessment of the GH response to a glucose load
Special Clinical Presentations
Cushing’s syndrome in children is most often due to adrenocortical neoplasms, with PPNAD and MAS accounting for a scattering of cases.281 Adrenal cancer is an extremely rare pediatric malignancy (0.3 to 0.4 per million children per year) except for a population in southern Brazil, where this tumor is approximately 10 times more frequent than in the rest of the world (3.4 to 4.2 per million children per year).282 Tumors arise mostly in preschool children (before 5 years of age) and present a slight female preponderance.29,282 In contrast to adult adrenocortical tumors, over 80% of childhood adrenal tumors are functional, and cortisol hypersecretion is almost invariably accompanied by enhanced production of androgens and thus virilization. Functioning adrenocortical tumors have even been encountered at birth.282 Children may present with temporal balding, acne, the appearance of pubic and axillary hair, precocious puberty, increased muscle mass, accelerated height, increased body weight (Fig. 7-17), moon facies, hypertension, and electrolyte imbalances.29,282 Bone age is usually advanced except in those rare cases of “pure” Cushing’s syndrome.29 Detection of increased 17-ketosteroids or DHEAS is virtually diagnostic for adrenocortical tumor, and imaging procedures such as sonography, CT, or MRI easily reveal its presence.29,282
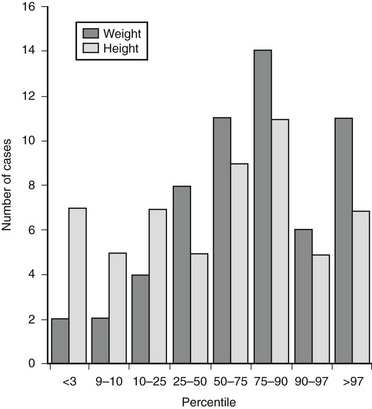
FIGURE 7-17 Distribution of weight and height in 58 children with adrenocortical tumors. (From Sandrini R, Ribeiro RC, de Lacerda L: Childhood adrenocortical tumors, J Clin Endocrinol Metab 82:2028, 1997. © Endocrine Society.)
Pathologic classification of tumors on the basis of commonly used algorithms (i.e., Weiss, Hough, van Slooten) does not provide a clear distinction between benign and malignant lesions in childhood adrenal cancer.283 Indeed, Weiss grade III or IV tumors exhibit an unusually benign course in children,284 and tumor size, weight, and invasion of adjacent organs appear to be more predictive of malignant behavior than classic histologic features.284,285 Postsurgical survival ranges between 56% and 70% at the 5-year landmark.282,284 Treatment with o,p’DDD has been carried out in several children, albeit with significant neurologic side effects and high risk of adrenal insufficiency, whereas reports on efficacy of chemotherapeutic regimens are few.281,285
PPNAD and MAS, the other possible causes of hypercortisolism in childhood and adolescence, are usually readily apparent because of the presence of associated features (see specific sections). Bilateral adrenalectomy provides ready relief from symptoms and usually allows catch-up growth.286
Pregnancy
Cushing’s syndrome is often associated with oligomenorrhea and anovulation, thus pregnancy very rarely occurs in hypercortisolemic women. However, several cases have been reported with considerable maternal morbidity and high fetal wastage, so prompt diagnosis and correction of hypercortisolism are therefore necessary. Urinary free cortisol is usually clearly elevated in pregnant hypercortisolemic women, and there is little risk of overlap with physiologically increased UFC levels during gestation.287 By contrast, cortisol suppression after low-dose dexamethasone is impaired during normal pregnancy, and cutoffs for diagnosis of Cushing’s syndrome have to be adapted.81,287 Establishment of the cause of hypercortisolism follows usual procedures, with ACTH levels normally suppressed in adrenal Cushing’s syndrome and imaging procedures (sonography or MR) usually enabling visualization of the adrenal lesion.288 Both malignant and benign lesions, the latter including also AIMAH and PPNAD, have been reported during pregnancy, thus none can be excluded a priori. Interestingly, among patients with Cushing’s syndrome diagnosed during gestation, adrenal causes account for over half of reported cases, which is considerably more than in the nonpregnant state.81 In some cases, cortisol levels increased after βhCG administration, and hypercortisolism resolved spontaneously after delivery,35,36 suggesting the involvement of aberrant LH/hCG receptors (see heading ACTH-Independent Adrenal Hyperplasia). To what extent these receptors account for the increased frequency of adrenal adenoma and hyperplasia during pregnancy remains to be established. Treatment of hypercortisolism during pregnancy is diversified according to the stage of gestation, with surgical removal of the adrenal lesion preferred during the first half of pregnancy and medical treatment with ketoconazole in the last trimester, although the converse approach also yielded favorable outcome.289
Treatment of Adrenal Cushing’s Syndrome
Surgery
Removal of the adrenal adenoma is followed by remission of hypercortisolism, and long-term survival becomes comparable to that of the general population.104,290 Exceptionally, Cushing’s syndrome may recur due to bilateral or ectopic adrenal adenomas290,291 or, as has also been reported, development of adrenal carcinoma on the same site of a previously resected adrenal adenoma.117 Alternative surgical approaches, such as percutaneous acetic acid injection, radiofrequency ablation, adrenal arterial embolization, and adrenal-preserving minimally invasive surgery have been attempted with variable success in cortisol-secreting adenomas.292–295 The drawback of these procedures is the absence of specimens for pathology, thus the preoperative diagnosis has to be certain.
In adrenal cancer, surgery is part of a multimodal approach also comprising chemotherapy and adrenolytic drugs. Only complete resection of the tumor offers potential for cure, although recurrence is not infrequent, sometimes after decades. On the whole, outcome is not encouraging, with less than 40% survival at the 5-year landmark25 without significant differences between functional and nonfunctional adrenal carcinomas. In hypersecreting malignant tumors, steroid hormone levels (e.g., DHEAS, UFC, estrogen, and 11-deoxycortisol) and plasma ACTH concentrations can be used as markers for residual disease and recurrence. For a detailed discussion of this topic, the reader is referred to Chapter 11.
Surgical procedures evolved from open surgery, via epigastric or subcostal transabdominal access or posterior lumbar extraperitoneal approach, to modern laparoscopic techniques, with a decrease in perioperative morbidity and mortality.296 This is nowadays the procedure of choice for removal of benign lesions in one or both adrenal glands, except for patients with coagulopathies, previous local surgery, or huge hyperplastic adrenals which necessitate long surgical procedures296,297 (see also Chapter 15). Adrenal cancer should be operated via laparoscopy only if this ensures wide margin resection of the tumor; most surgeons still prefer transabdominal access because this allows maximal exposure of the tumor and affected organs or vessels, in particular to tumor thrombi in the inferior vena cava, and minimizes the chance of tumor spillage.
Removal of the hyperfunctioning adrenal tissue is followed by a 1- to 2-year period of adrenal insufficiency due to pituitary-mediated inhibition of the normal adrenal gland.298,299 Steroid replacement therapy has to be administered (hydrocortisone 15 to 25 mg, two or three daily doses) and tapered over time according to recovery of the HPA axis. The latter, as well as the adequacy of steroid dosage, are largely established by clinical assessment rather than biochemical parameters.300 These include morning serum cortisol concentrations (levels greater than 10 µg/dL) or cortisol response to exogenous ACTH (peak response ≥20 µg/dL). Interestingly, several patients have been reported in whom the HPA axis never fully recovered and replacement therapy could not be discontinued.16
Bilateral adrenalectomy is the treatment of choice for AIMAH and PPNAD, with few exceptions (see related sections). Removal of the adrenal glands is easily performed using laparoscopy in PPNAD, whereas open surgery may be required if glands are extremely big in patients with AIMAH.182,266 Obviously, patients have to be started on lifelong steroid replacement therapy immediately after surgery, and usual precautions for adrenal insufficiency (see Chapter 6) are strenuously recommended; inappropriate treatment of adrenal crisis contributes significantly to mortality in these patients.
Medical Therapy
Drugs used for containment of hypercortisolism may be classed into three groups: adrenolytic, adrenostatic, and glucocorticoid receptor antagonists. Adrenolytic drugs—that is, compounds that destroy adrenocortical cells in addition to restraining steroid synthesis—are used mainly in adrenocortical cancer, whereas drugs that interfere with steroidogenesis without damaging adrenal cells (i.e., adrenostatic) can be used in any etiology of Cushing’s syndrome and also in pituitary-driven or ectopic ACTH-driven hypercortisolism. Lastly, the progesterone and glucocorticoid receptor antagonist mifepristone (RU486) has been employed to relieve symptoms related to severe hypercortisolism in a scattering of cases but has never entered mainstream antiglucocorticoid therapy. The effect of these drugs is not always predictable, thus expert handling, with close monitoring of efficacy and side effects, is necessary.301 On balance, metyrapone and ketoconazole are the drugs of choice for the medical containment of hypercortisolism, whereas other adrenostatic compounds are little used except for intravenous etomidate in severe Cushing’s syndrome. Mitotane (o,p’DDD) is indicated in adrenal cancer.
Medical therapy for illicit receptor–driven hypercortisolism has been discussed previously (see heading ACTH-Independent Adrenal Hyperplasia). On the whole, the use of specific receptor antagonists—while theoretically the most rational approach—is limited by its incomplete efficacy and the ease and feasibility of adrenalectomy. It may be employed in patients with mild hypercortisolism or after unilateral adrenalectomy to delay restrictions imposed by surgical adrenal insufficiency.
Chemotherapy, usually together with mitotane, is used in advanced-stage adrenal cancer, but experience is still limited.23,25 This and other topics related to oncologic aspects of adrenal cancer are discussed in Chapter 11.
Adrenolytic Drugs
Mitotane: Mitotane, 1,1-(dichlorodiphenyl)-2,2-dichloroethane; o,p′-DDD, derives from the organochloride insecticide DDT (dichloro-diphenyl-trichloroethane) and was first proven to induce adrenal cortical atrophy in 1949 and employed in adrenal cancer some 10 years later. Since then, scores of patients with functioning and nonfunctioning adrenocortical cancer have been treated with this compound, but its efficacy in stalling tumoral progression is unpredictable and often temporary.
Mitotane is a lipophilic compound that requires mitochondrial conversion into its active metabolites, o,p′-dichloroethene (o,p′-DDE) and o,p′-acetic acid (o,p′-DDA). Several facets of mitotane action have been defined, although the exact intracellular mechanisms have yet to be fully elucidated. The most significant action of mitotane is the inhibition of the first step in steroidogenesis, that is, conversion of cholesterol to pregnenolone, with inhibition of 11β-hydroxylase, 18-hydroxylase, and 3β-hydroxysteroid dehydrogenase occurring to a lesser extent.302 Mitotane also interferes with cortisol metabolism, favoring 6β-hydroxylation over 5β-reduction and accelerating hepatic steroid metabolism.303 At the same time, mitotane increases the synthesis of several hormone-binding proteins, most notably cortisol-binding globulin, sex hormone–binding globulin, and to a lesser extent thyroxin-binding globulin and vitamin D–binding globulin. The adrenolytic effect is achieved through several mechanisms, specifically, perturbation of lipid microenvironment with lipid accumulation, mitochondrial swelling and lysis, interference with ATPase activity and mitochondrial electron transport, superoxide formation, and covalent binding to proteins. Adrenal cell death occurs in the zonae fasciculata and reticularis as well as in metastases, with relative sparing of the zona glomerulosa. Mitotane also reverses the expression of the chemotherapy resistance gene, P-glycoprotein (Pgp), and enhances the efficacy of cytotoxic drugs in vitro.304 This effect, however, does not appear to translate into greater efficacy of cytotoxic regimens in vivo, and new mitotane analogues with greater suppression of adrenal cell growth and secretion are currently being developed.
Clinical Use.: Treatment with mitotane is initiated with 0.5 to 2 g daily and the dosage increased by 1 g every 4 weeks up to maximal daily doses of 4 to 12 g. Given its lipophilic properties, the drug accumulates in adipose tissue and may be discharged up to 2 years after discontinuation.305 Mitotane has been used in children282,285 but is contraindicated during pregnancy, owing to its teratogenic effects.305 The efficacy of mitotane in patients with either functioning or nonfunctioning adrenal cancer is actively debated, with some studies reporting objective tumor regression and lower recurrence rates,11,306 while others were not equally successful.7,25 Recent large studies provided new data demonstrating that administration of mitotane after surgery significantly prolonged relapse-free survival in patients with adrenal cancer.307,308 Serum mitotane levels should reach 14 µg/mL to achieve satisfactory response rates.309 Once these levels have been reached, usually 3 to 4 months after treatment with increasing doses, mitotane can be tapered down to avoid adverse effects. Combined mitotane-chemotherapy regimens have also been attempted in patients with advanced-stage adrenal cancer and will possibly be validated through prospective, randomized trials.310 Lastly, in individual patients, long-term treatment (up to 16 years) with low doses of mitotane has proven capable of controlling metastatic disease.311
Mitotane controls endocrine hypersecretion in the majority of patients.25 UFC levels, and in the long run also plasma ACTH, represent the best parameters to monitor the efficacy of mitotane treatment, since secretion of 17-hydroxysteroids is selectively reduced, and serum cortisol may be factitiously increased due to higher CBG levels.
Adverse Effects.: Untoward effects represent the main limitation to mitotane administration. Gastrointestinal distress (e.g., diarrhea, nausea, vomiting) may be counteracted by serotonin antagonists and administration at bedtime or meals. Neurologic toxicity symptoms (e.g., dizziness, lethargy, vertigo, speech disturbances, ataxia) usually accompany higher-dosage regimens.309 Optic nerve toxicity may result in complete blindness. Additional side effects are fatigue, gynecomastia, skin rashes, and hypoadrenalism. Iatrogenic adrenal insufficiency is counteracted by hydrocortisone and prednisone rather than dexamethasone administration, because the latter is metabolized at a greater rate by the hepatic microsomal machinery.303 As with adrenostatic compounds, concomitant administration of drugs such as rifampicin, warfarin, and phenytoin, which induce hepatic enzymes, thereby enhancing steroid metabolism, may require upscaling of steroid replacement dosage. Mineralocorticoid insufficiency may occur with long-term mitotane administration. Clinical chemistry evaluation usually reveals increased gamma glutamyl transferase, alkaline phosphatase, aminotransferases (rarely serious enough to require drug withdrawal), hyperlipidemia, hypouricemia, and reduced blood cell counts. Free and total thyroid hormone levels may decrease, but thyrotropin remains within the normal range. Symptomatic hypotestosteronemia requiring replacement therapy may ensue in male patients.11,312 Most side effects benefit from brief reduction or interruption of mitotane with a simultaneous increase in steroid replacement therapy.
Adrenostatic Drugs
These compounds reduce cortisol secretion by interfering with one or more steps of steroid synthesis (Fig. 7-18). With the exception of ketoconazole, all compounds of this class induce a compensatory ACTH increase which may partially override pharmacologic blockade. Higher drug doses are therefore necessary over time to maintain eucortisolism. If blockade of cortisol synthesis is complete, steroid replacement therapy is required to avoid symptoms of adrenal insufficiency.
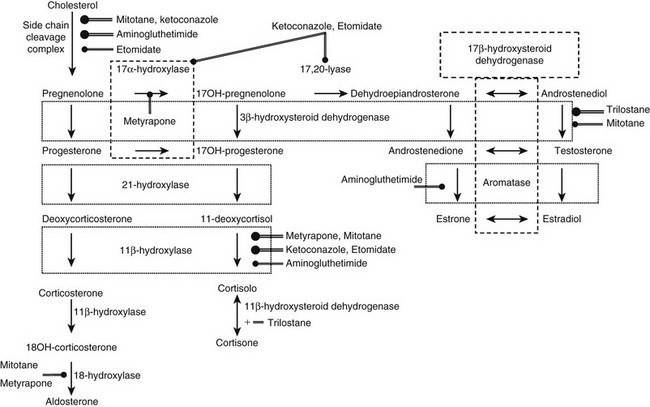
FIGURE 7-18 Blockade of steroidogenic enzymes by adrenolytic and adrenostatic compounds. Sites of major ( ) and minor (
) and minor ( ) inhibitory as well as stimulatory (
) inhibitory as well as stimulatory ( ) actions are shown.
) actions are shown.
Ketoconazole: Since it was discovered that this imidazole derivative, most commonly used as an antimycotic agent, inhibits cytochrome P450 enzymes, ketoconazole has taken center stage in the medical therapy of Cushing’s syndrome.313 Indeed, given its greater tolerability and ease of use, ketoconazole is the most widely used drug to control hypercortisolism.301,314
Ketoconazole inhibits several steps of steroidogenesis: in order of potency, the side-chain cleavage complex, 17,20-lyase, 17α-hydroxylase, and 11β-hydroxylase.315 Ketoconazole also interferes with ACTH-induced cAMP production and is a weak competitor for the glucocorticoid receptor. The inhibition of progestins to androgens conversion by blockade of 17,20-lyase explains the pronounced reduction of DHEA and androstenedione secretion.
Clinical Use.: Ketoconazole is usually administered starting with 200 mg daily and progressively increasing to 600 to 800 mg according to adrenal secretory parameters. Higher doses, up to 1200 mg daily, are rarely required. Cimetidine and other antacids interfere with gastric absorption of ketoconazole and thus should not be used together. Ketoconazole reduces UFC levels in patients with adrenal adenoma but is possibly less effective in adrenal carcinoma.315 Ketoconazole has also been reported to exert cytostatic effects in patients with secreting metastatic adrenocortical cancer.316 Of note, notwithstanding the considerable size of the adrenals, low doses of ketoconazole are usually sufficient to obtain eucortisolism in patients with AIMAH. Ketoconazole may safely be used in children and during pregnancy.
Adverse Effects.: Ketoconazole is usually well tolerated, with only a minority of patients (less than 5%) reporting adverse effects. These are mostly gastrointestinal (e.g., nausea, vomiting) or dermatologic (e.g., skin rash, pruritus) and disappear after drug suspension. Gynecomastia and impotence, most likely due to reduced androgen synthesis, have occasionally been reported. Hepatic dysfunction (cholestasis, hepatocellular injury) occurs in some 10% of patients treated with ketoconazole and typically resolves after drug discontinuation. If the increase in hepatic transaminases is modest (i.e., less than threefold increase over normal), ketoconazole may be continued with close monitoring of hepatic function. Fatal hepatitis is an exceptional occurrence. Similarly, liver metastases do not preclude the use of ketoconazole in adrenal cancer. Lastly, administration of ketoconazole at appropriate dosage does not necessitate adjunctive steroid replacement therapy.
Another nitroimidazole derivate, fluconazole, has recently been employed in a patient with cortisol-secreting adrenal cancer and achieved normalization of UFC and amelioration of symptoms for nearly 2 years.317
Metyrapone: This pyridine derivate—2-methyl-1,2-bis-(3-pyridyl)-1-propanone; SU-4885—inhibits 11β hydroxylase, the enzyme responsible for the final step in cortisol biosynthesis and, to a lesser extent, also 18-, 19-, and 17α-hydroxylases.318 In addition, metyrapone has been shown to suppress adrenal expression of the ACTH receptor MC2R.319 Metyrapone is used in both diagnosis and treatment of Cushing’s syndrome, although more accurate testing procedures have superseded its use in the former setting (see heading Diagnosis of Adrenal Cushing’s Syndrome). Metyrapone is usually administered together with other steroid synthesis inhibitors in patients with malignant lesions. Although efficacious, this compound is not universally available, which limits its use.
Clinical Use.: Metyrapone is administered in dosages ranging from 500 mg to 6 g daily, subdivided in three or four doses. Cortisol levels decrease promptly; indeed, one advantage of metyrapone treatment is its rapid cortisol-lowering effect (within 2 hours).320 Most patients with adrenal lesions respond well as regards both clinical signs and UFC concentrations.321 Hypoadrenalism may occur, and patients should be started on prednisone replacement. Additional side effects are worsening of hirsutism, acne, hypokalemia, and hypertension secondary to the increase in androgen and mineralocorticoids, as well as rash, nausea, and dizziness. The use of metyrapone in pregnancy is debated, with some reports describing amelioration of hypercortisolism without adverse fetal effects36 and others reporting worsening of hypertension and preeclampsia.322 Lastly, administration of metyrapone, together with aminoglutethimide, for 18 months obtained lasting normalization of HPA parameters and disappearance of Cushingoid features in an infant with MAS.323
Aminoglutethimide: Aminoglutethimide is an anticonvulsant drug which exerts a potent inhibitory action of P450 side chain–cleavage enzyme. Less relevant is its action on other P450 steroidogenic enzymes, on aromatase, and on the adrenal ACTH receptor.319 Aminoglutethimide also exerts an antiproliferative effect on adrenocortical cells in vitro.319
Clinical Use.: Successful palliation of hypercortisolism has been reported in over 60% of patients with adrenal carcinoma treated with aminoglutethimide.324 Starting dose is 250 mg daily, with progressive increases up to 1 to 2 g daily.324 Side effects include mild sedation, itchy rash, goiter, and upper gastrointestinal discomfort which may disappear spontaneously during treatment. Hypoadrenalism should be corrected with hydrocortisone rather than dexamethasone, because hepatic clearance of the latter is accelerated. Hypothyroidism may develop due to inhibition of thyroid hormone synthesis.
Trilostane: This androstane-carbonitrile derivative (WIN 24,540) selectively inhibits 3β-hydroxysteroid dehydrogenase and potentiates 11β-hydroxysteroid dehydrogenase type 2, thus yielding increased cortisone:cortisol ratios. Clinical efficacy in containing hypercortisolism is weak, even at high doses up to 1400 mg daily,44 and is accompanied by gastrointestinal side effects. Hypoadrenalism may ensue.
Etomidate: Etomidate is a short-acting hypnotic drug only effective when administered intravenously. This imidazole derivative potently inhibits 11β-hydroxylase and, to a lesser extent, also 17α- hydroxylase, 17,20-lyase, and side chain–cleavage enzymes.315 In addition, etomidate markedly inhibits adrenocortical cell proliferation and expression of the ACTH receptor.319 In vitro, etomidate is the most potent adrenostatic compound compared with other agents.315,319 It has been used successfully to reduce hypercortisolism in severely ill patients who are unresponsive or unable to ingest oral medications, and this is most likely the only indication for this compound,301 since sedation often occurs even when infused at nonhypnotic doses (1.2 to 2.5 mg/h). Adrenal insufficiency invariably occurs on etomidate therapy, thus replacement with dexamethasone or hydrocortisone is mandatory.
Glucocorticoid Receptor Antagonist
Mifepristone (RU486): RU486 is a steroid analogue [17β-hydroxy, 11β-(4-dimethylaminophenyl), 17α-(1-propynyl)-estra-4,9-diene-3-one] that competitively antagonizes binding to glucocorticoid and progesterone receptors and blocks steroid-induced peripheral effects. Administration of RU486 to small series of patients with Cushing’s syndrome induced a reversal of clinical manifestations of hypercortisolism, most notably acute psychosis.325 Dosage of RU486 is usually 5 to 25 mg/kg or 400 to 800 mg daily, with close monitoring to avoid adrenal insufficiency. Detection of the latter rests on indirect biochemical and clinical parameters such as hypoglycemia, hyperkalemia, and nausea, because cortisol levels remain elevated. The absence of a marker of peripheral glucocorticoid activity, the long half-life of the drug, and the difficulty in counteracting its antiglucocorticoid activity all hinder the clinical use of this compound.
References
1. Osler, W. An acute myxoedematous condition, with tachycardia, glycosuria, melena, mania, and death. J Nerv Ment Dis. 1899;26:65–71.
2. Cushing, H. The basophil adenomas of the pituitary body and their clinical manifestations. Johns Hopkins Bull. 1932;50:137–195.
3. Walters, W, Wilder, RM, Kepler, EJ. The suprarenal cortical syndrome with presentation of ten cases. Ann Surg. 1934;100:670–688.
4. Carpenter, PC. Diagnostic evaluation of Cushing’s syndrome. Endocrinol Metab Clin North Am. 1988;17:445–472.
5. Lindholm, J, Juul, S, Jørgensen, JOL, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86:117–123.
6. Demura, H, Takeda, R, Miyamori, I, et al. Cushing’s syndrome in Japan with special reference to adrenocortical nodular dysplasia or hyperplasia. In: Takeda R, Miyamori I, eds. Controversies in Disorders of Adrenal Hormones: Proceedings of the Open Symposium of Disorders of Adrenal Hormones. New York: Elsevier Science; 1988:3–17.
7. Bertagna, X, Orth, DN. Clinical and laboratory findings and results of therapy in 58 patients with adrenocortical tumors admitted to a single medical center (1951 to 1978). Am J Med. 1981;71:855–875.
8. Hsing, AW, Nam, JM, Co Chien, HT, et al. Risk factors for adrenal cancer: an exploratory study. Int J Cancer. 1996;65:432–436.
9. National Cancer Institute. Third National Cancer Survey: incidence data. In: Anonymous DHEW Publ. No. (NIH) 75–787. Bethesda: NCI Monograph; 1975:41.
10. Crucitti, F, Bellantone, R, Ferrante, A, et al. The Italian registry for adrenal cortical carcinoma: analysis of a multiinstitutional series of 129 patients. Surgery. 1996;119:161–170.
11. Luton, JP, Cerdas, S, Billaud, L, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 1990;322:1195–1201.
12. Carney, JA, Young, WF, Jr. Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist. 1992;2:6–21.
13. Lacroix, A, N’Diaye, N, Tremblay, J, et al. Ectopic and abnormal hormone receptors in adrenal Cushing’s syndrome. Endocrine Rev. 2001;22:75–110.
14. Lieberman, SA, Eccleshall, TR, Feldman, D. ACTH-independent massive bilateral adrenal disease (AIMBAD): a subtype of Cushing’s syndrome with major diagnostic and therapeutic implications. Eur J Endocrinol. 1994;131:67–73.
15. Perry, RR, Nieman, LK, Cutler, GB, Jr., et al. Primary adrenal causes of Cushing’s syndrome. Ann Surg. 1989;210:59–68.
16. Daitsch, JA, Goldfarb, DA, Novick, AC. Cleveland Clinic experience with adrenal Cushing’s syndrome. J Urol. 1997;158:2051–2055.
17. Kirk, JMW, Brain, CE, Carson, DJ, et al. Cushing’s syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J Pediatr. 1999;134:789–792.
18. Kasperlik-Zaluska, AA, Migdalska, BM, Zgliczynski, S, et al. Adrenocortical carcinoma. A clinical study and treatment results of 52 patients. Cancer. 1995;75:2587–2591.
19. Rockall, AG, Babar, S, Sohaib, SA, et al. CT and MR imaging of the adrenal glands in ACTH-independent Cushing’s syndrome. RadioGraphics. 2004;24:435–452.
20. Tenschert, W, Baumgart, P, Greminger, P, et al. Pathogenetic aspects of hypertension in Cushing’s syndrome. Cardiology. 1985;72(Suppl 1):84–90.
21. Ross, EJ, Linch, DC. The clinical response to treatment in adult Cushing’s syndrome following remission of hypercortisolaemia. Postgrad Med J. 1985;61:205–211.
22. Pikkarainen, L, Sane, T, Reunanen, A. The survival and well-being of patients treated for Cushing’s syndrome. J Intern Med. 1999;245:463–468.
23. Assié, G, Antoni, G, Tissier, F, et al. Prognostic parameters of metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2007;92:148–154.
24. Wajchenberg, BL, Albergaria Pereira, MA, Bilharinho de Mendonça, B, et al. Adrenocortical carcinoma. Clinical and laboratory observations. Cancer. 2000;88:711–736.
25. Allolio, B, Fassnacht, M. Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91:2027–2037.
26. Carney, JA, Gordon, H, Carpenter, PC, et al. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985;64:270–283.
27. Invitti, C, Pecori Giraldi, F, De Martin, M, et al. Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study. J Clin Endocrinol Metab. 1999;84:440–448.
28. O’Hare, MJ, Monaghan, P, Neville, AM. The pathology of adrenocortical neoplasia: a correlated structural and functional approach to the diagnosis of malignant disease. Hum Pathol. 1979;10:137–154.
29. Bilharinho de Mendonça, B, Lucon, AM, Menezes, CAV, et al. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J Urol. 1995;154:2004–2009.
30. Fukai, N, Hirono, Y, Yoshimoto, T, et al. A case of estrogen-secreting adrenocortical carcinoma with subclinical Cushing’s syndrome. Endocr J. 2006;53:237–246.
31. Adachi, J, Hirai, Y, Terui, K, et al. A report of 7 cases of adrenal tumors secreting both cortisol and aldosterone. Intern Med. 2003;42:714–718.
32. Balakumar, T, Perry, LA, Savage, MO. Adrenocortical adenoma—an unusual presentation with hypersecretion of oestradiol, androgens and cortisol. J Pediatr Endocrinol Metab. 1997;10:227–229.
33. Nieman, LK, Biller, BMK, Findling, JW, et al. Diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–1540.
34. Pecori Giraldi, F, Ambrogio, AG, De Martin, M, et al. Specificity of first-line tests for the diagnosis of Cushing’s syndrome: assessment in a large series. J Clin Endocrinol Metab. 2007;92:4123–4129.
35. Lacroix, A, Hamet, P, Boutin, J. Leuprolide acetate therapy in luteinizing hormone-dependent Cushing’s syndrome. N Engl J Med. 1999;341:1577–1581.
36. Hána, V, Dokoupilová, M, Marek, J, et al. Recurrent ACTH-independent Cushing’s syndrome in multiple pregnancies and its treatment with metyrapone. Clin Endocrinol. 2001;54:277–281.
37. Stratakis, CA, Sarlis, NJ, Kirschner, LS, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131:585–591.
38. Lim, LC, Tan, LHC, Rajasoorya, C. Unravelling the mystery in a case of persistent ACTH-independent Cushing’s syndrome. Ann Acad Med Singapore. 2006;35:892–896.
39. Orth, DN. Cushing’s syndrome. N Engl J Med. 1995;332:791–803.
40. Klose, M, Kofoed-Enevoldsen, A, Østergaard Kristensen, L. Single determination of plasma ACTH using an immunoradiometric assay with high detectability differentiates between ACTH-dependent and -independent Cushing’s syndrome. Scand J Clin Lab Invest. 2002;62:33–38.
41. Ruder, HJ, Loriaux, DL, Lipsett, MB. Severe osteopenia in young adults associated with Cushing’s syndrome due to micronodular adrenal disease. J Clin Endocrinol Metab. 1974;39:1138–1147.
42. Tillman, V, Mägi, ML, Metsvaht, T. Prenatal Cushing’s syndrome secondary to nodular adrenocortical hyperplasia with unsuppressed plasma ACTH levels. J Pediatr Endocrinol Metab. 2005;18:1027–1031.
43. Law, A, Hague, WM, Daly, JG, et al. Inappropriate ACTH concentrations in two patients with functioning adrenocortical carcinoma. Clin Endocrinol. 1988;29:53–62.
44. Komanicky, P, Spark, RF, Melby, JC. Treatment of Cushing’s syndrome with trilostane (WIN 24,540), an inhibitor of adrenal steroid biosynthesis. J Clin Endocrinol Metab. 1978;47:1042–1051.
45. Müller, OA, Stalla, GK, von Werder, K. Corticotropin releasing factor: a new tool for the differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 1983;57:227–229.
46. Hensen, J, Buhl, M, Bähr, V, et al. Endocrine activity of the “silent” adrenocortical adenoma is uncovered by response to corticotropin-releasing hormone. Klin Wochenschr. 1990;68:608–614.
47. Crapo, L. Cushing’s syndrome: a review of diagnostic tests. Metabolism. 1979;28:955–977.
48. Yamaji, T, Ishibashi, M, Sekihara, H, et al. Serum dehydroepiandrosterone sulfate in Cushing’s syndrome. J Clin Endocrinol Metab. 1984;59:1164–1168.
49. Terzolo, M, Alì, A, Osella, G, et al. The value of dehydroepiandrosterone sulfate measurement in the differentiation between benign and malignant adrenal masses. Eur J Endocrinol. 2000;142:611–617.
50. Tóth, M, Rácz, K, Varga, I, et al. Plasma dehydroepiandrosterone sulphate levels in patients with hyperfunctioning and non-hyperfunctioning adrenal tumors before and after adrenal surgery. Eur J Endocrinol. 1997;136:290–295.
51. Smals, AGH, Kloppenborg, PWC, Benraad, TJ. Plasma testosterone profiles in Cushing’s syndrome. J Clin Endocrinol Metab. 1977;45:240–245.
52. Doerr, P, Pirke, KM. Cortisol-induced suppression of plasma testosterone in normal adult males. J Clin Endocrinol Metab. 1976;43:622–629.
53. Levine, AC, Mitty, HA, Gabrilove, JL. Steroid content of the peripheral and adrenal vein in Cushing’s syndrome due to adrenocortical adenoma and carcinoma. J Urol. 1988;140:11–15.
54. Kikuchi, E, Yanaihara, H, Nakashima, J, et al. Urinary steroid profile in adrenocortical tumors. Biomed & Pharmacother. 2000;54(Suppl 1):194–197.
55. Gröndal, S, Erkisson, B, Hagenas, L, et al. Steroid profile in urine: a useful tool in the diagnosis and follow-up of adrenocortical carcinoma. Acta Endocrinol (Copenh). 1990;122:656–663.
56. Kelly, WF, Barnes, AJ, Cassar, J, et al. Cushing’s syndrome due to adrenocortical carcinoma—a comprehensive clinical and biochemical study of patients treated by surgery and chemotherapy. Acta Endocrinol (Copenh). 1979;91:303–318.
57. Homoki, J, Holl, R, Teller, WM. Urinary steroid profiles in Cushing’s syndrome and tumors of the adrenal cortex. Klin Wochenschr. 1987;65:719–726.
58. Haigh, SE, Tevaarwerk, JM. A rise in the glomerular filtration rate as the cause of a ‘paradoxical’ increase in urinary free cortisol during dexamethasone suppression in a patient with an adrenal adenoma: a case report. Clin Endocrinol. 1981;15:53–56.
59. Invitti, C, Pecori Giraldi, F, Cavagnini, F, et al. Unusual association of adrenal angiosarcoma and Cushing’s disease. Horm Res. 2001;56:124–129.
60. Barzon, L, Sonino, N, Fallo, F, et al. Prevalence and natural history of adrenal incidentalomas. Eur J Endocrinol. 2003;149:273–285.
61. Mantero, F, Terzolo, M, Arnaldi, G, et al. A survey on adrenal incidentaloma in Italy. J Clin Endocrinol Metab. 2000;85:637–644.
62. Tanabe, A, Naruse, M, Nishikawa, T, et al. Autonomy of cortisol secretion in clinically silent adrenal incidentaloma. Horm Metab Res. 2001;33:444–450.
63. Tsagarakis, S, Vassiliadi, D, Thalassinos, N. Endogenous subclinical hypercortisolism: diagnostic uncertainties and clinical implications. J Endocrinol Invest. 2006;29:471–482.
64. Bardet, S, Rohmer, V, Murat, A, et al. 131I-6β-Iodomethylnorcholesterol scintigraphy: an assessment of its role in the investigation of adrenocortical incidentalomas. Clin Endocrinol. 1996;44:587–596.
65. Reincke, M, Niecke, J, Krestin, GP, et al. Preclinical Cushing’s syndrome in adrenal “Incidentalomas”: comparison with adrenal Cushing’s syndrome. J Clin Endocrinol Metab. 1992;75:826–832.
66. Toniato, A, Merante-Boschin, I, Opocher, G, et al. Surgical versus conservative management for subclinical Cushing syndrome in adrenal incidentalomas: a prospective randomized study. Ann Surg. 2009;249:388–391.
67. Reznek, RH, Armstrong, P. Imaging in endocrinology: The adrenal gland. Clin Endocrinol. 1994;40:561–576.
68. Goldman, SM, Coelho, RD, Freire Filho, EdO, et al. Imaging procedures in adrenal pathology. Arq Bras Endocrinol Metab. 2004;48:592–611.
69. Bonnin, A, Abecassis, JP, Broussouloux, C, et al. La place de l’examen tomodensitométrique dans l’exploration des surrénales. Ann Endocrinol (Paris). 1988;49:332–336.
70. Korobkin, M, Brodeur, FJ, Francis, IR, et al. CT time-attenuation washout curves of adrenal adenomas and nonadenomas. Am J Roentgenol. 1998;170:747–752.
71. Prince, EA, Yoo, DC, DeLellis, RA, et al. CT and PET appearance of a pigmented “black” adrenal adenoma in a patient with lung cancer. Clin Radiol. 2007;62:1229–1231.
72. Newhouse, JH, Heffess, CS, Wagner, BJ, et al. Large degenerated adrenal adenomas: radiologic-pathologic correlation. Radiology. 1999;210:385–391.
73. Kouriefs, C, Mokbel, K, Choy, C. Is MRI more accurate than CT in estimating the real size of adrenal tumours? Eur J Surg Oncol. 2001;27:487–490.
74. Fig, LM, Gross, MD, Shapiro, B, et al. Adrenal localization in the adrenocorticotropin hormone-independent Cushing syndrome. Ann Intern Med. 1988;109:547–553.
75. Dunnick, NR, Schaner, EG, Doppman, JL, et al. Computed tomography in adrenal tumors. AJR. 1979;132:43–46.
76. Grant, CS, Carney, JA, Carpenter, PC, et al. Primary pigmented nodular adrenocortical disease: diagnosis and management. Surgery. 1986;100:1178–1184.
77. Doppman, JL, Travis, W, Nieman, LK, et al. Cushing syndrome due to primary pigmented nodular adrenocortical disease: findings at CT and MR imaging. Radiology. 1989;172:415–420.
78. Ilias, I, Sahdev, A, Reznek, RH, et al. The optimal imaging of adrenal tumours: a comparison of different methods. Endocr Relat Cancer. 2007;14:587–599.
79. Lee, MJ, Mayo-Smith, WW, Hahn, JA, et al. State-of-the-art MR imaging of the adrenal gland. Radio Graphics. 1994;14:1015–1029.
80. Outwater, EK, Siegelman, ES, Huang, AB, et al. Adrenal masses: correlation between CT attenuation value and chemical shift ratio at MR imaging with in-phase and opposed-phase sequences. Radiology. 1996;200:749–752.
81. Polli, N, Pecori Giraldi, F, Cavagnini, F. Cushing’s syndrome in pregnancy. J Endocrinol Invest. 2003;26:1045–1050.
82. Sarkar, SD, Cohen, EL, Beierwaltes, WH, et al. A new and superior adrenal imaging agent, 131I-6β-iodomethyl-19-Nor-cholesterol (NP-59): evaluation in humans. J Clin Endocrinol Metab. 1977;45:353–362.
83. Gross, MD, Shapiro, B, Thrall, JH, et al. The scintigraphic imaging of endocrine organs. Endocrine Rev. 1984;5:221–281.
84. Yoh, T, Hosono, M, Komeya, Y, et al. Quantitative evaluation of norcholesterol scintigraphy, CT attenuation value, and chemical-shift MR imaging for characterizing adrenal adenomas. Ann Nucl Med. 2008;22:513–519.
85. Gross, MD, Wong, KK, Rubello, D. Scintigraphic imaging of adrenal disease. Minerva Endocrinol. 2008;33:1–17.
86. Reschini, E, Baldini, M, Cantalamessa, L. A black adrenocortical adenoma causing Cushing’s syndrome not imaged by radiocholesterol scintigraphy. Eur J Nucl Med. 1990;17:185–187.
87. Barzon, L, Zucchetta, P, Boscaro, M, et al. Scintigraphic patterns of adrenocortical carcinoma: morpho-functional correlates. Eur J Endocrinol. 2001;145:743–748.
88. Thorin-Savouré, A, Tissier-Rible, F, Guignat, L, et al. Collision/composite tumors of the adrenal gland: a pitfall of scintigraphy imaging and hormone assays in the detection of adrenal metastasis. J Clin Endocrinol Metab. 2005;90:4924–4929.
89. Reschini, E, Peracchi, M. Uptake of 75Se-selenocholesterol by an adrenal cortical carcinoma and its metastases. Eur J Nucl Med. 1984;9:291–293.
90. Lumachi, F, Zucchetta, P, Marzola, MC, et al. Usefulness of CT scan, MRI and radiocholesterol scintigraphy for adrenal imaging in Cushing’s syndrome. Nucl Med Commun. 2002;23:469–473.
91. Sels, JP, Wouters, RM, Lamers, R, et al. Pitfall of the accessory spleen. Neth J Med. 2000;56:153–158.
92. Wulffraat, NM, Drexhage, HA, Wiersinga, WM, et al. Immmunoglobulins of patients with Cushing’s syndrome due to pigmented adrenocortical micronodular dysplasia stimulate in vitro steroidogenesis. J Clin Endocrinol Metab. 1988;66:301–307.
93. Yu, KC, Alexander, R, Ziessman, HA, et al. Role of preoperative iodocholesterol scintiscanning in patients undergoing adrenalectomy for Cushing’s syndrome. Surgery. 1995;118:981–987.
94. Garcia-Mayor, RV, Perez Mendez, L, Paramo, C, et al. Primary adrenocortical nodular dysplasia presented as adrenal adenoma by functional scintigraphy. Clin Nucl Med. 1993;18:220–222.
95. Young, WF, Jr., du Plessis, H, Thompson, GB, et al. The clinical conundrum of corticotropin-independent autonomous cortisol secretion in patients with bilateral adrenal masses. World J Surg. 2007;32:856–862.
96. Becherer, A, Vierhapper, H, Potzi, C, et al. FDG-PET in adrenocortical carcinoma. Cancer Biother Radiopharm. 2001;16:289–295.
97. Leboulleux, S, Dromain, C, Bonniaud, G, et al. Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. J Clin Endocrinol Metab. 2006;91:920–925.
98. Han, SJ, Kim, TS, Jeon, SW, et al. Analysis of adrenal masses by 18F-FDG positron emission tomography scanning. Int J Clin Pract. 2007;61:802–809.
99. Hennings, J, Lindhe, Ö, Bergström, M, et al. [11C]Metomidate positron emission tomography of adrenocortical tumors in correlation with histopathological findings. J Clin Endocrinol Metab. 2006;91:1410–1414.
100. Aiba, M, Hirayama, A, Iri, H, et al. Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia as a distinct subtype of Cushing’s syndrome. Am J Clin Pathol. 1991;96:334–340.
101. Saeger, W, Reinhard, K, Reinhard, C. Hyperplastic and tumorous lesions of the adrenals in an unselected autopsy series. Endocr Pathol. 1998;9:235–239.
102. Sasano, H, Suzuki, T, Irie, J, et al. Adrenal cortical disease: international case conference. Endocr Pathol. 2002;13:141–148.
103. Mackay, B, El-Naggar, A, Ordonez, NG. Ultrastructure of adrenal cortical carcinoma. Ultrastruct Pathol. 1994;18:181–190.
104. Välimäki, M, Pelkonen, R, Porkka, L, et al. Long-term results of adrenal surgery in patients with Cushing’s syndrome due to adrenocortical adenoma. Clin Endocrinol. 1984;20:229–236.
105. Desai, N, Kapoor, A, Sing, BK, et al. Bilateral adrenal adenomas and persistent leukocytosis: a unique case of Cushing’s syndrome. Am J Med. 2006;119:e3–e5.
106. Aiba, M, Kawakami, M, Ito, Y, et al. Bilateral adrenocortical adenomas causing Cushing’s syndrome. Report of two cases with enzyme histochemical and ultrastructural studies and a review of the literature. Arch Pathol Lab Med. 1992;116:146–150.
107. N’Diaye, N, Hamet, P, Tremblay, J, et al. Asynchronous development of bilateral nodular adrenal hyperplasia in gastric inhibitory polypeptide-dependent Cushing’s syndrome? J Clin Endocrinol Metab. 1999;84:2616–2622.
108. Sofat, N, Turner, J, Khoo, B, et al. An unusual case of Cushing’s syndrome: primary pigmented nodular adrenal dysplasia. Int J Clin Pract. 2000;54:269–271.
109. Aiba, M, Hirayama, A, Iri, H, et al. Primary adrenocortical micronodular dysplasia: enzyme histochemical and ultrastructural studies of two cases with a review of the literature. Hum Pathol. 1990;21:503–511.
110. Teding van Berkhout, F, Croughs, RJM, Kater, L, et al. Familial Cushing’s syndrome due to nodular adrenocortical dysplasia. A putative receptor-antibody disease? Clin Endocrinol. 1986;24:299–310.
111. Stratakis, CA, Carney, JA, Kirschner, LS, et al. Synaptophysin immunoreactivity in primary pigmented nodular adrenocortical disease: neuroendocrine properties of tumors associated with Carney complex. J Clin Endocrinol Metab. 1999;84:1122–1128.
112. D’Agata, R, Malozowski, S, Barkan, AL, et al. Steroid biosynthesis in human adrenal tumors. Horm Metab Res. 1987;19:38–388.
113. Weiss, LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol. 1984;8:163–169.
114. Hough, AJ, Hollifield, JW, Page, DL, et al. Prognostic factors in adrenal cortical tumors. A mathematical analysis of clinical and morphologic data. Am J Clin Pathol. 1979;72:390–399.
115. van Slooten, H, Schaberg, A, Smeenk, D, et al. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985;55:766–773.
116. Sasano, H, Suzuki, T, Moriya, T. Discerning malignancy in resected adrenocortical neoplasms. Endocr Pathol. 2001;12:397–406.
117. Tan, HS, Thai, AC, Nga, ME, et al. Development of ipsilateral adrenocortical carcinoma sixteen years after resection of an adrenal tumour causing Cushing’s syndrome. Ann Acad Med Singapore. 2005;34:271–274.
118. Klibanski, A, Stephen, AE, Greene, MF, et al. A 35-year-old pregnant woman with new hypertension. N Engl J Med. 2006;355:2237–2245.
119. Koch, CA, Pacák, K, Chrousos, GP. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab. 2002;87:5367–5384.
120. Brown, FM, Gaffey, TA, Wold, LE, et al. Myxoid neoplasms of the adrenal cortex: a rare histologic variant. Am J Surg Pathol. 2000;24:396–401.
121. Geramizadeh, B, Norouzzadeh, B, Bolandparvaz, S, et al. Functioning adrenocortical oncocytoma: a case report and review of the literature. Indian J Pathol Microbiol. 2008;51:237–239.
122. Hisamatsu, H, Sakai, H, Tsuda, S, et al. Combined adrenal adenoma and myelolipoma in a patient with Cushing’s syndrome: case report and review of the literature. Int J Urol. 2004;11:416–418.
123. Wieneke, JA, Thompson, LD, Heffess, CS. Corticomedullary mixed tumor of the adrenal gland. Ann Diagn Pathol. 2001;5:304–308.
124. Miettinen, M. Neuroendocrine differentiation in adrenocortical carcinoma. New immunohistochemical findings supported by electron microscopy. Lab Invest. 1992;66:169–174.
125. Haak, HR, Fleuren, GJ. Neuroendocrine differentiation of adrenocortical tumors. Cancer. 1995;75:860–864.
126. Geller, JL, Azer, PC, Weiss, LM, et al. Pigmented adrenocortical carcinoma: case report and review. Endocr Pathol. 2006;17:297–304.
127. Midorikawa, S, Hashimoto, S, Kuriki, M, et al. A patient with preclinical Cushing’s syndrome and excessive DHEA-S secretion having unilateral adrenal carcinoma and contralateral adenoma. Endocr J. 1999;46:59–66.
128. Venkatesh, S, Hickey, RC, Sellin, RV, et al. Adrenal cortical carcinoma. Cancer. 1989;64:765–769.
129. Gicquel, C, Leblond-Francillard, M, Bertagna, X, et al. Clonal analysis of human adrenocortical carcinomas and secreting adenomas. Clin Endocrinol. 1994;40:465–477.
130. Diaz-Cano, SJ, de Miguel, M, Blanes, A, et al. Clonality as expression of distinctive cell kinetics patters in nodular hyperplasia and adenomas of the adrenal cortex. Am J Pathol. 2000;156:311–319.
131. Bernard, MH, Sidhu, S, Berger, N, et al. A case report in favor of a multistep adrenocortical tumorigenesis. J Clin Endocrinol Metab. 2003;88:998–1001.
132. Stratakis, CA, Boikos, S. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. 2007;3:748–757.
133. Kjellman, M, Kallioniemi, OP, Karhu, R, et al. Genetic aberrations in adrenocortical tumors detected using comparative genomic hybridization correlate with tumor size and malignancy. Cancer Res. 1996;56:4219–4223.
134. Fogt, F, Vargas, MP, Zhuang, Z, et al. Utilization of molecular genetics in the differentiation between adrenal cortical adenomas and carcinoma. Hum Pathol. 1998;28:518–521.
135. Riberio, RC, Sandrini, F, Figueiredo, B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330–9335.
136. Beuschlein, F, Schulze, E, Mora, P, et al. Steroid 21-hydroxylase mutations and 21-hydroxylase messenger ribonucleic acid expression in human adrenocortical tumors. J Clin Endocrinol Metab. 1998;83:2585–2588.
137. Latronico, AC, Reincke, M, Bilharinho de Mendonça, B, et al. No evidence for oncogenic mutations in the adrenocorticotropin receptor gene in human adrenocortical neoplasms. J Clin Endocrinol Metab. 1995;80:875–877.
138. Syddall, HE, Baig, A, Malchoff, DM, et al. Impaired desensitization of a mutant adrenocorticotropin receptor associated with apparent constitutive activity. Mol Endocrinol. 2002;16:2746–2753.
139. Bertherat, J, Groussin, L, Sandrini, F, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003;63:5308–5319.
140. Bourdeau, I, Matyakhina, L, Stergiopoulos, SG, et al. 17q22–24 chromosomal losses and alterations of protein kinase A subunit expression and activity in adrenocorticotrophin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91:3626–3632.
141. Vincent-Dejean, C, Cazabat, L, Groussin, L, et al. Identification of a clinically homogeneous subgroup of benign cortisol-secreting adrenocortical tumors characterized by alterations of the protein kinase A (PKA) subunits and high PKA activity. Eur J Endocrinol. 2008;158:829–839.
142. Tissier, F, Cavard, C, Groussin, L, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7627.
143. Munro, LM, Kennedy, A, McNicol, AM. The expression of inhibin/activin subunits in the human adrenal cortex and its tumours. J Endocrinol. 1999;161:341–347.
144. Schulte, HM, Mengel, M, Heinze, M, et al. Complete sequencing and messenger ribonucleic acid expression analysis of the MEN1 gene in adrenal cancer. J Clin Endocrinol Metab. 2000;85:441–448.
145. Waldmann, J, Bartsch, DK, Kann, PH, et al. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbeck’s Arch Surg. 2007;392:437–443.
146. Alzahrani, AS, Al-Khaldi, N, Shi, Y, et al. Diagnosis by serendipity: Cushing syndrome attributable to cortisol-producing adrenal adenoma as the initial manifestation of multiple endocrine neoplasia type 1 due to a rare splicing site MEN1 gene mutation. Endocr Pract. 2008;14:595–602.
147. Weinstein, LS, Shenker, A, Gejman, OV, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695.
148. Boston, BA, Mandel, S, LaFranchi, SH, et al. Activating mutation in the stimulatory guanine nucleotide-binding protein in an infant with Cushing’s syndrome and nodular adrenal hyperplasia. J Clin Endocrinol Metab. 1994;79:890–893.
149. Dall’Asta, C, Ballarè, E, Mantovani, G, et al. Assessing the presence of abnormal regulation of cortisol secretion by membrane hormone receptors: in vivo and in vitro studies in patients with functioning and non-functioning adrenal adenoma. Horm Metab Res. 2004;36:578–583.
150. Bugalho, MJM, Li, X, Rao, CV, et al. Presence of a Gsα mutation in an adrenal tumor expressing LH/hCG receptor and clinically associated with Cushing’s syndrome. Gynecol Endocrinol. 2000;14:50–54.
151. Villares Fragoso, MCB, Domenice, S, Latronico, AC, et al. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of the GNAS1 gene. J Clin Endocrinol Metab. 2003;88:2147–2151.
152. Groen, EJ, Roos, A, Muntinghe, FL, et al. Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol. 2008;15:2439–2450.
153. Beuschlein, F, Reincke, M, Königer, M, et al. Cortisol producing adrenal adenoma: a new manifestation of Gardner’s syndrome. Endocrine Res. 2000;26:783–790.
154. Casey, M, Vaughan, CJ, He, J, et al. Mutations in the protein kinase A R1α regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest. 2000;106:R31–R38.
155. Kirschner, LS, Carney, JA, Pack, SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92.
156. Ogo, A, Haji, M, Ohashi, M, et al. Markedly increased expression of cytochrome P-450 17α-hydroxylase (P-450c17) mRNA in adrenocortical adenomas from patients with Cushing’s syndrome. Mol Cell Endocrinol. 1991;80:83–89.
157. Hornsby, PJ. Physiological and pathological effects of steroids on the function of the adrenal cortex. J Steroid Biochem. 1987;27:1161–1171.
158. Shibata, H, Suzuki, H, Ogishima, T, et al. Significance of steroidogenic enzymes in the pathogenesis of adrenal tumour. Acta Endocrinol (Copenh). 1993;128:235–242.
159. Sakai, Y, Yanase, T, Takayanagi, R, et al. High expression of cytochrome b5 in adrenocortical adenomas from patients with Cushing’s syndrome associated with high secretion of adrenal androgens. J Clin Endocrinol Metab. 1993;76:1286–1290.
160. Lamberts, SWJ, Zuiderwijk, J, Uitterlinden, P, et al. Characterization of adrenal autonomy in Cushing’s syndrome: a comparison between in vivo and in vitro responsiveness of the adrenal gland. J Clin Endocrinol Metab. 1990;70:192–199.
161. Sasano, H, Suzuki, T, Nagura, H, et al. Steroidogenesis in human adrenocortical carcinoma: biochemical activities, immunohistochemistry, and in situ hybridization of steroidogenic enzymes and histopathologic study in nine cases. Hum Pathol. 1993;24:397–404.
162. Barzon, L, Masi, G, Fincati, K, et al. Shift from Conn’s syndrome to Cushing’s syndrome in a recurrent adrenocortical carcinoma. Eur J Endocrinol. 2005;153:629–636.
163. Sasano, H. Localization of steroidogenic enzymes in adrenal cortex and its disorders. Endocr J. 1994;41:471–482.
164. Morita, H, Isomura, Y, Mune, T, et al. Plasma cortisol and cortisone concentrations in normal subjects and patients with adrenocortical disorders. Metabolism. 2004;53:89–94.
165. Mazzocchi, G, Aragona, F, Malendowicz, LK, et al. Cortisol-secreting adrenal adenomas express 11beta-hydroxysteroid dehydrogenase type-2 gene yet possess low 11beta-HSD2 activity. J Investig Med. 2001;49:191–194.
166. Imai, T, Sarkar, D, Shibata, A, et al. Expression of adrenocorticotropin receptor gene in adrenocortical adenomas from patients with Cushing syndrome: possible contribution for the autonomous production of cortisol. Ann Surg. 2001;234:85–91.
167. Reincke, M, Beuschlein, F, Menig, G, et al. Localization and expression of adrenocorticotropic hormone receptor mRNA in normal and neoplastic human adrenal cortex. J Endocrinol. 1998;156:415–423.
168. Penhoat, A, Jaillard, C, Saez, JM. Corticotropin positively regulates its own receptors and cAMP response in cultured bovine adrenal cells. Proc Natl Acad Sci USA. 1989;86:4978–4981.
169. Beuschlein, F, Fassnacht, M, Klink, A, et al. ACTH receptor expression, regulation and role in adrenocortical tumor formation. Eur J Endocrinol. 2001;144:199–206.
170. Ishizuka, T, Daidoh, H, Morita, H, et al. ACTH-induced cortisol secretion is mediated by cAMP and PKC in various adrenocortical adenomas. Endocr J. 1997;44:661–670.
171. Schorr, I, Ney, RL. Abnormal hormone responses of an adrenocortical cancer adenyl cyclase. J Clin Invest. 1971;50:1295–1300.
172. Hornsby, PJ. Regulation of adrenocortical cell proliferation in culture. Endocrine Res. 1984;10:259–281.
173. Mesiano, S, Jaffe, RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocrine Rev. 1997;18:378–403.
174. Fassnacht, M, Hahner, S, Hansen, IA, et al. N-terminal proopiomelanocortin acts as a mitogen in adrenocortical tumor cells and decreases adrenal steroidogenesis. J Clin Endocrinol Metab. 2003;88:2171–2179.
175. Coll, AP, Fassnacht, M, Klammer, S, et al. Peripheral administration of the N-terminal pro-opiomelanocortin fragment 1–28 to Pomc-/- mice reduces food intake and weight but does not affect adrenal growth or corticosterone secretion. J Endocrinol. 2006;190:515–525.
176. Darbeida, H, Durand, P. Mechanisms of glucocorticoid enhancement of the responsiveness of ovine adrenocortical cells to adrenocorticotropin. Biochem Biophys Res Commun. 1990;166:1183–1191.
177. Loose, DS, Do, YS, Chen, TL, et al. Demonstration of glucocorticoid receptors in the adrenal cortex: evidence for a direct dexamethasone suppressive effect on the rat adrenal gland. Endocrinology. 1980;107:137–146.
178. Kontula, K, Pomoell, UM, Gunsalus, GL, et al. Glucocorticoid receptors and responsiveness of normal and neoplastic human adrenal cortex. J Clin Endocrinol Metab. 1985;60:283–289.
179. Kirschner, MA, Powell, RD, Jr., Lipsett, MB. Cushing’s syndrome: nodular cortical hyperplasia of adrenal glands with clinical and pathological features suggesting adrenocortical tumor. J Clin Endocrinol Metab. 1964;24:947–955.
180. Malchoff, CD, Rosa, J, DeBold, RC, et al. Adrenocorticotropin-independent bilateral macronodular adrenal hyperplasia: an unusual cause of Cushing’s syndrome. J Clin Endocrinol Metab. 1989;68:855–860.
181. Shinojima, H, Kakizaki, H, Usuki, T, et al. Clinical and endocrinological features of adrenocorticotropic hormone-independent bilateral macronodular adrenocortical hyperplasia. J Urol. 2002;166:1639–1642.
182. Swain, JM, Grant, CS, Schlinkert, RT, et al. Corticotropin-independent macronodular adrenal hyperplasia: a clinicopathologic correlation. Arch Surg. 1998;133:541–545.
183. Verma, A, Mohan, S, Gupta, A. ACTH-independent macronodular adrenal hyperplasia: imaging findings of a rare condition—a case report. Abdom Imaging. 2008;33:225–229.
184. Morioka, M, Ohashi, Y, Watanabe, H, et al. ACTH-independent macronodular adrenocortical hyperplasia (AIMAH): report of two cases and the analysis of steroidogenic activity in adrenal nodules. Endocr J. 1997;44:65–72.
185. Josse, RG, Bear, R, Kovacs, K, et al. Cushing’s syndrome due to unilateral nodular adrenal hyperplasia: a new pathophysiological entity? Acta Endocrinol (Copenh). 1980;93:495–504.
186. Bourdeau, I, D’Amour, P, Hamet, P, et al. Aberrant membrane hormone receptors in incidentally discovered bilateral macronodular adrenal hyperplasia with subclinical Cushing’s syndrome. J Clin Endocrinol Metab. 2001;86:5534–5540.
187. Sasao, T, Itoh, N, Sato, Y, et al. Subclinical Cushing syndrome due to adrenocorticotropic hormone-independent macronodular adrenocortical hyperplasia: changes in plasma cortisol levels during long-term follow-up. Urology. 55, 2000. [145x–145xii].
188. Ohashi, A, Yamada, Y, Sakaguchi, K, et al. A natural history of adrenocorticotropin-independent adrenal macronodular hyperplasia (AIMAH) from preclinical to clinically overt Cushing’s syndrome. Endocr J. 2001;48:677–683.
189. Doppman, JL, Chrousos, GP, Papanicolaou, DA, et al. Adrenocorticotropin-independent macronodular adrenal hyperplasia: an uncommon cause of primary adrenal hypercortisolism. Radiology. 2000;216:797–802.
190. Smals, AGH, Pieters, GF, van Haelst, UJ, et al. Macronodular adrenocortical hyperplasia in long-standing Cushing’s disease. J Clin Endocrinol Metab. 1984;58:25–31.
191. Bourdeau, I, Antonini, SRR, Lacroix, A, et al. Gene array analysis of macronodular adrenal hyperplasia confirms clinical heterogeneity and identifies several candidate genes as molecular mediators. Oncogene. 2004;23:1575–1585.
192. Lampron, A, Bourdeau, I, Hamet, P, et al. Whole genome expression profiling of glucose-dependent insulinotropic peptide (GIP)- and adrenocorticotropin-dependent adrenal hyperplasias reveals novel targets for the study of GIP-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2006;91:3611–3618.
193. Hermus, AR, Pieters, GF, Smals, AGH, et al. Transition from pituitary-dependent to adrenal-dependent Cushing’s syndrome. N Engl J Med. 1988;318:966–970.
194. Hocher, B, Bähr, V, Dormüller, S, et al. Hypercortisolism with non-pigmented micronodular adrenal hyperplasia: transition from pituitary-dependent to adrenal-dependent Cushing’s syndrome. Acta Endocrinol (Copenh). 1993;128:120–125.
195. Vrezas, I, Willenberg, HS, Mansmann, G, et al. Ectopic adrenocorticotropin (ACTH) and corticotropin-releasing hormone (CRH) production in the adrenal gland: basic and clinical aspects. Microsc Res Tech. 2003;61:308–314.
196. Hiroi, N, Chrousos, GP, Kohn, B, et al. Adrenocortical-pituitary hybrid tumor causing Cushing’s syndrome. J Clin Endocrinol Metab. 2001;86:2631–2637.
197. Lefebvre, H, Duparc, C, Chartrel, N, et al. Intraadrenal adrenocorticotropin production in a case of bilateral macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab. 2003;88:3035–3042.
198. Feelders, RA, Lamberts, SWJ, Hofland, LJ, et al. Luteinizing hormone (LH)-responsive Cushing’s syndrome: the demonstration of LH receptor messenger ribonucleic acid in hyperplastic adrenal cells, which respond to chorionic gonadotropin and serotonin agonists in vitro. J Clin Endocrinol Metab. 2003;88:230–237.
199. Suri, D, Alonso, M, Weiss, RE. A case of ACTH-independent bilateral macronodular adrenal hyperplasia and severe congestive heart failure. J Endocrinol Invest. 2006;29:940–946.
200. Antonini, SRR, Baldacchino, V, Tremblay, J, et al. Expression of ACTH receptor pathway genes in glucose-dependent insulinotrophic peptide (GIP)-dependent Cushing’s syndrome. Clin Endocrinol. 2006;64:29–36.
201. Mazzucco, TL, Thomas, M, Martinie, M, et al. Cellular and molecular abnormalities of a macronodular adrenal hyperplasia causing beta-blocker-sensitive Cushing’s syndrome. Arq Bras Endocrinol Metab. 2007;51:1452–1462.
202. Hinshaw, HT, Ney, RL. Abnormal control in the neoplastic adrenal cortex. In: McKerns KW, ed. Hormones and Cancer. New York: Academic Press; 1974:309–327.
203. Reznik, Y, Allali-Zerah, V, Chayvialle, JA, et al. Food-dependent Cushing’s syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med. 1992;327:981–986.
204. Lacroix, A, Bolté, E, Tremblay, J, et al. Gastric inhibitory polypeptide-dependent cortisol hypersecretion—a new cause of Cushing’s syndrome. N Engl J Med. 1992;327:974–980.
205. Reznik, Y, Lefebvre, H, Rohmer, V, et al. Aberrant adrenal sensitivity to multiple ligands in unilateral incidentaloma with subclinical autonomous cortisol hypersecretion: a prospective clinical study. Clin Endocrinol. 2004;61:311–319.
206. Suzuki, S, Uchida, D, Koide, H, et al. Hyperresponsiveness of adrenal grand to vasopressin resulting in enhanced plasma cortisol in patients with adrenal nodules. Peptides. 2007;29:1767–1772.
207. Mircescu, H, Jilwan, J, N’Diaye, N, et al. Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing’s syndrome? J Clin Endocrinol Metab. 2000;85:3531–3536.
208. Groussin, L, Perlemoine, K, Contesse, V, et al. The ectopic expression of the gastric inhibitory polypeptide receptor is frequent in adrenocorticotropin-independent bilateral macronodular hyperplasia, but rare in unilateral tumors. J Clin Endocrinol Metab. 2002;87:1980–1985.
209. Mazzucco, TL, Chabre, O, Sturm, N, et al. Ectopic expression of the gastric inhibitory polypeptide receptor gene is a sufficient genetic event to induce benign adrenocortical tumor in a xenotransplantation model. Endocrinology. 2006;147:782–790.
210. Mazzucco, TL, Chabre, O, Feige, JJ, et al. Aberrant expression of human luteinizing hormone receptor by adrenocortical cells is sufficient to provoke both hyperplasia and Cushing’s syndrome features. J Clin Endocrinol Metab. 2006;91:196–203.
211. Miyamura, N, Tsutsumi, A, Senokuchi, H, et al. A case of ACTH-independent macronodular adrenal hyperplasia: simultaneous expression of several aberrant hormone receptors in the adrenal gland. Endocr J. 2003;50:333–340.
212. Louiset, E, Contesse, V, Groussin, L, et al. Expression of serotonin7 receptors and coupling of ectopic receptors to protein kinase A and ionic currents in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab. 2006;91:4578–4586.
213. Bertherat, J, Contesse, V, Louiset, E, et al. In vivo and in vitro screening for illegitimate receptors in adrenocorticotropin-independent macronodular adrenal hyperplasia causing Cushing’s syndrome: identification of two cases of gonadotropin/gastric inhibitory polypeptide-dependent hypercortisolism. J Clin Endocrinol Metab. 2005;90:1302–1310.
214. N’Diaye, N, Tremblay, J, Hamet, P, et al. Adrenocortical overexpression of gastric inhibitory polypeptide receptor underlies food-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1998;83:2781–2785.
215. Lebrethon, MC, Avallet, O, Reznik, Y, et al. Food-dependent Cushing’s syndrome: characterization and functional role of gastric inhibitory polypeptide receptor in the adrenal of three patients. J Clin Endocrinol Metab. 1998;83:4514–4519.
216. Swords, FM, Aylwin, SJB, Perry, L, et al. The aberrant expression of the gastric inhibitory polypeptide (GIP) receptor in adrenal hyperplasia: does chronic adrenocorticotropin exposure stimulate up-regulation of GIP receptors in Cushing’s disease? J Clin Endocrinol Metab. 2005;90:3009–3016.
217. Antonini, SRR, N’Diaye, N, Baldacchino, V, et al. Analysis of the putative regulatory region of the gastric inhibitory polypeptide receptor gene in food-dependent Cushing’s syndrome. J Steroid Biochem Mol Biol. 2004;91:171–177.
218. Baldacchino, V, Oble, S, Décarie, PO, et al. The Sp transcription factors are involved in the cellular expression of the human glucose-dependent insulinotropic polypeptide receptor gene and overexpressed in adrenals of patients with Cushing’s syndrome. J Mol Endocrinol. 2005;35:61–71.
219. Tsagarakis, S, Tsigos, C, Vassiliou, V, et al. Food-dependent androgen and cortisol secretion by a gastric inhibitory polypeptide-receptor expressive adrenocortical adenoma leading to hirsutism and subclinical Cushing’s syndrome: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86:583–589.
220. Albiger, N, Occhi, G, Mariniello, B, et al. Food-dependent Cushing’s syndrome: from molecular characterization to therapeutical results. Eur J Endocrinol. 2007;157:771–778.
221. Gwinup, G, Steinberg, T, King, CG, et al. Vasopressin-induced ACTH secretion in man. J Clin Endocrinol Metab. 1967;27:927–930.
222. Perraudin, V, Delarue, C, Lefebvre, H, et al. Vasopressin stimulates cortisol secretion from human adrenocortical tissue through activation of V1 receptors. J Clin Endocrinol Metab. 1993;76:1522–1528.
223. Hensen, J, Hader, O, Bähr, V, et al. Effects of incremental infusions of arginine vasopressin on adrenocorticotropin and cortisol secretion in man. J Clin Endocrinol Metab. 1988;66:668–671.
224. Lacroix, A, Tremblay, J, Touyz, R, et al. Abnormal adrenal and vascular responses to vasopressin mediated by a V1-vasopressin receptor in a patients with adrenocorticotropin-independent macronodular adrenal hyperplasia, Cushing’s syndrome, and orthostatic hypotension. J Clin Endocrinol Metab. 1997;82:2414–2422.
225. Perraudin, V, Delarue, C, De Keyzer, Y, et al. Vasopressin-responsive adrenocortical tumor in a mild Cushing’s syndrome: in vivo and in vitro studies. J Clin Endocrinol Metab. 1995;80:2661–2667.
226. Daidoh, H, Morita, H, Hanafusa, J, et al. In vivo and in vitro effects of AVP and V1a receptor antagonist on Cushing’s syndrome due to ACTH-independent bilateral macronodular adrenocortical hyperplasia. Clin Endocrinol. 1998;48:403–409.
227. Mune, T, Murase, H, Yamakita, N, et al. Eutopic overexpression of vasopressin V1a receptor in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87:5706–5713.
228. Vezzosi, D, Cartier, D, Régnier, C, et al. Familial adrenocorticotropin-independent macronodular adrenal hyperplasia with aberrant serotonin and vasopressin adrenal receptors. Eur J Endocrinol. 2007;156:21–31.
229. Louiset, E, Contesse, V, Groussin, L, et al. Expression of vasopressin receptors in ACTH-independent macronodular bilateral adrenal hyperplasia causing Cushing’s syndrome: molecular, immunohistochemical and pharmacological correlates. J Endocrinol. 2008;196:1–9.
230. Lee, S, Hwang, R, Lee, J, et al. Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin Endocrinol. 2006;63:625–630.
231. Nakamura, Y, Son, Y, Kohno, Y, et al. Case of adrenocorticotropic hormone-independent macronodular adrenal hyperplasia with possible adrenal hypersensitivity to angiotensin II. Endocrine. 2001;15:57–61.
232. Pabon, JE, Li, X, Lei, ZM, et al. Novel presence of luteinizing hormone/chorionic gonadotropin receptors in human adrenal glands. J Clin Endocrinol Metab. 1996;81:2397–2400.
233. Kero, J, Poutanen, M, Zhang, FP, et al. Elevated luteinizing hormone induces expression of its receptor and promotes steroidogenesis in the adrenal cortex. J Clin Invest. 2000;105:633–641.
234. Schoemaker, NJ, Teerds, KJ, Mol, JA, et al. The role of luteinizing hormone in the pathogenesis of hyperadrenocorticism in neutered ferrets. Mol Cell Endocrinol. 2002;197:117–125.
235. Hirata, Y, Uchihashi, M, Sueoka, S, et al. Presence of β-adrenergic receptors on human adrenocortical cortisol-producing adenomas. J Clin Endocrinol Metab. 1981;53:953–957.
236. Lacroix, A, Tremblay, J, Rousseau, G, et al. Propranolol therapy for ectopic β-adrenergic receptors in adrenal Cushing’s syndrome. N Engl J Med. 1997;337:1429–1434.
237. Lefebvre, H, Contesse, V, Delarue, C, et al. Effect of the serotonin-4 receptor agonist zacopride on aldosterone secretion from the human adrenal cortex: in vivo and in vitro studies. J Clin Endocrinol Metab. 1993;77:1662–1666.
238. Cartier, D, Lihrmann, I, Parmentier, F, et al. Overexpression of serotonin-4 receptors in cisapride-responsive adrenocorticotropin-independent bilateral macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab. 2003;88:248–254.
239. Mannelli, M, Ferruzzi, P, Luciani, P, et al. Cushing’s syndrome in a patient with bilateral macronodular adrenal hyperplasia responding to cisapride: an in vivo and in vitro study. J Clin Endocrinol Metab. 2003;88:4616–4622.
240. Willenberg, HS, Stratakis, CA, Marx, C, et al. Brief report: Aberrant interleukin-1 receptors in a cortisol- secreting adrenal adenoma causing Cushing’s syndrome. N Engl J Med. 1998;339:27–31.
241. Hanley, N, Williams, BC, Nicol, M, et al. Interleukin-1 beta stimulates growth of adrenocortical cells in primary culture. J Mol Endocrinol. 1992;8:131–136.
242. Caticha, O, Odell, WD, Wilson, DE, et al. Estradiol stimulates cortisol production by adrenal cells in estrogen-dependent primary adrenocortical nodular dysplasia. J Clin Endocrinol Metab. 1993;77:494–497.
243. Findlay, JC, Sheeler, LR, Engeland, WC, et al. Familial adrenocorticotropin-independent Cushing’s syndrome with bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 1993;76:189–191.
244. Kubo, N, Onoda, N, Ishikawa, T, et al. Simultaneous bilateral laparoscopic adrenalectomy for adrenocorticotropic hormone-independent macronodular adrenal hyperplasia: report of a case. Surg Today. 2006;36:642–646.
245. Kageyama, Y, Ishizaka, K, Iwashina, M, et al. A case of ACTH-independent bilateral macronodular adrenal hyperplasia successfully treated by subtotal resection of the adrenal glands: four-year follow-up. Endocr J. 2002;49:227–229.
246. Iacobone, M, Albiger, N, Scaroni, C, et al. The role of unilateral adrenalectomy in ACTH-independent macronodular adrenal hyperplasia (AIMAH). World J Surg. 2008;32:882–889.
247. Omori, N, Nomura, K, Omori, K, et al. Rational, effective metyrapone treatment of ACTH-independent bilateral macronodular adrenocortical hyperplasia (AIMAH). Endocr J. 2001;48:665–669.
248. Nagai, M, Narita, I, Omori, K, et al. Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia treated with mitotane. Intern Med. 1999;38:969–973.
249. Groussin, L, Jullian, E, Perlemoine, K, et al. Mutations of the PRKAR1A gene in Cushing’s syndrome due to sporadic primary pigmented nodular adrenocortical disease. J Clin Endocrinol Metab. 2002;27:4324–4329.
250. Schulz, S, Redlich, A, Köppe, I, et al. Carney complex—an unexpected finding during puerperium. Gynecol Obstet Invest. 2001;51:211–213.
251. Danoff, A, Jormark, S, Lorber, D, et al. Adrenocortical micronodular dysplasia, cardiac myxomas, lentigines, and spindle cell tumors. Report of a kindred. Arch Intern Med. 1987;147:443–448.
252. Young, WF, Jr., Carney, JA, Musa, BU, et al. Familial Cushing’s syndrome due to primary pigmented nodular adrenocortical disease. N Engl J Med. 1989;321:1659–1664.
253. Stratakis, CA, Carney, JA, Lin, JP, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996;97:699–706.
254. Oelkers, WKH, Bahr, V, Hensen, J, et al. Primary adrenocortical micronodular adenomatosis causing Cushing’s syndrome. Effects of ketoconazole on steroid production and in vitro performance of adrenal cells. Acta Endocrinol (Copenh). 1986;113:370–377.
255. Bourdeau, I, Lacroix, A, Schürch, W, et al. Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88:3931–3938.
256. Casey, M, Mah, C, Merliss, A, et al. Identification of a novel genetic locus for familial cardiac myxomas and Carney complex. Circulation. 1998;98:2560–2566.
257. Matyakhina, L, Pack, SD, Kirschner, LS, et al. Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet. 2003;40:268–277.
258. Horvath, A, Bossis, I, Giatzakis, C, et al. Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res. 2008;14:388–395.
259. Greene, EL, Horvath, A, Nesterova, M, et al. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat. 2008;29:633–639.
260. Amieux, PS, Cummings, DE, Motamed, K, et al. Compensatory regulation of RIalpha protein levels in protein kinase A mutant mice. J Biol Chem. 1997;272:3993–3998.
261. Groussin, L, Kirschner, LS, Vincent-Dejean, C, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet. 2002;71:1433–1442.
262. Groussin, L, Horvath, A, Jullian, E, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab. 2006;91:1943–1949.
263. Horvath, A, Giatzakis, C, Robinson-White, A, et al. Adrenal hyperplasia and adenomas are associated with inhibition of phosphodiesterase 11A in carriers of PDE11A sequence variants that are frequent in the population. Cancer Res. 2006;66:11571–11575.
264. Horvath, A, Mericq, MV, Stratakis, CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med. 2008;358:750–752.
265. Limone, P, Maccario, M, Vigliani, R, et al. Primary pigmented micronodular disease of the adrenals. J Endocrinol Invest. 1990;13:171–175.
266. Powell, AC, Stratakis, CA, Patronas, NJ, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143:750–758.
267. Sarlis, NJ, Chrousos, GP, Doppman, JL, et al. Primary pigmented nodular adrenocortical disease: re-evaluation of a patient with Carney complex 27 years after unilateral adrenalectomy. J Clin Endocrinol Metab. 1996;82:1274–1278.
268. Washecka, R, Dresner, MI, Honda, SAA. Testicular tumors in Carney’s complex. J Urol. 2002;167:1299–1302.
269. Pecori Giraldi, F, Fatti, LM, Bertola, G, et al. Carney’s complex with acromegaly as the leading clinical condition. Clin Endocrinol. 2008;68:322–324.
270. Pack, SD, Kirschner, LS, Pak, E, et al. Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “syndrome of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Clin Endocrinol Metab. 2000;85:3860–3865.
271. Watson, JC, Stratakis, CA, Bryant-Greenwood, PK, et al. Neurosurgical complications of Carney complex. J Neurosurg. 2000;92:413–418.
272. Ogo, A, Haji, M, Natori, S, et al. Acromegaly with hyperprolactinemia developed after bilateral adrenalectomy in a patient with Cushing’s syndrome due to adrenocortical nodular hyperplasia. Endocr J. 1993;40:17–25.
273. Raff, SB, Carney, JA, Krugman, D, et al. Prolactin secretion abnormalities in patients with the “syndrome of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Pediatr Endocrinol Metab. 2000;13:373–379.
274. Stratakis, CA, Courkoutsakis, NA, Abati, A, et al. Thyroid gland abnormalities in patients with the “syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J Clin Endocrinol Metab. 1997;82:2037–2043.
275. Stratakis, CA, Papageorgiou, T, Premkumar, A, et al. Ovarian lesions in Carney complex: clinical genetics and possible predisposition to malignancy. J Clin Endocrinol Metab. 2000;85:4359–4366.
276. Edwards, A, Bermudez, C, Piwonka, G, et al. Carney’s syndrome: complex myxomas. Report of four cases and review of the literature. Cardiovasc Surg. 2002;10:264–275.
277. Stratakis, CA, Kirschner, LS, Carney, JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–4046.
278. Carney, JA, Toorkey, BC. Ductal adenoma of the breast with tubular features. A probable component of the complex of myxomas, spotty pigmentation, endocrine overactivity, and schwannomas. Am J Surg Pathol. 1991;15:722–731.
279. Mateus, C, Palangié, A, Franck, N, et al. Heterogeneity of skin manifestations in patients with Carney complex. J Am Acad Dermatol. 2008;59:801–810.
280. Carney, JA, Boccon-Gibod, L, Jarka, DE, et al. Osteochondromyxoma of the bone. Am J Surg Pathol. 2001;25:164–176.
281. Sutter, JA, Grimberg, A. Adrenocortical tumors and hyperplasias in childhood—etiology, genetics, clinical presentation and therapy. Ped Endocrinol Rev. 2006;4:32–39.
282. Sandrini, R, Riberio, RC, DeLacerda, L. Childhood adrenocortical tumors. J Clin Endocrinol Metab. 1997;82:2027–2031.
283. Sbragia, L, Oliveria-Filho, AG, Vassallo, J, et al. Adrenocortical tumors in Brazilian children. Arch Pathol Lab Med. 2005;129:1127–1131.
284. Wieneke, JA, Thompson, LDR, Heffess, CS. Adrenal cortical neoplasms in the pediatric population. Am J Surg Pathol. 2003;17:867–881.
285. Teinturier, C, Pauchard, MS, Brugières, L, et al. Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Med Pediatr Oncol. 1999;32:106–111.
286. Storr, HL, Chan, LF, Grossman, AB, et al. Paediatric Cushing’s syndrome: epidemiology, investigation and therapeutic advances. Trends Endocrinol Metab. 2007;18:167–174.
287. Odagiri, E, Ishiwatari, N, Abe, Y, et al. Hypercortisolism and the resistance to dexamethasone suppression during gestation. Endocrinol Jpn. 1988;35:685–690.
288. Lindsay, JR, Jonklaas, J, Oldfield, EH, et al. Cushing’s syndrome during pregnancy: personal experience and review of the literature. J Clin Endocrinol Metab. 2005;90:3077–3083.
289. Bianco, C, Maqueda, E, Rubio, JA, et al. Cushing’s syndrome during pregnancy secondary to adrenal adenoma: metyrapone treatment and laparoscopic adrenalectomy. J Endocrinol Invest. 2006;29:164–167.
290. Imai, T, Kikumori, T, Funahashi, H, et al. Surgical management of Cushing’s syndrome. Biomed & Pharmacother. 2000;54(Suppl 1):140–145.
291. Tung, SC, Wang, PW, Huang, TL, et al. Bilateral adrenocortical adenomas causing ACTH-independent Cushing’s syndrome at different periods: a case report and discussion of corticosteroid replacement therapy following bilateral adrenalectomy. J Endocrinol Invest. 2004;27:375–379.
292. Minowada, S, Fujimura, T, Takahashi, N, et al. Computed tomography-guided percutaneous acetic acid injection therapy for functioning adrenocortical adenoma. J Clin Endocrinol Metab. 2003;88:5814–5817.
293. Ueno, K, Nakajo, M, Miyazono, N, et al. Transcatheter adrenal arterial embolization of cortisol-producing tumors. Acta Radiol. 1999;40:100–103.
294. Arima, K, Yamakado, K, Suzuki, R, et al. Image-guided radiofrequency ablation for adrenocortical adenoma with Cushing syndrome: outcomes after mean follow-up of 33 months. Urology. 2007;70:407–411.
295. Disick, GIS, Munver, R. Adrenal-preserving minimally invasive surgery: update on the current status of laparoscopic partial adrenalectomy. Curr Urol Rep. 2008;9:67–72.
296. Porterfield, JR, Thompson, GB, Young, WF, Jr., et al. Surgery for Cushing’s syndrome: an historical review and recent ten-year experience. World J Surg. 2008;32:659–677.
297. Porpiglia, F, Fiori, C, Bovio, S, et al. Bilateral adrenalectomy for Cushing’s syndrome: a comparison between laparoscopy and open surgery. J Endocrinol Invest. 2004;27:654–658.
298. Doherty, GM, Nieman, LK, Cutler, GB, Jr., et al. Time to recovery of the hypothalamic-pituitary-adrenal axis after curative resection of adrenal tumors in patients with Cushing’s syndrome. Surgery. 1990;108:1085–1090.
299. Schulick, RD, Brennan, MF. Long-term survival after complete resection and repeat resection in patients with adrenocortical carcinoma. Ann Surg Oncol. 1999;6:719–726.
300. Andrioli, M, Pecori Giraldi, F, De Martin, M, et al. Therapies for adrenal insufficiency. Exp Opin Ther Patents. 2007;17:1323–1329.
301. Pecori Giraldi, F, Cavagnini, F. Advances in the medical management of Cushing’s syndrome. Expert Opin Pharmacother. 2008;9:1–11.
302. Ojima, M, Saitoh, M, Itoh, N, et al. The effects of o,p′-DDD on adrenal steroidogenesis and hepatic steroid metabolism. Nippon Naibunpi Gakkai Zasshi. 1985;61:168–178.
303. Robinson, BG, Hales, IB, Henniker, AJ, et al. The effect of o,p′-DDD on adrenal steroid replacement therapy requirements. Clin Endocrinol. 1987;27:437–444.
304. Bates, SE, Shieh, CY, Mickely, LA, et al. Mitotane enhances cytotoxicity of chemotherapy in cell lines expression a multidrug resistance gene (mdr-1/P-glycoprotein) which is also expressed by adrenocortical carcinomas. J Clin Endocrinol Metab. 1991;73:18–29.
305. Leiba, S, Weinstein, R, Shindel, B, et al. The protracted effect of op’-DDD in Cushing’s disease and its impact on adrenal morphogenesis of young human embryo. Ann Endocrinol (Paris). 1989;50:49–53.
306. Kasperlik-Zaluska, AA. Clinical results of the use of mitotane for adrenocortical carcinoma. Braz J Med Biol Res. 2000;33:1191–1196.
307. Terzolo, M, Angeli, A, Fassnacht, M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372–2380.
308. Abiven, G, Coste, J, Groussin, L, et al. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91:2650–2655.
309. van Slooten, H, Moolenaar, AJ, van Seters, AP, et al. The treatment of adrenocortical carcinoma with o,p′-DDD: prognostic implications of serum level monitoring. Eur J Cancer Clin Oncol. 1984;20:47–53.
310. Berruti, A, Ferrero, A, Sperone, P, et al. Emerging drugs for adrenocortical carcinoma. Expert Opin Emerging Drugs. 2008;13:497–509.
311. Ilias, I, Alevizaki, M, Phlippou, G, et al. Sustained remission of metastatic adrenal carcinoma during long-term administration of low-dose mitotane. J Endocrinol Invest. 2001;24:532–535.
312. Daffara, F, De Francia, S, Reimondo, G, et al. Prospective evaluation of mitotane toxicity in adrenocortical cancer patients treated adjuvantly. Endocr Relat Cancer. 2008;15:1043–1053.
313. Sonino, N. The use of ketoconazole as an inhibitor of steroid production. N Engl J Med. 1987;317:812–818.
314. Castinetti, F, Morange, I, Jaquet, P, et al. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol. 2008;158:91–99.
315. Engelhardt, D, Weber, MM. Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J Steroid Biochem Mol Biol. 1994;49:261–267.
316. Contreras, P, Rojas, A, Biagini, L, et al. Regression of metastatic adrenal carcinoma during palliative ketoconazole treatment. Lancet. 1985;II:151–152.
317. Riedl, M, Maier, C, Zettinig, G, et al. Long term control of hypercortisolism with fluconazole: case report and in vitro studies. Eur J Endocrinol. 2006;154:519–524.
318. Gower, DB. Modifiers of steroid-hormone metabolism: a review of their chemistry, biochemistry and clinical applications. J Steroid Biochem. 1974;5:501–523.
319. Fassnacht, M, Hahner, S, Beuschlein, F, et al. New mechanisms of adrenostatic compounds in a human adrenocortical cancer cell line. Eur J Clin Invest. 2000;30:76–82.
320. Verhelst, JA, Trainer, PJ, Howlett, TA, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing’s syndrome. Clin Endocrinol. 1991;35:169–178.
321. Obinata, D, Yamaguchi, K, Hirano, D, et al. Preoperative management of Cushing’s syndrome with metyrapone for severe psychiatric disturbances. Int J Urol. 2008;15:361–362.
322. Connell, JMC, Cordiner, J, Davies, DL, et al. Pregnancy complicated by Cushing’s syndrome: potential hazard of metyrapone therapy. Case report. Br J Obstet Gynaecol. 1985;92:1192–1195.
323. Gillis, D, Rösler, A, Hannon, TS, et al. Prolonged remission of severe Cushing syndrome without adrenalectomy in an infant with McCune-Albright syndrome. J Pediatr. 2008;152:882–884.
324. Schteingart, DE, Cash, R, Conn, JW. Aminoglutethimide and metastatic adrenal cancer. Maintained reversal (six months) of Cushing’s syndrome. JAMA. 1966;198:1007–1010.
325. Johanssen, S, Allolio, B. Mifepristone (RU 486) in Cushing’s syndrome. Eur J Endocrinol. 2007;157:561–569.