[level-membership-for-pulmolory-and-respiratory-category]28
Acute Respiratory Distress Syndrome
This chapter continues the discussion of respiratory failure with more detailed consideration of one important type of acute respiratory failure: acute respiratory distress syndrome (ARDS). This entity was initially called adult respiratory distress syndrome, but it is not limited to adults, so acute rather than adult is now used to describe it. ARDS represents a major form of hypoxemic respiratory failure. Its clinical and pathophysiologic features differ considerably from those noted for acute-on-chronic respiratory failure. ARDS is characterized by the presence of severe arterial hypoxemia and diffuse bilateral pulmonary infiltrates not due exclusively to cardiogenic or hydrostatic causes. The full criteria for establishing the diagnosis of ARDS are shown in Table 28-1. This chapter describes in detail each of these criteria and the associated pathology and pathophysiology.

This chapter first considers the dynamics of fluid transfer between the pulmonary vessels and alveolar interstitium, because any alteration in this process is important in the pathogenesis of ARDS. Next is an outline of the many types of injury that can result in ARDS and some of the theories proposed to explain how such a diverse group of disorders can produce this syndrome. Following is a discussion of the pathologic, pathophysiologic, and clinical consequences. The chapter concludes with a general approach to treatment. More specific details about support of gas exchange are provided in Chapter 29.
Physiology of Fluid Movement in Alveolar Interstitium
Despite the diverse group of disorders that can cause ARDS, the net result of the syndrome is the same: a disturbance in the normal barrier that limits leakage of fluid out of the pulmonary capillaries and into the pulmonary parenchyma. Before a discussion of some of the theories explaining how this barrier is damaged, a brief consideration of the determinants of fluid transport among the pulmonary vessels, interstitial space, and alveolar lumen may be helpful. The pulmonary parenchyma (Fig. 28-1) consists of (1) small vessels coursing through the alveolar walls, which for simplicity are referred to as the pulmonary capillaries; (2) pulmonary capillary endothelium, the lining cells that normally limit but do not completely prevent fluid movement out of the capillaries; (3) pulmonary interstitium, which is considered here as the alveolar wall exclusive of vessels and the epithelial cells lining the alveolar lumen; (4) lymphatic channels, which are found mainly in perivascular connective tissue in the lungs; (5) alveolar epithelial cells, which line the surface of the alveolar lumen; and (6) alveolar lumen or alveolar space.
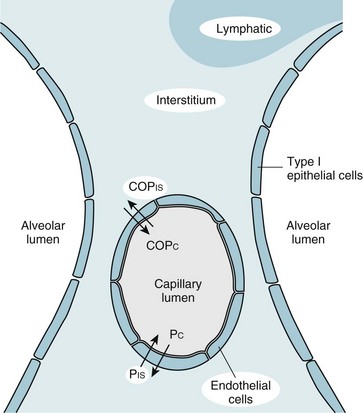
Movement of fluid out of the pulmonary capillaries and into the interstitial space is determined by a number of factors, including the hydrostatic pressures in the vessels and the pulmonary interstitium, the colloid osmotic pressures in these same two compartments, and the permeability of the endothelium. The effect of these factors in determining fluid transport is summarized in the Starling equation, examined in Chapter 15 with regard to fluid transport across the pleural space. The Starling equation is given as Equation 28-1:
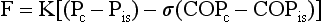
where F = fluid movement; Pc and Pis = pulmonary capillary and interstitial hydrostatic pressure, respectively; COPc and COPis = pulmonary capillary and interstitial colloid osmotic (oncotic) pressure, respectively; K = filtration coefficient; and σ = reflection coefficient (measure of permeability of endothelium for protein).
Two Mechanisms of Fluid Accumulation
In practice, the forces described in the Starling equation become altered in two main ways, producing interstitial and often alveolar edema (Table 28-2). The first occurs when hydrostatic pressure within the pulmonary capillaries (Pc) is increased, generally as a consequence of elevated left ventricular or left atrial pressure (e.g., in left ventricular failure or mitral stenosis). The resulting pulmonary edema is called cardiogenic or hydrostatic pulmonary edema, and the cause is essentially an imbalance between the hydrostatic and oncotic forces governing fluid movement. In this form of edema, the permeability barrier that limits movement of protein out of the intravascular space is intact, and the fluid that leaks out has a very low protein content.
Table 28-2
| Feature | Cardiogenic | Noncardiogenic |
| Major cause | Left ventricular failure, mitral stenosis | Acute respiratory distress syndrome |
| Pulmonary capillary pressure | Increased | Normal |
| Pulmonary capillary permeability | Normal | Increased |
| Protein content of edema fluid | Low | High |
Etiology
Numerous and varied disorders are associated with the potential to produce ARDS (Table 28-3). What these diverse etiologic factors in ARDS have in common is their ability to cause diffuse injury to the pulmonary parenchyma. Beyond that, defining other features that link the underlying causes is difficult on the basis of our present knowledge. Even the route of injury varies. Some etiologic factors involve inhaled injurious agents; others appear to mediate their effects on the lungs via the circulation rather than the airway.
Table 28-3
CAUSES OF DIFFUSE ALVEOLAR DAMAGE AND ARDS
Shock (accompanied by other etiologic factors)
Disseminated intravascular coagulation
Mechanical ventilation (overdistention and/or cyclic opening and closing of alveoli)*
ARDS = Acute respiratory distress syndrome.
*Generally not a primary cause of acute respiratory distress syndrome but a potential secondary contributor to alveolar damage (see Chapter 29).
Inhaled Injurious Agents
A number of inhaled gases have been identified as potential acute toxins and precipitants of ARDS. Nitrogen dioxide is one example, as are some chemical products of combustion inhaled in smoke. High concentrations of oxygen, particularly when given for prolonged periods, have been considered to contribute to alveolar injury. The mechanism of O2 toxicity is believed to be generation of free radicals and superoxide anions, byproducts of oxidative metabolism that are toxic to pulmonary epithelial and endothelial cells. It is ironic that O2 can contribute to lung injury, given that it is so important in supportive treatment of ARDS. Chapter 29 discusses an additional way in which treatment of ARDS may worsen alveolar damage through overdistention and/or cyclic opening and closing of alveoli induced by mechanical ventilation.
Pathophysiology
Most of the clinical consequences of ARDS follow in reasonably logical fashion from the presence of interstitial and alveolar edema. The most striking problem is alveolar flooding, which effectively prevents ventilation of affected alveoli even though perfusion may be relatively preserved. These alveoli, perfused but not ventilated, act as regions where blood is shunted from the pulmonary arterial to pulmonary venous circulation without ever being oxygenated. This type of shunting is one of the mechanisms of hypoxemia (see Chapter 1), and there is perhaps no better example of intrapulmonary shunting than ARDS.
 mismatch
mismatchIn addition to the direct effects of interstitial and alveolar fluid on oxygenation, other changes appear to be secondary to alterations in the production and effectiveness of surfactant. Chapter 8 refers to surfactant as a phospholipid responsible for decreasing surface tension and maintaining alveolar patency. When surfactant is absent, as is seen in the respiratory distress syndrome of neonates, there is extensive collapse of alveoli. In ARDS, surfactant production may be adversely affected by injury to alveolar type II epithelial cells. Additionally, evidence suggests that as a result of extensive fluid within the alveoli, surfactant is inactivated and therefore ineffective in preventing alveolar collapse.
Diagnostic Approach
The chest radiograph in patients with incipient ARDS does not necessarily reveal abnormal findings at the onset of clinical presentation. However, within a short period of time, evidence of interstitial and alveolar edema generally develops, the latter being the most prominent finding on chest radiograph. Edema appears diffuse, affecting both lungs relatively symmetrically. As an indication that fluid is filling alveolar spaces, air bronchograms often appear within the diffuse infiltrates. Unless the patient has prior heart disease and cardiac enlargement unrelated to the present problem, heart size remains normal. A characteristic example of a chest radiograph in a patient with severe ARDS is shown in Figure 3-7.
Treatment
Meticulous supportive management, particularly support of gas exchange and avoidance of hypervolemia, is critical for patients with ARDS to survive the acute illness. Given the life-threatening nature of ARDS, patients typically are intubated, ventilated with a mechanical ventilator, and managed in an intensive care unit. Failure of other organ systems besides the respiratory system is common, and patients often present some of the most complex and challenging management problems handled in intensive care units. Because of the importance of mechanical ventilation and ventilatory support in the management of respiratory failure associated with ARDS and with other disorders, Chapter 29 is devoted to a more detailed consideration of mechanical ventilation in the management of respiratory failure.
Arroliga, AC, Matthay, MA, Weidemann, HP. Acute respiratory distress syndrome. eds. Clin Chest Med. 2006;27:549–754.
Ferguson, ND, Fan, E, Camporota, L, et al. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med. 2012;38:1573–1582.
Leaver, SK, Evans, TW. Acute respiratory distress syndrome. BMJ. 2007;335:389–394.
Rubenfeld, GD, Herridge, MS. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554–562.
Ware, LB, Matthay, MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349.
Wheeler, AP, Bernard, GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369:1553–1564.
Bellingan, GJ. The pathogenesis of ALI/ARDS. Thorax. 2002;57:540–546.
Matthay, MA, Zimmerman, GA. Acute lung injury and the acute respiratory distress syndrome. Four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327.
Matthay, MA, Zimmerman, GA, Esmon, C, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167:1027–1035.
Suratt, BT, Parsons, PE. Mechanisms of acute lung injury/acute respiratory distress syndrome. Clin Chest Med. 2006;27:579–589.
Adhibari, N, Burns, KE, Meade, MO. Pharmacologic therapies for adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev. (4):2004. CD004477
Brodie, D, Bacchetta, M. Extracorporeal membrane oxygenation for ARDS in adults. N Engl J Med. 2011;365:1905–1914.
Calfee, CS, Matthay, MA. Nonventilatory treatments for acute lung injury and ARDS. Chest. 2007;131:913–920.
Diaz, JV, Brower, R, Calfee, CS, et al. Therapeutic strategies for severe acute lung injury. Crit Care Med. 2010;38:1644–1650.
Esan, A, Hess, DR, Raoof, S, et al. Severe hypoxemic respiratory failure: part 1–ventilatory strategies. Chest. 2010;137:1203–1216.
Fan, E, Needham, DM, Stewart, TE. Ventilatory management of acute lung injury and acute respiratory distress syndrome. JAMA. 2005;294:2889–2896.
Malhotra, A. Low-tidal-volume ventilation in the acute respiratory distress syndrome. N Engl J Med. 2007;357:1113–1120.
Pipeling, MR, Fan, E. Therapies for refractory hypoxemia in acute respiratory distress syndrome. JAMA. 2010;304:2521–2527.
Randolph, AG. Management of acute lung injury and acute respiratory distress syndrome in children. Crit Care Med. 2009;37:2448–2454.
Raoof, S, Goulet, K, Esan, A, et al. Severe hypoxemic respiratory failure: part 2–nonventilatory strategies. Chest. 2010;137:1437–1448.
Steinberg, KP, Hudson, LD, Goodman, RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354:1671–1684.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]28
Acute Respiratory Distress Syndrome
This chapter continues the discussion of respiratory failure with more detailed consideration of one important type of acute respiratory failure: acute respiratory distress syndrome (ARDS). This entity was initially called adult respiratory distress syndrome, but it is not limited to adults, so acute rather than adult is now used to describe it. ARDS represents a major form of hypoxemic respiratory failure. Its clinical and pathophysiologic features differ considerably from those noted for acute-on-chronic respiratory failure. ARDS is characterized by the presence of severe arterial hypoxemia and diffuse bilateral pulmonary infiltrates not due exclusively to cardiogenic or hydrostatic causes. The full criteria for establishing the diagnosis of ARDS are shown in Table 28-1. This chapter describes in detail each of these criteria and the associated pathology and pathophysiology.
This chapter first considers the dynamics of fluid transfer between the pulmonary vessels and alveolar interstitium, because any alteration in this process is important in the pathogenesis of ARDS. Next is an outline of the many types of injury that can result in ARDS and some of the theories proposed to explain how such a diverse group of disorders can produce this syndrome. Following is a discussion of the pathologic, pathophysiologic, and clinical consequences. The chapter concludes with a general approach to treatment. More specific details about support of gas exchange are provided in Chapter 29.
Physiology of Fluid Movement in Alveolar Interstitium
Despite the diverse group of disorders that can cause ARDS, the net result of the syndrome is the same: a disturbance in the normal barrier that limits leakage of fluid out of the pulmonary capillaries and into the pulmonary parenchyma. Before a discussion of some of the theories explaining how this barrier is damaged, a brief consideration of the determinants of fluid transport among the pulmonary vessels, interstitial space, and alveolar lumen may be helpful. The pulmonary parenchyma (Fig. 28-1) consists of (1) small vessels coursing through the alveolar walls, which for simplicity are referred to as the pulmonary capillaries; (2) pulmonary capillary endothelium, the lining cells that normally limit but do not completely prevent fluid movement out of the capillaries; (3) pulmonary interstitium, which is considered here as the alveolar wall exclusive of vessels and the epithelial cells lining the alveolar lumen; (4) lymphatic channels, which are found mainly in perivascular connective tissue in the lungs; (5) alveolar epithelial cells, which line the surface of the alveolar lumen; and (6) alveolar lumen or alveolar space.
Movement of fluid out of the pulmonary capillaries and into the interstitial space is determined by a number of factors, including the hydrostatic pressures in the vessels and the pulmonary interstitium, the colloid osmotic pressures in these same two compartments, and the permeability of the endothelium. The effect of these factors in determining fluid transport is summarized in the Starling equation, examined in Chapter 15 with regard to fluid transport across the pleural space. The Starling equation is given as Equation 28-1:
where F = fluid movement; Pc and Pis = pulmonary capillary and interstitial hydrostatic pressure, respectively; COPc and COPis = pulmonary capillary and interstitial colloid osmotic (oncotic) pressure, respectively; K = filtration coefficient; and σ = reflection coefficient (measure of permeability of endothelium for protein).
Two Mechanisms of Fluid Accumulation
In practice, the forces described in the Starling equation become altered in two main ways, producing interstitial and often alveolar edema (Table 28-2). The first occurs when hydrostatic pressure within the pulmonary capillaries (Pc) is increased, generally as a consequence of elevated left ventricular or left atrial pressure (e.g., in left ventricular failure or mitral stenosis). The resulting pulmonary edema is called cardiogenic or hydrostatic pulmonary edema, and the cause is essentially an imbalance between the hydrostatic and oncotic forces governing fluid movement. In this form of edema, the permeability barrier that limits movement of protein out of the intravascular space is intact, and the fluid that leaks out has a very low protein content.
Table 28-2
| Feature | Cardiogenic | Noncardiogenic |
| Major cause | Left ventricular failure, mitral stenosis | Acute respiratory distress syndrome |
| Pulmonary capillary pressure | Increased | Normal |
| Pulmonary capillary permeability | Normal | Increased |
| Protein content of edema fluid | Low | High |
Etiology
Numerous and varied disorders are associated with the potential to produce ARDS (Table 28-3). What these diverse etiologic factors in ARDS have in common is their ability to cause diffuse injury to the pulmonary parenchyma. Beyond that, defining other features that link the underlying causes is difficult on the basis of our present knowledge. Even the route of injury varies. Some etiologic factors involve inhaled injurious agents; others appear to mediate their effects on the lungs via the circulation rather than the airway.
Table 28-3
CAUSES OF DIFFUSE ALVEOLAR DAMAGE AND ARDS
Shock (accompanied by other etiologic factors)
Disseminated intravascular coagulation
Mechanical ventilation (overdistention and/or cyclic opening and closing of alveoli)*
ARDS = Acute respiratory distress syndrome.
*Generally not a primary cause of acute respiratory distress syndrome but a potential secondary contributor to alveolar damage (see Chapter 29).
Inhaled Injurious Agents
A number of inhaled gases have been identified as potential acute toxins and precipitants of ARDS. Nitrogen dioxide is one example, as are some chemical products of combustion inhaled in smoke. High concentrations of oxygen, particularly when given for prolonged periods, have been considered to contribute to alveolar injury. The mechanism of O2 toxicity is believed to be generation of free radicals and superoxide anions, byproducts of oxidative metabolism that are toxic to pulmonary epithelial and endothelial cells. It is ironic that O2 can contribute to lung injury, given that it is so important in supportive treatment of ARDS. Chapter 29
[/not-level-membership-for-pulmolory-and-respiratory-category]
