Acute Lung Injury, Pulmonary Edema, and Multiple System Organ Failure
After reading this chapter you will be able to:
 Identify the approximate incidence rate of acute respiratory distress syndrome (ARDS) and how the mortality rate has changed over the past several decades.
Identify the approximate incidence rate of acute respiratory distress syndrome (ARDS) and how the mortality rate has changed over the past several decades.
 State the risk factors associated with the onset of ARDS.
State the risk factors associated with the onset of ARDS.


 Describe the relationship between multiple organ dysfunction syndrome (MODS) and ARDS.
Describe the relationship between multiple organ dysfunction syndrome (MODS) and ARDS.

 Differentiate hydrostatic and nonhydrostatic pulmonary edema based on clinical setting.
Differentiate hydrostatic and nonhydrostatic pulmonary edema based on clinical setting.
 Describe the principles of supportive care followed for patients with ARDS.
Describe the principles of supportive care followed for patients with ARDS.


 State the approaches to the management of ARDS and MODS.
State the approaches to the management of ARDS and MODS.

 State the effect of prone positioning on oxygenation and mortality in a patient with ARDS.
State the effect of prone positioning on oxygenation and mortality in a patient with ARDS.

Acute hypoxemic respiratory failure may develop in many clinical settings and is a common reason for admission to the intensive care unit (ICU). Most cases of acute hypoxemic respiratory failure develop as a result of abnormal accumulations of fluid within the lung parenchyma and alveoli. These accumulations are collectively referred to as pulmonary edema. Pulmonary edema may arise from acute illnesses associated with increased pulmonary venous pressure (hydrostatic pulmonary edema or congestive heart failure [CHF]) or may result from conditions associated with acute lung injury (ALI), in which the normal barriers to fluid movement within the lungs are disrupted (nonhydrostatic pulmonary edema). ALI of sufficient severity to cause acute hypoxemic respiratory failure is commonly referred to as acute respiratory distress syndrome (ARDS). ALI and ARDS represent a spectrum of lung injury with many patients initially presenting with ALI and progressing to ARDS with more severe gas exchange abnormalities (Table 27-1).1,2
TABLE 27-1
Recommended Criteria for Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)
| Criteria Pressure | Timing | Oxygenation | Chest Radiograph | Pulmonary Artery Wedge |
| ALI | Acute onset | PaO2/FiO2 ≤ 300 mm Hg (regardless of PEEP level) | Bilateral infiltrates seen on frontal chest radiograph | ≤18 mm Hg when measured or no clinical evidence of left atrial hypertension |
| ARDS | Acute onset | PaO2/FiO2 ≤ 200 mm Hg (regardless of PEEP level) | Bilateral infiltrates seen on frontal chest radiograph | ≤18 mm Hg when measured or no clinical evidence of left atrial hypertension |

Modified from Bernard GR, Artigas A, Brigham KL, et al: The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 49(3 Pt 1):818–824, 1994.
Epidemiology
ARDS is a common cause of respiratory failure. It can occur as a consequence of critical illnesses of diverse causes. The exact incidence of ARDS varies depending on the population being studied, but in the United States it is thought to range from 13.5 to 64 cases per 100,000 person-years.3,4 ARDS may be present in 16% of mechanically ventilated patients on admission to the ICU.2 Despite uncertainties regarding the incidence of ARDS, the mortality rate associated with ARDS has seemed to decline over the past 3 decades from more than 90% to the present level of 30% to 40%.5,6
Risk Factors For Acute Respiratory Distress Syndrome
It has been proposed that ARDS can develop via different mechanisms and that the risk factors for ARDS should be categorized as either direct injury via damage directly to the alveolar space or indirect injury initiated by systemic disease (Box 27-1).1 However, all of these risk factors share the common ability to initiate a systemic inflammatory reaction, which, if sufficiently vigorous, may lead to diffuse lung injury (ARDS). In this regard, the probability of developing ARDS may depend in part on the severity and characteristics of the initial injury. Gastric aspiration and septic shock (sepsis with refractory hypotension) are associated with a greater than 25% risk of ARDS, whereas the administration of multiple blood transfusions is associated with an ARDS risk of less than 5%.7 The risk of ARDS seems to be additive when multiple risk factors are present.8
Pathophysiology
Normal Physiology
The lung structure is optimally designed to fulfill its physiologic functions. These are as follows:
• To deliver inhaled oxygen (O2) to the site of gas exchange—the alveoli
• To diffuse gases, mainly O2 and carbon dioxide (CO2), between the alveolar capillary membrane and the lumen of the alveolus
• To match alveolar ventilation with pulmonary capillary blood flow so that gas exchange is optimized
• To maintain a net flux of fluid through the lung parenchyma without inducing lung edema or alveolar consolidation
• To provide a barrier against toxic environmental exposures, including infectious agents, dusts, and fumes
Pulmonary Blood Flow
The intricate design of the pulmonary circulation provides for the efficient transfer of gases between the alveoli and the blood. On average, the entire blood volume of the body circulates through the lungs in 1 minute or less. This incredible feat is achieved through an ingenious design. Starting at the outflow tract of the right ventricle (pulmonary valve), the relatively thick-walled, smooth muscle–lined pulmonary artery branches successively and follows the divisions of the bronchi as far as the terminal bronchioles. Beyond the terminal bronchioles, the pulmonary vasculature divides further to form a fine capillary meshwork surrounding the alveoli. The large surface area of the capillary network provides for a low-pressure (5 to 12 mm Hg), high-volume system wherein large volumes of blood come into immediate contact with alveolar gases. At a capillary level, the vessel walls are composed solely of endothelial cells bound to a basal lamina. Because of the delicate nature of the alveolar-capillary interface, injury to the alveolar-capillary interface and high capillary blood pressure result in disruption of pulmonary gas exchange (see later section on Pulmonary Edema).
Lung Interstitium
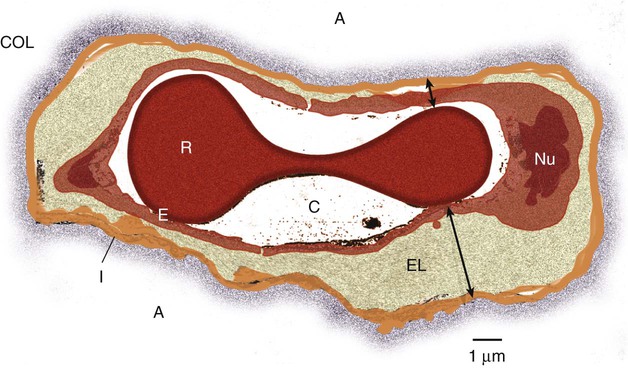
The interstitial space of the lung is the space between the alveolar epithelium and the capillaries. The interstitium is composed of several structural proteins (types I, III, and IV collagen and elastin) and proteoglycans. The proteoglycans make up the ground substance of the interstitium and are composed of 20% protein and 80% glycosaminoglycans. The alveolar-capillary interstitial space is composed of endothelial and epithelial cell membranes bound to a common basement membrane with a very thin (<0.5 µ) interstitial space. In contrast, the interstitium on the nonalveolar side of the capillaries contains separate basement membranes for both epithelial cells and endothelial cells and fibroblasts, structural collagen, elastin proteins, and mucopolysaccharides in a hyaluronic acid gel (Figure 27-1).
Liquid and Solute Transport in the Lungs
< ?xml:namespace prefix = "mml" />
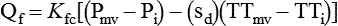
The lung protects itself from the devastating consequences of excessive fluid accumulation by several mechanisms. The lung lymphatic drainage system is the primary operant system under nonpathologic conditions. The lymphatic drainage system is the main conduit for the removal of filtered fluid and protein from the lungs. Fluid and solutes enter the lymphatic drainage channels from small lymphatic capillaries located around the respiratory bronchioles. This process is assisted by the presence of a modest pressure gradient within the lungs. That is, pressure is greatest within the dense alveolar interstitium and gradually decreases in the nonalveolar interstitium and terminal lymphatic vessels (Figure 27-2). Drainage is enhanced further by intrathoracic pressure alterations that occur with respiration, and retrograde flow is prevented by the presence of one-way lymphatic valves. Ultimately, lymphatic fluid drains into the superior vena cava through the thoracic duct.9
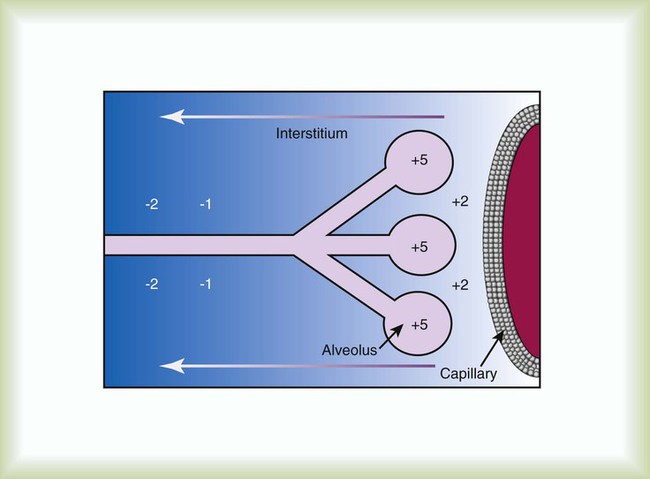
When fluid filtration exceeds the capacity for drainage through pulmonary lymphatic vessels, several “backup” systems exist for storing additional fluid and for protection against alveolar flooding. Loose connective tissue located along the peribronchovascular space and extending to the level of the bronchiole is capable of storing twice the normal fluid content of the lungs.9 Filling of these spaces or cuffs manifests radiographically as increased interstitial infiltrates (Figure 27-3), or Kerley’s lines, which are caused by increased fluid in interlobular septal spaces. The peribronchovascular spaces drain into the local blood vessels or follow the intrinsic pressure gradient in the lung and empty into pulmonary lymphatic vessels. As total lung fluid accumulation increases further, the gel-like matrix of the lung is capable of absorbing additional fluid without affecting interstitial pressure. The latter property is important because fluid is allowed to accumulate in the lungs without transmitting additional hydrostatic pressure to the alveolar barrier. Additional fluid movement into the lungs is avoided. The dense connective tissue making up the alveolar matrix resists edema formation in response to elevated hydrostatic or oncotic pressure.
Pulmonary Edema
Hydrostatic Pulmonary Edema
Hydrostatic pulmonary edema, also called cardiogenic pulmonary edema, is the result of increased venous hydrostatic pressure leading to fluid accumulation and alveolar flooding. The alveolar barrier is composed of the dense alveolar matrix (described earlier), the epithelial basement membrane, and the lining epithelial cells. The alveolar basement membrane is selectively permeable to only very small solutes such that osmotic forces favor the retention of fluid in the intravascular space. Alveolar pressure generally is slightly higher than interstitial pressure, and this difference provides further protection against alveolar fluid accumulation (see Figure 27-2). Under normal circumstances, there is little fluid movement into the alveoli across the alveolar barrier. Fluid that does form is composed of low-molecular-weight proteins and is actively transported back into the interstitium by type II pneumocytes.10 In contrast, when the vascular hydrostatic pressure increases and overwhelms the defense mechanisms against fluid accumulation in the lung, hydrostatic pressure increases sharply within the interstitium of the lung, and alveolar flooding ensues. The precise mechanism of alveolar flooding in hydrostatic pulmonary edema is unknown. It has been shown, however, that alveolar flooding occurs in an “all-or-none” manner. The alveolar fluid formed in the setting of increased hydrostatic pressure has characteristics identical to interstitial fluid even though the alveolar epithelium is normally impermeable to large proteins and molecules.9 This observation lends support to the findings of Conhaim,11 who showed experimentally that high interstitial fluid pressure leads to alveolar flooding through leakage of fluid from the epithelium of respiratory bronchioles, alveolar ducts, and their associated alveoli. The epithelium of the respiratory bronchioles and alveolar ducts may be particularly prone to hydrostatic injury because these locations represent the transition zone between respiratory and alveolar epithelium.
Nonhydrostatic Pulmonary Edema
Nonhydrostatic pulmonary edema, also called noncardiogenic pulmonary edema, is the result of the loss of microvascular membrane integrity. In contrast to hydrostatic pulmonary edema, nonhydrostatic pulmonary edema is associated with increased total lung water despite normal microvascular hydrostatic pressure. The mechanisms of nonhydrostatic pulmonary edema that ultimately lead to ARDS are more complex than the mechanisms responsible for hydrostatic pulmonary edema. Although many seemingly unrelated risk factors for ARDS have been identified, all causes of ARDS evoke disruption of endothelial and epithelial barriers and typically occur under conditions associated with widespread microvascular injury to the lungs. Vascular endothelial injury in the lungs results in increased microvascular permeability and fluid filtration, such that there is uninhibited entry of protein-rich fluid into the pulmonary interstitium. Alveolar flooding develops when the osmotic gradient between the capillary and the lung becomes essentially zero and no longer opposes the hydrostatic forces favoring fluid movement from the capillary into the lung: Kfc(Pmv − Pi) >> (sd)(TTmv − Tti) (see earlier equation). This process is likely facilitated both by damage to the normally impermeable alveolar epithelial barrier, which is a key feature of ARDS,12 and by impaired alveolar fluid clearance in ALI and ARDS.13 Investigations support a role for both necrotic epithelial cell death and programmed cell death (apoptosis) in the pathogenesis of alveolar wall damage.14,15 In addition, therapies aimed at improving the vascular barrier to fluid movement have shown protection against ALI animal models.
A common mechanism to explain how different acute illnesses (e.g., sepsis, gastric acid aspiration, pancreatitis) can lead to the development of ARDS was proposed by Weiland and colleagues.16 These investigators showed that ARDS, regardless of the cause, is associated with an influx of polymorphonuclear neutrophils (PMNs) and PMN-derived inflammatory by-products, such as neutrophil elastase and myeloperoxidase, into the lung. The PMN-activating cytokine interleukin (IL)-8 has been shown to be increased in the lungs of patients with ARDS, whereas a reduction in IL-8 and PMNs has been shown to correlate with recovery from ARDS.17 Taken together, the results of these studies suggest that the common mechanism for the development of ARDS is induction of lung inflammation leading to loss of membrane integrity, as a result of either local lung injury (direct injury) or systemic inflammation (indirect injury).
Although PMNs play a central role in the development of ARDS, other chemical (e.g., gastric aspiration) and immunologic pathways participate in the initiation and development of the systemic inflammatory response to critical illnesses associated with ARDS. Sepsis is associated with intense activation of systemic inflammatory pathways such that cytokines (e.g., tumor necrosis factor [TNF]-alpha, IL-1beta, IL-6, and IL-8), arachidonic acid metabolites (e.g., platelet-activating factor, leukotrienes), and nitric oxide (NO) all contribute to the hemodynamic and inflammatory events characteristic of this syndrome.18 The relative contributions and exact roles of these proinflammatory mediators in the pathogenesis of ARDS and MODS remain a topic of intense investigation and controversy.19,20 It is likely that there is no single dominant pathway in the development of ARDS because attempts to control the inflammatory response in sepsis by blocking specific mediators, such as anti-TNF antibodies, have not proved beneficial and may worsen outcome.
Gas Exchange and Lung Mechanics in Acute Respiratory Distress Syndrome
ARDS is associated with restrictive physiology and refractory hypoxemia, which are largely a result of pulmonary microvascular injury. Specifically, increased pulmonary capillary permeability facilitates the influx of inflammatory fluid into the lung interstitium and alveolar spaces and causes decreased lung compliance and alveolar consolidation. The presence of intraalveolar inflammatory fluid impairs surfactant synthesis and function so that pulmonary gas exchange (i.e., related to atelectasis) and compliance are further impaired. The negative effects of alveolar consolidation and atelectasis on pulmonary gas exchange are exacerbated by a loss of the normal vascular response to alveolar hypoxemia. The body is unable to shunt blood away from the diseased alveoli, and these unaerated alveoli receive excessive blood flow, which contributes to severe ventilation/perfusion mismatching and an effective intrapulmonary right-to-left shunting of blood flow in ARDS. The pulmonary manifestations of ARDS and CHF are summarized in Box 27-2.
Role of Organ-Organ Interactions in the Pathogenesis of Acute Respiratory Distress Syndrome and Multiple Organ Dysfunction Syndrome
The notion that factors operating outside the lungs may participate in the initiation and progression of ARDS has generated interest in the role of organ system interactions in the pathogenesis of ARDS and MODS. For example, injury to remote systemic organs is known to occur after ALI,21 apparently related to PMN-mediated mechanisms.22 In this way, ALI may perpetuate the systemic inflammatory response and lead to further lung and systemic organ injury. The gut-liver-lung axis may be most influential in causing the systemic inflammatory response associated with ARDS and MODS. The gastrointestinal (GI) tract contains large quantities of potentially pathogenic bacteria against which the host is normally protected by intact mucosal barriers and the reticuloendothelial system. However, the function of the GI tract and the liver is frequently compromised in critical illness. The widespread use of broad-spectrum antibiotics in the care of critically ill patients often leads to overgrowth of antibiotic-resistant bacteria within the GI tract. These bacteria and their toxic by-products (e.g., endotoxin) escape from the GI tract and are taken up by the reticuloendothelial cells of the liver, spleen, and regional lymph nodes. The resultant activation of the reticuloendothelial system may initiate and perpetuate a systemic inflammatory response that leads to systemic organ injury (i.e., ARDS and MODS).23 This sequence of events forms the basis for strategies designed to reduce the release of proinflammatory mediators from the GI tract, including selective decontamination, early enteral feeding, and other approaches designed to moderate the systemic inflammatory response in ARDS and MODS (see later section on Therapeutic Approach to Acute Respiratory Distress Syndrome).
Why ARDS and MODS develop in some patients with ALI and not in others is unknown. The determinants of ARDS may relate to the balance between proinflammatory and antiinflammatory factors within the body. In this regard, the liver plays a major role in both induction and modulation of the systemic inflammatory response to all kinds of initiating events and is primarily responsible for the breakdown of endogenous proinflammatory mediators, including TNF-alpha, leukotrienes, and others.24 Patients with liver disease have higher levels of circulating proinflammatory mediators, are more prone to bacteremia, and have a high incidence of ARDS compared with patients without liver disease.25
The liver is not the sole determinant of ARDS and MODS in critically ill patients. Other factors, such as the severity of the primary illness and comorbid diseases (e.g., cardiac disease, advanced age, renal failure, malignant disease) and perhaps the genetic profile of the patient, also seem to predispose patients to ARDS and MODS.23,26
Histopathology And Clinical Correlates Of Acute Respiratory Distress Syndrome
Exudative Phase (1 to 3 Days)
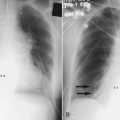
The exudative phase is characterized by diffuse damage to alveoli and blood vessels and the influx of inflammatory cells into the interstitium. Many of the alveolar spaces become filled with a proteinaceous, eosinophilic (on hematoxylin and eosin stain) material called hyaline membranes, which are composed of cellular debris and condensed plasma proteins. Pathologically, there is destruction of type I pneumocytes, which are normally the predominant cells lining the alveoli; type II pneumocytes are relatively resistant to injury.27,28 Patients with ARDS have profound dyspnea, tachypnea, and refractory hypoxemia. This phase of ARDS often is difficult to differentiate from respiratory failure related to hydrostatic pulmonary edema (CHF). The clinical presentations of these two forms of acute respiratory failure are discussed later (see the section on Differentiating Hydrostatic from Nonhydrostatic Pulmonary Edema in the Clinical Setting). The exudative phase may be self-limited or may progress to a fibroproliferative phase.
 Rule of Thumb
Rule of Thumb
A careful history and physical examination often are the most useful means by which CHF and ARDS can be initially differentiated in a patient who has refractory hypoxemia and bilateral infiltrates on chest radiographs. A history consistent with one of the common causes of CHF (Box 27-3) combined with physical examination findings of jugular venous distention, cardiac murmurs or gallops, bibasilar crackles, or peripheral edema suggests a diagnosis of CHF. ARDS is more likely when the history is positive for one of the established risk factors (see Box 27-1) and there is no clinical evidence in support of CHF.
Fibroproliferative Phase (3 to 7 Days)
After inflammatory injury to the lung is established and the initiating events are controlled, a process of lung repair begins. Pathologically, there is hyperplasia of alveolar type II pneumocytes and proliferation of fibroblasts within the alveolar basement membrane and intraalveolar spaces. Fibroblasts mediate the formation of intraalveolar and interstitial fibrosis.27 The extent of fibrosis determines the degree of pulmonary disability in patients who survive ARDS.
The exact mechanisms controlling lung remodeling in ALI are not well established but very likely involve by-products of inflammatory cells (e.g., proteases, antiproteases, IL-6) and various growth factors (transforming growth factor [TGF]-alpha, TGF-beta).29,30 However, the remodeling process after ARDS is quite variable. In some cases, patients have nearly complete normalization of lung compliance and oxygenation for 6 to 12 months after the illness. In other cases, the architecture of the lung never returns to normal, and patients experience severe respiratory disability related to extensive pulmonary fibrosis and obliteration of the pulmonary vasculature. The pattern of fibrosis after ALI suggests that, as in repair of the skin, an intact basement membrane is necessary for normal repair of the epithelium of the alveoli. It follows that disruption of the alveolar basement membrane is a prerequisite to the development of fibrosis after ALI. This line of reasoning is supported by the observation that the extent of recovery depends on the severity of the initial lung injury and on the influence of secondary forms of injury. Secondary forms of lung injury include nosocomial infection, O2 toxicity, and barotrauma (see later section on Therapeutic Approach to Acute Respiratory Distress Syndrome).
Differentiating Hydrostatic from Nonhydrostatic Pulmonary Edema in the Clinical Setting
The diagnostic criteria for ARDS are shown in Table 27-1. However, despite the existence of distinct pathophysiologic mechanisms of hydrostatic and nonhydrostatic pulmonary edema, differentiating these two forms of acute respiratory failure may be difficult because of similarities in their early clinical presentations. CHF is more common than ARDS and should be considered when any patient has pulmonary edema, in particular, when the history and physical examination findings suggest one of the causes of CHF listed in Box 27-3. Alveolar flooding by either hydrostatic or nonhydrostatic mechanisms results in diffuse radiographic infiltrates, altered gas exchange, and abnormal mechanical properties of the lung (see Box 27-2). A clinical history of infection, recent trauma, or risk factors for aspiration may be present in either patient group. Likewise, many patients with ARDS are older and have preexisting illnesses that place them at risk of CHF. In patients with acute hypoxemic respiratory failure, CHF must always be considered, even when the patient has obvious risk factors for ARDS.
Invasive hemodynamic monitoring may be unreliable in patients with high airway pressure. In particular, high levels of positive end expiratory pressure (PEEP) lead to expansion of non–zone 3 lung areas in which alveolar pressure exceeds venous pressure. PCWP measured in non–zone 3 lung reflects alveolar pressure and not left ventricular pressure.31 Hydrostatic forces may be overestimated or misdiagnosed in ARDS as hydrostatic pulmonary edema. These confounding variables may lead to misinterpretation of the hemodynamic status of the patient, resulting in mismanagement of his or her condition, and could explain why the routine use of pulmonary artery catheters in patients with ARDS is not beneficial.32 A similar estimate of pulmonary capillary hydrostatic pressure may be obtained via central venous pressure (CVP) obtained from a central line placed in the superior vena cava. CVP is less technically difficult to obtain and interpret compared with PCWP; in a large clinical trial, therapy guided by CVP estimation of the patient’s volume status was equivalent to therapy guided by PCWP.33
Another useful method of separating nonhydrostatic from hydrostatic pulmonary edema is based on differences in the characteristics of the edema fluid. As previously discussed (see the section on Pathophysiology), ARDS is associated with inflammatory injury to the pulmonary microvasculature, which allows the influx of inflammatory cells and proteinaceous fluid into the interstitium and alveolar spaces. The inflammatory nature of this exudative fluid is reflected by the presence of large quantities of inflammatory cells (predominantly neutrophils) in bronchoalveolar lavage fluid (BALF). The BALF findings provide diagnostic insights if infectious agents or signs of aspiration (e.g., food particles) are present. In contrast, although hydrostatic pulmonary edema is associated with alveolar flooding, the edema fluid typically is noninflammatory, and the protein content is much lower than the protein content of BALF.34 BALF often provides useful insight into the underlying cause of hypoxemic respiratory failure, but the role of BALF in the clinical management of patients with acute respiratory failure caused by pulmonary edema is not well established. The clinical characteristics that differentiate acute hypoxemic failure caused by hydrostatic pulmonary edema from acute hypoxemic failure caused by nonhydrostatic pulmonary edema are summarized in Box 27-2.
Mini Clini
Determination of “Optimal” PEEP
 Problem
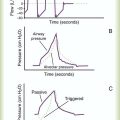
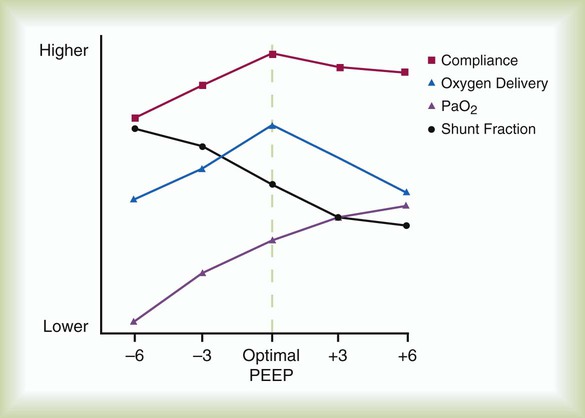
ProblemIn patients with ARDS, increasing levels of PEEP are associated with improved oxygenation and a reduction in the measured shunt fraction (PaO2/FiO2 ratio) (see Figure 27-5). Routine use of high levels of PEEP has not been shown to be beneficial for all patients. However, patients with severe ARDS may benefit from higher PEEP levels to maintain oxygenation.35 Despite initial increases in oxygenation above the level of PEEP designated the “optimal level” in Figure 27-5, there is a net decrease in systemic O2 delivery. Lung compliance may begin to decline at higher levels of PEEP, and the risk of volutrauma increases. Also, healthy alveoli may become overdistended, resulting in decreased perfusion and stretch-induced injury of the “good lung.” The pressure-volume relationships of the lungs may be used to adjust PEEP such that end expiratory alveolar collapse and end inspiratory overinflation are minimized (see Figure 27-6).
Therapeutic Approach To Acute Respiratory Distress Syndrome
The management of critical illness associated with ARDS previously was confined to supportive therapy (Box 27-4) designed to preserve systemic organ function and allow recovery from the underlying illness. Strategies have evolved for controlling the systemic inflammatory response that leads to lung and other organ injury. This section presents an overview of the current approach to supportive care and new potential therapies for ARDS and MODS (Figure 27-4).
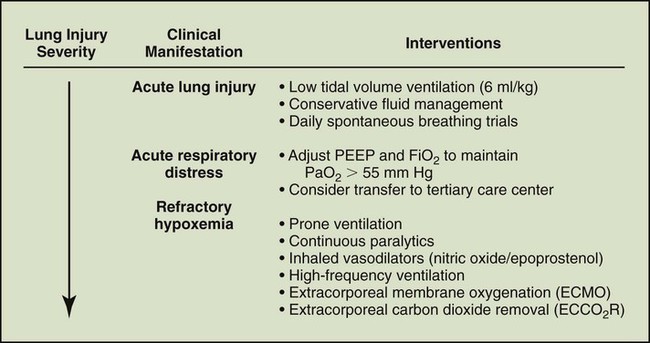
Hemodynamics and Fluid Management in Acute Respiratory Distress Syndrome
Preservation of vital organ integrity through the optimization of systemic O2 delivery is a principal goal of supportive management in all causes of respiratory failure. Ventilator strategies designed to improve arterial oxygenation must be weighed against any potential changes in hemodynamic values incurred as a result of these strategies. In view of the deleterious cardiopulmonary effects of high-pressure mechanical ventilation (e.g., PEEP) in ARDS, it is logical to imagine that invasive monitoring of both cardiac output and systemic O2 delivery (DO2) would be beneficial. As previously mentioned, the role of invasive hemodynamic monitoring (e.g., pulmonary artery catheters) in the management of ARDS was historically controversial. However, clarity was provided by the National Institutes of Health National Heart, Lung and Blood Institute–sponsored ARDS Clinical Trials Network, which evaluated the benefits and risks of the use of pulmonary artery catheters to guide treatment in the setting of ALI. The study involved 1000 participants and showed no benefit in terms of mortality, ventilator-free days, and duration of ICU admission and no difference in terms of overall fluid balance. The authors of the study concluded that pulmonary artery catheters “should not be routinely used for the management of ALI.”33
Gas exchange is highly dependent on total lung fluid during the exudative phase of ARDS, and small increases in hydrostatic forces (PCWP) lead to significant decreases in oxygenation consequent to alveolar flooding and associated right-to-left shunting. Measures that restrict intravascular volume are associated with improved oxygenation. However, as with increasing PEEP, improvements in arterial oxygenation attendant to reducing PCWP must be weighed against reduced cardiac output, as reflected by the measured DO2 or other measures of systemic tissue oxygenation. In this context, a randomized, multicenter study conducted by the ARDS Clinical Trials Network compared conservative and liberal fluid management strategies for the first 7 days of treatment of 1000 patients with ALI. In keeping with the concept of “leaky” pulmonary capillaries in the setting of ALI resulting in increased susceptibility to lung fluid accumulation in response to increased hydrostatic forces, conservative fluid management was associated with improved arterial oxygenation, increased ventilator-free days, and shorter stays in the ICU. There was no statistical improvement in 60-day mortality, and there was no difference between the groups in terms of vital organ failures. Nonetheless, the results of this study strongly favor the routine use of a conservative fluid management strategy in patients with ARDS.36
 Rule of Thumb
Rule of Thumb
The beneficial effects of PEEP are optimal at a pressure of 20 cm H2O or less in most patients with ARDS. Levels of PEEP greater than 20 cm H2O should not be routinely used unless the benefits of higher levels of PEEP are supported by objective end points, such as improved lung compliance (Figure 27-5) or optimal alveolar recruitment (see Figure 27-6).
With regard to tissue oxygenation in critical illness, various investigations have shown that an abnormal dependence of O2 consumption (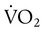 ) on DO2 exists throughout the physiologic range of DO2 in many critically ill patients.37 This abnormal dependence of
) on DO2 exists throughout the physiologic range of DO2 in many critically ill patients.37 This abnormal dependence of 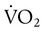 on DO2 is associated with impaired O2 extraction. These observations have been interpreted to imply that tissue hypoxia may exist and is contributing to organ failure (MODS) in these patients. This notion formed the basis for studies in which DO2 was increased to “supranormal” levels in patients at risk of sepsis and ARDS.7 The encouraging results of early trials using this strategy38,39 were not supported by larger, well-designed clinical trials in cohorts of patients with established sepsis.40,41 One study showed a higher mortality associated with the use of this strategy.40 Because of these findings, efforts to increase DO2 beyond normal values (3.5 L/min/kg) are not currently recommended. The “optimal DO2” for critically ill patients may never be established because the needs of individual patients must be factored into the decision to augment DO2. New techniques for detecting tissue hypoxia are needed for guiding the hemodynamic management of these patients. Until such tools become available, it seems prudent to prevent hypotension (systolic arterial blood pressure >90 mm Hg, mean arterial blood pressure >60 mm Hg), consider augmentation of DO2 in the setting of hyperlactatemia, optimize hemoglobin saturation (>90%), and ensure adequate organ function (e.g., urine output).
on DO2 is associated with impaired O2 extraction. These observations have been interpreted to imply that tissue hypoxia may exist and is contributing to organ failure (MODS) in these patients. This notion formed the basis for studies in which DO2 was increased to “supranormal” levels in patients at risk of sepsis and ARDS.7 The encouraging results of early trials using this strategy38,39 were not supported by larger, well-designed clinical trials in cohorts of patients with established sepsis.40,41 One study showed a higher mortality associated with the use of this strategy.40 Because of these findings, efforts to increase DO2 beyond normal values (3.5 L/min/kg) are not currently recommended. The “optimal DO2” for critically ill patients may never be established because the needs of individual patients must be factored into the decision to augment DO2. New techniques for detecting tissue hypoxia are needed for guiding the hemodynamic management of these patients. Until such tools become available, it seems prudent to prevent hypotension (systolic arterial blood pressure >90 mm Hg, mean arterial blood pressure >60 mm Hg), consider augmentation of DO2 in the setting of hyperlactatemia, optimize hemoglobin saturation (>90%), and ensure adequate organ function (e.g., urine output).
Mechanical Ventilation in Acute Respiratory Distress Syndrome
Despite the presence of widespread pulmonary injury and altered gas exchange in ARDS, more recent investigation has shown that aerated portions of the lung have near-normal mechanical characteristics. Gattinoni and Pesenti42 suggested that three distinct zones exist in the lungs of patients with ARDS. The most dependent lung zones are characterized by dense pulmonary infiltrates corresponding to nonventilated lung units. A second lung zone also has dense pulmonary infiltrates and nonventilated alveolar units but is distinct from the more dependent lung zones in that these areas may be made available for gas exchange through changes in the mode of mechanical ventilation. Finally, nondependent lung zones are fully inflated and receive most of the ventilation. The aerated lung zones have been shown to retain normal mechanical characteristics as reflected by the measured specific compliance (static compliance adjusted for the volume of ventilated lung). In ARDS, the lungs are effectively diminished in size to 20% to 30% of normal, but the aerated portions of the lungs retain near-normal physiologic properties.
Setting Tidal Volume
Because of the heterogeneous properties of the lungs in ARDS, mechanical ventilation with large tidal volumes is inappropriate for these patients. Tidal volumes of 10 to 15 ml/kg, when distributed primarily to the relatively small aerated lung zones, lead to hyperinflation and overdistention of the alveoli in these areas. In animal models, alveolar hyperinflation has been shown to result in altered alveolar capillary permeability identical to that associated with ARDS. The excessive volume, not high pressure, is responsible for lung injury.43 Lung tissue injury induced by alveolar hyperinflation has been termed volume trauma or volutrauma and can be avoided with the use of lower tidal volumes. In the original ARDSNet trial, patients with ARDS ventilated with an initial tidal volume of 6 ml/kg and a targeted plateau pressure of 30 cm H2O had a significantly reduced mortality compared with patients ventilated with 12 ml/kg.44
Mini Clini
Managing Hydrostatic Pressure in Patients With ARDS
 Problem
ProblemInaccuracies related to noninvasive estimates of total body fluid content are tolerable as long as the patient’s overall clinical status is improving and the hemodynamic status remains stable. However, in the setting of worsening pulmonary gas exchange or inadequate systemic DO2 despite appropriate supportive therapy, invasive measurement of intravascular volume status (e.g., CVP) often is necessary to optimize pulmonary and systemic gas exchange. In this regard, the goals of invasive monitoring should be to guide therapy so that the minimum intravascular volume associated with safe levels of inspired O2 (FiO2 < 0.6) and signs of effective circulation (e.g., urine output >0.5 ml/kg/hr, capillary refill <2 seconds) are attained. Invasive monitoring is particularly useful in the care of patients with ARDS, who have increased pulmonary capillary permeability and are consequently very sensitive to increases in pulmonary hydrostatic pressure. According to the protocol used by the National Institutes of Health National Heart, Lung and Blood Institute–sponsored ARDS Clinical Trials Network, in patients with signs of effective circulation, as described previously, a conservative fluid management strategy (i.e., CVP < 13 mm Hg) was associated with improved oxygenation and shorter duration of mechanical ventilation relative to a more liberal fluid management approach (i.e., CVP < 18 mm Hg).36
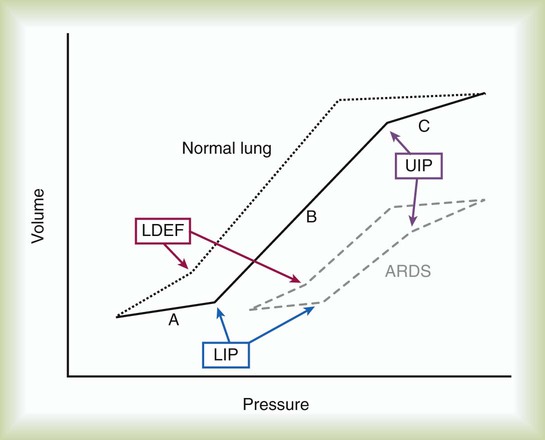
Tidal volume ideally would be selected on the basis of the patient’s individual pressure-volume relationships. Pressure-volume relationships can be established for each patient by measuring airway pressure changes over a wide range of tidal volumes. These measurements are used to describe the lower inflection point (LIP) and upper inflection point (UIP) (Figure 27-6). The UIP corresponds to the development of regional lung overdistention. Ventilatory pressure exceeding the pressure associated with the UIP is likely to cause lung injury. In contrast, the LIP represents the point in the pressure-volume curve at which dynamic collapse of alveolar units begins to occur. At ventilator pressures associated with lung volumes below the LIP, alveoli begin to collapse, and oxygenation is impaired. As airway pressure decreases to less than the LIP at end-expiration and increases to more than the LIP during the ventilatory cycle, the alveoli undergo repeated collapse and reexpansion. The shear stress induced by this cyclic opening and closing of the alveoli, particularly relating to forces generated when an alveolus opens adjacent to closed alveolar units creating tremendous distortion of the alveolar walls, represents another possible mechanism of ventilator-associated lung injury.45
Ventilator strategies designed to optimize pressure-volume relationships and minimize ventilator-associated lung injury were initially described by Amato and colleagues46 and were subsequently evaluated by the ARDS Clinical Trials Network. These strategies, termed volume-controlled ventilation (see later section on Innovative Ventilation Strategies for Acute Respiratory Distress Syndrome), have been an encouraging finding in an otherwise disappointing quest to discover effective treatments for patients with ARDS.
Adjusting Positive End Expiratory Pressure
The rationale behind the use of PEEP in the care of patients with ARDS is not directly related to its effect on lung injury. PEEP may contribute to further pulmonary injury (volutrauma) in these patients. The benefits of PEEP relate to recruitment of additional alveoli, which results in an increase in functional residual capacity (FRC) and improved oxygenation. By improving arterial oxygenation, PEEP may enable reduction of the fraction of inspired O2 (FiO2) and diminish the risk of O2 toxicity to the lungs. When patency of alveolar units is maintained throughout the ventilatory cycle, the damaging effects of opening and closing alveoli with each ventilatory cycle can be avoided. The form of lung injury occurring at lower lung volumes has been termed airway shear trauma, and the point at which it occurs is reflected by the LIP of the pressure-volume curve (see Figure 27-6).
The beneficial effects of PEEP must be balanced against the negative effects. Because the primary goal of mechanical ventilation is to provide adequate oxygenation at safe levels of FiO2 while maintaining adequate DO2 to the body, the inverse relationship between PEEP and cardiac output must be considered. One common clinical scenario involves increasing PEEP to improve arterial oxygenation at the expense of a reduction in the overall DO2. For this reason, patients who present with ARDS necessitating the use of PEEP may benefit from invasive hemodynamic monitoring. The more recent ARDS Clinical Trials Network study that evaluated low tidal volume with either high or low PEEP failed to show a survival advantage or shorter ventilator time with either measure.47 It is reasonable to use the lowest level of PEEP that maintains adequate oxygenation.
• Provide adequate oxygenation (PaO2 > 55 mm Hg) at a safe FiO2 (<0.6).
• Ensure adequate tissue oxygenation.
• Maintain the patency of alveolar units throughout the ventilatory cycle (see Figure 27-6).
• Avoid barotrauma by maintaining mean airway pressure less than 35 cm H2O or less than the pressure that corresponds to the UIP of the pressure-volume curve (see Figure 27-6).
 Rule of Thumb
Rule of Thumb
Administration of high levels of supplemental O2 (FiO2 > 0.6) for longer than 24 hours can cause lung injury as a result of O2 toxicity (oxidative stress), which advances ARDS and lung fibrosis. As the FiO2 becomes greater than 0.6, the time required to cause lung injury decreases. The FiO2 should be decreased to 0.6 as soon as possible via supportive therapy, such as positive pressure ventilation, PEEP, manipulations of pulmonary vascular pressure, or another recommended therapy (Tables 27-2 and 27-3).
TABLE 27-2
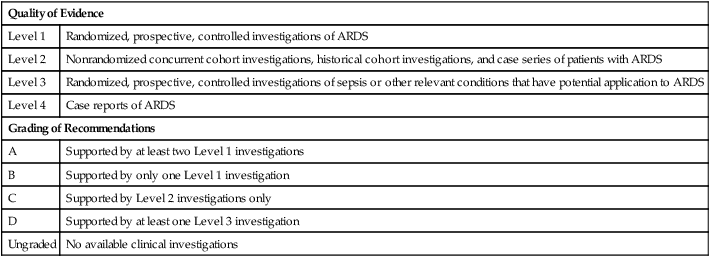
| Quality of Evidence | |
| Level 1 | Randomized, prospective, controlled investigations of ARDS |
| Level 2 | Nonrandomized concurrent cohort investigations, historical cohort investigations, and case series of patients with ARDS |
| Level 3 | Randomized, prospective, controlled investigations of sepsis or other relevant conditions that have potential application to ARDS |
| Level 4 | Case reports of ARDS |
| Grading of Recommendations | |
| A | Supported by at least two Level 1 investigations |
| B | Supported by only one Level 1 investigation |
| C | Supported by Level 2 investigations only |
| D | Supported by at least one Level 3 investigation |
| Ungraded | No available clinical investigations |
Modified from Kollef MH, Schuster D: Acute respiratory distress syndrome. Dis Mon 42:270–326, 1996.
TABLE 27-3
Recommendations for Nonpharmacologic Management of Acute Respiratory Distress Syndrome
| Treatment | Recommendation | Grade |
| Mechanical ventilation | Ungraded | |
| Initial settings: assist control mode; FiO2 1.0; PEEP ≤5 cm H2O; inspiratory flow, 60 L/min |
Yes | |
| Tidal volume, 6-10 ml/kg | Yes | C |
| Prophylactic PEEP (≤5 cm H2O) | No | B |
| Least PEEP with SaO2 ≥ 0.9 and FiO2 ≤ 0.6 | Yes | Ungraded |
| Permissive hypercapnia to maintain peak airway pressure <40-45 cm H2O and plateau pressure <35 cm H2O | Yes | C |
| Routine use of IRV | No | C |
| IRV for persistent hypoxemia or elevated airway pressure | Yes | C |
| APRV | No* | |
| HFV | No | B |
| Tracheal gas insufflation | * | |
| Partial liquid ventilation | * | |
| ECMO | No | B |
| ECCO2R | No | B |
| Patient repositioning (including prone position) | Yes | C |
| Early fluid restriction or diuresis | Yes | B |
| Supranormal DO2 goals | No | D |
*Pending results of ongoing clinical trials.
Modified from Kollef MH, Schuster D: Acute respiratory distress syndrome. Dis Mon 42:270–326, 1996.
Adjusting the Ventilatory Rate
ARDS is associated with alveolar consolidation and ventilation/perfusion mismatching, which results in a decrease in the number of normally functioning alveoli. Critically ill patients often have elevated metabolic rates so that CO2 production is increased. Compared with individuals with normal lungs, patients with ARDS require much higher minute ventilation to maintain PaCO2 in the normal range. In patients with ARDS, it is desirable to maintain lower tidal volumes and avoid volutrauma. The goal of reducing tidal volume and controlling ventilatory rate is achieved at the expense of considerable CO2 retention in patients with ARDS. In most cases, the PaCO2 increases to 60 to 80 mm Hg, and the arterial pH decreases to approximately 7.25. Subsequent metabolic compensation tends to correct the acidosis over several days.48 In some cases, the acidosis is more severe but appears to be well tolerated as long as tissue oxygenation is maintained. This ventilatory strategy has been designated permissive hypercapnia or controlled hypoventilation and often requires increased levels of sedation and, in some cases, paralysis to avoid patient discomfort owing to air hunger and a high respiratory rate.48
Animal models and human studies have confirmed the safety of controlled hypoventilation.49 Several investigations have shown a survival benefit of low-volume ventilation and permissive hypercapnia in patients with ARDS. This strategy is contraindicated in the care of patients with elevated intracranial pressure because this condition may be negatively affected by elevated PaCO2. Respiratory acidosis secondary to permissive hypercapnia may result in a further decrease in systolic blood pressure in patients with shock, especially patients receiving vasoactive medications; this generally occurs at an arterial pH less than 7.20. In these patients, parenteral replacement of bicarbonate may be considered to maintain an arterial pH of greater than 7.20 while still achieving the goal of low tidal volume ventilation.
Innovative Ventilation Strategies for Acute Respiratory Distress Syndrome
Volume-Controlled Mechanical Ventilation
Volume-controlled mechanical ventilation represents an exciting new area of ARDS research. A large, well-designed clinical trial sponsored by the National Institutes of Health ARDS Clinical Trials Network showed a significant reduction in mortality (approximately 20%) when lower tidal volumes were used in patients with ALI and ARDS.44 Volume-controlled ventilation is now considered a preferred ventilation strategy for patients with ARDS. This ventilation technique is typically adjusted to the specific pressure-volume relationships of each patient; however, a range of optimal initial ventilator settings can be derived from more recent experimental observations. A tidal volume of 10 ml/kg exceeds the volume at which the UIP is reached in more than 80% of patients with ARDS, and most patients need only 5 to 7 ml/kg tidal volume to reach this inflection point.50
On the basis of these observations, it is now recommended that the tidal volume for patients with ARDS be initiated at 5 to 7 ml/kg. Subsequent tidal volume adjustments should be made on the basis of each patient’s pressure-volume relationships (see Figure 27-6). Ideal PEEP should be determined as previously described (see Figure 27-5 and Mini Clini, “Determination of ‘Optimal’ PEEP”). Despite the convincing evidence that low-volume ventilation decreases mortality, institutional practices vary considerably, and overall compliance with this recommended approach to the management of patients with ALI is currently quite poor.51
High-Frequency Ventilation
High-frequency ventilation (HFV) was designed to maintain adequate ventilation and reduce alveolar collapse simultaneously through ventilation with high expiratory lung volumes and rapid (up to 300 breaths/min), small tidal volumes (3 to 5 ml/kg). This technique has been successfully applied to the ventilation of neonates with respiratory distress related to insufficient surfactant production.52 However, despite anecdotal evidence suggesting that HFV may be beneficial to adults with ARDS,53,54 larger clinical trials do not support the routine use of HFV in this patient population.55
Experience with the H1N1 influenza epidemic of 2009 has resulted in renewed interest in HFV. In many centers, high-frequency oscillatory ventilation (HFOV) is a rescue therapy applied to patients with inadequate response to conventional ARDS ventilator strategies.56 Although smaller trials seem to show equivalence with conventional ventilation,57 a more recent meta-analysis suggested that HFOV may provide a survival advantage relative to conventional modes of ventilation.58 Additionally, the combination of HFOV with pumpless interventional lung assist devices to remove CO2 has proven useful in small trials.59 Patients on HFOV usually have high mean airway pressures (≥30 cm H2O), and as with the use of high levels of PEEP, care must be taken that these elevated airway pressures do not overly reduce the cardiac output and the overall O2 delivery despite elevated arterial oxygenation.
Inverse-Ratio Ventilation
Inverse-ratio ventilation (IRV) is designed to recruit alveolar units through prolongation of the inspiratory phase of the ventilatory cycle and improve oxygenation. In conventional modes of mechanical ventilation, the respiratory cycle is characterized by inspiratory-to-expiratory (I : E) ratios exceeding 1 : 2. During IRV, the inspiratory time on the ventilator is prolonged so that the I : E ratio is reversed and may exceed 4 : 1. Initial reports of the use of this strategy showed significant improvement in oxygenation in patients with ARDS.60 However, these studies did not take into account other variables, such as the level of PEEP. After controlling for the level of PEEP, no change in oxygenation was observed in patients with ARDS who received IRV.61 No study has shown a significant survival benefit with this ventilator mode. Because of the discomfort associated with this mode of ventilation and the risk of asynchronous spontaneous ventilatory efforts, patients often demand heavy sedation or paralysis during IRV. The routine use of IRV in ARDS cannot be advocated at this time.
Mini Clini
Avoiding Ventilator-Induced Lung Injury
 Problem
ProblemAlveolar shear stress occurs when alveoli collapse during expiration and are reopened as the next tidal volume is delivered. Patients with ARDS are prone to development of this form of lung injury because surface tension increases as a result of impaired surfactant synthesis and function and decreased lung compliance. Alveolar collapse at end-expiration (the LIP of the pressure-volume curve) can be avoided with PEEP, which reduces alveolar shear stress by preventing the cyclic opening and closing of the alveoli during each ventilatory cycle. Because partially patent alveoli require less pressure to inflate than collapsed alveoli, PEEP also improves lung compliance. Improved compliance is reflected by an increase in the slope of the pressure-volume curve (see Figure 27-6).
Many newer ventilators can provide pressure-volume relationships. This information allows the clinician to estimate the PEEP needed to maintain alveolar patency (see Figure 27-6). Alternatively, the clinician can gauge the effects of PEEP on the basis of calculated changes in lung compliance (see Figure 27-5). In the clinical setting, changes in lung compliance are estimated from the compliance of the entire respiratory system, which includes the combined influences of lung compliance, chest wall compliance, and abdominal pressure:
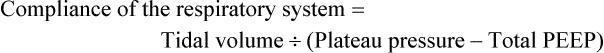
Just as alveolar collapse during end-expiration is associated with decreased lung compliance, alveolar overinflation during end-inspiration is reflected by decreasing compliance and corresponds to the UIP of the pressure-volume curve (see Figure 27-6). Tidal volumes should be adjusted so that maximal inspiratory pressure does not exceed the pressure corresponding to the UIP of the pressure-volume curve. In most patients, the UIP is attained at tidal volumes less than 10 ml/kg and plateau pressure less than 35 cm H2O.61
Pressure Control Ventilation
Pressure control ventilation (PCV) is designed to prevent ventilator-associated lung injury. The clinician sets the maximal inspiratory airway pressure. A maximal inspiratory pressure (30 to 35 cm H2O) is chosen that is likely to avert alveolar overdistention and prevent volume-associated lung injury. Minute ventilation is maintained by setting the respiratory rate. Tidal volume becomes a dynamic variable during PCV. That is, tidal volume primarily depends on the driving pressure (maximum inspiratory pressure—PEEP), airway resistance, inspiratory time, and lung compliance. Changes in intrathoracic pressure (e.g., owing to spontaneous inspiratory efforts) or airway resistance or changes in lung compliance influence tidal volume. Large swings in ventilation (increases or decreases in PaCO2) may be observed during PCV. Close monitoring of the ventilatory status of the patient must be maintained while the PCV mode is in use. Despite its theoretic benefits, PCV has not been shown to be superior to volume-controlled ventilation in clinical trials.62,63
Airway Pressure Release Ventilation
Comparisons of APRV with other forms of mechanical ventilation, including pressure support and synchronized intermittent mandatory ventilation, have shown APRV to be effective and well tolerated. In a study by Sydow and associates64 in which APRV was compared with IRV in patients with ARDS, APRV was better tolerated and was associated with lower peak airway pressures. Alveolar recruitment improved over time with APRV but not with IRV. Despite these potential advantages, APRV has not proved to be superior to conventional mechanical ventilation in large clinical trials in patients with ARDS.65
Adjunctive Strategies for Acute Respiratory Distress Syndrome
Patient Positioning
In view of the heterogeneous distribution of lung injury in patients with ARDS, it has been proposed that changing the position of the patient could result in improved ventilation/perfusion matching within the lungs. In this regard, it is known that alveolar consolidation tends to be most pronounced in the dependent lung zones in patients with ARDS where blood flow is greatest.66 These observations led investigators to experiment with positioning the patient so that aerated lung fields (nondependent lung zones) become dependent (by positioning of the patient in the prone position). Douglas and colleagues67 were the first to describe improved oxygenation with prone positioning of patients with ARDS. For unclear reasons, this ventilation strategy was not popularized until more recently, when other investigators showed similar beneficial effects in patients with ARDS.68
The mechanisms by which prone positioning improves oxygenation is the subject of ongoing debate. Some proposed mechanisms include improved matching of ventilation with perfusion, increased FRC, increased cardiac output, more effective drainage of upper and lower airway secretions, and improved diaphragmatic excursion. Investigations using animal models of ALI have shown that ventilation in the supine position causes compressive forces on the dorsal airspaces resulting in derecruitment of lung units. This phenomenon is reversed by ventilation in the prone position.69,70
A large multicenter, randomized clinical trial failed to show a survival benefit when ventilation in the prone position was used for at least 6 hours a day in the care of patients with ALI or ARDS, even though oxygenation was significantly improved in the prone position.69 A more recent trial that evaluated the implementation of prone ventilation very early in the course of ARDS and for prolonged periods appeared to show a survival benefit.71 It is also encouraging that a more recent meta-analysis suggested a survival advantage for prone ventilation, which was evident in patients with more severe hypoxemia, defined as a PaO2/FiO2 ratio less than 100 mm Hg.72
Extracorporeal Membrane Oxygenation and Extracorporeal Carbon Dioxide Removal
Extracorporeal membrane oxygenation (ECMO) was first introduced in 1972 as a form of respiratory support for patients with severe, acute hypoxemic respiratory failure. This modality involves the establishment of an arteriovenous circuit for diverting a large proportion of the cardiac output through an artificial gas exchange device, or “artificial lung,” to facilitate the exchange of CO2 and O2. Following an initial flurry of interest during the 1970s, a trial comparing ECMO plus conventional mechanical ventilation with conventional mechanical ventilation alone in ARDS showed no survival benefit with ECMO.73
Several more recent studies reported a significant improvement in survival74,75 and less severe disability at 6 months in patients who were managed with ECMO compared with patients who received conventional therapy.75 These promising results, along with others not mentioned here, have led many institutions to use ECMO as rescue therapy for patients with H1N1-induced ARDS who cannot be managed with conventional ventilatory modes or HFOV.74 The merits of ECMO for routine management of all patients with ARDS is an area of intense debate and conflicting opinions, but ECMO is currently a reasonable alternative to conventional ventilation strategies in the setting of refractory hypoxemia in patients with ARDS.76
Clinical trials comparing ECCO2R with conventional mechanical ventilation in the care of adults with ARDS have shown improved gas exchange, lower peak airway pressure, lower ventilatory rate, and reduced thoracic volume (less overinflation of the lungs) in patients treated with ECCO2R ventilation.77 This trial failed to show a survival benefit with ECCO2R at 30 days in 40 patients.
The H1N1 influenza outbreak of 2009 generated many reports supporting the use of ECCO2R employing technology, such as the Novalung (Heilbronn, Germany), in patients with ARDS failing to respond to conventional therapy.78 As experience grows in its use in specialized centers around the world, ECCO2R has become a therapy of last resort especially for viral-induced ARDS. Most hospitals have put in place contingency plans to move patients to these centers when deemed appropriate by critical care clinicians.
Pharmacologic Therapies for Acute Respiratory Distress Syndrome
Exogenous Surfactant Administration
The assumed role of surfactant deficiency in the pathogenesis of infant respiratory distress is well established. Exogenous surfactant administration is a cornerstone of therapy for infant respiratory distress syndrome. The pathogenesis of ARDS is more complex, however (see earlier section on Pathophysiology). Although surfactant is known to be qualitatively and quantitatively altered during ARDS, other factors, such as the severity of pulmonary microvascular injury, contribute to the gas exchange abnormalities associated with this condition. Nonetheless, surfactant has been shown to have immunomodulating (i.e., affecting immune function) properties that may reduce microvascular injury in the lungs. Surfactant dysfunction may contribute to the development of ARDS by promoting instability of the alveolar units (airway shear trauma, atelectasis, and right-to-left shunt) and by allowing inflammatory injury to alveoli to continue unchecked. This rationale explains the observed correlation between the degree of surfactant dysfunction and the severity of gas exchange abnormalities during adult ARDS and forms the basis for clinical trials of exogenous surfactant administration during ARDS.
Exogenous surfactant replacement is of greatest benefit in models of pure surfactant deficiency, such as infant respiratory distress or the aftermath of saline lavage. However, in adult ARDS, in which deactivation of surfactant relates to the influx of inflammatory cells and mediators into the alveolar space, the effects of exogenous surfactant are less apparent. Although initial studies showed a benefit in oxygenation with surfactant administration, either synthetic (Alveofact [Lyomark Pharma, Oberhaching, Germany])79 or a modified bovine lung surfactant (beractant [Survanta, Abbott Nutrition, Chicago, IL]),80 subsequent trials failed to show a survival benefit, and one trial showed a trend toward increasing mortality.81,82 Surfactant replacement therapy cannot be recommended for the routine management of patients with ARDS.
Neuromuscular Blockade
Neuromuscular blocking agents have been used for decades to enhance compliance with mechanical ventilation in patients with ARDS, especially when the ventilatory mode has involved low-volume ventilation, or inverse ratio ventilation. Although earlier studies showed improved oxygenation with neuromuscular blockade for the first 48 hours, they did not address any survival advantage gained from this intervention. A multicenter randomized trial considered 90-day in-hospital mortality as the outcome measure after therapeutic intervention with a bolus of the paralytic drug cisatracurium followed by a 48-hour infusion. The cisatracurium group showed an improvement in the adjusted 90-day survival, days off the ventilator, and incidence of barotrauma complications. In post hoc analysis of the study, the benefit seemed to be most evident in more severe disease as defined by a PaO2/FiO2 ratio less than 120. Although problems with muscle weakness related to neuromuscular blocker use were thought to be more likely in the intervention arm, this was not seen in practice in the study and may reflect the short duration of paralysis.83 At this time, it is difficult to make firm conclusions about the routine use of neuromuscular blockade, and further trials are needed to clarify this area.
Inhaled Nitric Oxide
The potential role of inhaled NO in ARDS is based on the idea that NO is preferentially distributed to well-ventilated portions of the lung, where it causes local vasodilation. In this way, well-ventilated areas of the lung receive a greater portion of the total pulmonary blood flow, which results in improved oxygenation by reducing ventilation/perfusion mismatch (Figure 27-7). In practice, the effects of inhaled NO in patients with ARDS have been variable. Generally, patients with high pulmonary vascular resistance, who presumably have more severe alterations in pulmonary vascular regulation, seem to benefit the most. The beneficial effects of NO can be achieved at low concentrations of inhaled NO, which explains the low incidence of systemic hypotension associated with its use. However, sudden discontinuation of inhaled NO may be associated with severe pulmonary vasoconstriction; slow weaning is necessary.
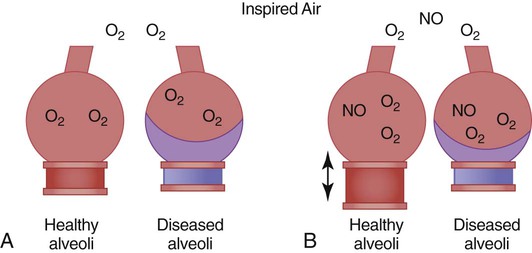
Despite encouraging results of animal studies showing tolerance of low doses of inhaled NO for 6 months, the safety and effectiveness of inhaled NO therapy for ARDS have not been established. The breakdown products of NO are potentially toxic and include highly reactive free radicals (e.g., peroxynitrite) and methemoglobin, potentially contributing to further lung injury in patients with ARDS or in individuals with normal lung function. The use of inhaled NO during mechanical ventilation involves close monitoring and special exhaust systems to prevent exposure of health care personnel to NO and its potentially toxic by-products. Many phase II and phase III studies have been completed with NO. No study has shown that NO improves outcome or mortality. Most recently, a large multicenter randomized trial involving more than 300 patients reported that NO at a dose of 5 ppm was ineffective in altering mortality, or duration of ventilator support, although there was a short-term benefit on oxygenation.84 Because of these uncertainties, inhaled NO, although promising, remains an experimental therapy for ARDS.
Inhaled Epoprostenol
Similar to NO, epoprostenol (Flolan) is a potent vasodilator with a short half-life. Epoprostenol is generally given intravenously to patients with pulmonary hypertension; however, because of the significantly lower costs compared with NO, there is interest in the use of inhaled epoprostenol in a nebulized form for refractory hypoxemia associated with ARDS. In patients with ARDS, nebulized epoprostenol is associated with improved oxygenation presumably secondary to preferential vasodilation of the good lung, as discussed earlier, leading to improvement in ventilation/perfusion matching.85 To date, there is no evidence of a mortality benefit with the use of inhaled epoprostenol to recommend its routine use in ARDS.86 Epoprostenol should be viewed as salvage therapy for refractory hypoxemia.
Corticosteroids for Late, Uncomplicated Acute Respiratory Distress Syndrome
Although most patients who survive ARDS have minimal residual pulmonary impairment, a small but significant number of patients cannot be weaned from mechanical ventilatory support because of abundant fibroproliferation during recovery from ARDS. The high mortality among these patients seems to relate to the extent and severity of pulmonary fibrosis.87 High-dose corticosteroids have been used to manage uncomplicated pulmonary fibrosis following ARDS. In a well-publicized but uncontrolled study by Meduri and coworkers,88 patients treated with corticosteroids for ARDS-related pulmonary fibrosis had improved gas exchange and low mortality (24%). Subsequently, the same investigators performed a randomized double-blind, placebo-controlled trial to determine the effect of prolonged methylprednisolone therapy for unresolving ARDS. The results were encouraging, showing improvement in lung injury scores and reduced mortality.89 The ARDS Clinical Trials Network conducted a large multicenter trial of corticosteroids for the treatment of patients with late, fibroproliferative ARDS (>7 days’ duration). The results showed no survival benefit with steroid use at 60 days and 180 days, and patients given steroids after 14 days from ARDS onset experienced a higher mortality. At this time, the routine use of corticosteroids for the treatment of established ARDS cannot be advocated and should be strictly avoided after 14 days from onset.90
Beta-2 Agonists
Although beta-2 agonists were first shown to decrease alveolar permeability to fluid in humans more than 20 years ago, the therapeutic implications of this class of drug were assessed in humans with ARDS only more recently. A cohort of patients with ALI or ARDS were randomly assigned to treatment with intravenous salbutamol (15 µg/kg/hr) or placebo for 7 days and assessed for the degree of extravascular lung water, measured by thermodilution (PiCCO) at day 7. Patients treated with salbutamol exhibited significantly lower lung water and lower plateau pressures, although there was no difference in the PaO2/FiO2 ratio at day 7, ventilator days, or 28-day mortality.91 It is not yet determined whether beta-2 agonists provide a mortality benefit or if this promising therapy will join the ever-increasing list of ineffective ARDS treatments.
Role Of The Respiratory Therapist In Acute Lung Injury And Acute Respiratory Distress Syndrome
In treating patients with ARDS and ALI, RTs are key members of the ICU team. In multiple randomized trials, RT-driven ventilator management protocols have outperformed usual care protocols in ventilator liberation.92 RT assistance in ventilator management, with regard to offering advice concerning ventilatory strategies; reinforcing the value of “low stretch” approaches to all members of the team; and performing the ventilator setups, adjustments, and checks, is essential to the care of patients with ALI and ARDS.