[level-membership-for-anesthesiology-category]
Chapter 38 Acute liver failure
DEFINITION AND AETIOLOGY
Acute liver failure (ALF) is a complex multisystemic illness that evolves after significant liver insult. The liver damage is manifest by the development of coagulopathy and encephalopathy within days or weeks of the liver injury. ALF is a heterogeneous condition incorporating a range of clinical syndromes. The dominant factors that give rise to this heterogeneity are the variable aetiology, the age of the patient and concomitant comorbidity and the duration of time over which the disease evolves. There are multiple definitions used in this disease but the one used by O’Grady et al. is most commonly used, utilising the description of acute, hyperacute and subacute.1 This system uses a trigger of jaundice and encephalopathy whilst others have used symptoms of encephalopathy. All the definitions used recognise that the syndrome may have a rapid presentation or a somewhat slower presentation, and the clinical features and outcomes are significantly different in the two presentations.
The O’Grady et al. definitions are given below.
SUBACUTE DISEASE
The aetiology of ALF must always be sought, not just for prognosis but for treatment options (Table 38.1).
| Cause | Agent responsible |
|---|---|
| Viral hepatitis | Hepatitis A, B, D, E, cytomegalovirus, herpes simplex virus, seronegative hepatitis (14–25% of cases in the UK) |
| Drug-related | Dose-related, e.g. paracetamol, and idiosyncratic reactions, e.g. antituberculous drugs, statins, recreational drugs, anticonvulsants, non-steroidal anti-inflammatory drugs, cyproterone and many others |
| Toxins | Carbon tetrachloride, Amanita phalloides |
| Vascular events | Ischaemic hepatitis, veno-occlusive disease, Budd–Chiari Heatstroke |
| Other | Pregnancy-related liver diseases, Wilson’s disease, lymphoma, carcinoma, trauma |
The relative incidence of aetiologies varies across the world. Paracetamol is common in the UK, USA and Denmark, whereas hepatitis B is more common in France. A recent paper from the USA Acute Liver Failure group highlights the increasing incidence of paracetamol toxicity and also the potential role it plays as a covert agent in patients with no history of excess ingestion and non-specific symptoms that would otherwise be attributed to the seronegative group.2 The rate of hepatitis B virus-induced ALF is decreasing with immunisation but is still highly prevalent and should always be considered.
Acute viral hepatitis accounts for 40–70% of patients with ALF worldwide. Clinical characteristics are shown in Table 38.2. Acute hepatitis A (HAV) infection rarely leads to ALF (0.35% of infections), but continues to account for up to 10% of cases; morbidity increases with the age of infection. It is hoped that the rate will decrease with improving hygiene standards generally and the uptake of vaccination. It is diagnosed by the presence of the immunoglobulin (Ig) M antibody to HAV. HAV-related ALF has a relatively good prognosis, although age and comorbidity are relevant.
Table 38.2 Aetiology of acute liver failure and initial investigations
| Hepatitis A (HAV) | Immunoglobulin M (IgM) anti-HAV |
| Hepatitis B + D (HBV, HDV) | HBsAg, IgM anti-core, HBeAg, HBeAb, HBV DNA, delta antibody |
| Hepatitis E (HEV) | IgM antibody |
| Seronegative hepatitis | All tests negative: diagnosis of exclusion |
| Paracetamol | Drug levels in blood and clinical pattern of disease – may be negative on third or subsequent days after overdose; markedly elevated aspartate and alanine serum transaminase (often > 10 000) |
| Idiosyncratic drug reactions | Eosinophil count may be elevated, although most diagnoses are based on temporal relationship |
| Ecstasy | Blood, urine, hair analysis and history |
| Autoimmune | Autoantibodies, immunoglobulin profile |
| Pregnancy-related syndromes | |
| Fatty liver | Uric acid elevated, neutrophilia, often first pregnancy, history, CT scan for rupture and assessment of vessels |
| HELLP syndrome | Platelet count, disseminated intravascular coagulation a prominent feature; CT scan as above |
| Liver rupture | May be seen in association with pre-eclampsia, fatty liver and HELLP |
| Wilson’s disease | Urinary copper, ceruloplasmin (although low in many causes of acute liver failure), present up to second decade of life, Kayser–Fleischer rings, low alkaline phosphate levels |
| Amanita phalloides | History of ingestion of mushrooms, diarrhoea |
| Budd–Chiari syndrome | Ultrasound of vessels (HV signal lost, reverse flow in portal vein), CT angiography, ascites, prominent caudate lobe on imaging, haematological assessment |
| Malignancy | Imaging and histology; increased alkaline phosphate and LDH; often imaging may be interpretated as normal |
| Ischemic hepatitis | Clinical context, marked elevation of transaminases (often > 5000); may demonstrate diated hepatic veins on ultrasound, echocardiogram |
| Heatstroke | Myoglobinuria and rhabdomyolysis are often prominent features |
CT, computed tomography; HELLP, haemolysis (microangiopathic haemolytic anaemia), elevated liver enzymes and low platelets; HIV, human immunodeficiency virus; LDH, lactate dehydrogenase.
Acute hepatitis B (HBV) has been the cause of 25–75% of instances of ALF from viral hepatitis. The liver injury is immunologically mediated with active destruction of infected hepatocytes. Diagnosis is by the presence of the IgM antibody (HBcAb) to hepatitis B core antigen. Hepatitis B surface antigen (HbsAg) is frequently negative by the time of presentation. Hepatitis B DNA should also be assayed. ALF may also be seen with hepatitis D, as either a coinfection or suprainfection. Reactivation of hepatitis B is an increasing cause of ALF and should always be considered in a patient who has received steroids or chemotherapy. High-risk patients should be screened for sAg and HBV DNA and treated with antiviral agents if they are positive. This is a recognised problem in oncology and haematology but it is also a potential risk to patients in intensive care where steroids may be administered.
Seronegative hepatitis (so called) is seen in patients in whom there are no identifiable viral causes nor obvious candidate drugs. Such patients may present with a prodromal illness and with acute or subacute maniifestations of the disease. Prognosis is less good than those with an identifiable virus and once they have poor prognostic criteria the chances of survival without liver transplant are exceptionally small. A subgroup may represent an acute autoimmune form of ALF, although many will not have any positive immmune markers such as elevated IgG or positive smooth-muscle or liver kidney antibodies. The pattern of markers shows an increased incidence of autoantibody positivity in seronegative cases and viral cases with elevated IgM in viral causes.2,3
Drug-induced hepatitis is responsible for approximately 15–25% of cases of ALF. In some patients there appears to be a true hypersensitivity reaction, and symptoms develop after a sensitisation period of 1–5 weeks, recur promptly with readministration of the drug and may be accompanied by fever, rash and eosinophilia. In others the clinical pattern is less acute. Some herbal remedies are implicated as putative hepatotoxins but their role is made more difficult to assess by the variable nature of the constituent parts. Halothane hepatitis is now almost unheard of.
FULMINANT WILSON’S DISEASE
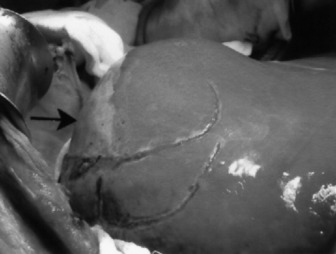
Pregnancy-related liver failure includes HELLP (haemolysis (microangiopathic haemolytic anaemia), elevated liver enzymes and low platelets), acute fatty liver of pregnancy and liver rupture, often in association with pre-eclampsia. The prognosis of pregnancy-related ALF is usually good, although some develop severe liver injury with small-vessel disease, liver ruptures may require packing and occasionally transplantation is required (Figure 38.1).
Heat shock injury is now relatively rarely seen but the ischaemic hepatitides remain relatively common. They are normally associated with a congested liver that is subjected to a secondary insult – hypoxia or decreased-flow arterial inflow. This is seen with hypoxaemic respiratory failure, cardiac arrhythmias and hypotension.
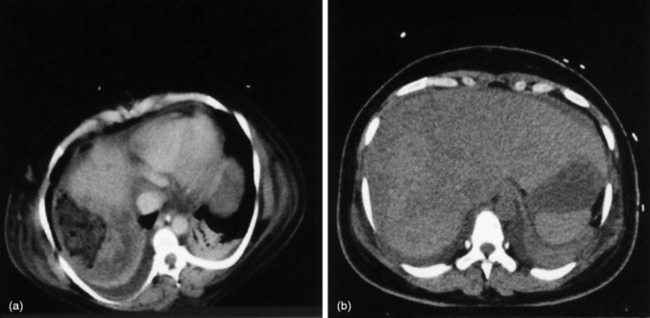
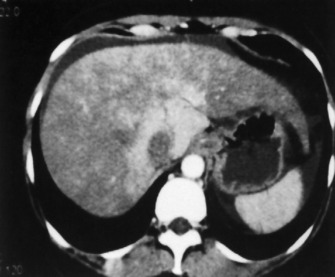
Hepatic venous obstruction (Budd–Chiari syndrome) may cause ALF. There are symptoms and signs of liver of necrosis, often with capsular pain from congestion and ascites. In Asia this may be associated with anatomical anomalies of the inferior vena cava, whereas in Europe and the USA the experience is normally of thrombosis of the hepatic veins, often with an underlying procoagulant condition (Figures 38.2 and 38.3).
DIAGNOSIS
The aetiology of ALF must be accurately identified; some specific investigations are outlined in Table 38.2. History and clinical examination are paramount in this disease and the clinical course of biochemical and haematological parameters is important in assessing course and management. All patients should have routine chemistry, haematology and coagulation assessment; viral and autoimmune profiles should similarly be undertaken in all.
The biopsy is normally undertaken by the transjugular route.
SPECIFIC TREATMENTS
SPECIFIC CONSIDERATIONS
Chelating agents are not of benefit in patients with established ALF secondary to Wilson’s disease but play an important role in chronic presentations. Withdrawal of such treatments, or indeed non-compliance, especially in teenagers, may precipitate ALF.
Steroid therapy is beneficial in acute autoimmune hepatitis but its role in autoimmune ALF has been less clear. A recent publication from the Paris group suggests that steroids for those with established ALF are potentially detrimental.4
CLINICAL COURSE, COMPLICATIONS AND MANAGEMENT
ENCEPHALOPATHY
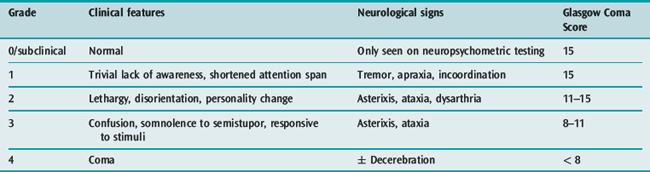
Encephalopathy is a clinical diagnosis, graded from 1 to 4 depending on clinical severity (Table 38.3). The development of encephalopathy is essential for the diagnosis of ALF. Patients with acute and hyperacute liver failure are at greatest risk of developing grade IV coma and cerebral oedema.
Clear aetiological factors in the development of encephalopathy remain under discussion but it is believed that there is a build-up of putative toxins, of which ammonia is thought to be most pertinent, resulting in glutamine intracerebral accumulation and the development of cerebral oedema.5–7 In ALF there is a clear relationship between the development of higher grades of coma and arterial ammonia levels (cut-off between 150 and 200 μmol/l) renal dysfunction and aetiology.8–10 The relationship in CLD exists but is less clear.
The development of cerebral oedema and intracranial hypertension in patients with ALF is thought to relate, amongst other things, to the speed of onset. In patients with CLD, adaptation occurs and there is improved control of intracellular osmolarity (myoinositol) so that cellular oedema is rare and of low grade.11
The association between the development of encephalopathy and markers of inflammation is well demonstrated, both in regard to systemic inflammatory response syndrome (SIRS) markers and inflammatory mediators such as tumour necrosis factor.12,13 Curiously, a similar relationship between inflammation and encephalopathy is seen in patients with CLD.14 What is not yet clear is whether treatments that modulate the inflammatory response will be of benefit. This and other avenues proposed from basic animal research may well result in several novel approaches to the treatment of cerebral oedema and possibly encephalopathy over the forthcoming years.
Cerebral blood flow shows marked variability in patients with ALF. Hyperaemia has increased prevalence in those who develop intracranial hypertension. The role of hyperventilation has not been shown to be beneficial in routine management but plays a role in the treatment of increased intracranial pressure (ICP) with associated hyperaemia.15
Cerebral perfusion pressure of greater than 60 mmHg has been suggested as optimal. It is of note that patients with ALF often do not autoregulate to pressure and consequently increases in blood pressure may be associated with increased cerebral blood flow and potentially increased ICP, particularly if they are at a critical point on their pressure–volume curve. This promotes the use of ICP monitoring in patients requiring pressor agents.8 Cardiovascular failure necessitates invasive monitoring to optimise treatment and to determine the optimal therapeutic agents. The issue of noradrenaline (norepinephrine) versus vasopressin or terlipressin is difficult. In one study terlipressin resulted in a significant increase in ICP but in another there were no such detrimental effects.14,16 It may be that the effects of terlipressin are highly individual and potentially time course-dependent, again supporting the role of ICP monitoring in these situations.
Carbon dioxide
Arterial ammonia – both the level (levels greater than 150–200 μmol/l) and the failure of this level to fall with treatment – may help in prognostication.8,10 The monitoring may comprise reverse jugular monitoring, middle cerebral artery Doppler measurements and ICP monitoring. ICP monitoring is more controversial as it has not been shown specifically to improve outcome (few monitoring devices in isolation have altered outcome). A recent retrospective study from the US Acute Liver Failure group showed that ICP monitoring carried a complication rate of 10%: half of the complications were serious and two-thirds possibly contributed to mortality. Monitoring did result in more treatment interventions being undertaken.17
Hyponatraemia and hyperammonaemia have been shown to be detrimental, whereas a randomised controlled trial showed benefit to the patients whose serum Na was maintained between 140 and 150 mmol/l. (Bolus hypertonic saline, 30%, was used for episodes of ICP greater than 25 mmHg.)18
Some patients remain resistant to treatment and in these cases other therapies may be considered. Hypothermia has been shown to decrease cerebral blood flow, decrease ICP and decrease cerebral ammonia uptake in one clinical study.19,20 The role of hypothermia as an early preventive intervention in grade III/IV coma is contentious and, given the problems of hypothermia in the neurosurgical arena, the results of controlled studies are awaited. Fever should be avoided in these patients and hypothermia reserved for intracranial hypertension that is resistant to standard treatment.
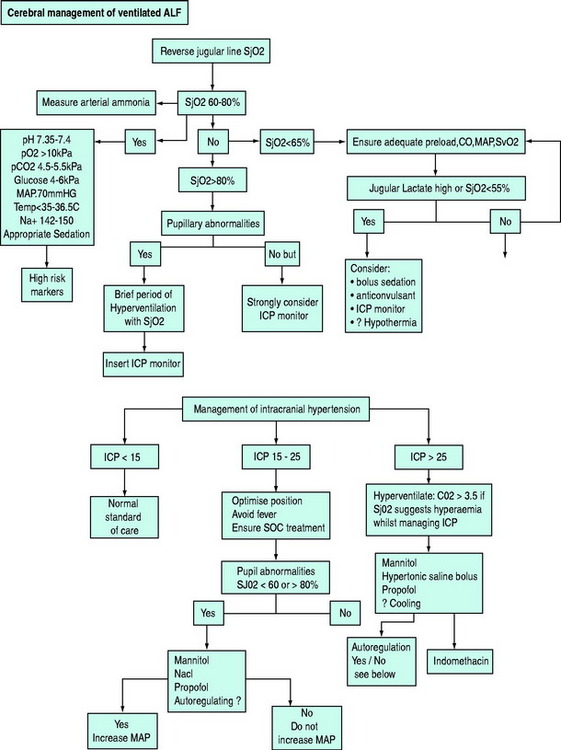
Other treatment options that have been shown to be potentially beneficial are thiopental and intravenous indometacin (0.5 mg/kg).21 Potential monitoring and treatment algorithms, as used by this unit, are seen in Figure 38.4.
SEPSIS
Sepsis is common in ALF and both culture-positive and negative SIRS are seen in ALF patients. They are functionally immunosuppressed in terms of impaired cell-mediated immunity, complement levels and phagocytosis.22 Functional immmunoparesis can only be observed and depression of human leukocyte antigen (HLA) DR expression correlates with prognosis and severity of liver injury.23,24 As such, scrupulous attention with regard to hand-washing and line care needs to be applied to decrease the risk of nosocomial infection. Regular culture screens are required and antimicrobials are indicated in patients with any clinical suggestion of sepsis. Prophylactic intravenous antifungals should be considered, especially in those listed for transplantation. The choice of antimicrobial agent should be driven by local resistance patterns. Antimicrobial therapy should be reviewed in the light of culture results on a daily basis.
CARDIOVASCULAR
The requirement for pressor agents should raise the possibility of adrenal dysfunction. Patients with ALF have been demonstrated to have impaired response to adrenocorticotrophic hormone (ACTH).25 26 The response at 30 and 60 minutes in terms of cortisol should be examined following 250 μg ACTH in all patients with ALF who are requiring pressor agents. A subnormal response should result in consideration of hydrocortisone replacement therapy, normally given for a period of 10 days. Interestingly, adrenal dysfunction has similarly been reported in acute-on-chronic liver failure and steroid replacement may result in improved outcome.26,27
Elevated troponin levels can be observed in patients with liver failure, especially those with cardiovascular failure.28
RENAL
ANTICOAGULATION AND COAGULOPATHY
A balance needs to be achieved between risk of bleeding and platelet protection across an extracorporeal filter. A prostaglandin such as epoprostenol may be advantageous in terms of decreasing bleeding and prolonging filter life. Alternatively, circuits may be run without anticoagulation, with regional heparin or citrate or low-dose systemic heparin. In patients with thrombocytopenia consideration should be given to the diagnosis of heparin-induced thrombocytopenia and, if confirmed, heparin should be withdrawn and circuits primed with an alternative agent, e.g. lepirudin.
METABOLIC AND FEEDING
Enteral nutrition should commence as soon as feasible after admission if there are no contraindications. In patients with large aspirates (> 200 ml/4 hours) a prokinetic agent should be commenced: erythromycin (250 mg intravenously 6-hourly) appears to be more effective than metoclopromide. Endoscopic placement of a postpyloric feeding tube should be considered in refractory cases, although this may need to delayed in patients with or at risk of cerebral oedema. In patients with profound coagulopathy, placement of a nasogastric tube may be associated with nasal/pharyngeal bleeding and oral tube placement may be preferred in ventilated patients. The optimal nature of enteral feed used has not been investigated but metabolic data on these patients demonstrate increased calorific requirements.27,29 Patients with ALF demonstrate both peripheral and hepatological insulin resistance.30 Tight glycaemic control would seem reasonable in this population.
Hyperlactataemia may be secondary to volume depletion and hence will resolve with appropriate fluid loading or may reflect the inability of the liver to metabolise the lactate produced. A failure of blood lactate to normalise following volume loading is associated with a poor prognosis.30,31 Metabolic acidosis may be a secondary effect of other drugs ingested as part of an episode of self-harm. Falls in serum phosphate levels are seen in ALF associated with liver regeneration and are associated with a good prognosis in paracetamol-induced ALF.32 Pancreatitis is a common complication of ALF and should be actively sought.
PROGNOSIS
Prognostication is important in the management of ALF. It is essential to identify those patients who will not survive without liver transplant but also to identify those who will succumb even if offered such a procedure.33 Several risk stratification systems are presented in Table 38.4. The most commonly used are those of O’Grady and Clichy. The model for end-stage liver disease (MELD) has also been examined with regard to prognosis in ALF and may be particularly useful in non-paracetamol cases.34 It is essential that such systems are rigorously applied and in the context of paracetamol are only utilised at least 24 hours postingestion and following appropriate volume resuscitation.
| O’Grady criteria |
| Paracetamol-related |
| Acidosis (pH < 7.3) or |
| Prothrombin time of > 100 seconds (INR > 6.5), creatinine > 300 µmol/l and grade III/IV encephalaopthy – all occurring within a 24-hour time frame |
| Non-paracetamol-related |
| Any three of the following in association with encephalopathy: |
| Age less than 10 or greater than 40 years |
| Bilirubin > 300 µmol/l |
| Time from jaundice to encephalopathy > 7 days |
| Aetiology: either non-A, non-B (seronegative hepatitis) or drug-induced |
| Prothrombin time > 50 seconds |
| or |
| Prothrombin time > 100 seconds/INR > 6.5 |
| French criteria (Clichy criteria) |
| The criteria are the presence of encephalopathy (coma or confusion) and |
| Age < 20 years with factor V level < 20% |
| or |
| Factor V levels < 30% if greater than 30 years of age59 |
INR, international normalised ratio.
CIRRHOSIS AND ACUTE-ON-CHRONIC LIVER DISEASE
The cause of decompensation should be sought in all cases when it is not clinically apparent. Common causes include sepsis, dehydration, drug therapies, e.g. opiates and sedatives, hepatocellular carcinoma (HCC) and portal vein thrombosis.
Sepsis should be actively sought and treated. In patients with ascites a diagnostic tap should always be undertaken for microbiological culture and cell count (a polymorphonuclear count > 250/mm3 is indicative of bacterial peritonitis).
Alcoholic hepatitis is a frequent cause of decompensation and requires aggressive treatment. The severity can be assessed using the Glasgow score or Madrey score and in high-risk patients steroid therapy may be considered.36,37 Response to steroids over the first 7 days is associated with improved outcome.38 The role of antioxidants has been examined but no benefit was seen.39,40 Another approach has been to look at the value of enteral feeding, which was comparable with steroid treatment over a 4-week period, albeit in a small study.41 The use of pentoxifylline has been reported in a single-centre study to decrease the risk of developing hepatorenal failure and hence impacting on outcome.42
ENCEPHALOPATHY
Therapies to alleviate encephalopathy aim to decrease ammonia levels and thus focus on bowel cleansing with agents such as lactulose. There is little evidence-based research to support any particular avenue of treatment. A recent trial examining the role of albumin dialysis in the management of encephalopathy has been recently completed.43
As noted previously, the role of inflammation seems of importance in the development of encephalopathy.44,45 The role of feeding and encephalopathy has always caused controversy. Recent guidance suggests that protein restriction is not appropriate and a study examining early versus slow introduction of protein into enteral nutrition showed no increase in encephalopathy and indeed appeared beneficial in respect of nitrogen balance.46,47
VARICEAL HAEMORRHAGE
The management of variceal haemorrhage remains that of basic resuscitation and care of the airway. Coagulation factors require appropriate supplementation along with other blood products. The role of sepsis in variceal haemorrhage has become clearer, with sepsis being a frequent precipitant of bleeding. Cultures should be taken and all patients with variceal haemorrhage should be given antibiotics and this has been shown to decrease the risk of rebleeding.48,49 Splanchnic vasoconstrictors, such as glypressin, are beneficial in controlling oesophageal haemorrhage but their role in gastric variceal haemorrhage per se has not been examined.50,51 Banding ligation therapy remains the treatment of choice for oesophageal haemorrhage, with tissue adhesives being utilised in gastric varices. Failure to control variceal bleeding after two endoscopic sessions should result in consideration of TIPS insertion.52,53 In patients in whom TIPS might be considered to be of benefit in controlling variceal bleeding, consideration needs to be given to the severity of the underlying liver disease that may in its own right make TIPS ill advised.54
RENAL FAILURE
Hepatorenal failure is seen in cirrhosis – the rapidly progressive form is more commonly seen in the critical care environment. It should be noted that this is a diagnosis of exclusion of other causes. The evidence base for colloid therapy (to ensure adequate central volume repletion) along with splanchnic vasoconstrictor therapy (glypressin) is reasonable but more recently data suggest that constrictors such as noradrenaline may be equally applied.51,55–57 Prevention of renal failure should also be considered with appropriate volume resuscitation in patients with sepsis and bacterial peritonitis. Ascitic drainage should be undertaken with appropriate albumin loading if large-volume drainage is undertaken or in patients at risk of central volume depletion.58
In acute Budd–Chiari syndrome the combination of encephalopathy and renal failure should lead to consideration for transplantation. Increasingly, TIPS shunts have a place in the earlier management of acute Budd–Chiari syndrome. Such an undertaking should be performed at a centre providing transplantation since a proportion of patients will decompensate with encephalopathy and severe ALF, requiring urgent transplantation.
1 O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342:273-275.
2 Lee WM. Acute liver failure in the United States. Semin Liver Dis. 2003;23:217-226.
3 Bernal W, Ma Y, Smith HM, et al. The significance of autoantibodies and immunoglobulins in acute liver failure: a cohort study. J Hepatol. 2007;47:664-670.
4 Ichai P, Duclos-Vallee JC, Guettier C, et al. Usefulness of corticosteroids for the treatment of severe and fulminant forms of autoimmune hepatitis. Liver Transpl. 2007;13:996-1003.
5 Jalan R, Olde Damink SW, Hayes PC, et al. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol. 2004;41:613-620.
6 Rose C, Ytrebo LM, Davies NA, et al. Association of reduced extracellular brain ammonia, lactate, and intracranial pressure in pigs with acute liver failure. Hepatology. 2007;46:1883-1892.
7 Tofteng F, Hauerberg J, Hansen BA, et al. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2006;26:21-27.
8 Bernal W, Hall C, Karvellas CJ, et al. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844-1852.
9 Bhatia V, Singh R, Acharya SK. Predictive value of arterial ammonia for complications and outcome in acute liver failure. Gut. 2006;55:98-104.
10 Clemmesen JO, Larsen FS, Kondrup J, et al. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648-653.
11 Shawcross DL, Balata S, Olde Damink SW, et al. Low myo-inositol and high glutamine levels in brain are associated with neuropsychological deterioration after induced hyperammonemia. Am J Physiol Gastrointest Liver Physiol. 2004;287:G503-G509.
12 Rolando N, Wade J, Davalos M, et al. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734-739.
13 Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755-764.
14 Shawcross DL, Davies NA, Mookerjee RP, et al. Worsening of cerebral hyperemia by the administration of terlipressin in acute liver failure with severe encephalopathy. Hepatology. 2004;39:471-475.
15 Strauss GI, Moller K, Holm S, et al. Transcranial Doppler sonography and internal jugular bulb saturation during hyperventilation in patients with fulminant hepatic failure. Liver Transpl. 2001;7:352-358.
16 Eefsen M, Dethloff T, Frederiksen HJ, et al. Comparison of terlipressin and noradrenalin on cerebral perfusion, intracranial pressure and cerebral extracellular concentrations of lactate and pyruvate in patients with acute liver failure in need of inotropic support. J Hepatol. 2007;47:381-386.
17 Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11:1581-1589.
18 Murphy N, Auzinger G, Bernel W, et al. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39:464-470.
19 Jalan R, Damink SW, Deutz NE, et al. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet. 1999;354:1164-1168.
20 Jalan R, Olde Damink SW, Deutz NE, et al. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001;34:50-54.
21 Tofteng F, Larsen FS. The effect of indomethacin on intracranial pressure, cerebral perfusion and extracellular lactate and glutamate concentrations in patients with fulminant hepatic failure. J Cereb Blood Flow Metab. 2004;24:798-804.
22 Wade J, Rolando N, Philpott-Howard J, et al. Timing and aetiology of bacterial infections in a liver intensive care unit. J Hosp Infect. 2003;53:144-146.
23 Antoniades CG, Berry PA, Davies ET, et al. Reduced monocyte HLA-DR expression: a novel biomarker of disease severity and outcome in acetaminophen-induced acute liver failure. Hepatology. 2006;44:34-43.
24 Clapperton M, Rolando N, Sandoval L, et al. Neutrophil superoxide and hydrogen peroxide production in patients with acute liver failure. Eur J Clin Invest. 1997;27:164-168.
25 Harry R, Auzinger G, Wendon J. The clinical importance of adrenal insufficiency in acute hepatic dysfunction. Hepatology. 2002;36:395-402.
26 Marik PE. Adrenal-exhaustion syndrome in patients with liver disease. Intens Care Med. 2006;32:275-280.
27 Fernandez J, Escorsell A, Zabalza M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: effect of treatment with hydrocortisone on survival. Hepatology. 2006;44:1288-1295.
28 Parekh NK, Hynan LS, De Lemos J, et al. Elevated troponin I levels in acute liver failure: is myocardial injury an integral part of acute liver failure? Hepatology. 2007;45:1489-1495.
29 Walsh TS, Wigmore SJ, Hopton P, et al. Energy expenditure in acetaminophen-induced fulminant hepatic failure. Crit Care Med. 2000;28:649-654.
30 Clark SJ, Shojaee-Moradie F, Croos P, et al. Temporal changes in insulin sensitivity following the development of acute liver failure secondary to acetaminophen. Hepatology. 2001;34:109-115.
31 Bernal W, Donaldson N, Wyncoll D, et al. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: a cohort study. Lancet. 2002;359:558-563.
32 Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology. 2002;36:659-665.
33 Blei AT. Selection for acute liver failure: have we got it right? Liver Transpl. 2005;11(Suppl. 2):S30-S34.
34 Yantorno SE, Kremers WK, Ruf AE, et al. MELD is superior to King’s College and Clichy’s criteria to assess prognosis in fulminant hepatic failure. Liver Transpl. 2007;13:822-828.
35 Schiodt FV, Rossaro L, Stravitz RT, et al. Gc-globulin and prognosis in acute liver failure. Liver Transpl. 2005;11:1223-1227.
36 Forrest EH, Evans CD, Stewart S, et al. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut. 2005;54:1174-1179.
37 Forrest EH, Morris J, Stewart S, et al. The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids. Gut. 2007;56:1743-1746.
38 Mathurin P, Abdelnour M, Ramond MJ, et al. Early change in bilirubin levels is an important prognostic factor in severe alcoholic hepatitis treated with prednisolone. Hepatology. 2003;38:1363-1369.
39 Phillips M, Curtis H, Portmann B, et al. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis – a randomised clinical trial. J Hepatol. 2006;44:784-790.
40 Stewart S, Prince M, Bassendine M, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47:277-283.
41 Cabre E, Rodriguez-Iglesias P, Caballeria J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology. 2000;32:36-42.
42 Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637-1648.
43 Hassanein TI, Tofteng F, Brown RS, et al. Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology. 2007;46:1853-1862.
44 Shawcross DL, Davies NA, Williams R, et al. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol. 2004;40:247-254.
45 Shawcross DL, Wright G, Olde Damink SW, et al. Role of ammonia and inflammation in minimal hepatic encephalopathy. Metab Brain Dis. 2007;22:125-138.
46 Marchesini G, Bianchi G, Rossi B, et al. Nutritional treatment with branched-chain amino acids in advanced liver cirrhosis. J Gastroenterol. 2000;35(Suppl. 12):7-12.
47 Alvarez MA, Cabre E, Lorenzo-Zuniga V, et al. Combining steroids with enteral nutrition: a better therapeutic strategy for severe alcoholic hepatitis? Results of a pilot study. Eur J Gastroenterol Hepatol. 2004;16:1375-1380.
48 Hou MC, Lin HC, Liu TT, et al. Antibiotic prophylaxis after endoscopic therapy prevents rebleeding in acute variceal hemorrhage: a randomized trial. Hepatology. 2004;39:746-753.
49 Fernandez J, Ruiz del Arbol L, Gomez C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology. 2006;131:1049-1056. quiz 285
50 Escorsell A, Ruiz del Arbol L, Planas R, et al. Multicenter randomized controlled trial of terlipressin versus sclerotherapy in the treatment of acute variceal bleeding: the TEST study. Hepatology. 2000;32:471-476.
51 Gluud LL, Kjaer MS, Christensen E. Terlipressin for hepatorenal syndrome. Cochrane Database Syst Rev. 4, 2006. CD005162
52 Khan S, Tudur Smith C, Williamson P, et al. Portosystemic shunts versus endoscopic therapy for variceal rebleeding in patients with cirrhosis. Cochrane Database Syst Rev. 4, 2006. CD000553
53 Thabut D, Bernard-Chabert B. Management of acute bleeding from portal hypertension. Best Pract Res Clin Gastroenterol. 2007;21:19-29.
54 Schepke M, Roth F, Fimmers R, et al. Comparison of MELD, Child–Pugh, and Emory model for the prediction of survival in patients undergoing transjugular intrahepatic portosystemic shunting. Am J Gastroenterol. 2003;98:1167-1174.
55 Alessandria C, Ottobrelli A, Debernardi-Venon W, et al. Noradrenalin vs terlipressin in patients with hepatorenal syndrome: a prospective, randomized, unblinded, pilot study. J Hepatol. 2007;47:499-505.
56 Moreau R, Durand F, Poynard T, et al. Terlipressin in patients with cirrhosis and type 1 hepatorenal syndrome: a retrospective multicenter study. Gastroenterology. 2002;122:923-930.
57 Moreau R, Lebrec D. The use of vasoconstrictors in patients with cirrhosis: type 1 HRS and beyond. Hepatology. 2006;43:385-394.
58 Moreau R, Asselah T, Condat B, et al. Comparison of the effect of terlipressin and albumin on arterial blood volume in patients with cirrhosis and tense ascites treated by paracentesis: a randomised pilot study. Gut. 2002;50:90-94.
59 Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology. 1986;6:648-651.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 38 Acute liver failure
DEFINITION AND AETIOLOGY
Acute liver failure (ALF) is a complex multisystemic illness that evolves after significant liver insult. The liver damage is manifest by the development of coagulopathy and encephalopathy within days or weeks of the liver injury. ALF is a heterogeneous condition incorporating a range of clinical syndromes. The dominant factors that give rise to this heterogeneity are the variable aetiology, the age of the patient and concomitant comorbidity and the duration of time over which the disease evolves. There are multiple definitions used in this disease but the one used by O’Grady et al. is most commonly used, utilising the description of acute, hyperacute and subacute.1 This system uses a trigger of jaundice and encephalopathy whilst others have used symptoms of encephalopathy. All the definitions used recognise that the syndrome may have a rapid presentation or a somewhat slower presentation, and the clinical features and outcomes are significantly different in the two presentations.
The O’Grady et al. definitions are given below.
SUBACUTE DISEASE
The aetiology of ALF must always be sought, not just for prognosis but for treatment options (Table 38.1).
| Cause | Agent responsible |
|---|---|
| Viral hepatitis | Hepatitis A, B, D, E, cytomegalovirus, herpes simplex virus, seronegative hepatitis (14–25% of cases in the UK) |
| Drug-related | Dose-related, e.g. paracetamol, and idiosyncratic reactions, e.g. antituberculous drugs, statins, recreational drugs, anticonvulsants, non-steroidal anti-inflammatory drugs, cyproterone and many others |
| Toxins | Carbon tetrachloride, Amanita phalloides |
| Vascular events | Ischaemic hepatitis, veno-occlusive disease, Budd–Chiari Heatstroke |
| Other | Pregnancy-related liver diseases, Wilson’s disease, lymphoma, carcinoma, trauma |
The relative incidence of aetiologies varies across the world. Paracetamol is common in the UK, USA and Denmark, whereas hepatitis B is more common in France. A recent paper from the USA Acute Liver Failure group highlights the increasing incidence of paracetamol toxicity and also the potential role it plays as a covert agent in patients with no history of excess ingestion and non-specific symptoms that would otherwise be attributed to the seronegative group.2 The rate of hepatitis B virus-induced ALF is decreasing with immunisation but is still highly prevalent and should always be considered.
Acute viral hepatitis accounts for 40–70% of patients with ALF worldwide. Clinical characteristics are shown in Table 38.2. Acute hepatitis A (HAV) infection rarely leads to ALF (0.35% of infections), but continues to account for up to 10% of cases; morbidity increases with the age of infection. It is hoped that the rate will decrease with improving hygiene standards generally and the uptake of vaccination. It is diagnosed by the presence of the immunoglobulin (Ig) M antibody to HAV. HAV-related ALF has a relatively good prognosis, although age and comorbidity are relevant.
Table 38.2 Aetiology of acute liver failure and initial investigations
| Hepatitis A (HAV) | Immunoglobulin M (IgM) anti-HAV |
| Hepatitis B + D (HBV, HDV) | HBsAg, IgM anti-core, HBeAg, HBeAb, HBV DNA, delta antibody |
| Hepatitis E (HEV) | IgM antibody |
| Seronegative hepatitis | All tests negative: diagnosis of exclusion |
| Paracetamol | Drug levels in blood and clinical pattern of disease – may be negative on third or subsequent days after overdose; markedly elevated aspartate and alanine serum transaminase (often > 10 000) |
| Idiosyncratic drug reactions | Eosinophil count may be elevated, although most diagnoses are based on temporal relationship |
| Ecstasy | Blood, urine, hair analysis and history |
| Autoimmune | Autoantibodies, immunoglobulin profile |
| Pregnancy-related syndromes | |
| Fatty liver | Uric acid elevated, neutrophilia, often first pregnancy, history, CT scan for rupture and assessment of vessels |
| HELLP syndrome | Platelet count, disseminated intravascular coagulation a prominent feature; CT scan as above |
| Liver rupture | May be seen in association with pre-eclampsia, fatty liver and HELLP |
| Wilson’s disease | Urinary copper, ceruloplasmin (although low in many causes of acute liver failure), present up to second decade of life, Kayser–Fleischer rings, low alkaline phosphate levels |
| Amanita phalloides | History of ingestion of mushrooms, diarrhoea |
| Budd–Chiari syndrome | Ultrasound of vessels (HV signal lost, reverse flow in portal vein), CT angiography, ascites, prominent caudate lobe on imaging, haematological assessment |
| Malignancy | Imaging and histology; increased alkaline phosphate and LDH; often imaging may be interpretated as normal |
| Ischemic hepatitis | Clinical context, marked elevation of transaminases (often > 5000); may demonstrate diated hepatic veins on ultrasound, echocardiogram |
| Heatstroke | Myoglobinuria and rhabdomyolysis are often prominent features |
CT, computed tomography; HELLP, haemolysis (microangiopathic haemolytic anaemia), elevated liver enzymes and low platelets; HIV, human immunodeficiency virus; LDH, lactate dehydrogenase.
Acute hepatitis B (HBV) has been the cause of 25–75% of instances of ALF from viral hepatitis. The liver injury is immunologically mediated with active destruction of infected hepatocytes. Diagnosis is by the presence of the IgM antibody (HBcAb) to hepatitis B core antigen. Hepatitis B surface antigen (HbsAg) is frequently negative by the time of presentation. Hepatitis B DNA should also be assayed. ALF may also be seen with hepatitis D, as either a coinfection or suprainfection. Reactivation of hepatitis B is an increasing cause of ALF and should always be considered in a patient who has received steroids or chemotherapy. High-risk patients should be screened for sAg and HBV DNA and treated with antiviral agents if they are positive. This is a recognised problem in oncology and haematology but it is also a potential risk to patients in intensive care where steroids may be administered.
Seronegative hepatitis (so called) is seen in patients in whom there are no identifiable viral causes nor obvious candidate drugs. Such patients may present with a prodromal illness and with acute or subacute maniifestations of the disease. Prognosis is less good than those with an identifiable virus and once they have poor prognostic criteria the chances of survival without liver transplant are exceptionally small. A subgroup may represent an acute autoimmune form of ALF, although many will not have any positive immmune markers such as elevated IgG or positive smooth-muscle or liver kidney antibodies. The pattern of markers shows an increased incidence of autoantibody positivity in seronegative cases and viral cases with elevated IgM in viral causes.2,3
Drug-induced hepatitis is responsible for approximately 15–25% of cases of ALF. In some patients there appears to be a true hypersensitivity reaction, and symptoms develop after a sensitisation period of 1–5 weeks, recur promptly with readministration of the drug and may be accompanied by fever, rash and eosinophilia. In others the clinical pattern is less acute. Some herbal remedies are implicated as putative hepatotoxins but their role is made more difficult to assess by the variable nature of the constituent parts. Halothane hepatitis is now almost unheard of.
FULMINANT WILSON’S DISEASE
Pregnancy-related liver failure includes HELLP (haemolysis (microangiopathic haemolytic anaemia), elevated liver enzymes and low platelets), acute fatty liver of pregnancy and liver rupture, often in association with pre-eclampsia. The prognosis of pregnancy-related ALF is usually good, although some develop severe liver injury with small-vessel disease, liver ruptures may require packing and occasionally transplantation is required (Figure 38.1).
Heat shock injury is now relatively rarely seen but the ischaemic hepatitides remain relatively common. They are normally associated with a congested liver that is subjected to a secondary insult – hypoxia or decreased-flow arterial inflow. This is seen with hypoxaemic respiratory failure, cardiac arrhythmias and hypotension.
Hepatic venous obstruction (Budd–Chiari syndrome) may cause ALF. There are symptoms and signs of liver of necrosis, often with capsular pain from congestion and ascites. In Asia this may be associated with anatomical anomalies of the inferior vena cava, whereas in Europe and the USA the experience is normally of thrombosis of the hepatic veins, often with an underlying procoagulant condition (Figures 38.2 and 38.3).
DIAGNOSIS
The aetiology of ALF must be accurately identified; some specific investigations are outlined in Table 38.2. History and clinical examination are paramount in this disease and the clinical course of biochemical and haematological parameters is important in assessing course and management. All patients should have routine chemistry, haematology and coagulation assessment; viral and autoimmune profiles should similarly be undertaken in all.
The biopsy is normally undertaken by the transjugular route.
SPECIFIC TREATMENTS
SPECIFIC CONSIDERATIONS
Chelating agents are not of benefit in patients with established ALF secondary to Wilson’s disease but play an important role in chronic presentations. Withdrawal of such treatments, or indeed non-compliance, especially in teenagers, may precipitate ALF.
Steroid therapy is beneficial in acute autoimmune hepatitis but its role in autoimmune ALF has been less clear. A recent publication from the Paris group suggests that steroids for those with established ALF are potentially detrimental.4
