Acute Kidney Injury
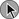
Definition
The primary function of the kidneys is the excretion of metabolic waste products and the maintenance of the composition of body fluids through regulation of water, electrolyte, and acid-base excretion. Acute kidney injury (AKI) describes a sudden decrease in glomerular filtration rate (GFR), resulting in the retention of metabolic waste products and dysregulation of fluid, electrolyte, and acid-base homeostasis.1–3 AKI, however, does not represent a discrete disease. Rather, AKI is a heterogeneous spectrum that includes hemodynamic perturbations that disrupt normal renal perfusion and decrease GFR without causing overt parenchymal injury; partial or complete obstruction to urine flow; and acute parenchymal injury resulting in glomerular, interstitial, tubular, or vascular dysfunction. Although all forms of AKI can occur in those who are critically ill, the most common causes in this population include hemodynamically mediated prerenal dysfunction and acute tubular necrosis (ATN) arising as a result of ischemia-reperfusion injury, nephrotoxin exposure, or sepsis.
The term acute kidney injury has largely replaced the older terminology of acute renal failure (ARF).3–6 Implicit in this older terminology was a focus on the most severe presentations of acute kidney dysfunction, generally characterized by overt organ failure. The term AKI attempts to broaden the focus to include less severe episodes that, although not resulting in overt organ failure, are associated with increased risks of morbidity and mortality, particularly in the critically ill. However, the use of the term injury may incorrectly connote the presence of parenchymal organ damage, which may be absent in a number of forms of AKI, particularly in hemodynamically mediated prerenal states and acute obstructive uropathy. In this chapter, the term AKI will be used to describe the entire spectrum of the syndrome, and the term ARF will be restricted to severe organ failure requiring renal replacement therapy. Often, the terms ATN and AKI have been used interchangeably; although ATN is the most common form of intrinsic AKI, particularly in critically ill patients, the terms are not synonymous, because ATN represents only one of the multiple forms of AKI.
The cardinal manifestation of AKI is the retention of metabolic waste products, most notably creatinine and urea.1,2 Decreased urine output may also be a manifestation of AKI; however, the urine output in AKI can be highly variable, ranging from virtual anuria (<100 mL/day) to polyuria (>3 L/day).1 In addition, transient oliguria (urine volume <400 mL/day) may occur in the absence of decreased kidney function in patients with intravascular volume depletion as the physiologic response to volume depletion due to an increase in tubular salt in water reabsorption.7 In contrast, persistent oliguria despite adequate volume resuscitation is virtually always a manifestation of AKI.7 Although the presence of oliguria complicates management, increasing the risk of volume overload, hyperkalemia, and other electrolyte disturbances, and is associated with a greater mortality risk, therapeutic interventions to augment urine output have not been shown to improve outcomes.
A wide array of operational definitions have been used to define AKI based primarily on relative or absolute changes in serum creatinine concentration.14 The first attempt at developing a consensus definition was undertaken by the Acute Dialysis Quality Initiative in 200215; the resultant RIFLE criteria consisted of three strata (“Risk,” “Injury,” and “Failure”) based on the magnitude of increase in serum creatinine and the duration of oliguria as well as two outcome stages (“Loss” of kidney function and “End-Stage” kidney disease) (Table 55.1). In the RIFLE criteria, the “Risk,” “Injury,” and “Failure” strata were defined based on an increase in serum creatinine developing over 7 days of 50%, 100%, or 200%, respectively, relative to baseline or oliguria, defined as a urine output of less than 0.5 mL/kg/hour for more than 6 hours (“Risk”) or 12 hours (“Injury”) or a urine output of less than 0.3 mL/kg/hour for more than 24 hours or anuria for more than 12 hours (“Failure”). These three strata were proposed as providing increasing specificity, although decreasing sensitivity, for the diagnosis of AKI. The outcome criteria were defined based on continued need for renal replacement therapy for more than 4 weeks (“Loss”) or more than 3 months (“Failure”). The RIFLE criteria were subsequently modified by the Acute Kidney Injury Network (AKIN) with the addition of an absolute increase in serum creatinine of more than 0.3 mg/dL to the definition of AKI, a decrease in the time frame for the increase in serum creatinine from 7 days to no more than 48 hours, and by dropping the two outcome criteria.4,5 More recently, the Kidney Disease Improving Global Outcomes (KDIGO) Clinical Practice Guideline for Acute Kidney Injury harmonized the RIFLE and AKIN definitions, maintaining the three stages, but specifying that the 0.3-mg/dL increase in serum creatinine was to have developed over no more than 48 hours but that the greater than 50% increase could occur over up to 7 days (see Table 55.1).3
Epidemiology
Estimates of the incidence of AKI are highly dependent on both the precise definition employed and the characteristics of the patient population studied. Many of the epidemiologic studies that have been published either preceded the development of the standardized definitions just described16–24 or have utilized administrative data that do not correlate with these definitions.25–28 In addition, there are important differences in the distribution of the different types of AKI among patients who develop AKI prior to hospitalization, on general medical-surgical wards, or in the intensive care unit. As a result, published estimates of the incidence of AKI in hospitalized patients have ranged from as high as 44%, using a 0.3-mg/dL change in serum creatinine as the definition to less than 1% when the definition was based on an increase is serum creatinine of more than 2 mg/L, with most estimates of the incidence of AKI ranging from 3% to 7% of the overall hospital population and 10% to 35% of critically ill patients, and 5% to 6% of the ICU (intensive care unit) population having AKI severe enough to require renal replacement therapy.16–24
The incidence of AKI has increased substantially over the past several decades. In an analysis of data from the National Hospital Discharge Survey in the United States conducted by the Centers for Disease Control and Prevention, the incidence of AKI among hospitalized patients increased from 18 per 100,000 population in 1980 to 365 per 100,000 population in 2005 with similar trends present in analyses of data from the U.S. National Inpatient Sample and a 5% sample of hospitalized Medicare beneficiaries.28 These data need to be interpreted with some caution; however, administrative coding for AKI is incomplete, capturing only 20% to 30% of all episodes, and trends over time may be subject to bias from changes in administrative coding practices.29 However, similar trends were observed in an analysis of administrative and clinical data from Kaiser Permanente of Northern California, an integrated health care delivery system.30 Using laboratory creatinine data to confirm the diagnosis of AKI, the incidence of AKI not requiring renal replacement therapy increased from 323 to 522 cases per 100,000 person-years between 1996 and 2003; over the same time period, the incidence of AKI requiring renal replacement therapy increased from 19.5 to 29.5 cases per 100,000 person-years. Epidemiologic studies have consistently found that AKI is more prevalent in men and in African Americans and increases in incidence with increasing age.25,26 Increasing risk of AKI is also associated with severity of baseline chronic kidney disease (CKD). In the aforementioned study from Kaiser Permanente of Northern California, the risk of developing AKI requiring dialysis was approximately double in patients with a baseline estimated GFR between 45 and 60 mL/minute/1.73 m2 as compared to patients with an estimated GFR greater than 60 mL/minute/1.73 m2, with the risk increasing to more than 40-fold among patients with a premorbid estimated GFR less than 15 mL/minute/1.73 m2.31 Other identified independent risk factors for the development of AKI include diabetes mellitus, hypertension, and the presence of proteinuria.31,32
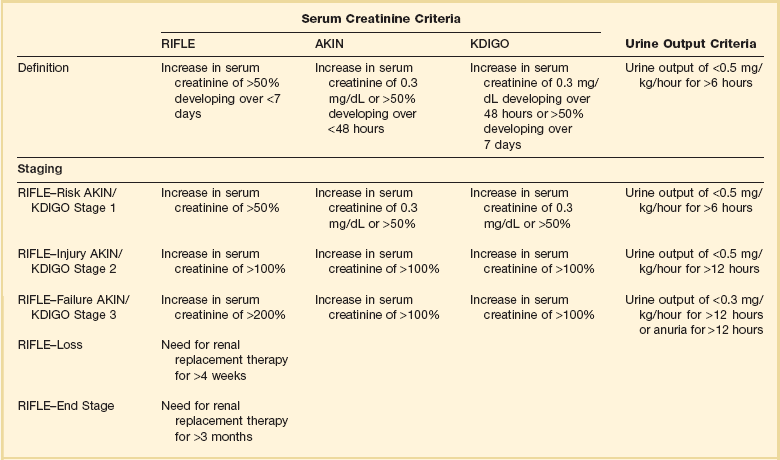
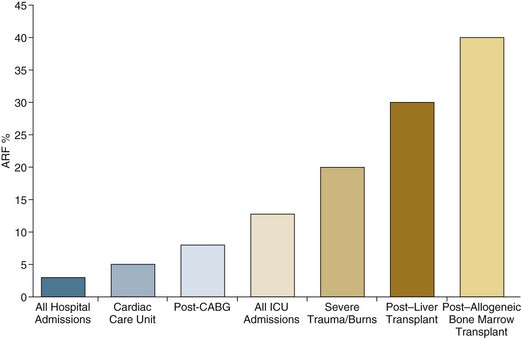
Although AKI is common in critically ill patients, robust epidemiologic data are lacking as the majority of published studies represent the experience at single centers or limited numbers of ICUs.19–23,33,34 In addition to varying criteria used to define AKI, the incidence of AKI is also highly dependent upon the characteristics of the ICU population. As shown in Figure 55.1, the incidence of AKI is lower in the cardiac care unit and in patients after coronary artery bypass grafting, being 5% to 10%,35,36 as compared to patients with severe trauma,37 burns,38 and following liver and allogeneic bone marrow transplant, in whom AKI occurs in 20% to 40%.39 The patterns of AKI in the ICU differ from those seen in the general hospital population (Table 55.2). Patients with ICU-associated AKI are younger, are more likely to be male, are more likely to have acute tubular necrosis as compared to prerenal AKI or obstructive disease, are more likely to have AKI in association with multisystem organ failure as opposed to isolated AKI, are more likely to require renal replacement therapy, and have markedly higher mortality rates.19 Sepsis is the most common predisposing condition for the development of AKI in critically ill patients, contributing to the development of AKI in as many as half of all cases.19,21,22,24 Other conditions associated with the development of AKI in ICU patients include major surgery, cardiogenic shock, hypovolemia, medication-associated toxicity, and advanced liver disease (Table 55.3).19,21,22,24 Two distinct patterns of ICU-associated AKI have been described: early-onset AKI is present on ICU admission or within the first 48 hours of ICU stay, and late-onset AKI develops after more than 48 hours of ICU care.23 Delayed-onset AKI is more likely to be due to postischemic or sepsis-associated AKI and is associated with higher mortality rates.
Table 55.2
Comparison of Intensive Care Unit (ICU)– and General Hospital Ward–Associated Acute Kidney Injury
From Liano F, Junco E, Pascual J, et al. The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. Kidney Int 1998;53(Suppl 66):S16-S24.
Table 55.3
Factors Associated with Acute Kidney Injury in the Intensive Care Unit Setting
| Factor | Frequency (%) |
| Multiorgan failure | 30-75 |
| Sepsis | 30-50 |
| Drugs/medications | 20-40 |
| Postoperative state | 15-35 |
| Cardiogenic shock | 15-30 |
| Hypovolemia | 15-30 |
| Liver failure | 5-10 |
Pathogenesis
AKI can be divided into three broad etiologic categories: prerenal AKI, intrinsic AKI, and postrenal (obstructive AKI). Prerenal states (Box 55.1) are characterized by effective hypoperfusion of the kidneys leading to a decrease in GFR in the absence of overt parenchymal damage to the kidneys. Postrenal or obstructive AKI is characterized by acute obstruction of the urinary tract, including ureters or bladder outlet (Box 55.2). In intrinsic AKI, acute injury to the renal parenchyma underlies the development of kidney dysfunction, as is seen in acute tubular necrosis (ATN), acute interstitial nephritis (AIN), and acute glomerulonephritis (AGN) (Box 55.3). Categorizing the forms of AKI in this fashion is useful for didactic purposes and is helpful in guiding the initial assessment of the patient with AKI, but substantial overlap may exist among these categories, particularly between prerenal and intrinsic causes of AKI. For example, renal hypoperfusion may cause a spectrum of renal dysfunction ranging from mild prerenal azotemia to overt ATN, depending upon its severity and duration. Even in patients with classic clinical presentations of volume depletion–associated prerenal AKI sensitive clinical markers of tubular injury may be elevated.40 Thus, precise categorization of the cause of AKI into one of these categories may not be possible, and overlap and transition among categories may occur.
Prerenal Acute Kidney Injury
Prerenal azotemia caused by extracellular fluid volume loss or volume sequestration, reduced cardiac output, systemic vasodilation, intrarenal vasoconstriction, or increased renal venous pressure is the most common cause of AKI (see Box 55.1), contributing to the development 30% to 60% of all cases.17–19,24 The primary pathogenesis of prerenal azotemia is a decrease in effective glomerular perfusion. This form of AKI, if treated early, is usually readily reversible; however, if left untreated, renal ischemia with resultant ATN may result.
In classic forms of prerenal azotemia, reduced renal perfusion pressure and afferent arteriolar constriction combine to lower glomerular capillary hydrostatic pressure and the formation of glomerular ultrafiltrate.1,41,42 Prerenal azotemia develops when the capacity of the usual physiologic responses to hypovolemia is exceeded and GFR falls. In response to hypovolemia there is a decrease in mean arterial pressure, triggering baroreceptors that ultimately lead to activation of the sympathetic nervous system, activation of the renin-angiotensin-aldosterone system (RAAS), and secretion of the antidiuretic hormone vasopressin. Activation of the renal sympathetic nerves constricts the afferent (preglomerular) arterioles and stimulates release of renin from the juxtaglomerular apparatus. Renin secretion is also directly stimulated in response to hypovolemia by changes in intrarenal hemodynamics. Secretion of renin activates a cascade in which angiotensinogen is cleaved to form angiotensin I, which is then cleaved by angiotensin-converting enzyme released by local endothelium to form angiotensin II. Angiotensin II stimulates both afferent and efferent (postglomerular) arteriolar vasoconstriction; however, the effect on the afferent vessel is opposed by vasodilatory prostaglandins, kallikrein, kinins, and nitric oxide.43,44 The net effect is to maintain the tone of the afferent arteriole while constricting the efferent vessel, returning intraglomerular pressure and glomerular ultrafiltration toward normal. In addition, angiotensin II stimulates proximal tubular sodium reabsorption and, through its action as an aldosterone secretagogue, increases plasma aldosterone levels and stimulates distal tubular sodium reabsorption. Increased vasopressin levels stimulate water and urea reabsorption in the collecting duct. The net effect of modest hypovolemia is maintenance of the GFR at near normal levels accompanied by production of concentrated urine with low sodium content and a decrease in the fractional excretion of urea. When effective volume depletion exceeds the normal physiologic compensatory mechanisms, the GFR falls and overt prerenal AKI ensues.
Multiple medications can exacerbate the development of prerenal AKI. The prerenal form of AKI complicating diuretic use is caused by extracellular volume depletion. With nonsteroidal anti-inflammatory drugs (NSAIDs), including both nonselective and cyclooxygenase 2 (COX-2)–selective agents, the inhibition of cyclooxygenase leads to depletion of renal vasodilatory prostaglandins that normally counteract the afferent arteriolar constricting effect of angiotensin II and increased renal adrenergic tone.45–47 In situations in which vasoconstrictor mechanisms are activated, such as volume depletion, heart failure, sepsis, cirrhosis, and nephrotic syndrome, and in patients with chronic kidney disease, the use of NSAIDs may result in severe afferent vasoconstriction, severely reducing glomerular capillary filtration pressure and causing AKI. If promptly discontinued, NSAID-induced AKI can be reversible; however, if NSAIDs continue to be administered ATN may ensue.
Angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and direct renin inhibitors act by inhibiting the RAAS. Inhibition of angiotensin II production by direct renin inhibitors and ACEIs or blockade of its action by ARBs prevents angiotensin II–mediated constriction of the efferent arteriole and diminishes glomerular capillary filtration pressure. Although this effect is desirable in preventing hyperfiltration injury in patients with CKD, the use of these agents may contribute to the development of AKI in the setting of renal hypoperfusion, when maintenance of glomerular filtration is dependent upon activation of renin and angiotensin II.48 This is particularly likely to happen when therapy is initiated or escalated and volume depletion or hypotension supervenes in patients with renal artery stenosis, CKD, heart failure, or liver disease. The prerenal azotemia that may develop in these settings is usually reversible if the offending agent is promptly discontinued; however, these agents have also been associated with an increased risk of ATN.49
Although the majority of forms of prerenal AKI are mediated by decreased renal perfusion, altered intrarenal hemodynamics may result in decreased GFR despite increased organ perfusion. Early in sepsis, renal perfusion may be increased; however, GFR may fall as the result of decreased efferent arteriolar tone, resulting in decreased glomerular capillary filtration pressure.50
Increases in renal venous pressure may also contribute to the development of prerenal AKI. As renal venous pressure increases, renal perfusion pressure falls; in addition, renal parenchymal pressure increases as venous pressure rises, narrowing the pressure gradient driving glomerular filtration.51,52 Two settings in which renal venous congestion results in a prerenal state are the abdominal compartment syndrome and heart failure. Abdominal compartment syndrome (ACS) typically develops in critically ill patients, most commonly in the setting of trauma with abdominal hemorrhage, abdominal surgery, massive fluid resuscitation, liver transplantation, and gastrointestinal conditions including peritonitis and pancreatitis. ACS is defined by an intra-abdominal pressure of 20 mm Hg or higher associated with dysfunction of one or more organ systems.53 However, intra-abdominal pressures lower than 20 mm Hg may be associated with ACS, while values higher than this threshold do not universally lead to the ACS.54–57 Oliguria, which can lead to anuria, often develops and, as is true for other forms of AKI associated with impaired renal perfusion, urine sodium concentration is commonly reduced. Renal venous congestion associated with modest increases in intra-abdominal pressure has also been shown to contribute to the acute renal dysfunction accompanying acute decompensated heart failure.58,59
Postrenal Acute Kidney Injury
AKI resulting from obstruction usually accounts for fewer than 5% of ICU-associated AKI.19 Urinary tract obstruction may occur at any level of the renal collecting system, beginning at the renal pelvis and ending at the urethra (see Box 55.2). Obstruction above the level of the bladder is referred to as upper tract obstruction and may result from either intrinsic (intraluminal) or extrinsic disease. The development of AKI from upper tract obstruction requires the presence of bilateral obstruction or unilateral obstruction in the setting of a single functioning kidney or dysfunction of the contralateral kidney. Common causes for upper tract obstruction include luminal obstruction from ureteric calculi, blood clots, urothelial tumors, sloughed renal papillae or ureteral strictures, and external compression from retroperitoneal and pelvic tumors, adenopathy, hematomas, or retroperitoneal fibrosis. More commonly, obstruction occurs at the level of the bladder neck or urethra, as may occur with benign prostatic hypertrophy or prostate cancer, bladder tumors, and bladder stones. Neurogenic bladder dysfunction associated with diabetes mellitus, autonomic neuropathy, and spinal cord disease may also contribute to the development of obstructive disease. Treatment with narcotics and agents with anticholinergic action may exacerbate subclinical voiding dysfunction and precipitate acute urinary retention in the setting of acute illness.
Patients with obstructive disease may present with anuria if obstruction is complete, with normal or even increased urine volume in the setting of partial obstruction, or with fluctuating urine output with periods of anuria alternating with rapid passage of urine as the pressure in the collecting system rises and overcomes the obstructing disease. In the acute phase of obstruction, intratubular pressure rises, resulting in a reduction in glomerular filtration pressure. As the obstruction persists, renal blood flow diminishes, intratubular pressures fall toward normal, but GFR remains severely depressed. Relief of the obstruction with placement of a bladder catheter for lower tract obstruction or ureteral stents or percutaneous nephrostomy tubes for upper tract obstruction usually results in prompt return of GFR if the duration of obstruction has not been excessive.60
Intrinsic Acute Kidney Injury
The causes of intrinsic AKI can be categorized based on the anatomic compartments of the kidney that are involved (see Box 55.3). Using this approach, intrinsic AKI is commonly divided into tubular, interstitial, glomerular, and vascular processes. In addition, in a variety of settings the primary pathophysiology consists of intratubular deposition of crystals or protein. These latter syndromes could be considered as a subset of acute tubular disease, but given the striking difference in pathogenesis, these disorders will be discussed separately in this chapter. It is also important to recognize that although this approach provides a construct for understanding the multitude of causes of intrinsic AKI, there are substantial overlaps among categories. Thus, small vessel vasculitides with predominant glomerular involvement could be classified either with acute glomerulonephritis or as a renal vascular disorder. Similarly, interstitial inflammation may play a substantial role in the pathogenesis of ATN, blurring the distinction with acute interstitial nephritis (AIN).
Acute Tubular Necrosis
The most common intrinsic cause of AKI is ATN, accounting for 85% to 90% of intrinsic ICU-associated AKI.19,24 The causes of ATN can be broken down into three major categories: ischemia-reperfusion injury, nephrotoxins, and sepsis (see Box 55.3). There is, however, significant overlap between septic and ischemic ATN; many, although not all, cases of sepsis-associated ATN develop in the setting of septic shock, and sepsis-associated ATN has frequently been classified as a subcategory of ischemic ATN.61 Data suggest, however, that sepsis-associated ATN has unique features and may develop in the absence of overt renal ischemia and should be considered as a separate etiologic category.62–64 Nephrotoxic ATN can result from toxicity from the endogenous heme pigments myoglobin and hemoglobin or may be caused by exogenous toxins, most commonly medications such as aminoglycosides, amphotericin B, and cisplatin. In many patients, multiple etiologic factors are present and the ATN must be characterized as multifactorial.
A description of the pathophysiologic mechanisms thought to underlie ischemic, septic, and nephrotoxic causes of ATN are provided in the online supplement at www.expertconsult.com.
Although the characteristic histologic lesion in ATN is epithelial cell injury with tubular cell necrosis or apoptosis, the pathophysiology of ATN also includes endothelial cell damage and activation of inflammatory pathways. Although all segments of the nephron may be affected, proximal tubular epithelial cells at the corticomedullary junction and outer stripe of the medulla are most susceptible to ischemic injury.65 More proximal tubular segments are involved in toxic nephropathy as a result of their transport and endocytotic pathways that lead to increased cellular uptake of the toxin.
Ischemic injury results in depletion of cellular adenosine triphosphate (ATP) in the tubular epithelial cells leading to disruption of the cytoskeleton.66 Maintenance of the cytoskeleton is critical to the functional integrity of the tubular epithelium; its loss leads to disruption of the luminal (apical) brush border membrane and of the adhesion proteins that are critical for maintaining attachment to the basement membrane and for the integrity of the tight junctions between adjacent cells. With their loss, transport proteins such as Na+/K+-ATPase that are normally restricted to the apical or basolateral domains can migrate and normal vectorial transport of sodium and other solutes from tubular lumen to blood is disrupted.67 This may account, in part, for the elevated urine sodium and fractional excretion of sodium often seen in patients with ATN. The loss of attachment to the basement membrane leads to the sloughing of cells into the tubular lumen. Severe ATP depletion may lead to cell necrosis, but less severe injury activates apoptotic pathways, which appear to be the predominant mechanism of cell death in ATN.68 In addition, ATP depletion leads to a rise in intracellular calcium, which serves as a further mediator of cell damage.
In addition to epithelial injury, endothelial damage and microvascular dysfunction play a central role in the pathogenesis of ischemic and septic ATN. Endothelial damage results in increased expression of adhesion molecules, leukocyte and platelet adhesion, activation of coagulation pathways, vascular congestion, and obstruction resulting in diminished blood flow through the renal microvasculature.69 The endothelial and epithelial cell injury also leads to recruitment of leukocytes and activation of inflammatory pathways.70,71 The combination of endothelial damage and inflammation may amplify the tubular injury, even after the inciting insult has resolved.69
The reduction in GFR in ATN is the result of several factors.1 Early in ischemic ATN, the combined influence of a decrease in renal perfusion pressure and afferent arteriolar constriction initiates the reduction in glomerular filtration. Later, endothelial damage and microvascular obstruction contribute to the persistence of decreased glomerular filtration. Sloughed debris from necrotic and apoptotic tubular epithelial cells and nonadherent viable cells form obstructing intratubular casts, increasing intratubular hydrostatic pressure and further decreasing glomerular filtration. When these casts pass into the urine they are the “muddy brown” granular casts that are characteristic of ATN. Finally, “back-leak” of filtrate across the denuded tubular basement membrane contributes to the loss of effective GFR.
The clinical course of AKI can be conceptualized as consisting of four phases.72 The initial inciting injury is followed by an extension phase during which the endothelial and inflammatory processes result in extension of the tubular injury despite resolution of the triggering insult. The extension phase is followed by a maintenance phase of variable duration during which the GFR remains very low. During this phase, tubular epithelial cells are believed to undergo a process of dedifferentiation and proliferation, repopulating the tubular basement membrane. Finally there is a recovery phase, in which the tubular epithelial cells redifferentiate and reestablish a normal tubular epithelium and GFR recovers. To the extent that repair to the tubular epithelium is complete, kidney function can recover to near normal levels; however, to the extent that fibrotic pathways are activated and there is rarefaction of the capillary bed, recovery of kidney function will be incomplete.73–75
ATN also exhibits distant organ effects. In experimental models, renal ischemia reperfusion injury is associated with alterations in cardiac, pulmonary, hepatic, and brain function.76–78 It is postulated that these distant organ effects are mediated by systemic inflammatory changes, activation of proapoptotic pathways, increases in leukocyte trafficking, and dysregulated channel expression and that these distant effects may contribute to nonrenal organ dysfunction in patients with ATN.
The precise mechanisms by which sepsis causes ATN have not been elucidated. In septic shock, renal hypoperfusion likely predominates.61 In less severe sepsis the pathophysiology is thought to include a combination of intrarenal hemodynamic changes; endothelial dysfunction; infiltration of the renal interstitium by lymphocytes, neutrophils, and mononuclear cells; and activation of both pro- and anti-inflammatory mechanisms.62 The precise role of individual mediators in the pro- and anti-inflammatory pathways, including tumor necrosis factor-α interleukin (IL) 1, IL-6, and IL-10, is uncertain.
The pathophysiologic process by which nephrotoxins induce renal damage is incompletely understood and most likely differs from agent to agent.46,79 Several of the more commonly encountered nephrotoxins in the ICU setting are aminoglycoside antibiotics, amphotericin B, and iodinated radiocontrast agents. Aminoglycosides generally induce mild (mean rise in serum creatinine of 1 to 3 mg/dL), nonoliguric ATN in 5% to 20% of patients receiving a 7- to 10-day course of therapy.80,81 However, even brief exposure in the setting of impaired renal perfusion can induce some renal damage. Aminoglycosides are avidly taken up by the proximal tubular epithelial brush border via a megalin-dependent mechanism. Once within the cell they are transported to the endoplasmic reticulum and then enter the cytosol. Once in the cytosol, the aminoglycoside disperses to various intracellular organelles, such as mitochondria, where it mediates organelle-specific toxicity.82 Risk factors for aminoglycoside nephrotoxicity include the use of high doses or prolonged courses of therapy, volume depletion, diabetes mellitus, advanced age, baseline CKD and the presence of hypovolemia, renal ischemia, or concomitant administration of other nephrotoxins.81,83
Radiocontrast agents are relatively nontoxic in healthy individuals with an incidence of contrast nephropathy of less than 1%.84,85 Risk factors for the development of contrast-induced AKI include preexistent kidney disease, diabetes mellitus, effective intravascular volume depletion, concomitant therapy with NSAIDs, and exposure to high doses and repeated doses of the contrast agent. The precise mechanism of toxicity is not understood. Administration of radiocontrast agents induces marked renal vasoconstriction, mediated in part by endothelin. The osmotic load and altered flow through the microvasculature due to the high viscosity of the contrast media induces medullary hypoxia. The hypoxia as well as direct effects of the contrast agents induces the generation of reactive oxygen species that may cause membrane and DNA damage. In addition, direct cytotoxicity to renal epithelial cells can be demonstrated. The risk of contrast nephropathy can be mitigated by the use of relatively less toxic low- and isosmolar agents, as compared to the older and more nephrotoxic high-osmolar agents and by administration of intravenous fluids to correct volume depletion and maintain high urine flow rates.
Myoglobin and hemoglobin are endogenous heme-containing proteins that are associated with the development of ATN.86,87 Myoglobinuric AKI is much more common than hemoglobinuric disease. Myoglobin is released during muscle injury and is freely filtered by the glomerulus. In contrast, hemoglobin released during intravascular hemolysis is normally bound by haptoglobin; hemoglobinuria will only develop when hemolysis is severe enough to overwhelm the binding capacity of haptoglobin, such as may occur with an ABO-incompatible blood transfusion. Renal injury in pigment nephropathy is due to a combination of factors including volume depletion, particularly in rhabdomyolysis, in which there is sequestration of fluid in the injured muscle, renal vasoconstriction, direct heme-pigment mediated cytotoxicity, and intraluminal cast formation. Despite the toxicity of the heme pigments, AKI is uncommon in the absence of volume depletion, acidosis, and possibly mild ischemia. Prevention of nephropathy in patients with rhabdomyolysis or hemolysis focuses on maintaining adequate extracellular fluid volume and renal perfusion.
Acute Interstitial Nephritis
AIN constitutes approximately 5% to 10% of intrinsic AKI in the ICU setting and is defined by the presence of lymphocytic infiltration of the kidney (see Box 55.3). AIN is most commonly related to medication use but may also be associated with a wide variety of infections, autoimmune disorders, and malignancy.88 The most common offending medications include penicillin, cephalosporin, sulfonamide, and quinolone antibiotics; proton pump inhibitors; phenytoin; allopurinol; and NSAIDs. The classic presentation of medication-associated AIN includes fever, rash, and eosinophilia; however, this complete triad is present in fewer than one third of patients and is unusual in NSAID-associated AIN. Characteristic urinary findings include hematuria and pyuria and white blood cell casts in the absence of infection. Eosinophiluria may also be present but is neither a sensitive nor a specific finding. Although the diagnosis may be made based on clinical setting, history of medication use, and urinary findings, definitive diagnosis usually requires kidney biopsy.
Acute Vascular Syndromes
Acute vascular syndromes are divided into large vessel (renal artery and renal vein) and small vessel disease. Large vessel diseases include thrombosis, thromboembolism and dissection of the renal arteries, and renal vein thrombosis.89,90 Acute arterial or venous occlusion may present with renal infarction. With unilateral or subtotal disease, the presentation usually consists of pain and hematuria. Lactate dehydrogenase levels will be elevated and the diagnosis made by contrast-enhanced computed tomography (CT) or nuclear scintigraphy. As with obstructive disease, AKI is the presenting manifestation only when there is bilateral involvement or unilateral disease with an absent or nonfunctional contralateral kidney.
Multiple conditions can affect the renal microvasculature and present with AKI, including the systemic vasculitides (discussed earlier) that present with primarily glomerular involvement. Other diseases with predominant renal microvascular involvement leading to AKI include malignant hypertension, scleroderma renal crisis, toxemia of pregnancy, and the microangiopathic hemolytic diseases, hemolytic-uremic syndrome, and thrombotic thrombocytopenic purpura. Atheroembolic disease represents another important disease of the renal microvasculature that may present as acute or subacute kidney disease.91,92 Atheroembolic disease is characterized by the systemic showering of atheromatous debris to the distal branches of the arterial tree. Although atheroembolism may occur spontaneously, it develops most commonly after cardiac or aortic angiography or surgery, or in association with systemic anticoagulation or thrombolytic therapy. Involvement is generally systemic with common manifestations including cutaneous livedo reticularis and digital ischemia and gastrointestinal, hepatic, and nervous system involvement. Because the kidneys receive approximately 20% of the cardiac output, renal involvement is common, ranging from renal infarction to AKI to a subacute and stuttering decline in kidney function.
Intratubular Obstruction
AKI may also occur as the result of crystalline or proteinaceous obstruction of the renal tubules. In acute tumor lysis syndrome, AKI often results from intratubular deposition of uric acid.93,94 Following ethylene glycol ingestion and in certain inborn errors of metabolism, the major mechanism for development of AKI is intratubular deposition of calcium oxalate. A number of medications have also been associated with acute crystalline nephropathy, including acyclovir, methotrexate, indinavir, triamterene, and sulfadiazine.95,96 In multiple myeloma, tubular obstruction by monoclonal light chains (Bence-Jones protein) is the cause of acute myeloma kidney disease.97,98
Clinical Manifestations
Retention of Filtration Markers
Urea
Urea is the major end product of nitrogen metabolism. Blood urea concentration, commonly assayed as blood urea nitrogen (BUN), is dependent upon the balance between hepatic synthesis and renal excretion. Increased urea generation resulting in elevations in the BUN in the absence of a marked decrease in GFR may result from high dietary protein intake, amino acid loading during parenteral nutrition, and the endogenous protein load from gastrointestinal hemorrhage as well as in hypercatabolic states associated with fever, sepsis, and glucocorticoid administration or inhibition of protein synthesis as may be seen with tetracycline antibiotics. Hypercatabolic states may also result in increases in BUN during AKI that exceed the increase of 10 to 20 mg/dL/day that usually is typically associated with absence of glomerular filtration.99 Normally, filtered urea is partially reabsorbed along the length of the nephron, with increased reabsorption in states of low urine flow. In volume depletion, severe heart failure, and obstructive uropathy, increased tubular reabsorption of urea often results in increases in the BUN that are disproportionate to the fall in GFR. This variability in the synthesis of urea and the non-GFR-related factors that influence its urinary excretion render the BUN a less reliable marker of GFR than the serum creatinine concentration. However, the level of BUN generally correlates with symptoms of renal failure, with uremic manifestations usually absent until the BUN is greater than 100 mg/dL.
Creatinine
Creatinine is derived from the nonenzymatic hydrolysis of creatine, which is usually released at a constant rate from skeletal muscle and is excreted primarily by filtration at the glomerulus. In patients with normal kidney function, less than 10% of creatinine excretion occurs by tubular secretion, although this percentage increases in CKD. There is essentially no tubular reabsorption of creatinine. This relationship allows creatinine to serve as a reliable endogenous marker of glomerular filtration when the creatinine concentration is in steady state. Because much of the interindividual variability in creatinine production can be accounted for based on demographic and clinical variables including age, gender, race, and weight, it is possible to reliably estimate creatinine clearance or GFR from the serum creatinine concentration.8–10,12 The confidence intervals around these estimates are wide, however, and these estimates should not be interpreted as precise measures of kidney function. In the absence of glomerular filtration, serum creatinine typically increases by 1 to 2 mg/dL/day.100 This increase is influenced by numerous factors including the magnitude of decrement in GFR, the rate of creatinine production, and changes in the volume of distribution.100 For example, creatinine production may be reduced in sepsis,101 and increased in the setting of skeletal muscle injury (rhabdomyolysis). Aggressive volume resuscitation may also mask any increase in serum creatinine through dilution.102 Thus, although a sudden increase in serum creatinine concentration is the most common parameter triggering recognition of AKI, the use of estimating equations for GFR are not reliable in critically ill patients with AKI. There are several other factors that may also impair the reliability of serum creatinine as a marker of kidney function in critical illness. Some medications, most notably trimethoprim and cimetidine, block tubular secretion of creatinine, leading to an increase in serum creatinine concentration in the absence of decreased kidney function.103 This effect is generally minimal in patients with normal kidney function, but the increase in serum creatinine may exceed 30% in patients with underlying CKD. Reported creatinine concentrations may also be increased as a result of chemical interference with some assay methods by ketone bodies or by medications, such as cefoxitin.103 One final drawback to the use of creatinine as a marker of kidney function in patients with AKI is the inverse relationship between serum creatinine concentrations and GFR. Thus, significant reductions in GFR may occur prior to the serum creatinine concentration being recognized as increasing.
Cystatin C
Given the drawbacks to urea and creatinine as markers of kidney function, other readily available markers of GFR have been sought. The use of exogenous filtration markers such as inulin, iothalamate, or iohexol is cumbersome and not practical for the recognition of abrupt changes in kidney function. Cystatin C has been proposed as a more sensitive endogenous marker of GFR, including in ICU patients.104–106 Cystatin C is a cysteine protease inhibitor that is released into the bloodstream at a constant rate from all nucleated cells. It is readily filtered at the glomerulus and reabsorbed and catabolized by renal proximal tubular epithelial cells such that virtually no cystatin C appears in the urine. The interindividual variability in cystatin C production appears to be less than that for creatinine. Thus, in steady-state situations cystatin C may be a more reliable marker of GFR.107 In addition, the serum half-life of cystatin C is shorter than that of creatinine, making it a more sensitive marker for acute changes in GFR.104–106 However, cystatin C assays are not currently readily available in the acute setting, and the optimal place for cystatin C in the detection of AKI in at-risk critically ill patients remains to be determined.
Markers of Tubular Injury
A number of markers of tubular injury have been proposed as novel diagnostic tests for the early diagnosis of AKI. These markers include kidney injury molecule-1 (KIM-1),108,109 neutrophil gelatinase-associated lipocalin (NGAL),110–116 interleukin 18 (IL-18),114–118 liver fatty acid binding protein (L-FABP)40,119 and α- and π-glutathione-S-transferase (GST),120,121 among others. Even though these markers have shown promise, their role in the clinical care of patients at risk for AKI remains uncertain.
Oligoanuria
Decreased urine output represents a second major reason for recognition of AKI. Sustained oliguria, which is usually defined as a urine output of less than 400 to 500 mL/day, or a sustained urine output of less than 20 mL/hour, in the absence of overt volume depletion almost always indicates the presence of AKI.7 Although oliguria is often considered to be a cardinal feature of AKI, most cases of AKI in the critically ill are nonoliguric.122 Thus, although sustained oliguria should suggest prompt the evaluation for AKI, the presence of a well-maintained urine output should not be construed to represent the presence of adequate kidney function. Anuria (the absence of urine output) always demands prompt attention. True anuria is most often caused by complete urinary obstruction but may also be seen with vascular catastrophes with bilateral renal infarction and less commonly with severe rapidly progressive glomerulonephritis. Rarely, severe ATN may result in a short period of complete anuria.
Diagnostic Approach
General Aspects
The first step in evaluating the patient with AKI is to determine whether it is primarily prerenal or postrenal in origin or is due to intrinsic renal disease. Although this division is useful for conceptualizing and categorizing the causes of AKI, it is important to recognize that many patients may not fall neatly into a single category. For example, prerenal azotemia and ischemic ATN fall on a continuum; a process that may start out as pure prerenal azotemia may evolve over time into ischemic ATN. This blurring between these broad categories is further evidenced by the fact that markers of tubular epithelial cell injury are elevated in patients with classic presentations of prerenal azotemia.40 Moreover, most cases of AKI encountered in ICU patients have more than a single cause,* with the single most common condition predisposing to AKI in the critically ill patient being sepsis and with the majority of patients having AKI in the setting of multiple organ failure.24,61,125 The evaluation of the patient should begin with a careful review of the history and physical examination with particular attention to hemodynamic status, episodes of hypotension and infection, and the record of medication administration. Assessment of voiding function and postvoiding residual bladder volume should be performed to exclude bladder outlet obstruction. This step should then be followed by microscopic examination of the urine and evaluation of urine electrolytes. Renal imaging may be necessary to evaluate for upper tract obstruction or to evaluate for patency of the renal vasculature. In a minority of patients, a renal biopsy will be required for definitive diagnosis.
History, Physical Examination, and Record Review
The initial step in attempting to identify the cause of AKI should be a thorough history, physical examination, and review of the medical record. The overall clinical setting, recent events in the patient’s illness, use of medications, and possible toxic exposures should be noted, with particular attention to events during the 1- to 2-day interval prior to the onset of AKI. A history of vomiting, blood loss, diarrhea, diuretic use, burns, or symptoms compatible with decompensation of heart failure or of liver disease suggests potential prerenal azotemia. A history of prostatism with intermittency, hesitancy, or decrease in the force of the urinary stream; history of urologic, gynecologic, or other pelvic or retroperitoneal malignancy; flank or suprapubic pain; or hematuria or pyuria may suggest obstructive (postrenal) disease. The history of a systemic disorder, fever, rash, vascular disease, or musculoskeletal complaints is compatible with a renovascular, glomerular, or interstitial disorder. Review of the medical record should focus on indices of volume status including intake and output records, serial weights, and serial measurements of blood pressure to help assess for risk factors for intravascular volume depletion and prerenal azotemia. Delineation of the pattern of urine output is often helpful. Finally, a careful review of medication records to assess for exposure to potential nephrotoxins is critical.46,79
The physical examination should focus on assessing for evidence of intravascular volume depletion, such as orthostatic changes in pulse and blood pressure, dry mucous membranes, decreased skin turgor, longitudinal tongue furrows, and absence of sweat in the axilla and inguinal regions.126 A fall in systolic blood pressure of more than 20 mm Hg and in diastolic blood pressure of more than 10 mm Hg accompanied by an increase in heart rate of more than 30 beats per minute is suggestive of intravascular volume depletion. However, these findings are neither sensitive nor specific. Orthostatic changes in blood pressure may be absent in some patients despite significant intravascular volume depletion and may be observed in the absence of volume depletion in patients with autonomic dysfunction and in the elderly. Examination for neck vein distention, pulmonary rales, ventricular gallops, and pedal edema may indicate the presence of heart failure. Occasionally, a chest radiograph, cardiac function testing (i.e., an echocardiogram or nuclear gated blood pool scan), or assessment of plasma brain natriuretic peptide (BNP) or N-terminal pro-BNP (NT-proBNP) levels may assist in the diagnosis of heart failure. Accurate assessment of extracellular fluid volume status and cardiac function may be difficult on clinical grounds, particularly in immobilized, mechanically ventilated ICU patients, and invasive monitoring of central venous pressure, or less commonly, pulmonary artery and pulmonary capillary occlusion pressure, and assessment of cardiac output may be helpful. Assessment for ascites and abdominal distention may assist in the diagnosis of AKI associated with liver disease or the abdominal compartment syndrome. Abdominal palpation to detect flank, suprapubic or central abdominal masses may be helpful in assessing the presence of obstructive uropathy or for an abdominal aortic aneurysm with possible renovascular compromise. A bladder scan or placement of a bladder catheter to assess postvoid bladder volume in patients who do not have an indwelling bladder catheter should be performed to help exclude bladder outlet obstruction as a cause of obstructive uropathy. A rectal and pelvic examination may also be helpful in assessing for possible causes of obstructive uropathy. Careful examination of the skin may detect rashes compatible with a drug-induced eruption, which may suggest drug-induced acute interstitial nephritis; palpable purpura, suggesting a microangiopathic hemolytic anemia such as hemolytic-uremic syndrome or thrombotic thrombocytopenic purpura or a systemic vasculitis, such as cryoglobulinemia; and livedo reticularis or digital ischemia, suggesting atheroembolic disease.
Urinalysis and Urine Indices
Assessment of urine composition and examination of the urine sediment can assist in determining the cause of AKI (Table 55.4). The electrolyte composition of the urine may be helpful in differentiating between prerenal azotemia and ATN.127–130 In prerenal azotemia, renal tubular function is intact and the tubules avidly reabsorb sodium in an effort to restore extracellular fluid volume and renal perfusion to normal. Thus, in prerenal azotemia, urine sodium concentrations are usually less than 20 mmol/L and the fractional excretion of sodium [calculated as (UNa/PNa) ÷ (UCr/PCr), where UNa is the urine sodium concentration, PNa is the plasma or serum sodium concentration, UCr is the urine creatinine concentration, and PCr is the plasma or serum creatinine concentration] is less than 1%. In contrast, in ATN, the damaged renal tubular cells fail to reabsorb sodium normally, perhaps because of loss of cellular polarity, with a resultant urine sodium concentration greater than 40 mmol/L and a fractional excretion of sodium above 2%. It needs to be recognized, however, that these urinary electrolyte findings are not absolute. Thus, increased fractional excretion of sodium may occur in prerenal states when diuretics have been administered, in patients with underlying CKD who may take several days after the onset of volume depletion to maximize sodium conservation, in the presence of glycosuria or other osmotic diuresis, or in the presence of bicarbonaturia. A fractional excretion of sodium of less than 1% may be present in multiple settings other than prerenal azotemia, including in the absence of any kidney disease and in many forms of glomerulonephritis, in microangiopathic hemolytic anemia, and in some forms of ATN, including radiocontrast-associated AKI, myoglobinuric ATN, early sepsis-associated ATN, and nonoliguric ATN.131,132 The fractional excretion of urea [calculated as (Uurea/Purea) ÷ (UCr/PCr), where Uurea is the urine urea nitrogen concentration, Purea is the blood urea nitrogen concentration, UCr is the urine creatinine concentration, and PCr is the plasma or serum creatinine concentration] has been proposed as an alternative index of tubular function for the diagnosis of prerenal azotemia in patients who have received diuretic therapy.133–136 The normal fractional excretion of urea is greater than 60%; in reversible prerenal states, values typically fall to less than 35%. Urinary chemical indices are not of value in establishing the diagnosis of obstructive uropathy.
Table 55.4
Urinalysis Findings in Acute Kidney Injury (AKI)
| Etiologic Disorder | Urine Chemistry | Urine Sediment |
| Prerenal | UNa <20 mmol/L FENa <1% FEurea <35% |
Normal or nearly normal (hyaline casts and rare granular casts) |
| Acute tubular necrosis | UNa >40 mmol/L FENa >2% FEurea >60% |
Renal tubular epithelial cells, epithelial cell casts, coarse pigmented (muddy brown) casts |
| Acute interstitial nephritis | Variable; UNa may be >40 mmol/L, FENa may be >2% | Red blood cells, white blood cells, white blood cell casts, eosinophils |
| Acute glomerulonephritis | Variable; UNa may be <20 mmol/L, FENa may be <1% | Red blood cells (dysmorphic), red blood cell casts |
| Acute vascular disease | Variable; UNa may be <20 mmol/L, FENa may be <1% | Red blood cells, red blood cell casts in HUS/TTP, eosinophils in atheroembolic disease |
| Crystal-associated AKI | Variable | Crystalluria Uric acid crystals in tumor lysis syndrome Calcium oxalate crystals in ethylene glycol ingestion Drug crystals (acyclovir, methotrexate, indinavir, triamterene, sulfadiazine) |
| Obstructive | Variable; early UNa may be <20 mmol/L, FENa may be <1%; late UNa may be >40 mmol/L, FENa may be >2% | Normal or red blood cells, white blood cells and crystals |
Urine microscopy is also often very helpful in determining the cause of AKI.137–141 A normal urine sediment suggests the presence of either a prerenal or postrenal cause of AKI, although obstructive uropathy may be associated with hematuria, pyuria, or crystalluria. In the presence of proteinuria a urinary sediment containing abundant cells or casts suggests an intrinsic cause of AKI. Specifically, the presence of many renal tubular epithelial cells, epithelial cell casts, or pigmented (muddy brown) granular casts suggests the diagnosis of ATN and has been associated with increased risk for greater severity of disease.141,142 The presence of white blood cells and white blood cell casts suggests AIN. Although eosinophiluria may be seen in association with interstitial nephritis, it is also associated with atheroembolic disease and with urinary tract infections, and the specificity and sensitivity of this finding are limited.143 The presence of dysmorphic red blood cells (which are best seen using phase contrast microscopy) and red blood cell casts suggests the presence of an acute glomerulonephritis or vasculitis involving the kidney. A urine dipstick test that is positive for red blood cells in the absence of red blood cells on microscopy suggests the presence of either hemoglobinuria or myoglobinuria. Heavy crystalluria suggests the presence of crystalline intratubular obstruction with uric acid crystals suggestive of tumor lysis syndrome, oxalate crystals suggestive of ethylene glycol ingestion, and drug crystals associated with drug-induced nephropathy. The presence of heavy proteinuria with negative or only trace protein by dipstick and only minimal albuminuria suggests the presence of light chains in the urine and a diagnosis of acute myeloma kidney.
Radiologic Imaging
Imaging of the kidneys and bladder is required for the diagnosis of obstructive kidney disease.144,145 The usual initial test is a renal ultrasound. Rarely, the presence of extensive retroperitoneal disease may give a false-negative ultrasonographic result (so-called nondilated obstructive uropathy).146 Non-contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI) may be helpful in this setting to delineate the presence or absence of retroperitoneal disease. Non-contrast-enhanced CT scanning is also the initial diagnostic test of choice for the evaluation of obstruction from nephrolithiasis. Intravenous radiocontrast administration should be avoided in patients with AKI due to the risk of worsening kidney function from superimposed contrast nephropathy. Similarly, the use of gadolinium-containing contrast agents is contraindicated in AKI due to the risk of inducing nephrogenic systemic fibrosis.147,148 Cystoscopic retrograde or percutaneous antegrade pyelography is occasionally necessary both diagnostically and for the treatment of obstructive disease. Imaging studies may also be helpful for the diagnosis of renovascular disease, renal vein thrombosis, and renal infarction.
Other Laboratory Testing
A careful review of other standard laboratory tests may be helpful in identifying the cause of AKI. A markedly elevated creatinine phosphokinase (CPK) suggests the diagnosis of rhabdomyolysis. A disproportionately elevated serum potassium, phosphate, and uric acid accompanied by elevated lactate dehydrogenase (LDH) suggests diffuse tissue destruction, as occurs in AKI complicating both rhabdomyolysis and the tumor lysis syndrome. An elevated LDH may also be seen in patients with renal infarction.89 Findings on the hemogram and peripheral blood smear may also be informative. The presence of anemia and rouleaux formation, particularly if accompanied by an increase in serum globulins and hypercalcemia may suggest the presence of multiple myeloma. Eosinophilia is associated with allergic interstitial nephritis, atheroembolic disease and polyarteritis nodosa. Thrombocytopenia and microangiopathic hemolytic anemia may be seen in vasculitis; hemolytic-uremic syndrome; thrombotic thrombocytopenic purpura; disseminated intravascular coagulation, which is often seen in association with sepsis; scleroderma renal crisis; and malignant hypertension. If a glomerulonephritis or vasculitis is suspected, specific serologic testing may be indicated, including serum complement levels, antinuclear antibody (ANA), anti-double-stranded DNA, antineutrophilic cytoplasmic antibodies (ANCA), anti–glomerular basement membrane (anti-GBM) antibodies, hepatitis B and C virus, rheumatoid factor, and serum cryoglobulins.
Novel Biomarkers
A number of novel biomarkers, including kidney injury molecule-1 (KIM-1),108,109 neutrophil gelatinase-associated lipocalin (NGAL),110–116 interleukin 18 (IL-18),114–118 liver fatty acid binding protein (L-FABP),40,119 and α- and π-glutathione-S-transferase (GST),120,121 have been proposed both as a means for early diagnosis of AKI and as a means for differentiating between prerenal and intrinsic causes of AKI. For example, elevations in NGAL have been shown to predict progression of AKI and ultimate need for renal replacement therapy.149 However, these biomarkers may also be increased in prerenal disease.40 At present, the optimal role of these biomarkers in the evaluation of patients with AKI remains uncertain.
Renal Biopsy
Renal biopsy is usually reserved for patients in whom prerenal and obstructive causes of AKI have been excluded and the cause of intrinsic AKI remains unclear.150,151 Kidney biopsy is particularly useful for the diagnosis of acute glomerulonephritis, vasculitis, and interstitial nephritis, in which definitive diagnosis is required to guide specific therapy with corticosteroids or immunosuppresion. The precise indications for a kidney biopsy in the critically ill patient with AKI are not established. Factors such as the lack of clinical or laboratory clues to the cause of the AKI and the presence of features atypical for ATN (such as heavy proteinuria, dysmorphic red blood cells, and red blood cell casts on urinalysis, or the presence of an unexplained pulmonary-renal syndrome) are reasonable indications for proceeding with a biopsy. The primary risk of kidney biopsy is postbiopsy hemorrhage, with increased risk seen in patients with poorly controlled hypertension, coagulopathy, thrombocytopenia, or uremic platelet dysfunction. Experience has demonstrated, however, that kidney biopsy can be safely performed even in ICU patients undergoing mechanical ventilation.152
Prevention
Maintenance of Renal Perfusion
Intravascular volume expansion for the maintenance of renal perfusion in high-risk patients is one of the simplest interventions for the prevention of AKI. Volume administration has been shown to decrease the risk of AKI in a variety of clinical settings including myoglobinuric and hemoglobinuric AKI,153–155 radiocontrast nephropathy,156,157 and aminoglycoside- and amphotericin-associated nephrotoxicity.158,159 Fluid administration has also been shown to decrease the risk of intratubular obstruction from crystal deposition in the tumor lysis syndrome160 and in the drug-induced crystalline nephropathies.95,96 The role of volume administration for the prevention of AKI in sepsis is more challenging.61,161 Septic shock is associated with relative vasodilation and increased vascular permeability leading to sequestration of extracellular fluid volume and renal hypoperfusion. In addition, although sepsis is a hyperdynamic state with increased cardiac output, relative myocardial depression is common. The use of goal-directed fluid resuscitation in early sepsis has been advocated, and data suggest a modest reduction in the incidence of AKI.162,163 However, fluid resuscitation in this setting is not without risk. Although fluid challenges can result in stabilization and improvement in renal function, persistent fluid challenges should be avoided if either no improvement in kidney function or worsening oxygenation from pulmonary vascular congestion occurs. The selection of fluid has also been subject to considerable scrutiny. The use of synthetic colloids as compared to saline is thought to provide greater expansion of the intravascular space. Recent studies, however have suggested that hydroxyethyl starch is associated with an increased risk of AKI and mortality in critically ill patients, and in particular, in patients with severe sepsis.164–167
The role of renal vasodilators, including dopamine and fenoldopam, to maintain renal perfusion has also been advocated in the past. Multiple studies have demonstrated, however, that this strategy does not reduce the risk of AKI.168–177 Thus the use of these agents cannot be advocated for the prevention of AKI.
Avoidance of Nephrotoxicity
Many of the medications used for the treatment of critically ill patients carry a risk for nephrotoxicity, with pharmacologic agent–associated nephrotoxicity accounting for approximately one quarter of ICU-associated AKI.19,46,79,178 Common nephrotoxins include radiocontrast agents, ACEIs, ARBs, direct renin inhibitors, NSAIDs, and aminoglycosides. Other nephrotoxins that are less frequently encountered or are used in highly selective populations include amphotericin B, pentamidine, vancomycin, colistin, cisplatin, and high doses of methotrexate and acyclovir, intravenous immunoglobulin (IVIG), and nucleoside inhibitors.
Several general principles are involved in avoiding drug-induced nephrotoxicity: (1) recognizing the nephrotoxic potential of selected pharmacologic agents; (2) knowing which patient populations are at high risk; (3) weighing the risk-benefit ratio for use of a potential nephrotoxin in each patient; (4) considering the use of alternative non-nephrotoxic agents; (5) using the smallest effective dose of each potential nephrotoxin for the briefest interval possible; (6) monitoring blood levels of nephrotoxic agents, when available; (7) frequently monitoring kidney function; and (8) having a surveillance system for early detection of drug-related nephrotoxicity (Box 55.4).
Risk factors for aminoglycoside nephrotoxicity include the use of high doses, prolonged duration of therapy, decreased renal perfusion, associated liver disease, concomitant nephrotoxin administration, and possibly, advanced age.80,81,83 In patients in whom aminoglycoside therapy is considered, clear goals for the use of the agent and plans for duration of therapy need to be formulated to ensure that treatment duration is no longer than absolutely necessary. Euvolemia and adequate renal perfusion should be maintained, concomitant nephrotoxins should be avoided, dosage should be adjusted based on measurement of plasma levels, and kidney function should be monitored on an ongoing basis. The observation that antimicrobial efficacy of aminoglycosides persists even after the drug levels in the blood have fallen below the minimal bactericidal concentration (postantibiotic killing) has led to the development of once-daily dosing regimens that provide higher peak levels but with less frequent dosing.81,179–181 These regimens have been shown to result in rates of bacteriologic cure comparable with standard dosing regimens but are associated with lower rates of AKI.
Vancomycin is an antibiotic that has been associated with an increasing incidence of AKI as a result of both increased utilization and the targeting of higher drug levels.182,183 Although many cases of putative vancomycin nephrotoxicity merely represent elevated drug levels due to decreased renal clearance, the use of higher doses has been associated with an increase in nephrotoxicity. Adjustment of dosing in response to close monitoring of blood levels, especially in patients with fluctuating kidney function, should minimize the risk of drug-induced AKI. Nephrotoxicity of drugs may be reduced through changes in formulation. For example, the use of lipid emulsions of amphotericin B is associated with reduced risk of amphotericin-induced AKI.184
NSAIDs often induce AKI by causing renal vasoconstriction, especially in settings where the renal circulation is dependent on angiotensin.45,185 Less frequently, NSAIDs cause an interstitial nephritis, often accompanied by glomerular involvement with heavy proteinuria. Patients at risk for the development of NSAID-induced AKI include individuals with volume depletion, shock, hypotension, cardiac or liver disease, advanced age, and underlying CKD. Any patient with one or more of these risk factors should be carefully monitored for changes in kidney function associated with NSAID therapy. The same patient population at risk for NSAID-induced AKI is also at greatest risk for an abrupt decrease in kidney function following initiation or adjustment in the dose of ACEIs, ARBs, or direct renin inhibitors.48 In addition, patients with bilateral renal artery stenosis or unilateral renal artery stenosis with a nonfunctional or absent contralateral kidney are also at increased risk for AKI with inhibition of the renin-angiotensin-aldosterone system.
Prevention of Contrast-Induced Acute Kidney Injury
Risk factors for CI-AKI are well defined, with the single most important risk factor being preexisting CKD. In patients with normal kidney function, the risk of CI-AKI is much lower than 1% but increases in a graded fashion with severity of CKD.186,187 Other risk factors include increasing age, diabetes mellitus, congestive heart failure, volume depletion, and concomitant NSAID use.85 Utilization of alternative imaging modalities is the most effective means of preventing CI-AKI; however, when that is not possible strategies should be implemented to minimize the risk of nephrotoxicity. The primary intervention for prevention of CI-AKI is administration of isotonic intravenous fluids prior to and following the administration of contrast medium,157,188 although the optimal composition and rate of fluid administration are not known. Studies comparing the relative benefits of isotonic saline and isotonic sodium bicarbonate have yielded conflicting results.189–192 In hospitalized patients the administration of either isotonic saline or isotonic sodium bicarbonate at a rate of 1 mL/kg/hour for 12 hours prior to and 12 hours following the procedure is recommended3; however, alternative regimens involving more rapid fluid administration for a shorter duration may be equally efficacious.
Multiple pharmacologic agents have been evaluated, but most of them are not beneficial for preventing CI-AKI, including mannitol, furosemide, dopamine, fenoldopam, and calcium channel blockers.176,193–197 Trials of other agents, including natriuretic peptides, aminophylline and theophylline, statins, and ascorbic acid have yielded conflicting results.198–207 N-acetylcysteine (NAC), an antioxidant with vasodilatory properties, has been evaluated for the prevention of CI-AKI in multiple clinical trials. The rationale for the use of NAC relates to its capacity to scavenge reactive oxygen species, reduce the depletion of glutathione, and stimulate the production of vasodilatory mediators including nitric oxide.208,209 These clinical trials of both oral and intravenous NAC have yielded contradictory findings.210–218 Although it is uncertain whether NAC is beneficial in preventing CI-AKI, in its oral form it is both safe and inexpensive. For this reason, some, but not all, practice guidelines have recommended its use, with the important caveat that it should not be employed in lieu of intravenous fluids for the prevention of CI-AKI.3,219
Renal replacement therapies for the prevention of CI-AKI are ineffective, and the use of “prophylactic” hemodialysis has been associated with harm.220–222 Although hemofiltration has been suggested to decrease the postprocedural increase in serum creatinine, the interpretation of these studies is confounded by the lowering of serum creatinine by the intervention.223,224 Given the risks associated with intravenous line placement along with a lack of clear benefit, the use of dialysis or hemofiltration to prevent CI-AKI is not recommended.225
Management of Established Acute Kidney Injury
There is no specific management for the vast majority of patients with established AKI. Patients with prerenal azotemia should have intravascular volume deficits corrected and cardiac function optimized. Patients with hepatorenal syndrome may respond to vasoconstrictor therapy to reverse the splanchnic and peripheral vasodilation that underlies the development of the syndrome.226,227 Abdominal compartment syndrome is treated with abdominal decompression.53,54 Obstructive (postrenal) kidney disease is treated by mechanical relief of obstruction. The primary management of acute interstitial nephritis is discontinuation of the inciting agent; in patients with persistent AKI there may be a role for treatment with glucocorticoids. The treatment of AKI developing as a result of acute glomerulonephritis or renal involvement by vasculitis is dependent upon the precise cause and may include glucocorticoids and immunomodulatory therapy. Once AKI has developed and treatable or reversible causes have been excluded, the general therapeutic approach outlined in Box 55.5 is followed. Optimization of extracellular fluid volume status, cardiac index, and maintenance of perfusion and oxygenation of vital organs are considered first.
Management of Volume Status
Once volume status and cardiac output have been optimized, if the patient remains oliguric the use of a short trial of diuretics to establish urine output can be considered. Although nonoliguric forms of ATN are associated with significantly lower risk of morbidity and mortality than oliguric forms, the primary rationale for a trial of diuretic therapy is to facilitate volume management.122 There is no evidence that conversion of ATN from an oliguric to a nonoliguric form alters risk of morbidity and mortality,228,229 and if a diuretic response is not seen after one or two doses, diuretic agents should be discontinued. The utility of diuretic therapy is supported by a post hoc analysis of data from 306 patients from the Acute Respiratory Distress Syndrome Network’s Fluid and Catheter Treatment Trial (FACTT) who developed AKI.230 Positive fluid balance after development of AKI was associated with increased mortality rate; higher doses of furosemide had a protective effect on mortality rate, although this effect was attenuated after adjustment for fluid balance. Thus, the benefit of diuretics was thought to be mediated by fluid balance. No threshold dose of furosemide was observed above which the mortality rate increased.
In contrast to the use of diuretics, the use of renal vasodilators, including dopamine, fenoldopam, and atrial natriuretic peptide (ANP), has not been shown to be beneficial in AKI. Low-dose dopamine increases renal blood flow and to a lesser extent GFR in experimental animals and in healthy human volunteers and inhibits sodium reabsorption in the proximal tubule. However, in prospective clinical trials, low-dose dopamine has not been demonstrated to prevent or alter the course of ischemic or nephrotoxic ATN.168–175 In a meta-analysis, the use of low-dose dopamine was associated with a transient increase in urine output on day 1 but had no impact on the development or progression of AKI, need for renal replacement therapy, or mortality rate.174 Thus, the use of low-dose dopamine in the treatment of AKI is not justified.231,232 Fenoldopam is a selective dopamine (DA1) receptor agonist.233 In a randomized controlled trial of ICU patients with early ATN, fenoldopam failed to reduce mortality rate or the need for renal replacement therapy.177 Similarly, anaritide, a synthetic analog of ANP, failed to show clinically significant improvement in dialysis-free survival rate or overall mortality rate in patients with ATN.234 Although there was an improvement in dialysis-free survival rate in the subset of patients who were oliguric, this benefit was not confirmed in a subsequent study.235
Preventing, Monitoring, and Treating Complications
A detailed discussion of these complications can be found in the online supplement at www.expertconsult.com.
Medication Dosing Considerations
Most pharmacologic agents are eliminated, at least in part, by the kidneys. Thus, use of standard doses and dosing intervals of many pharmacologic agents in patients with AKI can lead to accumulation of active drug or metabolites, with subsequent drug-induced morbidity and death. To avoid medication-induced toxicity in AKI, scrutiny of the medication list is essential. All medications not essential for care should be eliminated. A thorough understanding of the pathways of drug metabolism and elimination and the potential side effects of each drug used is mandatory. Excellent, up-to-date resources of this information are readily available.265 Careful monitoring of clinical and biochemical parameters and drug levels is indicated, especially for agents with narrow therapeutic indices that are eliminated by the kidneys. In addition, further dosage adjustments may be required when renal replacement therapy is provided, especially for antibiotics to ensure that adequate therapeutic levels are achieved.
Nutritional Considerations
Nutritional support for patients with AKI must be considered in the overall context of associated comorbid conditions and other aspects of the management of critically ill patients.266–269 The optimal nutritional support in critically ill patients with AKI is often influenced more by the nature of underlying illness and the type and frequency of renal replacement therapy rather than the AKI per se. The presence of oliguria and of high protein catabolic rates becomes an important consideration in the nutritional management of patients with severe ICU-associated AKI.266–269 When possible, nutrition should be provided via the enteral route, as even small amounts of luminal nutrition help maintain intestinal integrity and decrease the risk of bacterial translocation. However, enteral nutrition is often insufficient to meet patient requirements and parenteral nutrition is often necessary. Caloric intake should generally be 25 to 30 kcal/kg/day and even in hypercatabolic patients, energy intake should not exceed 35 kcal/kg/day.270 In noncatabolic patients, a protein intake of 1 to 1.3 g/kg/day is generally sufficient to achieve positive nitrogen balance. In hypercatabolic patients and patients receiving continuous renal replacement therapy, protein catabolic rates may reach 1.5 to 1.7 g/kg/day and higher protein intakes are ineffective in achieving positive nitrogen balance.271 Current recommendations are that catabolic patients with AKI should receive 1.2 to 1.5 g/kg of protein and amino acids on a daily basis, even after adjusting for amino acid losses that occur during renal replacement therapy.269,272 Careful attention must be paid to fluid volume and electrolyte content of nutritional formulations so as to minimize the risk of exacerbating volume overload or causing serious mineral and electrolyte disturbances.
Hyperkalemia
Hyperkalemia is an ever-present threat in patients with AKI. Plasma potassium concentrations of more than 5 mmol/L occur in up to 50% and more than 6 mmol/L in up to 30% of patients with AKI.236 Hyperkalemia occurs because of continued accumulation of potassium in extracellular fluid associated with diminished ability of the kidney to eliminate potassium. Usually the potassium concentration increases by approximately 0.2 to 0.4 mmol/L/day. Greater increases occur in the presence of continued potassium intake (e.g., intravenous fluids, blood products, and dietary sources) or continued release of potassium from body stores (e.g., hemolysis, tumor lysis, and rhabdomyolysis).
Hyperkalemia increases the resting electrical membrane potential of myocardial conducting tissue. As the resting potential nears the threshold potential, electrocardiographic (ECG) changes (loss of P waves, peaked T waves, bradycardia, left-axis deviation, left bundle branch block, wide QRS complex) can occur.237 These ECG changes are seen most prominently in the anterior precordial (V2 to V4) leads. With severe hyperkalemia, a sine wave and ventricular fibrillation or asystole can result.
The therapeutic approach for treating hyperkalemia in AKI are listed in Table 55.E1. Mild elevations (<5.5 mmol/L without ECG changes) can best be treated by withdrawing all exogenous sources of potassium and continuing close ECG and laboratory follow-up. If potassium concentration increases to more than 6.0 mmol/L, and especially if ECG changes are present, then more active therapy is indicated (see Table 55.E1). Intravenous calcium is immediately effective for hyperkalemic emergencies, stabilizing excitable membranes, but does not lower the serum potassium concentration. While calcium is indicated when serious ECG manifestations (more than peaked T waves) are present, calcium administration is not appropriate in the absence of these changes. Administration of intravenous calcium in patients with significant hyperphosphatemia may cause precipitation of calcium and phosphate in vascular and soft tissues. Intravenous insulin and β-adrenergic agonists (inhaled albuterol) will cause potassium to shift into the intracellular compartment, lowering the extracellular potassium concentration.238,239 Although sodium bicarbonate also drives potassium from the extracellular fluid into cells, the slow time course of this response makes intravenous bicarbonate ineffective in the acute treatment of hyperkalemia in the absence of severe acidemia.239,240 The ultimate treatment of hyperkalemia is increased excretion. In AKI, the use of diuretics to increase renal potassium excretion is unlikely to be effective. Therapies therefore include the use of gastrointestinal sodium polystyrene sulfonate, and potassium-binding resin to increase gastrointestinal potassium losses or the initiation of dialysis.
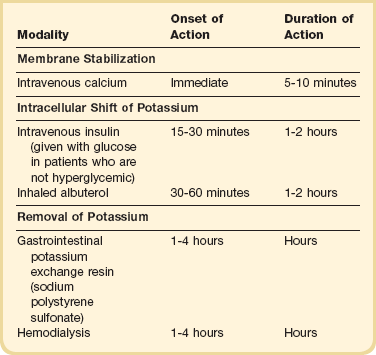
Hyponatremia
Hyponatremia from excess free water administration in the presence of impaired renal ability to excrete water is common in AKI.241 This hyponatremia is generally mild and asymptomatic, although a marked reduction in serum sodium concentration that occurs quickly can lead to confusion, disorientation, and seizures. Treatment consists of water restriction. In unusual circumstances, dialytic therapy may be necessary.
Calcium, Phosphorus, and Magnesium Disorders
Hypocalcemia and hyperphosphatemia are nearly uniform occurrences in AKI.236,242,243 The hyperphosphatemia is caused primarily by decreased urinary excretion of phosphorus with continued release of phosphorus from intracellular stores. Usually the increase in serum phosphorus is modest, with peak values in the 4- to 6-mg/dL range. In the presence of diffuse tissue injury (i.e., rhabdomyolysis, tumor lysis syndrome), marked (>9 mg/dL) hyperphosphatemia can occur. Significant hyperphosphatemia can lead to hypohypocalcemia and tissue deposition of calcium phosphate salts. Therefore, monitoring of serum phosphorus concentration is necessary. Mild elevations can be treated with oral phosphate binders such as aluminum hydroxide, calcium salts, sevelamer, or lanthanum carbonate. More marked elevations are usually treated with dialysis.
Hypocalcemia in AKI is usually modest, with values rarely falling below 6 to 7 mg/dL.236,242,243 Hypocalcemia is rarely of clinical significance because the ionized calcium is maintained by metabolic acidemia. However, aggressive correction of acidemia can precipitate tetany and other symptoms of hypocalcemia. The cause of the hypocalcemia is multifactorial. It may be partially attributed to hyperphosphatemia with a presumed reciprocal lowering of calcium as a result of calcium phosphate tissue precipitation. The most significant hypocalcemia usually occurs in the presence of profound hyperphosphatemia. Other factors can also contribute to hypocalcemia of AKI. Despite very high parathyroid hormone levels in AKI, parathyroid hormone does not mobilize calcium from bone as it does in normal individuals. This skeletal resistance to parathyroid hormone may result in part from an impaired ability of the diseased kidney to render vitamin D active by converting the inactive 25-(OH)-vitamin D to the active 1,25-(OH)2-vitamin D. Thus, low levels of the active form of vitamin D are frequently seen in patients with AKI. Usually, lowering serum phosphate concentration when appropriate is the only treatment needed to improve the serum calcium concentration in AKI. If symptoms of hypocalcemia are present, the infusion of calcium gluconate to alleviate these symptoms may be indicated.
Rarely, hypercalcemia complicates the course of AKI. This hypercalcemia usually occurs during the diuretic phase of rhabdomyolysis-associated AKI.243 The mechanisms of this hypercalcemia are unclear. Mobilization of calcium salts deposited in damaged tissue combined with normalization of 1,25-(OH)2-vitamin D levels and loss of skeletal resistance to parathyroid hormone may all contribute. This hypercalcemia can be treated by dialysis with low-calcium dialysate if necessary.
Hypermagnesemia of modest degree (2 to 4 mg/dL) is frequently seen in AKI.236,242 This degree of hypermagnesemia is almost always asymptomatic. However, significant symptomatic hypermagnesemia can occur if magnesium-containing antacids are given to patients with kidney failure.
Hyperuricemia
Hyperuricemia from continued production of uric acid concomitant with decreased renal excretion of uric acid usually accompanies AKI. The usual peak uric acid concentration is 9 to 10 mg/dL. Much higher levels of uric acid are seen when AKI accompanies diffuse cell injury, as in rhabdomyolysis, in which case the mean uric acid concentration averages 14 to 15 mg/dL and not infrequently exceeds 20 mg/dL.86,244 Despite high concentrations of uric acid in the blood, there is little evidence that this temporary hyperuricemia results in harm. The GFR in AKI is usually so low that little uric acid is filtered, and thus intratubular precipitation does not occur. Acute gout is rarely seen in patients with AKI, despite frequent hyperuricemia. Thus, the secondary hyperuricemia of AKI is usually not specifically treated. One setting in which hyperuricemia is harmful is acute uric acid nephropathy, which typically occurs after therapy for bulky tumors when there was no pretreatment with rasburicase (recombinant uricase) or xanthine oxidase inhibitors (allopurinol) and aggressive hydration.244–247 Almost always the plasma uric acid concentration exceeds 20 mg/dL. Sometimes it is difficult to distinguish whether hyperuricemia is a cause or a consequence of AKI. Hyperuricemia as a cause of AKI usually requires an acute elevation of plasma uric acid to more than 20 mg/dL and results in oligoanuric AKI with abundant uric acid crystals in the urine. Measurement of the urinary uric acid/creatinine ratio on a random specimen may also help distinguish between hyperuricemia caused by overproduction and impaired excretion. This ratio is typically greater than 1.0 when uric acid production is increased and less than 0.75 in normal individuals and patients with impaired kidney function.248
Metabolic Acidosis
Metabolic acidosis, from continued production of nonvolatile acid combined with inability of the injured kidney to generate new bicarbonate, occurs frequently in AKI. The decline in plasma bicarbonate concentration that occurs from metabolic acidosis in AKI is usually about 0.5 to 1 mmol/L/day.122 Approximately 20% of AKI patients develop a bicarbonate concentration of less than 15 mmol/L. This metabolic acidosis is usually associated with an increase in anion gap. In some cases of rhabdomyolysis-associated AKI, a greater anion gap will be seen.86 The mild nature of the metabolic acidosis that can be attributed to AKI per se usually does not require therapy. However, AKI often occurs in a setting in which excess lactic acid accumulates and following ingestions (e.g., ethylene glycol) that result in marked acidosis.
Anemia
Some degree of anemia usually occurs with AKI. In general, with uncomplicated severe AKI, the hemoglobin concentration slowly declines to a nadir of about 10 to 12 g/dL, where it remains until renal function improves. The mechanisms underlying the anemia are related, at least in part, to impaired red blood cell production caused by deficiency of erythropoietin, which is synthesized by the kidney.249,250 In a recent study of 20 patients with AKI, radioimmunoassay values for erythropoietin were very low. Several weeks and in some cases months were required for normalization of serum erythropoietin in AKI patients.250 In one patient with AKI, exogenous recombinant erythropoietin resulted in a brisk reticulocytosis and amelioration of anemia, suggesting a lack of endogenous inhibitors of erythropoiesis in AKI.250 In other studies, erythropoietin use has not been associated with either a decrease in transfusion requirements or with enhanced renal recovery when given in the context of AKI.251 Other factors often contribute to the anemia of AKI including frequent blood drawing for laboratory testing, gastrointestinal blood loss, loss of blood during procedures, marrow suppression caused by medications, and loss of red blood cells in dialyzers. Of note, several disease processes that can produce AKI may have prominent hematologic abnormalities such as anemia (plasma cell dyscrasia); eosinophilia (allergic interstitial nephritis, polyarteritis nodosa, atheroembolic disease); and thrombocytopenia (systemic lupus erythematosus, thrombotic thrombocytopenia purpura, hemolytic-uremia syndrome, malignant hypertension, disseminated intravascular coagulation). In general the anemia of AKI develops slowly and is usually well tolerated. Thus, not only is specific treatment often not indicated, but it is not likely to be effective.250–252
Bleeding
Clinically significant bleeding may complicate the course of 10% to 30% of patients with AKI. In a study of patients developing AKI after contrast-enhanced imaging studies, 15% of patients with mild CI-AKI had a bleeding disorder before they developed AKI, and this increased to 27% after the development of AKI.253 In a large multicenter study, 32% to 42% of patients with AKI had some degree of thrombocytopenia, and 13% to 27% had a coagulopathy.234 Usually the coagulopathy results from associated diseases (e.g., liver disease, sepsis, disseminated intravascular coagulation) or treatments (e.g., multiple transfusions). When AKI results from rhabdomyolysis, particularly in association with cocaine abuse, thrombocytopenia and disseminated intravascular coagulopathy are especially likely.254 The AKI state per se may result in a functional defect in platelets, leading to prolongation of the bleeding time and perhaps contributing to clinically relevant coagulopathy.255–259 Several therapeutic maneuvers can improve the bleeding time and the coagulopathy that is occasionally seen in AKI. Intravenous administration of desmopressin is the easiest, safest method of shortening the bleeding time.256 Unfortunately, the effect of desmopressin is short-lived, and patients rapidly become tachyphylactic to repetitive doses. Cryoprecipitate also shortens the bleeding time in renal failure states.255 Again, the duration is relatively brief. The precise mechanisms whereby desmopressin and cryoprecipitate shorten bleeding time in renal failure are not known. Most data suggest that these agents result in induction (desmopressin) or infusion (cryoprecipitate) of large, multimeric forms of factor VIII–von Willebrand factor aggregates, which enhances binding of platelets to endothelial surfaces.259 Repeated doses of conjugated estrogen can result in prolonged normalization of the bleeding time in uremia, although the mechanism is not clear.258 Restoration of the hemoglobin concentration toward normal also shortens the bleeding time in uremia.257 Finally, dialysis may shorten the bleeding time, but the magnitude of effect and amount of dialysis required to achieve improvement are highly variable.
Infections
Infectious complications are among the most common causes of death in AKI. For example, in an older study infections were judged to be the primary cause of death in 30% to 50% of seriously ill patients with AKI.260 In most studies of ICU patients with AKI, more than 50% to 75% of patients experience at least one infectious complication. In a study of CI-AKI, the frequency of sepsis before and after the development of AKI was 22% and 45%, respectively.253 Common sources of infections are operative sites, intravascular access lines, the urinary tract, and the lungs. Because of the frequent presence of infection, most ICU patients with AKI are treated with multiple antibiotics for several days. Clinical signs such as fever and leukocytosis are often less pronounced when infection is present in patients with AKI compared with those with normal renal function.261 To reduce the risk of infection, efforts should be directed at possible prevention of nosocomial infections by removing invasive lines and bladder catheters as soon as possible. Strict aseptic technique and clinical monitoring of line sites are necessary. When infection is suspected, prompt culturing and selective use of antimicrobial agents are indicated.
Cardiovascular Complications
Because most patients with ICU-associated AKI are severely ill, hemodynamic instability is commonly encountered. In an oliguric patient with AKI, volume overload with resultant pulmonary edema, peripheral edema, and hypertension is an ever-present threat to well-being and survival. Although pulmonary edema is usually caused by positive fluid balance, decreased colloid oncotic pressure caused by hypoalbuminemia and perhaps increased pulmonary capillary permeability can contribute. Volume overload has been shown to be an independent risk factor for death in AKI.262,263 Also, many patients at high risk for developing AKI have underlying chronic heart disease. Careful serial clinical evaluation and monitoring of volume status, body weight, oxygenation, hemodynamic measurements, and chest radiographs are often necessary to help assess volume status in patients with AKI. Several cardiovascular complications such as myocardial infarction; arrhythmias; and, rarely, pericarditis can complicate the course of patients with AKI. In a multicenter study, 22% to 29% of patients with AKI experienced cardiac arrhythmias requiring therapy and 13% to 19% had evidence of myocardial ischemia.234 The development of any of these cardiovascular complications is a major risk factor for adverse outcomes from AKI. Treatment of fluid overload in the setting of AKI depends on the degree of renal impairment. If it is mild nonoliguric AKI, loop diuretics are often effective in reducing positive fluid balance. If the patient has severe oliguric AKI, some type of extracorporeal method of removing volume is necessary.
Pulmonary Complications
In addition to volume overload, several other processes can involve the lungs in patients with AKI. Thus aspiration, infection, and adult respiratory distress syndrome (ARDS) all occur commonly in patients with AKI. Patients with AKI have about a 30% chance of developing an infiltrate on chest radiograph.122 In many series of patients with severe AKI, up to 50% also have respiratory failure sufficient to require mechanical ventilation. In a clinical trial of ANP for the treatment of AKI, 48% to 55% of AKI patients were intubated and ventilated at the time they developed AKI; In the Acute Renal Failure Trial Network (ATN) study, More than 80% of patients were on mechanical ventilation at the time of initiation of renal replacement therapy.125 Mechanical ventilation is clearly a significant, independent factor associated with increased mortality in patients with AKI.24 Several disease processes can cause simultaneous renal and respiratory failure and include acute glomerulonephritis with volume overload, Goodpasture’s syndrome, granulomatosis with polyangiitis or microscopic polyangiitis, systemic lupus erythematosus, polyarteritis nodosa, cryoglobulinemia, and renal vein thrombosis with pulmonary emboli.
Gastrointestinal Complications
Anorexia, nausea, and vomiting are symptoms commonly seen in patients with advanced renal failure. In many series, gastrointestinal hemorrhage, often ascribed to stress gastritis or ulcers, has been reported to occur in 5% to 30% of patients with AKI. Usually gastrointestinal hemorrhage is loosely defined, relatively mild, and easily controlled, but on occasion it may be the cause of death in patients with AKI. Whether the presence of AKI per se is an indication for prophylactic therapy for gastrointestinal bleeding, in the absence of well-recognized indications such as respiratory failure and coagulopathy, is debated.264 Careful prospective studies are necessary for defining the frequency of occurrence and delineating the cause of gastrointestinal hemorrhage in AKI. Also, extreme caution in using magnesium-containing antacids is necessary in the setting of AKI because of the almost exclusive renal route of elimination of magnesium.
Jaundice frequently occurs during the course of AKI in ICU patients. In one prospective study, 28% of patients with ICU-associated AKI were jaundiced at some time during the course of their illness.19 The etiologic picture in jaundice is usually multifactorial, with passive hepatic congestion, multiple blood transfusions, sepsis, hypotension, and many medications contributing. A mild increase in amylase also occurs in AKI, and pancreatitis is present in 2% to 10% of patients at the onset of AKI. Because of this amylase elevation, careful clinical assessment and determination of serum lipase levels are often necessary to ascertain whether pancreatitis is present in a patient with AKI.
Neurologic Complications
Neurologic complications such as confusion, disorientation, lethargy, somnolence, coma, myoclonus, asterixis, and seizures can occur as a result of AKI. In a study of CI-AKI, the frequency of mental status changes increased from 41% before to 68% after development of AKI.253 Abnormal consciousness at the time of first visit in patients with AKI was found in 30% of patients in another prospective series.19 Several causes of neurologic dysfunction are often present in patients with AKI. Most prominent among these are medication-induced encephalopathy, other metabolic disturbances, and primary neurologic disorders such as stroke. Also, several systemic disorders such as vasculitis, endocarditis, thrombotic thrombocytopenic purpura, and malignant hypertension can present with simultaneous AKI and neurologic dysfunction.
Renal Replacement Therapy
Modalities of Renal Replacement Therapy
The available modalities of RRT include conventional intermittent hemodialysis (IHD); the various forms of continuous renal replacement therapy (CRRT) including continuous hemodialysis, continuous hemofiltration, and continuous hemodiafiltration; the hybrid modalities of prolonged intermittent renal replacement therapy (PIRRT; also known as extended duration dialysis273–275 [EDD] or sustained low-efficiency dialysis [SLED])276,277; and peritoneal dialysis.278,279
In IHD and PIRRT, vascular access is generally achieved using large-bore double lumen venous catheters in the internal jugular, subclavian, or femoral veins. Because of concern of causing subclavian stenoses that might impair future angioaccess for chronic dialysis in patients who do not recover kidney function, internal jugular and femoral catheters are generally preferred over subclavian catheters.3 CRRT was initially described using an arteriovenous extracorporeal circuit with inflow through a large-bore arterial catheter and return through a venous catheter. Although these arteriovenous modalities allowed for technical simplicity, they have been largely replaced by pumped venovenous extracorporeal circuits that allow for blood flow rates that are higher and are independent of mean arterial pressure and avoid the complications of prolonged arterial cannulation.273–275
Prolonged intermittent renal replacement therapy is a modification of conventional IHD, utilizing lower blood and dialysate flow rates while prolonging the treatment duration to 8 to 16 hours as compared to the usual 3- to 5-hour IHD treatment.276,277 As with the continuous therapies, by prolonging the treatment duration and decreasing the rate of net ultrafiltration, the hemodynamic stress on the patient is minimized.
There has been considerable debate regarding which modality is most appropriate for use in critically ill patients with AKI. Current data suggest that no individual modality of RRT provides either better patient survival or recovery of kidney function.3,280 Comparing outcomes between modalities in observational studies is complicated by the fact that patients treated with continuous or extended duration therapy are more likely to have greater severity of illness and be hemodynamically unstable. Not unexpectedly, observational studies have generally found higher unadjusted mortality rates when comparing CRRT to conventional IHD.281–287 Randomized controlled trials have generally found no survival benefit when comparing continuous and intermittent therapies.288–293 For example, in the Hemodiafe study, the largest of the randomized controlled trials comparing IHD and CRRT, which enrolled 360 patients who were well matched with regard to severity of illness across 21 ICUs in France, there was no difference in either survival or recovery of kidney function.292 Recent systematic reviews and meta-analyses have confirmed the absence of differences in mortality rate or recovery of kidney function across modalities294–296 but have suggested that continuous therapy is more effective at attaining negative fluid balance263 but is more expensive.295 Based on these data, the Kidney Disease Improving Global Outcomes (KDIGO) Clinical Practice Guidelines for Acute Kidney Injury has recommended that continuous and intermittent modalities of RRT be used as complementary therapies, although with the suggestion that CRRT be used preferentially for hemodynamically unstable patients.297 In patients with acute brain injury or increased intracranial pressure, CRRT is associated with better preservation of cerebral perfusion than IHD and is the more appropriate modality of RRT.298–302
Only limited comparisons between PIRRT and the other modalities of therapy are available and have demonstrated similar hemodynamic stability and metabolic control303–305 and comparable clinical outcomes.306 Peritoneal dialysis (PD) has long been used as a dialytic therapy in AKI, although its use has declined as the use of CRRT has increased. There are only a limited number of studies that have compared outcomes with PD to outcomes with other modalities of renal support in AKI.307–310 Although one study demonstrated poorer survival with PD than with continuous hemofiltration,307 others have demonstrated biochemical and patient outcomes with high-volume peritoneal dialysis that are comparable to those seen with intermittent hemodialysis.308–310
In the absence of data suggesting better survival or recovery of kidney function with any one modality of RRT, these modalities should be considered to be complementary.3 In general, the slower continuous (CRRT) and prolonged intermittent (PIRRT) modalities may be more optimal therapies for patients who are hemodynamically unstable. The choice of modality, however, should also reflect the local availability and expertise of the practitioners prescribing RRT and the nursing staff delivering the therapy.
Timing of Initiation of Renal Replacement Therapy
Standard indications for initiation of renal replacement therapy include volume overload unresponsive to diuretic therapy; electrolyte and acid-base disturbances refractory to medical management, particularly severe hyperkalemia and metabolic acidosis; and overt uremia, characterized pericarditis or encephalopathy (Box 55.6). Common practice, however, is to initiate RRT preemptively, well before the development of these advanced complications, in patients with severe AKI in whom imminent recovery of kidney function is unlikely. Uncertainty in predicting if and when kidney function will recover creates a clinical conundrum: the earlier the initiation of therapy, the greater the probability of dialyzing patients who, if managed conservatively, might have recovered kidney function and survived without needing RRT.
The clinical data guiding current practice regarding the optimal timing of initiation of RRT is weak, derived primarily from retrospective and observational cohort studies and small, underpowered prospective trials. Several observational studies published in the 1960s and early 1970s concluded that mortality rates were higher when dialysis was initiated late, when the BUN was above 160 to 200 mg/dL, as compared to earlier initiation, when the BUN was approximately 90 to 150 mg/dL.311–313 Two subsequent small prospective studies compared more intensive and earlier initiation of therapy to more “conventional” management.314,315 In the first, a study of 18 patients with posttraumatic AKI, 5 of 8 patients (64%) assigned to the more intensive regimen which maintained the predialysis BUN below 70 mg/dL and serum creatinine below 5 mg/dL survived, as compared to 2 of 10 patients (20%) assigned to the less intensive strategy in which dialysis was not performed until the BUN approached 150 mg/dL, the creatinine reached 10 mg/dL, or other indications for dialysis were present (p = 0.14).314 In the subsequent study, 34 patients with severe AKI were assigned to either an intensive regimen, designed to maintain the predialysis BUN less than 60 mg/dL and serum creatinine less than 5 mg/dL or to a delayed and less intensive regimen, in which the BUN was allowed to reach 100 mg/dL and the serum creatinine 9 mg/dL when their serum creatinine reached 8 mg/dL.315 Initiation of dialysis was 2 days earlier in the more intensive regimen. The mortality rate was slightly higher (59% vs. 47%) with the earlier and more intensive strategy; however, this difference did not reach statistical significance. On the basis of these data, the conventional approach to management of RRT in AKI has been to initiate therapy prophylactically, in the absence of specific metabolic indications or symptoms, when the BUN rises to approximately 80 to 100 mg/dL.
A number of more recent retrospective and observational studies have suggested that even earlier initiation of RRT may be associated with better survival.316–328 For example, in a retrospective analysis of timing of initiation of CRRT in 100 consecutive patients with posttraumatic AKI, 39% of patients who were started on CRRT when their BUN was less than 60 mg/dL survived as compared to 20% of patients in whom CRRT was not begun until their BUN was more than 60 mg/dL.316 Only one small randomized trial has attempted to rigorously evaluate the timing of initiation of therapy. In this study of 106 critically ill patients with AKI randomized to early high-volume, early low-volume, and late low-volume continuous venovenous hemodiafiltration (CVVHDF), there was no difference in survival between early and late initiation of therapy.329 A recent systematic review and meta-analysis of studies comparing early and late initiation of renal support published between 1985 and 2010, which included 15 unique studies, found an odds ratio for 28-day mortality rate of 0.45 associated with early initiation of renal support but noted that the methodologic quality of the included studies was low.330 However, there is an important methodologic flaw underlying the majority of studies that have evaluated the timing of RRT. In restricting their analyses to patients who actually received RRT, either “early” or “late,” these studies excluded the large number of patients who did not receive “early” RRT and either recovered kidney function and survived or died prior to reaching the criteria for “late” initiation of RRT.
The issue of severity of volume overload as an indication for initiation of renal support has garnered considerable attention and deserves special mention. The severity of volume overload at initiation of RRT is a strong predictor of fatality.263,331–333 For example, in a cohort of pediatric patients, there was an increase in mortality rate from 29% in patients whose fluid gain was less than 10% of premorbid body weight at the time of initiation of CRRT as opposed to 66% in patients with 20% or greater fluid overload at initiation of therapy and an adjusted odds ratio of death of 8.5 associated with 20% or greater fluid overload.333 Similarly, in a cohort of 396 critically ill adult patients, the presence of greater than 10% fluid overload at initiation of RRT was associated with an odds ratio of death of 2.1.263 Although these data provide a strong caution regarding overly aggressive volume administration in patients with AKI, the hypothesis that earlier initiation of renal support to prevent or reverse volume overload still needs to be tested in prospective clinical trials.
Intensity of Renal Support in Acute Kidney Injury
Assessment of the dose of both IHD and PIRRT is dependent on both the intensity of therapy delivered with each individual treatment, usually quantified in terms of urea reduction ratio or the fractional clearance of urea (Kt/Vurea), and the frequency with which the treatments are provided. For the continuous therapies, dose is usually expressed in terms of the total effluent flow (dialysate plus ultrafiltrate) normalized to body weight. Several small clinical trials suggested that an augmented dose of RRT, either as intermittent hemodialysis or CRRT, improves survival.334–336 These results were not consistent, however, across all studies329,337 and were not confirmed in two large multicenter randomized controlled trials.125,338 The Veterans Administration/National Institutes of Health (VA/NIH) Acute Renal Failure Trial Network (ATN) study randomized 1124 critically ill patients to lower- or higher-intensity RRT using a strategy that allowed patients to shift between modalities as hemodynamic status changed over time.125 In the intensive arm, continuous venovenous hemodiafiltration (CVVHDF) was provided with a total effluent flow of 35 mL/kg/hour and conventional and prolonged intermittent hemodialysis were provided six times per week (daily, except Sunday); in the less intensive arm, the dose of CVVHDF was 20 mL/kg/hour and conventional and prolonged intermittent hemodialysis was provided three times per week (every other day, except Sunday). In the more intensive arm, 60-day all-cause mortality rate was 54% as compared to 52% in the less intensive arm. The Randomized Evaluation of Normal Versus Augmented Level (RENAL) Replacement Therapy study randomized 1508 patients in 35 ICUs in Australia and New Zealand to two doses of CVVHDF (25 or 40 mL/kg/hour) during their ICU stay. The survival rate to 90 days was 55.3% in both treatment arms.
The results of these two studies do not support the concept that more intensive renal support results in improved outcomes. It is recommended that intermittent hemodialysis be provided to deliver a total weekly therapy equivalent to that used in the ATN study (a target Kt/Vurea of 1.2-1.4 per treatment delivered three times per week).3 More frequent IHD may be necessary, however, for fluid management in extremely hypercatabolic patients and for control of severe hyperkalemia. For patients treated with CRRT, the recommended delivered dose of therapy is an effluent flow rate of 20 to 25 mL/kg/hour.3 Achieving this target dose may require the prescription of a slightly higher dose (25 to 30 mL/kg/hour) and careful attention to minimization of interruptions of therapy.
Prognosis and Outcomes
The mortality rate associated with AKI is high, although there is significant variation based on underlying cause, with much higher mortality rates associated with intrinsic forms of AKI than with prerenal or postrenal disease. In intrinsic AKI short-term mortality rates are approximately 50% and have changed little over the past 3 decades.* It is speculated that this lack of improvement despite significant advances in supportive care most likely reflects a decrease in the proportion of patients presenting with isolated AKI, and a corresponding increase in the percentage of patients with AKI accompanied by multiple-organ dysfunction.22,24,351,352 In patients with sepsis-associated AKI, mortality rates are as high as 60% to 90%.17,61,341,351
Multiple factors are associated with mortality risk in AKI and include male gender, advanced age, oliguria (<400 mL/day), a rise in the serum creatinine value of greater than 3 mg/dL, and coexistent sepsis or nonrenal organ failure. In general, these factors reflect severity of renal injury and overall severity of illness. However, even less severe AKI is associated with an increase in mortality risk. In a study of patients undergoing cardiothoracic surgery, increases in serum creatinine less than 0.6 mg/dL were independently associated with a nearly twofold increase in the 30-day mortality rate.353 Similarly, small increments in serum creatinine following radiocontrast agent administration, even if transient, are associated with increased rates of short- and long-term mortality.187,253,354,355 It remains unclear, however, whether such transient increases in serum creatinine actually mediate the adverse long-term outcomes or merely represent a biochemical marker of risk.356
In addition to the very high short-term mortality rate associated with AKI, there is also an increased risk of longer-term morbidity and mortality. It was previously believed that if a patient with AKI survived the acute episode the prognosis was good, with approximately 90% of patients having complete recovery of kidney function, 5% remaining dialysis dependent, and an additional 5% developing progressive deterioration in kidney function after initially recovering function. More recent data suggested a less optimistic prognosis. In recent clinical trials of RRT in AKI, rates of recovery have been highly variable.125,338,357 In the RENAL study, fewer than 10% of patients remained dialysis dependent as compared to 25% of surviving patients in the ATN study and 40% of patients in the Hanover Dialysis Outcomes study. The reason for this variability in recovery of kidney function across studies is unclear and may reflect differences in patterns of care or may reflect differences in the study population.
Approximately half of patients who recover renal function can be demonstrated to have subclinical kidney disease including modest decrements in GFR, defects in tubular function and urinary concentration, and tubulointerstitial scarring on kidney biopsy.358–361 More importantly, patients who recover from AKI are at increased risk of progressive CKD and are at increased risk for the development of end-stage renal disease (ESRD).31,362–365 For example, in a 5% representative sample of elderly Medicare beneficiaries AKI in the absence of underlying CKD was independently associated with a 13-fold hazard for the development of ESRD within 2 years while AKI that developed in the setting of preexistent CKD was associated with a greater than 41-fold hazard for the development of ESRD.362 Similarly, in a population-based cohort study using data from Ontario, Canada, 3769 patients who developed AKI that required temporary dialysis had a more than threefold increased risk for the development of ESRD over 3 years as compared to 13,598 matched control subjects who were hospitalized but did not develop AKI.363 In a similar analysis using data from 562,799 patients hospitalized in the Kaiser-Permanente of Northern California health system, AKI requiring transient dialysis was associated with a twofold increased risk of death over 6 years of follow-up and a 28-fold increased risk of developing progressive CKD.31 Mild AKI has also been associated with an increased risk of AKI. For example, increases in serum creatinine of as little as 0.3 to 0.5 mg/dL following an acute MI were independently associated with a greater than twofold increased hazard for ESRD and an approximately 25% increased hazard for long-term mortality.364
AKI is associated with prolongation of the length of hospitalization and substantial health resource utilization.284,345,366–368 In 1999, the cost of treating an episode of AKI was estimated to be $50,000 per Quality Adjusted Life Year (QALY).369 In a more recent analysis, quality adjusted survival was poor among 153 ICU survivors who had recovered from AKI as compared to an age- and gender-matched community population.370 Although quality adjusted survival in this cohort was poor (15 quality adjusted years per 100 patient-years in the first year following discharge), the survivors’ self-perceived health satisfaction was not significantly different from that of the general population.370 Similar poor health-related quality of life was also observed in a follow-up of 415 subjects who participated in the ATN study and survived at least 60 days, with 27% of respondents’ health status corresponding to levels considered by the general population to be equivalent to or worse than death.371
References
1. Thadhani, R, Pascual, M, Bonventre, JV. Acute renal failure. N Engl J Med. 1996; 334:1448–1460.
2. Lameire, N, Van Biesen, W, Vanholder, R. Acute renal failure. Lancet. 2005; 365:417–430.
3. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012; 2:1–138.
4. Mehta, RL, Kellum, JA, Shah, SV, et al. Acute Kidney Injury Network (AKIN): Report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007; 11:R31.
5. Molitoris, BA, Levin, A, Warnock, DG, et al. Improving outcomes from acute kidney injury. J Am Soc Nephrol. 2007; 18:1992–1994.
6. Kellum, JA, Bellomo, R, Ronco, C. The concept of acute kidney injury and the RIFLE criteria. Contrib Nephrol. 2007; 156:10–16.
7. Klahr, S, Miller, SB. Acute oliguria. N Engl J Med. 1998; 338:671–675.
8. Stevens, LA, Coresh, J, Greene, T, Levey, AS. Assessing kidney function—Measured and estimated glomerular filtration rate. N Engl J Med. 2006; 354:2473–2483.
9. Cockcroft, DW, Gault, MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976; 16:31–41.
10. Levey, AS, Bosch, JP, Lewis, JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999; 130:461–470.
11. Levey, AS, Coresh, J, Greene, T, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006; 145:247–254.
12. Levey, AS, Stevens, LA, Schmid, CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009; 150:604–612.
13. Palevsky, PM. Off-pump cardiac surgery and acute kidney injury. Crit Care Med. 2006; 34:3052–3053.
14. Mehta, RL, Chertow, GM. Acute renal failure definitions and classification: Time for change? J Am Soc Nephrol. 2003; 14:2178–2187.
15. Bellomo, R, Ronco, C, Kellum, JA, et al. Acute renal failure—Definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004; 8:R204–212.
16. Hou, SH, Bushinsky, DA, Wish, JB, et al. Hospital-acquired renal insufficiency: A prospective study. Am J Med. 1983; 74:243–248.
17. Nash, K, Hafeez, A, Hou, S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002; 39:930–936.
18. Liano, F, Pascual, J. Epidemiology of acute renal failure: A prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int. 1996; 50:811–818.
19. Liano, F, Junco, E, Pascual, J, et al. The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The Madrid Acute Renal Failure Study Group. Kidney Int Suppl. 1998; 66:S16–S24.
20. Vivino, G, Antonelli, M, Moro, ML, et al. Risk factors for acute renal failure in trauma patients. Intensive Care Med. 1998; 24:808–814.
21. Brivet, FG, Kleinknecht, DJ, Loirat, P, Landais, PJ. Acute renal failure in intensive care units—Causes, outcome, and prognostic factors of hospital mortality. A prospective, multicenter study. French Study Group on Acute Renal Failure. Critical Care Med. 1996; 24:192–198.
22. Mehta, RL, Pascual, MT, Soroko, S, et al. Spectrum of acute renal failure in the intensive care unit: The PICARD experience. Kidney Int. 2004; 66:1613–1621.
23. Guerin, C, Girard, R, Selli, JM, et al. Initial versus delayed acute renal failure in the intensive care unit. A multicenter prospective epidemiological study. Rhone-Alpes Area Study Group on Acute Renal Failure. Am J Respir Crit Care Med. 2000; 161:872–879.
24. Uchino, S, Kellum, JA, Bellomo, R, et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA. 2005; 294:813–818.
25. Liangos, O, Wald, R, O’Bell, JW, et al. Epidemiology and outcomes of acute renal failure in hospitalized patients: A national survey. Clin J Am Soc Nephrol. 2006; 1:43–51.
26. Xue, JL, Daniels, F, Star, RA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006; 17:1135–1142.
27. Waikar, SS, Curhan, GC, Wald, R, et al. Declining mortality in patients with acute renal failure, 1988 to 2002. J Am Soc Nephrol. 2006; 17:1143–1150.
28. Hospitalization discharge diagnoses for kidney disease—United States, 1980-2005. MMWR Morb Mortal Wkly Rep. 2008; 57:309–312.
29. Waikar, SS, Wald, R, Chertow, GM, et al. Validity of International Classification of Diseases, Ninth Revision, Clinical Modification Codes for Acute Renal Failure. J Am Soc Nephrol. 2006; 17:1688–1694.
30. Hsu, CY, McCulloch, CE, Fan, D, et al. Community-based incidence of acute renal failure. Kidney Int. 2007; 72:208–212.
31. Lo, LJ, Go, AS, Chertow, GM, et al. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009; 76:893–899.
32. Grams, ME, Astor, BC, Bash, LD, et al. Albuminuria and estimated glomerular filtration rate independently associate with acute kidney injury. J Am Soc Nephrol. 2010; 21:1757–1764.
33. Hoste, EA, Lameire, NH, Vanholder, RC, et al. Acute renal failure in patients with sepsis in a surgical ICU: Predictive factors, incidence, comorbidity, and outcome. J Am Soc Nephrol. 2003; 14:1022–1030.
34. Bernieh, B, Al Hakim, M, Boobes, Y, et al. Outcome and predictive factors of acute renal failure in the intensive care unit. Transplant Proc. 2004; 36:1784–1787.
35. Thakar, CV, Liangos, O, Yared, JP, et al. ARF after open-heart surgery: Influence of gender and race. Am J Kidney Dis. 2003; 41:742–751.
36. Thakar, CV, Arrigain, S, Worley, S, et al. A clinical score to predict acute renal failure after cardiac surgery. J Am Soc Nephrol. 2005; 16:162–168.
37. Gomes, E, Antunes, R, Dias, C, et al. Acute kidney injury in severe trauma assessed by RIFLE criteria: A common feature without implications on mortality? Scand J Trauma Resusc Emerg Med. 2010; 18:1.
38. Lopes, JA, Jorge, S, Neves, FC, et al. Acute renal failure in severely burned patients. Resuscitation. 2007; 73:318.
39. Lopes, JA, Jorge, S, Silva, S, et al. Acute renal failure following myeloablative autologous and allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2006; 38:707.
40. Doi, K, Katagiri, D, Negishi, K, et al. Mild elevation of urinary biomarkers in prerenal acute kidney injury. Kidney Int. 2012; 82:1114–1120.
41. Badr, KF, Ichikawa, I. Prerenal failure: A deleterious shift from renal compensation to decompensation. N Engl J Med. 1988; 319:623–629.
42. Blantz, RC. Pathophysiology of pre-renal azotemia. Kidney Int. 1998; 53:512–523.
43. Yared, A, Kon, V, Ichikawa, I. Mechanism of preservation of glomerular perfusion and filtration during acute extracellular fluid volume depletion. Importance of intrarenal vasopressin-prostaglandin interaction for protecting kidneys from constrictor action of vasopressin. J Clin Invest. 1985; 75:1477–1487.
44. Oliver, JA, Sciacca, RR, Cannon, PJ. Renal vasodilation by converting enzyme inhibition. Role of renal prostaglandins. Hypertension. 1983; 5:166–171.
45. Cheng, HF, Harris, RC. Renal effects of non-steroidal anti-inflammatory drugs and selective cyclooxygenase-2 inhibitors. Curr Pharm Des. 2005; 11:1795–1804.
46. Joannidis, M. Drug-induced renal failure in the ICU. Int J Artif Organs. 2004; 27:1034–1042.
47. Bennett, WM, Henrich, WL, Stoff, JS. The renal effects of nonsteroidal anti-inflammatory drugs: Summary and recommendations. Am J Kidney Dis. 1996; 28:S56–S62.
48. Schoolwerth, AC, Sica, DA, Ballermann, BJ, Wilcox, CS. Renal considerations in angiotensin converting enzyme inhibitor therapy: A statement for healthcare professionals from the Council on the Kidney in Cardiovascular Disease and the Council for High Blood Pressure Research of the American Heart Association. Circulation. 2001; 104:1985–1991.
49. Arora, P, Rajagopalam, S, Ranjan, R, et al. Preoperative use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol. 2008; 3:1266–1273.
50. Bellomo, R, Wan, L, Langenberg, C, et al. Septic acute kidney injury: The glomerular arterioles. Contrib Nephrol. 2011; 174:98–107.
51. Doty, JM, Saggi, BH, Sugerman, HJ, et al. Effect of increased renal venous pressure on renal function. J Trauma. 1999; 47:1000–1003.
52. Doty, JM, Saggi, BH, Blocher, CR, et al. Effects of increased renal parenchymal pressure on renal function. J Trauma. 2000; 48:874–877.
53. Malbrain, ML, Cheatham, ML, Kirkpatrick, A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med. 2006; 32:1722–1732.
54. Sugrue, M. Abdominal compartment syndrome. Curr Opin Crit Care. 2005; 11:333–338.
55. Malbrain, ML, Chiumello, D, Pelosi, P, et al. Incidence and prognosis of intraabdominal hypertension in a mixed population of critically ill patients: A multiple-center epidemiological study. Critical Care Med. 2005; 33:315–322.
56. Cheatham, ML, White, MW, Sagraves, SG, et al. Abdominal perfusion pressure: A superior parameter in the assessment of intra-abdominal hypertension. J Trauma. 2000; 49:621–626.
57. Moore, AF, Hargest, R, Martin, M, Delicata, RJ. Intra-abdominal hypertension and the abdominal compartment syndrome. Br J Surg. 2004; 91:1102–1110.
58. Mullens, W, Abrahams, Z, Skouri, HN, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: A potential contributor to worsening renal function? J Am Coll Cardiol. 2008; 51:300–306.
59. Mullens, W, Abrahams, Z, Francis, GS, et al. Prompt reduction in intra-abdominal pressure following large-volume mechanical fluid removal improves renal insufficiency in refractory decompensated heart failure. J Card Fail. 2008; 14:508–514.
60. Mustonen, S, Ala-Houhala, IO, Tammela, TL. Long-term renal dysfunction in patients with acute urinary retention. Scand J Urol Nephrol. 2001; 35:44–48.
61. Schrier, RW, Wang, W. Acute renal failure and sepsis. N Engl J Med. 2004; 351:159–169.
62. Zarjou, A, Agarwal, A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011; 22:999–1006.
63. Bagshaw, SM, Uchino, S, Bellomo, R, et al. Septic acute kidney injury in critically ill patients: Clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007; 2:431–439.
64. Wan, L, Bagshaw, SM, Langenberg, C, et al. Pathophysiology of septic acute kidney injury: What do we really know? Crit Care Med. 2008; 36:S198–S203.
65. Venkatachalam, MA, Bernard, DB, Donohoe, JF, Levinsky, NG. Ischemic damage and repair in the rat proximal tubule: Differences among the S1, S2, and S3 segments. Kidney Int. 1978; 14:31–49.
66. Molitoris, BA. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004; 66:871–883.
67. Molitoris, BA. Na(+)-K(+)-ATPase that redistributes to apical membrane during ATP depletion remains functional. Am J Physiol. 1993; 265:F693–F697.
68. Lieberthal, W, Koh, JS, Levine, JS. Necrosis and apoptosis in acute renal failure. Semin Nephrol. 1998; 18:505–518.
69. Molitoris, BA, Sutton, TA. Endothelial injury and dysfunction: Role in the extension phase of acute renal failure. Kidney Int. 2004; 66:496–499.
70. Burne-Taney, MJ, Rabb, H. The role of adhesion molecules and T cells in ischemic renal injury. Curr Opin Nephrol Hypertens. 2003; 12:85–90.
71. Li, L, Okusa, MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010; 30:268–277.
72. Sutton, TA, Fisher, CJ, Molitoris, BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002; 62:1539–1549.
73. Basile, DP, Donohoe, D, Roethe, K, Osborn, JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001; 281:F887–F899.
74. Venkatachalam, MA, Griffin, KA, Lan, R, et al. Acute kidney injury: A springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010; 298:F1078–F1094.
75. Wynn, TA. Fibrosis under arrest. Nat Med. 2010; 16:523–525.
76. Kelly, KJ. Acute renal failure: Much more than a kidney disease. Semin Nephrol. 2006; 26:105–113.
77. Kelly, KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003; 14:1549–1558.
78. Grams, ME, Rabb, H. The distant organ effects of acute kidney injury. Kidney Int. 2012; 81:942–948.
79. Perazella, MA. Drug use and nephrotoxicity in the intensive care unit. Kidney Int. 2012; 81:1172–1178.
80. Swan, SK. Aminoglycoside nephrotoxicity. Semin Nephrol. 1997; 17:27–33.
81. Hatala, R, Dinh, T, Cook, DJ. Once-daily aminoglycoside dosing in immunocompetent adults: A meta-analysis. Ann Intern Med. 1996; 124:717–725.
82. Sandoval, RM, Molitoris, BA. Gentamicin traffics retrograde through the secretory pathway and is released in the cytosol via the endoplasmic reticulum. Am J Physiol Renal Physiol. 2004; 286:F617–F624.
83. Rea, RS, Capitano, B. Optimizing use of aminoglycosides in the critically ill. Semin Respir Crit Care Med. 2007; 28:596–603.
84. Weisbord, SD, Palevsky, PM. Radiocontrast-induced acute renal failure. J Intensive Care Med. 2005; 20:63–75.
85. Weisbord, SD, Palevsky, PM. Strategies for the prevention of contrast-induced acute kidney injury. Curr Opin Nephrol Hypertens. 2010; 19:539–549.
86. Gabow, PA, Kaehny, WD, Kelleher, SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982; 61:141–152.
87. Bosch, X, Poch, E, Grau, JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009; 361:62–72.
88. Kodner, CM, Kudrimoti, A. Diagnosis and management of acute interstitial nephritis. Am Fam Physician. 2003; 67:2527–2534.
89. Domanovits, H, Paulis, M, Nikfardjam, M, et al. Acute renal infarction. Clinical characteristics of 17 patients. Medicine (Baltimore). 1999; 78:386–394.
90. Shumei, S, Ling, X, Yanxia, W, et al. Acute kidney injury as the first sign of spontaneous renal vein thrombosis: Report of 2 cases. J Thromb Thrombolysis. 2012; 33:129–132.
91. Modi, KS, Rao, VK. Atheroembolic renal disease. J Am Soc Nephrol. 2001; 12:1781–1787.
92. Scolari, F, Tardanico, R, Zani, R, et al. Cholesterol crystal embolism: A recognizable cause of renal disease. Am J Kidney Dis. 2000; 36:1089–1109.
93. Lameire, N, Van Biesen, W, Vanholder, R. Acute renal problems in the critically ill cancer patient. Curr Opin Crit Care. 2008; 14:635–646.
94. Givens, ML, Wethern, J. Renal complications in oncologic patients. Emerg Med Clin North Am. 2009; 27:283–291.
95. Perazella, MA. Crystal-induced acute renal failure. Am J Med. 1999; 106:459–465.
96. Yarlagadda, SG, Perazella, MA. Drug-induced crystal nephropathy: An update. Expert Opin Drug Saf. 2008; 7:147–158.
97. Gertz, MA. Managing myeloma kidney. Ann Intern Med. 2005; 143:835–837.
98. Kyle, RA, Gertz, MA, Witzig, TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003; 78:21–33.
99. Bullock, ML, Umen, AJ, Finkelstein, M, Keane, WF. The assessment of risk factors in 462 patients with acute renal failure. Am J Kidney Dis. 1985; 5:97–103.
100. Moran, SM, Myers, BD. Course of acute renal failure studied by a model of creatinine kinetics. Kidney Int. 1985; 27:928–937.
101. Doi, K, Yuen, PS, Eisner, C, et al. Reduced production of creatinine limits its use as marker of kidney injury in sepsis. J Am Soc Nephrol. 2009; 20:1217–1221.
102. Macedo, E, Bouchard, J, Soroko, SH, et al. Fluid accumulation, recognition and staging of acute kidney injury in critically-ill patients. Crit Care. 2010; 14:R82.
103. Muther, RS. Drug interference with renal function tests. Am J Kidney Dis. 1983; 3:118–120.
104. Villa, P, Jimenez, M, Soriano, MC, et al. Serum cystatin C concentration as a marker of acute renal dysfunction in critically ill patients. Crit Care. 2005; 9:R139–143.
105. Ahlstrom, A, Tallgren, M, Peltonen, S, Pettila, V. Evolution and predictive power of serum cystatin C in acute renal failure. Clin Nephrol. 2004; 62:344–350.
106. Herget-Rosenthal, S, Marggraf, G, Husing, J, et al. Early detection of acute renal failure by serum cystatin C. Kidney Int. 2004; 66:1115–1122.
107. Inker, LA, Schmid, CH, Tighiouart, H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012; 367:20–29.
108. Han, WK, Bailly, V, Abichandani, R, et al. Kidney injury molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002; 62:237–244.
109. Han, WK, Bonventre, JV. Biologic markers for the early detection of acute kidney injury. Curr Opin Crit Care. 2004; 10:476–482.
110. Cruz, DN, de Cal, M, Garzotto, F, et al. Plasma neutrophil gelatinase-associated lipocalin is an early biomarker for acute kidney injury in an adult ICU population. Intensive Care Med. 2010; 36:444–451.
111. Haase, M, Bellomo, R, Haase-Fielitz, A. Neutrophil gelatinase-associated lipocalin: A superior biomarker for detection of subclinical acute kidney injury and poor prognosis. Biomark Med. 2011; 5:415–417.
112. Mishra, J, Dent, C, Tarabishi, R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005; 365:1231–1238.
113. Mishra, J, Ma, Q, Prada, A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003; 14:2534–2543.
114. Chuang, P, Parikh, C, Reilly, RF. A case review: Anticoagulation in hemodialysis patients with heparin-induced thrombocytopenia. Am J Nephrol. 2001; 21:226–231.
115. Parikh, CR, Coca, SG, Thiessen-Philbrook, H, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol. 2011; 22:1748–1757.
116. Parikh, CR, Devarajan, P, Zappitelli, M, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J Am Soc Nephrol. 2011; 22:1737–1747.
117. Parikh, CR, Abraham, E, Ancukiewicz, M, Edelstein, CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005; 16:3046–3052.
118. Parikh, CR, Mishra, J, Thiessen-Philbrook, H, et al. Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2006; 70:199–203.
119. Portilla, D, Dent, C, Sugaya, T, et al. Liver fatty acid-binding protein as a biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2008; 73:465–472.
120. Koyner, JL, Vaidya, VS, Bennett, MR, et al. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol. 2010; 5:2154–2165.
121. McMahon, BA, Koyner, JL, Murray, PT. Urinary glutathione S-transferases in the pathogenesis and diagnostic evaluation of acute kidney injury following cardiac surgery: A critical review. Curr Opin Crit Care. 2010; 16(6):550–555.
122. Anderson, RJ, Linas, SL, Berns, AS, et al. Nonoliguric acute renal failure. N Engl J Med. 1977; 296:1134–1138.
123. Kanagasundaram, NS, Paganini, P. Acute renal failure on the intensive care unit. Clin Med. 2005; 5:435–440.
124. Behrend, T, Miller, SB. Acute renal failure in the cardiac care unit: Etiologies, outcomes, and prognostic factors. Kidney Int. 1999; 56:238–243.
125. Palevsky, PM, Zhang, JH, O’Connor, TZ, et al. Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med. 2008; 359:7–20.
126. McGee, S, Abernethy, WB, 3rd., Simel, DL. The rational clinical examination. Is this patient hypovolemic? JAMA. 1999; 281:1022–1029.
127. Espinel, CH. The FENa test. Use in the differential diagnosis of acute renal failure. JAMA. 1976; 236:579–581.
128. Espinel, CH, Gregory, AW. Differential diagnosis of acute renal failure. Clin Nephrol. 1980; 13:73–77.
129. Rabb, H. Evaluation of urinary markers in acute renal failure. Curr Opin Nephrol Hypertens. 1998; 7:681–685.
130. Miller, TR, Anderson, RJ, Linas, SL, et al. Urinary diagnostic indices in acute renal failure: A prospective study. Ann Intern Med. 1978; 89:47–50.
131. Bagshaw, SM, Langenberg, C, Bellomo, R. Urinary biochemistry and microscopy in septic acute renal failure: A systematic review. Am J Kidney Dis. 2006; 48:695–705.
132. Bagshaw, SM, Langenberg, C, Wan, L, et al. A systematic review of urinary findings in experimental septic acute renal failure. Crit Care Med. 2007; 35:1592–1598.
133. Kaplan, AA, Kohn, OF. Fractional excretion of urea as a guide to renal dysfunction. Am J Nephrol. 1992; 12:49–54.
134. Carvounis, CP, Nisar, S, Guro-Razuman, S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002; 62:2223–2229.
135. Pepin, MN, Bouchard, J, Legault, L, Ethier, J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007; 50:566–573.
136. Diskin, CJ, Stokes, TJ, Dansby, LM, et al. The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract. 2010; 114:c145–150.
137. Perazella, MA, Coca, SG, Kanbay, M, et al. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2008; 3:1615–1619.
138. Perazella, MA, Parikh, CR. How can urine microscopy influence the differential diagnosis of AKI? Clin J Am Soc Nephrol. 2009; 4:691–693.
139. Perazella, MA, Coca, SG. Traditional urinary biomarkers in the assessment of hospital-acquired AKI. Clin J Am Soc Nephrol. 2012; 7:167–174.
140. Kanbay, M, Kasapoglu, B, Perazella, MA. Acute tubular necrosis and pre-renal acute kidney injury: Utility of urine microscopy in their evaluation—A systematic review. Int Urol Nephrol. 2010; 42:425–433.
141. Chawla, LS, Dommu, A, Berger, A, et al. Urinary sediment cast scoring index for acute kidney injury: A pilot study. Nephron Clin Pract. 2008; 110:c145–150.
142. Perazella, MA, Coca, SG, Hall, IE, et al. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol. 2010; 5:402–408.
143. Rossert, J. Drug-induced acute interstitial nephritis. Kidney Int. 2001; 60:804–817.
144. Mucelli, RP, Bertolotto, M. Imaging techniques in acute renal failure. Kidney Int Suppl. 1998; 66:S102–S105.
145. Pozzi Mucelli, R, Bertolotto, M, Quaia, E. Imaging techniques in acute renal failure. Contrib Nephrol. 2001; 132:76–91.
146. Bhandari, S. The patient with acute renal failure and non-dilated urinary tract. Nephrol Dial Transplant. 1998; 13:1888.
147. Cowper, SE. Nephrogenic systemic fibrosis: A review and exploration of the role of gadolinium. Adv Dermatol. 2007; 23:131–154.
148. Cowper, SE. Nephrogenic systemic fibrosis: An overview. J Am Coll Radiol. 2008; 5:23–28.
149. Singer, E, Elger, A, Elitok, S, et al. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int. 2011; 80:405–414.
150. Andreucci, VE, Fuiano, G, Stanziale, P, Andreucci, M. Role of renal biopsy in the diagnosis and prognosis of acute renal failure. Kidney Int Suppl. 1998; 66:S91–S95.
151. Solez, K, Racusen, LC. Role of the renal biopsy in acute renal failure. Contrib Nephrol. 2001; 132:68–75.
152. Conlon, PJ, Kovalik, E, Schwab, SJ. Percutaneous renal biopsy of ventilated intensive care unit patients. Clin Nephrol. 1995; 43:309–311.
153. Better, OS. Post-traumatic acute renal failure: Pathogenesis and prophylaxis. Nephrol Dial Transplant. 1992; 7:260–264.
154. Sever, MS, Vanholder, R, Lameire, N. Management of crush-related injuries after disasters. N Engl J Med. 2006; 354:1052–1063.
155. Brown, CV, Rhee, P, Chan, L, et al. Preventing renal failure in patients with rhabdomyolysis: Do bicarbonate and mannitol make a difference? J Trauma. 2004; 56:1191–1196.
156. Mueller, C, Prevention of contrast-induced nephropathy with volume supplementation. Kidney Int Suppl. 2006(100):S16–S19.
157. Trivedi, HS, Moore, H, Nasr, S, et al. A randomized prospective trial to assess the role of saline hydration on the development of contrast nephrotoxicity. Nephron Clin Pract. 2003; 93:C29–34.
158. Llanos, A, Cieza, J, Bernardo, J, et al. Effect of salt supplementation on amphotericin B nephrotoxicity. Kidney Int. 1991; 40:302–308.
159. Guo, X, Nzerue, C. How to prevent, recognize, and treat drug-induced nephrotoxicity. Cleve Clin J Med. 2002; 69:289–297.
160. Conger, JD, Falk, SA. Intrarenal dynamics in the pathogenesis and prevention of acute urate nephropathy. J Clin Invest. 1977; 59:786–793.
161. Van Biesen, W, Yegenaga, I, Vanholder, R, et al. Relationship between fluid status and its management on acute renal failure (ARF) in intensive care unit (ICU) patients with sepsis: A prospective analysis. J Nephrol. 2005; 18:54–60.
162. Rivers, E, Nguyen, B, Havstad, S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001; 345:1368–1377.
163. Lin, SM, Huang, CD, Lin, HC, et al. A modified goal-directed protocol improves clinical outcomes in intensive care unit patients with septic shock: A randomized controlled trial. Shock. 2006; 26:551–557.
164. Schortgen, F, Lacherade, JC, Bruneel, F, et al. Effects of hydroxyethylstarch and gelatin on renal function in severe sepsis: A multicentre randomised study. Lancet. 2001; 357:911–916.
165. Dart, AB, Mutter, TC, Ruth, CA, Taback, SP. Hydroxyethyl starch (HES) versus other fluid therapies: Effects on kidney function. Cochrane Database Syst Rev. (1):2010.
166. Perner, A, Haase, N, Guttormsen, AB, et al. Hydroxyethyl starch 130/0. 42 versus Ringer’s acetate in severe sepsis. N Engl J Med. 2012; 367:124–134.
167. Myburgh, JA, Finfer, S, Bellomo, R, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med. 2012; 367:1901–1911.
168. Graziani, G, Cantaluppi, A, Casati, S, et al. Dopamine and frusemide in oliguric acute renal failure. Nephron. 1984; 37:39–42.
169. Conger, JD, Falk, SA, Hammond, WS. Atrial natriuretic peptide and dopamine in established acute renal failure in the rat. Kidney Int. 1991; 40:21–28.
170. Parker, S, Carlon, GC, Isaacs, M, et al. Dopamine administration in oliguria and oliguric renal failure. Crit Care Med. 1981; 9:630–632.
171. Kellum, JA, Decker, JM. Use of dopamine in acute renal failure: A meta-analysis. Critical Care Med. 2001; 29:1526–1531.
172. Denton, MD, Chertow, GM, Brady, HR. “Renal-dose” dopamine for the treatment of acute renal failure: Scientific rationale, experimental studies and clinical trials. Kidney Int. 1996; 50:4–14.
173. Marik, PE, Iglesias, J. Low-dose dopamine does not prevent acute renal failure in patients with septic shock and oliguria. NORASEPT II Study Investigators. Am J Med. 1999; 107:387–390.
174. Friedrich, JO, Adhikari, N, Herridge, MS, Beyene, J. Meta-analysis: Low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann Intern Med. 2005; 142:510–524.
175. Bellomo, R, Chapman, M, Finfer, S, et al. Low-dose dopamine in patients with early renal dysfunction: A placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000; 356:2139–2143.
176. Stone, GW, McCullough, PA, Tumlin, JA, et al. Fenoldopam mesylate for the prevention of contrast-induced nephropathy: A randomized controlled trial. JAMA. 2003; 290:2284–2291.
177. Tumlin, JA, Finkel, KW, Murray, PT, et al. Fenoldopam mesylate in early acute tubular necrosis: A randomized, double-blind, placebo-controlled clinical trial. Am J Kidney Dis. 2005; 46:26–34.
178. Henrich, WL. Nephrotoxicity of several newer agents. Kidney Int Suppl. 2005; 94:S107–S109.
179. Blaser, J, Konig, C. Once-daily dosing of aminoglycosides. Eur J Clin Microbiol Infect Dis. 1995; 14:1029–1038.
180. Craig, WA. Once-daily versus multiple-daily dosing of aminoglycosides. J Chemother (Florence). 1995; 7(Suppl 2):47–52.
181. Gilbert, DN. Once-daily aminoglycoside therapy. Antimicrob Agents Chemother. 1991; 35:399–405.
182. Pritchard, L, Baker, C, Leggett, J, et al. Increasing vancomycin serum trough concentrations and incidence of nephrotoxicity. Am J Med. 2010; 123:1143–1149.
183. Gupta, A, Biyani, M, Khaira, A. Vancomycin nephrotoxicity: Myths and facts. Neth J Med. 2011; 69(9):379–383.
184. Deray, G. Amphotericin B nephrotoxicity. J Antimicrob Chemother. 2002; 49(Suppl 1):37–41.
185. Huerta, C, Castellsague, J, Varas-Lorenzo, C, Garcia Rodriguez, LA. Nonsteroidal anti-inflammatory drugs and risk of ARF in the general population. Am J Kidney Dis. 2005; 45:531–539.
186. Mehran, R, Aymong, ED, Nikolsky, E, et al. A simple risk score for prediction of contrast-induced nephropathy after percutaneous coronary intervention: Development and initial validation. J Am Coll Cardiol. 2004; 44:1393–1399.
187. Bartholomew, BA, Harjai, KJ, Dukkipati, S, et al. Impact of nephropathy after percutaneous coronary intervention and a method for risk stratification. Am J Cardiol. 2004; 93:1515–1519.
188. Mueller, C, Buerkle, G, Buettner, HJ, et al. Prevention of contrast media-associated nephropathy: Randomized comparison of 2 hydration regimens in 1620 patients undergoing coronary angioplasty. Arch Intern Med. 2002; 162:329–336.
189. Brar, SS, Shen, AY, Jorgensen, MB, et al. Sodium bicarbonate vs sodium chloride for the prevention of contrast medium-induced nephropathy in patients undergoing coronary angiography: A randomized trial. JAMA. 2008; 300:1038–1046.
190. Maioli, M, Toso, A, Leoncini, M, et al. Sodium bicarbonate versus saline for the prevention of contrast-induced nephropathy in patients with renal dysfunction undergoing coronary angiography or intervention. J Am Coll Cardiol. 2008; 52:599–604.
191. Merten, GJ, Burgess, WP, Gray, LV, et al. Prevention of contrast-induced nephropathy with sodium bicarbonate: A randomized controlled trial. JAMA. 2004; 291:2328–2334.
192. Vasheghani-Farahani, A, Sadigh, G, Kassaian, SE, et al. Sodium bicarbonate in preventing contrast nephropathy in patients at risk for volume overload: A randomized controlled trial. J Nephrol. 2010; 23:216–223.
193. Cacoub, P, Deray, G, Baumelou, A, Jacobs, C. No evidence for protective effects of nifedipine against radiocontrast-induced acute renal failure. Clin Nephrol. 1988; 29:215–216.
194. Kellum, JA. The use of diuretics and dopamine in acute renal failure: A systematic review of the evidence. Crit Care (Lond). 1997; 1:53–59.
195. Khoury, Z, Schlicht, JR, Como, J, et al. The effect of prophylactic nifedipine on renal function in patients administered contrast media. Pharmacotherapy. 1995; 15:59–65.
196. Solomon, R, Werner, C, Mann, D, et al. Effects of saline, mannitol, and furosemide to prevent acute decreases in renal function induced by radiocontrast agents. N Engl J Med. 1994; 331:1416–1420.
197. Weinstein, JM, Heyman, S, Brezis, M. Potential deleterious effect of furosemide in radiocontrast nephropathy. Nephron. 1992; 62:413–415.
198. Jo, SH, Koo, BK, Park, JS, et al. Prevention of radiocontrast medium-induced nephropathy using short-term high-dose simvastatin in patients with renal insufficiency undergoing coronary angiography (PROMISS) trial—A randomized controlled study. Am Heart J. 2008; 155:499.
199. Spargias, K, Alexopoulos, E, Kyrzopoulos, S, et al. Ascorbic acid prevents contrast-mediated nephropathy in patients with renal dysfunction undergoing coronary angiography or intervention. Circulation. 2004; 110:2837–2842.
200. Boscheri, A, Weinbrenner, C, Botzek, B, et al. Failure of ascorbic acid to prevent contrast-media induced nephropathy in patients with renal dysfunction. Clin Nephrol. 2007; 68:279–286.
201. Spargias, K, Adreanides, E, Demerouti, E, et al. Iloprost prevents contrast-induced nephropathy in patients with renal dysfunction undergoing coronary angiography or intervention. Circulation. 2009; 120:1793–1799.
202. Spargias, K, Adreanides, E, Giamouzis, G, et al. Iloprost for prevention of contrast-mediated nephropathy in high-risk patients undergoing a coronary procedure. Results of a randomized pilot study. Eur J Clin Pharmacol. 2006; 62:589–595.
203. Kurnik, BR, Allgren, RL, Genter, FC, et al. Prospective study of atrial natriuretic peptide for the prevention of radiocontrast-induced nephropathy. Am J Kidney Dis. 1998; 31:674–680.
204. Morikawa, S, Sone, T, Tsuboi, H, et al. Renal protective effects and the prevention of contrast-induced nephropathy by atrial natriuretic peptide. J Am Coll Cardiol. 2009; 53:1040–1046.
205. Ix, JH, McCulloch, CE, Chertow, GM. Theophylline for the prevention of radiocontrast nephropathy: A meta-analysis. Nephrol Dial Transplant. 2004; 19:2747–2753.
206. Shammas, NW, Kapalis, MJ, Harris, M, et al. Aminophylline does not protect against radiocontrast nephropathy in patients undergoing percutaneous angiographic procedures. J Invasive Cardiol. 2001; 13:738–740.
207. Erley, CM, Duda, SH, Rehfuss, D, et al. Prevention of radiocontrast-media-induced nephropathy in patients with pre-existing renal insufficiency by hydration in combination with the adenosine antagonist theophylline. Nephrol Dial Transplant. 1999; 14:1146–1149.
208. DiMari, J, Megyesi, J, Udvarhelyi, N, et al. N-acetyl cysteine ameliorates ischemic renal failure. Am J Physiol. 1997; 272:F292–F298.
209. Fishbane, S. N-Acetylcysteine in the prevention of contrast-induced nephropathy. Clin J Am Soc Nephrol. 2008; 3:281–287.
210. Carbonell, N, Blasco, M, Sanjuan, R, et al. Intravenous N-acetylcysteine for preventing contrast-induced nephropathy: A randomised trial. Int J Cardiol. 2007; 115:57–62.
211. Durham, JD, Caputo, C, Dokko, J, et al. A randomized controlled trial of N-acetylcysteine to prevent contrast nephropathy in cardiac angiography. Kidney Int. 2002; 62:2202–2207.
212. Fung, JW, Szeto, CC, Chan, WW, et al. Effect of N-acetylcysteine for prevention of contrast nephropathy in patients with moderate to severe renal insufficiency: A randomized trial. Am J Kidney Dis. 2004; 43:801–808.
213. Gomes, VO, Poli de Figueredo, CE, Caramori, P, et al. N-acetylcysteine does not prevent contrast induced nephropathy after cardiac catheterisation with an ionic low osmolality contrast medium: A multicentre clinical trial. Heart. 2005; 91:774–778.
214. Rashid, ST, Salman, M, Myint, F, et al. Prevention of contrast-induced nephropathy in vascular patients undergoing angiography: A randomized controlled trial of intravenous N-acetylcysteine. J Vasc Surg. 2004; 40:1136–1141.
215. Webb, JG, Pate, GE, Humphries, KH, et al. A randomized controlled trial of intravenous N-acetylcysteine for the prevention of contrast-induced nephropathy after cardiac catheterization: Lack of effect. Am Heart J. 2004; 148:422–429.
216. Tepel, M, van der Giet, M, Schwarzfeld, C, et al. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med. 2000; 343:180–184.
217. Marenzi, G, Assanelli, E, Marana, I, et al. N-Acetylcysteine and contrast-induced nephropathy in primary angioplasty. N Engl J Med. 2006; 354:2773–2782.
218. Trivedi, H, Daram, S, Szabo, A, et al. High-dose N-acetylcysteine for the prevention of contrast-induced nephropathy. Am J Med. 2009; 122:874.
219. Joannidis, M, Druml, W, Forni, LG, et al. Prevention of acute kidney injury and protection of renal function in the intensive care unit. Expert opinion of the Working Group for Nephrology, ESICM. Intensive Care Med. 2010; 36:392–411.
220. Cruz, DN, Perazella, MA, Bellomo, R, et al. Extracorporeal blood purification therapies for prevention of radiocontrast-induced nephropathy: A systematic review. Am J Kidney Dis. 2006; 48:361–371.
221. Reinecke, H, Fobker, M, Wellmann, J, et al. A randomized controlled trial comparing hydration therapy to additional hemodialysis or N-acetylcysteine for the prevention of contrast medium-induced nephropathy: The Dialysis-versus-Diuresis (DVD) Trial. Clin Res Cardiol. 2007; 96:130–139.
222. Lehnert, T, Keller, E, Gondolf, K, et al. Effect of haemodialysis after contrast medium administration in patients with renal insufficiency. Nephrol Dial Transplant. 1998; 13:358–362.
223. Marenzi, G, Lauri, G, Campodonico, J, et al. Comparison of two hemofiltration protocols for prevention of contrast-induced nephropathy in high-risk patients. Am J Med. 2006; 119:155–162.
224. Marenzi, G, Marana, I, Lauri, G, et al. The prevention of radiocontrast-agent-induced nephropathy by hemofiltration. N Engl J Med. 2003; 349:1333–1340.
225. Stacul, F, Adam, A, Becker, CR, et al. Strategies to reduce the risk of contrast-induced nephropathy. Am J Cardiol. 2006; 98:59K–77K.
226. Gines, P, Cardenas, A, Arroyo, V, Rodes, J. Management of cirrhosis and ascites. N Engl J Med. 2004; 350:1646–1654.
227. Gines, P, Schrier, RW. Renal failure in cirrhosis. N Engl J Med. 2009; 361:1279–1290.
228. Brown, CB, Ogg, CS, Cameron, JS. High dose frusemide in acute renal failure: A controlled trial. Clin Nephrol. 1981; 15:90–96.
229. Mehta, RL, Pascual, MT, Soroko, S, Chertow, GM. Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA. 2002; 288:2547–2553.
230. Grams, ME, Estrella, MM, Coresh, J, et al. Fluid balance, diuretic use, and mortality in acute kidney injury. Clin J Am Soc Nephrol. 2011; 6:966–973.
231. Jones, D, Bellomo, R. Renal-dose dopamine: From hypothesis to paradigm to dogma to myth and, finally, superstition? J Intensive Care Med. 2005; 20:199–211.
232. Bellomo, R. Has renal-dose dopamine finally been relegated to join the long list of medical myths? Crit Care Resusc. 2001; 3:7–10.
233. Singer, I, Epstein, M. Potential of dopamine A-1 agonists in the management of acute renal failure. Am J Kidney Dis. 1998; 31:743–755.
234. Allgren, RL, Marbury, TC, Rahman, SN, et al. Anaritide in acute tubular necrosis. Auriculin Anaritide Acute Renal Failure Study Group. N Engl J Med. 1997; 336:828–834.
235. Lewis, J, Salem, MM, Chertow, GM, et al. Atrial natriuretic factor in oliguric acute renal failure. Anaritide Acute Renal Failure Study Group. Am J Kidney Dis. 2000; 36:767–774.
236. Dolson, GM. Electrolyte abnormalities before and after the onset of acute renal failure. Miner Electrolyte Metab. 1991; 17:133–140.
237. Esposito, C, Bellotti, N, Fasoli, G, et al. Hyperkalemia-induced ECG abnormalities in patients with reduced renal function. Clin Nephrol. 2004; 62:465–468.
238. Allon, M, Copkney, C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int. 1990; 38:869–872.
239. Greenberg, A. Hyperkalemia: Treatment options. Semin Nephrol. 1998; 18:46–57.
240. Blumberg, A, Weidmann, P, Shaw, S, Gnadinger, M. Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure. Am J Med. 1988; 85:507–512.
241. Anderson, RJ, Chung, HM, Kluge, R, Schrier, RW. Hyponatremia: A prospective analysis of its epidemiology and the pathogenetic role of vasopressin. Ann Intern Med. 1985; 102:164–168.
242. Massry, SG, Arieff, AI, Coburn, JW, et al. Divalent ion metabolism in patients with acute renal failure: Studies on the mechanism of hypocalcemia. Kidney Int. 1974; 5:437–445.
243. Llach, F, Felsenfeld, AJ, Haussler, MR. The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure. Interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. N Engl J Med. 1981; 305:117–123.
244. Haas, M, Ohler, L, Watzke, H, et al. The spectrum of acute renal failure in tumour lysis syndrome. Nephrol Dial Transplant. 1999; 14:776–779.
245. Cairo, MS. Prevention and treatment of hyperuricemia in hematological malignancies. Clin Lymphoma. 2002; 3(Suppl 1):S26–S31.
246. Davidson, MB, Thakkar, S, Hix, JK, et al. Pathophysiology, clinical consequences, and treatment of tumor lysis syndrome. Am J Med. 2004; 116:546–554.
247. Wang, LY, Shih, LY, Chang, H, et al. Recombinant urate oxidase (rasburicase) for the prevention and treatment of tumor lysis syndrome in patients with hematologic malignancies. Acta Haematol. 2006; 115:35–38.
248. Tungsanga, K, Boonwichit, D, Lekhakula, A, Sitprija, V. Urine uric acid and urine creatine ratio in acute renal failure. Arch Internal Med. 1984; 144:934–937.
249. Nielsen, OJ, Thaysen, JH. Erythropoietin deficiency in acute renal failure. Lancet. 1989; 1:624–625.
250. Nielsen, OJ, Thaysen, JH. Erythropoietin deficiency in acute tubular necrosis. J Intern Med. 1990; 227:373–380.
251. du Cheyron, D, Parienti, JJ, Fekih-Hassen, M, et al. Impact of anemia on outcome in critically ill patients with severe acute renal failure. Intensive Care Med. 2005; 31:1529–1536.
252. Park, J, Gage, BF, Vijayan, A. Use of EPO in critically ill patients with acute renal failure requiring renal replacement therapy. Am J Kidney Dis. 2005; 46:791–798.
253. Levy, EM, Viscoli, CM, Horwitz, RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA. 1996; 275:1489–1494.
254. Roth, D, Alarcon, FJ, Fernandez, JA, et al. Acute rhabdomyolysis associated with cocaine intoxication. N Engl J Med. 1988; 319:673–677.
255. Janson, PA, Jubelirer, SJ, Weinstein, MJ, Deykin, D. Treatment of the bleeding tendency in uremia with cryoprecipitate. N Engl J Med. 1980; 303:1318–1322.
256. Mannucci, PM, Remuzzi, G, Pusineri, F, et al. Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. N Engl J Med. 1983; 308:8–12.
257. Fernandez, F, Goudable, C, Sie, P, et al. Low haematocrit and prolonged bleeding time in uraemic patients: Effect of red cell transfusions. Br J Haematol. 1985; 59:139–148.
258. Vigano, G, Gaspari, F, Locatelli, M, et al. Dose-effect and pharmacokinetics of estrogens given to correct bleeding time in uremia. Kidney Int. 1988; 34:853–858.
259. Sagripanti, A, Barsotti, G. Bleeding and thrombosis in chronic uremia. Nephron. 1997; 75:125–139.
260. McMurray, SD, Luft, FC, Maxwell, DR, et al. Prevailing patterns and predictor variables in patients with acute tubular necrosis. Arch Intern Med. 1978; 138:950–955.
261. Wolk, PJ, Apicella, MA. The effect of renal function on the febrile response to bacteremia. Arch Intern Med. 1978; 138:1084–1085.
262. Sutherland, SM, Zappitelli, M, Alexander, SR, et al. Fluid overload and mortality in children receiving continuous renal replacement therapy: The prospective pediatric continuous renal replacement therapy registry. Am J Kidney Dis. 2010; 55:316–325.
263. Bouchard, J, Soroko, SB, Chertow, GM, et al. Fluid accumulation, survival and recovery of kidney function in critically ill patients with acute kidney injury. Kidney Int. 2009; 76:422–427.
264. Saint, S, Matthay, MA. Risk reduction in the intensive care unit. Am J Med. 1998; 105:515–523.
265. Brier, ME, Aronoff, GR. Drug Prescribing in Renal Failure, 5th ed. Philadelphia: American College of Physicians; 2007.
266. Heyland, DK, MacDonald, S, Keefe, L, Drover, JW. Total parenteral nutrition in the critically ill patient: A meta-analysis. JAMA. 1998; 280:2013–2019.
267. Wooley, JA, Btaiche, IF, Good, KL. Metabolic and nutritional aspects of acute renal failure in critically ill patients requiring continuous renal replacement therapy. Nutr Clin Pract. 2005; 20:176–191.
268. Strejc, JM. Considerations in the nutritional management of patients with acute renal failure. Hemodial Int. 2005; 9:135–142.
269. Druml, W. Nutritional management of acute renal failure. J Ren Nutr. 2005; 15:63–70.
270. Schneeweiss, B, Graninger, W, Stockenhuber, F, et al. Energy metabolism in acute and chronic renal failure. Am J Clin Nutr. 1990; 52:596–601.
271. Macias, WL, Alaka, KJ, Murphy, MH, et al. Impact of the nutritional regimen on protein catabolism and nitrogen balance in patients with acute renal failure. JPEN J Parenter Enteral Nutr. 1996; 20:56–62.
272. Druml, W. Nutritional management of acute renal failure. Am J Kidney Dis. 2001; 37:S89–S94.
273. Manns, M, Sigler, MH, Teehan, BP. Continuous renal replacement therapies: An update. Am J Kidney Dis. 1998; 32:185–207.
274. Mehta, RL. Continuous renal replacement therapy in the critically ill patient. Kidney Int. 2005; 67:781–795.
275. Ronco, C, Cruz, D, Bellomo, R. Continuous renal replacement in critical illness. Contrib Nephrol. 2007; 156:309–319.
276. Marshall, MR, Golper, TA, Shaver, MJ, et al. Sustained low-efficiency dialysis for critically ill patients requiring renal replacement therapy. Kidney Int. 2001; 60:777–785.
277. Marshall, MR, Ma, T, Galler, D, et al. Sustained low-efficiency daily diafiltration (SLEDD-f) for critically ill patients requiring renal replacement therapy: Towards an adequate therapy. Nephrol Dial Transplant. 2004; 19:877–884.
278. Ash, SR. Peritoneal dialysis in acute renal failure of adults: The under-utilized modality. Contrib Nephrol. 2004; 144:239–254.
279. Passadakis, PS, Oreopoulos, DG. Peritoneal dialysis in patients with acute renal failure. Adv Perit Dial. 2007; 23:7–16.
280. Abi Antoun, T, Palevsky, PM. Selection of modality of renal replacement therapy. Semin Dial. 2009; 22:108–113.
281. van Bommel, E, Bouvy, ND, So, KL, et al. Acute dialytic support for the critically ill: Intermittent hemodialysis versus continuous arteriovenous hemodiafiltration. Am J Nephrol. 1995; 15:192–200.
282. Swartz, RD, Messana, JM, Orzol, S, Port, FK. Comparing continuous hemofiltration with hemodialysis in patients with severe acute renal failure. Am J Kidney Dis. 1999; 34:424–432.
283. Guerin, C, Girard, R, Selli, JM, Ayzac, L. Intermittent versus continuous renal replacement therapy for acute renal failure in intensive care units: Results from a multicenter prospective epidemiological survey. Intensive Care Med. 2002; 28:1411–1418.
284. Manns, B, Doig, CJ, Lee, H, et al. Cost of acute renal failure requiring dialysis in the intensive care unit: Clinical and resource implications of renal recovery. Critical Care Med. 2003; 31:449–455.
285. Swartz, RD, Bustami, RT, Daley, JM, et al. Estimating the impact of renal replacement therapy choice on outcome in severe acute renal failure. Clin Nephrol. 2005; 63:335–345.
286. Jacka, MJ, Ivancinova, X, Gibney, RT. Continuous renal replacement therapy improves renal recovery from acute renal failure. Can J Anaesth. 2005; 52:327–332.
287. Cho, KC, Himmelfarb, J, Paganini, E, et al. Survival by dialysis modality in critically ill patients with acute kidney injury. J Am Soc Nephrol. 2006; 17:3132–3138.
288. Mehta, RL, McDonald, B, Gabbai, FB, et al. A randomized clinical trial of continuous versus intermittent dialysis for acute renal failure. Kidney Int. 2001; 60:1154–1163.
289. Gasparovic, V, Filipovic-Grcic, I, Merkler, M, Pisl, Z. Continuous renal replacement therapy (CRRT) or intermittent hemodialysis (IHD)—What is the procedure of choice in critically ill patients? Ren Fail. 2003; 25:855–862.
290. Augustine, JJ, Sandy, D, Seifert, TH, Paganini, EP. A randomized controlled trial comparing intermittent with continuous dialysis in patients with ARF. Am J Kidney Dis. 2004; 44:1000–1007.
291. Uehlinger, DE, Jakob, SM, Ferrari, P, et al. Comparison of continuous and intermittent renal replacement therapy for acute renal failure. Nephrol Dial Transplant. 2005; 20:1630–1637.
292. Vinsonneau, C, Camus, C, Combes, A, et al. Continuous venovenous haemodiafiltration versus intermittent haemodialysis for acute renal failure in patients with multiple-organ dysfunction syndrome: A multicentre randomised trial. Lancet. 2006; 368:379–385.
293. Lins, RL, Elseviers, MM, Van der Niepen, P, et al. Intermittent versus continuous renal replacement therapy for acute kidney injury patients admitted to the intensive care unit: Results of a randomized clinical trial. Nephrol Dial Transplant. 2009; 24:512–518.
294. Rabindranath, K, Adams, J, Macleod, AM, Muirhead, N. Intermittent versus continuous renal replacement therapy for acute renal failure in adults. Cochrane Database Syst Rev. (3):2007.
295. Pannu, N, Klarenbach, S, Wiebe, N, et al. Renal replacement therapy in patients with acute renal failure: A systematic review. JAMA. 2008; 299:793–805.
296. Bagshaw, SM, Berthiaume, LR, Delaney, A, Bellomo, R. Continuous versus intermittent renal replacement therapy for critically ill patients with acute kidney injury: A meta-analysis. Crit Care Med. 2008; 36:610–617.
297. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012; 2:1–138.
298. Davenport, A, Will, EJ, Davison, AM. Continuous vs. intermittent forms of haemofiltration and/or dialysis in the management of acute renal failure in patients with defective cerebral autoregulation at risk of cerebral oedema. Contrib Nephrol. 1991; 93:225–233.
299. Davenport, A, Will, EJ, Davison, AM. Early changes in intracranial pressure during haemofiltration treatment in patients with grade 4 hepatic encephalopathy and acute oliguric renal failure. Nephrol Dial Transplant. 1990; 5:192–198.
300. Lin, CM, Lin, JW, Tsai, JT, et al. Intracranial pressure fluctuation during hemodialysis in renal failure patients with intracranial hemorrhage. Acta Neurochir Suppl. 2008; 101:141–144.
301. Davenport, A. Continuous renal replacement therapies in patients with liver disease. Semin Dial. 2009; 22:169–172.
302. Davenport, A. Continuous renal replacement therapies in patients with acute neurological injury. Semin Dial. 2009; 22:165–168.
303. Kielstein, JT, Kretschmer, U, Ernst, T, et al. Efficacy and cardiovascular tolerability of extended dialysis in critically ill patients: A randomized controlled study. Am J Kidney Dis. 2004; 43:342–349.
304. Kumar, VA, Yeun, JY, Depner, TA, Don, BR. Extended daily dialysis vs. continuous hemodialysis for ICU patients with acute renal failure: A two-year single center report. Int J Artif Organs. 2004; 27:371–379.
305. Fieghen, HE, Friedrich, JO, Burns, KE, et al. The hemodynamic tolerability and feasibility of sustained low efficiency dialysis in the management of critically ill patients with acute kidney injury. BMC Nephrol. 2010; 11:32.
306. Abe, M, Okada, K, Suzuki, M, et al. Comparison of sustained hemodiafiltration with continuous venovenous hemodiafiltration for the treatment of critically ill patients with acute kidney injury. Artif Organs. 2010; 34:331–338.
307. Phu, NH, Hien, TT, Mai, NT, et al. Hemofiltration and peritoneal dialysis in infection-associated acute renal failure in Vietnam. N Engl J Med. 2002; 347:895–902.
308. Gabriel, DP, Nascimento, GV, Caramori, JT, et al. High volume peritoneal dialysis for acute renal failure. Perit Dial Int. 2007; 27:277–282.
309. Gabriel, DP, Caramori, JT, Martin, LC, et al. Continuous peritoneal dialysis compared with daily hemodialysis in patients with acute kidney injury. Perit Dial Int. 2009; 29(Suppl 2):S62–S71.
310. Gabriel, DP, Caramori, JT, Martim, LC, et al, High volume peritoneal dialysis vs daily hemodialysis: A randomized, controlled trial in patients with acute kidney injury. Kidney Int Suppl. 2008(108):S87–S93.
311. Parsons, FM, Hobson, SM, Blagg, CR, McCracken, BH. Optimum time for dialysis in acute reversible renal failure. Description and value of an improved dialyser with large surface area. Lancet. 1961; 1:129–134.
312. Fischer, RP, Griffen, WO, Jr., Reiser, M, Clark, DS. Early dialysis in the treatment of acute renal failure. Surg Gynecol Obstet. 1966; 123:1019–1023.
313. Kleinknecht, D, Jungers, P, Chanard, J, et al. Uremic and non-uremic complications in acute renal failure: Evaluation of early and frequent dialysis on prognosis. Kidney Int. 1972; 1:190–196.
314. Conger, JD. A controlled evaluation of prophylactic dialysis in post-traumatic acute renal failure. J Trauma. 1975; 15:1056–1063.
315. Gillum, DM, Dixon, BS, Yanover, MJ, et al. The role of intensive dialysis in acute renal failure. Clin Nephrol. 1986; 25:249–255.
316. Gettings, LG, Reynolds, HN, Scalea, T. Outcome in post-traumatic acute renal failure when continuous renal replacement therapy is applied early vs. late. Intensive Care Med. 1999; 25:805–813.
317. Elahi, MM, Lim, MY, Joseph, RN, et al. Early hemofiltration improves survival in post-cardiotomy patients with acute renal failure. Eur J Cardiothorac Surg. 2004; 26:1027–1031.
318. Demirkilic, U, Kuralay, E, Yenicesu, M, et al. Timing of replacement therapy for acute renal failure after cardiac surgery. J Card Surg. 2004; 19:17–20.
319. Liu, KD, Himmelfarb, J, Paganini, E, et al. Timing of initiation of dialysis in critically ill patients with acute kidney injury. Clin J Am Soc Nephrol. 2006; 1:915–919.
320. Piccinni, P, Dan, M, Barbacini, S, et al. Early isovolaemic haemofiltration in oliguric patients with septic shock. Intensive Care Med. 2006; 32:80–86.
321. Andrade, L, Cleto, S, Seguro, AC. Door-to-dialysis time and daily hemodialysis in patients with leptospirosis: Impact on mortality. Clin J Am Soc Nephrol. 2007; 2:739–744.
322. Wu, VC, Ko, WJ, Chang, HW, et al. Early renal replacement therapy in patients with postoperative acute liver failure associated with acute renal failure: Effect on postoperative outcomes. J Am Coll Surg. 2007; 205:266–276.
323. Manche, A, Casha, A, Rychter, J, et al. Early dialysis in acute kidney injury after cardiac surgery. Interact Cardiovasc Thorac Surg. 2008; 7:829–832.
324. Sabater, J, Perez, X, Albertos, R, et al. Acute renal failure in septic shock. Should we consider different continuous renal replacement therapies on each RIFLE score stage? Intensive Care Med. 2009; 35:S239.
325. Bagshaw, SM, Uchino, S, Bellomo, R, et al. Timing of renal replacement therapy and clinical outcomes in critically ill patients with severe acute kidney injury. J Crit Care. 2009; 24:129–140.
326. Iyem, H, Tavli, M, Akcicek, F, Buket, S. Importance of early dialysis for acute renal failure after an open-heart surgery. Hemodial Int. 2009; 13:55–61.
327. Shiao, CC, Wu, VC, Li, WY, et al. Late initiation of renal replacement therapy is associated with worse outcomes in acute kidney injury after major abdominal surgery. Crit Care. 2009; 13:R171.
328. Carl, DE, Grossman, C, Behnke, M, et al. Effect of timing of dialysis on mortality in critically ill, septic patients with acute renal failure. Hemodial Int. 2010; 14:11–17.
329. Bouman, CS, Oudemans-Van Straaten, HM, Tijssen, JG, et al. Effects of early high-volume continuous venovenous hemofiltration on survival and recovery of renal function in intensive care patients with acute renal failure: A prospective, randomized trial. Crit Care Med. 2002; 30:2205–2211.
330. Karvellas, CJ, Farhat, MR, Sajjad, I, et al. A comparison of early versus late initiation of renal replacement therapy in critically ill patients with acute kidney injury: A systematic review and meta-analysis. Crit Care. 2011; 15:R72.
331. Payen, D, de Pont, AC, Sakr, Y, et al. A positive fluid balance is associated with a worse outcome in patients with acute renal failure. Crit Care. 2008; 12:R74.
332. Goldstein, SL, Somers, MJ, Baum, MA, et al. Pediatric patients with multi-organ dysfunction syndrome receiving continuous renal replacement therapy. Kidney Int. 2005; 67:653–658.
333. Sutherland, SM, Zappitelli, M, Alexander, SR, et al. Fluid overload and mortality in children receiving continuous renal replacement therapy: The prospective pediatric continuous renal replacement therapy registry. Am J Kidney Dis. 2010; 55:316–325.
334. Schiffl, H, Lang, SM, Fischer, R. Daily hemodialysis and the outcome of acute renal failure. N Engl J Med. 2002; 346:305–310.
335. Ronco, C, Bellomo, R, Homel, P, et al. Effects of different doses in continuous veno-venous haemofiltration on outcomes of acute renal failure: A prospective randomised trial. Lancet. 2000; 356:26–30.
336. Saudan, P, Niederberger, M, De Seigneux, S, et al. Adding a dialysis dose to continuous hemofiltration increases survival in patients with acute renal failure. Kidney Int. 2006; 70:1312–1317.
337. Tolwani, AJ, Campbell, RC, Stofan, BS, et al. Standard versus high-dose CVVHDF for ICU-related acute renal failure. J Am Soc Nephrol. 2008; 19:1233–1238.
338. Bellomo, R, Cass, A, Cole, L, et al. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med. 2009; 361:1627–1638.
339. Abosaif, NY, Tolba, YA, Heap, M, et al. The outcome of acute renal failure in the intensive care unit according to RIFLE: Model application, sensitivity, and predictability. Am J Kidney Dis. 2005; 46:1038–1048.
340. Silvester, W, Bellomo, R, Cole, L. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Critical Care Med. 2001; 29:1910–1915.
341. Soubrier, S, Leroy, O, Devos, P, et al. Epidemiology and prognostic factors of critically ill patients treated with hemodiafiltration. J Crit Care. 2006; 21:66–72.
342. Clermont, G, Acker, CG, Angus, DC, et al. Renal failure in the ICU: Comparison of the impact of acute renal failure and end-stage renal disease on ICU outcomes. Kidney Int. 2002; 62:986–996.
343. Metnitz, PG, Krenn, CG, Steltzer, H, et al. Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Critical Care Med. 2002; 30:2051–2058.
344. Benoit, DD, Hoste, EA, Depuydt, PO, et al. Outcome in critically ill medical patients treated with renal replacement therapy for acute renal failure: Comparison between patients with and those without haematological malignancies. Nephrol Dial Transplant. 2005; 20:552–558.
345. Chertow, GM, Burdick, E, Honour, M, et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005; 16:3365–3370.
346. Ympa, YP, Sakr, Y, Reinhart, K, Vincent, JL. Has mortality from acute renal failure decreased? A systematic review of the literature. Am J Med. 2005; 118:827–832.
347. Tonelli, M, Manns, B, Feller-Kopman, D. Acute renal failure in the intensive care unit: A systematic review of the impact of dialytic modality on mortality and renal recovery. Am J Kidney Dis. 2002; 40:875–885.
348. Palevsky, PM, Metnitz, PG, Piccinni, P, Vinsonneau, C. Selection of endpoints for clinical trials of acute renal failure in critically ill patients. Curr Opin Crit Care. 2002; 8:515–518.
349. Chertow, GM, Lazarus, JM, Paganini, EP, et al. Predictors of mortality and the provision of dialysis in patients with acute tubular necrosis. The Auriculin Anaritide Acute Renal Failure Study Group. J Am Soc Nephrol. 1998; 9:692–698.
350. Rasmussen, HH, Pitt, EA, Ibels, LS, McNeil, DR. Prediction of outcome in acute renal failure by discriminant analysis of clinical variables. Arch Intern Med. 1985; 145:2015–2018.
351. Turney, JH, Marshall, DH, Brownjohn, AM, et al. The evolution of acute renal failure, 1956-1988. Q J Med. 1990; 74:83–104.
352. Biesenbach, G, Zazgornik, J, Kaiser, W, et al. Improvement in prognosis of patients with acute renal failure over a period of 15 years: An analysis of 710 cases in a dialysis center. Am J Nephrol. 1992; 12:319–325.
353. Lassnigg, A, Schmid, ER, Hiesmayr, M, et al. Impact of minimal increases in serum creatinine on outcome in patients after cardiothoracic surgery: Do we have to revise current definitions of acute renal failure? Crit Care Med. 2008; 36:1129–1137.
354. Goldenberg, I, Chonchol, M, Guetta, V. Reversible acute kidney injury following contrast exposure and the risk of long-term mortality. Am J Nephrol. 2009; 29:136–144.
355. Gruberg, L, Mintz, GS, Mehran, R, et al. The prognostic implications of further renal function deterioration within 48 h of interventional coronary procedures in patients with pre-existent chronic renal insufficiency. J Am Coll Cardiol. 2000; 36:1542–1548.
356. Weisbord, SD, Palevsky, PM. Acute kidney injury: Kidney injury after contrast media: Marker or mediator? Nat Rev. 2010; 6:634–636.
357. Faulhaber-Walter, R, Hafer, C, Jahr, N, et al. The Hannover Dialysis Outcome study: Comparison of standard versus intensified extended dialysis for treatment of patients with acute kidney injury in the intensive care unit. Nephrol Dial Transplant. 2009; 24:2179–2186.
358. Edwards, KD. Recovery of renal function after acute renal failure. Australas Ann Med. 1959; 8:195–199.
359. Price, JD, Palmer, RA. A functional and morphological follow-up study of acute renal failure. Arch Intern Med. 1960; 105:90–98.
360. Briggs, JD, Kennedy, AC, Young, LN, et al. Renal function after acute tubular necrosis. Br Med J. 1967; 3:513–516.
361. Lewers, DT, Mathew, TH, Maher, JF, Schreiner, GE. Long-term follow-up of renal function and histology after acute tubular necrosis. Ann Intern Med. 1970; 73:523–529.
362. Ishani, A, Xue, JL, Himmelfarb, J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009; 20:223–228.
363. Wald, R, Quinn, RR, Luo, J, et al. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA. 2009; 302:1179–1185.
364. Newsome, BB, Warnock, DG, McClellan, WM, et al. Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med. 2008; 168:609–616.
365. James, MT, Ghali, WA, Tonelli, M, et al. Acute kidney injury following coronary angiography is associated with a long-term decline in kidney function. Kidney Int. 2010; 78:803–809.
366. Hoste, EA, Kellum, JA. Acute renal failure in the critically ill: Impact on morbidity and mortality. Contrib Nephrol. 2004; 144:1–11.
367. Bates, DW, Su, L, Yu, DT, et al. Mortality and costs of acute renal failure associated with amphotericin B therapy. Clin Infect Dis. 2001; 32:686–693.
368. Fischer, MJ, Brimhall, BB, Lezotte, DC, et al. Uncomplicated acute renal failure and hospital resource utilization: A retrospective multicenter analysis. Am J Kidney Dis. 2005; 46:1049–1057.
369. Hamel, MB, Phillips, RS, Davis, RB, et al. Outcomes and cost-effectiveness of initiating dialysis and continuing aggressive care in seriously ill hospitalized adults. SUPPORT Investigators. Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments. Ann Intern Med. 1997; 127:195–202.
370. Ahlstrom, A, Tallgren, M, Peltonen, S, et al. Survival and quality of life of patients requiring acute renal replacement therapy. Intensive Care Med. 2005; 31:1222–1228.
371. Johansen, KL, Smith, MW, Unruh, ML, et al. Predictors of health utility among 60-day survivors of acute kidney injury in the Veterans Affairs/National Institutes of Health Acute Renal Failure Trial Network Study. Clin J Am Soc Nephrol. 2010; 5:1366–1372.