CHAPTER 7 Abnormalities of the red cell membrane
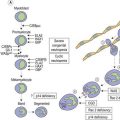
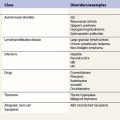
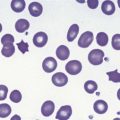
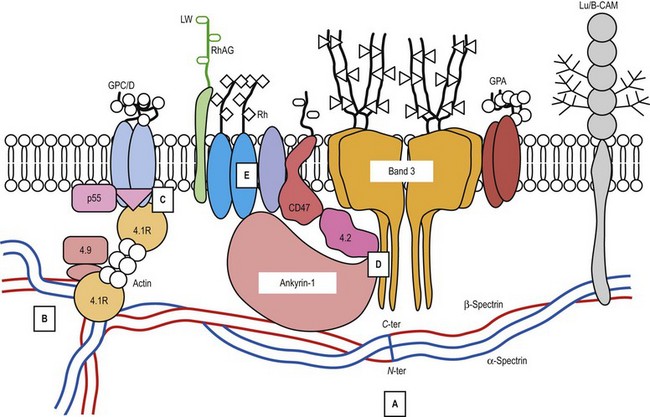
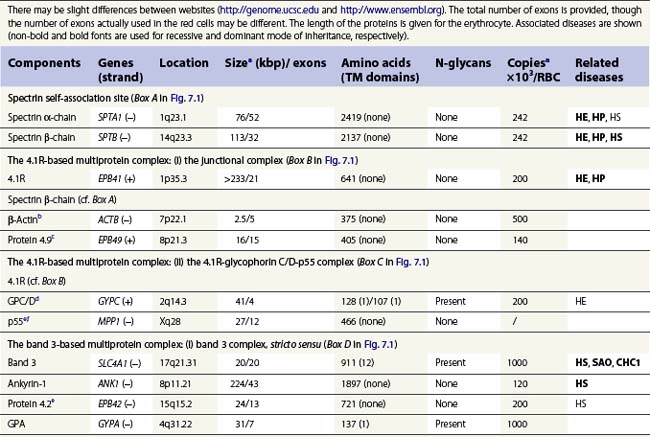
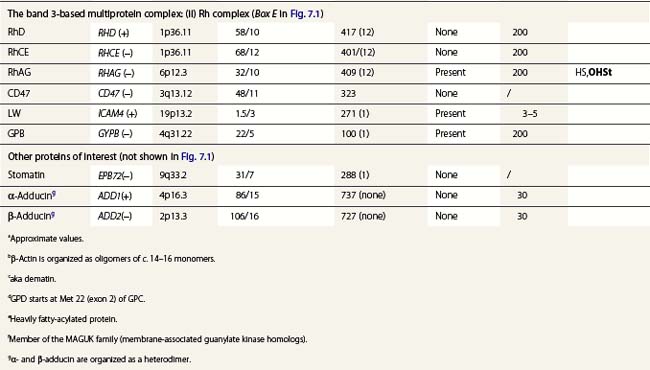
A number of hereditary hemolytic anemias result from mutations affecting the quality and/or amount of proteins that belong to the red cell membrane, its skeleton, or the attachment systems (nexuses) of the latter to the former. Most proteins participate in complexes (Fig. 7.1). They play a role in erythrocyte resilience and elastic deformability, either mechanically, through the skeleton and its attaching systems, or osmotically, through a variety of transporters and pumps. Major proteins and their genes are listed in Table 7.1. The ever increasing number of mutations, too numerous to detail here, have given insight into the function of such protein domains and the regulatory regions of some genes. A selection of abnormally-shaped red cells is shown in Fig. 7.2.
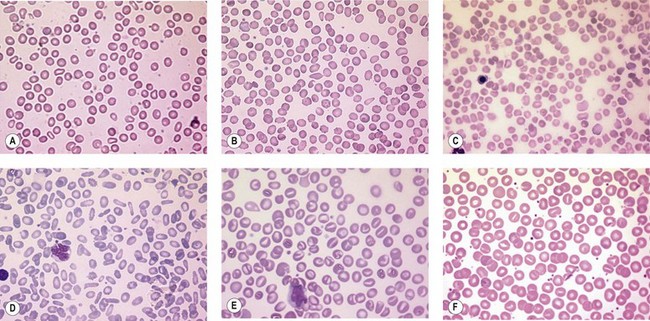
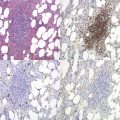
Hereditary spherocytosis
Hereditary spherocytosis (HS) is the most common genetic disorder of the red cell membrane in Western countries. Its incidence has been estimated as 1 in 2000 live births and there is a wide spectrum of clinical severity. In typical cases the hemolytic anemia is moderate, with an increased reticulocyte count a reticulocytosis, intermittent jaundice, gallstones and splenomegaly. Severe cases are rare and may cause death in utero or shortly after birth. In contrast, patients with mild HS may be over 60 years of age when diagnosed. Parvovirus B19 infection commonly occurs. Blood films show a variable percentage of spherocytes. The diagnosis relies on an increased percentage of hyperdense cells and a reduction in osmotic resistance, and on a number of tests, including polyacrylamide gel electrophoresis of the red cell membrane proteins in the presence of sodium dodecylsulfate (SDS-PAGE). The main treatment is splenectomy, though the need for this should be carefully weighed owing to its complications, namely severe infections and a statistically significant increase in thromboembolic accidents.1 Transfusions may be necessary.
ANK1 gene mutations
Ankyrin-1 is encoded by ANK1.2 It connects the skeleton to band 3, i.e. the anion exchanger-1. Approximately 60% of HS are due to ANK1 gene mutations and have reduced ankyrin-1. HS due to ANK1 mutations is relatively severe and has a dominant inheritance pattern, although de novo mutations may occur. Homozygosity is bound to be lethal. (One case has been recently described, however.) This may not be evident due to the elevated reticulocyte count, as young cells have a higher ankyrin-1 content. Spectrin α- and β-chains, and protein 4.2, interacting with ankyrin-1, are secondarily decreased.
SLC4A1 gene mutations
Band 3, encoded by SLC4A1, is the pillar of the band 3 complex stricto sensu, which is itself attached to the Rh complex. Both complexes are linked through protein 4.2-CD473 and Rh/RhAG-ankyrin-1 contacts.4 In the heterozygous state, mutations in SLC4A1 produce a mild, dominantly inherited HS (approximately 20% of HS cases). Band 3 is uniformly reduced, along with a proportional decrease in protein 4.2. Scores of mutations have been reported since the first report.5 More severe cases are seen in compound heterozygotes. Two homozygous cases, leading to missing or strongly reduced band 3, have been reported. They were associated with a dramatic picture. Spherocytes were replaced in part by poikilocytes (see below).6,7 The absence of band 3 is likely to be lethal unless intensive care is provided prior to and following birth. An early subtotal splenectomy, to be completed later, is indicated. These cases are accompanied by distal renal tubular acidosis, due to the fact that α-intercalated cells of the distal tubule basolateral membrane contain an isoform of band 3, lacking the 65 first amino acids of the erythroid isoform. The prognosis is obscured by nephrocalcinosis. A third case of homozygosity was described free of renal disorders because the mutation lay in the missing region of the renal isoform of band 3.
SPTB gene mutations
SPTB encodes spectrin β-chain. Mutations generate a dominantly inherited, relatively severe form of HS (20% of HS in Europe).8 There is an isolated reduction in spectrin (α- and β-chains) on SDS-PAGE. De novo mutations may occur. Homozygous cases have never been reported.
EPB42 gene mutations
Mutations in EPB42, encoding protein 4.2, engender a relatively rare recessively inherited form of HS which is not absolutely typical. Spherocytes are bulky. A mutation has shown some frequency in Japan (Ala142Thr).9
SPTA1 gene mutations
SPTA1 encodes spectrin α-chain. Mutations in SPTA1 are rare and show a recessive inheritance pattern. Homozygosity, or compound heterozygosity (involving peremptory mutations: stop codon or frameshift), has never been reported. In contrast, compound heterozygosity for a peremptory HS mutation and a rather common weak allele of the SPTA1 gene, allele αLEPRA, has been observed, causing a severe picture with poikilocytosis.10
Hereditary elliptocytosis and poikilocytosis
SPTA1 gene mutations
SPTA1 mutations account for approximately 60% of HE and HP. Their mode of inheritance is dominant. Mutations are located from the N-terminal region of the spectrin α-chain down to repeat α9.11 As a consequence, the self-association process is impaired, loosening the meshwork at a critical junction. Homozygosity or compound heterozygosity has a moderate to severe presentation depending on the mutation(s). Severe forms display an HP phenotype. The same situation happens when an HE mutation lies in trans to a frequent, worldwide, low-expression allele of the SPTA1 gene, allele αLELY.12 Splenectomy should only be considered in the most severe cases.
SPTB gene mutations
SPTB mutations are rarer than SPTA1 mutations and are associated with HE or HP. They are sporadic and dominantly transmitted. Mutations are situated in the C-terminal region (repeat β17) of the spectrin β-chain, in the self-association site, and with the same consequences as the facing mutations in the α-chain.11 Homozygosity may be life-threatening.
EPB41 gene mutations
The EPB41 gene encodes protein 4.1R. Mutations in EPB41 account for 20–30% of HE in Caucasians. In the heterozygous state, 20–30% of 4.1R is missing. The 4.1R (−) trait, which is dominantly transmitted, is symptomless. In the homozygous state,13 in which 4.1R is absent, HP is severe in the newborn but tends to improve later on. Early subtotal splenectomy may accelerate the improvement.
Genetic disorders of the monovalent cation leak across the membrane
Disorders of monovalent cation leak across the membrane, or cation leak, encompass conditions that remain poorly understood. Most include hemolytic anemia, jaundice and splenomegaly, erythrocyte shape abnormalities and macrocytosis. They have a major tendency to iron overload, an alteration of intra-erythrocytic cation concentrations, and an increase in the cation leak. The leak is defined as the fluxes that remain when the Na+, K+ ATPase and the Na+, K+, 2Cl− co-transporter are inhibited by ouabain and bumetanide, respectively. The leak assumes various curves as a function of temperature. In a subset of cases, the leak diminishes with a fall in temperature from 37°C to 20°C, and then resumes, sometimes dramatically, at lower temperatures, warranting the prefix ‘cryo-’. The inheritance pattern is dominant. Splenectomy is contraindicated in most of these conditions due to the risk of thromboembolic accidents, which may be lethal.14
A pleiotropic syndrome revolving around dehydrated hereditary stomatocytosis
Dehydrated hereditary stomatocytosis (DHSt) may occur alone, as first reported by Oski et al.15 The presentation is mild and sometimes revealed only at a late stage by its major complication, an iron overload (hemochromatosis). Anemia is well compensated, and the reticulocyte count is elevated. Stomatocytes are scarce and incompletely formed. There is a borderline macrocytosis. The osmotic resistance is increased. De novo mutations may occur. In the homozygous state it is lethal. The curve : cation leak as a function of temperature is monophasic and has a ‘shallow’ slope. One responsible gene has been mapped to 16q23-ter in a large Irish kindred.16 The region of interest was recently narrowed down to 16q24.1-ter in an extended French kindred.17 However, there must be another or other gene(s) involved. DHSt is associated with pseudohyperkalemia and/or perinatal fluid effusions, resulting in a pleiotropic syndrome.18
Pseudohyperkalemia designates an increase in potassium when collected blood is left at room temperature for a few hours. DHSt-related pseudohyperkalemia is similar to familial pseudohyperkalemia (FP), a dominantly inherited trait.19 Although extremely rare, it has been mapped to 16q23-ter20 and to 2q35-3621 in distinct (Scottish and Flemish) extended families. The first location coincides with that of some cases of DHSt, strengthening the idea that FP is a truncated form of the pleiotropic syndrome.
Overhydrated hereditary stomatocytosis
Overhydrated hereditary stomatocytosis (OHSt) is an exceedingly rare form of stomatocytosis. The first case of stomatocytosis ever described was an OHSt.22 The presentation is quite pronounced with anemia (sometimes requiring transfusions), marked macrocytosis, a strong tendency to iron overload and a reduced osmotic resistance. Stomatocytes are numerous and fully fledged. De novo mutations are frequent. A salient feature is the sharp decrease in, or the absence of, stomatin (not shown in Fig. 7.1). The responsible gene is RHAG.23
Hereditary cryohydrocytosis
Hereditary cryohydrocytosis with normal stomatin (CHC1) is also an exceedingly rare disorder, first described by Miller et al.24 CHC 1 presents like a stomatocytosis (though of the dehydrated type, with elevated mean corpuscular hemoglobin concentration (MCHC)). Pseudohyperkalemia may be present. The salient feature is the resumption of the leak at low temperature. Mutations have been found in the SLC4A1 gene.25 Rare, atypical forms of HS (on the basis of the curve : leak as a function of temperature) displayed mutations in the involved region of band 3 (but with no reduction, as seen above); they were eventually related to CHC1.25
Hereditary cryohydrocytosis with low or missing stomatin (CHC2) presents as OHSt. Only two cases have been reported worldwide.26 Various neurological signs were found to be associated with the condition. There was a soaring resumption of the leak at low temperatures. No mutations were found in the RHAG gene.
Southeast Asian ovalo-stomatocytosis
Southeast Asian ovalo-stomatocytosis is a dominantly inherited condition. It is a symptomless trait. The red cells are oval and show a longitudinal slit or two transverse ridges. This condition is classified with disorders of cation leak because an increased leak is present at low temperatures. The red cell membrane is very rigid and it is this feature which accounts for the associated resistance to malaria. The genetic alteration is a deletion of 27 nucleotides in SLC4A1, generating a deletion of 9 amino acid at the junction of the cytoplasmic and membrane domains of band 3.27 Homozygosity is bound to be lethal, in keeping with the other disorders of cation leak.
Paroxysmal exertion-induced dyskinesia
Paroxysmal exertion-induced dyskinesia is characterized by disorders induced by prolonged exercise. They include epilepsy, mild developmental delay and reduced cerebrospinal fluid glucose level. Some forms are associated with a hemolytic anemia and echinocytosis, altered intra-erythrocytic cation concentrations and an increased cation leak. In one family, a deletion of four highly conserved amino acids was found GLUT1 encoded by SLC2A1.28
1 Schilling RF, Gangnon RE, Traver MI. Delayed adverse vascular events after splenectomy in hereditary spherocytosis. J Thromb Haemost. 2008;6:1289-1295.
2 Eber SW, Gonzalez JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nature Genetics. 1996;13:214-218.
3 Bruce LJ, Beckmann R, Ribeiro ML, et al. Evidence for a band 3-based membrane macrocomplex: a potential gas exchange metabolon. Blood. 2003;101:4180-4188.
4 Nicolas V, Le Van Kim C, Gane P, et al. Rh-RhAG/ankyrin-R, a new interaction site between the membrane bilayer and the red cell skeleton, is impaired by Rh(null)-associated mutation. Journal of Biological Chemistry. 2003;278:25526-25533.
5 Jarolim P, Rubin HL, Liu SC, et al. Duplication of 10 nucleotides in the erythroid band 3 (AE1) gene in a kindred with hereditary spherocytosis and band 3 protein deficiency (band 3 PRAGUE). Journal of Clinical Investigation. 1994;93:121-130.
6 Ribeiro L, Alloisio N, Almeida H, et al. Near lethal hereditary spherocytosis and distal tubular acidosis associated with the total absence of band 3. Blood. 2000;96:1602-1604.
7 Toye AM, Williamson RC, Khanfar M, et al. Band 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild type band 3 and glycophorin A. Blood. 2008;111:5380-5389.
8 Hassoun H, Vassiliadis JN, Murray J, et al. Molecular basis of spectrin deficiency in β spectrin Durham. A deletion within β spectrin adjacent to the ankyrin-binding site precludes spectrin attachment to the membrane in hereditary spherocytosis. Journal of Clinical Investigation. 1995;96:2623-2629.
9 Bouhassira EE, Schwartz RS, Yawata Y, et al. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood. 1992;79:1846-1854.
10 Wichterle H, Hanspal M, Palek J, et al. Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia. Journal of Clinical Investigation. 1996;98:2300-2307.
11 Maillet P, Alloisio N, Morlé L, Delaunay J. Spectrin mutations in hereditary elliptocytosis and hereditary spherocytosis. Human Mutation. 1996;8:97-107.
12 Wilmotte R, Maréchal J, Morlé L, et al. Low expression allele αLELY of red cell spectrin is associated with mutations in exon 40 (αV/41 polymorphism) and intron 45 and with partial skipping of exon 46. Journal of Clinical Investigation. 1993;91:2091-2096.
13 Dalla Venezia N, Gilsanz F, Alloisio N, et al. Homozygous 4.1(−) hereditary elliptocytosis associated with a point mutation in the downstream initiation codon of protein 4.1 gene. Journal of Clinical Investigation. 1992;90:1713-1717.
14 Stewart GW, Amess JAL, Eber SW, et al. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. British Journal of Haematology. 1996;93:303-310.
15 Oski FA, Naiman JL, Blum SF, et al. Congenital hemolytic anemia with high-sodium, low-potassium red cells. Studies of three generations of a family with a new variant. New England. Journal of Medicine. 1969;280:909-916.
16 Carella M, Stewart G, Ajetunmobi JF, et al. Genomewide search for dehydrated hereditary stomatocytosis (hereditary xerocytosis): mapping of locus to chromosome 16 (16q23-qter). American Journal of Human Genetics. 1998;63:810-816.
17 Beaurain G, Mathieu F, Grootenboer S, et al. Dehydrated hereditary stomatocytosis mimicking familial hyperkalaemic hypertension: clinical and genetic investigation. European Journal of Haematology. 2007;78:253-259.
18 Grootenboer S, Schischmanoff PO, Laurendeau I, et al. Pleiotropic syndrome of dehydrated hereditary stomatocytosis, pseudohyperkalemia and perinatal edema maps to 16q23-q24. Blood. 2000;96:2599-2605.
19 Stewart GW, Corral RJ, Fyffe JA, et al. Familial pseudohyperkalemia. A new syndrome. Lancet. 1979;2(8135):175-177.
20 Iolascon A, Stewart G, Ajetunmobi JF, et al. Familial pseudohyperkalemia maps to the same locus as dehydrated hereditary stomatocytosis (hereditary xerocytosis). Blood. 1999;93:3120-3123.
21 Carella M, Pio d’Adamo A, Grootenboer-Mignot S, et al. A second locus mapping to 2q35-36 for familial pseudohyperkalaemia. European. Journal of Human Genetics. 2004;12:1073-1076.
22 Lock SP, Sephton Smith R, Hardisty RM. Stomatocytosis: a hereditary red cell anomaly associated with haemolytic anaemia. British Journal of Haematology. 1961;7:303-314.
23 Bruce LJ, Guizouarn H, Burton NM, et al. The monovalent cation leak in over-hydrated stomatocytic red blood cells results from amino acid substitutions in the Rh associated glycoprotein (RhAG). Blood. 2009;113:1350-1357.
24 Miller G, Townes PL, MacWhinney JB. A new congenital hemolytic anemia with deformed erythrocytes (? ‘stomatocytes’) and remarkable susceptibility of erythrocytes to cold hemolysis in vitro. I. Clinical and hematologic studies. Pediatrics. 1965;35:906-915.
25 Bruce LJ, C Robinson H, Guizouarn H, et al. Monovalent cation leaks in human red cells caused by single amino-acid substitutions in the transport domain of the band 3 chloride-bicarbonate exchanger, AE1. Nature Genetics. 2005;37:1258-1263.
26 Fricke B, Jarvis HG, Reid CDL, et al. Four new cases of stomatin-deficient hereditary stomatocytosis syndrome. Association of the stomatin-deficient cryohydrocytosis variant with neurological dysfunction. British Journal of Haematology. 2004;125:796-803.
27 Jarolim P, Palek J, Amato D, et al. Deletion of erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proceedings of the National Academy of Sciences USA. 1991;88:11022-11026.
28 Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. Journal of Clinical Investigation. 2008;118:2157-2168.