[level-membership-for-pathology-category]
CHAPTER 16 Abnormalities in leukocyte morphology and number
Neutrophils
In the peripheral blood (PB) of healthy white adults the mean neutrophil count is 4.4 (4.3–4.6) × 109/l,1,2 while in healthy black adults it is 3.6 (3.3–3.9) × 109/l.3
Neutropenia
Neutropenia is defined as the absolute neutrophil count (ANC) in PB lower than 1.5 × 109/l. Referring to white population, a useful classification in predicting risk infection indicates neutropenia as mild, moderate or severe according to the ANC value of 1.0–1.5 × 109/l, 0.5–1.5 × 109/l or less than 0.5 × 109/l respectively. Neutropenia may arise from various disorders of bone marrow (BM) function resulting in a decreased rate of release of neutrophils from the BM into the circulation. It is a common manifestation in several BM failures, such as aplasia, leukemia or myelodysplasia (see Chapters 13, 18, 20 for details). The conditions that may be associated with a neutropenia are listed in Box 16.1. Clinically neutropenia is considered as acute when it occurs over hours to a few days, usually developing from rapid neutrophil use/destruction or from impaired production. Neutropenia is considered chronic, when it lasts months to years, usually arising from reduced production or excessive splenic sequestration. Neutropenia can be caused by an intrinsic defect in BM myeloid cells, the congenital and idiopathic neutropenias, or by factors extrinsic to BM, the acquired neutropenias.
Box 16.1 Main causes of neutropenia
Congenital and idiopathic neutropenias
Severe congenital neutropenia (Kostmann’s disease). Kostmann disease4 is a rare autosomal recessive disorder of neutrophil number. The ANC is characteristically less than 0.2 × 109/l. Severe persistent neutropenia results in an increased susceptibility to bacterial infections. Granulocyte colony-stimulating factor (G-CSF) receptors are expressed on myeloid cells in slightly increased numbers, while the binding affinity for G-CSF to its receptor is normal. The neutrophil elastase gene mutations (ELA2) have been found in a subgroup of patients with Kostmann disease, although these mutations are also found in some healthy family members.5 Autosomal recessive inheritance6 of homozygous hematopoietic cell-specific protein-1 (HS1)-associated protein X-1 (HAX-1) mutations appear to lead to the increased apoptosis of myeloid precursors observed in patients with severe congenital neutropenia.7 HAX-1 functions include signal transduction, cytoskeletal control and regulation of apoptosis and it is reported to play a role in suppression of apoptosis in lymphocytes and neurons, resulting in prolonged survival of these cells.8 While the G-CSF receptor gene mutations have not been detected at birth, patients with Kostmann disease who develop leukemia have been found to have acquired these mutations.9 Point mutations in the gene for the G-CSF receptor CSF3R have been implicated in the progression of severe congenital neutropenia to leukemia. These mutations lead to the production of truncated CSF3R receptors determining hyper-responsive forms (G-CSFR-hyper) with increased proliferation and highly decreased maturation of myeloid cells.10,11
Congenital aleukia (reticular dysgenesis). Reticular dysgenesis12 is a rare inherited disorder characterized by the failure of hematopoietic stem cells committed to myeloid and lymphoid development. Red blood cell count (RBC) is normal, while platelet count may be decreased. There are no lymphocytes in the thymus, which is hypoplastic, and there is complete absence of peripheral lymphoid tissues. Allogenic BM transplant (BMT) remains the sole therapeutic option.13
Benign chronic neutropenias
Familial benign chronic neutropenia. This disorder,14,15 inherited as an autosomal dominant trait, is characterized by chronic moderate to severe neutropenia, monocytosis, lymphocytosis and occasionally moderate eosinophilia. Patients with mild or moderate neutropenia are asymptomatic. The BM is normocellular and usually shows few neutrophil precursors beyond the myelocyte stage. Patients have a reduced reserve myeloid pool in the BM, which makes G-CSF therapy not appropriate.16
Non-familial benign chronic neutropenia. Cases of chronic benign neutropenia without any identified familial pattern are grouped in this category. In several patients, the presence of autoantibodies suggests the immune aetiology.17 Even though neutropenia is usually severe in patients with chronic benign neutropenia of infancy and childhood, the risk of infection is very low. BM is hypercellular and the granulopoietic differentiation is present until the stage of band form. A reduced neutrophil chemotaxis from BM to PB is a possible explanation. In situations causing acute stress such as infection or cortisol administration, the number of circulating neutrophils increases.18 G-CSF therapy in the forms with a benign course is not indicated.
Familial cyclic neutropenia. This is a rare disorder,19–21 in which neutropenia of 4–10 days duration occurs at intervals of 15–35 days (average 21 days). Most cases present in infancy or childhood with periodic bouts of fever, malaise, headache, sore throat, oral ulceration, skin infections or, occasionally, infections of the lungs and other organs. Neutropenia results from cyclic changes in neutrophil granulocytopoiesis. Cyclic neutropenia has been transferred by BM transplantation to a histocompatible sibling with acute lymphoblastic leukemia.22 In about 25% of patients, the condition is inherited as a dominant trait with point mutations involving the neutrophil elastase gene ELA-2 on chromosome 19p13.3. Sporadic, not familial, as well as acquired cases associated with large granular lymphocyte (LGL) expansion are reported in the literature.23,24
Chronic idiopathic neutropenia of adult. This is a cytokine-mediated syndrome characterized by varying degrees of neutropenia associated with a low number of lineage-specific CD34+ cells and increased production of inhibitors of hematopoiesis (including tumor growth factor (TGF)-β1 and tumor necrosis factor (TNF)-α) and lymphopenia due to selective loss of primed/memory T-cells and natural killer (NK) cells. Other symptoms, reported in about 50% of patients, include splenomegaly, osteopenia and/or osteoporosis. The presence of features of chronic antigenic stimulation and increased concentrations of a variety of macrophage-derived pro-inflammatory cytokines25 are suggestive of the existence of an unrecognized low-grade chronic inflammatory process, which may be involved in the pathogenesis of the disorder. Neutropenia in these patients is probably resulting from a combination of at least three factors:26 reduced production of BM neutrophils, enhanced neutrophil extravasation and increased sequestration and/or extravasation of neutrophils into the spleen.
Neutropenia in patients with congenital immune defects
Neutropenia can be associated with congenital immunologic defects and patients with combined defects are at very high risk of infection. Neutropenia is reported in about one fourth of patients with Bruton agammaglobulinemia (X-linked hypogammaglobulinemia),27 in about 50% of patients with dysgammaglobulinemia,28 and in several patients with hyper IgM syndrome or with isolated IgA syndrome.29 Treatment with intravenous infusion of immunoglobulin is required, possibly associated with G-CSF.
Neutropenia in patients with congenital chromosomal abnormalities
Shwachman–Diamond syndrome. Shwachman–Diamond30 syndrome is a rare autosomal recessive disorder characterized by pancreatic exocrine insufficiency, metaphyseal chondrodysplasia, mental retardation, thrombocytopenia and defective neutrophil motility. The mutations involve a single gene, the Shwachman-Bodian-Diamond-Syndrome (SBDS) gene on chromosome 7q11. Neutropenia is determined by an increased apoptosis of myeloid precursors caused by the SBDS. About 20% of patients evolve into acute leukemia.
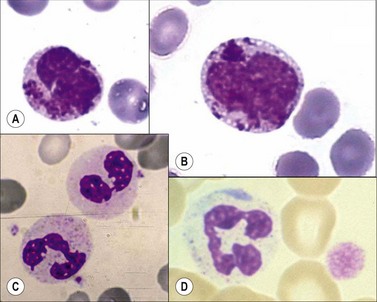
Chediak–Higashi syndrome. Chediak–Higashi31 syndrome is a rare autosomal recessive disorder characterized by recurrent infections, albinism, and by the presence of a reduced number of granules and/or formation of some abnormally large granules (by progressive fusion of normally formed granules) in most granule-containing cells. The giant granules present in neutrophils are peroxidase-positive and represent primary granules that have fused together. The abnormal granule fusion and function is determined by mutations of the CHS1 gene coding for the lysosomal trafficking regulator (LYST) protein on chromosome 1q42. Neutropenia is present in about 75% of patients. BMT provides a supportive cure for hematological signs but it does not prevent the neurologic deterioration.32 The affected cells include the neutrophils, eosinophils, lymphocytes, monocytes, melanocytes, Schwann cells of peripheral nerves, fibroblasts, vascular endothelial cells, renal tubular cells, and the parenchymal cells of the adrenal and pituitary glands. There is a deficiency of platelet dense granules, leading to easy bruising and bleeding. The large granules in leukocytes vary from a pale slate-gray to a dark reddish colour (Romanowsky stain) (Fig. 16.1A,B). There is a marked reduction in circulating NK cells.33 The leukocyte abnormalities are associated with recurrent infections. Some affected children, particularly those who live beyond early childhood, develop a terminal accelerated phase characterized by a lymphoma-like picture with lymphadenopathy, hepatosplenomegaly, neuropathy, widespread infiltration of tissues by non-clonal lymphatic and histiocytic cells, and pancytopenia. Death results from the complications of pancytopenia.
WHIM syndrome. Wart, hypogammaglobulinemia, infection and myelokathexis (WHIM) syndrome is a rare congenital autosomal dominant disorder determined by mutation in the gene coding for the neutrophil chemokine receptor CXCR.34 It is characterized by chronic non-cyclic neutropenia and increased susceptibility to bacterial and viral infections, especially from common serotype human papilloma virus, resulting in warts on the hands and feet starting in childhood. There is myeloid hyperplasia with degenerating granulocytes in the BM (myelokathexis).
Glycogen storage disease type 1b. It is a rare congenital autosomal recessive condition, determined by mutations on chromosome 11q23. These mutations cause glycogen accumulation in tissues due to deficiency of intracellular enzymes, which normally catalyze reactions that convert glycogen compounds to glucose. Severe neutropenia with consequent high risk of infections is present only in the type 1b and is determined by a disturbed myeloid maturation.35
Other rare conditions
Congenital dysgranulopoietic neutropenia. Congenital dysgranulopoietic neutropenia38,39is characterized by ineffective myelopoiesis and morphological abnormalities in the neutrophilic series. The term has been used to describe a disorder affecting six unrelated children presenting recurrent severe bacterial infections since birth, marked neutropenia, impaired neutrophil migration, normal numbers of BM colony-forming cells, normal or slightly increased colony-stimulating activity in the serum, and prominent morphologic and ultrastructural abnormalities in granulopoietic cells beginning from the neutrophil promyelocyte/myelocyte stage. The abnormalities of these cells included numerous autophagic vacuoles, abnormally electron-lucent primary granules, myelinization of primary granules, granule fusion, absence or marked decrease of secondary granules, and maturation of the cytoplasm ahead of the nucleus. Stem cell involvement in the pathogenesis of this disorder is postulated.40
Acquired neutropenias
Neonatal alloimmune neutropenia. In this syndrome, neutrophils and neutrophil precursors of the fetus are destroyed by the transplacental passage of maternal IgG alloantibodies formed against HLA or neutrophil-specific antigens (NA1 and NA2) present on fetal as well as on paternal neutrophils but not on maternal ones.41,42 The incidence is estimated at 1 in 500 live births. Most patients recover spontaneously usually within 2 months, depending on the life span of the maternal immunoglobulins. The neutropenia is occasionally severe and may cause life-threatening neonatal infections. Such patients respond well to intravenous immunoglobulins and G-CSF. The antibodies against neutrophil-specific antigens transferred to the fetus by the transplacental passage may be present either because the mother is affected by an autoimmune disease or as consequence of maternal sensibilization against the antigens of the fetal neutrophils of paternal origin. Despite a very low number of ANC, there is usually a spontaneous resolution within a few months. Supportive treatment with antibiotics is required in presence of infections, such as onphalitis or cellulitis.
Autoimmune neutropenia. Autoimmune neutropenia as a consequence of antineutrophil antibodies is a common finding associated with various collagen-vascular disorders, including rheumatoid arthritis (30% of patients with Felty’s syndrome), Systemic lupus erythematodes (SLE, 50% of patients), Sjögrens syndrome, scleroderma, polymyositis and polymyalgia rheumatica. It has also been described in association with autoimmune hemolytic anemia, immune thrombocytopenic purpura or both. In adults, autoimmune neutropenia is less likely to resolve spontaneously as compared to infants and children. In a subset of patients who have antibodies against the granulocyte precursors, the clinical course is more aggressive.43 Severe autoimmune neutropenias respond to treatment with metothrexate, steroids, G-CSF and granulocyte-macrophage colony stimulating factor (GM-CSF). Chronic autoimmune neutropenia44,45 is usually a relatively mild disease not associated with any other autoimmune disorders and characterized by recurrent infections, particularly of the oropharynx and skin. In some patients, the antineutrophil antibody has the specificity anti-NA2 or pan-Fcγ RIII (CD16, NA1/NA2). Some patients requiring treatment have responded to G-CSF. The LGL leukemia is often associated with neutropenia (see Chapter 28).
Infections associated with neutropenia
Infections are the most common cause of acquired neutropenia with different pathogenetic mechanisms (Box 16.1). In particular, some viruses (EBV, HAV, HBV, HCV and HIV) induce a severe and prolonged neutropenia by direct damage of the granulocytic precursors.46,47 Fungal, rickettsial and protozoal infections damage the BM endothelial cells causing vasculitis and severe cytopenias.48 Bacterial septicemia, in particular with Gram-negative agents, may induce neutropenia through an endotoxin-induced shift of neutrophils from the circulating to the marginated cell pools, a reduced survival time of circulating granulocytes (due either to destruction of cells within the circulation or to an accelerated rate of egress of cells from the blood), and a BM failure to adequately increase the rate of effective granulocytopoiesis. In certain circumstances, infection may be associated both with a failure adequately to increase effective granulocytopoiesis and with an exhaustion of the marrow granulocyte pool so that neutropenia is seen together with a left shift. This is particularly common in very severe bacterial infections and in bacterial infections in neonates and alcoholics (who have a reduced BM granulocyte pool). In several infections associated with spleen enlargement, such as typhoid fever, malaria and kala-azar, neutropenia results from splenic trapping and destruction of neutrophils in the spleen. In most of the latter cases, neutropenia does not require treatment, which should be focused on the underlying disease.
Neutropenia is the most common drug-induced blood dyscrasia.49,50 Some drugs (e.g. alkylating agents or antifolate drugs), certain chemicals (e.g. benzene) and irradiation induce neutropenia in all affected individuals in a dose-dependent manner. Other drugs cause neutropenia only occasionally and this phenomenon is usually at least partly based on a genetic polymorphism of drug metabolism. Drugs of the latter category may either cause neutropenia as part of an aplastic anemia (AA) or induce a selective neutropenia. Furthermore, several of the drugs causing AA in susceptible individuals initially cause a neutropenia that subsequently progresses to AA. The agranulocytosis induced by amidopyrin51 (and possibly also by other drugs) is often attributed to the destruction of circulating neutrophils and neutrophil precursors by drug-related immune mechanisms. There is also evidence that some drug metabolites may directly damage neutrophils and their precursors through the myeloperoxidase (MPO) system.52 Drugs that may occasionally cause selective neutropenia are listed in Box 16.2. The relatively high-risk drugs include amidopyrine and the antithyroid drugs. Some of the listed drugs usually induce a mild to moderate neutropenia that is asymptomatic and does not progress despite continuation of the drug. Other drugs usually cause a complete or almost complete absence of neutrophils (agranulocytosis).
Box 16.2 Main drugs which may cause neutropenia
Idiosyncratic drug-induced agranulocytosis (incidence 2.4–15.4 cases per million) results in a life-threatening clinical syndrome53 characterized by fever, sweating, vomiting, sore throat, dysphagia due to necrotic ulceration of the mouth and pharynx, extreme prostration and, frequently, death from overwhelming infection. At autopsy, necrotic ulcers are found in the mucous membranes of the entire alimentary tract as well as in the vagina. The prognosis is considerably improved if the offending drug is stopped and infection is adequately controlled by antibiotic therapy. To date, drug-induced agranulocytosis remains a serious adverse event due to the high frequency of sepsis with severe deep infections (such as pneumonia), septicemia, and septic shock in about two-thirds of all patients. Old age (>65 years), septicemia or shock, metabolic disorders such as renal failure, and a neutrophil count below 0.1 × 109/l are poor prognostic factors. Drugs or their metabolites might induce neutropenia by one or both of the following mechanisms: direct toxic effect on myeloid precursors or marrow environmental, dose-dependent inhibition of granulopoiesis, and immune-mediated destruction of granulocytes or their precursors. The classic example of a drug that causes agranulocytosis by a toxic effect on the BM is chlorpromazine, which causes a transient and moderate neutropenia in one-third of treated individuals. It causes agranulocytosis in about 1 in 1200 individuals, usually after a cumulative dose of 10–20 g over 20–30 days. Chlorpromazine appears to cause agranulocytosis by inhibiting DNA synthesis and cell proliferation in the granulocyte precursors of susceptible individuals. At the time of the agranulocytosis, the BM shows few or no neutrophil granulocytopoietic cells. The susceptibility of occasional individuals to develop agranulocytosis following therapy with chlorpromazine or other sulphur-containing drugs such as carbimazole or metiamide may depend on their genetically determined ability to oxidize the drug to highly-reactive myelotoxic metabolites (sulphoxides) more rapidly than most individuals.54 It is likely that many other drugs that cause neutropenia do so by directly or indirectly impairing biochemical processes within granulocytopoietic cells or by causing some form of immune destruction of the cells. The best understood example of a drug that causes agranulocytosis by destroying circulating neutrophils is amidopyrine. Individuals with amidopyrine-induced agranulocytosis have a history of previous exposure to the drug and may, after recovery from the initial agranulocytosis, show a recurrence of agranulocytosis within 12 h of a test dose. The serum of such individuals contains an antibody that causes agglutination of neutrophils followed by an acute neutropenia in the presence of the drug. It has been suggested that the antibody may be directed against complexes between drug metabolites and cellular components. The BM shows increased granulocytopoietic activity and a depletion of the more mature precursors. The dynamics of drug-induced neutropenia vary depending on the underlying mechanism: it has rapid onset in the immune-mediated mechanism, especially if the patient was previously exposed, while it is delayed when drugs exert direct marrow toxicity. Recombinant human G-CSF is indicated in patients who do not improve their ANC levels after the drug discontinuation.
Miscellaneous neutropenias
Neutropenia and nutritional deficiencies. Vitamin B12, folic acid or copper deficiency can result in suppressed or ineffective granulopoiesis. In various series, neutropenia associated with a large number of degenerated dysmorphic myeloid cells in the marrow was seen in 17–49% of cases, suggesting abnormal release and/or maturation failure.55 In PB, hypersegmented granulocytes with more than five nuclear lobes are the morphologic hallmark of megaloblastic granulopoiesis. Replenishment of nutrients generally corrects the neutropenia. Starvation, anorexia or cachexia may also induce neutropenia.
Dialysis neutropenia. Dialysis neutropenia56 is the result of pulmonary sequestration of neutrophils after complement activation by the dialyzer membrane. Increased expression of neutrophil adhesion receptors, such as CD11b/CD18, suggests that neutrophil adhesion to the capillary endothelium is a possible mechanism. An alternative hypothesis is that the complement fragment C5a modulates neutrophil mechanical properties via the cytoskeleton – largely filamentous actin (F-actin) – stiffening them and thereby slowing their passage through the pulmonary capillaries. Acute cardiopulmonary failure may occur. After some hours, neutropenia is usually reversed and a rebound neutrophilia occurs.
Pseudoneutropenia. A false reduced neutrophil count provided by routine hematology analyzers is mainly caused by in vitro agglutination of neutrophils,57 usually EDTA-dependent. This artifact is completely solved after addition of kanamycin in the tube.58 The presence of paraproteinemia, a prolonged storage after the withdrawal and the asymmetric distribution of circulating neutrophils to the marginate pool are reported too as possible causes of pseudoneutropenia.
Neutrophilia
Neutrophilia is defined as an increased absolute neutrophil count (ANC) in PB above 2SD of the mean value for healthy individuals, i.e. above 9.5 × 109/l. Some of the causes of a neutrophil leukocytosis are listed in Box 16.3. Physiologic neutrophilia is seen in pregnancy, labor, and newborns. Neutrophilia can result from altered rates of neutrophil release from the BM, changes in margination and egress from the blood or both. Shift neutrophilia (pseudoneutrophilia) results from a shift of cells from marginal to the circulating granulocyte pool without any quantitative change in the total blood granulocyte pool (TBGP), while in true neutrophilia TBGP is increased. Both pools are approximately equal in size and are in constant equilibrium.59 Strenuous exercise, electric shocks, emotional states, vomiting, convulsions, paroxysmal tachycardia and epinephrine injection are the most frequent causes of shift neutrophilia. In the true neutrophilia, an increased rate of release of neutrophils from the BM into the blood occurs acutely as a result of the emptying of the BM granulocyte pool into the blood or more slowly in association with an increased rate of granulocytopoiesis. The first of these mechanisms is responsible for the initial acute neutrophilia in response to bacterial endotoxins, corticosteroids and etiocholanolone. The second mechanism operates when there is a sustained neutrophil leukocytosis, in conditions such as infections, inflammation and malignant diseases. The level of neutrophilia is the result of the balance between the production rate and the death rate of neutrophils.
Box 16.3 Main causes of neutrophilia
Neutrophilia in acute infection or inflammation
During an average acute infection the ANC rises to levels of 15–25 × 109/l. In certain infections such as pneumococcal pneumonia or childhood infections, ANC ranges from 20 to 40 × 109/l (but may reach levels >80 × 109/l), while the eosinophil and basophil counts are reduced. The most common cause of neutrophil leukocytosis is an acute infection with a pyogenic organism. However, neutrophil leukocytosis also occurs in infections with certain non-pyogenic organisms (e.g. in poliomyelitis, herpes zoster, typhus). Tuberculosis does not usually cause a neutrophil leukocytosis but it may occur when there is rapid local spread of tubercle bacilli as in tuberculosis meningitis. Severe infections may be accompanied by a left-shifted differential count and toxic granulation or Döhle bodies within neutrophils. Young children may respond to acute infections with a lymphocytosis rather than a neutrophil leukocytosis. A persistent chronic neutrophilia is associated with inflammatory non-infectious conditions such as obstructive bronchopneumonia, burns, postoperative states, acute myocardial infarction, acute attack of gout, acute glomerulonephritis, rheumatic fever, collagen vascular diseases, hypersensitivity reaction, auto-inflammatory syndromes, and cigarette smoking.60 The neutrophilic activation is mediated by circulating cytokines, such as TNF-α, interleukin (IL)-1β, IL-6 and IL-8. A leukemoid reaction is defined by a WBC count more than 50 × 109/l and it resembles leukemia but arises from other causes such as infection or cancer.
Neutrophilia and cancer
Neutrophilia can occur in association with rapidly growing metastatic lung and gastrointestinal neoplasms. This process is thought to be due to superinfections, high levels of TNF-α or to direct BM stimulation by neutrophilic growth factors produced by cancer cells in some tumor types such as squamous cell cancers of the head and neck.61 In some patients with carcinoma or sarcoma, the increase of neutrophil granulocytopoiesis results from the production of proteins, which stimulate the growth of CFU-GM.62 In Cushing’s syndrome, pheochromocytoma and in other catecholamine-secreting tumors, neutrophilia is associated with ectopic secretion of adrenocorticotropic hormone or corticotropin-releasing hormone.63 The high ANC in clonal hematological malignancies is mainly determined by an impairment of apoptosis with an abnormally long survival time in the blood.
Miscellaneous neutrophilias
Drug-related neutrophilia. Drugs very rarely induce neutrophilia, except for glucocorticoids, catecholamine, etiocholanolone and lithium. Neutrophilia seen in patients on long-term corticosteroid therapy is associated with a normal rate of release of neutrophils from the BM into the blood, a decreased rate of egress from the blood into the tissues as well as a decreased rate of apoptosis, due to drug-induced reduction of adhesion molecules on neutrophils and endothelial cells.64 Moreover neutrophilia can result from poisoning with lead, mercury, digitalis, camphor, antipyrine, phenacetin, quinidine, pyrogallol, turpentine, arsphenamine and insect venoms.
Hereditary chronic neutrophilia. Familial cases of hereditary neutrophilia mimicking a myeloproliferative disorder are reported in the literature. An activating mutation in the CSF3R gene favors dimerization of the G-CSF receptor transmembrane domain, strongly promoting activation and hypersensitivity for proliferation and differentiation.65
Qualitative and morphological abnormalities
Inherited abnormalities
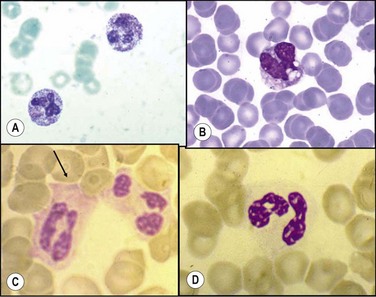
Pelger–Huët anomaly is a benign, dominantly inherited anomaly of granulocytes66 marked by failure of normal nuclear lobe development during terminal differentiation, due to mutations in the lamin B receptor (LBR) gene.67 The incidence at birth in different studies ranges from as high as 1 in 1000, to 1 in 4000, 6000 or even in 10 000 persons. Pelger–Huët anomaly does not impair neutrophil function and is readily identified by two morphologic hallmarks: the bilobated ‘pince-nez’ nuclei and the excessively coarse clumping of nuclear chromatin. Heterozygotes have bilobed nuclei in 69–93% of neutrophils (Fig. 16.1C); cells with three lobes are <10% while only rare neutrophils have four lobes. Some individuals display eosinophils with round condensed nuclei and excessively coarse clumping of nuclear chromatin in lymphocytes and monocytes. In homozygote states, round or oval unsegmented nuclei are present in >95% of neutrophils.
May–Hegglin anomaly. Macrothrombocytopenia with leukocyte inclusions (May–Hegglin anomaly, MHA) is a rare autosomal dominant disorder characterized by thrombocytopenia, giant platelets, and Döhle body-like inclusions in leukocytes. The MHA gene has been localized to a 13.6-cM region on chromosome 22.68 The cytoplasmic inclusions are 2–5 µm in diameter, appear grayish-blue when stained by a Romanowsky method (Fig. 16.1D) and are composed of stacks of rough endoplasmic reticulum, similar to Döhle bodies but larger.
Alder–Reilly anomaly is associated with the genetic mucopolysaccharidoses (lack of lysosomal enzymes necessary to break down mucopolysaccharides) except Morquio syndrome and is characterized by the presence of dense azurophilic granules, resembling toxic granulation in all leukocytes including neutrophils. It apparently does not interfere with the leukocyte function. Associations with a mutation of the MPO structural gene69 and with a myelodysplastic syndrome (MDS)70 have been reported in the literature.
Jordans’ anomaly (familial vacuolization of leukocytes) is characterized by the presence of fatty inclusions in all neutrophils and most monocytes and lymphocytes (Fig. 16.2A). These inclusions measure from 2 to 5 µm and appear as unstained vacuoles in panoptical stains while they strongly stain with Sudan II and Nile blue sulphate. This anomaly has only been reported in a few families.71,72
Hereditary hypersegmentation of neutrophils is a rare condition inherited in an autosomal dominant character.73 The proportion of neutrophils with five or more lobes exceeds a tenth and one-third in heterozygotes and homozygotes, respectively.
Hereditary giant neutrophilia is a rare and asymptomatic autosomal dominant anomaly, in which a fraction of neutrophils (1–2%) is nearly double in size and has 6–10 nuclear lobes, in the absence of chromatin abnormalities, dysplastic features and circulating precursors.74
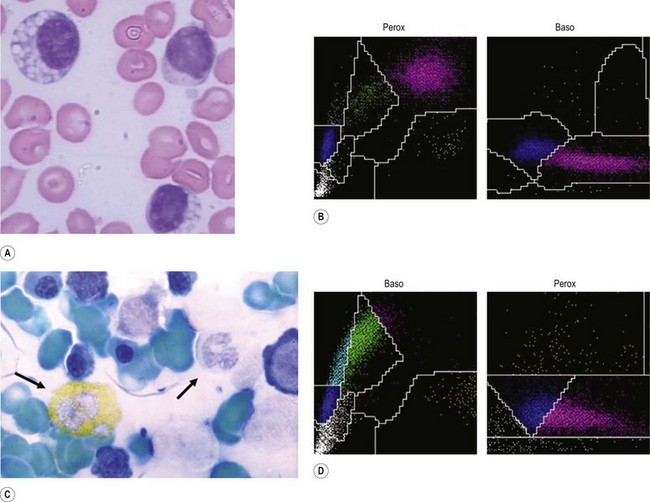
Myeloperoxidase deficiency (Alius–Grignaschi anomaly). First described in 1969, MPO deficiency is a benign autosomal recessive disorder that leads to absence of this enzyme in neutrophils and monocytes, but not in eosinophils. After the introduction of automated differential counting based on cytochemical reactions for MPO (Fig. 16.2B, D), this condition has a reported incidence of 1 in 2000 and became the most common disorder of the neutrophil function, as complete (Fig. 16.2C) or partial deficiency. In a recent report, a significantly higher occurrence of severe infections and chronic inflammatory processes was noted among MPO-deficient patients.75
Specific granule deficiency. Neutrophil-specific granule deficiency (SGD) is a rare congenital disorder,76 presenting with increased susceptibility to pyogenic infections. Neutrophils have bilobated nuclei and contain more primary than secondary (specific) granules. Leukocyte alkaline phosphatase activity is reduced. Lactoferrin deficiency77 in these patients is confined to myeloid cells and is secondary to a deficiency of RNA transcripts.
Chronic granulomatous disease of childhood. Chronic granulomatous disease (CGD) is a congenital disorder, mostly with X-linked inheritance78 and comprises a rare group of genetic disorders, in which neutrophils, monocytes and eosinophils can ingest but cannot kill catalase-positive bacteria and fungi due to a deficiency of NDPH oxidase or to an abnormality in its activation.
Acquired abnormalities
Toxic granulations (Fig. 16.3A) are purple or dark-blue staining azurophilic granules in the cytoplasm of neutrophils, bands and metamyelocytes resulting from an abnormality in the maturation of the primary granules with a consequent retention of their azurophilic property,79 while toxic vacuolizations (Fig. 16.3B) are vacuoles representing phagocytosis and depletion of toxic granules. Both are seen in severe infections and in other toxic conditions.
Döhle bodies are single or multiple blue or grayish-blue cytoplasmic inclusions, representing free ribosomes or rough endoplasmic reticulum,80 usually situated at the periphery and protruding beyond the normal contour of the cell (Fig. 16.3C). They are associated with myeloid ‘left shifts’ and are found in normal pregnancy, in various infections, in patients with various neoplasms and after severe burns.
Macropolycytes are very large polymorphonuclear leukocytes, with a diameter >16 µm, often showing hypersegmented nuclei with 6–14 nuclear segments (Fig. 16.4A). They may be found in the blood in vitamin B12 or folate deficiency and in infections, myeloproliferative disorders and drug-induced BM damage. The macropolycytes seen in vitamin B12 deficiency have tetraploid or hypotetraploid DNA contents and appear to result from nuclear segmentation in giant metamyelocytes that are not phagocytized by BM macrophages. The occurrence of macropolycytes has been reported as an inherited condition in a single family.
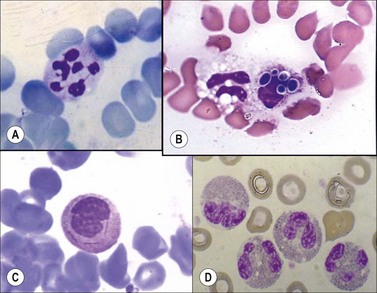
Hypogranular neutrophils (acquired ‘specific granule’ deficiency) are found especially in MDS (Fig. 16.3D) but may also be seen in other conditions such as Ph-negative chronic myeloid leukemia (CML), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML). In addition, they may be found in neonates and in patients with burns.
Pseudo Pelger–Huët anomaly. The morphological abnormalities similar to the hereditary form are commonly seen in MDS, in AML, in chronic myeloproliferative disorders and, more rarely, chronic lymphatic leukemia. The acquired Pelger–Huët anomaly develops transiently in patients treated with paclitaxel and docetaxel81 and the anomaly has also been found in patients with severe hematological toxicity caused by valproic acid.82 It may also be seen after mononucleosis.
Botryoid nuclei are small, moderately pyknotic nuclear segments which are clustered like grapes on a stem, observed in over 50% of neutrophils in patients with heat stroke.83
Intracellular organisms. Ingested microrganisms, such as bacteria Rickettsiae, fungi (Fig. 16.4B), etc., may be rarely seen in the cytoplasm of neutrophils and sometimes of monocytes.
Auer rods are pink or red-stained needle-shaped structures seen in the cytoplasm of myeloid cells, containing agglomeration of azurophilic granules containing enzymes such as acid phosphatase, MPO and esterase, and may represent abnormal derivatives of cytoplasmic granules. Auer rods can be seen in myeloid neoplasms ranging from AML to MDS, but not in normal or non-neoplastic reactive states (Fig. 16.4C).
Eosinophils
Eosinophils are quite rare in PB, while tissues of the pulmonary and gastrointestinal systems are their normal environment. In healthy adults the reference interval of eosinophils in PB is 0.02–0.5 ± 2SD × 109/l.2 The number is physiologically slightly increased in newborns and in the presence of environmental stimuli (mainly allergens), while it may be decreased in normal pregnancy and during labor.
Eosinopenia
Drug-induced eosinopenia
Eosinopenia develops promptly after the administration of corticotropin, corticosteroids, epinephrine or histamine, but disappears within hours unless repeated doses are given.84 In patients treated by corticosteroids, eosinopenia appears to result both from an impairment of the release of eosinophils from the BM and from the increased margination (or sequestration) of blood eosinophils. β-blockers inhibit adrenaline-induced eosinopenia and favor an increase in the number of circulating eosinophils.
Eosinophilia
Eosinophilia (Fig. 16.4D) is commonly seen in a wide spectrum of allergic responses, medication usage, and many skin diseases such as dermatitis, parasitic infestations, some autoimmune disorders and malignancy (Box 16.4). Eosinophilia is defined as an increase of circulating eosinophils >0.6 × 109/l, although an increase in eosinophils (with or without concurrent peripheral eosinophilia) can be observed in body fluids, such as CSF and urine, and in many tissues, such as skin, lung, heart, liver, intestine, bladder, BM, muscle or nerve. Eosinophilia may be defined as mild, moderate or severe when the number of circulating eosinophils is 0.6–1.5 × 109/l, 1.5–5.0 × 109/l or >5 × 109/l, respectively. IL-5, IL-3 and GM-CSF are the primary stimuli for eosinophil production. High eosinophilia persistent for >6 months carries significant risk of end-organ damage. According to the 2008 WHO classification, primary eosinophilia include chronic eosinophilic leukemia not otherwise specified and neoplasms associated with rearrangements of with PDGFRA, PDGFRAB or FGFR1 genes87 (see Chapter 25). Secondary eosinophilia is a reactive phenomenon driven by eosinophilopoietic cytokines released by non-myeloid cells and is usually solved after specific treatment of the underlying condition.
Box 16.4 Main causes of eosinophilia
Eosinophilia and asthma
An increased number of eosinophils is observed in patients with asthma, both atopic and non-atopic. A hallmark of allergic disease is eosinophil infiltration of the tissues. Selective traffic of eosinophils into allergic tissues is caused by high levels of IL-5, CCR3 (CD193) binding chemokines and of the adhesion molecules P-selectin and vascular cells adhesion molecule-1. The T-helper 2 (Th2) allergen specific lymphocytes take a relevant part in this process. Eosinophil-mediated damage to the respiratory epithelium is a major pathogenetic mechanism in asthma,88 playing an important role in the maintenance of bronchial inflammation and also in tissue injury.
Eosinophilia in tumors
The eosinophilia associated with malignant tumors may be caused by a tumor-derived glycoprotein, which stimulates eosinophil granulocytopoiesis.89 Lung tumors release a factor chemotactic for eosinophils, which resembles eosinophil chemotactic factor for anaphylaxis (ECF-A). In Hodgkin’s lymphoma, eosinophilic proliferation is caused by IL-5 produced by tumor cells.
Qualitative and morphological abnormalities
Eosinophilic peroxidase deficiency is a very rare familial disorder, not correlated with any particular disease, characterized by the presence of morphologically normal eosinophils, partially or totally lacking the dimethylaminoazobenzene-positive specific granules.90 Pseudoeosinopenia is the characteristics of these individuals, when differential count is performed with analyzers based on cytochemical reactions for MPO (Fig. 16.5A).
Basophils
Basophils are the least frequent leukocytes. In healthy adults the reference intervals of basophils in PB is 0.02–0.1 × 109/l.2
Basopenia
Basopenia91 is defined as a basophil count less than 0.01 × 109/l and may be found in patients with acute hypersensitivity reactions, autoimmune urticaria, acute stress, hyperthyroidism, Cushing’s syndrome and pregnancy as well as in response to the administration of progesterone, corticosteroids or corticotrophin. The blood basophil count falls on the day of ovulation.
Basophilia
A reactive basophil leukocytosis may occur in hypersensitivity reactions, hypothyroidism, ulcerative colitis, smallpox, chickenpox and the idiopathic hypereosinophilic syndrome as well as after exposure to ionic radiations. Basophil leukocytosis also occurs in mastocytosis, CML, polycythemia vera, essential thrombocythemia, myelofibrosis, basophilic leukemia, eosinophilic leukemia, and Ph-positive acute leukemia. In CML, a further and considerable increase in both immature and mature basophils may precede transformation of the disease to a more malignant phase (see Chapter 24). Basophilia is also reported after splenectomy.
Monocytes
Monocytes are the largest leukocytes. In healthy adults, the reference interval for monocytes in the PB is 0.2–1.0 × 109/l.2 In newborns, the levels are physiologically increased, while in the elderly monocyte levels are decreased.
Monocytopenia
Monocytopenia is defined as level of PB monocytes <0.2 × 109/l. Early monocytopenia after chemotherapy is a risk factor for neutropenia. Monocytopenia is seen in cases of aplastic anemia.92 Cyclic neutropenia is notable for intermittent periods of monocytopenia. Transient, significant monocytopenia occurs during the first 30 min of hemodialysis: monocyte counts return to normal within hours after the procedure ends without any rebound monocytosis.93 Monocytopenia also occurs in severe thermal injuries, in acquired immunodeficiency syndrome and in hairy cell leukemia.
Monocytosis
Monocytosis is defined by the presence of circulating monocytes ≥1.0 × 109/l. The various causes are listed in Box 16.5. The spleen is a site for storage and rapid deployment of monocytes and splenic monocytes are a resource that the body exploits to regulate inflammation.94 Monocytosis occurs as a compensatory event in association with congenital as well as drug-induced neutropenia. It represents a benign non-prognostic epiphenomenon in lymphomas and other solid tumors, without any predictive significance for metastasis. In patients with vascular disorders, increasing monocytosis correlates with an increased risk of heart attack.
Lymphocytes
In healthy adults, the reference intervals of PB lymphocytes are 1.0–3.0 × 109/l.2 About 3% of circulating lymphocytes are LGL, which are NK or CD8+ T-cells.
Lymphopenia
Congenital lymphopenias
DiGeorge syndrome is a rare congenital disorder where symptoms vary greatly between individuals, but commonly include a history of recurrent infection, heart defects and characteristic facial features. Often the parathyroid glands may have failed to develop, leading to low serum levels of calcium, resulting in tetany and seizures. The thymus may also be underdeveloped or absent, resulting in a pure deficiency of T-lymphocytes (CD3+ <0.5 × 109/l). The heterogeneity in the symptoms is related to the amount of genetic material lost in the chromosomal 22 deletion. Although 90% of cases have been attributed to a 22q11.2 deletion, other chromosome defects (e.g. chromosome 10 or 18) have been identified. Therapy is aimed at correcting the defects in the affected organs or tissues.95
Severe combined immunodeficiency (SCID) is a life-threatening syndrome of recurrent infections, diarrhea, dermatitis, and failure to thrive. It is caused by a variety of mutations that interfere with the differentiation or function of T-cells, B-cells, and occasionally NK cells. The most common autosomal recessive form is due to absence of the enzyme ADA (adenosine deaminase), while the X-linked form is due to the absence of a cell-surface receptor protein γc, which is a component of the receptor for IL-2, IL-4 and IL-7. The latter patients have low levels of T-lymphocytes but normal to elevated numbers of virgin B-lymphocytes. Clinically, most patients present before age 3 months with unusually severe and frequent infections by common or opportunistic pathogens. Presently, hematopoietic stem cell gene therapy represents a therapeutic opportunity.96
Lymphocytosis
In adults, lymphocytosis is defined by an increased absolute number of circulating lymphocytes more than 4 × 109/l. The various causes of lymphocytosis are listed in Box 16.6. For details of lymphocytosis due to chronic lymphoid leukemias and lymphomas see Chapters 28 and 29.
Box 16.6 Main causes of lymphocytosis
X-linked lymphoproliferative syndrome (Duncan’s syndrome). This is an X-linked recessive condition97 in which there is a mutation in the gene encoding SLAM (signaling-lymphocyte activation molecule) associated protein, located on Xq25. In affected boys, primary EBV infection causes a life-threatening illness, with fulminant hepatitis, hemophagocytic syndrome and 50% mortality. Some patients subsequently develop AA, acquired hypogammaglobulinemia, or lymphoproliferative disorders including B-cell lymphoma (diffuse large B-cell lymphoma or Burkitt-like lymphoma). AA may respond to corticosteroids, anti-thymocyte globulin or immunosuppression followed by syngeneic BMT and appears to have an immunologic basis.
Lymphocytosis and infections
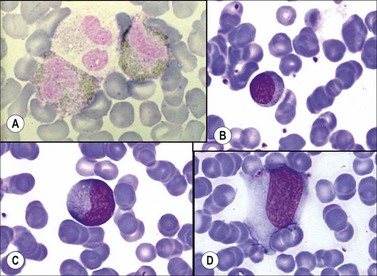
Tuberculosis, syphilis and brucellosis may be associated with an increase in both lymphocytes and granulocytes, while the only acute bacterial infection frequently associated with a lymphocytosis is whooping cough (pertussis).98 In some children with this disorder, the lymphocyte count may exceed 50 × 109/l, which is caused by the lymphocytosis-promoting factor (LPF), a protein toxin from Bordetella pertussis. Lymphocytosis around 20–30 × 109/l (but sometimes up to 100 × 109/l), may be found in several viral infections. The most common is Epstein–Barr virus (EBV) that induces the presence of a polyclonal population of CD8+, NK and T-γδ circulating lymphocytes, while CD4+ subpopulation is normal. Large, atypical, reactive type lymphocytes are commonly detected in PB. Infectious mononucleosis99 is a clinical syndrome resulting from an acute self-limiting primary infection by EBV, characterized by fever, pharyngitis, the presence of increased numbers of highly atypical lymphocytes in PB, and the production of heterophile antibodies. EBV infects and replicates in the B-lymphocytes and epithelial cells of the pharynx and also in a small proportion of T-cells: all three cell types have the specific receptor for EBV, namely CD21 (C3d receptor). Morphologic features of atypical lymphocytes include large size, irregular shape, increased cytoplasmic basophilia, and diffuse chromatin pattern, prominent nucleoli and a scalloped margin at points of contact with other cells (Fig.16.5B, C, D). The atypical lymphocytes are not the virus-infected B-lymphocytes, but T-lymphocytes with CD8+ cytotoxic/suppressor phenotype. These cells may contain tartrate-resistant acid phosphatase and may show block-positivity when stained by the periodic acid-Schiff (PAS) method. Hematological complications include autoimmune hemolytic anemia due to a cold antibody with anti-I specificity, thrombocytopenia due to peripheral destruction of platelets, and rarely granulocytopenia or AA, the latter occurring several weeks after infectious mononucleosis. Another rare complication is severe pancytopenia secondary to hemophagocytosis. When infectious mononucleosis is associated with a hemophagocytic syndrome (see Chapter 5) the BM shows increased numbers of promonocytes and phagocytic histiocytes. BM aplasia is a rare complication while disseminated intravascular coagulation has been observed in association with the hemophagocytic syndrome. Changes very similar to those of infectious mononucleosis may be seen in cytomegalovirus infection (CMV), while lesser changes may be seen in toxoplasmosis, malaria, herpes simplex, varicella zoster, rubella or other viral infections such as infectious hepatitis or HIV infection (see Chapter 5).
1 Cronkite EP, Fliedner TM. Granulocytopoiesis. N Engl J Med. 1964;270:1347-1351.
2 Dacie JV, Lewis SM. Practical Haematology, Ninth Edition. Edinburgh: Churchill Livingstone; 2001.
3 Reed WW, Diehl L. Leukopenia, neutropenia, and reduced hemoglobin levels in healthy American blacks. Arch Intern Med. 1991;151:501-505.
4 Kostmann R. Infantile genetic agranulocytosis (agranulocytosis infantilis hereditaria): a new recessive lethal disease in man. Acta Pediatr Scand. 1956;45:1-18.
5 Dror Y, Sung L. Update on childhood neutropenia: molecular and clinical advances. Hematol Oncol Clin North Am. 2004;18:1439-1458.
6 Ward AC, Dale DC. Genetic and molecular diagnosis of severe congenital neutropenia. Current Opinion in Hematology. 2009;16:9-13.
7 Germeshausen M, Grudzien M, Zeidler C, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111:4954-4957.
8 Chao JR, Parganas E, Boyd K, et al. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98-102.
9 Weinblatt ME, Scimeca P, James-Herry A, et al. Transformation of congenital neutropenia into monosomy 7 and acute nonlymphoblastic leukemia in a child treated with granulocyte colony-stimulating factor. J Pediatr. 1995;126:263-265.
10 Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: results of a long-term survey. Blood. 2007;109:93-99.
11 Beel K, Vanderberghe P. G-CSF receptor (CSF3R) mutations in X-linked neutropenia evolving to acute myeloid leukemia or myelodysplasia. Haematologica. 2009;94:1449-1452.
12 De Vaal OM, Seynhaeve V. Reticular dysgenesis. Lancet. 1959;11:1123.
13 Emile JF, Geissmann F, de la Calle O, et al. Langerhans cell deficiency in reticular dysgenesis. Blood. 2000;96:58-62.
14 Cutting HO, Lang JE. Familial benign chronic neutropenia. Annals of Internal Medicine. 1964;61:876.
15 Busch FH. Familial benign chronic neutropenia in a Danish family. Ugeskrift for Laeger. 1990;152:2565-2566.
16 Joyce RA, Boggs DR, Chervenick PA. Neutrophil kinetics in hereditary and congenital neutropenias. N Engl J Med. 1976;295:1385-1390.
17 Bux J, Kissei K, Nowak K, et al. Autoimmune neutropenia: clinical and laboratory studies in 143 patients. Ann Hematol. 1991;63:249-252.
18 Jonsson OG, Buchanan GR. Chronic neutropenia during childhood: a 13-year experience in a single institution. Am J Dis Child. 1991;145:232-235.
19 Reimann HA, De Berardinis CT. Periodic (cyclic) neutropenia, an entity. Blood. 1949;4:1109.
20 von Schulthess GK, Fehr J, Dahinden C. Cyclic neutropenia: amplification of granulocyte oscillations by lithium and long-term suppression of cycling by plasmapheresis. Blood. 1983;62:320-326.
21 Dale DC, Hammond WPIV. Cyclic neutropenia: a clinical review. Blood Reviews. 1988;2:178.
22 Krance RA, Spruce WE, Forman SJ, et al. Human cyclic neutropenia transferred by allogeneic bone marrow grafting. Blood. 1982;60:1263-1266.
23 Morley AA, Carew JP, Baikie AG. Familial cyclical neutropenia. Brit J Haemat. 1967;13:719-738.
24 Loughran TP, Clark EA, Price TH, et al. Adult onset cyclic neutropenia is associated with increased large granular lymphocyte. Blood. 1986;68:1082-1087.
25 Papadaki HA, Coulocheri S, George D, Eliopoulos GD. Patients with chronic idiopathic neutropenia of adults have increased serum concentrations of inflammatory cytokines and chemokines. Am J Hematol. 2000;65:271-277.
26 Papadaki HA, Palmblad J, Eliopoulos GD. Non-immune chronic idiopathic neutropenia of adult: an overview. Eur J Haematol (2001). 2001;67:35-44.
27 Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9:722-728.
28 Kozlowski C, Evans DI. Neutropenia associated with X-linked agammaglobulinaemia. J Clin Pathol. 1991;44:388-390.
29 Ng RP, Prankerd TA. IgA deficiency and neutropenia. Br Med J. 1976;1:563.
30 Shwachman H, Diamond L, Oski F, et al. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645-663.
31 Root RK, Rosenthal AS, Bales DJ. Abnormal bactericidal, metabolic, and lysosomal functions of Chediak-Higashi syndrome leukocytes. J Clin Invest. 1972;51:649-665.
32 Tardieu M, Lacroix C, Neven B. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome. Blood. 2005;106:40-42.
33 Virelizier J-L, Lagrue A, Durandy A, et al. Reversal of natural killer defect in a patient with Chediak–Higashi syndrome after bone marrow transplantation. New England Journal of Medicine. 1982;306:1055-1056.
34 Hernandez PA, Gorlin RJ, Gelb B, et al. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91:368-376.
35 Schaub J, Heyne K. Glycogen storage disease type Ib. Eur J Pediatr. 1983;140:283-288.
36 Lux SE, Johnston RBJr, August CS, et al. Chronic neutropenia and abnormal cellular immunity in cartilage-hair hypoplasia. New England Journal of Medicine. 1970;282:231-236.
37 Miller ME, Oski FA, Harris MB. Lazy-leucocyte syndrome. A new disorder of neutrophil function. Lancet. 1971;i:665-669.
38 Parmley RT, Crist WM, Ragab AH, et al. Congenital dysgranulopoietic neutropenia: clinical, serologic, ultrastructural, and in vitro proliferative characteristics. Blood. 1980;56:465-475.
39 Lightsey AL, Parmley RT, Marsh WL, et al. Severe congenital neutropenia with unique features of dysgranulopoiesis. Am JHematol. 1985;18:59-71.
40 Olcay L, Yetgin S, Erdemli E, et al. Congenital dysgranulopoietic neutropenia. Pediatr Blood Cancer. 2008;50:115-119.
41 Maheshwari A, Christensen RD, Calhoun DA. Immune-mediated neutropenia in the neonate. Acta Paediatr Suppl. 2002;91:98-103.
42 Verheugt FW, van Noord-Bokhorst JC, von dem Borne AEG, et al. A family with allo-immune neonatal neutropenia: group-specific pathogenicity of maternal antibodies. Vox Sanguinis. 1979;36:1-8.
43 Capsoni F, Sarzi-Puttini P, Zanella A. Primary and secondary autoimmune neutropenia. Arthritis Res Ther. 2005;7:208-214.
44 Boxer LA, Greenberg MS, Boxer GJ, Stossel TP. Autoimmune neutropenia. New England Journal of Medicine. 1975;293:748-753.
45 Verheugt FWA, von dem Borne AEGKr, van Noord-Bokhorst JC, Engelfriet CP. Autoimmune granulocytopenia: the detection of granulocyte antibodies with the immunofluorescence test. British Journal of Haematology. 1978;39:339-350.
46 Lee WM. Hepatitis B Virus infection. NEJM. 1997;337:1733-1745.
47 Levine AM, Karim R, Mack V, et al. Neutropenia in human immunodeficiency virus. Infection Arch Intern Med. 2006;166:405-410.
48 Cines DB, Lyss AP, Bina M, et al. Fc and C3 Receptors induced by herpes simplex virus on cultured human endothelial cells. J Clin Invest. 1982;69:123-128.
49 van der Klauw MM, Goudsmit R, Halie MR, et al. A population-based case-cohort study of drug-associated agranulocytosis. Arch Intern Med. 1999;159:369-374.
50 Van der Klauw MM, Goudsmit R, Halie MR, et al. Agranulocytosis induced by nonchemotherapy drugs. Ann Int Med. 2008;148:320-321.
51 Dameshek W, Colmes A. The effect of drugs in the production of agranulocytosis with particular reference to amidopyrine hypersensitivity. J Cl Inv. 1936;15:85.
52 Uetrecht JP. Reactive metabolites and agranulocytosis. Eur J Haemat. 1996;57(suppl):83-88.
53 Andrès E, Zimmer J, Affenberger S, et al. Idiosyncratic drug-induced agranulocytosis: update of an old disorder. Eur J of Int Med. 2006;17(8):529-535.
54 Pisciotta AV. Immune and toxic mechanisms in drug-induced agranulocytosis. Sem in Hemat. 1973;10:279-310.
55 M.Crist WM, Parmley RT, Holbrook CT, et al. Dysgranulopoietic neutropenia and abnormal monocytes in childhood vitamin B12 deficiency. Am J Hematol. 1980;9:89-107.
56 Tabor B, Geissler B, Odell R, et al. Dialysis neutropenia: the role of the cytoskeleton. Kidney International. 1998;53:783-789.
57 Glasser L. Pseudo-neutropenia secondary to leukoagglutination. Am J Hematol. 2005;80:147.
58 Hoffmann JJ. EDTA-induced pseudo-neutropenia resolved with kanamycin. Cl Lab Haemat. 2001;23:193-196.
59 von Vietinghoff S, Ley K. Homeostatic regulation of blood neutrophil counts. J Immunology. 2008;181:5183-5188.
60 Galeazzi M, Gasbarrini G, Ghirardello A, et al. Autoinflammatory syndromes. Clin Exp Rheumatol. 2006;24:79-85.
61 Hocking W, Goodman J, Golde D. Granulocytosis associated with tumor cell production of colony-stimulating activity. Blood. 1983;61:600-603.
62 Kimura N, Niho Y, Yanase T. A high level of colony-stimulating activity in a lung cancer patient with extensive leucocytosis, and the establishment of a CSA producing cell line (KONT). Scand J Haemat. 1982;28:417-424.
63 Sawka AM, Kudva YC, Singh R, et al. Persistent neutrophilia as a preceding symptom of pheochromocytoma. J Clin Endocrinol Metab. 2005;90:2472-2473.
64 Ngakawa M, Terashima T, D’yachkova Y, et al. Glucocorticoid-induced granulocytosis: contribution of marrow release and demargination of intravascular granulocytes. Circulation. 1998;98:2307-2313.
65 Plo I, Zhang Y, Le Couédic JP, Nakatake M, et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J Exp Med. 2009;206:1701-1707.
66 Cunningham JM, Patnaik MM, Hammerschmidt DE, et al. Historical perspective and clinical implications of the Pelger–Huet cell. Am J Hematol. 2009;84:116-119.
67 Cohen TV, Klarmann KD, Sakchaisri K, et al. The lamin B receptor under transcriptional control of C/EBPepsilon is required for morphological but not functional maturation of neutrophils. Hum Mol Genet.. 2008;17:2921-2933.
68 Martignetti JA, Heath KE, Harris J, et al. The gene for May–Hegglin anomaly localizes to a <1-mb region on chromosome 22q12.3-13.1. Am J Hum Genet. 2000;66:1449-1454.
69 Presentey B. Alder anomaly accompanied by a mutation of the myeloperoxidase structural gene. Acta Haematol. 1986;75:157-9159.
70 Ghandi MK, Howard MR, Hamilton PJ. The Alder–Reilly anomaly in association with the myelodysplastic syndrome. Clin Lab Haematol. 1996;18:39-40.
71 Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordans’ anomaly in white blood cells. Blood. 1966;28:258.
72 Piva E, Pajola R, Binotto G, et al. Jordans’ anomaly in a new neutral lipid storage disease. Am J Hematol. 2009;84:254-255.
73 Undritz VE. Eine neue Sippe mit erblich-konstitutionellen Hochsegmentierung der Neutrophilenkerne Undritz. Schweizerische Medizinische Wochenschrift. 1964;94:1365.
74 Davidson WM, Milner RDG, Lawler SD, et al. Giant neutrophil leucocytes: an inherited anomaly. Br J Haematol. 1960;6:339-343.
75 Kutter D, Devaquet P, Vanderstocken G, et al. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit ? Acta Haemat. 2000;104:10-15.
76 Ambruso DR, Sasada M, Nishiyama H, et al. Defective bactericidal activity and absence of specific granules in neutrophils from a patient with recurrent bacterial infections. J Clin Immunol. 1894;4:23-26.
77 Breton-Gorius J, Mason DY, Buriot D, et al. Lactoferrin deficiency as a consequence of a lack of specific granules in neutrophils from a patient with recurrent infections. Detection by immunoperoxidase staining for lactoferrin and cytochemical electron microscopy. Am J Pathol. 1980;99:413-416.
78 Di Matteo G, Giordani L, Finocchi A, et al. Molecular characterization of a large cohort of patients with chronic granulomatous disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. 2009;46:1935-1941.
79 Schofield KP, Stone PC, Beddall AC, Stuart J. Quantitative cytochemistry of the toxic granulation blood neutrophil. British Journal of Haematology. 1983;53:15-22.
80 Itoga T, Laszlo J. Döhle bodies and other granulocytic alterations during chemotherapy with cyclophosphamide. Blood. 1962;20:668-674.
81 Juneja SK, Matthews JP, Luzinat R, et al. Association of acquired Pelger–Huët anomaly with taxoid therapy. Br J Haemat. 1996;93:139-141.
82 Ganick DJ, Sunder T, Finley JL. Severe hematologic toxicity of valproic acid. A report of four patients. Am J Ped Hematol Oncol. 1990;12:80-85.
83 Hernandez JA, Aldred SW, Bruce JR, et al. ‘Botryoid’ nuclei in neutrophils of patients with heatstroke. Lancet. 1980;ii:642-643.
84 Krause JR, Boggs DR. Search for eosinopenia in hospitalized patients with normal blood leukocyte concentration. Am J Hematol. 2006;24:55-63.
85 Bass DA, Gonwa TA, Szejda P. Eosinopenia of acute infection. J Clin Invest. 1980;65:1265-1271.
86 Gleich GG. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immun. 2000;105:651-663.
87 Tefferi A. Modern diagnosis and treatment of primary eosinophilia. Acta Haematol. 2005;114:52-60.
88 Wardlaw AJ. Eosinophil trafficking in asthma. Clin Med. 2001 May–Jun;1(3):214-218.
89 Slungaard A, Ascensao J, Zanjani E, Jacob HS. Pulmonary carcinoma with eosinophilia. Demonstration of a tumor-derived eosinophilopoietic factor. New England Journal of Medicine. 1983;309:778-781.
90 Cappelletti P, Doretto P, Signori D, et al. Eosinophilic peroxidase deficiency. Cytochemical and ultrastructural characterization of 21 new cases. Am J Clin Pathol. 1992;98:615-622.
91 Dvorak HF, Dvorak AM. Basophil leukocytes: structure, function and role in disease. Clinics in Haematology. 1975;4:651-683.
92 Twomey JJ, Douglass CC, Sharkey OJr. The monocytopenia of aplastic anemia. Blood. 1973;41:187-195.
93 Nockher WA, Wieme JJr, Scherberich JE. Haemodialysis monocytopenia: differential sequestration kinetics of CD14+CD16+ and CD14++ blood monocyte subsets. Clin Exp Immunol. 2001;123:49-55.
94 Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612-616.
95 Minier F, Carles D, Pelluard F, et al. DiGeorge syndrome, a review of 52 patients. Arch Pediatr. 2005;12:254-257.
96 Aiuti A, Brigida I, Ferrua F, et al. Hematopoietic stem cell gene therapy for adenosine deaminase deficient-SCID. Immunol Res. 2009;44:150-159.
97 Sullivan JL. The abnormal gene in X-linked lymphoproliferative syndrome. Curr Opin Imm. 1999;11:431-434.
98 Horwitz MS, Moore GT. Acute infectious lymphocytosis: an etiologic and epidemiologic study of outbreak. N Engl J Med. 1968;279:399-404.
99 Okano M. Haematological associations of Epstein–Barr virus infection. Baillière’s Clinical Haematology. 2000;13:199-214.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 16 Abnormalities in leukocyte morphology and number
Neutrophils
In the peripheral blood (PB) of healthy white adults the mean neutrophil count is 4.4 (4.3–4.6) × 109/l,1,2 while in healthy black adults it is 3.6 (3.3–3.9) × 109/l.3
Neutropenia
Neutropenia is defined as the absolute neutrophil count (ANC) in PB lower than 1.5 × 109/l. Referring to white population, a useful classification in predicting risk infection indicates neutropenia as mild, moderate or severe according to the ANC value of 1.0–1.5 × 109/l, 0.5–1.5 × 109/l or less than 0.5 × 109/l respectively. Neutropenia may arise from various disorders of bone marrow (BM) function resulting in a decreased rate of release of neutrophils from the BM into the circulation. It is a common manifestation in several BM failures, such as aplasia, leukemia or myelodysplasia (see Chapters 13, 18, 20 for details). The conditions that may be associated with a neutropenia are listed in Box 16.1. Clinically neutropenia is considered as acute when it occurs over hours to a few days, usually developing from rapid neutrophil use/destruction or from impaired production. Neutropenia is considered chronic, when it lasts months to years, usually arising from reduced production or excessive splenic sequestration. Neutropenia can be caused by an intrinsic defect in BM myeloid cells, the congenital and idiopathic neutropenias, or by factors extrinsic to BM, the acquired neutropenias.
Box 16.1 Main causes of neutropenia
Congenital and idiopathic neutropenias
Severe congenital neutropenia (Kostmann’s disease). Kostmann disease4 is a rare autosomal recessive disorder of neutrophil number. The ANC is characteristically less than 0.2 × 109/l. Severe persistent neutropenia results in an increased susceptibility to bacterial infections. Granulocyte colony-stimulating factor (G-CSF) receptors are expressed on myeloid cells in slightly increased numbers, while the binding affinity for G-CSF to its receptor is normal. The neutrophil elastase gene mutations (ELA2) have been found in a subgroup of patients with Kostmann disease, although these mutations are also found in some healthy family members.5 Autosomal recessive inheritance6 of homozygous hematopoietic cell-specific protein-1 (HS1)-associated protein X-1 (HAX-1) mutations appear to lead to the increased apoptosis of myeloid precursors observed in patients with severe congenital neutropenia.7 HAX-1 functions include signal transduction, cytoskeletal control and regulation of apoptosis and it is reported to play a role in suppression of apoptosis in lymphocytes and neurons, resulting in prolonged survival of these cells.8 While the G-CSF receptor gene mutations have not been detected at birth, patients with Kostmann disease who develop leukemia have been found to have acquired these mutations.9 Point mutations in the gene for the G-CSF receptor CSF3R have been implicated in the progression of severe congenital neutropenia to leukemia. These mutations lead to the production of truncated CSF3R receptors determining hyper-responsive forms (G-CSFR-hyper) with increased proliferation and highly decreased maturation of myeloid cells.10,11
Congenital aleukia (reticular dysgenesis). Reticular dysgenesis12 is a rare inherited disorder characterized by the failure of hematopoietic stem cells committed to myeloid and lymphoid development. Red blood cell count (RBC) is normal, while platelet count may be decreased. There are no lymphocytes in the thymus, which is hypoplastic, and there is complete absence of peripheral lymphoid tissues. Allogenic BM transplant (BMT) remains the sole therapeutic option.13
Benign chronic neutropenias
Familial benign chronic neutropenia. This disorder,14,15 inherited as an autosomal dominant trait, is characterized by chronic moderate to severe neutropenia, monocytosis, lymphocytosis and occasionally moderate eosinophilia. Patients with mild or moderate neutropenia are asymptomatic. The BM is normocellular and usually shows few neutrophil precursors beyond the myelocyte stage. Patients have a reduced reserve myeloid pool in the BM, which makes G-CSF therapy not appropriate.16
Non-familial benign chronic neutropenia. Cases of chronic benign neutropenia without any identified familial pattern are grouped in this category. In several patients, the presence of autoantibodies suggests the immune aetiology.17 Even though neutropenia is usually severe in patients with chronic benign neutropenia of infancy and childhood, the risk of infection is very low. BM is hypercellular and the granulopoietic differentiation is present until the stage of band form. A reduced neutrophil chemotaxis from BM to PB is a possible explanation. In situations causing acute stress such as infection or cortisol administration, the number of circulating neutrophils increases.18 G-CSF therapy in the forms with a benign course is not indicated.
Familial cyclic neutropenia. This is a rare disorder,19–21 in which neutropenia of 4–10 days duration occurs at intervals of 15–35 days (average 21 days). Most cases present in infancy or childhood with periodic bouts of fever, malaise, headache, sore throat, oral ulceration, skin infections or, occasionally, infections of the lungs and other organs. Neutropenia results from cyclic changes in neutrophil granulocytopoiesis. Cyclic neutropenia has been transferred by BM transplantation to a histocompatible sibling with acute lymphoblastic leukemia.22 In about 25% of patients, the condition is inherited as a dominant trait with point mutations involving the neutrophil elastase gene ELA-2 on chromosome 19p13.3. Sporadic, not familial, as well as acquired cases associated with large granular lymphocyte (LGL) expansion are reported in the literature.23,24
Chronic idiopathic neutropenia of adult. This is a cytokine-mediated syndrome characterized by varying degrees of neutropenia associated with a low number of lineage-specific CD34+ cells and increased production of inhibitors of hematopoiesis (including tumor growth factor (TGF)-β1 and tumor necrosis factor (TNF)-α) and lymphopenia due to selective loss of primed/memory T-cells and natural killer (NK) cells. Other symptoms, reported in about 50% of patients, include splenomegaly, osteopenia and/or osteoporosis. The presence of features of chronic antigenic stimulation and increased concentrations of a variety of macrophage-derived pro-inflammatory cytokines25 are suggestive of the existence of an unrecognized low-grade chronic inflammatory process, which may be involved in the pathogenesis of the disorder. Neutropenia in these patients is probably resulting from a combination of at least three factors:26 reduced production of BM neutrophils, enhanced neutrophil extravasation and increased sequestration and/or extravasation of neutrophils into the spleen.
Neutropenia in patients with congenital immune defects
Neutropenia can be associated with congenital immunologic defects and patients with combined defects are at very high risk of infection. Neutropenia is reported in about one fourth of patients with Bruton agammaglobulinemia (X-linked hypogammaglobulinemia),27 in about 50% of patients with dysgammaglobulinemia,28 and in several patients with hyper IgM syndrome or with isolated IgA syndrome.29 Treatment with intravenous infusion of immunoglobulin is required, possibly associated with G-CSF.
Neutropenia in patients with congenital chromosomal abnormalities
Shwachman–Diamond syndrome. Shwachman–Diamond30 syndrome is a rare autosomal recessive disorder characterized by pancreatic exocrine insufficiency, metaphyseal chondrodysplasia, mental retardation, thrombocytopenia and defective neutrophil motility. The mutations involve a single gene, the Shwachman-Bodian-Diamond-Syndrome (SBDS) gene on chromosome 7q11. Neutropenia is determined by an increased apoptosis of myeloid precursors caused by the SBDS. About 20% of patients evolve into acute leukemia.
Chediak–Higashi syndrome. Chediak–Higashi31 syndrome is a rare autosomal recessive disorder characterized by recurrent infections, albinism, and by the presence of a reduced number of granules and/or formation of some abnormally large granules (by progressive fusion of normally formed granules) in most granule-containing cells. The giant granules present in neutrophils are peroxidase-positive and represent primary granules that have fused together. The abnormal granule fusion and function is determined by mutations of the CHS1 gene coding for the lysosomal trafficking regulator (LYST) protein on chromosome 1q42. Neutropenia is present in about 75% of patients. BMT provides a supportive cure for hematological signs but it does not prevent the neurologic deterioration.32 The affected cells include the neutrophils, eosinophils, lymphocytes, monocytes, melanocytes, Schwann cells of peripheral nerves, fibroblasts, vascular endothelial cells, renal tubular cells, and the parenchymal cells of the adrenal and pituitary glands. There is a deficiency of platelet dense granules, leading to easy bruising and bleeding. The large granules in leukocytes vary from a pale slate-gray to a dark reddish colour (Romanowsky stain) (Fig. 16.1A,B). There is a marked reduction in circulating NK cells.33 The leukocyte abnormalities are associated with recurrent infections. Some affected children, particularly those who live beyond early childhood, develop a terminal accelerated phase characterized by a lymphoma-like picture with lymphadenopathy, hepatosplenomegaly, neuropathy, widespread infiltration of tissues by non-clonal lymphatic and histiocytic cells, and pancytopenia. Death results from the complications of pancytopenia.
WHIM syndrome. Wart, hypogammaglobulinemia, infection and myelokathexis (WHIM) syndrome is a rare congenital autosomal dominant disorder determined by mutation in the gene coding for the neutrophil chemokine receptor CXCR.34 It is characterized by chronic non-cyclic neutropenia and increased susceptibility to bacterial and viral infections, especially from common serotype human papilloma virus, resulting in warts on the hands and feet starting in childhood. There is myeloid hyperplasia with degenerating granulocytes in the BM (myelokathexis).
Glycogen storage disease type 1b. It is a rare congenital autosomal recessive condition, determined by mutations on chromosome 11q23. These mutations cause glycogen accumulation in tissues due to deficiency of intracellular enzymes, which normally catalyze reactions that convert glycogen compounds to glucose. Severe neutropenia with consequent high risk of infections is present only in the type 1b and is determined by a disturbed myeloid maturation.35
Other rare conditions
Congenital dysgranulopoietic neutropenia. Congenital dysgranulopoietic neutropenia38,39is characterized by ineffective myelopoiesis and morphological abnormalities in the neutrophilic series. The term has been used to describe a disorder affecting six unrelated children presenting recurrent severe bacterial infections since birth, marked neutropenia, impaired neutrophil migration, normal numbers of BM colony-forming cells, normal or slightly increased colony-stimulating activity in the serum, and prominent morphologic and ultrastructural abnormalities in granulopoietic cells beginning from the neutrophil promyelocyte/myelocyte stage. The abnormalities of these cells included numerous autophagic vacuoles, abnormally electron-lucent primary granules, myelinization of primary granules, granule fusion, absence or marked decrease of secondary granules, and maturation of the cytoplasm ahead of the nucleus. Stem cell involvement in the pathogenesis of this disorder is postulated.40
Acquired neutropenias
Neonatal alloimmune neutropenia. In this syndrome, neutrophils and neutrophil precursors of the fetus are destroyed by the transplacental passage of maternal IgG alloantibodies formed against HLA or neutrophil-specific antigens (NA1 and NA2) present on fetal as well as on paternal neutrophils but not on maternal ones.41,42 The incidence is estimated at 1 in 500 live births. Most patients recover spontaneously usually within 2 months, depending on the life span of the maternal immunoglobulins. The neutropenia is occasionally severe and may cause life-threatening neonatal infections. Such patients respond well to intravenous immunoglobulins and G-CSF. The antibodies against neutrophil-specific antigens transferred to the fetus by the transplacental passage may be present either because the mother is affected by an autoimmune disease or as consequence of maternal sensibilization against the antigens of the fetal neutrophils of paternal origin. Despite a very low number of ANC, there is usually a spontaneous resolution within a few months. Supportive treatment with antibiotics is required in presence of infections, such as onphalitis or cellulitis.
Autoimmune neutropenia. Autoimmune neutropenia as a consequence of antineutrophil antibodies is a common finding associated with various collagen-vascular disorders, including rheumatoid arthritis (30% of patients with Felty’s syndrome), Systemic lupus erythematodes (SLE, 50% of patients), Sjögrens syndrome, scleroderma, polymyositis and polymyalgia rheumatica. It has also been described in association with autoimmune hemolytic anemia, immune thrombocytopenic purpura or both. In adults, autoimmune neutropenia is less likely to resolve spontaneously as compared to infants and children. In a subset of patients who have antibodies against the granulocyte precursors, the clinical course is more aggressive.43 Severe autoimmune neutropenias respond to treatment with metothrexate, steroids, G-CSF and granulocyte-macrophage colony stimulating factor (GM-CSF). Chronic autoimmune neutropenia44,45 is usually a relatively mild disease not associated with any other autoimmune disorders and characterized by recurrent infections, particularly of the oropharynx and skin. In some patients, the antineutrophil antibody has the specificity anti-NA2 or pan-Fcγ RIII (CD16, NA1/NA2). Some patients requiring treatment have responded to G-CSF. The LGL leukemia is often associated with neutropenia (see Chapter 28).
Infections associated with neutropenia
Infections are the most common cause of acquired neutropenia with different pathogenetic mechanisms (Box 16.1). In particular, some viruses (EBV, HAV, HBV, HCV and HIV) induce a severe and prolonged neutropenia by direct damage of the granulocytic precursors.46,47 Fungal, rickettsial and protozoal infections damage the BM endothelial cells causing vasculitis and severe cytopenias.48 Bacterial septicemia, in particular with Gram-negative agents, may induce neutropenia through an endotoxin-induced shift of neutrophils from the circulating to the marginated cell pools, a reduced survival time of circulating granulocytes (due either to destruction of cells within the circulation or to an accelerated rate of egress of cells from the blood), and a BM failure to adequately increase the rate of effective granulocytopoiesis. In certain circumstances, infection may be associated both with a failure adequately to increase effective granulocytopoiesis and with an exhaustion of the marrow granulocyte pool so that neutropenia is seen together with a left shift. This is particularly common in very severe bacterial infections and in bacterial infections in neonates and alcoholics (who have a reduced BM granulocyte pool). In several infections associated with spleen enlargement, such as typhoid fever, malaria and kala-azar, neutropenia results from splenic trapping and destruction of neutrophils in the spleen. In most of the latter cases, neutropenia does not require treatment, which should be focused on the underlying disease.
Neutropenia is the most common drug-induced blood dyscrasia.49,50 Some drugs (e.g. alkylating agents or antifolate drugs), certain chemicals (e.g. benzene) and irradiation induce neutropenia in all affected individuals in a dose-dependent manner. Other drugs cause neutropenia only occasionally and this phenomenon is usually at least partly based on a genetic polymorphism of drug metabolism. Drugs of the latter category may either cause neutropenia as part of an aplastic anemia (AA) or induce a selective neutropenia. Furthermore, several of the drugs causing AA in susceptible individuals initially cause a neutropenia that subsequently progresses to AA. The agranulocytosis induced by amidopyrin51 (and possibly also by other drugs) is often attributed to the destruction of circulating neutrophils and neutrophil precursors by drug-related immune mechanisms. There is also evidence that some drug metabolites may directly damage neutrophils and their precursors through the myeloperoxidase (MPO) system.52 Drugs that may occasionally cause selective neutropenia are listed in Box 16.2. The relatively high-risk drugs include amidopyrine and the antithyroid drugs. Some of the listed drugs usually induce a mild to moderate neutropenia that is asymptomatic and does not progress despite continuation of the drug. Other drugs usually cause a complete or almost complete absence of neutrophils (agranulocytosis).
Box 16.2 Main drugs which may cause neutropenia
Idiosyncratic drug-induced agranulocytosis (incidence 2.4–15.4 cases per million) results in a life-threatening clinical syndrome53 characterized by fever, sweating, vomiting, sore throat, dysphagia due to necrotic ulceration of the mouth and pharynx, extreme prostration and, frequently, death from overwhelming infection. At autopsy, necrotic ulcers are found in the mucous membranes of the entire alimentary tract as well as in the vagina. The prognosis is considerably improved if the offending drug is stopped and infection is adequately controlled by antibiotic therapy. To date, drug-induced agranulocytosis remains a serious adverse event due to the high frequency of sepsis with severe deep infections (such as pneumonia), septicemia, and septic shock in about two-thirds of all patients. Old age (>65 years), septicemia or shock, metabolic disorders such as renal failure, and a neutrophil count below 0.1 × 109/l are poor prognostic factors. Drugs or their metabolites might induce neutropenia by one or both of the following mechanisms: direct toxic effect on myeloid precursors or marrow environmental, dose-dependent inhibition of granulopoiesis, and immune-mediated destruction of granulocytes or their precursors. The classic example of a drug that causes agranulocytosis by a toxic effect on the BM is chlorpromazine, which causes a transient and moderate neutropenia in one-third of treated individuals. It causes agranulocytosis in about 1 in 1200 individuals, usually after a cumulative dose of 10–20 g over 20–30 days. Chlorpromazine appears to cause agranulocytosis by inhibiting DNA synthesis and cell proliferation in the granulocyte precursors of susceptible individuals. At the time of the agranulocytosis, the BM shows few or no neutrophil granulocytopoietic cells. The susceptibility of occasional individuals to develop agranulocytosis following therapy with chlorpromazine or other sulphur-containing drugs such as carbimazole or metiamide may depend on their genetically determined ability to oxidize the drug to highly-reactive myelotoxic metabolites (sulphoxides) more rapidly than most individuals.54
[/not-level-membership-for-pathology-category]
