[level-membership-for-pathology-category]
CHAPTER 30 Abnormalities in immunoglobulin synthesizing cells
Multiple myeloma
Epidemiology and etiology
MM represents 10–15% of all hematologic malignancies and 1% of all cancers, with an incidence of 2/100 000.1 The incidence increases with age, with approximately 40% of patients presenting under the age of 60 years and only 2% of cases occurring before the age of 40 years. There is a moderate excess in males. Geographic and racial differences play an important role, as the disease is more common in black people than Caucasians and has a low incidence in Chinese people. These rates are retained after migration to new countries, suggesting an inherited rather than an environmental explanation for the differences. Epidemiological studies have been carried out to identify environmental risk factors.2,3 An association with radiation exposure is seen in survivors of the World War II atomic bombs, as well as in occupationally and therapeutically exposed groups. There is also a suggestion of an association with farming, paper production, woodwork and exposure to a variety of chemicals including petroleum, benzene and materials associated with plastic and rubber manufacture. In addition to traditional epidemiology studies, there is much interest in determining whether inherited polymorphic variation can influence the development of MM or a patient’s response to treatment.2,3 To date most studies have been small and have concentrated on single nucleotide polymorphisms (SNPs) that affect the function of genes already known to be important in MM pathogenesis (e.g. immune response and cytokine genes). The introduction of newer high-throughput technologies with near complete genome coverage will enable this important area to be investigated further over the next few years.
Biology
The cell of origin
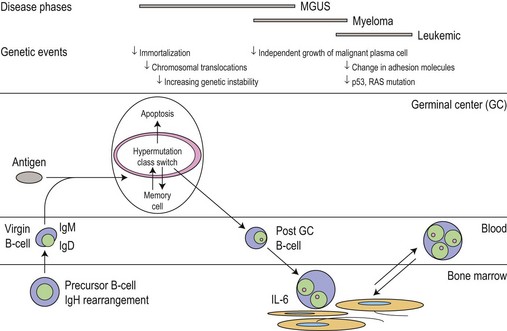
The main phenotypic features of myeloma PC include abnormal localization within the BM, replacement of normal BM elements, and dysregulation of Ig secretion. Normal PC in BM are derived from cells that have passed through a germinal center in a lymph node or other organ. Within the germinal center, cells undergo somatic hypermutation, class switching of the Ig gene, and selection by antigen-binding affinity; only cells with high binding affinity survive to become PC. In myeloma the Ig genes from individual plasma cells show the same pattern of somatic hypermutation, consistent with the clonal expansion of a single postgerminal center B-cell.4,5 The high incidence of translocations involving the switch region on chromosome 14 would also indicate that the final molecular oncogenic event occurs late in B-cell development. This contrasts with monoclonal gammopathy of unknown significance (MGUS) where there is intraclonal variation in the pattern of mutation, suggesting transformation of a virgin or memory B-cell with progeny which continue to pass through the normal process of germinal center selection before becoming plasma cells (Fig. 30.1).
Biology and growth signaling
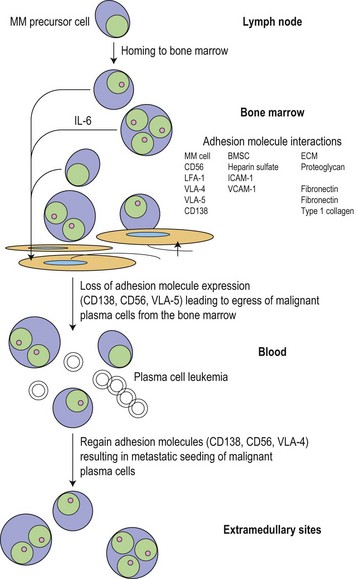
Following the transformation of the proliferative ‘plasmablastic’ cell located in the germinal centre, adhesion molecules mediate homing of the immortalized progeny of this cell from the lymph node to specialized niches within the BM, where maturation into a malignant PC occurs (Fig. 30.2). Further genetic hits lead to loss of tumor suppressor genes, expression of oncogenes and alteration in cell cycle control, resulting in a proliferative advantage for the MM cell and disease progression. Binding of myeloma cells to the BM stroma occurs and localizes tumor cells within the BM microenvironment. This binding to stroma results in an increase in the paracrine transcription and secretion of cytokines (particularly interleukin (IL)-6, insulin-like growth factor 1 (IGF1) and vascular endothelial growth factor (VEGF)), mediating myeloma cell growth and survival, and protection from drug-induced apoptosis (Fig. 30.3).6,7
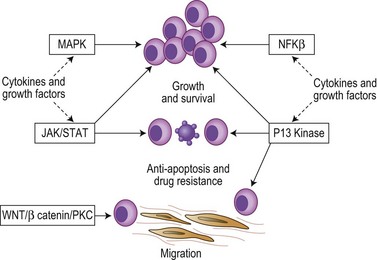
The cytokines IL6, IGF1 and VEGF together with direct myeloma cell to cell contact trigger signaling via the Ras/MEK/MAPK pathway resulting in myeloma cell growth, survival and drug resistance.7–9 Mutations affecting these pathways result in cytokine independent myeloma cell growth, the development of drug resistance and extramedullary disease. IL6 and IGF1 also signal via the PI3kinase-AKT-mTOR pathway, mediating myeloma growth, cell cycle and apoptosis.7–9 Activation of mTOR results in phosphorylation of P70S6 and 4E-BP1, which plays a key role in regulating the translation of cyclin D and c-myc, two proteins known to be central to myeloma pathogenesis. IL6 also triggers signaling via the JAK/STAT3 pathway and triggers drug resistance via activation of RAFTK and the mitochondrial release of Smac.
Nuclear factor kappa B (NFκB) signaling is important in B-cell biology and most myeloma cell lines demonstrate activation of NFκB leading to increased myeloma cell growth and survival. The pathway is also the target of multiple mutational events with 20% of cases harboring mutations or deletions of key inhibitory members of both the canonical and non-canonical pathways.10,11 A further key pathway is the TNFα superfamily (SDF1, CD40, BAFF, APRIL). Although the direct effect of TNFα on cell proliferation is modest, it markedly up-regulates the secretion of IL6 from BM stromal cells leading to dramatic increases in myeloma cell growth. TNFα also induces NFκB dependent expression of adhesion molecules increasing binding between myeloma cells and stromal cells resulting in protection from drug-induced apoptosis. In addition, CD40 mediates a p53 dependent increase in myeloma cell growth and PI3kinase/AKT/NFκB dependent migration.
Cytogenetic and molecular abnormalities
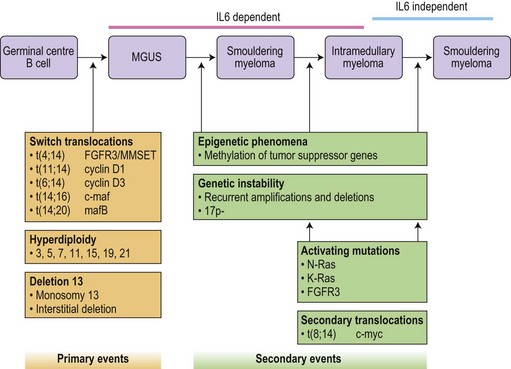
Translocations occurring as a result of aberrant class switch recombination events are the earliest known genetic events in MM (Fig. 30.4).12 The molecular characterization of the common recurrent translocations, t(4;14), t(11;14), t(14;16) and t(6;14), has identified a number of deregulated oncogenes including FGFR3/MMSET, cyclin D1, c-MAF and cyclin D3, respectively. In addition to switch translocations, secondary events such as chromosomal copy number alterations are common and genetic instability occurs resulting in deletions (e.g. 13q-, 17p-/p53, 1p-/CDKN2C, 16q-/CYLD/WWOX, NFκB inactivation/BIRC/TRAF3), activating mutations (e.g. NRas, FGFR3) and secondary translocations (e.g. t(8;14)). This has led to a molecular classification of myeloma based on the presence of switch translocations, hyperdiploidy and deregulation of the D group cyclins (Table 30.1).13 Two broad groups of patients can be recognized: a hyperdiploid group where there is a low incidence of switch translocations (<30%) and a non-hyperdiploid group where the incidence is high (>85%). Chromosome 1 abnormalities, usually 1q gain and 1p loss, are among the most prevalent cytogenetic abnormalities. The majority involve rearrangements located in the pericentromeric regions of the chromosomes and form jumping translocations. The actual gene responsible for the biological effects is uncertain although CSK1B and CDKN2C have been suggested as candidates. Recent studies have also suggested epigenetic changes contribute to the disease phenotype with patients showing overexpression of MMSET, a protein with histone methyl transferase activity and mutations in UTX, a histone demethylase.15
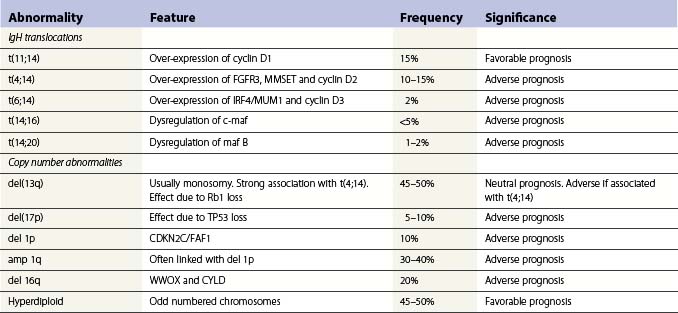
Advances in technology have now enabled the correlation of these genetic features with clinical outcome and has identified a series of distinct clinical subgroups. One such group are patients with the t(4;14)(p16.3;q32), which is present in 10–15% of myeloma cases.16,17 The translocation leads to dysregulation of two potential oncogenes, fibroblast growth factor receptor 3 (FGFR3) and multiple myeloma SET domain (MMSET). Myeloma carrying t(4;14) has a distinct gene expression profile and clinical profile with a short duration of response to chemotherapy, resistance to conventional alkylating agents and poor overall prognosis compared with other translocation groups. Patients with a t(11;14), present in 15% of cases, also have a distinct clinical phenotype. This translocation results in the up-regulation of cyclin D1, is associated with lymphoplasmacytic morphology, CD20 expression, λ light chain usage and low CD56.18 In addition, rare IgM myeloma often carry t(11;14).19 The prognostic significance depends on the series examined but ranges from neutral to favorable.
Patients with deletion of 17p also have a distinct clinical phenotype with a high incidence of extramedullary disease and aggressive course, short remissions and a short overall survival.20,21 Abnormalities of both the long and short arm of chromosome 1 have been linked with short survival, and gene expression profiles identifying patients with high risk disease are highly enriched for genes located on this chromosome. The prognostic significance of chromosome 13 deletion is more controversial with some studies showing a strong prognostic significance whereas other studies demonstrate little effect. This appears to depend on the detection method used to determine the presence of the abnormality, chromosome banding versus interphase FISH. In addition, all patients with a t(4;14) demonstrate deletion of chromosome 13, and as t(4;14) patients tend to have a poor prognosis this ‘linked effect’ may account for some of the differences.
Bone disease
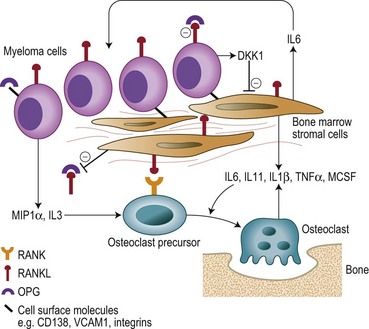
Bone destruction in MM is a prominent feature and causes considerable morbidity. Bone remodeling is a continuous process of resorption by osteoclasts and the subsequent formation of new bone by osteoblasts. In myeloma there is an increase in the number of osteoclasts and bone resorption in areas of the marrow adjacent to abnormal PC, but not in those areas adjacent to normal BM cells. New bone formation is also reduced when the tumor burden in the BM is high, and the combination of increased resorption and decreased formation leads to an uncoupling of normal bone remodeling.22,23 The central players involved in this process include: the receptor activator of NFκB (RANK); RANKL, the ligand for RANK; and osteoprotegerin (OPG) (Fig. 30.5). RANKL exists in a membrane and soluble form and via its receptor RANK increases bone resoprtion by increasing osteoclast formation and activity. OPG prevents bone resorption by acting as a decoy receptor preventing the binding of RANKL to RANK thereby inhibiting the up-regulation proliferation and fusion of osteoclast precursors to produce mature osteoclasts. A number of other cytokines and chemokines modify the BM microenvironment leading to an upregulation of RANKL by both stroma and osteoblasts including IL6, IL1β, IL11, lymphotoxin, Tumor necrosis factor (TNF)-α, and macrophage inflammatory protein 1α (MIP-1α), further perpetuating the cycle of bone destruction. In addition the increased osteoclast activity results in the secretion of tumor growth factor (TGF)-β, IL-6, β-fibroblast growth factor (FGF) and IGF-1 from the BM matrix in turn leading to further myeloma cell growth.
Diagnostic criteria
An international classification system has recently replaced a number of different diagnostic criteria to aid in the classification of the monoclonal gammopathies.24 Due to overlapping features, myeloma must be distinguished from the other disorders characterized by the presence of a monoclonal protein including MGUS, Waldenström’s macroglobulinemia, non-Hodgkin lymphoma, light-chain amyloid, idiopathic cold agglutinin disease, essential cryoglobulinemia, and heavy-chain disease. Some of these disorders are discussed later in this chapter, while non-Hodgkin lymphomas are discussed in Chapter 29. The majority of MM patients will have an M protein in the serum >30 g/l and/or BM clonal plasma cells >10% (Table 30.2). Patients are then classified depending on the presence or absence of end organ damage related to the plasma cell proliferative process (Table 30.3). Symptomatic patients have evidence of related organ or tissue impairment (end organ damage) (ROTI). Examples include raised calcium levels, renal insufficiency, anemia and bone lesions. Generally these patients require urgent therapy. Asymptomatic patients have no evidence of ROTI and usually undergo close monitoring with treatment initiated at disease progression. The term asymptomatic myeloma tends to include patients previously classified as having smoldering myeloma or Durie–Salmon stage I disease.
| Type | Incidence | |
|---|---|---|
| Serum M protein | Detectable in over 90% of patients using immunofixation | |
| Urinary M protein | Present in over 75% of patients | |
| M protein type | IgG | >50% |
| IgA | 20% | |
| IgD | 2% | |
| IgE | 1% | |
| Light chain only | 20% | |
| Non-secretory disease | 3% | |
Table 30.3 Myeloma-related organ or tissue impairment (end organ damage)24
| *Calcium | >0.25 mmol/l above the upper limit of normal or >2.75 mmol/l |
| *Renal insufficiency | Creatinine >173 mmol/l |
| *Anemia | Hemoglobin 2 g/dl below the lower limit of normal or hemoglobin <10 g/dl |
| *Bone lesions | Lytic lesions or osteoporosis with compression fractures |
| Other | Symptomatic hyperviscosity, amyloidosis recurrent bacterial infections (>2 episodes in 12 months) |
* CRAB, calcium, renal insufficiency, anemia or bone lesions.
Clinical features
Bone disease
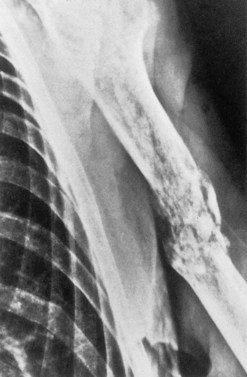
The accumulation of myeloma cells within the cavity of bones in the axial skeleton produces bone pain and destruction. The pain arises in the axial skeleton, and loss of height due to collapse of vertebrae and kyphosis are common. Although bone pain may be gradual in onset, pathologic fractures are frequent and usually indicated by the sudden onset of local tenderness and pain. Seventy per cent of patients will have evidence of bone disease at presentation and in almost all cases the bone lesions are osteolytic (Fig. 30.6), but a minority of patients (2%) have osteosclerotic lesions. The majority of patients also have diffuse osteopenia. Bone resorption leads to increased calcium in 20–40% of patients.
Staging and prognostic factors
The international staging system (ISS) reliably separates patients into prognostic groups using simple laboratory measurements.25 The system is widely used in the clinic to determine which patients should receive therapy, although few physicians would use it to direct specific choice of therapy. The system was derived from clinical and laboratory data from over 10 000 newly diagnosed patients and highlighted beta 2 microglobulin, serum albumin, platelet count, serum creatinine and age as powerful predictors of survival (Table 30.4). In addition to the cytogenetic abnormalities already discussed, other factors with prognostic significance include the extent and type of BM infiltration and measures of tumor-cell proliferation such as the plasma cell labeling index (PCLI). Patients with a plasmablastic morphology have a median survival of 16 months, compared to a median survival of 35 months for patients with other morphologic subtypes. PCLI is usually low (<1%) at diagnosis, higher at relapse, and lower in patients with MGUS. In addition, a high PCLI correlates with a shorter survival time independent of tumor-cell mass. The percentage of circulating PC at diagnosis has also been shown to be an independent prognostic variable.26 Some of the newer targeted therapies may be able to overcome the negative prognostic effects of the genetic abnormalities. This highlights the importance of performing FISH/gene array analysis at diagnosis, and suggests that making treatment decisions based on these results is a real option in the near future.
Table 30.4 International staging system25 for myeloma patients
| Factors | Median survival | |
|---|---|---|
| Stage I | β2m <3.5 mg/l albumin >3.5 g/dl |
62 months |
| Stage II | Neither I or III | 44 months |
| Stage III | β2m >3.5 mg/l albumin <3.5 g/dl |
29 months |
β2m, serum β2 microglobulin.
Pathology
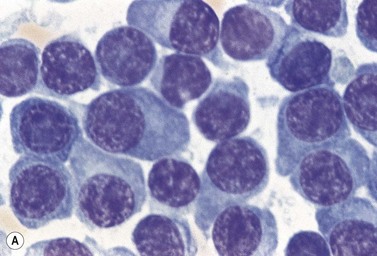
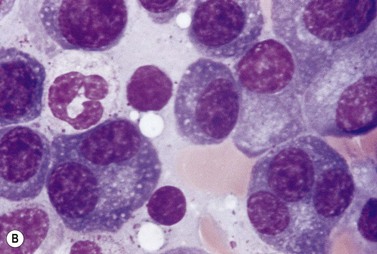
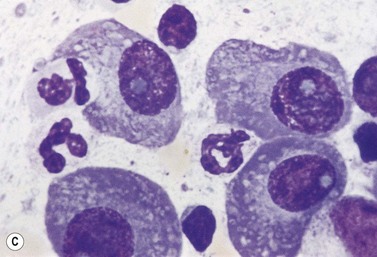
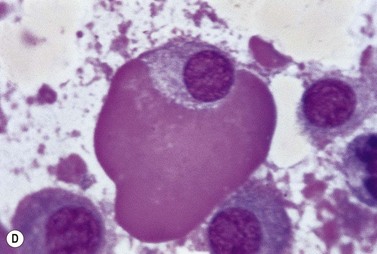
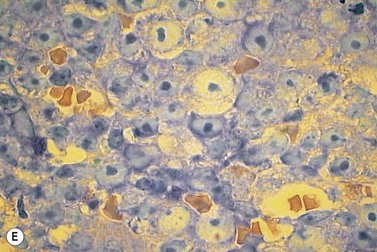
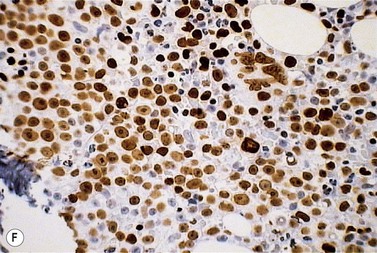
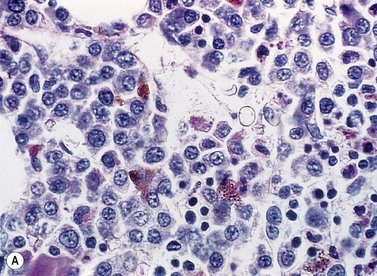
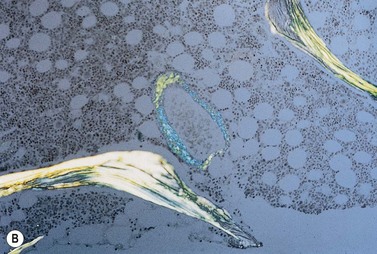
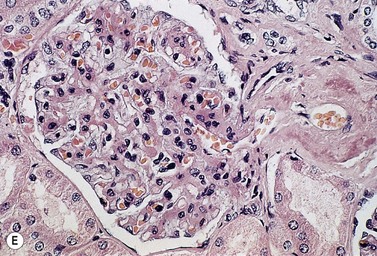
A normochromic normocytic anemia is often present, with rouleaux formation and a high nonspecific background staining on the blood smear due to the presence of circulating paraprotein. In patients with more advanced disease, thrombocytopenia and neutropenia may also be present. Occasional circulating PC with a phenotype similar to those within the marrow can be demonstrated. In the majority of cases, PC will exceed 10% of the nucleated cells within the BM. PC usually appear moderately to severely dysplastic with large eccentrically placed nuclei, which may be either multiple or cleaved (Fig. 30.7A, B). Nucleoli are prominent and nuclear inclusions may be present, including Dutcher bodies. Cytoplasm may be sparse or foamy or vacuolated and show Ig inclusions such as Russell bodies (Fig. 30.7C). Plasma cells from IgA myeloma often have a characteristic flame-cell appearance (Fig. 30.7D). Approximately 8% of myeloma cases demonstrate plasmablastic features, with more than 2% of cells having a plasmablastic morphology (Fig. 30.7E). Plasmablasts have a fine reticular chromatin pattern, large nucleoli and less abundant cytoplasm (less than half of the nuclear area). This morphologic subset is associated with a high PCLI, more advanced and aggressive disease, and a worse prognosis (Fig. 30.7F). A reactive plasmacytosis due to chronic inflammation or infection may also present with an increase in BM plasma cells. This can be readily distinguished from MM by the normal morphology as well as polyclonal phenotype of the plasma cells and an increase in eosinophils, mast cells and megakaryocytes.
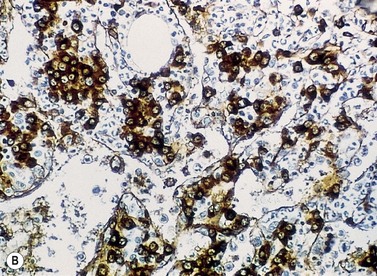
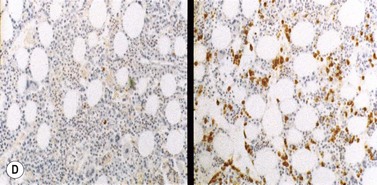
Plasma cells are terminally differentiated B-cells and hence express a number of B-cell antigens as well as myeloma-associated antigens. Both normal and malignant plasma cells express CD38 and CD138, but usually lack CD10, CD20, CD23, CD34 and CD45RO. The most reliable antibodies used to detect plasma cells by flow cytometry are therefore CD38, CD138 and CD45RO. It is possible to distinguish myeloma plasma cells from their normal counterparts, since the former usually express significantly higher levels of the important adhesion molecules CD56 and CD138 and significantly lower levels of CD19, CD38 and CD45 than the latter. Plasma cells from normal individuals are consistently CD19+ CD56 low; whereas 65% of myeloma cases have a plasma cell phenotype of CD19− CD56+, 30% CD19− CD56 low, and 5% CD19+ CD56+.26 Other immunophenotypic features allowed stratification of patients with MM into three risk categories: poor risk (CD28+ CD117−), intermediate (either both markers negative or both positive), and good risk (CD28− CD117+).27 Immunocytochemical studies confirm the monoclonality of the plasma cells, and the type of secreted Ig should correlate with the serum paraprotein and urinary light chain.
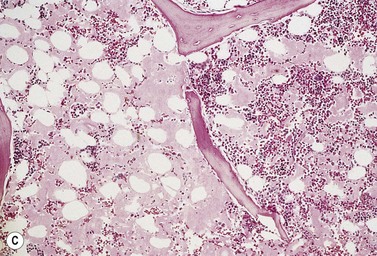
On BM biopsy, plasma cells normally accumulate around blood vessels. However, in MM this pattern is lost and the myeloma cells are found as single cells or small clusters between adipocytes. As the disease progresses, diffuse marrow replacement occurs, resulting in a packed marrow with complete loss of normal architecture (Fig. 30.8A, B). Occasionally, there is a paratrabecular distribution similar to follicular center lymphoma, and in some patients with aggressive disease tumor nodules containing abnormal PC may be seen. Assessing the degree of marrow infiltration is often difficult due both to the patchy nature of the disease and the presence of either hypoplastic or hyperplastic areas. There may be a discrepancy between the percentage of PC in the aspirate compared to the biopsy, due either to the presence of fibrosis or nodular infiltration resulting in a low number of PC on the smear.
Treatment
Myeloma has a relapsing remitting course that requires serial courses of treatment, with periods of durable remissions in between. With each successive relapse, the treatment free period often becomes shorter and eventually the myeloma cells become resistant to therapy and the disease rapidly progresses leading to death. During the last decade the introduction of a number of targeted therapies for myeloma has resulted in a significant increase in the number of patients achieving a response to therapy, an improvement in quality of life and increased survival. In view of the increased response rates seen with these targeted therapies the response criteria have been standardized and updated.28 The targeted therapies include the immunomodulatory drugs, thalidomide and lenalidomide, and the proteasome inhibitor bortezomib.29–35 These now form the backbone of therapy to which corticosteroids or conventional chemotherapy are often added. The mechanisms of action of the commonly used drugs are summarized in Table 30.5. A number of other novel biologically based treatment approaches are currently being evaluated which will be used either alone or in combination with existing treatments to further improve response and outcome. Importantly, in vitro and in vivo studies of the mechanisms of action of these novel approaches are also providing new insights into the biology of myeloma.
Table 30.5 Mechanisms of action of commonly used antimyeloma drugs29–35
| Drug | Mechanism of action |
|---|---|
| Alkylating agents (melphalan, cyclophosphamide, BCNU) | Cross-linking DNA strands in resting and dividing cells |
| Glucocorticoids (dexamethasone, prednisolone) | Increased apoptosis of myeloma plasma cells with associated phosphorylation of RAFTK |
| Vinca alkaloids (vincristine) | Inhibition of microtubule formation resulting in the arrest of dividing cells in metaphase |
| Anthracyclines (adriamycin) | Binds to nucleic acids by intercalation with base pairs of the DNA double helix interfering with DNA synthesis |
| Bisphosphonates (clodronate, pamidronate, zoledronic acid) | Reduced osteoclastic activity and decreased bone absorption Myeloma plasma cell and bone-marrow apoptosis Decreased production of IL-6 and metalloproteinases Activation of γδ T-cells |
| Immunomodulatory drugs – IMiDs (thalidomide and lenalidomide) | Growth arrest or increased apoptosis of myeloma plasma cells Alteration in adhesion molecule profile Alteration in cytokine secretion and/or bioavailability (IL6 and VEGF) Decreased angiogenesis Altered immune response |
| Proteasome inhibitors | Decreased myeloma cell growth and survival signaling (e.g. NFκB) Decreased expression of key anti-apoptotic molecules Dysregulation of intracellular calcium metabolism Down-regulation of genes involved in mismatch repair and DNA double strand break repair Decreased myeloma cell adhesion Stimulation of osteoblast activity |
BCNU, carmustine (bis-chloronitrosourea); IL6, interleukin 6; RAFTK, related adhesion focal tyrosine kinase; VEGF, vascular endothelial growth factor.
Monoclonal gammopathy of undetermined significance
Monoclonal gammopathy of undetermined significance (MGUS) describes a condition characterized by the presence of a low level of paraprotein in the absence of other clinical features of MM, Waldenström’s macroglobulinemia or other B-cell lymphoproliferative disorders.24 The previous term ‘benign monoclonal gammopathy’ is a misleading description of the disease, since a proportion of patients will develop a more aggressive plasma cell disorder.
Clinical features
The incidence of MGUS increases with age, with 1% of the population under the age of 60 years and between 4–5% of the population over the age of 80 years being affected. The prevalence in African Americans and Africans is approximately double that in white people.36,37 The demonstration of a paraprotein in the serum or urine is often an incidental finding and patients are typically asymptomatic. A recent prospective study indicated that MM is always preceded by MGUS.38 The serum paraprotein is usually less than 3 g/dl, with little or no urinary component, and may be present despite normal total protein or globulin levels. Immune paresis may occur in 30% of cases. There should be no evidence of end organ damage; the presence of bone lesions, hypercalcemia, renal impairment, lymphadenopathy or organomegaly should suggest a diagnosis of myeloma or Waldenström’s macroglobulinemia. There is an association of MGUS with a number of disorders including chronic lymphocytic leukemia, polyneuropathy and myopathy, connective tissue disorders, dermatologic disorders, hepatitis C and immunosuppression (AIDS and transiently post bone marrow or renal transplantation).
Biology
The main features of the biology of the disease have been discussed in detail above. Immunoglobulin heavy chain sequence analysis demonstrates an intraclonal variation in the pattern of mutation, suggesting transformation of a virgin or memory B-cell with progeny continuing to pass through the normal process of germinal center selection before becoming plasma cells.5 Cytogenetic analysis reveals findings similar to myeloma, including a complex karyotype with trisomies and monosomies, structural abnormalities, and translocations involving chromosome 14, although the prognostic significance is not known at present.12
Pathology
The peripheral blood picture is normal, with no evidence of circulating plasma cells or anemia. Clonal plasma cells are present within the bone marrow in an interstitial distribution and represent less than 5–10% of the total nucleated cells. Using flow cytometry it is possible to identify plasma cells with both a normal and ‘myeloma’ phenotype (CD38++, CD19+ and CD56− vs. CD38+, CD19− and CD56+).26
Treatment/management/disease progression
There is no specific treatment for patients with MGUS. Long-term surveillance is recommended since approximately 25% of cases will develop an overt plasma cell disorder. Data regarding disease progression have been derived from a cohort of 241 patients with MGUS at the Mayo Clinic who have been followed for 24–38 years.36,37 The risk of progression was approximately 1% per year with the majority of patients developing myeloma, although some show symptoms or signs of amyloidosis or Waldenström’s macroglobulinemia. There are currently no predictive factors to determine which patients will progress. Cases with M proteins of IgA or IgM type are associated with an increased risk of progression, as are patients with higher M protein levels or an abnormal serum free light chain ratio.39 Overall, 50% of patients with MGUS will die from an unrelated cause; the remaining 25% of patients will continue with stable paraprotein levels, but may develop disease over a longer follow-up period.
Plasma cell leukemia
Plasma cell leukemia (PCL) is a rare plasma cell disorder characterized by the presence of more than 2 × 109/1 circulating plasma cells which constitute at least 20% of all PB cells.1 The majority (60%) of cases are de novo or primary, in which a leukemic picture develops in the absence of a documented preceding plasma cell disorder. Forty per cent of cases are secondary and occur in a minority of myeloma patients (1%) with terminal disease.
Clinical features
The median age at diagnosis of primary PCL is 55 years, approximately 10 years younger than for myeloma.40 Although patients with primary PCL present with symptoms similar to those of myeloma, the disease course is often more aggressive with symptoms relating to extramedullary disease including plasmacytomas and hepatosplenomegaly. Anemia, hypercalcemia and renal failure are also more common. Patients with secondary disease usually have advanced myeloma that is refractory to treatment and may present with worsening symptoms related to bone marrow failure including anemia, infections, bleeding or plasmacytomas.
Biology
The adverse biological prognostic factors usually associated with end-stage myeloma are invariably present at diagnosis in cases of primary PCL. These include a high PCLI; frequent translocations involving 11q (cyclin D1); complex cytogenetic abnormalities including amplification of c-myc; as well as mutations of RAS and p53; and changes in adhesion-molecule profile.41,42
Pathology
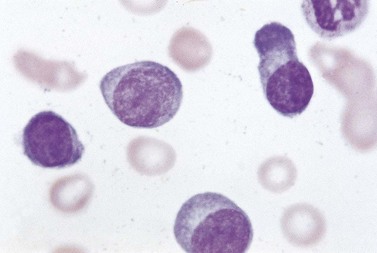
The peripheral blood is characterized by the presence of a large number of circulating PC, which may be morphologically normal or have blastic features (Fig. 30.9). Anemia is invariably present, and both neutropenia and thrombocytopenia are common. Rouleaux formation is usually also present with a high nonspecific background staining, especially in secondary PCL cases where the level of paraprotein is often high. The BM is heavily infiltrated with plasma cells morphologically similar to those within the PB. Other normal hematopoietic elements are reduced. Occasionally cells can be of lymphocyte size without obvious evidence of plasma cell morphology. In these cases other phenotypic features of plasma cell differentiation must be sought, such as loss of B-cell markers and high expression of CD38 and CD138.
Solitary bone plasmacytoma
Clinical features
The bone lesions are commonly in the axial skeleton, particularly in the vertebrae. The median age at presentation is 60 years and a monoclonal protein may be present, although at a level lower than is usual in myeloma.1,24,43 Full clinical staging is required in order to differentiate solitary plasmacytoma from MM and reveals a negative skeletal survey, absence of clonal plasma cells on BM examination, and lack of anemia, hypercalcemia or renal involvement.
Treatment/management/disease progression
The treatment of choice is radiotherapy. Local control is achieved in 90% of patients, with an accompanying fall in paraprotein level. The majority of patients do develop myeloma over time, with projected 5- and 10-year probabilities of 50% and 75% respectively. Magnetic resonance imaging (MRI) has revealed unsuspected bone lesions in approximately 30% of patients, and there is a suggestion that this technique may be able to distinguish patients who will progress to MM from patients with a more benign clinical course.44 Other potential prognostic markers include an abnormal serum free light chain ratio and the persistence of the serum monoclonal band for more than a year post radiotherapy.45,46 There is no definitive evidence that systemic treatment delays the onset of myeloma.
Extramedullary plasmacytoma
Clinical features
Isolated extramedullary plasmacytoma may also occur.1,47,48 Over 80% of lesions are in the upper respiratory tract and present with epistaxis, nasal discharge, hoarseness or sore throat. Lesions may also occur in the gastrointestinal tract, thyroid, thymus and skin (Fig. 30.10A, B). Twenty-five per cent of patients will have a monoclonal protein present in the serum or urine. It is important to distinguish patients with solitary extramedullary plasmacytoma from patients with soft tissue spread of advanced myeloma due to differences in the clinical disease course.
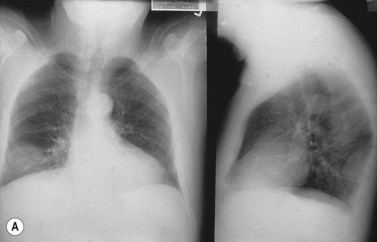
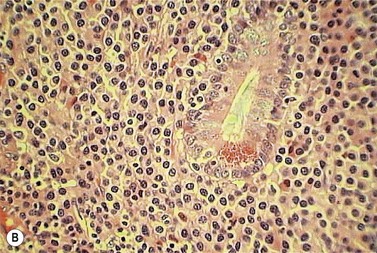
POEMS syndrome
The acronym polyneuropathy (P), organomegaly (O), endocrinopathy (E), M-protein (M) and skin changes (S) describes a syndrome associated with plasma cell disorders, particularly but not exclusively osteosclerotic myeloma.1,49,50 Although extremely rare, it is most frequently reported in Japanese males. A monoclonal protein is demonstrable in the plasma or urine in 75% of patients, usually of IgAl isotype. Clinical features include a demyelinating and axonal mixed neuropathy (more motor than sensory); hepatomegaly, splenomegaly and lymphadenopathy; diabetes mellitus, primary gonadal failure, hypothyroidism and Addison’s disease; and skin hyperpigmentation, thickening and hypertrichosis. Sixty per cent of patients will have pathologic changes consistent with multiple myeloma, although the extent of BM infiltration by plasma cells may be low. The increase in PC is usually associated with osteosclerosis. Lymph node involvement is common, and pathologic features resemble the hyaline-vascular variant of Castleman’s disease with follicular hyperplasia, vascular proliferation and an interfollicular infiltration of lymphocytes, plasma cells and immunoblasts. The pathogenesis is not well understood but the overproduction of VEGF secreted by PC is likely to be responsible for the characteristic symptoms. The median survival depends on the pattern of organ involvement and ranges from 2 to 10 years. Treatment is of the underlying plasma cell disorder.
Lymphoproliferative disorders associated with an IgM paraprotein
Waldenström’s macroglobulinemia (WM) is a chronic B-cell lymphoproliferative disorder in which most of the clinical manifestations are due to the presence of an IgM paraprotein. The disorder is characterized by bone marrow infiltration with small lymphocytes, lymphoplasmacytoid cells and plasma cells, and a high level of IgM paraprotein. The World Health Organization (WHO) classification considers WM as a clinical syndrome associated with the diagnosis of lymphoplasmacytoid lymphoma/immunocytoma.1 The morphologic and immunophenotypic features are described in Chapter 29. However, the presence of an IgM paraprotein is not specific for WM. The differential diagnosis includes other indolent lymphoproliferative disorders, for example chronic lymphocytic leukemia (CLL) with IgM paraprotein, splenic lymphoma with villous lymphocytes (SLVL), and splenic marginal zone lymphoma (SMZL).51,52 Differences in the level of paraprotein, lymphocytic morphology, and degree of marrow involvement in relation to spleen size may help to distinguish WM from SLVL and SMZL, since the malignant cells within these disorders share the same phenotype. Other conditions associated with an IgM paraprotein include MGUS and, rarely, true IgM myeloma.
Clinical features
WM is 10–20% less common than multiple myeloma. It is predominantly a disease of the elderly, with a median age at presentation of 65 years. It is more common in white people than black people, with a slight male predominance. Symptoms may occur due to tumor cell infiltration of BM (cytopenia, increased infections and bleeding), splenomegaly, hepatomegaly and lymphadenopathy.51 Other clinical features include hyperviscosity in 20% of patients and type 1 cryoglobulinemia, although clinically relevant cryoglobulinemia causing Raynaud’s phenomenon, purpura and glomerulonephritis occurs in less than 5% of patients. Cold agglutinin anemia may occur in 10% of patients when the monoclonal IgM behaves as a cold reactive antibody that interacts with erythrocyte antigens at low temperatures and results in the development of acrocyanosis, Raynaud’s phenomenon, and episodic or chronic hemolysis. Neurological manifestations are present in 10% of patients related to infiltration of peripheral nerves with paraprotein, antibodies against various glycoproteins and glycolipids of the peripheral nerves, and amyloid deposition. In contrast to other plasma cell disorders, there is an absence of bony changes and lytic lesions. Occasionally, renal involvement may be present, due either to glomerular abnormalities or to amyloid deposition resulting in a non-selective proteinuria. Diagnosis requires the presence of an IgM paraprotein in the serum and the infiltration of characteristic cells within the BM (see Chapter 29). As in myeloma, the presence of an IgM paraprotein may be an incidental finding, and the distinction between MGUS and WM is often controversial. MGUS is defined as IgM paraprotein <3 g/dl associated with no constitutional symptoms, organomegaly or anemia. Approximately 10% of patients with MGUS will develop WM at a median follow-up of 8 years.52 The International Prognostic Staging System for WM (IPSSWM) defines three prognostic groups for patients based on five adverse features (age, hemoglobin, platelets, β2m and level of monoclonal protein).53 The low risk group is characterized by age less than 65 years and the presence of one adverse feature, whereas the high risk group is characterized by two or more adverse features. The 5-year survival rates are 87%, 68% and 38% respectively.
Treatment/management
Patients with disease related symptoms require treatment. The initiation of therapy should not be based on the level of serum paraprotein alone and asymptomatic patients should be observed. The appropriate choice of therapy depends on a number of patient-related factors including the presence of cytopenias, need for rapid disease control, age and comorbidities.54,55 Initial management includes the correction of the raised plasma viscosity using plasmapheresis. This treatment is offered as a short-term measure while concomitant chemotherapy to reduce the tumor burden becomes effective. Combination chemotherapy includes the use of alkylating agents, nucleoside analogs and/or rituximab. Patients in resistant relapse are candidates for treatment with novel therapeutic agents.
Light-chain-associated amyloidosis
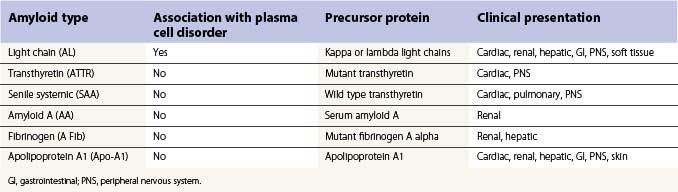
Amyloidosis is a spectrum of diseases associated with the deposition of extracellular protein in major organs to form characteristic fibrillar sheets that disrupt organ structure and function. Light-chain amyloid (AL), previously called primary amyloid, is characterized by the extracellular deposition of fibrillar protein derived from monoclonal light chains. The light chains are cleaved into fragments that consist of the whole or part of the variable domain of the molecule, although occasionally intact light chains may also be deposited. The fragments form beta-pleated sheets which become insoluble and resistant to degradation following the deposition of glycosaminoglycans and the normal protein serum amyloid P component (SAP). A number of other types of proteins may also be deposited; however, these types of amyloid are not associated with plasma cell abnormalities. The proteins deposited include mutant or wild type transthyretin, the circulating acute phase reactant protein serum amyloid A, and apolipoprotein A1 (Table 30.6).1,56–58
Pathology
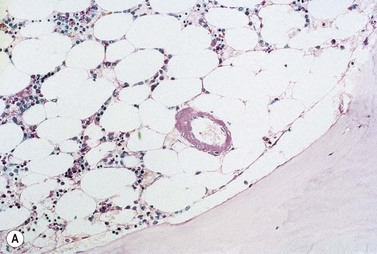
The peripheral blood picture is usually normal, although Howell–Jolly bodies are present in 25% of cases suggesting hyposplenism due to amyloid infiltration of spleen. A low-level plasmacytosis is present on the BM aspirate, and in some cases PC may account for more than 20% of the total nucleated cells. Even if the PC number is normal there is often an imbalance in κ:λ ratio suggesting the presence of abnormal cells, and immunoglobulin heavy chain PCR will confirm a clonal picture. When amyloid occurs in association with WM, lymphoplasmacytoid cells will be present. The bone biopsy reveals similar findings to those in the aspirate, but in addition demonstrates amyloid deposits within the small blood vessel walls or extravascularly. Examination with bipolarized light following staining with Congo red demonstrates apple green birefringence. This is pathognomonic for light-chain amyloid and is the standard for diagnosis. Amyloid also stains metachromically with crystal violet, fluoresces after staining with thioflavine-T, and is pink and homogenous on H&E staining (Fig. 30.11 A–D). Immunohistochemistry staining for fibrinogen may also be useful. Light-chain amyloid may be distinguished from other types of amyloid immunochemically using type-specific anti-light-chain sera, although this may be negative since a number of antibodies react with parts of the molecule that have been degraded during formation of the protein deposit. It may also be distinguished from other types of amyloid, as there is no abolition of Congo red staining by prior treatment with potassium permanganate. As immunohistochemistry staining for amyloid typing is unreliable immunogold electron microscopy may be required. The characteristic amyloid deposition can be demonstrated in all affected organs on biopsy (Fig. 30.11E). If biopsy of the primary organ is dangerous, then a fine-needle aspirate of abdominal fat, or a gingiva or rectal biopsy are less invasive ways to demonstrate the presence of disease and are commonly positive (80% of cases). Scintigraphy using radio-labeled SAP is useful to determine the extent of disease and organ involvement.
Treatment/management
Supportive therapy is directed toward alleviating symptoms and improving the function of affected organs (i.e. controlling heart failure, renal failure). Specific treatment is also required to reduce or eliminate the plasma cell clone, as the amyloid deposition may regress when the primary source of protein is removed. Oral melphalan and dexamethasone or high dose melphalan and stem cell transplant are considered the standard therapies for AL amyloid.58,59 Owing to an increased risk of peri-transplant related mortality (10–25%), current guidelines suggest that this therapy should be offered only to patients with less than three-organ involvement and reasonable cardiac function. Given the impressive improvement in response rates and survival with the introduction of novel therapies in myeloma, ongoing studies are investigating the potential use of thalidomide, lenalidomide and bortezomib in amyloid.60,61 Splenectomy may be helpful in ameliorating coagulation deficiencies and patients with predominantly cardiac or renal involvement should be considered for organ transplantation.
Heavy-chain disorders
This group of rare plasma cell disorders is characterized by the production of a monoclonal Ig that is formed from truncated heavy chains with no associated light chains.1,62–64 The diagnosis depends on the detection of structurally abnormal immunoglobulin in patient serum or urine which consists mainly of the Fc region of the molecule. The actual length of the chain varies between patients but is usually one-half to three-quarters of the normal counterpart, with the majority of cases having complete deletion of the CHl domain. The mechanisms leading to the production of the abnormal protein are not clearly understood. The analysis of rearranged gene sequences demonstrates a high level of somatic mutation with deletions and insertions of sequences of unknown origin. This suggests that cells producing the abnormal heavy chains arise during somatic hypermutation in the germinal center, and that further genetic alterations are required at a later stage in the developmental process for malignant transformation to occur. The abnormal Ig is not evident by serum electrophoresis in a high proportion of cases, and identification may therefore require more sensitive techniques. The presenting clinical features depend on the type of heavy chain involved.
α-Heavy-chain disease
Clinical features
Over 400 cases have been reported to date with the majority from the Mediterranean or the Middle East.65 Presentation is usually in the third decade of life with a slight male predominance. Environmental factors in early infancy are important in the etiology of the disease, especially low socioeconomic status and poor hygiene leading to the development of recurrent infectious diarrhea and chronic parasitic infections. The main clinical features include diarrhea, weight loss, abdominal pain, vomiting and evidence of malabsorption. Abdominal masses may also be present. Occasionally, the disease may present with respiratory symptoms due to infiltrations in the respiratory tract.
γ-Heavy-chain disease
Pathology
There is no consistent morphologic pattern corresponding to the serologic diagnosis of γHCD.66 The BM is usually infiltrated with lymphocytes, lymphoplasmacytoid cells or plasma cells. Lymph node biopsy may show evidence of a lymphoplasmacytoid proliferation, a PC infiltrate, or non-Hodgkin’s lymphoma. Unusual pathologic features include eosinophils or multinucleated giant cells, suggesting the presence of an atypical granulomatous lesion. The PB may show evidence of a moderate normochromic normocytic anemia, lymphocytosis with atypical lymphoplasmacytoid cells or plasma cells, and eosinophilia.
Cryoglobulinemia
The presence of an underlying plasma cell disorder may lead to the development of cryoglobulinemia. In type I cryoglobulinemia the paraprotein secreted, usually IgG or IgM, has the characteristics of a cryoglobulin. In type II and type III cryoglobulinemia the paraprotein, usually IgM, has antibody activity against another immunoglobulin, often polyclonal IgG, and results in immune complex formation. In 75% of patients, no pathologic manifestations other than those due to the characteristic of the immunoglobulin are present at the time of diagnosis and the term ‘essential cryoglobulinemia’ is appropriate.67 Over a 10-year period of follow-up, approximately 25% of patients will develop symptoms and signs associated with a lymphoproliferative disorder. In the remaining 25% of cases, the cryoglobulinemia is a manifestation of an overt lymphoproliferative disorder including myeloma, WM, immunocytoma, MALT lymphoma and follicular lymphoma. In this subset of patients 25% will have type I, 70% type II and 5% type III cryoglobulinemia.
1 McKenna RW, Kyle RA, Khuel WM, et al. cell neoplasms. In: Swerdlow SH, Campo C, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2008:194-195.
2 Morgan GJ, Davies FE, Linet M. Myeloma aetiology and epidemiology. Biomedicine and Pharmacotherapy. 2002;56:223-234.
3 Davies FE, Avet-Loiseau H, Bergsagel PL. Epidemiology, etiology and molecular pathogenesis. Richardson P, editor. Multiple Myeloma: State of the Art. Remedica, London. 2010.
4 Bakkus MHC, Heirman C, Van Riet I, et al. Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood. 1992;80:2326-2335.
5 Sahota SS, Leo R, Hamblin TJ, et al. Ig VH gene mutational patterns indicate different tumour cell status in human myeloma and monoclonal gammopathy of undetermined significance. Blood. 1996;87:746-755.
6 Teoh G, Anderson KC. Interaction of tumour and host cells with adhesion and extracellular matrix molecules in the development of multiple myeloma. Hematology and Oncology Clinics of North America. 1997;11:27-42.
7 Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009;23:10-24.
8 Hideshima T, Mitsiades C, Tonon G, et al. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nature Cancer Reviews. 2007;7:585-598.
9 Podar K, Hideshima T, Chauhan D, et al. Targeting signaling pathways for the treatment of mulitple myeloma. Expert Opinions in Therapeutic Targets. 2005;9:359-381.
10 Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NFkappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115-130.
11 Keats JJ, Fonesca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NFkappaB pathways in multiple myeloma. Cancer Cell. 2007;12:131-144.
12 Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210-2221.
13 Bergsagel PL, Kuehl WM, Zhan F, et al. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296-303.
14 Walker BA, Morgan GJ. Use of single nucleotide polymorphism-based mapping arrays to detect copy number changes and loss of heterozygosity in multiple myeloma. Clinical Lymphoma and Myeloma. 2006;7:186-191.
15 Smith E, Boyd K, Davies FE. The potential role of epigenetic therapy in multiple myeloma. British Journal of Haematology. 2010;148:702-713.
16 Chesi M, Nardini E, Brents LA, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma: association with increased expression and activating mutations of fibroblast growth factor receptor 3. Nature Genetics. 1997;16:260-264.
17 Dring AM, Davies FE, Fenton JAL, et al. A global expression based analysis of the consequences of the t(4;14) in myeloma. Clinical Cancer Research. 2004;10:5692-5701.
18 Robillard N, Avet-Loiseau H, Garand R, et al. CD20 is associated with a small mature plasma cell morphology and t(11;14) in multiple myeloma. Blood. 2003;102:1070-1071.
19 Feyler S, O’Connor SJ, Rawstron AC, et al. IgM myeloma: a rare entity characterised by a CD20-CD56-CD117- immunophenotype and the t(11;14). British Journal of Haematology. 2008;140:545-551.
20 Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109:3489-3495.
21 Avet-Loiseau H, Li C, Magrangeas F, et al. Prognostic significance of copy-number alterations in multiple myeloma. Journal of Clinical Oncology. 2009;27:4585-4590.
22 Ashcroft AJ, Davies FE, Morgan GJ. Aetiology of bone disease and the role of bisphosphonates in multiple myeloma. Lancet Oncology. 2003;4:284-292.
23 Lentzsch S, Ehrlich LA, Roodman GD. Pathophysiology of multiple myeloma bone disease. Hematology and Oncology Clinics of North America. 2007;21:1035-1049.
24 Kyle RA, Child JA, Anderson KC, et al. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. British Journal of Haematology. 2003;121:749-757.
25 Greipp PR, San Miguel Dure BGM, et al. International staging system for multiple myeloma. Journal of Clinical Oncology. 2005;23:3412-3420.
26 Rawstron AC, Owen RG, Davies FE, et al. Circulating plasma cells in multiple myeloma: characterisation and correlation with disease status. British Journal of Haematology. 1997;97:46-55.
27 Mateo G, Montalban MA, Vidriales MB, et al. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. Journal of Clinical Oncology. 2008;26:2737-2744.
28 Durie BGM, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467-1473.
29 Raab MS, Podar K, Breitkreutz I, et al. Multiple myeloma. Lancet. 2009;374:324-339.
30 Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. New England Journal of Medicine. 2007;357:2133-2142.
31 Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. New England Journal of Medicine. 2007;357:2123-2132.
32 Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. New England Journal of Medicine. 2005;352:2487-2498.
33 Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. New England Journal of Medicine. 1999;18:1565-1571.
34 Hideshima T, Chauhan D, Shima Y, et al. Thalidomide and its analogues overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood. 2000;96:2943-2950.
35 Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Research. 2001;61:3071-3076.
36 Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. New England Journal of Medicine. 2006;354:1362-1369.
37 Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. British Journal of Haematology. 2006;134:573-580.
38 Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113:5412-5417.
39 Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in MGUS. Blood. 2005;106:812-817.
40 Ramsingh G, Mehan P, Luo J, et al. Primary plasma cell leukemia: a surveillance, epidemiology, and end results database analysis between 1973 and 2004. Cancer. 2009;115:5734-5739.
41 Garcia-Sanz R, Orfao A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy and cytogenetic characteristics. Blood. 1999;93:1032-1037.
42 Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Blood. 2008;22:1044-1052.
43 British Committee for Standards in Haematology. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. British Journal of Haematology. 2004;124:717-726.
44 Moulopouos LA, Dimopoulos MA, Weber D, et al. Magnetic resonance imaging in the staging of solitary plasmacytoma of bone. Journal of Clinical Oncology. 1993;11:1311-1315.
45 Dingli D, Kyle RA, Rajkumar S, et al. Immunoglobulin free light chains and solitary plasmacytoma of bone. Blood. 2006;108:1979-1983.
46 Ozsahin M, Tsang R, et al. Outcomes and patterns of failure in solitary plasmacytoma. International Journal of Radiation Oncology. 2006;64:210-217.
47 Dimopoulos MA, Kiamouris C, Moulopoulos LA. Solitary plasmacytoma of bone and extramedullary plasmacytoma. Hematology and Oncology Clinics of North America. 1999;13:1249-1257.
48 Knowling MA, Harwood AR, Bergsagel DE. Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. Journal of Clinical Oncology. 1983;1:255-262.
49 Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long term outcome. Blood. 2003;101:2496-2506.
50 Dispenzieri A. POEMS syndrome. Blood Reviews. 2007;21:285-299.
51 Dimopoulos MA, Panayiotidis P, Moulopoulos LA, et al. Waldenströms macroglobulinemia: clinical features, complications and management. Journal of Clinical Oncology. 2000;18:214-226.
52 Kyle RA, Garton JP. The spectrum of IgM monoclonal gammopathy in 430 cases. Mayo Clinic Proceedings. 62, 1987. 719–711
53 Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood. 2009;113:4163-4170.
54 Dimopoulos MA, Gertz MA, Kastritis E, et al. Update on treatment recommendations from the fourth international workshop on Waldenström’s macroglobulinemia. Journal Clinical Oncology. 2009;27:120-126.
55 Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2009;114:2375-2385.
56 Gilmore JD, Hawkins PN, Pepys MB. Amyloidosis: a review of recent diagnostic and therapeutic developments. British Journal of Haematology. 1997;99:245-256.
57 Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. New England Journal of Medicine. 1997;337:898-909.
58 Comenzo RL. How I treat amyloidosis. Blood. 2009;114:3147-3157.
59 Jaccard A, Moreau P, Leblond V, et al. High dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. New England Journal of Medicine. 2007;357:1083-1093.
60 Dispenzieri A, Lacy MQ, Zeldenrust SR, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109:465-470.
61 Kastritis E, Enagnostopoulos A, Roussou M, et al. Treatment of light chain AL amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92:135-1358.
62 Cogne M, Silvain C, Khamlichi AA, et al. Structurally abnormal immunoglobulins in human immunoproliferative disorders. Blood. 1992;79:2181-2195.
63 Fermand JP, Brouet JC. Heavy chain diseases. Hematology and Oncology Clinics of North America. 1999;13:1281-1294.
64 Seligmann M, Mihaesco E, Preud’homme JL, et al. Heavy chain diseases: current findings and concepts. Immunology Reviews. 1979;48:145-167.
65 Rambaud JC, Brouet JC Seligmann M, et al. Alpha chain disease and related lymphoproliferative disorders. In: Ogra P, Mestecky J, Lamm ME, et al, editors. Handbook of Mucosal Immunology. San Diego: Academic Press; 1994:425.
66 Wester SM, Banks PM, Li CY. The histopathology of gamma heavy chain disease. American Journal of Clinical Pathology. 1982;78:427-436.
67 Monti G, Galli M, Invernizzi F, et al. Cryoblobulinaemias: a multicentre study of the early clinical and laboratory manifestations of primary and secondary disease. Q J M. 1995;88:115-126.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 30 Abnormalities in immunoglobulin synthesizing cells
Multiple myeloma
Epidemiology and etiology
MM represents 10–15% of all hematologic malignancies and 1% of all cancers, with an incidence of 2/100 000.1 The incidence increases with age, with approximately 40% of patients presenting under the age of 60 years and only 2% of cases occurring before the age of 40 years. There is a moderate excess in males. Geographic and racial differences play an important role, as the disease is more common in black people than Caucasians and has a low incidence in Chinese people. These rates are retained after migration to new countries, suggesting an inherited rather than an environmental explanation for the differences. Epidemiological studies have been carried out to identify environmental risk factors.2,3 An association with radiation exposure is seen in survivors of the World War II atomic bombs, as well as in occupationally and therapeutically exposed groups. There is also a suggestion of an association with farming, paper production, woodwork and exposure to a variety of chemicals including petroleum, benzene and materials associated with plastic and rubber manufacture. In addition to traditional epidemiology studies, there is much interest in determining whether inherited polymorphic variation can influence the development of MM or a patient’s response to treatment.2,3 To date most studies have been small and have concentrated on single nucleotide polymorphisms (SNPs) that affect the function of genes already known to be important in MM pathogenesis (e.g. immune response and cytokine genes). The introduction of newer high-throughput technologies with near complete genome coverage will enable this important area to be investigated further over the next few years.
Biology
The cell of origin
The main phenotypic features of myeloma PC include abnormal localization within the BM, replacement of normal BM elements, and dysregulation of Ig secretion. Normal PC in BM are derived from cells that have passed through a germinal center in a lymph node or other organ. Within the germinal center, cells undergo somatic hypermutation, class switching of the Ig gene, and selection by antigen-binding affinity; only cells with high binding affinity survive to become PC. In myeloma the Ig genes from individual plasma cells show the same pattern of somatic hypermutation, consistent with the clonal expansion of a single postgerminal center B-cell.4,5 The high incidence of translocations involving the switch region on chromosome 14 would also indicate that the final molecular oncogenic event occurs late in B-cell development. This contrasts with monoclonal gammopathy of unknown significance (MGUS) where there is intraclonal variation in the pattern of mutation, suggesting transformation of a virgin or memory B-cell with progeny which continue to pass through the normal process of germinal center selection before becoming plasma cells (Fig. 30.1).
Biology and growth signaling
Following the transformation of the proliferative ‘plasmablastic’ cell located in the germinal centre, adhesion molecules mediate homing of the immortalized progeny of this cell from the lymph node to specialized niches within the BM, where maturation into a malignant PC occurs (Fig. 30.2). Further genetic hits lead to loss of tumor suppressor genes, expression of oncogenes and alteration in cell cycle control, resulting in a proliferative advantage for the MM cell and disease progression. Binding of myeloma cells to the BM stroma occurs and localizes tumor cells within the BM microenvironment. This binding to stroma results in an increase in the paracrine transcription and secretion of cytokines (particularly interleukin (IL)-6, insulin-like growth factor 1 (IGF1) and vascular endothelial growth factor (VEGF)), mediating myeloma cell growth and survival, and protection from drug-induced apoptosis (Fig. 30.3).6,7
The cytokines IL6, IGF1 and VEGF together with direct myeloma cell to cell contact trigger signaling via the Ras/MEK/MAPK pathway resulting in myeloma cell growth, survival and drug resistance.7–9 Mutations affecting these pathways result in cytokine independent myeloma cell growth, the development of drug resistance and extramedullary disease. IL6 and IGF1 also signal via the PI3kinase-AKT-mTOR pathway, mediating myeloma growth, cell cycle and apoptosis.7–9 Activation of mTOR results in phosphorylation of P70S6 and 4E-BP1, which plays a key role in regulating the translation of cyclin D and c-myc, two proteins known to be central to myeloma pathogenesis. IL6 also triggers signaling via the JAK/STAT3 pathway and triggers drug resistance via activation of RAFTK and the mitochondrial release of Smac.
Nuclear factor kappa B (NFκB) signaling is important in B-cell biology and most myeloma cell lines demonstrate activation of NFκB leading to increased myeloma cell growth and survival. The pathway is also the target of multiple mutational events with 20% of cases harboring mutations or deletions of key inhibitory members of both the canonical and non-canonical pathways.10,11 A further key pathway is the TNFα superfamily (SDF1, CD40, BAFF, APRIL). Although the direct effect of TNFα on cell proliferation is modest, it markedly up-regulates the secretion of IL6 from BM stromal cells leading to dramatic increases in myeloma cell growth. TNFα also induces NFκB dependent expression of adhesion molecules increasing binding between myeloma cells and stromal cells resulting in protection from drug-induced apoptosis. In addition, CD40 mediates a p53 dependent increase in myeloma cell growth and PI3kinase/AKT/NFκB dependent migration.
Cytogenetic and molecular abnormalities
Translocations occurring as a result of aberrant class switch recombination events are the earliest known genetic events in MM (Fig. 30.4).12 The molecular characterization of the common recurrent translocations, t(4;14), t(11;14), t(14;16) and t(6;14), has identified a number of deregulated oncogenes including FGFR3/MMSET, cyclin D1, c-MAF and cyclin D3, respectively. In addition to switch translocations, secondary events such as chromosomal copy number alterations are common and genetic instability occurs resulting in deletions (e.g. 13q-, 17p-/p53, 1p-/CDKN2C, 16q-/CYLD/WWOX, NFκB inactivation/BIRC/TRAF3), activating mutations (e.g. NRas, FGFR3) and secondary translocations (e.g. t(8;14)). This has led to a molecular classification of myeloma based on the presence of switch translocations, hyperdiploidy and deregulation of the D group cyclins (Table 30.1).13 Two broad groups of patients can be recognized: a hyperdiploid group where there is a low incidence of switch translocations (<30%) and a non-hyperdiploid group where the incidence is high (>85%). Chromosome 1 abnormalities, usually 1q gain and 1p loss, are among the most prevalent cytogenetic abnormalities. The majority involve rearrangements located in the pericentromeric regions of the chromosomes and form jumping translocations. The actual gene responsible for the biological effects is uncertain although CSK1B and CDKN2C have been suggested as candidates. Recent studies have also suggested epigenetic changes contribute to the disease phenotype with patients showing overexpression of MMSET, a protein with histone methyl transferase activity and mutations in UTX, a histone demethylase.15
Advances in technology have now enabled the correlation of these genetic features with clinical outcome and has identified a series of distinct clinical subgroups. One such group are patients with the t(4;14)(p16.3;q32), which is present in 10–15% of myeloma cases.16,17 The translocation leads to dysregulation of two potential oncogenes, fibroblast growth factor receptor 3 (FGFR3) and multiple myeloma SET domain (MMSET). Myeloma carrying t(4;14) has a distinct gene expression profile and clinical profile with a short duration of response to chemotherapy, resistance to conventional alkylating agents and poor overall prognosis compared with other translocation groups. Patients with a t(11;14), present in 15% of cases, also have a distinct clinical phenotype. This translocation results in the up-regulation of cyclin D1, is associated with lymphoplasmacytic morphology, CD20 expression, λ light chain usage and low CD56.18 In addition, rare IgM myeloma often carry t(11;14).19 The prognostic significance depends on the series examined but ranges from neutral to favorable.
Patients with deletion of 17p also have a distinct clinical phenotype with a high incidence of extramedullary disease and aggressive course, short remissions and a short overall survival.20,21 Abnormalities of both the long and short arm of chromosome 1 have been linked with short survival, and gene expression profiles identifying patients with high risk disease are highly enriched for genes located on this chromosome. The prognostic significance of chromosome 13 deletion is more controversial with some studies showing a strong prognostic significance whereas other studies demonstrate little effect. This appears to depend on the detection method used to determine the presence of the abnormality, chromosome banding versus interphase FISH. In addition, all patients with a t(4;14) demonstrate deletion of chromosome 13, and as t(4;14) patients tend to have a poor prognosis this ‘linked effect’ may account for some of the differences.
Bone disease
Bone destruction in MM is a prominent feature and causes considerable morbidity. Bone remodeling is a continuous process of resorption by osteoclasts and the subsequent formation of new bone by osteoblasts. In myeloma there is an increase in the number of osteoclasts and bone resorption in areas of the marrow adjacent to abnormal PC, but not in those areas adjacent to normal BM cells. New bone formation is also reduced when the tumor burden in the BM is high, and the combination of increased resorption and decreased formation leads to an uncoupling of normal bone remodeling.22,23 The central players involved in this process include: the receptor activator of NFκB (RANK); RANKL, the ligand for RANK; and osteoprotegerin (OPG) (Fig. 30.5). RANKL exists in a membrane and soluble form and via its receptor RANK increases bone resoprtion by increasing osteoclast formation and activity. OPG prevents bone resorption by acting as a decoy receptor preventing the binding of RANKL to RANK thereby inhibiting the up-regulation proliferation and fusion of osteoclast precursors to produce mature osteoclasts. A number of other cytokines and chemokines modify the BM microenvironment leading to an upregulation of RANKL by both stroma and osteoblasts including IL6, IL1β, IL11, lymphotoxin, Tumor necrosis factor (TNF)-α, and macrophage inflammatory protein 1α (MIP-1α), further perpetuating the cycle of bone destruction. In addition the increased osteoclast activity results in the secretion of tumor growth factor (TGF)-β, IL-6, β-fibroblast growth factor (FGF) and IGF-1 from the BM matrix in turn leading to further myeloma cell growth.
Diagnostic criteria
An international classification system has recently replaced a number of different diagnostic criteria to aid in the classification of the monoclonal gammopathies.24 Due to overlapping features, myeloma must be distinguished from the other disorders characterized by the presence of a monoclonal protein including MGUS, Waldenström’s macroglobulinemia, non-Hodgkin lymphoma, light-chain amyloid, idiopathic cold agglutinin disease, essential cryoglobulinemia, and heavy-chain disease. Some of these disorders are discussed later in this chapter, while non-Hodgkin lymphomas are discussed in Chapter 29. The majority of MM patients will have an M protein in the serum >30 g/l and/or BM clonal plasma cells >10% (Table 30.2). Patients are then classified depending on the presence or absence of end organ damage related to the plasma cell proliferative process (Table 30.3). Symptomatic patients have evidence of related organ or tissue impairment (end organ damage) (ROTI). Examples include raised calcium levels, renal insufficiency, anemia and bone lesions. Generally these patients require urgent therapy. Asymptomatic patients have no evidence of ROTI and usually undergo close monitoring with treatment initiated at disease progression. The term asymptomatic myeloma tends to include patients previously classified as having smoldering myeloma or Durie–Salmon stage I disease.
| Type | Incidence | |
|---|---|---|
| Serum M protein | Detectable in over 90% of patients using immunofixation | |
| Urinary M protein | Present in over 75% of patients | |
| M protein type | IgG | >50% |
| IgA | 20% | |
| IgD | 2% | |
| IgE | 1% | |
| Light chain only | 20% | |
| Non-secretory disease | 3% | |
Table 30.3 Myeloma-related organ or tissue impairment (end organ damage)24
| *Calcium | >0.25 mmol/l above the upper limit of normal or >2.75 mmol/l |
| *Renal insufficiency | Creatinine >173 mmol/l |
| *Anemia | Hemoglobin 2 g/dl below the lower limit of normal or hemoglobin <10 g/dl |
| *Bone lesions | Lytic lesions or osteoporosis with compression fractures |
| Other | Symptomatic hyperviscosity, amyloidosis recurrent bacterial infections (>2 episodes in 12 months) |
* CRAB, calcium, renal insufficiency, anemia or bone lesions.
Clinical features
Bone disease
The accumulation of myeloma cells within the cavity of bones in the axial skeleton produces bone pain and destruction. The pain arises in the axial skeleton, and loss of height due to collapse of vertebrae and kyphosis are common. Although bone pain may be gradual in onset, pathologic fractures are frequent and usually indicated by the sudden onset of local tenderness and pain. Seventy per cent of patients will have evidence of bone disease at presentation and in almost all cases the bone lesions are osteolytic (Fig. 30.6), but a minority of patients (2%) have osteosclerotic lesions. The majority of patients also have diffuse osteopenia. Bone resorption leads to increased calcium in 20–40% of patients.
