[level-membership-for-basic-science-category]
Chapter 25 Renal and Genitourinary Systems
Prototype and Common Drugs


MOA (Mechanism of Action)









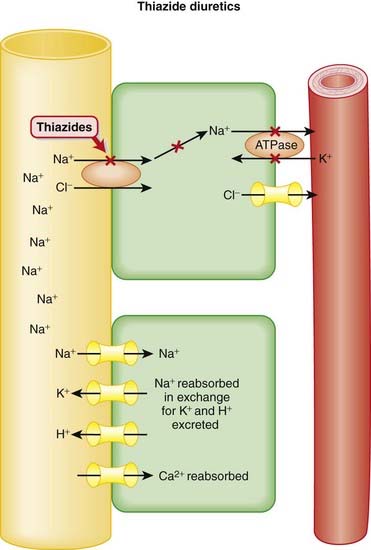
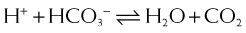
Pharmacokinetics














Important Notes






Evidence
As First-Line Agents in Hypertension
 A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year. Thiazides (19 trials) reduced mortality (relative risk [RR] 0.89), stroke (RR 0.63), and coronary heart disease (RR 0.84) versus placebo.
A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year. Thiazides (19 trials) reduced mortality (relative risk [RR] 0.89), stroke (RR 0.63), and coronary heart disease (RR 0.84) versus placebo.

Diuretics result in increased urine production and also lower blood pressure.


 Generally speaking, diuretics manipulate a solute (usually Na+), and water passively follows:
Generally speaking, diuretics manipulate a solute (usually Na+), and water passively follows:
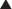







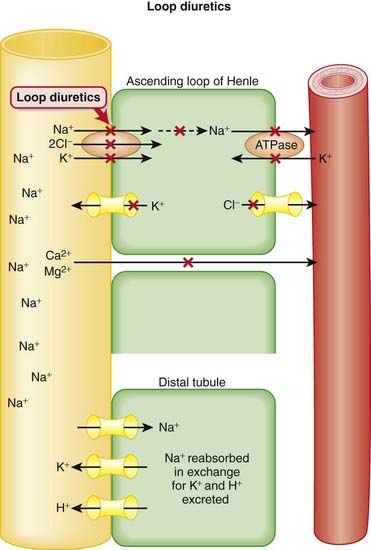


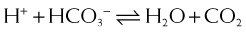











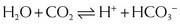 and that decreased H+ will drive the equation to the right and cause increased HCO3−.
and that decreased H+ will drive the equation to the right and cause increased HCO3−.



Diuretics versus Placebo or Other Interventions for Treatment of Heart Failure
 A 2006 Cochrane review (14 trials, N = 525 participants) compared diuretics with placebo (seven trials) and other interventions (seven trials) for treatment of heart failure. The diuretics included were a mixture of loop diuretics, potassium-sparing diuretics, and thiazides. Based on three trials (N = 202 participants), diuretic treatment reduced the odds of death versus placebo (OR 0.24), and in two trials (N = 169 participants) it reduced the odds of admission because of worsening heart failure (OR 0.07). Diuretics improved exercise capacity in four trials (N = 91 participants) versus active controls.
A 2006 Cochrane review (14 trials, N = 525 participants) compared diuretics with placebo (seven trials) and other interventions (seven trials) for treatment of heart failure. The diuretics included were a mixture of loop diuretics, potassium-sparing diuretics, and thiazides. Based on three trials (N = 202 participants), diuretic treatment reduced the odds of death versus placebo (OR 0.24), and in two trials (N = 169 participants) it reduced the odds of admission because of worsening heart failure (OR 0.07). Diuretics improved exercise capacity in four trials (N = 91 participants) versus active controls.Blood-Pressure–Lowering Effect versus Placebo or No Treatment
 A 2009 Cochrane review (nine trials, N = 460 participants) compared the blood-pressure–lowering efficacy, tolerability, and biochemical effects of a variety of loop diuretics versus placebo or no treatment. The authors found a mean reduction (systolic/diastolic blood pressure) of −7.9/−4.4 mmHg and no differences between drugs in this class with respect to blood-pressure–lowering efficacy. This is considered to be a modest antihypertensive effect. Withdrawals because of adverse effects and biochemical changes were not significantly different from control.
A 2009 Cochrane review (nine trials, N = 460 participants) compared the blood-pressure–lowering efficacy, tolerability, and biochemical effects of a variety of loop diuretics versus placebo or no treatment. The authors found a mean reduction (systolic/diastolic blood pressure) of −7.9/−4.4 mmHg and no differences between drugs in this class with respect to blood-pressure–lowering efficacy. This is considered to be a modest antihypertensive effect. Withdrawals because of adverse effects and biochemical changes were not significantly different from control. Diuretics that exert their effects by acting on the renal tubules require functioning kidneys to induce diuresis. Patients with advanced renal dysfunction will not respond to these diuretics.
Diuretics that exert their effects by acting on the renal tubules require functioning kidneys to induce diuresis. Patients with advanced renal dysfunction will not respond to these diuretics.












Spironolactone and Eplerenone (Figure 25-3)

Other Effects
 Spironolactone also binds to androgen and progesterone receptors. This is not by design but reflects the similarities in chemical structure between androgens (e.g., testosterone) progesterone, and aldosterone. Because these receptors are also similar in structure, spironolactone, which was designed before these receptors were mapped, binds to all three.
Spironolactone also binds to androgen and progesterone receptors. This is not by design but reflects the similarities in chemical structure between androgens (e.g., testosterone) progesterone, and aldosterone. Because these receptors are also similar in structure, spironolactone, which was designed before these receptors were mapped, binds to all three.
 Spironolactone has an active metabolite, canrenone, which is also marketed in some jurisdictions. Canrenone has a much longer elimination half-life (16 hours) than spironolactone (2 hours).
Spironolactone has an active metabolite, canrenone, which is also marketed in some jurisdictions. Canrenone has a much longer elimination half-life (16 hours) than spironolactone (2 hours). As adjunct (add-on) therapy to a regular (potassium-depleting) diuretic such as a thiazide or loop diuretic, in the treatment of:
As adjunct (add-on) therapy to a regular (potassium-depleting) diuretic such as a thiazide or loop diuretic, in the treatment of:
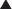





 K+-sparing diuretics all produce a fairly modest diuresis and are typically given as adjunctive (add-on) therapy with more potent diuretics, with the intention of mitigating the hypokalemia associated with those agents.
K+-sparing diuretics all produce a fairly modest diuresis and are typically given as adjunctive (add-on) therapy with more potent diuretics, with the intention of mitigating the hypokalemia associated with those agents.


 All three prototypical K+-sparing diuretics (spironolactone, triamterene, and amiloride) are available in fixed-dose combinations (i.e., in the same pill) with hydrochlorothiazide.
All three prototypical K+-sparing diuretics (spironolactone, triamterene, and amiloride) are available in fixed-dose combinations (i.e., in the same pill) with hydrochlorothiazide.Carbonic Anhydrase Inhibitors (CAIs)
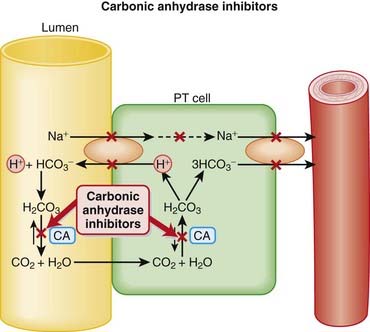
 Carbonic Anhydrase (CA) is classified as a diuretic, but its important clinical effects are related to its effect on acid-base balance and on intraocular fluid formation.
Carbonic Anhydrase (CA) is classified as a diuretic, but its important clinical effects are related to its effect on acid-base balance and on intraocular fluid formation.

 CA catalyzes the following reaction:
CA catalyzes the following reaction:
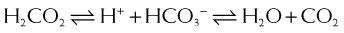








 Acetazolamide is cleared by the kidneys, whereas methazolamide is cleared primarily by the kidneys but also partially by the liver.
Acetazolamide is cleared by the kidneys, whereas methazolamide is cleared primarily by the kidneys but also partially by the liver. Dorzolamide and brinzolamide are administered topically, as eye drops in the management of glaucoma.
Dorzolamide and brinzolamide are administered topically, as eye drops in the management of glaucoma.






 Acting early in the nephron, the diuretic actions of CAIs are countered by a number of sites for Na+ reabsorption that appear distally in the tubule. That, coupled with the complication of metabolic acidosis, has limited the therapeutic usefulness of CAIs in the management of edema, and certainly in hypertension. In fact, CAIs are not even used for the treatment of edema or hypertension currently.
Acting early in the nephron, the diuretic actions of CAIs are countered by a number of sites for Na+ reabsorption that appear distally in the tubule. That, coupled with the complication of metabolic acidosis, has limited the therapeutic usefulness of CAIs in the management of edema, and certainly in hypertension. In fact, CAIs are not even used for the treatment of edema or hypertension currently.

 One systematic review found that acetazolamide was more efficacious than placebo in preventing acute mountain sickness (number needed to treat [NNT], two to three). A lower dose was not found to be more effective than placebo.
One systematic review found that acetazolamide was more efficacious than placebo in preventing acute mountain sickness (number needed to treat [NNT], two to three). A lower dose was not found to be more effective than placebo. How do CAIs work in the treatment of glaucoma? Glaucoma is a condition of the eye characterized by increased intraocular pressure (IOP). CA located in the ciliary processes of the eye results in the formation of HCO3− in aqueous humor. By decreasing the rate of formation of aqueous humor, CAIs reduce IOP.
How do CAIs work in the treatment of glaucoma? Glaucoma is a condition of the eye characterized by increased intraocular pressure (IOP). CA located in the ciliary processes of the eye results in the formation of HCO3− in aqueous humor. By decreasing the rate of formation of aqueous humor, CAIs reduce IOP.Osmotic diuretics increase the oncotic pressure of fluids and carry water with them.

 Osmotic agents increase the oncotic pressure of the blood; this pulls water from tissues and increases the volume of the blood acutely. The increased blood volume will inhibit renin release, thus increasing renal blood flow.
Osmotic agents increase the oncotic pressure of the blood; this pulls water from tissues and increases the volume of the blood acutely. The increased blood volume will inhibit renin release, thus increasing renal blood flow.




 Raised intracranial pressure (ICP) and cerebral edema. The concept behind using osmotic manipulation for treating raised ICP is that water will get sucked out of the brain and into the blood (which has high osmotic pressure from the mannitol); this will reduce brain swelling and ICP.
Raised intracranial pressure (ICP) and cerebral edema. The concept behind using osmotic manipulation for treating raised ICP is that water will get sucked out of the brain and into the blood (which has high osmotic pressure from the mannitol); this will reduce brain swelling and ICP.

 Active cranial bleeding: If bleeding is occurring into the brain, then the mannitol will be directly added to the brain and will raise the osmotic pressure in the brain and lead to water being added to the brain, which is the opposite of what the mannitol is being given for.
Active cranial bleeding: If bleeding is occurring into the brain, then the mannitol will be directly added to the brain and will raise the osmotic pressure in the brain and lead to water being added to the brain, which is the opposite of what the mannitol is being given for.
 Extracellular volume expansion: Osmotic diuretics rapidly distribute to the extracellular compartment, extracting water from cells. Before onset of the diuresis, this can lead to expansion of the extracellular space. Frank pulmonary edema can arise in patients with heart failure or pulmonary congestion.
Extracellular volume expansion: Osmotic diuretics rapidly distribute to the extracellular compartment, extracting water from cells. Before onset of the diuresis, this can lead to expansion of the extracellular space. Frank pulmonary edema can arise in patients with heart failure or pulmonary congestion.
 Osmotic diuretics increase the excretion of nearly all electrolytes (Na+, K+, Ca+2, Mg+2, Cl−, HCO3−, and phosphate).
Osmotic diuretics increase the excretion of nearly all electrolytes (Na+, K+, Ca+2, Mg+2, Cl−, HCO3−, and phosphate).
 A 2007 Cochrane review (four trials, N = 197 participants) compared different mannitol regimens with other interventions, including placebo and no treatment, in patients with acute traumatic brain injury. There was no difference in the incidence of death between mannitol and “standard care,” pentobarbital, hypertonic saline, or placebo.
A 2007 Cochrane review (four trials, N = 197 participants) compared different mannitol regimens with other interventions, including placebo and no treatment, in patients with acute traumatic brain injury. There was no difference in the incidence of death between mannitol and “standard care,” pentobarbital, hypertonic saline, or placebo. Mannitol is a six-carbon sugar alcohol. It was first discovered in the sap of a plant and was thought to resemble biblical food; therefore the plant was called manna ash, after manna (biblical reference to food).
Mannitol is a six-carbon sugar alcohol. It was first discovered in the sap of a plant and was thought to resemble biblical food; therefore the plant was called manna ash, after manna (biblical reference to food).Antidiuretic Hormone (Vasopressin) Analogues


 Vasopressin is produced in the hypothalamus and stored in the posterior pituitary gland. Factors that stimulate its physiologic release include increased serum osmolarity and hypotension. Its primary actions are therefore to increase body water to control osmolality and to increase blood pressure. A third action that it exerts is to help stop bleeding through platelet stimulation.
Vasopressin is produced in the hypothalamus and stored in the posterior pituitary gland. Factors that stimulate its physiologic release include increased serum osmolarity and hypotension. Its primary actions are therefore to increase body water to control osmolality and to increase blood pressure. A third action that it exerts is to help stop bleeding through platelet stimulation.

 V1 receptors are located on vascular smooth muscle and produce a potent vasoconstrictor effect when stimulated. This occurs only at higher serum levels of vasopressin.
V1 receptors are located on vascular smooth muscle and produce a potent vasoconstrictor effect when stimulated. This occurs only at higher serum levels of vasopressin.

 Filtrate that reaches the collecting duct in the kidney has been diluted by previous tubules. Therefore filtrate that is reabsorbed in the collecting duct is quite dilute and low in sodium.
Filtrate that reaches the collecting duct in the kidney has been diluted by previous tubules. Therefore filtrate that is reabsorbed in the collecting duct is quite dilute and low in sodium.










 Excessive vasoconstriction. The use of vasopressin should be restricted to physicians trained in critical care.
Excessive vasoconstriction. The use of vasopressin should be restricted to physicians trained in critical care. Water intoxication (hyponatremia): This is caused by the antidiuretic action and results in water retention leading to dilution of electrolytes, specifically Na+.
Water intoxication (hyponatremia): This is caused by the antidiuretic action and results in water retention leading to dilution of electrolytes, specifically Na+.





Desmopressin and Bedwetting (Enuresis) in Children
 A Cochrane review in 2002 (47 studies, 3448 children) demonstrated that desmopressin is effective at reducing bedwetting but that the effects do not persist when the drug is stopped. Using a wake-up alarm (the alarm goes off when the child gets wet) was just as effective as desmopressin but had better long-lasting effects.
A Cochrane review in 2002 (47 studies, 3448 children) demonstrated that desmopressin is effective at reducing bedwetting but that the effects do not persist when the drug is stopped. Using a wake-up alarm (the alarm goes off when the child gets wet) was just as effective as desmopressin but had better long-lasting effects.Desmopressin and Surgical Blood Loss in Patients without Bleeding Disorders
 A Cochrane review in 2004 (19 studies, 1387 patients) demonstrated that bleeding with desmopressin was statistically less compared with placebo (mean difference = 241 mL less). However, there was no difference in blood transfusions, and the volume of blood loss reduction is not a clinically significant amount.
A Cochrane review in 2004 (19 studies, 1387 patients) demonstrated that bleeding with desmopressin was statistically less compared with placebo (mean difference = 241 mL less). However, there was no difference in blood transfusions, and the volume of blood loss reduction is not a clinically significant amount. From Latin:
From Latin:








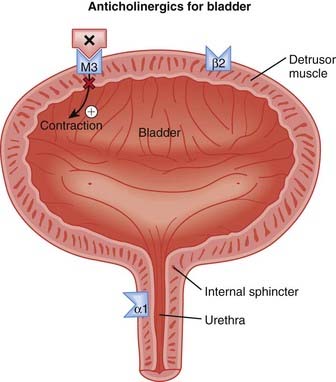
 The human bladder contains all five subtypes (M1 to M5) of muscarinic (M) receptors. Although M2 receptors are more abundant in this region, M3 receptors mediate contraction of the detrusor muscle in the bladder.
The human bladder contains all five subtypes (M1 to M5) of muscarinic (M) receptors. Although M2 receptors are more abundant in this region, M3 receptors mediate contraction of the detrusor muscle in the bladder. Detrusor muscle contraction facilitates emptying of the bladder (micturition). Abnormal contractions of the detrusor, also known as hyperreflexia, can lead to incontinence, specifically urge incontinence. Urge incontinence is defined as the inability to reach the toilet in time following the urge to urinate (Figure 25-5).
Detrusor muscle contraction facilitates emptying of the bladder (micturition). Abnormal contractions of the detrusor, also known as hyperreflexia, can lead to incontinence, specifically urge incontinence. Urge incontinence is defined as the inability to reach the toilet in time following the urge to urinate (Figure 25-5).

 M1 receptors are found in the central nervous system (CNS), on glands (enteric and salivary), and on enteric nerves.
M1 receptors are found in the central nervous system (CNS), on glands (enteric and salivary), and on enteric nerves.

 Most of the anticholinergics for OAB are lipophilic, meaning that they readily cross the blood-brain barrier. The exception is trospium, a polar molecule that does not cross into the brain.
Most of the anticholinergics for OAB are lipophilic, meaning that they readily cross the blood-brain barrier. The exception is trospium, a polar molecule that does not cross into the brain.


 Conditions in which cholinergic blockade would exacerbate an already serious condition, or patients at risk for the following:
Conditions in which cholinergic blockade would exacerbate an already serious condition, or patients at risk for the following:
 Typical anticholinergic side effects:
Typical anticholinergic side effects:
 CNS side effects are a concern with the use of anticholinergics, particularly in seniors and the elderly. Attempts have been made to develop compounds that either have limited penetration into the CNS (trospium) or that bind with lower affinity to M1 receptors (darifenacin), receptors that are thought to play an important role in cognition. However, although there is some evidence of reduced CNS side effects in older, otherwise-healthy patients, there is no definitive proof that these approaches are able to reduce the burden of CNS side effects in patients with OAB.
CNS side effects are a concern with the use of anticholinergics, particularly in seniors and the elderly. Attempts have been made to develop compounds that either have limited penetration into the CNS (trospium) or that bind with lower affinity to M1 receptors (darifenacin), receptors that are thought to play an important role in cognition. However, although there is some evidence of reduced CNS side effects in older, otherwise-healthy patients, there is no definitive proof that these approaches are able to reduce the burden of CNS side effects in patients with OAB.

Anticholinergics for Overactive Bladder
 A 2008 systematic review (83 trials) compared various anticholinergics in treating OAB. All the included anticholinergics (oxybutynin, tolterodine, fesoterodine, propiverine, solifenacin, darifenacin, and trospium) demonstrated efficacy in a variety of outcome measures related to incontinence (incontinence episodes, frequency, urgency) versus placebo.
A 2008 systematic review (83 trials) compared various anticholinergics in treating OAB. All the included anticholinergics (oxybutynin, tolterodine, fesoterodine, propiverine, solifenacin, darifenacin, and trospium) demonstrated efficacy in a variety of outcome measures related to incontinence (incontinence episodes, frequency, urgency) versus placebo.
 Controversy still exists over the relative benefit to risk of anticholinergics for OAB, particularly in seniors and the elderly. Although these agents have demonstrated improved symptoms versus placebo, the clinical significance of these benefits is constantly being weighed against a considerable list of side effects.
Controversy still exists over the relative benefit to risk of anticholinergics for OAB, particularly in seniors and the elderly. Although these agents have demonstrated improved symptoms versus placebo, the clinical significance of these benefits is constantly being weighed against a considerable list of side effects.


 Antagonism of α1 receptors on vascular smooth muscle induces vasodilation and decreases systemic vascular resistance (SVR).
Antagonism of α1 receptors on vascular smooth muscle induces vasodilation and decreases systemic vascular resistance (SVR).




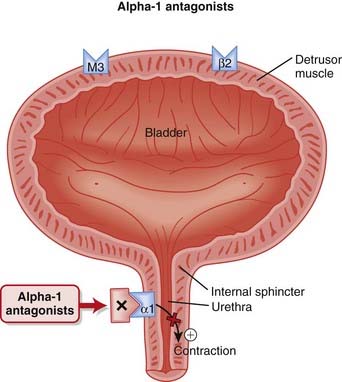
 Prazosin has a short half-life and thus must be given twice daily. Longer-acting agents such as terazosin and doxazosin were developed to provide the advantage of once-daily administration.
Prazosin has a short half-life and thus must be given twice daily. Longer-acting agents such as terazosin and doxazosin were developed to provide the advantage of once-daily administration.

Phentolamine and Phenoxybenzamine Only


 Orthostatic hypotension is common and can be quite significant, usually occurring within 90 minutes of the first dose of the drug. It can lead to dizziness and falls.
Orthostatic hypotension is common and can be quite significant, usually occurring within 90 minutes of the first dose of the drug. It can lead to dizziness and falls.



 Cardiovascular events: Although these agents are typically well tolerated, results of a recent study suggest that they may be associated with an increased risk, summarized in the Advanced section.
Cardiovascular events: Although these agents are typically well tolerated, results of a recent study suggest that they may be associated with an increased risk, summarized in the Advanced section. Compensatory sympathetic nervous system responses such as renin release and tachycardia are significant in the short term. However, in many cases these reflex responses subside, leaving a vasodilatory effect that predominates in the long term.
Compensatory sympathetic nervous system responses such as renin release and tachycardia are significant in the short term. However, in many cases these reflex responses subside, leaving a vasodilatory effect that predominates in the long term.
 The use of α antagonists in hypertension was dealt a severe blow after treatment in the doxazosin arm of the landmark ALLHAT study had to be halted prematurely after observations of a significantly increased incidence of congestive heart failure, angina, and stroke in subjects treated with this α antagonist.
The use of α antagonists in hypertension was dealt a severe blow after treatment in the doxazosin arm of the landmark ALLHAT study had to be halted prematurely after observations of a significantly increased incidence of congestive heart failure, angina, and stroke in subjects treated with this α antagonist.Tamsulosin versus Placebo for Treatment of Benign Prostatic Hypertrophy
 A 2002 Cochrane review (14 studies, N = 4122 participants) assessed tamsulosin for treatment of BPH. Tamsulosin improved symptoms and peak urine flow relative to placebo and was as effective as nonselective α antagonists. Men receiving a low dose of tamsulosin (0.2 mg) were less likely to discontinue treatment compared with men receiving terazosin. The adverse effects of terazosin increased markedly with increasing dose, compared with placebo. The most common adverse effects associated with tamsulosin were dizziness, rhinitis, and abnormal ejaculation.
A 2002 Cochrane review (14 studies, N = 4122 participants) assessed tamsulosin for treatment of BPH. Tamsulosin improved symptoms and peak urine flow relative to placebo and was as effective as nonselective α antagonists. Men receiving a low dose of tamsulosin (0.2 mg) were less likely to discontinue treatment compared with men receiving terazosin. The adverse effects of terazosin increased markedly with increasing dose, compared with placebo. The most common adverse effects associated with tamsulosin were dizziness, rhinitis, and abnormal ejaculation.Terazosin versus Other α-Blockers for Treatment of Benign Prostatic Obstruction
 A 2000 Cochrane review (17 studies, N = 5151 participants) assessed terazosin versus placebo (10 trials), α-blockers (seven trials), or the 5α-reductase inhibitor finasteride (one trial) for benign prostatic obstruction. Terazosin improved symptom scores and urine flow rates more than placebo or finasteride and similarly to other α antagonists. The proportion of men discontinuing treatment was higher than the proportion of men receiving other α antagonists and was comparable to what was seen with placebo and finasteride. Adverse effects occurring more often than with placebo included dizziness, asthenia, headache, and postural hypotension.
A 2000 Cochrane review (17 studies, N = 5151 participants) assessed terazosin versus placebo (10 trials), α-blockers (seven trials), or the 5α-reductase inhibitor finasteride (one trial) for benign prostatic obstruction. Terazosin improved symptom scores and urine flow rates more than placebo or finasteride and similarly to other α antagonists. The proportion of men discontinuing treatment was higher than the proportion of men receiving other α antagonists and was comparable to what was seen with placebo and finasteride. Adverse effects occurring more often than with placebo included dizziness, asthenia, headache, and postural hypotension. Phenoxybenzamine is an irreversible antagonist of α1 receptors, whereas other α1 antagonists are reversible.
Phenoxybenzamine is an irreversible antagonist of α1 receptors, whereas other α1 antagonists are reversible.



 The 5α-reductase enzyme is the last step in the synthesis of DHT, the active form of testosterone (Figure 25-7).
The 5α-reductase enzyme is the last step in the synthesis of DHT, the active form of testosterone (Figure 25-7).


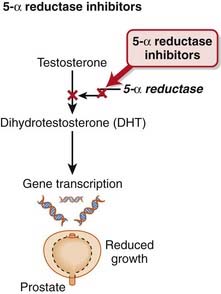
 No drug interactions of clinical significance have been detected with finasteride. Dutasteride is metabolized by CYP3A4; thus the potential for drug interactions with inhibitors or inducers of this isozyme does exist.
No drug interactions of clinical significance have been detected with finasteride. Dutasteride is metabolized by CYP3A4; thus the potential for drug interactions with inhibitors or inducers of this isozyme does exist.

 Pregnancy: Women of childbearing age should not use 5α-reductase inhibitors. They are strictly contraindicated in pregnancy. Testosterone is essential for development of genitalia in males; therefore these agents can lead to abnormal development. Pregnant women are advised not only to avoid taking the medication but also to avoid even handling the pills.
Pregnancy: Women of childbearing age should not use 5α-reductase inhibitors. They are strictly contraindicated in pregnancy. Testosterone is essential for development of genitalia in males; therefore these agents can lead to abnormal development. Pregnant women are advised not only to avoid taking the medication but also to avoid even handling the pills. Sexual dysfunction: Decreased libido, ejaculation disorders, and erectile dysfunction are caused by the antiandrogenic effects.
Sexual dysfunction: Decreased libido, ejaculation disorders, and erectile dysfunction are caused by the antiandrogenic effects.Finasteride versus Placebo for Prevention of Prostate Cancer
 A 2008 Cochrane review (nine trials, N = 34,410 males) examined the effectiveness and harms of 5α-reductase inhibitors in preventing prostate cancer. Finasteride reduced the risk of prostate cancer by 2.9% (incidence of 6.3% in finasteride versus 9.2% in placebo). Impaired erectile function or endocrine effects were more common with finasteride than with placebo. However, the risk of high-grade disease may be increased with finasteride therapy. The authors recommended that future studies examine the impact of 5α-reductase inhibitors on mortality and also further examine the risk associated with development of high-grade cancers.
A 2008 Cochrane review (nine trials, N = 34,410 males) examined the effectiveness and harms of 5α-reductase inhibitors in preventing prostate cancer. Finasteride reduced the risk of prostate cancer by 2.9% (incidence of 6.3% in finasteride versus 9.2% in placebo). Impaired erectile function or endocrine effects were more common with finasteride than with placebo. However, the risk of high-grade disease may be increased with finasteride therapy. The authors recommended that future studies examine the impact of 5α-reductase inhibitors on mortality and also further examine the risk associated with development of high-grade cancers. Because of their lowering effects on prostate serum antigen (PSA), there is concern that 5α-reductase inhibitors may mask elevations in PSA associated with a malignancy of the prostate. This could lead to a delay in the diagnosis of prostate cancer because of false-negative screening test results.
Because of their lowering effects on prostate serum antigen (PSA), there is concern that 5α-reductase inhibitors may mask elevations in PSA associated with a malignancy of the prostate. This could lead to a delay in the diagnosis of prostate cancer because of false-negative screening test results.



[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 25 Renal and Genitourinary Systems
Prototype and Common Drugs
MOA (Mechanism of Action)
Pharmacokinetics
Important Notes
Evidence
As First-Line Agents in Hypertension
A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year. Thiazides (19 trials) reduced mortality (relative risk [RR] 0.89), stroke (RR 0.63), and coronary heart disease (RR 0.84) versus placebo.Diuretics result in increased urine production and also lower blood pressure.
Generally speaking, diuretics manipulate a solute (usually Na+), and water passively follows:
Diuretics versus Placebo or Other Interventions for Treatment of Heart Failure
A 2006 Cochrane review (14 trials, N = 525 participants) compared diuretics with placebo (seven trials) and other interventions (seven trials) for treatment of heart failure. The diuretics included were a mixture of loop diuretics, potassium-sparing diuretics, and thiazides. Based on three trials (N = 202 participants), diuretic treatment reduced the odds of death versus placebo (OR 0.24), and in two trials (N = 169 participants) it reduced the odds of admission because of worsening heart failure (OR 0.07). Diuretics improved exercise capacity in four trials (N = 91 participants) versus active controls.Blood-Pressure–Lowering Effect versus Placebo or No Treatment
A 2009 Cochrane review (nine trials, N = 460 participants) compared the blood-pressure–lowering efficacy, tolerability, and biochemical effects of a variety of loop diuretics versus placebo or no treatment. The authors found a mean reduction (systolic/diastolic blood pressure) of −7.9/−4.4 mmHg and no differences between drugs in this class with respect to blood-pressure–lowering efficacy. This is considered to be a modest antihypertensive effect. Withdrawals because of adverse effects and biochemical changes were not significantly different from control. Diuretics that exert their effects by acting on the renal tubules require functioning kidneys to induce diuresis. Patients with advanced renal dysfunction will not respond to these diuretics.