[level-membership-for-basic-science-category]
Chapter 11 Cardiology
Angiotensin-Converting Enzyme Inhibitors (ACEIs)


MOA (Mechanism of Action)

 Through inhibition of ACE with an ACEI, the following effects occur:
Through inhibition of ACE with an ACEI, the following effects occur:
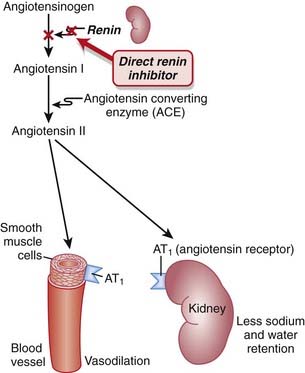
 Net result: Because angiotensin II levels are lower and bradykinin levels are higher, there is more vasodilation; SVR (systemic vascular resistance) and afterload are lowered. Because aldosterone levels are lower, less Na and water are reabsorbed in the kidney; therefore preload is reduced (Figure 11-1).
Net result: Because angiotensin II levels are lower and bradykinin levels are higher, there is more vasodilation; SVR (systemic vascular resistance) and afterload are lowered. Because aldosterone levels are lower, less Na and water are reabsorbed in the kidney; therefore preload is reduced (Figure 11-1).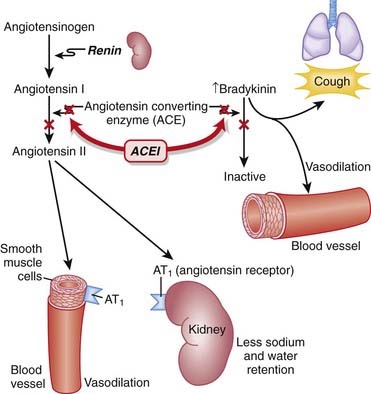
Pharmacokinetics





Contraindications


Side Effects





Important Notes
 The renin-angiotensin system (RAS) plays an important role in the body’s compensation for a failing heart. Activation of the sympathetic nervous system (SNS) leads to the release of renin, which in turn increases vascular tone and sodium and water retention.
The renin-angiotensin system (RAS) plays an important role in the body’s compensation for a failing heart. Activation of the sympathetic nervous system (SNS) leads to the release of renin, which in turn increases vascular tone and sodium and water retention.
Advanced
 In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.
In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.
Evidence
Hypertension
 A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year in patients with hypertension. ACEIs (three trials) reduced mortality (relative risk [RR] 0.83), stroke (RR 0.65), coronary heart disease (RR 0.81), and cardiovascular events (RR 0.76).
A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year in patients with hypertension. ACEIs (three trials) reduced mortality (relative risk [RR] 0.83), stroke (RR 0.65), coronary heart disease (RR 0.81), and cardiovascular events (RR 0.76).



Angiotensin Receptor Blockers (ARBs)


MOA (Mechanism of Action)
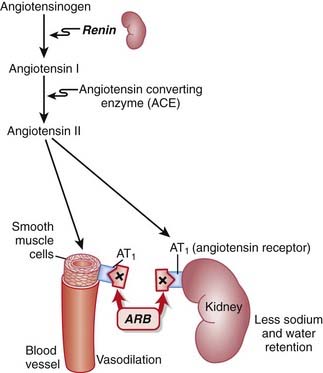
 ARBs are antagonists of the angiotensin-1 (AT1) receptor. Therefore they block the actions of angiotensin II.
ARBs are antagonists of the angiotensin-1 (AT1) receptor. Therefore they block the actions of angiotensin II.




Pharmacokinetics











Important Notes
 Perhaps because of the lack of increased bradykinin levels, ARBs are not typically associated with the side effect of cough, which can be a significant limitation to the use of ACEIs.
Perhaps because of the lack of increased bradykinin levels, ARBs are not typically associated with the side effect of cough, which can be a significant limitation to the use of ACEIs.
Advanced
 In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.
In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.





Direct Renin Inhibitors

MOA (Mechanism of Action)
 Renin is an enzyme released from the kidneys that converts angiotensinogen to angiotensin I. It is considered to be the rate-limiting step in the eventual formation of angiotensin II.
Renin is an enzyme released from the kidneys that converts angiotensinogen to angiotensin I. It is considered to be the rate-limiting step in the eventual formation of angiotensin II.












Evidence
Blood-Pressure Lowering Efficacy versus Placebo
 A 2008 Cochrane review (six trials, 3694 participants) compared the blood-pressure–lowering efficacy of renin inhibitors versus placebo in primary HTN. The authors found that aliskiren elicits a dose-dependent reduction in both systolic and diastolic pressure similar to that seen with ACEIs or ARBs. In the included trials, aliskiren did not increase withdrawals due to adverse events versus placebo.
A 2008 Cochrane review (six trials, 3694 participants) compared the blood-pressure–lowering efficacy of renin inhibitors versus placebo in primary HTN. The authors found that aliskiren elicits a dose-dependent reduction in both systolic and diastolic pressure similar to that seen with ACEIs or ARBs. In the included trials, aliskiren did not increase withdrawals due to adverse events versus placebo.FYI Notes
 Renin was first identified in 1898, when it was extracted from kidneys and discovered to have pressor properties. It would be another 40 years before it was determined that renin was an enzyme that catalyzed the formation of a pressor substance (angiotensin II), rather than being the pressor itself.
Renin was first identified in 1898, when it was extracted from kidneys and discovered to have pressor properties. It would be another 40 years before it was determined that renin was an enzyme that catalyzed the formation of a pressor substance (angiotensin II), rather than being the pressor itself.

Sodium Channel Blockers (Class I Antiarrhythmics)
Description
Na channel blockers are Vaughan Williams class I antiarrhythmics. There are three subclasses: Ia, Ib, and Ic. The use of Na channel blockers as local anesthetics is discussed in the discussion of local anesthetics in Chapter 21.



MOA (Mechanism of Action)
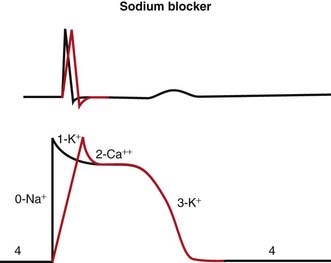
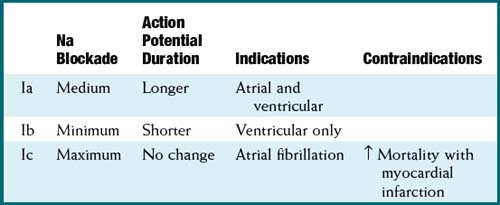
 Na channels are blocked, so Na ion movement during phase 0 of the action potential is inhibited. The result is a “slow” phase 0, which results in a wider (and slower) QRS wave on the electrocardiogram (ECG). The net result is slower conduction (Figure 11-4).
Na channels are blocked, so Na ion movement during phase 0 of the action potential is inhibited. The result is a “slow” phase 0, which results in a wider (and slower) QRS wave on the electrocardiogram (ECG). The net result is slower conduction (Figure 11-4).







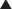



Pharmacokinetics



Side Effects




Evidence
 Atrial fibrillation and prevention of recurrence: A Cochrane review in 2007 (45 studies, 12,559 patients) evaluated the efficacy and safety of multiple different antiarrhythmics in patients who had previously experienced atrial fibrillation (a very common arrhythmia). Class Ia antiarrhythmics were associated with increased mortality compared with controls (odds ratio [OR] 2.39; number needed to harm [NNH] 109). Class Ia and Ic were associated with reduced occurrences of atrial fibrillation (OR 0.19 to 0.6). There were many withdrawals from treatment because of side effects for all antiarrhythmics (NNH 17 to 36).
Atrial fibrillation and prevention of recurrence: A Cochrane review in 2007 (45 studies, 12,559 patients) evaluated the efficacy and safety of multiple different antiarrhythmics in patients who had previously experienced atrial fibrillation (a very common arrhythmia). Class Ia antiarrhythmics were associated with increased mortality compared with controls (odds ratio [OR] 2.39; number needed to harm [NNH] 109). Class Ia and Ic were associated with reduced occurrences of atrial fibrillation (OR 0.19 to 0.6). There were many withdrawals from treatment because of side effects for all antiarrhythmics (NNH 17 to 36).

β Antagonists (β-Blockers)
Prototype and Common Drugs



MOA (Mechanism of Action)
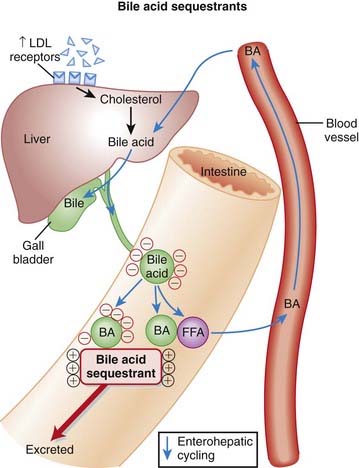
To understand β-blockers, you must understand the effects of the adrenergic system and which effects are mediated via β receptors. β-Blockers competitively antagonize the action of catecholamines at β receptors. There are many cardiac and noncardiac consequences of β-blockade. More details on the autonomic nervous system are described in Chapter 3.

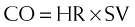

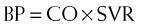




Tachycardia and Arrhythmia
The properties of β-blockers that make them antitachycardics include the following:
 Catecholamine β1 stimulation results in an increase in the slow Na+ current (If) of the action potential phase 4 in the SA node. This results in a faster rising (and shorter) phase 4, a shorter time to the next heartbeat, and thus a faster HR. β-Blockers will oppose this action, slowing the SA pacemaker rate.
Catecholamine β1 stimulation results in an increase in the slow Na+ current (If) of the action potential phase 4 in the SA node. This results in a faster rising (and shorter) phase 4, a shorter time to the next heartbeat, and thus a faster HR. β-Blockers will oppose this action, slowing the SA pacemaker rate.
Myocardial Ischemia and Infarction




Pharmacokinetics









Contraindications














Important Notes





Evidence

After Myocardial Infarction
 Evidence for the role of β-blockers in secondary prevention after an MI comes from several trials and was summarized in a 1999 systematic review (82 trials, N = 54,234 patients). There was a 23% reduction in the odds of death in long-term trials but only a 4% reduction in short-term trials. The review found that the number needed to treat (NNT) to avoid a fatality over the course of 2 years is 42. The greatest amount of evidence available was for propranolol, timolol, and metoprolol.
Evidence for the role of β-blockers in secondary prevention after an MI comes from several trials and was summarized in a 1999 systematic review (82 trials, N = 54,234 patients). There was a 23% reduction in the odds of death in long-term trials but only a 4% reduction in short-term trials. The review found that the number needed to treat (NNT) to avoid a fatality over the course of 2 years is 42. The greatest amount of evidence available was for propranolol, timolol, and metoprolol.Hypertension and Associated Stroke and Coronary Artery Disease
 A Cochrane review in 2007 (13 studies, N = 91,561 patients) compared β-blockers with other agents for HTN. Atenolol was the β-blocker most frequently used. The authors found that β-blockers had only weak effects in reducing stroke and no effect on coronary heart disease versus placebo. There was also a trend toward worse outcomes when compared with calcium channel blockers (CCBs), RAS inhibitors, and thiazides, prompting the authors to suggest that β-blockers should not be considered as first-line agents for HTN.
A Cochrane review in 2007 (13 studies, N = 91,561 patients) compared β-blockers with other agents for HTN. Atenolol was the β-blocker most frequently used. The authors found that β-blockers had only weak effects in reducing stroke and no effect on coronary heart disease versus placebo. There was also a trend toward worse outcomes when compared with calcium channel blockers (CCBs), RAS inhibitors, and thiazides, prompting the authors to suggest that β-blockers should not be considered as first-line agents for HTN.Obstructive Airway Disease (Asthma and Chronic Obstructive Pulmonary Disease)
 A Cochrane review in 2002 (29 studies, N = 381 patients) examined the impact of single-dose or short-term selective β1-blockers in patients with mild to moderate obstructive airway disease. There were no differences in pulmonary flow measurements compared with placebo except for a small decrease in FEV1 after the first treatment—an effect that disappeared with subsequent doses.
A Cochrane review in 2002 (29 studies, N = 381 patients) examined the impact of single-dose or short-term selective β1-blockers in patients with mild to moderate obstructive airway disease. There were no differences in pulmonary flow measurements compared with placebo except for a small decrease in FEV1 after the first treatment—an effect that disappeared with subsequent doses.

Potassium Channel Blockers (Class III Antiarrhythmics)


MOA (Mechanism of Action)
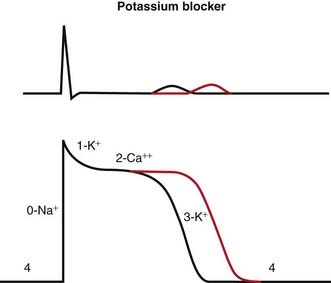
 Blocking K channels in phase 3 of the action potential slows the efflux of K back out of the myocyte, which slows the rate at which the cell repolarizes and therefore lengthens the plateau phase of the action potential. This increases the refractory period of atrial, ventricular, and Purkinje cells. This also increases the QT interval on the ECG (Figure 11-5).
Blocking K channels in phase 3 of the action potential slows the efflux of K back out of the myocyte, which slows the rate at which the cell repolarizes and therefore lengthens the plateau phase of the action potential. This increases the refractory period of atrial, ventricular, and Purkinje cells. This also increases the QT interval on the ECG (Figure 11-5).
Pharmacokinetics

 Drug interactions:
Drug interactions:





Side Effects



Evidence
 Atrial fibrillation (AF) and multiple outcomes: A Cochrane review in 2007 (45 studies, 12,559 patients) evaluated the efficacy and safety of multiple different antiarrhythmics in patients who had previously experienced atrial fibrillation (the most common arrhythmia).
Atrial fibrillation (AF) and multiple outcomes: A Cochrane review in 2007 (45 studies, 12,559 patients) evaluated the efficacy and safety of multiple different antiarrhythmics in patients who had previously experienced atrial fibrillation (the most common arrhythmia).

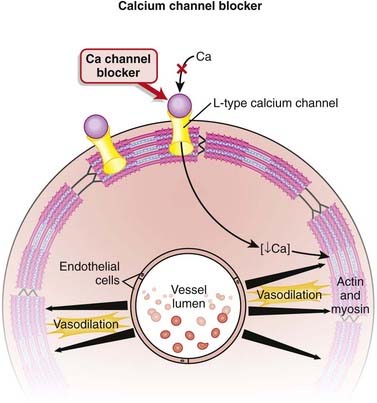
Calcium Channel Blockers



MOA (Mechanism of Action)
Hypertension
 Calcium channels are designated as L-, T-, P/Q-, N-, and R-type, depending on kinetics and receptor specificity. L means long duration, and T means transient.
Calcium channels are designated as L-, T-, P/Q-, N-, and R-type, depending on kinetics and receptor specificity. L means long duration, and T means transient.




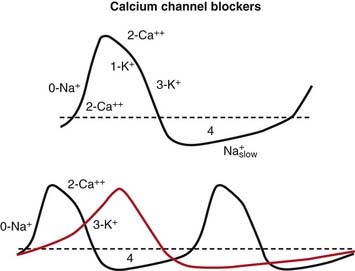
Tachycardia and Arrhythmia
The non-DHPs (but not the DPHs) are also class IV antiarrhythmics.
Blocking Ca+2 channels in phase 0 of the action potential lengthens the depolarizing current in SA and AV nodal cells. This results in more time before the next action potential. In the SA node, the result is a slower pacemaker. In the AV node, the result is a longer refractory period (Figure 11-7).
Pharmacokinetics












Contraindications
Nondihydropyridines
 Wolff-Parkinson-White (WPW) syndrome: In WPW the accessory pathway (bundle of Kent) is not blocked by Ca+2 channel blockers, but the AV node is. Therefore the use of Ca+2 channel blockers will promote conduction through the accessory pathway. Because the accessory pathway does not have a conduction delay as the AV node does, conduction through the accessory pathway can result in very high ventricular rates in the presence of atrial fibrillation or atrial flutter.
Wolff-Parkinson-White (WPW) syndrome: In WPW the accessory pathway (bundle of Kent) is not blocked by Ca+2 channel blockers, but the AV node is. Therefore the use of Ca+2 channel blockers will promote conduction through the accessory pathway. Because the accessory pathway does not have a conduction delay as the AV node does, conduction through the accessory pathway can result in very high ventricular rates in the presence of atrial fibrillation or atrial flutter.







Important Notes
 Vasodilation results in lower BP, and low BP often induces a baroreceptor-mediated reflex tachycardia. An important clinical consequence of DHPs is that they can indirectly induce tachycardia, which can be very dangerous in conditions in which tachycardia is contraindicated (e.g., coronary artery disease).
Vasodilation results in lower BP, and low BP often induces a baroreceptor-mediated reflex tachycardia. An important clinical consequence of DHPs is that they can indirectly induce tachycardia, which can be very dangerous in conditions in which tachycardia is contraindicated (e.g., coronary artery disease).

Evidence
As First-Line Agents in Hypertension
 A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year. Only one randomized controlled trial (RCT) was found that included a CCB, and in this trial it reduced stroke (RR 0.58) but not coronary heart disease or mortality. Thiazides (19 trials) reduced mortality (RR 0.89), stroke (RR 0.63), and coronary heart disease (RR 0.84).
A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year. Only one randomized controlled trial (RCT) was found that included a CCB, and in this trial it reduced stroke (RR 0.58) but not coronary heart disease or mortality. Thiazides (19 trials) reduced mortality (RR 0.89), stroke (RR 0.63), and coronary heart disease (RR 0.84).For Aneurysmal Subarachnoid Hemorrhage
 A 2007 Cochrane review (16 studies, N = 3361 patients) examined whether DHP calcium antagonists alone or combined with magnesium sulfate (three studies) improved outcomes in patients with subarachnoid hemorrhage. Overall, calcium antagonists reduced the risk of poor outcomes (death or disability), with an NNT of 19 (1 to 51). When separated by individual calcium antagonists, only oral nimodipine was statistically significant versus control. Calcium antagonists also reduced the risk of secondary ischemia.
A 2007 Cochrane review (16 studies, N = 3361 patients) examined whether DHP calcium antagonists alone or combined with magnesium sulfate (three studies) improved outcomes in patients with subarachnoid hemorrhage. Overall, calcium antagonists reduced the risk of poor outcomes (death or disability), with an NNT of 19 (1 to 51). When separated by individual calcium antagonists, only oral nimodipine was statistically significant versus control. Calcium antagonists also reduced the risk of secondary ischemia.FYI Notes
 Patients have been known to take advantage of the interaction between felodipine and grapefruit juice to save themselves a few dollars on the drug cost. They will reduce their dose of felodipine by consuming grapefruit juice on a consistent basis. The bioflavonoids are found in the peel, however, so patients who grind their own juice may find they are not getting adequate drug levels if they do not include the peel.
Patients have been known to take advantage of the interaction between felodipine and grapefruit juice to save themselves a few dollars on the drug cost. They will reduce their dose of felodipine by consuming grapefruit juice on a consistent basis. The bioflavonoids are found in the peel, however, so patients who grind their own juice may find they are not getting adequate drug levels if they do not include the peel.
Anticholinergics


MOA (Mechanism of Action)
 Anticholinergics block the activity of acetylcholine (ACh) of the parasympathetic system at muscarinic receptors (M).
Anticholinergics block the activity of acetylcholine (ACh) of the parasympathetic system at muscarinic receptors (M).


| Heart | Increased heart rate |
| Airways | Bronchodilation |
| Secretions | Reduced |
| Pupils | Dilated |
| Urinary function | Decreased |
| Gastrointestinal motility | Decreased |
| Central nervous system | Increased temperature, confusion |













FYI Notes
 One chemical used in a form of chemical warfare is an anticholinesterase. An anticholinesterase blocks cholinesterase, which is an enzyme that degrades ACh. The result is too much ACh, which mimics an overactive parasympathetic system and can even cause paralysis from overstimulation of the nicotinic receptor in the neuromuscular junction. Atropine is the antidote.
One chemical used in a form of chemical warfare is an anticholinesterase. An anticholinesterase blocks cholinesterase, which is an enzyme that degrades ACh. The result is too much ACh, which mimics an overactive parasympathetic system and can even cause paralysis from overstimulation of the nicotinic receptor in the neuromuscular junction. Atropine is the antidote.




Adenosine
MOA (Mechanism of Action)




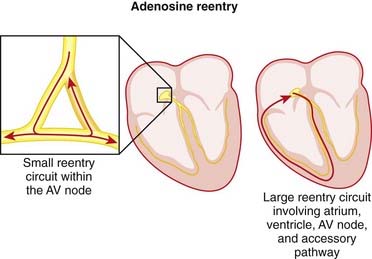
Pharmacokinetics









Important Notes





Evidence
Adenosine versus Verapamil for Supraventricular Tachycardia
 A Cochrane review in 2006 (eight trials, N = 577 patients) found that both drugs were 90% effective in treating SVTs and that there were no differences in efficacy; however, adenosine converted heart rhythm to sinus rhythm a little bit faster than verapamil did. Adenosine caused unpleasant side effects in about 10% of patients, and verapamil caused hypotension in 2% of patients.
A Cochrane review in 2006 (eight trials, N = 577 patients) found that both drugs were 90% effective in treating SVTs and that there were no differences in efficacy; however, adenosine converted heart rhythm to sinus rhythm a little bit faster than verapamil did. Adenosine caused unpleasant side effects in about 10% of patients, and verapamil caused hypotension in 2% of patients.


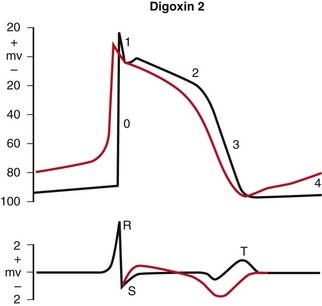
Digoxin


MOA (Mechanism of Action)
Digoxin has two mechanisms of action:
 Through the action of Na/K/ATP ion pump blockade (Figure 11-9), the following sequence of ionic events occurs:
Through the action of Na/K/ATP ion pump blockade (Figure 11-9), the following sequence of ionic events occurs:
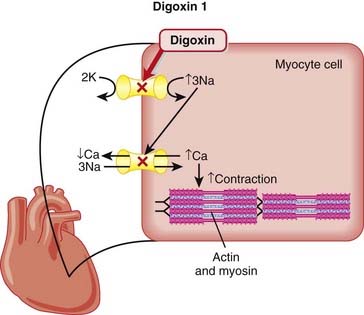
Pharmacokinetics
 The therapeutic index is very low. The therapeutic range for digoxin is 0.5 to 2.5 nmol/L (in the blood). The probability of toxicity is significant when the level is > 2.6, and toxicity is virtually guaranteed when the level is >3. Contrast this to acetaminophen (Tylenol), in which the toxic oral dose is 10 times the normal dose.
The therapeutic index is very low. The therapeutic range for digoxin is 0.5 to 2.5 nmol/L (in the blood). The probability of toxicity is significant when the level is > 2.6, and toxicity is virtually guaranteed when the level is >3. Contrast this to acetaminophen (Tylenol), in which the toxic oral dose is 10 times the normal dose.









Side Effects (Toxicity)












Inhibitors of Cholesterol Synthesis: Statins
Description
Statins are designed to reduce low-density–lipoprotein (LDL) cholesterol and raise HDL cholesterol.


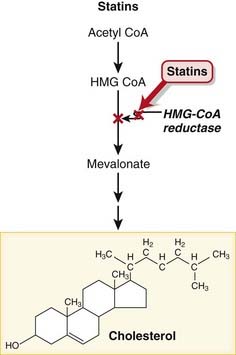
MOA (Mechanism of Action)
 These drugs are HMG-CoA reductase inhibitors. HMG-CoA reductase mediates the first step in the biosynthesis of cholesterol.
These drugs are HMG-CoA reductase inhibitors. HMG-CoA reductase mediates the first step in the biosynthesis of cholesterol. Reduced biosynthesis of cholesterol causes an increase in the number of LDL receptors, through the actions of sterol regulatory element binding proteins (SREBPs). Additional LDL receptors increase the catabolic rate of LDL, and the liver’s extraction of LDL precursors. These actions reduce the plasma pool of LDL (Figure 11-11).
Reduced biosynthesis of cholesterol causes an increase in the number of LDL receptors, through the actions of sterol regulatory element binding proteins (SREBPs). Additional LDL receptors increase the catabolic rate of LDL, and the liver’s extraction of LDL precursors. These actions reduce the plasma pool of LDL (Figure 11-11).




Side Effects


Rare, Serious Side Effects


Important Notes
 Statins can be used in combination with other agents such as bile acid sequestrants or inhibitors of cholesterol absorption. Fibrates and niacin (other drugs used to achieve a more favorable lipid profile) should be used with caution, as both can interact with statins.
Statins can be used in combination with other agents such as bile acid sequestrants or inhibitors of cholesterol absorption. Fibrates and niacin (other drugs used to achieve a more favorable lipid profile) should be used with caution, as both can interact with statins.
Advanced
 Statins are being studied for their potential role in neuroprotection, with indications such as Alzheimer’s disease, Parkinson’s disease, stroke, and multiple sclerosis (MS). Statins appear to activate neuroprotective pathways and also may play a role in neurogenesis. The mechanisms of all these effects are still being worked out; however, some researchers believe that the use of statins may become even more widespread than it is today.
Statins are being studied for their potential role in neuroprotection, with indications such as Alzheimer’s disease, Parkinson’s disease, stroke, and multiple sclerosis (MS). Statins appear to activate neuroprotective pathways and also may play a role in neurogenesis. The mechanisms of all these effects are still being worked out; however, some researchers believe that the use of statins may become even more widespread than it is today.
Evidence
Patients with Cardiac Risk Factors But Without Cardiac Disease (Primary Prevention)
 A 2009 systematic review (10 studies, N = 70,388 participants) examined the use of statins in people without established cardiovascular disease but with cardiovascular risk factors. The included studies had a mean follow-up of 4.1 years. Statins reduced the risk of all-cause mortality (OR 0.88), major coronary events (OR 0.70), and major cerebrovascular events (OR 0.81).
A 2009 systematic review (10 studies, N = 70,388 participants) examined the use of statins in people without established cardiovascular disease but with cardiovascular risk factors. The included studies had a mean follow-up of 4.1 years. Statins reduced the risk of all-cause mortality (OR 0.88), major coronary events (OR 0.70), and major cerebrovascular events (OR 0.81).Prevention of Stroke Recurrence (Secondary Prevention)
 A 2009 Cochrane review (eight studies, N ≈10,000 participants) examined the effect of altering serum lipids for prevention of subsequent cardiovascular disease and stroke in patients with a history of stroke. Statins marginally reduced subsequent cerebrovascular events (OR 0.88) but not all-cause mortality or sudden deaths. Three trials showed a subsequent reduction in serious vascular events (OR 0.74).
A 2009 Cochrane review (eight studies, N ≈10,000 participants) examined the effect of altering serum lipids for prevention of subsequent cardiovascular disease and stroke in patients with a history of stroke. Statins marginally reduced subsequent cerebrovascular events (OR 0.88) but not all-cause mortality or sudden deaths. Three trials showed a subsequent reduction in serious vascular events (OR 0.74).

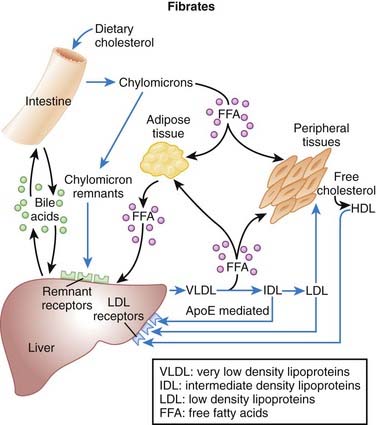
Fibrates


MOA (Mechanism of Action) (Figure 11-12)
 Transcription factors are proteins that bind DNA and increase or decrease transcription (production of RNA and thus proteins) of selected genes.
Transcription factors are proteins that bind DNA and increase or decrease transcription (production of RNA and thus proteins) of selected genes.

Pharmacokinetics


Drug Interactions
 Gemfibrozil competes for the same glucuronosyltransferases that metabolize statins. This is important because statins are used for the same indication (high cholesterol) as fibrates are, and both drugs can cause muscle breakdown and muscle pain (rhabdomyositis). Coadministration can result in increased levels of both drugs and an increase in the risk for this side effect.
Gemfibrozil competes for the same glucuronosyltransferases that metabolize statins. This is important because statins are used for the same indication (high cholesterol) as fibrates are, and both drugs can cause muscle breakdown and muscle pain (rhabdomyositis). Coadministration can result in increased levels of both drugs and an increase in the risk for this side effect.








Important Notes





Evidence

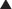



Bile Acid Sequestrants


MOA (Mechanism of Action)





Pharmacokinetics








Side Effects
 GI: constipation is the most common side effect. Others can include bloating, gas, nausea, and upper abdominal pain.
GI: constipation is the most common side effect. Others can include bloating, gas, nausea, and upper abdominal pain.Serious Side Effects
 Hyperchloremic acidosis (rare): The bile acid binding resins are chloride salts and in susceptible individuals (typically children) may increase chloride levels enough to induce acidosis. Chloride and bicarbonate are the primary anions in the blood, and if one goes up, the other usually goes down to maintain electrical neutrality. Dropping bicarbonate results in metabolic acidosis.
Hyperchloremic acidosis (rare): The bile acid binding resins are chloride salts and in susceptible individuals (typically children) may increase chloride levels enough to induce acidosis. Chloride and bicarbonate are the primary anions in the blood, and if one goes up, the other usually goes down to maintain electrical neutrality. Dropping bicarbonate results in metabolic acidosis.Important Notes
 To compensate for the actions of the bile acid binding resins, upregulation occurs in the activity of the HMG-CoA reductase enzyme, which is the rate-limiting enzyme in the production of cholesterol. This is the rationale for using the statins, which are HMG-CoA reductase inhibitors, in combination with the bile acid binding resins.
To compensate for the actions of the bile acid binding resins, upregulation occurs in the activity of the HMG-CoA reductase enzyme, which is the rate-limiting enzyme in the production of cholesterol. This is the rationale for using the statins, which are HMG-CoA reductase inhibitors, in combination with the bile acid binding resins.




Inotropes and Pressors






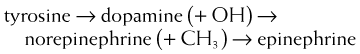


MOA (Mechanism of Action)
 These drugs all exert their actions via the autonomic nervous system α and β receptors. In addition to these receptors, dopamine (and only dopamine) also acts via dopaminergic (DA) receptors.
These drugs all exert their actions via the autonomic nervous system α and β receptors. In addition to these receptors, dopamine (and only dopamine) also acts via dopaminergic (DA) receptors.




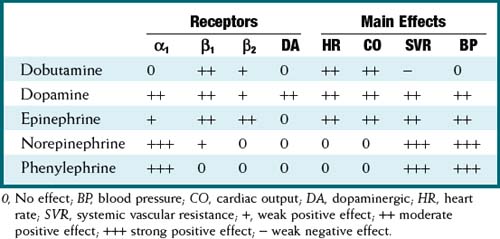
Pharmacokinetics







Important Notes
 A thorough assessment to determine the specific cause(s) of hypotension or shock must always be performed, and the primary cause of the problem must be corrected. For example, a patient who is bleeding and has hypotension has a preload (volume) problem, and therefore the preload should be corrected before the contractility or afterload is manipulated.
A thorough assessment to determine the specific cause(s) of hypotension or shock must always be performed, and the primary cause of the problem must be corrected. For example, a patient who is bleeding and has hypotension has a preload (volume) problem, and therefore the preload should be corrected before the contractility or afterload is manipulated.




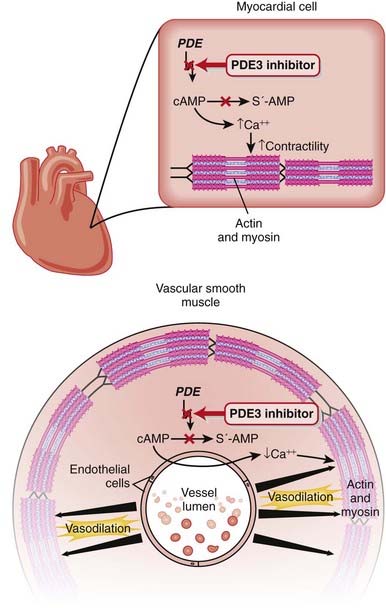
Phosphodiesterase-3 Inhibitors


MOA (Mechanism of Action) (Figure 11-14)




 PDE3 inhibitors also have intracellular actions that should theoretically improve diastolic function:
PDE3 inhibitors also have intracellular actions that should theoretically improve diastolic function:

Pharmacokinetics
 The half-life is about 6 hours. This is important because it is administered as an intravenous infusion. Most cardiovascular drugs (e.g., dopamine, nitroglycerin [NTG]) that are administered as intravenous infusions (to optimize BP and CO) have half-lives of 3 to 5 minutes. Therefore, milrinone is less titratable than these other drugs, and the drug levels can become unexpectedly high.
The half-life is about 6 hours. This is important because it is administered as an intravenous infusion. Most cardiovascular drugs (e.g., dopamine, nitroglycerin [NTG]) that are administered as intravenous infusions (to optimize BP and CO) have half-lives of 3 to 5 minutes. Therefore, milrinone is less titratable than these other drugs, and the drug levels can become unexpectedly high.


Side Effects
 Hypotension secondary to vasodilation: This is a very important side effect. Because of the relatively long half-life of milrinone and the fact that it is administered as an infusion, the potential for steadily increasing serum levels of drug over a period of many hours after the start of the infusion, especially in patients with renal dysfunction, results in hypotension being a frequent side effect. Hypotension induced by PDE3 infusions is treated with vasoconstrictors (usually norepinephrine) and by decreasing the infusion rate. With the longer half-life, hypotension usually persists for many hours even after the infusion is decreased.
Hypotension secondary to vasodilation: This is a very important side effect. Because of the relatively long half-life of milrinone and the fact that it is administered as an infusion, the potential for steadily increasing serum levels of drug over a period of many hours after the start of the infusion, especially in patients with renal dysfunction, results in hypotension being a frequent side effect. Hypotension induced by PDE3 infusions is treated with vasoconstrictors (usually norepinephrine) and by decreasing the infusion rate. With the longer half-life, hypotension usually persists for many hours even after the infusion is decreased.
Important Notes
 Milrinone is not used for chronic heart failure. It is used only in select patients with severe acute heart failure.
Milrinone is not used for chronic heart failure. It is used only in select patients with severe acute heart failure.
 However, there are important clinical differences between the two PDE3 inhibitors and β agonists:
However, there are important clinical differences between the two PDE3 inhibitors and β agonists:


Advanced
 PDE3 breaks down both cAMP and cGMP. The subtype of PDE determines the ratio of activity against cAMP versus cGMP. PDE3 preferentially acts on cAMP 10 times more than it does cGMP. The changes in cAMP and cGMP in different cells dictate the clinical effects seen in either the heart or the vasculature.
PDE3 breaks down both cAMP and cGMP. The subtype of PDE determines the ratio of activity against cAMP versus cGMP. PDE3 preferentially acts on cAMP 10 times more than it does cGMP. The changes in cAMP and cGMP in different cells dictate the clinical effects seen in either the heart or the vasculature.





Nitrates


MOA (Mechanism of Action)

 NO, also known as endothelium-derived relaxing factor (EDRF), induces vasodilation primarily on the venous side of the circulation (venodilation). It does this by activating guanylyl cyclase, an enzyme that produces cGMP, which decreases Ca+ entry into cells and causes relaxation of vascular smooth muscle (Figure 11-15).
NO, also known as endothelium-derived relaxing factor (EDRF), induces vasodilation primarily on the venous side of the circulation (venodilation). It does this by activating guanylyl cyclase, an enzyme that produces cGMP, which decreases Ca+ entry into cells and causes relaxation of vascular smooth muscle (Figure 11-15).
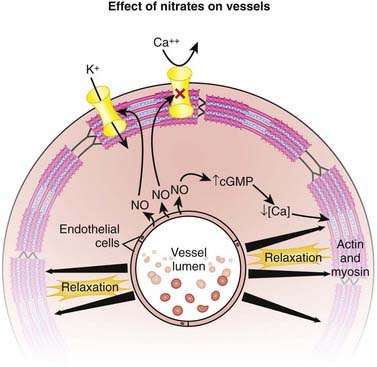


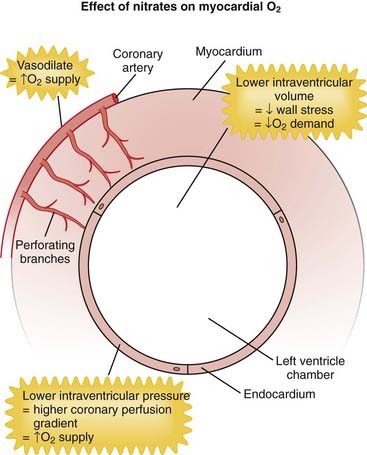

















Important Notes
 The drug interaction with PDE5 inhibitors (vasodilators used for treatment of erectile dysfunction) is significant. Many men with erectile dysfunction are older and have heart problems, such as angina. Both classes of drugs increase intracellular cGMP, and the hypotension that develops from the profound vasodilation can be severe and refractory to treatment. Hence there is great potential for this interaction to occur and for potentially serious consequences if it does occur.
The drug interaction with PDE5 inhibitors (vasodilators used for treatment of erectile dysfunction) is significant. Many men with erectile dysfunction are older and have heart problems, such as angina. Both classes of drugs increase intracellular cGMP, and the hypotension that develops from the profound vasodilation can be severe and refractory to treatment. Hence there is great potential for this interaction to occur and for potentially serious consequences if it does occur.


Evidence






Vasodilators


MOA (Mechanism of Action)
 The underlying mechanism of vasodilation of arteriolar smooth muscle by the direct vasodilators is still not confirmed, but theories include interference with the release of Ca+2 from the SR and activation of potassium channels.
The underlying mechanism of vasodilation of arteriolar smooth muscle by the direct vasodilators is still not confirmed, but theories include interference with the release of Ca+2 from the SR and activation of potassium channels.



Pharmacokinetics





Contraindications













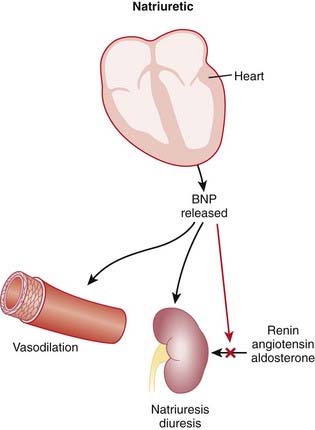
Natriuretic Peptides

MOA (Mechanism of Action)
 A number of hormonal and neurologic mechanisms are activated in heart failure, of which natriuretic factors (peptides) are one.
A number of hormonal and neurologic mechanisms are activated in heart failure, of which natriuretic factors (peptides) are one.


Pharmacokinetics






Important Notes
 Endogenous BNP is elevated in patients with heart failure. Most systems that are activated in heart failure (such as the renin-angiotensin-aldosterone system and the SNS) result in vasoconstriction and water retention, which is the opposite effect of BNP. Therefore BNP acts as a counterbalancing hormone system in heart failure.
Endogenous BNP is elevated in patients with heart failure. Most systems that are activated in heart failure (such as the renin-angiotensin-aldosterone system and the SNS) result in vasoconstriction and water retention, which is the opposite effect of BNP. Therefore BNP acts as a counterbalancing hormone system in heart failure.



Evidence
 A meta-analysis in 2005 (three trials, N = 862 patients) showed a possibility of increased death and renal dysfunction in patients with acute decompensated heart failure treated with nesiritide. All studies were double blinded. However, another meta-analysis in 2006 (seven trials, N = 1717 patients) did not show increased mortality or renal failure in patients with acute heart failure treated with nesiritide. Two of the trials were not blinded.
A meta-analysis in 2005 (three trials, N = 862 patients) showed a possibility of increased death and renal dysfunction in patients with acute decompensated heart failure treated with nesiritide. All studies were double blinded. However, another meta-analysis in 2006 (seven trials, N = 1717 patients) did not show increased mortality or renal failure in patients with acute heart failure treated with nesiritide. Two of the trials were not blinded.







[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 11 Cardiology
Angiotensin-Converting Enzyme Inhibitors (ACEIs)
MOA (Mechanism of Action)
Through inhibition of ACE with an ACEI, the following effects occur:
Net result: Because angiotensin II levels are lower and bradykinin levels are higher, there is more vasodilation; SVR (systemic vascular resistance) and afterload are lowered. Because aldosterone levels are lower, less Na and water are reabsorbed in the kidney; therefore preload is reduced (Figure 11-1).Pharmacokinetics
Contraindications
Side Effects
Important Notes
The renin-angiotensin system (RAS) plays an important role in the body’s compensation for a failing heart. Activation of the sympathetic nervous system (SNS) leads to the release of renin, which in turn increases vascular tone and sodium and water retention.Advanced
In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.Evidence
Hypertension
A 2009 Cochrane review (24 trials, N = 58,040 participants) compared benefits and harms of first-line antihypertensives with those of placebo or no treatment over a minimum of 1 year in patients with hypertension. ACEIs (three trials) reduced mortality (relative risk [RR] 0.83), stroke (RR 0.65), coronary heart disease (RR 0.81), and cardiovascular events (RR 0.76).Angiotensin Receptor Blockers (ARBs)
MOA (Mechanism of Action)
ARBs are antagonists of the angiotensin-1 (AT1) receptor. Therefore they block the actions of angiotensin II.Pharmacokinetics
Important Notes
Perhaps because of the lack of increased bradykinin levels, ARBs are not typically associated with the side effect of cough, which can be a significant limitation to the use of ACEIs.Advanced
In addition to the beneficial effects of RAS inhibitors in diabetic nephropathy, there is emerging evidence that RAS inhibitors may reduce the incidence of new-onset diabetes. Potential mechanisms for this effect include improvements in blood flow that improve the delivery of insulin and glucose to skeletal muscle, as well as effects on glucose transport and insulin signaling. If this preventative effect of RAS inhibition in diabetes becomes established, it could change the way these agents are used.Direct Renin Inhibitors
MOA (Mechanism of Action)
Renin is an enzyme released from the kidneys that converts angiotensinogen to angiotensin I. It is considered to be the rate-limiting step in the eventual formation of angiotensin II.Evidence
Blood-Pressure Lowering Efficacy versus Placebo
A 2008 Cochrane review (six trials, 3694 participants) compared the blood-pressure–lowering efficacy of renin inhibitors versus placebo in primary HTN. The authors found that aliskiren elicits a dose-dependent reduction in both systolic and diastolic pressure similar to that seen with ACEIs or ARBs. In the included trials, aliskiren did not increase withdrawals due to adverse events versus placebo.FYI Notes
Renin was first identified in 1898, when it was extracted from kidneys and discovered to have pressor properties. It would be another 40 years before it was determined that renin was an enzyme that catalyzed the formation of a pressor substance (angiotensin II), rather than being the pressor itself.Sodium Channel Blockers (Class I Antiarrhythmics)
Description
Na channel blockers are Vaughan Williams class I antiarrhythmics. There are three subclasses: Ia, Ib, and Ic. The use of Na channel blockers as local anesthetics is discussed in the discussion of local anesthetics in Chapter 21.
MOA (Mechanism of Action)
Na channels are blocked, so Na ion movement during phase 0 of the action potential is inhibited. The result is a “slow” phase 0, which results in a wider (and slower) QRS wave on the electrocardiogram (ECG). The net result is slower conduction (Figure 11-4).Pharmacokinetics
Side Effects
Evidence
Atrial fibrillation and prevention of recurrence: A Cochrane review in 2007 (45 studies, 12,559 patients) evaluated the efficacy and safety of multiple different antiarrhythmics in patients who had previously experienced atrial fibrillation (a very common arrhythmia). Class Ia antiarrhythmics were associated with increased mortality compared with controls (odds ratio [OR] 2.39; number needed to harm [NNH] 109). Class Ia and Ic were associated with reduced occurrences of atrial fibrillation (OR 0.19 to 0.6). There were many withdrawals from treatment because of side effects for all antiarrhythmics (NNH 17 to 36).β Antagonists (β-Blockers)
Prototype and Common Drugs
MOA (Mechanism of Action)
To understand β-blockers, you must understand the effects of the adrenergic system and which effects are mediated via β receptors. β-Blockers competitively antagonize the action of catecholamines at β receptors. There are many cardiac and noncardiac consequences of β-blockade. More details on the autonomic nervous system are described in Chapter 3.
Tachycardia and Arrhythmia
The properties of β-blockers that make them antitachycardics include the following:
Catecholamine β1 stimulation results in an increase in the slow Na+ current (If) of the action potential phase 4 in the SA node. This results in a faster rising (and shorter) phase 4, a shorter time to the next heartbeat, and thus a faster HR. β-Blockers will oppose this action, slowing the SA pacemaker rate.Myocardial Ischemia and Infarction
Pharmacokinetics
Contraindications
Important Notes
Evidence
After Myocardial Infarction
Evidence for the role of β-blockers in secondary prevention after an MI comes from several trials and was summarized in a 1999 systematic review (82 trials, N = 54,234 patients). There was a 23% reduction in the odds of death in long-term trials but only a 4% reduction in short-term trials. The review found that the number needed to treat (NNT) to avoid a fatality over the course of 2 years is 42. The greatest amount of evidence available was for propranolol, timolol, and metoprolol.Hypertension and Associated Stroke and Coronary Artery Disease
A Cochrane review in 2007 (13 studies, N = 91,561 patients) compared β-blockers with other agents for HTN. Atenolol was the β-blocker most frequently used. The authors found that β-blockers had only weak effects in reducing stroke and no effect on coronary heart disease versus placebo. There was also a trend toward worse outcomes when compared with calcium channel blockers (CCBs), RAS inhibitors, and thiazides, prompting the authors to suggest that β-blockers should not be considered as first-line agents for HTN.Obstructive Airway Disease (Asthma and Chronic Obstructive Pulmonary Disease)
A Cochrane review in 2002 (29 studies, N = 381 patients) examined the impact of single-dose or short-term selective β1-blockers in patients with mild to moderate obstructive airway disease. There were no differences in pulmonary flow measurements compared with placebo except for a small decrease in FEV1 after the first treatment—an effect that disappeared with subsequent doses.Potassium Channel Blockers (Class III Antiarrhythmics)
MOA (Mechanism of Action)
Blocking K channels in phase 3 of the action potential slows the efflux of K back out of the myocyte, which slows the rate at which the cell repolarizes and therefore lengthens the plateau phase of the action potential. This increases the refractory period of atrial, ventricular, and Purkinje cells. This also increases the QT interval on the ECG (Figure 11-5).Pharmacokinetics
Drug interactions:
