Defects of Adrenal Steroidogenesis
11β-Hydroxylase Deficiency Congenital Adrenal Hyperplasia
Corticosterone Methyl Oxidase Deficiency
Dexamethasone (Glucocorticoid)-Suppressible Hyperaldosteronism
Disorders of 3β-Hydroxysteroid Dehydrogenase
Disorders of 17α-Hydroxylase/17,20-Lyase
Isolated 17α-Hydroxylase Deficiency
Isolated 17,20-Lyase Deficiency
Combined 17α-Hydroxylase/17,20-Lyase Deficiency
Combined Disorders of 21- and 17α-Hydroxylase/17,20-Lyase Deficiency
Adrenal Failure With Male-Limited Gonadal Failure and XY Gender Reversal
Steroidogenic Factor-1 Deficiency
DSS-AHB Critical Region on the X Chromosome 1
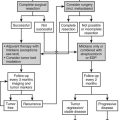
The human adrenal gland is composed of the cortex and the medulla. The medulla produces bioamines, and the adrenal cortex secretes several classes of steroids (corticosteroids). The adrenal cortex can be considered to be made up of three distinct subunits, each having a characteristic steroid profile. The outermost unit, the zona glomerulosa, produces mineralocorticoids, principally aldosterone (a salt-retaining hormone), which serve to maintain sodium and fluid balance. Glucocorticoids, primarily cortisol, arise from the central zona fasciculata and maintain glucose homeostasis and vascular integrity. The innermost subunit, the zona reticularis, secretes sex steroids (androgens). Disorders of adrenal steroidogenesis may involve overproduction, underproduction, or both the simultaneous overproduction and underproduction of corticosteroids (Fig. 8-1). In this chapter, the following conditions are discussed:
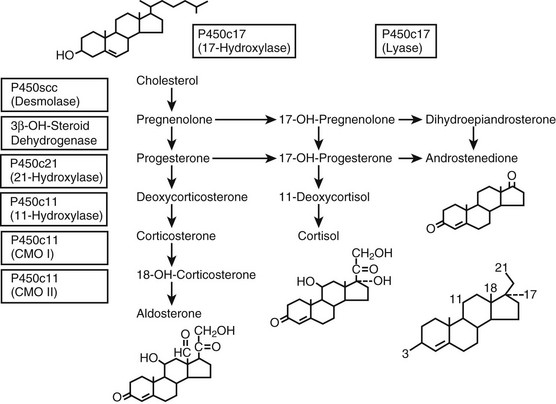
FIGURE 8-1 Adrenal steroidogenesis. Biosynthetic pathways from cholesterol to mineralocorticoids (aldosterone), glucocorticoids (cortisol), and androgens (androstenedione) are shown and the cellular locations of enzyme activities indicated. CMO, Corticosterone methyl oxidase; OH, hydroxylase. (From White PC, New MI, Dupont B: Medical progress: congenital adrenal hyperplasia, N Engl J Med 316:1519–1524, 1987. © 1987, Massachusetts Medical Society.)
1. Disorders of P450c21 resulting in the 21-hydroxylase deficiency form of congenital adrenal hyperplasia (CAH) (salt-wasting, simple virilizing, and nonclassic forms)
2. Disorders of P450c11, including (a) 11β-hydroxylase deficiency form of CAH (classic and nonclassic forms), (b) corticosterone methyl oxidase (CMO) deficiency types I and II, (c) dexamethasone-suppressible hyperaldosteronism
3. 3β-Hydroxysteroid dehydrogenase (3β-HSD) deficiency form of CAH (classic and nonclassic forms)
4. Disorders of P450c17 activity, including (a) isolated 17α-hydroxylase deficiency, (b) isolated 17,20-lyase deficiency, and (c) combined 17α-hydroxylase deficiency/17,20-lyase deficiency
5. Lipoid CAH: P450scc deficiency and steroidogenic acute response (StAR) deficiency
6. Adrenal failure with male-limited gonadal failure and XY gender reversal: steroidogenic factor-1 deficiency
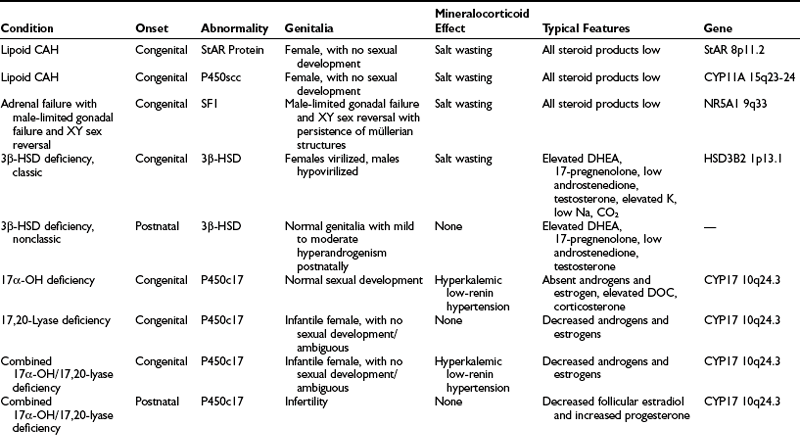
A summary of the clinical, hormonal, and genetic features of these steroidogenic defects appears in Table 8-1.

Distinction is made between classic forms of disease, defined by significantly reduced enzyme activity manifesting clinically at birth, and nonclassic forms, in which the enzyme defect is less severe and symptoms are not present at birth, and when they do appear are generally milder. The classification of CAH subtypes has significant clinical implications for treatment and prenatal diagnosis, but it should be appreciated that patients exist on a clinical continuum rather than within absolute, discrete, easily defined conditions. Approximately 90% to 95% of cases of classic CAH are due to 21-hydroxylase deficiency,1 whereas defects in the enzymes 11β-hydroxylase and 3β-HSD account for almost all the rest. 17α-Hydroxylase deficiency and lipoid CAH are rare causes of CAH.
The 21-hydroxylase and 11β-hydroxylase deficiencies, which occur late in cortisol synthesis, cause shunting of accumulated precursor steroids into pathways of androgen biosynthesis, which do not require these enzymes. Because the external genitalia of the fetus are sensitive to androgens,2 excess androgen secretion by the adrenal masculinizes the female genitalia, causing genital ambiguity in utero in affected females, but no genital alterations in affected males. Postnatal hyperandrogenism affects both genders.
The nonclassic forms of 21-hydroxylase and 11β-hydroxylase deficiencies may be extremely common (and treatable) causes of hyperandrogenism.3–8
History
The observation of hyperplastic adrenal glands in association with internal female gonads and ductal structures in a phenotypic male appeared in the anatomic literature in 1865 with De Crecchio’s9 description of his autopsy of a Neapolitan pseudohermaphrodite. Fibiger,10 Apert,11 and Gallais12 amassed case histories involving precocious puberty, hirsutism, pseudohermaphroditism, and obesity in the early 1900s, and attempted to classify them. The term adrenogenital syndrome was used for many years to describe conditions characterized by elevated adrenal androgens due to virilizing adrenal tumors or to CAH.
The first comprehensive view of CAH was based on the biochemical discoveries of the 1940s and 1950s.13 Among the pioneers who subsequently characterized the variants of CAH in the late 1950s and early 1960s were Bongiovanni,14,15 Eberlein and Bongiovanni,16 Prader and Siebenmann,17 Biglieri and colleagues,18 and New.19 CAH now is more appropriately referred to by the names of the specific enzyme deficiencies that characterize it.
In 1977, the discovery of the association between the well-studied human leukocyte antigens (HLAs) and the 21-hydroxylase trait opened a window to a new form of definition of CAH through molecular genetics.20 Since the gene responsible for classic 21-hydroxylase deficiency (found within the HLA complex) was isolated in 1984,21 knowledge of the specific mutations that cause the different forms of CAH has grown rapidly, much the way that biochemical discoveries of the earlier era led to construction of the scheme for steroidogenesis. Mutations in the genes encoding the steroidogenic enzymes have been confirmed as the basis for all the CAHs. Additionally, steroidogenic defects may be due to mutations in transcription factors or other cofactors (e.g., StAR protein).22
Disorders of 21-Hydroxylase
Epidemiology and Population Genetics
Results of newborn screening in a number of localities around the world23–27 yield a worldwide incidence of classic 21-hydroxylase deficiency of approximately 1 in 14,500 live births, ranging from 1 in 858628 to 23,000.29 It has been estimated that 75% have the salt-wasting phenotype.30 Applying the Hardy-Weinberg formula for a population at equilibrium gives a computed heterozygote frequency for classic 21-hydroxylase deficiency of 1 in 61 persons. In areas where patient retesting is difficult/inefficient, it may be necessary to lower the treatment threshold to save lives of affected individuals who die before the diagnosis is confirmed and treatment is started.31
Nonclassic 21-hydroxylase deficiency, one of the most common autosomal recessive diseases, is more frequent than cystic fibrosis. The frequency is ethnic specific, as first determined by Speiser and colleagues.32 Incidences are 1 in 27 Ashkenazi Jews, 1 in 40 Hispanics, 1 in 50 Yugoslavs, 1 in 300 Italians, and 1 in 100 in a heterogeneous New York population.32–34 In an attempt to determine an earliest date for the appearance within the Ashkenazi Jewish population of a founder mutation, DNA analysis was undertaken for representative individuals from the Roman Jewish ghetto, a community already established by the time of the second Diaspora (70 ce). No evidence was seen in Roman Jews of the B14-related nonclassic 21-hydroxylase deficiency mutation, thus suggesting a date after 70 ce for the appearance of this mutation among Ashkenazi Jews.35 Further genetic characterization of this population in terms of its affinity to the general European population and to other Jewish groups continues.36 The nonclassic 21-hydroxylase deficiency mutation can be dated to between 70 ce and the second millennium, based on the significantly higher frequency observed in Ashkenazi Jews compared with Sephardic Jews.
Classic 21-Hydroxylase Deficiency
External Genitalia: Adrenocortical cell differentiation occurs early in embryogenesis, and although the biochemical schedule of steroid synthesis has not been completely elucidated, it is clear that genital development in the fetus takes place in the setting of active fetal adrenal steroid synthesis. Because differentiation of the external genitalia is sensitive to androgen, excess adrenal androgen produces genital ambiguity in females affected by classic 21-hydroxylase deficiency. In utero masculinization consists of mild to pronounced clitoral enlargement, varying degrees of labioscrotal fusion, and a urogenital sinus with the type and degree of virilization proportional to the onset of hyperandrogenism (Figs. 8-2 and 8-3). The 21-hydroxylase form of CAH is the most common cause of genital ambiguity in females. Every phenotypic male with hypospadias and bilateral cryptorchidism should be considered a female with CAH and should be evaluated immediately for classic 21-hydroxylase deficiency.
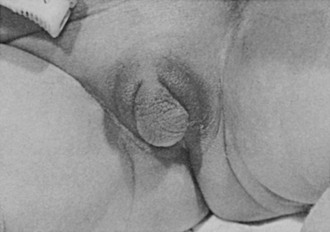
FIGURE 8-2 Ambiguous genitalia in a newborn female with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Note the enlarged clitoris, the single orifice on the perineum, and scrotalization of the labia majora. (Modified from New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K [eds]: Advances in Human Genetics, vol 4. New York, Plenum, 1973, pp 251–326.)
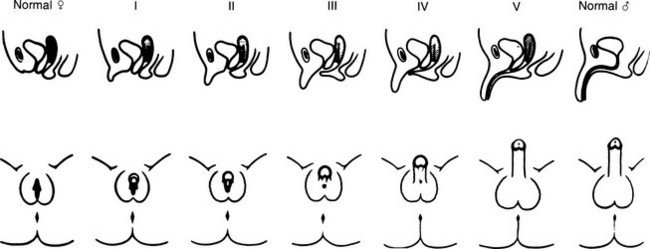
FIGURE 8-3 Prader characterization of the range of genital malformations found in females with classic 21-hydroxylase deficiency. In type I, the only abnormality is enlargement of the clitoris; in type II, partial labioscrotal fusion occurs; in type III, a funnel-shaped urogenital sinus is seen at the posterior end of a small vulva; in type IV, a very small urogenital sinus is found at the base of an enlarged phallus; and in type V, a penile urethra is evident. (From Prader A: Die Haufigkeit der Kongenitalen Adrenogenitalen Syndromes, Helv Paediatr Acta 13:5, 1958.)
Internal Genitalia: Gonadal differentiation and internal genital morphogenesis are not affected by the enzyme abnormalities of classic steroid 21-hydroxylase deficiency. Because there is no anomalous secretion of antimüllerian hormone (AMH), which is synthesized by the Sertoli cells of the fetal testis, müllerian duct development in the female proceeds normally into the uterus and fallopian tubes.37 Thus, normal child-bearing capacity exists in females. Wolffian duct stabilization and differentiation normally take place in the context of local testosterone levels in the male, and this process appears to be unaffected by elevated systemic prenatal adrenal androgens.
Growth
Postnatal somatic growth in both genders is markedly affected by the chronic hyperandrogenism of untreated 21-hydroxylase deficiency. High levels of androgens cause accelerated growth in early childhood, producing an unusually tall and often quite muscular child, an “infant Hercules” (Fig. 8-4). This early growth spurt, however, is followed by premature epiphyseal maturation and closure, resulting in a final height that is short relative to that expected on the basis of midparental target height. Exposure to glucocorticoids used in replacement therapy at doses that may exceed the physiologic requirement has been postulated to be another important factor in the poor growth of these patients.38
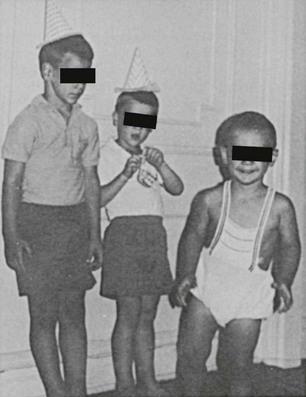
FIGURE 8-4 Untreated congenital adrenal hyperplasia in two brothers: a 4-year-old on the left and a 2-year-old on the right. In the center is their normal 6-year-old brother. (Modified from New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K [eds]: Advances in Human Genetics, vol 4. New York, Plenum, 1973, pp 251–326.)
We have previously shown that final height is one of the features of CAH least amenable to glucocorticoid replacement therapy.39 Analysis of 47 patients with classic CAH separated into two groups defined by the degree of hormonal control failed to show a significant difference in height outcome with the use of standard treatment.40 A pilot study combining recombinant human growth hormone with a gonadotropin-releasing hormone superagonist to improve final height in those patients with CAH with advanced skeletal maturation and poor predicted final adult height demonstrated that this significantly improved final adult height in children with CAH.41
Hair and Skin Gland Abnormalities
Bone Health: Bone mineral density (BMD) is affected by the competing actions of androgen excess (from undertreatment) and glucocorticoid excess (from overtreatment), which can occur in patients simultaneously. The relative extent of undertreatment and overtreatment determines the net effect on BMD. Two studies found that BMD was lower in young adults with CAH than in controls42,43; one of these studies showed a trend toward increased fracture rate of wrist and vertebrae in young women with CAH.43 Dosing regimens, beginning in the young adult years, clearly must take bone health into consideration.
Fertility
Excess adrenal sex steroids inhibit the pubertal changes in gonadotropin secretion directed by the hypothalamic-pituitary axis, probably via a negative feedback effect at the hypothalamus or the pituitary. This inhibition is reversible by suppression of adrenal androgen production. In most untreated or poorly treated adolescent girls and in some adolescent boys, spontaneous pubertal development does not occur until proper glucocorticoid treatment is instituted. Menstrual irregularity and secondary or even primary amenorrhea with or without hirsutism can occur in untreated and poorly controlled women. An association of virilizing CAH with polycystic ovary syndrome has also been noted.44,45 Reproductive capacity in females with classic CAH is reduced. Meyer-Bahlburg46 compared the fertility of women with different forms of classic 21-hydroxylase deficiency. In those women with an adequate introitus and heterosexual activity, he found that fertility was approximately 60% with the simple virilizing form (n = 25), whereas in the salt-wasting form (n = 15), fertility was only 7%.
Traditionally, well-treated patients were thought to have the onset of puberty occur at the expected chronologic age; however, a recent analysis suggests that puberty does in fact occur early than expected—and height is compromised—especially in salt-wasting males.47 The gonadotropin response to gonadotropin-releasing hormone (gonadotropin-releasing hormone or luteinizing hormone-releasing hormone) is appropriate for age in well-controlled prepubertal and pubertal female patients unless ovarian disease is present. Poor control of the disease in males with classic CAH has been associated with small testes and azoospermia. However, cases of normal testicular maturation and spermatogenesis and fertility in untreated men have been reported.48 Well-treated males are normally fertile and have normal pubertal development, spermatogenesis, and testicular function. Later in life, males may develop nodular hyperplasia of adrenal rest tissue, causing enlargement of the testes, which may respond to high-dose glucocorticoid treatment. With the use of ultrasonography, several studies have identified testicular “tumors” in many or even most adolescent and adult male patients with CAH (ages 16 to 40 years).49,50 Tumors ranged in size from 0.2 to 4.0 cm, and in many of these patients, the tumors were palpable. The degree of hormonal control did not correlate with the presence of tumor; indeed, many adequately or even overtreated subjects had identifiable tumors. Genotype was predictive of tumor size, with the most severe mutations correlating with the largest tumors.49 Leydig cell function also was commonly impaired in these subjects.50 The adrenal glands of subjects with mutations in the 21-hydroxylase gene, both homozygous and heterozygous, are known to be larger than those of controls. This increased surface area has been associated with an increased risk of adrenal tumor formation, and indeed adrenal “incidentalomas” are more common in these groups.51 However, a retrospective analysis of “true” adrenal tumors failed to demonstrate an increased incidence of mutations, either germline or somatic.52
Salt Wasting
Salt wasting results from inadequate secretion of salt-retaining steroids, especially aldosterone. In addition, hormone precursors elevated in 21-hydroxylase deficiency may act as mineralocorticoid antagonists (e.g., 17-hydroxyprogesterone). Newborns are especially prone to the development of a salt-wasting crisis because the sodium-conserving mechanism of their renal tubules is only marginally competent. The adrenal medulla of patients with CAH may be incompletely formed in some patients. Merke and coworkers53 noted that the epinephrine and metanephrine concentrations of subjects manifesting a salt-wasting crisis were approximately 50% of those of controls; pathologic evaluation of adrenal medullary tissue (removed during adrenalectomy) demonstrated depletion of secretory vesicles.
A single enzyme is responsible for 21-hydroxylation in both the zona fasciculata and the zona glomerulosa. The pathogene-tic difference between salt wasting and simple virilizing 21-hydroxylase deficiency may simply be the result of a quantitative difference in enzyme activity caused by conformational changes induced by the specific mutations. In vitro expression studies show that as little as 1% of normal activity of 21-hydroxylase allows adequate aldosterone synthesis to prevent significant salt wasting.54,55 The issue is complicated by the occasional finding of discordance for salt-wasting expression among siblings with identical mutations in their inherited 21-hydroxylase genes, and by the observed improvement in salt wasting in some individuals over time.48,56–58 In one case, a girl born with male genitalia and salt wasting, presumably with severe enzyme deficiency, was no longer a salt waster by age 4. Another important case finding was that of a girl with a homozygous deletion of the 21-hydroxylase gene and a history of multiple salt-wasting crises in infancy, who discontinued her therapy in adolescence and was found at that time to be secreting aldosterone.59
Nonclassic 21-Hydroxylase Deficiency
Nonclassic 21-hydroxylase deficiency is distinguished from the classic form symptomatically by its age at onset and biochemically by its less severe impairment of steroid 21-hydroxylation; because of these two factors, it does not result in ambiguous genitalia in the newborn female. It often initially manifests just before the age of normal puberty, although it may present at any age after birth. Patients may be homozygous for a mild genetic defect or may have compound heterozygotes for one severe mutation and one mild mutation. Nonclassic 21-hydroxylase deficiency was first defined in the course of family studies of patients with classic CAH. Family members who should have been carriers for 21-hydroxylase deficiency were found to have overt hormonal disturbances that were associated with distinct alleles at the 21-hydroxylase locus. These alleles were determined to be in linkage disequilibrium with the HLA locus on chromosome 6. It thus was found through these studies that the gene locus for 21-hydroxylase lies between HLA-B and HLA-DR.60
In addition to linkage of the 21-hydroxylase locus with the neighboring HLA-B and HLA-DR antigen loci, 21-hydroxylase deficiency alleles are found in linkage disequilibrium with other HLA genes or haplotypic combinations that may include specific alleles of the neighboring genes C4A and C4B encoding the fourth component of serum complement.61 The two most prominent such cases are linkage disequilibrium of the extended haplotype HLA-A3,Bw47,DR7 with the classic salt-wasting form, and HLA-B14,DR1 with nonclassic 21-hydroxylase deficiency.62
Clinical Features
The clinical features of nonclassic 21-hydroxylase deficiency range widely, begin at any age, and commonly wax and wane over time. Although genital ambiguity in the newborn period is never a feature of this disorder, the appearance of any of the other signs and symptoms of hyperandrogenism can prompt the patient to seek medical attention, or patients may be identified through family or population screening. Individuals commonly present just before the time of expected pubarche. Underlying biochemical abnormalities are always demonstrable at any age by ACTH stimulation testing, regardless of whether symptoms are evident.63
Nonclassic 21-hydroxylase deficiency can result in the premature development of pubic hair (pubarche), which may occur as early as 6 months of age.64 In one study, nonclassic 21-hydroxylase deficiency was found in 30% of children with premature pubarche,65 whereas a lower prevalence was reported by another group.66 Severe cystic acne refractory to oral antibiotics and retinoic acid has been associated with nonclassic 21-hydroxylase deficiency by some.67,68 In one young woman, male pattern alopecia was the sole presenting symptom. Menarche in females can be normal or delayed, and secondary amenorrhea or oligomenorrhea is frequent. Final height, as in classic 21-hydroxylase deficiency, is less than predicted based on the midparental target height and linear growth percentiles, even though excess androgen secretion may not have been apparent.40,50
A number of women with polycystic ovary disease have been found on ACTH testing to have a primary adrenal defect, for example, nonclassic 21-hydroxylase deficiency, 3β-HSD deficiency, or 11β-hydroxylase deficiency.45,69–72 The reported prevalence of nonclassic 21-hydroxylase deficiency as a cause of these endocrine symptoms in women ranges from 1.2% to 30%. The scope of the range may relate to differences in the ethnic makeup of the groups studied. Women heterozygous for a 21-hydroxylase gene mutation have been shown to have higher free testosterone levels than controls (i.e., hyperandrogenemia), but, it is important to note, not an increased incidence of hyperandrogenism.73
Fertility in Nonclassic 21-Hydroxylase Deficiency
It has been recognized for 30 years that infertility in women may be reversed by glucocorticoid therapy. In one report, five patients with irregular menses and high 17-keto(oxo)steroids (urinary androgen metabolites) resumed regular menses and demonstrated adequate suppression of 17-ketosteroids and pregnanetriol (a urinary metabolite of 17-OHP) within 2 months after beginning therapy with glucocorticoids alone, suggesting the presence of an adrenal 21-hydroxylating defect.74 In another report of 18 infertile women with acne and/or facial hirsutism and hormonal criteria consistent with 21-hydroxylase deficiency, seven conceived shortly after initiating prednisone treatment; an additional four women conceived within 2 months of the addition of clomiphene to the prednisone.75 Hormonal profiles after initiation of therapy were not reported in this study. As mentioned earlier, oligospermia and subfertility have been reported in men with nonclassic 21-hydroxylase deficiency.76 We recommend screening for nonclassic 21-hydroxylase deficiency during every evaluation for infertility.
Molecular Genetics
The molecular genetics basis of 21-hydroxylase deficiency has been studied extensively. The 21-hydroxylase enzyme is a microsomal cytochrome P-450 enzyme termed P450c21. The structural genes encoding P450c21, CYP21, and a pseudogene, CYP21P, are located on chromosome 6p21.3, adjacent to the genes C4B and C4A, encoding the two isoforms of the fourth component of serum complement in the class III region of the HLA complex.77,78 CYP21 and CYP21P each contains 10 exons. Their nucleotide sequences are 98% identical in exons and approximately 96% identical in introns.79
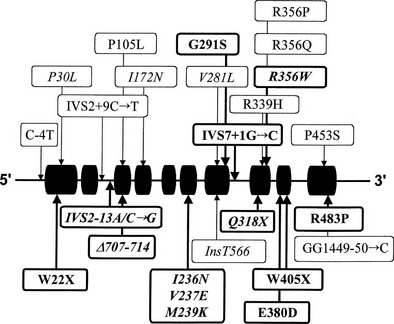
Many of the mutations known to cause 21-hydroxylase deficiency are apparently the result of either of two types of recombination between CYP21 and CYP21P: (1) chromatid misalignment and unequal crossing over, resulting in large-scale DNA deletions, and (2) gene conversion events that result in the transfer to CYP21 of smaller scale deleterious mutations normally present in the CYP21P pseudogene. Seven of the eight possible exonic differences between CYP21 and CYP21P have been observed and confirmed to be causative of 21-hydroxylase deficiency. At least 25 mutations causing 21-hydroxylase deficiency have been identified; one single-nucleotide mutation altering the splicing of an intron is particularly common and is associated with both simple virilizing and salt-wasting phenotypes (Fig. 8-5). Deletions and gene conversion events may be common findings in 21-OHD, owing to the chromosomal arrangement of the gene and pseudogene (i.e., homologous genes in a tandem array),80,81 and to the presence of multiple chi-like sequences.82
Correlation of Genotype With Phenotype
In general, the functional consequence of each DNA alteration corresponds with the clinical severity of the inherited disease: total deletion of the functional gene, stop codon (nonsense) mutations, frameshifts, and several amino acid substitutions (missense mutations) have been shown to result in salt-wasting classic alleles; one nonconservative amino acid substitution has been associated exclusively with simple virilizing disease, and a single conservative amino acid substitution (V281L) is the mutation associated with the HLA-B14,DR1 haplotype found so frequently in nonclassic disease.83
However, comparison of the clinical characteristics and molecular genetics data in 532 affected individuals at one site reveals that the genotype correctly predicted the phenotype in 89% of cases, a high figure but significantly shy of the 100% that might be expected. Rocha et al.84 suggested that much of the variability in the external virilization of CAH women is due to the genotype (i.e., number of CAG repeats) of the androgen receptor.84 This analysis underlines the need for careful ongoing clinical assessment of all persons affected by 21-hydroxylase deficiency.
Biochemical Characterization
In classic CAH, baseline serum cortisol levels are at the lower limits of detection or in the low to normal range. Baseline concentrations of serum 17-OHP and adrenal androgens are elevated, with serum 17-OHP levels often several hundred times normal.85 In nonclassically affected persons, because of pulsatile and diurnal variations in 17-OHP secretion, midmorning and afternoon concentrations may be normal.86 Of all single measurements, early morning measurement of serum 17-OHP concentration is the most likely to show an elevation. However, we caution against relying on baseline 17-OHP levels to exclude nonclassic 21-hydroxylase deficiency; mild defects may be only minimally elevated, especially in the afternoon, leading to many false-negative diagnoses.
The standard diagnostic procedure for diagnosing all forms of CAH is the 60 minute (synthetic) ACTH stimulation test. At 08:00 hours, when cortisol secretion is at its normal diurnal peak, blood is drawn before and then 60 minutes after intravenous injection of a 0.25 mg bolus of ACTH. A nomogram (Fig. 8-6) that relates baseline to ACTH-stimulated serum concentrations of 17-OHP has been constructed that can be used to identify individuals with classic and nonclassic forms, as well as heterozygote carriers of the 21-hydroxylase deficiency form of CAH.
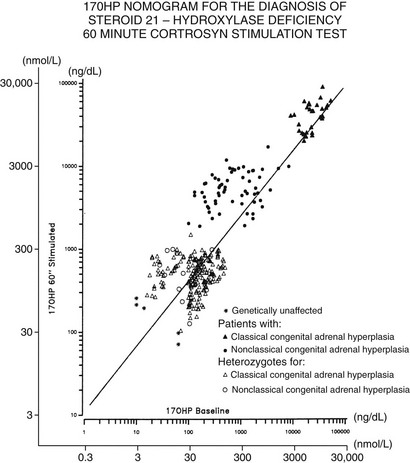
FIGURE 8-6 Nomogram relating baseline to adrenocorticotropic hormone–stimulated serum concentrations of 17-hydroxyprogesterone. Scales are logarithmic. The regression line shown is for all data points. Data points cluster as shown into three nonoverlapping groups: classic (congenital adrenal hyperplasia) and nonclassic forms of 21-hydroxylase deficiency are readily distinguished from each other and from heterozygote/unaffected. Distinguishing unaffected from heterozygote responses is difficult.
Neonatal screening via heel-stick capillary blood has been available since 197787 and is mandated in many states in the United States. The 17-OHP content of the dried blood, which is sampled on filter paper, analogous to the phenylketonuria test standard for all newborns, can be determined by a qualified laboratory. The value of newborn screening has been demonstrated repeatedly by the finding that most (biochemically proven) affected children were not identified on clinical grounds.88–90
The 08:00 hours salivary level of 17-OHP correlates extremely well with serum assays and is highly recommended as a screening test.86 It is especially useful when venipuncture is difficult.
Diagnosis at Birth
The following data should be collected in the evaluation of a newborn with possible CAH:
1. Karyotype or other genetic analysis should be performed to establish the genetic gender in cases of ambiguous genitalia.
2. ACTH stimulation test: measuring serum 17-OHP concentration before and after ACTH stimulation. This test should NOT be performed during the initial 24 hours of life because samples from this time period are typically elevated in all infants and may yield false-positive results.
3. Aldosterone and plasma renin should be measured during the ACTH test.
4. Urinary sodium and potassium should be measured to assess salt-preserving ability.
5. Evidence of suppression of steroid secretion by glucocorticoid administration should be sought.
Further characterization of DNA by molecular genetic analysis is desirable when available.
Disorders of 11β-Hydroxylase
Steroid 11β-hydroxylase activity in the adrenal cortex is required for the synthesis of both glucocorticoids and mineralocorticoids. It has been shown that distinct isozymes of P450c11 participate in cortisol and aldosterone synthesis in humans.91 The structure of the two CYP11B genes (nine exons and eight introns) is also identical to that of the CYP11A gene, which encodes the cholesterol desmolase protein. The genes for the two CYP11B isozymes are 93% identical in predicted amino acid sequence and 90% identical in the intronic region and are encoded by two vicinal genes on chromosome 8q24.3.92–94 It is important to note that the upstream regions of the two genes are quite different, suggesting functionally different control. Disorders of the two enzymes may manifest as hypocortisolism (i.e., CAH), hypoaldosteronism, or hyperaldosteronism.
11β-Hydroxylase Deficiency Congenital Adrenal Hyperplasia
Abnormal steroid secretion attributed to defective 11β-hydroxylation was first described by Eberlein and Bongiovanni16 in 1955. It has proved to be the second most common form of CAH (5% to 8% of all cases in the general population).95 In the cortisol pathway, conversion of 11-deoxycortisol to cortisol is reduced. As a result, 11-deoxysteroids (11-deoxycortisol and 11-DOC) accumulate. The hyperandrogenism resulting from shunting of cortisol precursors is similar to that of 21-hydroxylase deficiency, including genital ambiguity in classically affected females.
Approximately two thirds of patients with 11β-hydroxylase deficiency become hypertensive, with or without hypokalemic alkalosis, sometime early in life. Hypertension does not correlate with the presence or degree of hypokalemia or with the extent of virilization.96
In addition to the classic form (i.e., present at birth), milder nonclassic forms of 11β-hydroxylase deficiency have been reported3–8 and may represent allelic variants, analogous to 21-hydroxylase deficiency. No biochemical defect has been demonstrated consistently in the obligate heterozygote parents of patients with the classic form, either in the baseline state or with ACTH stimulation.97 As noted earlier, such a biochemical defect can be demonstrated in 21-hydroxylase heterozygotes. As in 21-hydroxylase deficiency, persons with identical 11β-hydroxylase mutations may differ in the severity of their signs and symptoms of androgen and mineralocorticoid excess, suggesting a role for epigenetic or nongenetic factors in the expression of clinical phenotype.98
Hypertension in 11β-Hydroxylase Deficiency
Hypertension in 11β-hydroxylase deficiency CAH is commonly attributed to DOC-induced sodium retention, which results in volume expansion. However, it has not been consistently proven that DOC causes hypertension.91 In 1970, New and Seaman99 showed that the large amounts of DOC produced in 11β-hydroxylase deficiency are glucocorticoid (dexamethasone) suppressible and therefore originate in the zona fasciculata, rather than in the zona glomerulosa. When DOC is suppressed with dexamethasone treatment, renin levels rise, causing secretion of aldosterone in the zona glomerulosa, in which the 11β-hydroxylase enzyme (P450c11B2) is normal.
Suppression of DOC by glucocorticoid treatment, however, may not lower blood pressure in hypertensive 11β-hydroxylase–deficient patients. Normotensive patients with markedly elevated DOC levels,100,101 hypertensive patients with normal or only mildly elevated DOC,102,103 and atypical 11β-hydroxylase–deficient patients with normal PRA104 present a challenge to the DOC-centered explanation of hypertension. Of course, the lack of response to treatment is a feature of long-standing hypertension of many causes.
Epidemiology
Classic 11β-hydroxylase CAH (due to defects in the CYP11B1 gene) occurs in approximately 1 in 100,000 births in the general white population.105 A large number of cases have been reported in Israel. The incidence there now is estimated to be 1 in 5000 to 1 in 7000 births, with a gene frequency of between 1 in 71 and 1 in 83.106 This unexpected clustering of cases was traced to Jewish families of North African origin, particularly from Morocco and Tunisia. Turkish Jews also have been found to carry the identical 11β-hydroxylase gene mutation with high frequency.96,105 The incidence of the nonclassic form is not known at present but would be predicted to be considerably higher than the classic form, as in 21-hydroxylase deficiency.
Molecular Genetics
One of the 11β-hydroxylase genes, CYP11B1, is expressed at high levels in normal adrenal glands.93 Transcription of this gene is regulated by cyclic adenosine monophosphate (the second messenger for ACTH). Low levels of transcripts of the second gene, CYP11B2, have been detected in normal adrenal with the use of reverse transcriptase polymerase chain reaction. The enzymes encoded by the CYP11B1 and CYP11B2 genes have been studied by expressing the corresponding complementary DNA in cultured cells and after actual purification from aldosterone-secreting tumors.107–109 The high degree of similarity in both gene and protein structures between CYP11B1 and CYP11B2 suggested that the two 11β-hydroxylase genes were members of the same gene family.93,110
Mutations in the zona fasciculata enzyme (P450c11B1) result in defective synthesis of cortisol, producing hypertension from precursor (e.g., DOC) accumulation. Mutations in the zona glomerulosa gene (P450c11B2) result in defective synthesis of aldosterone and consequent salt wasting. An interesting molecular defect of the 11β-hydroxylase genes results in glucocorticoid-responsive hyperaldosteronism and resultant hypertension (see “Dexamethasone [Glucocorticoid]-Suppressible Hyperaldosteronism”).
Almost all affected alleles in the Moroccan-Israeli population carry the same mutation, the substitution of histidine for arginine at codon 448111 (R448H) in the CYP11B1 gene. This mutation is incompatible with normal enzymatic activity112 and appears to represent a founder effect. At least 31 mutations causing 11β-hydroxylase deficiency CAH have been identified and are shown in Fig. 8-7. Mutations are spread across the gene, including at several identified hot spots.113
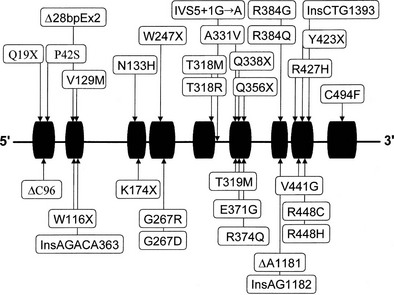
Diagnosis
Elevated serum 11-deoxycortisol (compound S) and DOC, confirmed by marked elevation of their urinary tetrahydrometabolites, is diagnostic. Further confirmation can be found in a complete absence of any 11-oxygenated C19 or C21 steroids in blood or urine.114 Diagnosis is made as for 21-hydroxylase deficiency with the additions of baseline and ACTH-stimulated serum 11-deoxycortisol (compound S) and DOC. Further characterization by molecular genetics analysis should be pursued in families anticipating additional children, with prenatal diagnosis now also available. Diagnosis in a newborn is quite difficult because the characteristic hypertension generally does not appear during the newborn period, and distinction from 21-hydroxylase deficiency based on steroid patterns is also problematic at this age.115 Infantile gynecomastia has been described and may offer a clue to the diagnosis.116 Treatment consists of glucocorticoid replacement. Determination of PRA is the standard test for mineralocorticoid excess. Although a nonelevated DOC is desirable, follow-up usually is accomplished more easily by following the renin (or PRA) level. Conversely, suppressed renin is typically a sign of insufficient control of 11β-hydroxylase deficiency.
Corticosterone Methyl Oxidase Deficiency
The conversion of corticosterone to 18-hydroxycorticosterone is referred to as CMO I activity, whereas the subsequent 18-oxidation to aldosterone is known as CMO II activity. In the zona glomerulosa, these steps are catalyzed by distinct domains within a single enzyme, P450c11B2, also referred to as P450c18, aldosterone synthase, and P450aldo. Deficient CMO I activity typically results in (practically) no aldosterone production and phenotypically is more severe than CMO II deficiency. Defects in CMO I activity have been reported but appear to be less common than those of CMO II.117,118 Defects in both CMO I and CMO II activity are inherited as autosomal recessive conditions, and because cortisol synthesis is not affected in either condition, defects of P450aldo are not considered to be forms of CAH.
Infants with CMO II deficiency are subject to potentially fatal electrolyte abnormalities; in childhood, however, recurrent dehydration, a variable degree of hyponatremia and hyperkalemia, and poor growth are characteristic.119,120 Adults may be asymptomatic, their affected status coming to light only in the course of family studies of a symptomatic child. CMO II deficiency has been found at an increased frequency among Jews of Iranian origin.121 Molecular genetic analyses of such families have demonstrated two missense mutations in the CYP11B2 genes of affected individuals: the replacement of arginine by tryptophan at position 181 (R181W) and the replacement of valine by alanine at position 386 (V386A).122
CMO I deficiency is characterized biochemically by essentially unmeasurable aldosterone levels with low or normal 18-hydroxycorticosterone levels. Patients with CMO II deficiency typically have normal (or low) aldosterone levels with elevated 18-hydroxycorticosterone levels (i.e., an elevated serum 18-hydroxycorticosterone-to-aldosterone ratio), along with a low corticosterone-to-18-hydroxycorticosterone ratio. Alternatively, CMO II deficiency can be identified by an elevated ratio of the major metabolites of these steroids (tetrahydro-18-hydroxy-11-dehydrocorticosterone and tetrahydroaldosterone) in urine.120
Dexamethasone (Glucocorticoid)-Suppressible Hyperaldosteronism
Dexamethasone-suppressible hyperaldosteronism (DSH; also known as glucocorticoid-remediable aldosteronism) is a rare form of hypertension123,124 in which aldosterone synthesis appears to be regulated abnormally by ACTH.125 Dexamethasone-suppressible hyperaldosteronism is characterized by (1) rapid and complete suppression of aldosterone secretion on dexamethasone administration, (2) continued increase in aldosterone with long-term administration of ACTH, (3) suppressed PRA, and (4) a dominant mode of inheritance.126 Urinary excretion of 18-hydroxycortisol and 18-oxocortisol is increased.127,128
Individuals with DSH have been shown to have an intergenic recombination, juxtaposing the promoter of CYP11B1 with the coding sequence of CYP11B2.129,130 The fusion or chimeric gene creates an alteration in the regulation of synthesis of aldosterone, making the zona glomerulosa sensitive to ACTH rather than to renin-angiotensin, resulting in glucocorticoid-responsive hyperaldosteronism and hypertension.
Disorders of 3β-Hydroxysteroid Dehydrogenase
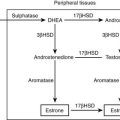
The enzyme 3β-HSD is necessary for the synthesis of bioactive Δ4 adrenal and gonadal steroids from the relatively inactive Δ5 precursors. In addition to 3β-hydroxysteroid dehydrogenation, this enzyme performs 3-oxosteroid isomerization, giving rise to the alternate term of Δ4/Δ5 isomerase. 3β-HSD is present not only in the adrenal cortex, gonads, and placenta but also in the liver and in nearly all peripheral tissues. The placental form is distinct from the adrenal/gonadal form. Differences in the levels of 3β-HSD activity in different tissues suggest tissue-specific isoforms or tissue-specific regulation of one or more shared isoforms of this protein.131–134
Classic 3β-Hydroxysteroid Dehydrogenase Deficiency
3β-HSD deficiency is an autosomal recessive form of CAH that was first described by Bongiovanni135 in 1962. The exact frequency of this rare disorder is unknown. It has been postulated that individuals with defects early in the pathway of steroidogenesis, which severely impair cortisol synthesis (i.e., 3β-HSD and StAR), have poor survival rates.
Owing to reduced 3β-HSD enzyme activity in the gonads, genetic males are incompletely masculinized and exhibit genital ambiguity at birth. In affected females, conversely, very high levels of circulating DHEA converted peripherally to active androgens produce a limited androgen effect. Clitoral enlargement occurs and, rarely, labial fusion. As with the 21-hydroxylase and 11β-hydroxylase enzymes, the severity of the enzyme defect may not be determined on the basis of the appearance of the external genitalia at birth. Deficient steroid production in 3β-HSD deficiency may result in salt wasting, although this clearly is not true in every case.136,137
Nonclassic 3β-Hydroxysteroid Dehydrogenase Deficiency
As with nonclassic 21-hydroxylase deficiency, nonclassic 3β-HSD deficiency is an attenuated enzyme defect with no major developmental abnormalities.69,138 Signs of virilization appear in females after adrenarche or at the time of puberty.
The possibility that nonclassic 3β-HSD deficiency may be a significantly underdiagnosed cause of excess androgen symptoms, including hirsutism and infertility, has been suggested.139 Review of clinical data on more than 700 women with signs of androgen excess demonstrated decreased 3β-HSD activity in 16%.140 It has been suggested that nonclassic 3β-HSD deficiency may be more frequent than nonclassic 21-hydroxylase deficiency in women with androgen excess syndrome.140 In a group of 25 menarchal women with 3β-HSD deficiency, an improvement in regulation of menses and in acne was observed to result from glucocorticoid therapy of at least 3 months’ duration.139 Hirsutism was less well reversed by treatment. Polycystic ovary disease was noted in 50% of these women.
Biochemistry and Molecular Genetics
Two genes encoding 3β-HSD have been localized to chromosome 1p13.1 and cloned.134,141 These genes, HSD3B1 and HSD3B2, encode the skin-placental form (referred to as the type I isoform) and the adrenal-gonadal form (type II isoform) (Fig. 8-8). These isozymes are 372 and 371 amino acids in length, respectively, and 93.5% similar. A representation of mutations in the 3β-HSD type II gene is shown in Fig. 8-8.142–144
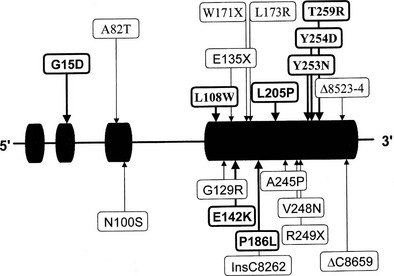
FIGURE 8-8 Mutations in the HSD3B2 gene causing the 3β-hydroxysteroid dehydrogenase/Δ5/Δ4 isomerase deficiency form of congenital adrenal hyperplasia. Alleles associated with salt wasting are printed in bold; non–salt-wasting alleles are in regular type.
Caution should be exercised when the nonclassic form of 3β-HSD deficiency is diagnosed because the diagnostic hormonal profile in this condition has been shown to “disappear” in previously confirmed cases.69 In addition, the paucity of confirmed mutation analysis/enzyme expression studies has made suspect the diagnosis of nonclassic 3β-HSD deficiency as a genetic disorder.
Diagnosis
The external genitalia of affected newborn males with classic 3β-HSD are incompletely masculinized, and females are masculinized to a variable degree. A high ratio of Δ5-to-Δ4 steroids, characterized specifically by elevated serum levels of the Δ5 steroids pregnenolone, 17-hydroxypregnenolone, and DHEA, along with increased excretion of the Δ5 metabolites pregnenetriol and 16-pregnenetriol in the urine, is diagnostic for this enzyme disorder. These criteria should not be used during the newborn period when Δ5 steroids are universally elevated, representing a “physiologic 3β-HSD deficiency.”136 The 3β-HSD defect is diagnosed by 60 minute ACTH testing that produces elevations of 2 or more standard deviations above the mean in all the following: (1) serum Δ5 17-hydroxypregnenolone, (2) DHEA concentration, (3) serum ratios of Δ5 17-hydroxypregnenolone/17-hydroxyprogesterone, and (4) serum ratios of Δ5 17-hydroxypregnenolone/cortisol.137 Using genotypically proven subjects, Lutfallah and colleagues145 suggested biochemical criteria using ACTH-stimulated 17-hydroxypregnenolone levels alone. In the general population, findings limited to modestly elevated Δ5 17-hydroxypregnenolone and/or DHEA are nonspecific and do not establish the diagnosis of partial or mild 3β-HSD deficiency. As stated above, although nonclassic 3β-HSD deficiency may represent a true abnormality in endocrine physiology, it has very rarely been shown to have a genetic basis.
To rule out an adrenal or ovarian steroid–producing tumor, a dexamethasone suppression test (0.5 mg every 6 hours for 2 to 3 days) should be performed with subsequent measurement of serum hormone concentrations. An ovarian source of the androgens is excluded by the addition of the progestogen norethindrone acetate (5 mg every 8 hours for 3 days) to the dexamethasone. Both adrenal and ovarian hormones should be suppressed under this regimen. Ovarian sonography and either adrenal computed tomography or magnetic resonance imaging may be performed when there is a high index of suspicion for an adrenal or ovarian tumor, such as rapid progression or failure to suppress steroid production with dexamethasone or dexamethasone and norethindrone. As an aid to diagnosis, one can use published nomograms (Fig. 8-9A and B). Reference data established by Temeck and associates65 on ACTH-stimulated hormonal values are used for the diagnosis of nonclassic 3β-HSD in children with precocious adrenarche (see Fig. 8-9C and D).
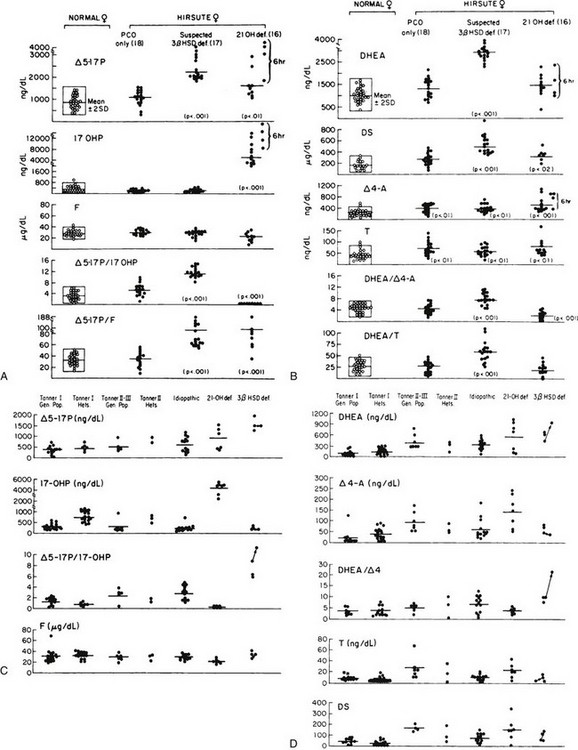
FIGURE 8-9 A, Serum 17-hydroxysteroid responses to adrenocorticotropic hormone (ACTH) stimulation (1 hour after a 0.25 mg IV bolus dose) in normal women and women with hirsutism. B, Serum androgen levels after ACTH stimulation in normal women and women with hirsutism. C, ACTH-stimulated serum 17-hydroxysteroid levels in normal children from the general population and from proven heterozygotes for 21-hydroxylase deficiency and in children with premature pubarche. The children with premature pubarche are classified as idiopathic (no steroidogenic defect) or as patients with 21-hydroxylase deficiency or 3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency. D, ACTH-stimulated serum androgen levels in normal children from the general population and from proven heterozygotes for 21-hydroxylase deficiency and in children with premature pubarche. Children with premature pubarche are classified as idiopathic (no steroidogenic defect) or as patients with 21-hydroxylase deficiency or 3β-HSD deficiency. The testosterone (T) levels in males are represented by filled squares. (A and B, From Pang SY, Lerner AJ, Stoner E, et al: Late-onset adrenal steroid 3 beta-hydroxysteroid dehydrogenase deficiency. I. A cause of hirsutism in pubertal and postpubertal women, J Clin Endocrinol Metab 60:428–439, 1985. © 1985, The Endocrine Society. C and D, From Temeck JW, Pang S, Nelson C, New MI: Genetic defects of steroidogenesis in premature pubarche, J Clin Endocrinol Metab 64:609–617, 1987. © 1987, The Endocrine Society.)
Disorders of 17α-Hydroxylase/17,20-Lyase
A single enzyme (P450c17) catalyzes both the conversion of mineralocorticoids to glucocorticoids (17α-hydroxylase activity) and the conversion of glucocorticoids to sex steroids (17,20-lyase activity). The enzyme P450c17 is encoded by the gene CYP17, which is located on chromosome 10q24-25. Abnormal enzyme function can, however, present as isolated 17α-hydroxylase deficiency, isolated 17,20-lyase deficiency, or combined 17α-hydroxylase/17,20-lyase deficiency. Based on the total number of reported cases (in both genders), 17α-hydroxylase with or without 17,20-lyase deficiency appears to be a rare steroidogenic defect.146
Isolated 17α-Hydroxylase Deficiency
The enzyme domain that performs the 17α-hydroxylase activity is functionally distinct from the domain that performs the 17,20-lyase function. Therefore, it is reasonable to expect that isolated deficiency of either function may be found. However, because the product of 17α-hydroxylation is the precursor for 17,20-lyase action, isolated 17α-hydroxylase deficiency may be difficult to identify because it should present as the more common combined 17α-hydroxylase/17,20-lyase deficiency. Individuals with “pure 17α-hydroxylase deficiency” would be expected to present with hypokalemic hypertension with alkalosis, possibly with normal gender development. These individuals could be identified by expression studies of mutant enzymes in cell culture, and it has been suggested that more than 20% residual 17,20-lyase function would not be apparent. Clinically, at least one such description has been reported,147 but in reality, these cases represent a quantitative rather than a qualitative difference. Individuals with isolated 17α-hydroxylase deficiency would be phenotypically identical to patients with DSH.
Isolated 17,20-Lyase Deficiency
Deficiency of 17,20-lyase activity impedes synthesis of C19 sex steroids in the adrenal and gonads148 without affecting cortisol synthesis in the adrenal gland and therefore is not a form of CAH but is a potential cause of abnormal gender development. Urinary pregnanetriolone, a metabolite of 17-OHP, is increased and increases further after ACTH and human chorionic gonadotropin stimulation. DHEA and testosterone excretion does not increase appreciably. At birth, such individuals all have normal female genitalia, regardless of chromosomal gender, and generally present at adolescence with primary amenorrhea and/or lack of gender development. Other oversecreted steroids (e.g., corticosterone) subserve the glucocorticoid function. It is important to note that one longitudinal study of a 46,XY individual who began receiving estrogen replacement at puberty revealed apparently progressive extinction of 17α-hydroxylating activity from ages 20 to 26.149 Thus, this patient, who started life with an isolated 17,20-lyase defect, converted to a combined 17α-hydroxylase/17,20-lyase defect at puberty, suggesting the need for continued vigilance in cases of isolated 17,20-lyase deficiency.
Combined 17α-Hydroxylase/17,20-Lyase Deficiency
A defect in 17α-hydroxylase/17,20-lyase will result in diminished production of cortisol and sex steroids, the production of which requires the 17,20-lyase function. The enzyme defect affects steroid synthesis in both the adrenals and the gonads and reduces production of all androgens and estrogens. Because of high corticosterone (B) levels, patients with P450c17 abnormalities do not manifest adrenal crises and therefore often go undiagnosed, often until evaluated for abnormal pubertal development.150 In the genetic male, there is pseudohermaphroditism, whereas in the female, infantile genitalia are present.151 At puberty, gonadotropins increase to very high concentrations, the very low sex steroid production failing to provide adequate regulatory feedback. Breast development may occur in males.19 In the female at pubertal age, no secondary gender characteristics develop, and primary amenorrhea may be noted. Phenotypically, such subjects may be similar to individuals with androgen insensitivity syndrome. 17,20-Lyase deficiency (isolated or as combined 17α-hydroxylase/17,20-lyase deficiency) occurs approximately 65% as often as androgen insensitivity syndrome.152
Nonclassic combined 17α-hydroxylase/17,20-lyase deficiency has been described in a subset of women undergoing evaluation for infertility. These women had normal potassium levels, blood pressure, and female external genitalia but high follicular phase progesterone combined with low estrogen levels.153
Hypertension
Hypertension is observed in the 17α-hydroxylase deficiency form of CAH. The serum concentration of DOC is markedly elevated (30 to 60 times normal), but, as in 11β-hydroxylase deficiency,105 circulating levels of DOC do not correlate entirely with blood pressure values, suggesting that other factors contribute to hypertension in this condition.154,155 The serum aldosterone level in some individuals is also elevated, but usually the aldosterone is very low, secondary to the volume expansion caused by excess mineralocorticoid secretion and the resultant suppressed renin. Individuals may develop hypertension in childhood.
As in 21-hydroxylase deficiency, sibling pairs expected to have a common genetic defect do not always exhibit the same biochemical findings.146
Partial Defects
The heterozygous state has been identified by ACTH stimulation testing156; however, a nonclassic form has not been identified. The steroid pattern of some cases of low-renin hypertension without abnormalities of gender157 raises the possibility that an isolated 17α-hydroxylase deficiency may be a more common cause of low renin hypertension than is currently recognized.146
Molecular Genetics
Human complementary DNA corresponding to P450c17 has been characterized,158 and the single-copy CYP17 gene locus is situated on chromosome 10q24-25.159 Many structural and gene-regulatory mutations in CYP17 have been identified in patients with combined 17α-hydroxylase/17,20-lyase deficiency (Fig. 8-10). It is interesting to note that phenotypic variation occurs even among mutation-identical subjects.152 An interesting finding reported by Imai and colleagues160 is the discovery of a mutation (a four-base duplication) common to two unrelated Canadian Mennonite pedigrees and six families (eight individuals) living in the Friesland region of the Netherlands. This mutation almost certainly represents a founder effect because the Mennonite sect originated in Friesland.
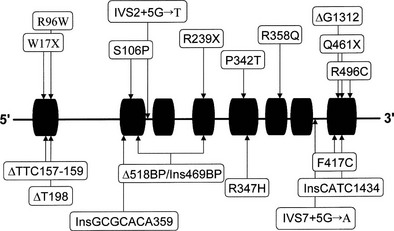
Diagnosis
Testing for 17α-hydroxylase deficiency should be performed in anyone with hypokalemic (low-renin, low-aldosterone) hypertension,150 whereas 17,20-lyase deficiency (isolated or combined) should be sought in (apparent) females presenting at pubertal age with sexual infantilism. The disorder may be revealed earlier in 46,XY subjects who present in infancy or childhood with a hernia or inguinal mass and the finding of normal male internal structures with female external genitalia. Consideration also should be given in women with unexplained infertility. As described previously, the hormonal picture will vary depending on the relative deficiencies of 17α-hydroxylase and 17,20-lyase activities. Plasma levels of corticosterone and 18-hydroxy-DOC and an elevated ratio of 18-hydroxycorticosterone to aldosterone are diagnostic of 17α-hydroxylase deficiency, whereas low androgens to estrogens indicates the addition of 17,20-lyase deficiency.161 In long-standing untreated cases, refractory hypertension of considerable severity can develop.
Combined Disorders of 21- and 17α-Hydroxylase/17,20-Lyase Deficiency
The combination of prenatal androgen excess with postnatal androgen deficiency has been reported. In some of these patients (postnatal), steroid profiles are consistent with combined (partial) 21-hydroxylase and 17α-hydroxylase/17,20-lyase deficiencies. Female newborns may have severe virilization (Prader III or IV), and males may be normal or undervirilized. Many of these subjects also have skeletal and other abnormalities consistent with Antley-Bixler syndrome (type 2). Steroid analysis of such subjects (especially after the neonatal period) typically demonstrates elevation of progesterone, 17-hydroxyprogesterone, and corticosterone. Pregnancies are commonly marked by maternal virilization—with both male and female fetuses—and low (maternal) estriol levels. Mild forms may be detected by the low maternal estriol level, with completely normal ultrasound.162 Subjects with combined 21-hydroxylase and 17α-hydroxylase/17,20-lyase deficiencies, with or without Antley-Bixler syndrome (type 2), have been shown to have mutations in the P450 oxidoreductase gene, POR.163,164 P450 oxidoreductase is an important cofactor for electron transfer from nicotinamide adenine dinucleotide phosphate (NADPH) to the 21-hydroxylase and 17α-hydroxylase/17,20-lyase (as well as many other) enzymes.164 Maternal gestational virilization is likely due to the P450 oxidoreductase effect on P450 aromatase.163,165
Adrenal Failure With Male-Limited Gonadal Failure and XY Gender Reversal
Lipoid Congenital Adrenal Hyperplasia
Lipoid CAH is a rare form of CAH that was originally described in 1955 by Prader and associates17,166 and represents the most extreme form of CAH with only negligible production of all steroids. Massive accumulations of cholesterol in the adrenocortical tissue (but not in the Leydig cells of the testis, where the enzyme is also active) lead to the characteristic fatty appearance of the glands and the descriptive name used for this disorder.
The cholesterol side-chain cleavage enzyme (P450scc, also known as cholesterol desmolase and encoded by CYP11A) catalyzes the conversion of cholesterol to pregnenolone in the first and rate-limiting step in the production of corticosteroids, and therefore was the most logical but not the only genetic candidate proposed for lipoid CAH. Mutations leading to lipoid CAH occur in CYP11A and in the gene coding for the StAR protein, encoded by the StAR gene on chromosome 8p11.2. StAR serves to shuttle cholesterol to the inner mitochondrial membrane, where the P450scc enzyme is located.22 Mutations in StAR appear to be more common than those in CYP11A.
Affected individuals exhibit hypogonadism, severe fluid and electrolyte disturbances, hyperpigmentation, and susceptibility to infection. They often do not survive infancy. Lipoid CAH is rare in most populations but appears to occur at a somewhat higher frequency and with less severity among Japanese, Koreans, and Palestinian Arabs.167,168 Phenotypic variability exists, with some patients described as having small adrenal glands without significant intracellular lipid deposits.
Both dominant and recessive mutations have been described in CYP11A. Tajima and colleagues169 described an apparent somatic mutation present in multiple tissues in the affected subject but not found in either parent. This child was born with normal weight and length and was assigned a female gender. She was clinically unaffected (and untreated) until after 4 years of age, when she developed acute adrenal failure. No uterus was identified, and a karyotype at that time revealed 46,XY. This mutation appears to lead to a protein with essentially no enzymatic activity.
Another child with adrenal insufficiency was found to be a compound heterozygote for mutations in CYP11A. One mutation arose de novo, whereas the other was inherited from the mother, who was clinically unaffected.170
Baker et al described two families with mutations in the StAR gene, with a mild phenotype. The affected children presented with adrenal failure at the relatively late age of 2 to 4 years, but with normal genital development; the mutant StAR protein was shown to retain 20% of wild-type activity. They referred to this phenotype as “nonclassical lipoid CAH.”171
The pathology of lipoid CAH appears to result from two distinct but related events. The first event results from deficient steroid hormone production. The second event is increasing lipid deposition inside the cell, which results in increasing intracellular damage/death. This latter event is theorized to lead to the salt-wasting adrenal crisis that often results in death in infancy.172 The case report by Tajima and colleagues also would appear to confirm this hypothesis.169 Histologic examination of a StAR knockout mouse supports this theory, with significant lipid deposits noted in the adrenals and mild accumulation in the testes but none in the ovaries.173
Steroidogenic Factor-1 Deficiency
The human homologue of the fushi tarazu factor is the steroidogenic factor-1 (SF1), also known as adrenal 4–binding protein. This protein is a nuclear receptor that regulates the transcription of numerous genes, including AMH, DAX1, CYP11A, CYP11B2, CYP21, and StAR; the gonadotropins; and aromatase174 and itself is regulated by the upstream stimulatory factor-1175 and by mitogen-activated protein kinase–dependent phosphorylation.176,177
Independent of its steroidogenic actions, SF1 is an active determinant of gender differentiation, chiefly via upregulation of AMH (or müllerian-inhibiting substance),178 in conjunction with the product of the Wilms tumor gene (WT-1). DAX1 serves to block male differentiation by binding to SF1 and preventing this interaction with AMH.179 SF1 also combines with SRY and SOX9 in advancing male differentiation of the gonad. Mutations of SF1 (and DAX1) therefore can be associated with persistence of müllerian structures in XY individuals.180
Subjects with mutations in NR5A1 have been described. The first report described a 2-week-old, phenotypically female infant with adrenal failure. She was found to have normal müllerian structures but a 46,XY karyotype. As noted by Achermann and colleagues,181 the failure of müllerian regression may have been directly due to the loss of SF1-directed AMH production or secondarily through Sertoli cell maldevelopment. A second subject with adrenal failure and XY gender reversal was found to have a homozygous mutation of NR5A1. Both parents (and a sister) were shown to be carriers but were unaffected.182 In contrast to this, a 46,XX female with a heterozygous mutation of NR5A1 had adrenal failure but normal ovarian function, consistent with the male-limited nature of the gonadal failure but demonstrating that the adrenal requirement is not gender specific.183 The conclusion that can be drawn from most SF1/NR5A1 mutations is that SF1 is critical to adrenal gland development but is not critical to ovarian or testicular development. However, reports have described humans with SF1 mutations with genital abnormalities but normal adrenal function,184 showing that the above statement is also not absolute.
DSS-AHB Critical Region on the X Chromosome 1
The DAX1 gene (also called NR0B1) is located on the short arm of the X chromosome and, when mutated, leads to X-linked congenital adrenal hypoplasia (also referred to as adrenal hypoplasia, congenita [AHC]), as well as hypogonadotropic hypogonadism. DAX1 duplication, however, leads to XY-limited gender reversal with ambiguous or completely female phenotypic development, regardless of chromosomal gender.185
The DAX1 protein is an important negative modulator of transcription, which decreases SF1 and StAR expression (and therefore general steroid production).186 DAX1 also downregulates AMH, leading to müllerian duct development/internal female differentiation. DAX1 is expressed in the adrenal gland, the gonad, the pituitary, and the hypothalamus; its expression in the developing gonad decreases as testicular differentiation increases but continues in the case of ovarian differentiation.187 Mutations in DAX1 (and SF1) should be sought in children with unexplained adrenal failure and especially in boys with hypogonadotropic hypogonadism.188
Treatment for Steroidogenic Defects
The fundamental aim of endocrine therapy for CAH is to provide replacement of the deficient hormones. Since 1949, when Wilkins and colleagues189 and Bartter190 discovered the efficacy of cortisone therapy for CAH due to 21-hydroxylase deficiency, glucocorticoid therapy has been the cornerstone of treatment for all forms of cortisol deficiency. Glucocorticoid administration replaces the deficient cortisol and suppresses ACTH overproduction; a concomitant reduction in adrenal cortex activity occurs, thereby reducing the production of other adrenal steroids (i.e., precursor by-products) and remission of (most) symptoms over time. Patients with nonclassical 21-hydroxylase deficiency may have resolution of symptoms within 3 months of initiating corticosteroid treatment.191
Adrenal suppression in 21-hydroxylase, 11β-hydroxylase, and 3β-HSD deficiency reduces the excess production of androgens, averting further inappropriate virilization, slowing accelerated growth and bone age advancement to a more normal rate, and allowing normal onset of puberty. An improved body habitus is seen with a progressively earlier start of treatment (Fig. 8-11). Individuals with the salt-wasting type of 21-hydroxylase or 3β-HSD deficiency require the administration of a salt-retaining steroid to maintain adequate sodium balance. Suppression of adrenal activity in 11β-hydroxylase and 17α-hydroxylase deficiency normalizes DOC/mineralocorticoid secretion and often results in remission of hypertension. In lipoid CAH and adrenal failure, total hormone replacement is required.
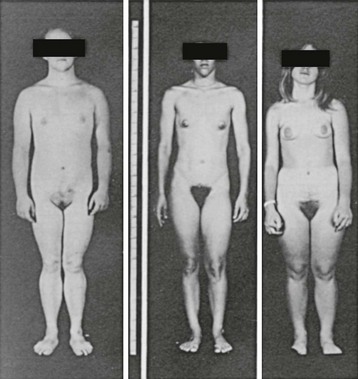
FIGURE 8-11 Habitus of pubertal girls with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The patient on the left was untreated until age 16 years; the patient in the center was started on treatment at age 9 years; and the patient on the right was treated from age 4 years. Note the progressively more feminine habitus with earlier treatment. (Modified from New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K [eds]: Advances in Human Genetics, vol 4. New York, Plenum, 1973, pp 251–326.)
If poor hormonal control with hydrocortisone is noted at the standard dose, the dose may be increased temporarily to 20 (or even 30) mg/m2 per day, or the regimen may be changed to a synthetic hormone analogue such as prednisone or dexamethasone. These agents are more potent and longer acting, although their relative glucocorticoid and mineralocorticoid effects differ, and the smaller amounts used make dose adjustment more crucial. Because of individual variations in the activity of hepatic enzymes metabolizing 11-oxosteroids, and thus in plasma clearance and half-life, in some patients, prednisolone (the 11β-hydroxy analogue of prednisone, also called Δ1 cortisol) is more effective than prednisone (Δ1 cortisone) as glucocorticoid replacement. Individuals without overt salt wasting have been shown to have elevated renin levels,192–198 and its fluctuation may correlate with ACTH. Addition of a mineralocorticoid to the therapeutic regimen in individuals with the simple virilizing form of 21-hydroxylase deficiency has been shown to result in improved hormonal control at the same glucocorticoid dose. A longer-term benefit of a reduced glucocorticoid requirement is improved statural growth.197,198
Monitoring Treatment
Glucocorticoid doses should be titrated to optimize biochemical control balanced against physiologic parameters (e.g., in a growing child with 21-hydroxylase deficiency, a 17-OHP of between 500 and 1000 ng/dL and suppressed androgens while maintaining a good growth velocity). The goal, with respect to corticosteroid replacement, is to give the minimal dose required for optimal control. Serum 17-OHP and Δ4 androstenedione concentrations determined by radioimmunoassay can be used to monitor biochemical control in patients with 21-hydroxylase deficiency.199–201 In females and prepubertal males (but not in newborn and pubertal males), the serum testosterone level is also a useful index.200 Combined determinations of PRA, 17-OHP, and serum androgens, as well as clinical assessment of growth and pubertal status, all must be considered when the doses of glucocorticoid and salt-retaining steroid are adjusted for optimal therapeutic control. A combination of hydrocortisone and 9α-FF has proved to be a highly effective treatment modality.199 Measurement of PRA can be used to monitor efficacy of treatment in most forms of CAH. PRA is elevated in the salt-losing state and is suppressed in the volume-overloaded state. Its normalization indicates improved hormonal control.
Although glucocorticoid treatment has been available since 1950, little agreement has been reached as to which regimen gives the best outcome in terms of height. Recent studies suggest that even the most compliant patient may not achieve a final height compatible with parental stature. It is not known whether this is due to overtreatment, to failure of oral glucocorticoid therapy given twice or even three times daily to suppress excess androgen production, or to simultaneous overtreatment and undertreatment. A simple home-based system to monitor the hormonal status of patients at frequent intervals may improve hormonal control and final growth. Salivary 17-OHP concentration may provide an easy means for monitoring hormone levels on a daily basis.86 Similar considerations may apply to the preservation of fertility.
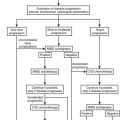
Management of the Child with Ambiguous Genitalia
The newborn with ambiguous genitalia represents a medical emergency. Determination of genetic gender by karyotype and accurate diagnosis of the specific underlying defect are essential for initial management, but these factors do not address questions of gender identity and sexual orientation—questions that often are raised by confused parents at an extremely vulnerable time. It is well established that steroids influence aspects of central nervous system development,202,203 but the data are controversial regarding specific androgen effects resulting from CAH.204–206 It has been proposed that a masculinized gender role in girls, with behavioral manifestations such as tomboyishness, results from prenatal androgen excess in CAH. In a controlled play situation, Nordenstrom and colleagues207 observed that girls with CAH played with masculine toys more than controls, and they demonstrated that the degree of masculine play correlated with the degree of virilization. Girls with CAH were shown to score similarly to control males on tests of spatial cognition.208 In adult women, Long et al209 found that self-reported femininity decreased and masculinity increased with prenatal androgen exposure. Similarly considered, behavioral changes resulting from alterations in the androgen milieu are seen in studies on male pseudohermaphrodites with 5α-reductase or 17β-hydroxysteroid dehydrogenase deficiency, who often elect a male gender identity at puberty.210, 211 However, a great body of psychological studies indicate that in humans, unlike other mammals, the gender of rearing overrides prenatal hormonal effects.212,213
When a gender of rearing is assigned to a pseudohermaphrodite, the genetic gender is of less consideration than the physiologic and anatomic character of the genitalia, their potential for development and function, and the psychosocial milieu of the infant. Gender assignment of female pseudohermaphrodites with 21- or 11β-hydroxylase deficiency or 3β-HSD deficiency in the newborn period should be female. In fact, Dessens et al214 found that gender dysphoria in 46,XX CAH patients was 12.1% when the children were raised as males, compared with 5.2% when they were raised as females.
Wide individual variability has been noted in the presentation of ambiguous genitalia in these patients. When medical treatment is begun early in life, the initially large and prominent clitoris may shrink slightly. As the surrounding structures grow normally, the clitoris becomes much less prominent, and surgical revision may not be required. When clitoral enlargement is conspicuous enough to interfere with parent-child bonding or formation of female gender identity in the patient or raises doubts in the parents about the true gender of the infant, corrective surgery of the genitalia should be carried out as early as possible, and certainly when the child is younger than 2 years of age.213 Psychoendocrinologically trained psychologists and/or psychiatrists provide a vital component of the treatment regimen because one of the major goals of therapy is to ensure that gender role, gender behavior, and gender identity are isosexual with the gender of assignment.215,216
Clitoral surgery must aim to preserve erectile tissue along with the dorsal neurovascular bundle and thus clitoral erotic sensation. It must be determined before menarche that vaginal formation permits adequate outflow of blood, and an early procedure ensuring this may have to be performed in rare cases. Vaginoplasty performed before regular sexual intercourse may require continual mechanical dilatation of the vagina. Surgery before the patient is able to take responsibility for this risks recurrent stenosis, formation of adhesions, and scarring, with a need for further surgery and permanent harm to the vaginal orifice. Too long a delay, conversely, risks harm to the patient’s sense of self as a normal female. All these factors are taken into account when the age for the necessary procedure(s) is chosen; this generally is done during the patient’s early to middle adolescence. In some patients, mechanical dilatation may be sufficient. In the hands of an experienced surgeon, vaginoplasty can yield excellent results.217 Because of the normal internal genitalia and gonads in these patients, normal puberty, fertility, and child-bearing are possible with early and proper therapeutic intervention.
Prenatal Diagnosis
Each pregnancy that occurs in a family in which steroid 21-hydroxylase deficiency has been identified has a 25% chance of resulting in an affected newborn. Since 1965, when Jeffcoate and associates218 correlated a clearly elevated value for amniotic fluid pregnanetriol with the diagnosis of an affected child (confirmed at term), amniotic fluid steroid hormone assay has been done in pregnancies at risk.219–221 Radioimmunoassay of amniotic fluid for 17-OHP has shown that in all patients affected with the salt-wasting form, the amniotic fluid 17-OHP concentration is unambiguously elevated222–226; however, in simple virilizing 21-hydroxylase deficiency, the 17-OHP level may not be elevated above normal.227 The amniotic fluid Δ4 androstenedione and testosterone levels also have been measured, but the latter value is less useful because testosterone is normally high in the amniotic fluid of a male fetus.225,228
Fetal cells (mostly fibroblasts) in the amniotic fluid may be cultured for DNA analysis. HLA serotyping in conjunction with hormonal assay was begun in 1979229 but is less sensitive than direct mutation analysis. Specific probes for 21-hydroxylase mutations allow direct and rapid identification of known mutations through the use of polymerase chain reaction (i.e., allele specific). Panels of oligonucleotide probes currently available for use in prenatal diagnosis230 are expected to identify well more than 95% of current 21-hydroxylase mutations.
According to the population studied, deletions of the active 21-hydroxylase gene (CYP21) occur in from 20% to more than 40% of patients (Northwest United Kingdom)231,232; the remaining 60% to 80% of cases represent missense and nonsense mutations, as well as small deletions, sometimes even complicated noncontiguous deletions. These mutations are commonly the result of gene conversions and nonreciprocal transfers of the nucleotide sequence of longer or shorter segments of the pseudogene (CYP21P) to the active gene, with deleterious results.
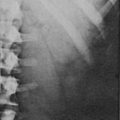
11β-Hydroxylase Deficiency
Levels of 11-deoxysteroids and metabolites in amniotic fluid and maternal urine have been found to be increased in pregnancies with a fetus affected with 11β-hydroxylase deficiency,233,234 suggesting that prenatal diagnosis of this disorder by hormonal measurement may be feasible,235 although the reliability of this method has not been reported on by other groups. However, the 11β-hydroxylase genes have been cloned, and many such mutations have been described. Where the mutations are known, prenatal diagnosis by DNA analysis of chorionic villus sampling (CVS) is possible and recommended. Experience with prenatal diagnosis in 11β-hydroxylase deficiency CAH is limited.
Prenatal Treatment
Dexamethasone crosses the placenta without undergoing significant metabolism and therefore has been used in the treatment of various fetal abnormalities. For the fetus at risk of 21- or 11β-hydroxylase deficiency, prophylactic treatment should be started as soon as pregnancy is confirmed.236–245 Chorionic villus sampling is currently advocated to permit diagnosis in the first trimester and to allow discontinuation of unnecessary steroid treatment in the case of a male fetus or an unaffected female fetus (Fig. 8-12). Initiation of steroid therapy by the sixth or seventh week of gestation should effectively suppress fetal adrenal androgen production in time to allow normal separation of the vaginal and urethral orifices and continued suppression through gestation, to prevent or reduce the degree of clitoromegaly (Fig. 8-13). The current recommendation is dexamethasone at 20 µg/kg of maternal prepregnancy weight, given in three divided doses. To date, no fetus of a mother treated with low-dose dexamethasone has been found to have any congenital malformations other than genital ambiguity. Specifically, no reports have described increases in the number of cases of cleft palate, placental degeneration, or fetal death, all of which are observed in rodent models of in utero exposure to high-dose glucocorticoids.246,247 Hirvikoski et al.248 reported that psychoneurological testing in prenatally treated children revealed no differences in psychopathology, behavioral problems, or adaptive functioning. It is interesting to note that children were described by their parents as being more sociable than controls (P = .042).248 We reported a study of all prenatal diagnoses between 1978 and 2002 that confirms the safety and efficacy of this treatment protocol. Of 595 pregnancies evaluated, 126 fetuses were found to be affected, 108 of these with classic forms and 64 of which were female. In 13 of these cases, the families declined dexamethasone treatment. Female newborns in this cohort were highly virilized at birth, with a mean Prader score of 3.77. Of 51 pregnant women who received treatment, 27 began dexamethasone early (before 9 weeks’ gestation) and continued to term. Female newborns in this group were only mildly virilized, with a mean Prader score of 1.04. The remaining 13 pregnant women began treatment late or were treated only partially. Female newborns in this group had an intermediate degree of virilization, with a mean Prader score of 3.00.245 Statistically significant adverse effects of dexamethasone treatment (relative to the untreated group) included an excess weight gain of 7.1 lb (3.2 kg) and the presence of edema and striae. No correlation was noted between prenatal dexamethasone treatment and fetal demise, birth weight, hypertension, or gestational diabetes mellitus.245 These data are in close agreement with those of the retrospective analysis of the European Society of Pediatric Endocrinology (ESPE).249 Normal birth weight and length and normal physical and psychological development were reported in treated fetuses (affected and unaffected) in the French multicenter study241 and in our series.245 These studies reflect the experience with prenatal treatment of pregnancies at risk for 21-hydroxylase deficiency; prenatal treatment for pregnancies at risk for 11β-hydroxylase deficiency has been similarly successful and is predicted to have the same outcome and degree of safety.
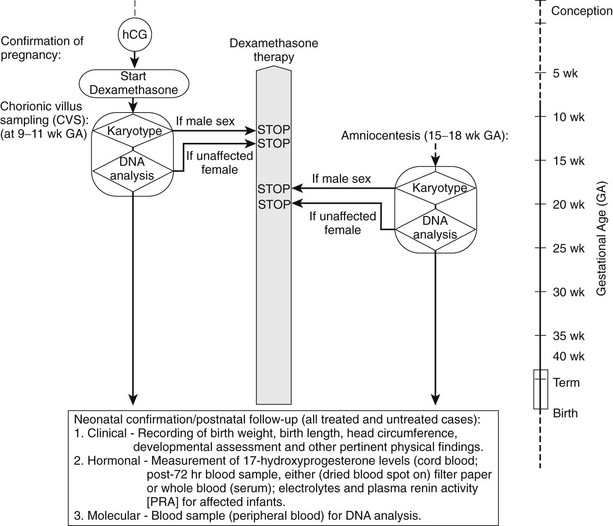
FIGURE 8-12 Protocol for prenatal diagnosis and treatment. hCG, Human chorionic gonadotropin. (From Mercado AB, Wilson RC, Cheng KC, et al: Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiency, J Clin Endocrinol Metab 80:2014–2020, 1995. © 1995, The Endocrine Society.)
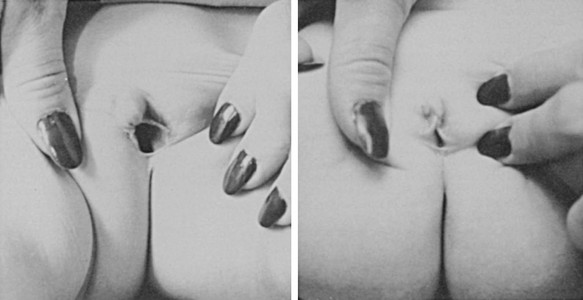
FIGURE 8-13 Untreated (left) and prenatally treated (right) female newborns. (From Speiser PW, Laforgia N, Kato K, et al: First trimester prenatal treatment and molecular genetic diagnosis of congenital adrenal hyperplasia [21-hydroxylase deficiency], J Clin Endocrinol Metab 70:838–848, 1990. © 1990, The Endocrine Society.)
Dexamethasone treatment should be initiated early in all at-risk pregnancies and maintained until confirmation is obtained that such treatment is not necessary, that is, in a genetic male or an unaffected female. This usually can be done by chorionic villus sampling at 9 to 10 weeks, or by amniocentesis at 16 to 18 weeks. The increase in risk for miscarriage after chorionic villus sampling is very low250 and may be offset by the benefit of early diagnosis (shortened time of exposure to steroid treatment). The pregnant woman must be made aware of the available options and led through these to understand the possible outcomes.
References
1. New, MI, White, PC, Pang, S, et al. The adrenal hyperplasias. In: Scriver CR, et al, eds. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1989:1881–1917.
2. Grumbach, MM, Conte, FA. Disorders of sex differentiation. In: Wilson JD, Foster DW, eds. Williams’ Textbook of Endocrinology. Philadelphia: WB Saunders; 1992:853–951.
3. Gabrilove, J, Sharma, D, Dorfman, R. Adrenocortical 11B-hydroxylase deficiency and virilism first manifest in the adult woman. N Engl J Med. 1965;272:1189–1194.
4. Newmark, D, Dluhy, R, Williams, G, et al. Partial 11-hydroxylase and 21-hydroxylase deficiencies in hirsute women. Am J Obstet Gynecol. 1977;127:594–598.
5. Cathelineau, G, Brerault, J, Fiet, W. Adrenocortical 11β-hydroxylation defect in adult women with postmenarchal onset of symptoms. J Clin Endocrinol Metab. 1980;51:287–291.
6. Birnbaum, M, Rose, L. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. 1984;63:445–451.
7. Hurwitz, A, Brautbar, C, Milwidsky, A. Combined 21- and 11β-hydroxylase deficiency in familial congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1985;60:631–638.
8. Clark, PA. Nonclassic 11-beta-hydroxylase deficiency: report of two patients and review. J Pediatr Endocr Metab. 2000;13:105–109.
9. De Crecchio, L. Sopra un caso di apparenze virile in una donna. Morgagni. 1865;7:1951.
10. Fibiger, J. Beiträge zur Kenntnis des weiblichen Scheinzwittertums. Virchows Arch Pathol Anat. 1905;181:1–51.
11. Apert, A. Dystrophies en relation avec des lésions de capsules surrénales. Hirsutisme et progeria. Bull Soc Pediatr (Paris). 1910;12:501–518.
12. Migeon, CJ. Diagnosis and treatment of adrenogenital disorders. In: DeGroot L, et al, eds. Endocrinology. Philadelphia: WB Saunders; 1989:1676–1704.
13. Wilkins, L. Adrenal cortex: virilizing adrenal hyperplasia; virilizing and feminizing tumors; and primary hyperaldosteronism. In: Wilkins L, ed. The Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence. Springfield, IL: Charles C Thomas; 1965:368–381.
14. Bongiovanni, AM. Detection of pregnanediol and pregnanetriol in urine of patients with adrenal hyperplasia: suppression with cortisone: a preliminary report. Bull Johns Hopkins Hosp. 1953;92:244–251.
15. Bongiovanni, AM, Root, AW. The adrenogenital syndrome. N Engl J Med. 1963;268:1283–1289.
16. Eberlein, WR, Bongiovanni, AM. Congenital adrenal hyperplasia with hypertension: unusual steroid pattern in blood and urine. J Clin Endocrinol Metab. 1955;15:1531–1534.
17. Prader, A, Siebenmann, RE. Nebenniereninsuffizienz bei kongenitaler Lipoid-hyperplasie der Nebennieren. Helv Paediatr Acta. 1957;12:569–595.
18. Biglieri, EG, Herron, MA, Brust, N. 17-Hydroxylation deficiency in man. J Clin Invest. 1966;45:1946–1954.
19. New, MI. Male pseudohermaphroditism due to 17 alpha-hydroxylase deficiency. J Clin Invest. 1970;49:1930–1941.
20. Dupont, B, Oberfield, SE, Smithwick, EM, et al. Close genetic linkage between HLA and congenital adrenal hyperplasia (21-hydroxylase deficiency). Lancet. 1977;2:1309–1312.
21. White, PC, New, MI, Dupont, B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci U S A. 1984;81:7505–7509.
22. Lin, D, Sugawara, T, Strauss, JF, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995;267:1828–1831.
23. Suwa, S, Shimozawa, K, Kitagawa, T, et al. Collaborative study on regional neonatal screening for congenital adrenal hyperplasia in Japan. In: Therell BLJ, ed. Advances in Neonatal Screening. New York: Elsevier; 1987:279–286.
24. Wallace, AM, Beastall, GH, Kennedy, R, et al. Congenital adrenal hyperplasia screening in 120,000 Scottish neonates. In: Therell BLJ, ed. Advances in Neonatal Screening. New York: Elsevier; 1987:293–295.
25. Sólyom, J, Hughes, IA. Value of selective screening for congenital adrenal hyperplasia in Hungary. Arch Dis Child. 1989;64:338–342.
26. Pang, SY, Wallace, MA, Hofman, L, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 1988;81:866–874.
27. Cutfield, WS, Webster, D. Newborn screening for congenital adrenal hyperplasia in New Zealand. J Pediatr. 1995;126:118–121.
28. Cacciari, E, Balsamo, A, et al. Neonatal screening for congenital adrenal hyperplasia. Arch Dis Child. 1983;58:803–806.
29. Cutfield, WS, Webster, D. Newborn screening for congenital adrenal hyperplasia in New Zealand. J Pediatr. 1995;126:118–121.
30. Allen, DB, Hoffman, GL, Fitzpatrick, P, et al. Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. J Pediatr. 1997;130:128–133.
31. Janejai, N, Krasao, P, et al. Congenital adrenal hyperplasia: should nationwide screening be implemented in Thailand? Southeast Asian J Trop Med Public Health. 2003;34(suppl 3):170–173.
32. Speiser, PW, Dupont, B, Rubinstein, P, et al. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Genet. 1985;37:650–667.
33. Sherman, SL, Aston, CE, Morton, NE, et al. A segregation and linkage study of classical and nonclassical 21-hydroxylase deficiency. Am J Hum Genet. 1988;42:830–838.
34. Dumic, M, Brkljacic, L, Speiser, P, et al. An update on the frequency of nonclassic deficiency of adrenal 21-hydroxylase in the Yugoslav population. Acta Endocrinol (Copenh). 1990;122:703–710.
35. Bonne-Tamir, B, Bodmer, JG, Bodmer, WF, et al. HLA polymorphism in Israel: an overall comparative analysis. Tissue Antigens. 1978;11:235–250.
36. Kidd, KK, Kidd, JR, Bonne-Tamir, B, et al. Nuclear DNA polymorphisms and population relationships. In: Bonne-Tamir B, Adam A, eds. Genetic Diversity among Jews. Diseases and Markers at the DNA Level. London: Oxford University Press; 1992:33–44.
37. Josso, N. Antimullerian hormone: new perspectives for a sexist molecule. Endocr Rev. 1986;7:421–433.
38. Klingensmith, G, Garcia, S, Jones, H, et al. Glucocorticoid treatment of girls with congenital adrenal hyperplasia: effects on height, sexual maturation, and fertility. J Pediatr. 1977;90:996–1004.
39. DiMartino-Nardi, J, Stoner, E, O’Connell, A, et al. The effect of treatment of final height in classical congenital adrenal hyperplasia (CAH). In: Illig R, Visser HKA, eds. Paediatric Endocrinology. Copenhagen: Acta Endocrinologica Copenhagen; 1986:305–314.
40. New, MI, Gertner, JM, Speiser, PW, et al. Growth and final height in classical and nonclassical 21-hydroxylase deficiency. Acta Paediatr Jpn. 1988;30:79–88.
41. Vogiatzi, MG, Lin-Su, K, New, MI. Treatment with growth hormone and GnRH analogue improves final height in children with congenital adrenal hyperplasia. In: Endocrine Society 2003. Philadelphia: The Endocrine Society; 2003.
42. Sciannamblo, MR, Cuccato, D, Chiumello, G, et al. Reduced bone mineral density and increased bone metabolism rate in young adult patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:4453–4458.
43. Falhammar, H, Filipsson, H, et al. Fractures and Bone Mineral Density in Adult Women with 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. 2007;92:4643–4649.
44. Barnes, RB, Rosenfeld, RL, Ehrmann, DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–1333.
45. Lobo, R, Goebelsmann, U. Adult manifestation of congenital adrenal hyperplasia due to incomplete 21-hydroxylase deficiency mimicking polycystic ovarian disease. Am J Obstet Gynecol. 1980;138:720–726.
46. Meyer-Bahlburg, HFL. What causes low rates of child-bearing in congenital adrenal hyperplasia? J Clin Endocrinol Metab. 1999;84:1844–1847.
47. Trinh, L, Nimkarn, S, et al. Growth and pubertal characteristics in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Pediatr Endocrinol Metab. 2007;20:883–891.
48. Prader, A. Vollkommen männliche äussere Genitalentwicklung und Salzverlustsyndrom bei Madchen mit kongenitalem Adrenogenitalem Syndrom. Helv Paediatr Acta. 1958;13:5–14.
49. Stikkelbroeck, NM, Otten, BJ, Pasic, A, et al. High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86:5721–5728.
50. Cabrera, M, Vogiatzi, M, New, M. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86:3070–3080.
51. Ravichandran, R, Lafferty, F, McGinniss, MJ, et al. Congenital adrenal hyperplasia presenting as massive adrenal incidentalomas in the sixth decade of life: report of two patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1996;81:1776–1779.
52. Beuschlein, F, Schulze, E, Mora, P, et al. Steroid 21-hydroxylase mutations and 21-hydroxylase messenger ribonucleic acid expression in human adrenocortical tumors. J Clin Endocrinol Metab. 1998;83:2585–2588.
53. Merke, DP, Chrousos, GP, Eisenhofer, G, et al. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med. 2000;343:1362–1368.
54. Tusie-Luna, M, Traktman, P, White, PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265:20916–20922.
55. Chiou, SH, Hu, MC, Chung, BC. A missense mutation at Ile172-Asn or Arg356-Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265:3549–3552.
56. Horner, JM, Hintz, RL, Luetscher, JA. The role of renin and angiotensin in salt-losing, 21-hydroxylase-deficient congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1979;48:776–783.
57. Stoner, E, Dimartino-Nardi, J, Kuhnle, U, et al. Is salt-wasting in congenital adrenal hyperplasia due to the same gene as the fasciculata defect? Clin Endocrinol (Oxf). 1986;24:9–20.
58. Morel, Y, David, M, Forest, MG, et al. Gene conversions and rearrangements cause discordance between inheritance of forms of 21-hydroxylase deficiency and HLA types. J Clin Endocrinol Metab. 1989;68:592–599.
59. Speiser, PW, Agdere, L, Ueshiba, H, et al. Aldosterone synthesis in salt-wasting congenital adrenal hyperplasia with complete absence of adrenal 21-hydroxylase. N Engl J Med. 1991;324:145–149.
60. Dupont, B, Pollack, M, Levine, L, et al. Congenital adrenal hyperplasia and HLA: joint report from the Eighth International Histocompatibility Workshop. In: Terasaki P, ed. Histocompatibility Testing 1980. Los Angeles: UCLA Tissue Typing Laboratory; 1981:693–706.
61. Trowsdale, J, Ragoussis, J, Campbell, RD, et al. Map of the human MHC. Immunol Today. 1991;12:443–446.
62. Dupont, B, Virdis, R, Lerner, AJ, et al. Distinct HLA-B antigen associations for the salt-wasting and simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. In: Albert ED, Baur M, Mayr W, eds. Histocompatibility Testing 1984. Heidelberg: Springer-Verlag; 1984:660.
63. Levine, LS, Dupont, B, Lorenzen, F, et al. Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1980;51:1316–1324.
64. Kohn, B, Levine, LS, Pollack, MS, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1982;55:817–827.
65. Temeck, JW, Pang, SY, Nelson, C, et al. Genetic defects of steroidogenesis in premature pubarche. J Clin Endocrinol Metab. 1987;64:609–617.
66. Granoff, A, Chasalow, F, Blethen, S. 17-Hydroxyprogesterone responses to adrenocorticotropin in children with premature adrenarche. J Clin Endocrinol Metab. 1985;60:409–415.
67. Lucky, A, Rosenfield, R, McGuire, J, et al. Adrenal androgen hyperresponsiveness to adrenocorticotropin in women with acne and/or hirsutism: adrenal enzyme defects and exaggerated adrenarche. J Clin Endocrinol Metab. 1986;62:840–848.
68. Rose, L, Newmark, S, Strauss, J, et al. Adrenocortical hydroxylase deficiencies in acne vulgaris. J Invest Dermatol. 1976;66:324–326.
69. Pang, SY, Lerner, AJ, Stoner, E, et al. Late-onset adrenal steroid 3 beta-hydroxysteroid dehydrogenase deficiency. I. A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab. 1985;60:428–439.
70. Child, D, Bullock, D, Anderson, D. Adrenal steroidogenesis in hirsute women. Clin Endocrinol (Oxf). 1980;12:595–601.
71. Gibson, M, Lackritz, R, Schiff, I, et al. Abnormal adrenal responses to adrenocorticotropic hormone in hyperandrogenic women. Fertil Steril. 1980;33:43–48.
72. Chrousos, G, Loriaux, D, Mann, D, et al. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. 1982;96:143–148.
73. Knochenhauer, ES, Cortet-Rudelli, C, Cunningham, RD, et al. Carriers of 21-hydroxylase deficiency are not at increased risk for hyperandrogenism. J Clin Endocrinol Metab. 1997;82:479–485.
74. Riddick, D, Hammond, C. Adrenal virilism due to 21-hydroxylase deficiency in the postmenarchial female. Obstet Gynecol. 1975;45:21–24.
75. Birnbaum, M, Rose, L. The partial adrenocortical hydroxylase deficiency syndrome in infertile women. Fertil Steril. 1979;32:536–541.
76. Chrousos, GP, Loriaux, DL, Sherines, RJ, et al. Bilateral testicular enlargement resulting from inapparent 21-hydroxylase deficiency. J Urol. 1981;126:127–128.
77. White, PC, Grossberger, D, Onufer, BJ, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci U S A. 1985;82:1089–1093.
78. Carroll, MC, Campbell, RD, Porter, RR. Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci U S A. 1985;82:521–525.
79. White, PC, New, MI, Dupont, B. Structure of the human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A. 1986;83:5111–5115.
80. Higashi, Y, Yoshioka, H, et al. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Sci U S A. 1986;83:2841–2845.
81. Harada, F, Kimura, A, et al. Gene conversion-like events cause steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. Proc Natl Acad Sci U S A. 1987;84:8091–8094.
82. Amor, M, Parker, KL, et al. Mutation in the CYP21B gene (Ile-172-Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85:1600–1607.
83. Speiser, PW, New, MI, White, PC. Molecular genetic analysis of nonclassic steroid 21-hydroxylase deficiency associated with HLA-B14,DR1. N Engl J Med. 1988;319:19–23.
84. Rocha, RO, Billerbeck, AE, et al. The degree of external genitalia virilization in girls with 21-hydroxylase deficiency appears to be influenced by the CAG repeats in the androgen receptor gene. Clin Endocrinol (Oxf). 2008;68:226–232.
85. Bongiovanni, AM, Eberlein, WR, Goldman, AS, et al. Disorders of adrenal steroid biogenesis. Recent Prog Horm Res. 1967;23:375–449.
86. Zerah, M, Pang, SY, New, MI. Morning salivary 17-hydroxyprogesterone is a useful screening test for nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1987;65:227–232.
87. Pang, S, Hotchkiss, J, Drash, AL, et al. Microfilter paper method for 17 alpha-hydroxyprogesterone radioimmunoassay: its application for rapid screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1977;45:1003–1008.
88. Therrell, BL, Jr., Berenbaum, SA, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101(4):583–590.
89. Cavarzere, P, Camilot, M, et al. Neonatal screening for congenital adrenal hyperplasia in North-Eastern Italy: a report three years into the program. Horm Res. 2005;63:180–186.
90. Gruñieiro-Papendieck, L, Chiesa, A, et al. Neonatal screening for congenital adrenal hyperplasia: experience and results in Argentina. J Pediatr Endocrinol Metab. 2008;21:73–78.
91. Levine, LS, Rauh, W, Gottesdiener, K, et al. New studies of the 11 beta-hydroxylase and 18-hydroxylase enzymes in the hypertensive form of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1980;50:258–263.
92. Chua, S, Szabo, P, Vitek, A, et al. Cloning of cDNA encoding steroid 11 beta-hydroxylase (P450c11). Proc Natl Acad Sci U S A. 1987;84:7193–7197.
93. Mornet, E, Dupont, J, Vitek, A, et al. Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450(11) beta). J Biol Chem. 1989;264:20961–20967.
94. Taymans, SE, Pack, S, Pak, E, et al. Human CYP11B2 (aldosterone synthase) maps to chromosome 8q24.3. J Clin Endocrinol Metab. 1998;83:1033–1036.
95. Rösler, A, Leiberman, E. Enzymatic defects of steroidogenesis: 11 beta-hydroxylase deficiency congenital adrenal hyperplasia. In: New MI, Levine LS, eds. Adrenal Diseases in Childhood: Pathophysiologic and Clinical Aspects. Basel: S. Karger; 1984:47–71.
96. Rösler, A, Leiberman, E, Sack, J. Clinical variability of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Horm Res. 1982;16:133–141.
97. Pang, S, Levine, LS, Lorenzen, F, et al. Hormonal studies in obligate heterozygotes and siblings of patients with 11 beta-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1980;50:586–589.
98. White, PC, Curnow, KM, Pascoe, L. Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev. 1994;15:421–438.
99. New, MI, Seaman, MP. Secretion rates of cortisol and aldosterone precursors in various forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1970;30:361–371.
100. Gandy, HM, Keutmann, EH, Izzo, AJ. Characterization of urinary steroids in adrenal hyperplasia: isolation of metabolites of cortisol, compound S, and deoxycorticosterone from a normotensive patient with adrenogenital syndrome. J Clin Invest. 1960;39:364–377.
101. Blunck, W. Die Beta-ketolischen Cortisol und Corticosteronmetaboliten sowie die 11-oxy-und 11-desoxy-17-ketosteroide im urin von Kindern. Acta Endocrinol. 1968;59(Suppl 134):9–112.
102. Green, OC, Migeon, CJ, Wilkins, L. Urinary steroids in the hypertensive form of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1960;20:929–946.
103. Glenthoj, A, Nielsen, MD, Starup, J. Congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency: final diagnosis in adult age in three patients. Acta Endocrinol (Copenh). 1980;93:94–99.
104. New, M, Nemery, R, Chow, D, et al. The adrenal and hypertension: from cloning to clinic. In: Ares-Serona Symposium. New York: Raven Press; 1989.
105. Zachmann, M, Tassinari, D, Prader, A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983;56:222–229.
106. Rösler, A. Classic and nonclassic congenital adrenal hyperplasia among non-Ashkenazi Jews. In: Bonne-Tamir B, Adam A, eds. New perspectives on genetic markers and diseases among the Jewish people. Oxford: Oxford University Press; 1992:488.
107. Kawamoto, T, Mitsuuchi, Y, Ohnishi, T, et al. Cloning and expression of a cDNA for human cytochrome P-450aldo as related to primary aldosteronism. Biochem Biophys Res Commun. 1990;173:309–316.
108. Curnow, KM, Tusie-Luna, MT, Pascoe, L, et al. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Mol Endocrinol. 1991;5:1513–1522.
109. Ogishima, T, Shibata, H, Shimada, H, et al. Aldosterone synthase cytochrome P-450 expressed in the adrenals of patients with primary aldosteronism. J Biol Chem. 1991;266:10731–10734.
110. Nebert, DW, Adesnik, M, Coon, MJ, et al. The P450 gene superfamily: recommended nomenclature. DNA. 1987;6:1–13.
111. White, PC, Dupont, J, New, MI, et al. A mutation in CYP11B1 (Arg-448-His) associated with steroid 11 beta-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest. 1991;87:1664–1667.
112. Curnow, KM, Slutsker, L, Vitek, J, et al. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc Natl Acad Sci U S A. 1993;90:4552–4556.
113. Geley, S, Kapelari, K, Johrer, K, et al. CYP11B1 mutations causing congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab. 1996;81:2896–2901.
114. Eberlein, W, Bongiovanni, A. Plasma and urinary corticosteroids in the hypertensive form of congenital adrenal hyperplasia. J Biol Chem. 1956;223:85–94.
115. Mimouni, M, Kaufman, H, Roitman, A, et al. Hypertension in a neonate with 11 beta-hydroxylase deficiency. Eur J Pediatr. 1985;143:231–233.
116. Hochberg, Z, Schechter, J, et al. Growth and pubertal development in patients with congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency. Arch J Dis Child. 1985;139:771–776.
117. Mitsuuchi, Y, Kawamoto, K, Ulick, S, et al. Congenitally defective aldosterone biosynthesis in humans: inactivation of the P450c18 gene CYP11B2 due to nucleotide deletion in CMO I deficient patients. Biochem Biophys Res Commun. 1993;190:864–869.
118. Portrat-Doyen, S, Tourniaire, J, Richard, O, et al. Isolated aldosterone synthetase deficiency caused by simultaneous E98D and V386A mutations in the CYP11B2 gene. J Clin Endocrinol Metab. 1998;83:4156–4161.
119. Hauffa, BP, Sólyom, J, Gláz, E, et al. Severe hypoaldosteronism due to corticosterone methyl oxidase type II deficiency in two boys: metabolic and gas chromatography-mass spectrometry studies. Eur J Pediatr. 1991;150:149–153.
120. Picco, P, Garibaldi, L, Cotellessa, M, et al. Corticosterone methyl oxidase type II deficiency: a cause of failure to thrive and recurrent dehydration in early infancy. Eur J Pediatr. 1992;151:170–173.
121. Globerman, H, Rosler, A, Theodor, R, et al. An inherited defect in aldosterone biosynthesis caused by a mutation in or near the gene for steroid 11-hydroxylase. N Engl J Med. 1988;319:1193–1197.
122. Pascoe, L, Curnow, KM, Slutsker, L, et al. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc Natl Acad Sci U S A. 1992;89:4996–5000.
123. New, MI, Peterson, RE. A new form of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1967;27:300–305.
124. Sutherland, DJ, Ruse, JL, Laidlaw, JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. CMAJ. 1966;95:1109–1119.
125. Oberfield, SE, Levine, LS, Stoner, E, et al. Adrenal glomerulosa function in patients with dexamethasone suppressible hyperaldosteronism. J Clin Endocrinol Metab. 1981;53:158–164.
126. New, MI, Oberfield, SE, Levine, LS, et al. Demonstration of autosomal dominant transmission and absence of HLA linkage in dexamethasone suppressible hyperaldosteronism. Lancet. 1980;1:550–551.
127. Ulick, S, Chu, M. Hypersecretion of a new corticosteroid, 18-hydroxycortisol in two types of adrenocortical hypertension. Clin Exp Hypertens A. 1982;4:1771–1777.
128. Gomez-Sanchez, C, Montgomery, M, Ganguly, A, et al. Elevated urinary excretion of 18-oxocortisol in glucocorticoid-suppressible aldosteronism. J Clin Endocrinol Metab. 1984;59:1022–1024.
129. Lifton, RP, Dluhy, RG, Powers, M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355:262–265.
130. Pascoe, L, Curnow, KM, Slutsker, L, et al. Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal crossovers between CYP11B1 and CYP11B2. Proc Natl Acad Sci U S A. 1992;89:8327–8331.
131. Lachance, Y, Luu-The, V, Labrie, C, et al. Characterization of human 3β-hydroxysteroid dehydrogenase/D5-D4 isomerase gene and its expression in mammalian cells. J Biol Chem. 1990;265:20469–20475.
132. Lachance, Y, Luu-The, V, Verreault, H, et al. Structure of the human type II 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase (3 beta-HSD) gene: adrenal and gonadal specificity. DNA Cell Biol. 1991;10:701–711.
133. Rhéaume, E, Simard, J, Morel, Y, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat Genet. 1992;1:239–245.
134. Simard, J, Rheaume, E, Sanchez, R, et al. Molecular basis of congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol. 1993;7:716–728.
135. Bongiovanni, A. Adrenogenital syndrome with deficiency of 3beta-hydroxysteroid dehydrogenase. J Clin Invest. 1962;41:2086–2092.
136. Bongiovanni, AM. Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase. In: New MI, Levine LS, eds. Adrenal Diseases in Childhood. Basel: Karger; 1984:72–82.
137. Pang, S, Levine, LS, Stoner, E, et al. Non-salt-losing congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency with normal glomerulosa function. J Clin Endocrinol Metab. 1983;56:808–818.
138. Rosenfield, RL, Rich, BH, Wolfsdorf, JI, et al. Pubertal presentation of congenital delta 5–3 beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1980;51:345–353.
139. Schram, P, Zerah, M, Mani, P, et al. Nonclassical 3 beta-hydroxysteroid dehydrogenase deficiency: a review of our experience with 25 female patients. Fertil Steril. 1992;58:129–136.
140. Zerah, M, Schram, P, New, M. The diagnosis and treatment of nonclassical 3β-HSD deficiency. Endocrinologist. 1991;1:75–81.
141. Lorence, MC, Corbin, CJ, Kamimura, N, et al. Structural analysis of the gene encoding human 3β-hydroxysteroid dehydrogenase/D5,4-isomerase. Mol Endocrinol. 1990;4:1850–1855.
142. Katsumata, N, Tanae, A, Yasunaga, T, et al. A novel missense mutation in the type II 3 beta-hydroxysteroid dehydrogenase gene in a family with classical salt-wasting congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency. Hum Mol Genet. 1995;4:745–746.
143. Tajima, T, Fujieda, K, Nakae, J, et al. Molecular analysis of type II 3β-hydroxysteroid dehydrogenase gene in Japanese patients with classical 3β-hydroxysteroid dehydrogenase deficiency. Hum Mol Genet. 1995;4:969–971.
144. Morel, Y, Mebarki, F, Rheaume, E, et al. 3β-Hydroxysteroid dehydrogenase: Contribution made by the molecular genetics of 3β-hydroxysteroid dehydrogenase deficiency. Steroids. 1997;62:176–184.
145. Lutfallah, C, Wang, W, Mason, JI, et al. Newly proposed hormonal criteria via genotypic proof for type II 3-beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611–2622.
146. Yanase, T, Simpson, E, Waterman, M. 17 Alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991;12:91–108.
147. Miura, K, Yasuda, K, Yanase, T, et al. Mutation of cytochrome P-45017a gene (CYP17) in a Japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: with a review of Japanese patients with mutations of CYP17. J Clin Endocrinol Metab. 1996;81:3797–3801.
148. Zachmann, M, Prader, A. 17,20-Desmolase deficiency. In: New M, Levine L, eds. Adrenal Disease in Childhood. Basel: Karger, 1984.
149. Zachmann, M, Kempken, B, Manella, B, et al. Conversion from pure 17,20-desmolase- to combined 17,20-desmolase/17 alpha-hydroxylase deficiency with age. Acta Endocrinol (Copenh). 1992;127:97–99.
150. Martin, RM, Lin, CJ, Costa, EMF, et al. P450c17 deficiency in Brazilian patients: biochemical diagnosis through progesterone levels confirmed by CYP17 genotyping. J Clin Endocrinol Metab. 2003;88:5739–5746.
151. Mantero, F, Scaroni, C, Pasini, CV, et al. No linkage between HLA and congenital adrenal hyperplasia due to 17b-hydroxylase deficiency [letter]. N Engl J Med. 1980;303:530.
152. Boehmer, ALM, Brinkmann, AO, Sandkuijl, LA, et al. 17-Beta-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J Clin Endocrinol Metab. 1999;84:4713–4721.
153. Levran, D, Ben-Shlomo, I, Pariente, C, et al. Familial partial 17,20-desmolase and 17alpha-hydroxylase deficiency presenting as infertility. J Assist Reprod Genet. 2003;20:21–28.
154. Ulick, S. Diagnosis and nomenclature of the disorders of the terminal portion of the aldosterone biosynthetic pathway. J Clin Endocrinol Metab. 1976;43:92–96.
155. Griffing, GT, Wilson, TE, Holbrook, MM, et al. Plasma and urinary 19-nor-deoxycorticosterone in 17 alpha-hydroxylase deficiency syndrome. J Clin Endocrinol Metab. 1984;59:1011–1015.
156. Wit, JM, van Roermund, HPC, Oostdik, W, et al. Heterozygotes for 17α-hydroxylase deficiency can be detected with a short ACTH test. Clin Endocrinol. 1988;28:657–664.
157. Miura, K, Yoshinaga, K, Goto, K, et al. A case of glucocorticoid-responsive hyperaldosteronism. J Clin Endocrinol Metab. 1968;28:1807–1815.
158. Chung, B, Picado-Leonard, J, Haniu, M, et al. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci U S A. 1987;84:407–411.
159. Matteson, K, Picado-Leonard, J, Chung, B, et al. Assignment of the gene for adrenal P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase) to human chromosome 10. J Clin Endocrinol Metab. 1986;63:789–791.
160. Imai, T, Yanase, T, Waterman, M, et al. Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17 alpha-hydroxylase deficiency. Hum Genet. 1992;89:95–96.
161. D’Armiento, M, Reda, G, Kater, C, et al. 17a-Hydroxylase deficiency: mineralocorticoid hormone profiles in an affected family. J Clin Endocrinol Metab. 1983;56:697–701.
162. Williamson, L, Arlt, W, et al. Linking Antley-Bixler syndrome and congenital adrenal hyperplasia: a novel case of P450 oxidoreductase deficiency. Am J Med Genet. 2006;140A:1797–1803.
163. Flück, CE, Tajima, T, Pandey, AV, et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet. 2004;36:228–230.
164. Arlt, W, Walker, EA, Draper, N, et al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet. 2004;363:2128–2135.
165. Shackleton, C, Marcos, J, Malunowicz, EM, et al. Biochemical diagnosis of Antley-Bixler syndrome by steroid analysis. Am J Med Genet (Part A). 2004;128A:223–231.
166. Prader, A, Gurtner, HP. Das Syndrom des Pseudohermaphroditismus masculinus bei kongenitaler Nebennierenden-Hyperplasie ohne Androgenüberproduktion (Adrenaler Pseudohermaphroditismus masculinus). Helv Paediatr Acta. 1955;10:397–412.
167. Yoo, HW, Kim, GH. Molecular and clinical characterization of Korean patients with congenital lipoid adrenal hyperplasia. J Pediatr Endocrinol Metab. 1998;11:707–711.
168. Bose, HS, Sato, S, Aisenberg, J, et al. Mutations in the steroidogenic acute regulatory protein (StAR) in six patients with congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab. 2000;85:3636–3639.
169. Tajima, T, Fujieda, K, Kouda, N, et al. Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocrinol Metab. 2001;86:3820–3825.
170. Katsumata, N, Ohtake, M, Hojo, T, et al. Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J Clin Endocrinol Metab. 2002;87:3808–3813.
171. Baker, BY, Lin, L, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab. 2006;91:4781–4785.
172. Bose, HS, Sugawara, T, Strauss, JF, III., et al. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med. 1996;335:1870–1878.
173. Caron, KM, Soo, S-C, Wetsel, WC, et al. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Nat Acad Sci U S A. 1997;94:11540–11545.
174. Luo, X, Ikeda, Y, Parker, KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490.
175. Harris, AN, Mellon, PL. The basic helix-loop-helix, leucine zipper transcription factor, USF (upstream stimulatory factor), is a key regulator of SF-1 (steroidogenic factor-1) gene expression in pituitary gonadotrope and steroidogenic cells. Mol Endocrinol. 1998;12:714–726.
176. Hammer, GD, Krylova, I, Zhang, Y, et al. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: integration of hormone signaling in reproduction and stress. Mol Cell. 1999;3:521–526.
177. Tremblay, A, Tremblay, GB, Labrie, F, et al. Ligand-independent recruitment of SRC-1 to estrogen receptor beta through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–519.
178. Shen, W-H, Moore, CCD, Ikeda, Y, et al. Nuclear receptor steroidogenic factor 1 regulates the Mullerian inhibiting substance gene: a link to the sex determination cascade. Cell. 1994;77:651–661.
179. Nachtigal, M, Hirokawa, Y, Enyeart-Van, HD, et al. Wilms’ tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell. 1998;93:445–454.
180. Sekido, R, Lovell-Badge, R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453:930–934.
181. Achermann, JC, Ito, M, Ito, M, et al. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans [letter]. Nat Genet. 1999;22:125–126.
182. Achermann, JC, Ozisik, G, Ito, M, et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002;87:1829–1833.
183. Biason-Lauber, A, Schoenle, EJ. Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet. 2000;67:1563–1568.
184. Biason-Lauber, A, Schoenle, EJ. Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet. 2000;67:1563–1568.
185. McCabe, ERB. Sex and the single DAX1: too little is bad, but can we have too much? J Clin Invest. 1996;98:881–882.
186. Zanaria, E, Muscatelli, F, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. 1994;372:635–641.
187. Swain, A, Zanaria, E, et al. Mouse Dax1 expression is consistent with a role in sex determination as well as in adrenal and hypothalamus function. Nat Genet. 1996;12:404–409.
188. Lin, L, Gu, W-X, et al. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: ten years’ experience. J Clin Endocrinol Metab. 2006;91:3048–3054.
189. Wilkins, L, Lewis, R, Klein, R, et al. The suppression of androgen secretion by cortisone in a case of congenital adrenal hyperplasia. Bull Johns Hopkins Hosp. 1950;86:249–252.
190. Bartter, F. Adrenogenital syndromes from physiology to chemistry, 1950–1975. In: Lee P, ed. Congenital Adrenal Hyperplasia. Baltimore: University Park Press; 1977:9–18.
191. New, MI. An update of congenital adrenal hyperplasia. Ann N Y Acad Sci. 2004;1038:14–43.
192. Godard, C, Riondel, A, Veyrat, R. Plasma renin activity and aldosterone secretion in congenital adrenal hyperplasia. Pediatrics. 1968;41:883–904.
193. Simopoulos, A, Marshall, J, Delea, C, et al. Studies on the deficiency of the deficiency of 21-hydroxylation in patients with congenital adrenal hyperplasia. J Clin Endocrinol. 1971;32:438–443.
194. Strickland, A, Kotchen, T. A study of the renin-aldosterone system in congenital adrenal hyperplasia. J Pediatr. 1972;81:962–969.
195. Dillon, M. Plasma renin activity and aldosterone concentrations in children: results in salt-wasting states. Arch Dis Child. 1975;50:330.
196. Edwin, C, Lanes, R, Migeon, C. Persistence of the enzymatic block in adolescent patients with salt-losing congenital adrenal hyperplasia. J Pediatr. 1979;95:534.
197. Rösler, A, Levine, LS, Schneider, B, et al. The interrelationship of sodium balance, plasma renin activity and ACTH in congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1977;45:500–512.
198. Kuhnle, U, Rosler, A, Pareira, JA, et al. The effects of long-term normalization of sodium balance on linear growth in disorders with aldosterone deficiency. Acta Endocrinol (Copenh). 1983;102:577–582.
199. Winter, J. Current approaches to the treatment of congenital adrenal hyperplasia [editorial]. J Pediatr. 1980;97:81–82.
200. Korth-Schutz, S, Virdis, R, Saenger, P, et al. Serum androgens as a continuing index of adequacy of treatment of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1978;46:452–458.
201. Golden, M, Lippe, B, Kaplan, S. Management of congenital adrenal hyperplasia using serum dehydroepiandrosterone sulfate and 17-hydroxyprogesterone concentrations. Pediatrics. 1978;61:867–871.
202. Döhler, K. The special case of hormonal imprinting, the neonatal influence of sex. Experientia. 1986;42:759–769.
203. Döhler, K, Hancke, J, Srivastava, S. Participation of estrogens in female sexual differentiation of the brain: Neuroanatomical, neuroendocrine, and behavioral evidence. Prog Brain Res. 1984;61:99–117.
204. Berenbaum, SA. Congenital adrenal hyperplasia: intellectual and psychosexual functioning. In: Holmes CPS, ed. Psychoneuroendocrinology: Brain, Behavioral, and Hormonal Interactions. New York: Springer-Verlag; 1990:227–260.
205. Nass, R, Baker, S. Learning disabilities in children with congenital adrenal hyperplasia. J Child Neurol. 1991;6:306–312.
206. Nass, R, Heier, L, Moshang, T, et al. Magnetic resonance imaging in the congenital adrenal hyperplasia population: increased frequency of white-matter abnormalities and temporal lobe atrophy. J Child Neurol. 1997;12:181–186.
207. Nordenstrom, A, Servin, A, Bohlin, G, et al. Sex-typed toy play behavior correlates with the degree of prenatal androgen exposure assessed by CYP21 genotype in girls with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87:5119–5124.
208. Mueller, SC, Temple, V, et al. Early androgen exposure modulates spatial cognition in congenital adrenal hyperplasia (CAH). Psychoneuroendocrinology. 2008;33:973–980.
209. Long, DN, Wisniewski, CJ. Gender role across development in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Pediatr Endocrinol Metab. 2004;17:1367–1373.
210. Herdt, G, Davidson, J. The Sambia “turnim-man”: sociocultural and clinical aspects of gender formation in male pseudohermaphrodites with 5-alpha-reductase deficiency in Papua New Guinea. Arch Sex Behav. 1988;17:33–56.
211. Price, P, Wass, J, Griffin, J, et al. High dose androgen therapy in male pseudohermaphroditism due to 5 alpha-reductase deficiency and disorders of the androgen receptor. J Clin Invest. 1984;74:1496–1508.
212. Money, J, Hampson, JG, Hampson, JL. Hermaphroditism: recommendations concerning assignment of sex, change of sex, and psychologic management. Bull Johns Hopkins Hosp. 1955;96:284–300.
213. Money, J, Ehrhardt, AA. Differentiation and dimorphism of gender identity from conception to maturity. In: Man and Woman, Boy and Girl. Baltimore: Johns Hopkins University Press; 1972.
214. Dessens, AB, Slijper, FM, et al. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav. 2005;34:389–397.
215. Baker, S. Psychological management of intersex children. In: Josso N, ed. The Intersex Child. Basel: Karger, 1981.
216. Meyer-Bahlburg, HFL. Gender identity development in intersex patients. Child Adolesc Psychiatry Clin North Am. 1993;2:501–512.
217. Nihoul-Fekete, C. Feminizing genitoplasty in the intersex child. In: Josso N, ed. The Intersex Child. Basel: Karger, 1981.
218. Jeffcoate, T, Fleigner, J, Russell, S, et al. Diagnosis of the adrenogenital syndrome before birth. Lancet. 1965;2:553–555.
219. Merkatz, IR, New, MI, Peterson, RE, et al. Prenatal diagnosis of adrenogenital syndrome by amniocentesis. J Pediatr. 1969;75:977–982.
220. New, MI, Levine, LS. Congenital adrenal hyperplasia. In: Harris H, Hirschhorn K, eds. Advances in Human Genetics. New York: Plenum; 1973:251–326.
221. Levine, L. Prenatal detection of congenital adrenal hyperplasia. In: Milunsky A, ed. Genetic Disorders and the Fetus. New York: Plenum; 1986:369–385.
222. Frasier, S, Thorneycroft, I, Weiss, B, et al. Elevated amniotic fluid concentration of 17 alpha-hydroxyprogesterone in congenital adrenal hyperplasia [letter]. J Pediatr. 1975;86:310–312.
223. Nagamani, M, McDonough, P, Ellegood, J, et al. Maternal and amniotic fluid 17 alpha-hydroxyprogesterone levels during pregnancy: diagnosis of congenital adrenal hyperplasia in utero. Am J Obstet Gynecol. 1978;130:791–794.
224. Hughes, I, Laurence, K. Antenatal diagnosis of congenital adrenal hyperplasia. Lancet. 1979;2:7–9.
225. Pang, S, Levine, LS, Cederqvist, LL, et al. Amniotic fluid concentrations of delta 5 and delta 4 steroids in fetuses with congenital adrenal hyperplasia due to 21 hydroxylase deficiency and in anencephalic fetuses. J Clin Endocrinol Metab. 1980;51:223–229.
226. Hughes, I, Laurence, K. Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency by amniotic fluid steroid analysis. Prenat Diagn. 1982;2:97–102.
227. Pang, S, Pollack, MS, Loo, M, et al. Pitfalls of prenatal diagnosis of 21-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1985;61:89–97.
228. Frasier, S, Weiss, B, Horton, R. Amniotic fluid testosterone: implications for the prenatal diagnosis of congenital adrenal hyperplasia. J Pediatr. 1974;84:738–741.
229. Pollack, MS, Maurer, D, Levine, LS, et al. Prenatal diagnosis of congenital adrenal hyperplasia (21-hydroxylase deficiency) by HLA typing. Lancet. 1979;1:1107–1108.
230. Speiser, PW, Dupont, J, Zhu, D, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90:584–595.
231. Werkmeister, JW, New, MI, Dupont, B, et al. Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Hum Genet. 1986;39:461–469.
232. Collier, S, Sinnott, PJ, Dyer, PA, et al. Pulsed field gel electrophoresis identifies a high degree of variability in the number of tandem 21-hydroxylase and complement C4 gene repeats in 21-hydroxylase deficiency. EMBO J. 1989;8:1393–1402.
233. Rösler, A, Leiberman, E, Rosenmann, A. Prenatal diagnosis of 11beta hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1979;49:546–551.
234. Schumert, Z, Rosenmann, A, Landau, H, et al. 11-Deoxycortisol in amniotic fluid: prenatal diagnosis of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Clin Endocrinol. 1980;12:257–260.
235. Rösler, A, Weshler, N, Leiberman, E, et al. 11β-Hydroxylase deficiency congenital adrenal hyperplasia: update of prenatal diagnosis. J Clin Endocrinol Metab. 1988;66:830–838.
236. David, M, Forest, MG. Prenatal treatment of congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. J Pediatr. 1984;105:799–803.
237. Evans, M, Chrousos, G, Mann, D, et al. Pharmacologic suppression of the fetal adrenal gland in utero. Attempted prevention of abnormal external genital masculinization in suspected congenital adrenal hyperplasia. JAMA. 1985;253:1015–1020.
238. Dorr H, Sippell W, Haack D, et al: Pitfalls of prenatal treatment of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Paper presented at the 25th Annual Meeting of the European Society for Paediatric Endocrinology, 1986, Zurich.
239. Forest, M, Betuel, H, David, M. Traitement antenatal de l’hyperplasie congenitale des surrenales par deficit en 21-hydroxylase: Etude multicentrique. Ann Endocrinol. 1987;48:31–34.
240. Speiser, PW, Laforgia, N, Kato, K, et al. First trimester prenatal treatment and molecular genetic diagnosis of congenital adrenal hyperplasia (21-hydroxylase deficiency). J Clin Endocrinol Metab. 1990;70:838–848.
241. Forest, MG, David, M, Morel, Y. Prenatal diagnosis and treatment of 21-hydroxylase deficiency. J Steroid Biochem Mol Biol. 1993;45:75–82.
242. Mercado, AB, Wilson, RC, Cheng, KC, et al. Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1995;80:2014–2020.
243. Carlson, AD, Obeid, JS, Kanellopoulou, N, et al. Congenital adrenal hyperplasia: update on prenatal diagnosis and treatment. J Steroid Biochem Mol Biol. 1999;69:19–29.
244. New, MI, Carlson, A, Obeid, J, et al. Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab. 2001;86:5651–5657.
245. New, MI, Carlson, A, Obeid, J, et al. Update: prenatal diagnosis for congenital adrenal hyperplasia in 595 pregnancies. Endocrinologist. 2003;13:233–239.
246. Goldman, A, Sharpior, B, Katsumata, M. Human foetal palatal corticoid receptors and teratogens for cleft palate. Nature. 1978;272:464–466.
247. Lajic, S, Levo, A, et al. A cluster of missense mutations at Arg356 of human steroid 21-hydroxylase may impair redox partner interaction. Hum Genet. 1997;99:704–709.
248. Hirvikoski, T, Nordenstrom, A, et al. Long-term follow-up of prenatally treated children at risk for congenital adrenal hyperplasia: does dexamethasone cause behavioural problems? Eur J Endocrinol. 2008;159:309–316.
249. Forest, MG, Dörr, HG. Prenatal therapy in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: retrospective follow-up study of 253 treated pregnancies in 215 families. Endocrinologist. 2003;13:252–259.
250. Canadian Collaborative CVS-Amniocentesis Clinical Trial Group. Multicentre randomised clinical trial of chorion villus sampling and amniocentesis: first report. Lancet. 1989;333:1–6.