[level-membership-for-cardiovascular-category]
Chapter 25 Ventricular Tachycardia in Nonischemic Dilated Cardiomyopathy
Pathophysiology
The diagnosis of nonischemic dilated CMP is established by the absence of significant (>75% stenosis) coronary artery disease and prior myocardial infarction (MI). Nonischemic dilated CMP is not a single disease entity. Valvular heart disease, hypertension, sarcoidosis, amyloidosis, Chagas disease, alcohol abuse, infections, and pregnancy, among others, need to be considered as possible etiologies. An underlying etiology for adult dilated CMP is found in only 50% of patients. The remaining 50% are considered idiopathic. Idiopathic dilated CMP is characterized by an increase in myocardial mass and a reduction in ventricular wall thickness. The heart assumes a globular shape, and there is a pronounced ventricular chamber dilation and atrial enlargement.1
To date, 33 genes have been linked to nonsyndromic familial dilated CMP. Notably, the frequencies of dilated CMP mutations in any one gene are low (<1% to 8%), and a genetic cause is identified in only 30% to 35% of familial dilated CMP cases. Although usually nonsyndromic, dilated CMP can be included in syndromic disease involving various organ systems, most commonly skeletal muscle disease (muscular dystrophy).2
As compared with sporadic cases of idiopathic dilated CMP, familial CMP patients are younger and tend to have higher left ventricular ejection fraction (LVEF) and more significant myocardial fibrosis. In patients with idiopathic dilated CMP, the proposed diagnostic criteria for the familial form of the disease are the existence of two or more affected family members, or of one first-degree relative with a documented history of unexplained sudden death before 35 years of age. In most cases, proof of a genetic cause of a CMP has a limited impact on the treatment of the index patient, but can have important implications in regard to family screening and genetic counseling. Dilated CMP in patients who do not have a known family history may also have a genetic basis.3
In contrast to ischemic heart disease, the electrophysiological (EP) substrate for sustained monomorphic ventricular tachycardia (VT) in patients with nonischemic dilated CMP is not clearly defined. Although bundle branch reentry (BBR) VT is identified as the VT mechanism in a significant percentage of patients with monomorphic VT in the setting of nonischemic CMP, the majority (80%) of VTs appear to originate from the myocardium and are due to scar-related reentry rather than BBR.4 BBR VT is discussed separately in Chapter 26.
Myocardial fibrosis, myocyte disarray, and membrane abnormalities are important factors in the substrate causing VT in patients with dilated CMP. Sustained VT is associated with more extensive myocardial fibrosis and nonuniform anisotropy involving both the endocardium and epicardium, compared with patients without sustained reentry. The reentry circuits are typically associated with regions of low-voltage electrograms, consistent with scar. Catheter mapping studies of patients with nonischemic CMP point to reentry around scar deep in the myocardium, near the ventricular base and in the perivalvular region, as the underlying mechanism for VT. Studies of explanted hearts with dilated nonischemic CMP have found inexcitable fibrosis creating regions of conduction block and surviving myocardium providing the substrate for potential reentry circuits. Slow conduction through muscle bundles separated by interstitial fibrosis can cause a zigzag path and promote reentry. Furthermore, patients with nonischemic CMP and predominance of scar distribution involving 26% to 75% of wall thickness (as quantified by magnetic resonance [MR] imaging) are more likely to have inducible VT. Delayed-enhancement MR imaging typically reveals nontransmural scar areas often distributed in the basal portion of the ventricular free wall or basal to midportion of the septum. Sustained VTs are observed more frequently in patients having a greater volume of hyperenhanced areas and greater number of hyperenhanced segments, and nontransmural scar tissue was present at the VT circuit exit site in the majority of patients.5,6
The scar and fibrosis resulting from nonischemic etiologies are distinctly different from post-MI scar; hence, the reentrant circuit may have different anatomical and functional properties that affect propagation. Compared with post-MI VT, the scar tends to be smaller and less confluent, the total number of the transmural scar segments is significantly smaller, and with less endocardial involvement in nonischemic CMP.4 Whereas ischemia produces a predictable wavefront of necrosis progressing from subendocardium to epicardium (and scar areas larger endocardially than epicardially), usually confined to a specific coronary vascular territory, scars in nonischemic CMP have been shown to have a predilection for the midmyocardium and epicardium. In contrast to the dense scar with isolated surviving myocardial bundles, scar in nonischemic CMP is patchy and may have fewer fixed boundaries and protected channels or isthmuses, which can alter the extent of local conduction slowing.7
Clinical Considerations
Epidemiology
Ventricular arrhythmias, both symptomatic and asymptomatic, are common in patients with nonischemic dilated CMP. Nonsustained VT can be observed in 30% to 50% of patients, but its incidence decreases significantly after optimization of medical treatment.8 However, syncope and SCD are infrequent initial manifestations of the disease. The incidence of SCD is highest among patients with indicators of more advanced cardiac disease who are also at highest risk of all-cause mortality. Although VT, ventricular fibrillation (VF), or both are considered the most common mechanism of SCD, bradycardia, pulmonary embolism, electromechanical dissociation, and other causes account for up to 50% of SCDs in patients with advanced heart failure.
Risk Stratification
LVEF has remained the most studied and the most powerful predictor and is the primary method currently used in clinical decisions for the prevention of SCD in patients with heart failure. Depressed LVEF is also a powerful predictor of cardiac mortality. On the basis of the results of large clinical trials, in clinical practice an LVEF of 35% or less has become the primary criterion used for prophylactic ICD placement. The use of LVEF as the predominant risk stratifier has serious limitations, however, because LVEF lacks sensitivity for prediction of SCD. Even a low LVEF (<20%) may not have high positive predictive value for SCD. Clinical factors such as functional class, history of heart failure, nonsustained VT, age, LV conduction abnormalities, inducible sustained VT, and atrial fibrillation influence arrhythmic death and total mortality risk and, consequently, potentially influence the prognostic value of a depressed LVEF. Therefore, patients with an LVEF greater than 30% and other risk factors may have a higher mortality and a higher risk of SCD than those with an LVEF less than 30% but no other risk factors.9
PVCs and nonsustained VT correlate with the severity of cardiac disease and occur in the majority of patients with severe LV dysfunction. This limits the usefulness of ventricular arrhythmias as risk stratifiers as they would be expected to be sensitive but not specific. Additionally, the presence and characteristics (frequency, length, and rate) of nonsustained VT do not appear to predict increased risk of subsequent life-threatening ventricular arrhythmias in patients with severe LV impairment receiving optimal medical treatment. Nevertheless, it has been suggested that the presence of nonsustained VT may be more specific in the individual with better LV systolic function. Nonsustained VT significantly increases the risk of malignant ventricular arrhythmias in the subgroup with an LVEF greater than 35%. In these patients, even without worsening LV systolic function and symptoms, survival free from malignant ventricular arrhythmias is similar to that of patients with an LVEF less than 35% with or without nonsustained VT.8
Cardiac MR can be used to evaluate the presence and magnitude of nontransmural scar tissue. Patients with nonischemic CMP and sustained VT typically have a greater volume and number of hyperenhanced (scar) areas compared with those without sustained VT.5
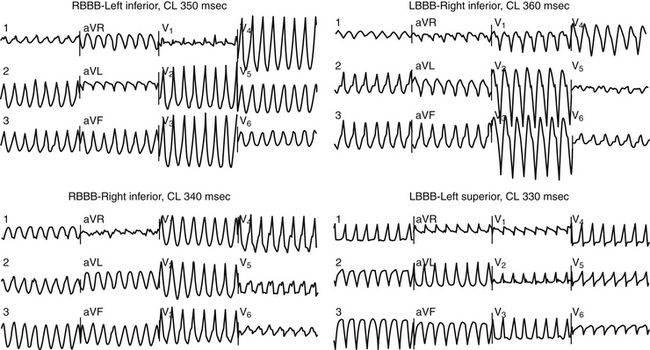
Patients typically have wide QRS complexes during the baseline rhythm, often with left bundle branch block or a nonspecific intraventricular conduction defect. Prolonged QRS duration has been associated with increased mortality in heart failure patients, but association with SCD has not been proven. Recently, fragmentation of the QRS complex on the 12-lead surface electrocardiogram (ECG) (filter range, 0.15 to 100 Hz; AC filter, 60 Hz, 25 mm/sec, 10 mm/mV) has been found to potentially predict increased risk of appropriate ICD therapies as well as a higher combined endpoint of ICD therapy and mortality in nonischemic CMP patients who received an ICD for primary and secondary prevention. The usefulness of this parameter needs further evaluation.10 During VT, QRS complexes are typically very wide and fragmented; most patients have multiple QRS morphologies of VT (Fig. 25-1).11
Prolongation of the QT interval, QT dispersion, and QT variability have had mixed predictive results with limited clinical applicability at present. Furthermore, studies evaluating the association of abnormal heart rate turbulence and reduced heart rate variability and SCD in heart failure patients have had conflicting results.9
Microvolt T-wave alternans has relatively modest (0.22) positive predictive value for SCD in patients with dilated CMP. Previous studies suggested a high negative predictive value for primary prevention of SCD, and T-wave alternans was hypothesized to be a useful tool to differentiate between patients who would benefit from ICD implantation and those who would not. However, results from recent studies failed to support this hypothesis and strongly suggested that a negative microvolt T-wave alternans result should not be used to withhold ICD therapy among patients who meet standard criteria.9
EP testing plays a minor role in risk stratification because of low VT inducibility, low reproducibility, and poor predictive value of induced VT. Although induction of VT by EP testing has been shown to predict SCD, unfortunately failure to induce VT misses most individuals destined to die suddenly.1
Principles of Management
Pharmacological Therapy
Drug therapy, such as beta blockers and angiotensin-converting enzyme inhibitors, improves overall mortality in patients with heart failure and reduces the risk of SCD. In contrast, the use of antiarrhythmic drugs for primary prevention in patients with nonischemic CMP does not improve survival and is not recommended.1
In patients with symptomatic ventricular arrhythmias, amiodarone is generally the preferred antiarrhythmic agent because of the absence of significant negative hemodynamic effects and low proarrhythmic potential, although controlled comparative trials of drugs are not available. Antiarrhythmic drug therapy can help improve quality of life in ICD patients receiving frequent appropriate shocks. Although amiodarone can potentially improve mortality and reduce the incidence of SCD in patients with nonischemic dilated CMP, it is inferior to ICD therapy for secondary prevention of VT and VF.1,12
Implantable Cardioverter-Defibrillator
Vigorous efforts have been made in developing noninvasive stratification methods to identify the subgroup of nonischemic CMP patients at high risk for SCD. However, the best approach to identifying patients and the value of various risk stratification tools are not entirely clear. Currently, there is no coherent strategy for intervention based on data integrating the results of these techniques. Many of the identified risk factors are also associated with increased risk for nonsudden death. At the present time, LVEF remains the single most important risk stratification tool to identify individuals with a high risk of SCD, again emphasizing that it predicts all-cause mortality and not necessarily arrhythmic risk. Despite some uncertainty regarding ICD benefit for nonischemic CMP patients without heart failure, regardless of LVEF, the cumulative information available from clinical trials and observational data, in conjunction with opinions of experts in the field, supports prophylactic ICD therapy among the subgroup of patients with nonischemic CMP and LVEF less than 35% who remain in NYHA functional class II or III heart failure on optimal medical therapy.12
The appropriate timing to perform ICD implantation in dilated CMP is still controversial. It is important to understand that medical management with angiotensin-converting enzyme inhibitors and beta blockers with or without aldosterone antagonists has proven mortality benefit in these patients and should be optimized as much as possible before ICD placement. Many patients significantly improve their clinical status, and may be excluded as candidates for an ICD after optimization of medical treatment. The most recent European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA) guidelines suggest ICD implantation for primary prevention only in patients with dilated CMP receiving chronic optimal medical therapy. How long it takes to reach optimal medical treatment can be debatable.8
Mapping
After exclusion of BBR, mapping of sustained VT in nonischemic CMP employs the approaches for scar-related VT (see Chap. 22). QRS morphology during VT can be used to regionalize the site of origin of the VT. Endocardial activation mapping is performed in patients with stable VTs. Most VTs are localized to the area around the mitral annulus. Entrainment and pace mapping are used to define the relationship of different endocardial sites to the circuit of the VT. Furthermore, the relationship of reentry circuits to regions of scar supports the feasibility of a substrate mapping approach, targeting the abnormal area based on voltage mapping during sinus rhythm, to guide ablation of unstable VT, similar to that described for patients with post-MI VT.
Substrate Mapping
Patients with nonischemic CMP frequently have VTs that are unstable or unsustainable and therefore not approachable by conventional entrainment maneuvers and point-by-point activation mapping. Therefore, mapping during the baseline rhythm (substrate mapping) rather than during VT is of significant value. Substrate mapping refers to delineation of the VT substrate based on the identification of abnormal local electrogram configuration (fractionated electrograms, multipotential electrograms, and/or electrograms with isolated delayed components) and the identification of abnormal local electrogram amplitude during sinus rhythm (voltage mapping). Substrate mapping is also of value in ablation of stable VTs, because it can help focus activation-entrainment mapping efforts on a small region harboring the VT substrate, and therefore help minimize how long the patient is actually in VT.4
Isolated potentials during sinus rhythm likely reflect fixed scar tissue. Although large areas of scar are uncommon in patients with nonischemic CMP, extensive interstitial fibrosis is frequently seen histologically. Strands of fibrous tissue may serve as electrical barriers and result in electrogram fragmentation. Isolated potentials can be identified in many, but not all, patients with nonischemic CMP. Additionally, isolated potentials are not present to the same extent in all forms of nonischemic CMP. They could be identified in all patients with ARVD and most patients with cardiac sarcoidosis but not in all patients with idiopathic dilated CMP. Patients in whom isolated potentials can be identified at critical ablation sites appear to have a better short- and mid-term prognosis after catheter ablation than patients in whom the arrhythmogenic substrate is not characterized by isolated potentials.8
Electroanatomical Voltage Mapping
Frequently, the VT substrate may be solely or predominantly intramural or epicardial. Because of the frequent presence of an epicardial substrate, some experienced operators use a simultaneous endocardial and epicardial mapping approach.13
Activation Mapping
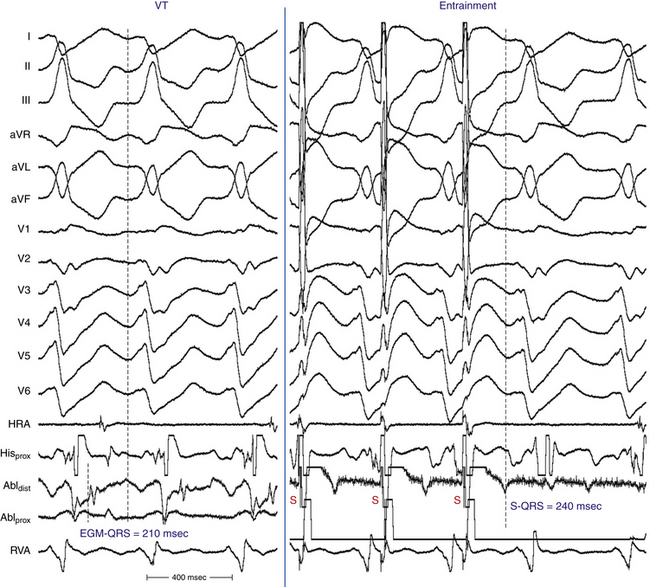
After construction of an endocardial voltage map during the baseline rhythm, VT is induced by programmed ventricular stimulation. The average number of induced VT morphologies is 3 ± 1 per patient, with a range of one to six VTs. Because surface ECG documentation of the presenting arrhythmia is often not available, clinical versus nonclinical VT is difficult to define with certainty. Mapping is therefore directed at all sustained monomorphic VTs induced by programmed stimulation. For hemodynamically tolerated sustained VT, activation mapping is performed to identify sites with early presystolic or mid-diastolic activity (Fig. 25-2).14
Entrainment Mapping
Entrainment can be demonstrated in most patients with sustained monomorphic VT. The critical isthmus for hemodynamically tolerated VT is defined as the site of entrainment with concealed fusion and a return cycle length equal to the VT cycle length (see Fig. 25-2). For unstable VT, limited entrainment mapping can be feasible with brief induction and termination of the tachycardia.14
Pace Mapping
Pace mapping has been shown to be an effective corroborative method to regionalize the VT circuit and define potential exit sites along the border zone of any low-voltage region. Pace mapping with the paced QRS morphology mimicking that of VT on the 12-lead ECG can help identify the exit site of the VT circuit. Additionally, pace mapping can identify regions of slow conduction with a stimulus-QRS interval (S-QRS) greater than 40 milliseconds and a pace map match consistent with the exit region from an isthmus. For the unmappable VT (because of hemodynamic intolerance, inconsistent induction, altering QRS morphology, and/or nonsustained duration), pace mapping is the predominant mapping technique and is directed to the scar border zone as defined by voltage mapping.14
Epicardial Mapping
When endocardial mapping and ablation fail, the epicardial approach should be considered. Mapping is initially attempted within the coronary venous system, and if no suitable ablation sites are identified, mapping within the pericardial space is then performed. However, if there are clues on the ECG that suggest an epicardial VT or if there is documentation by MR imaging of largely epicardial scar, performing percutaneous epicardial mapping as the first step (before the administration of heparin) may be considered.15
The importance of epicardial reentry circuits in CMP has been demonstrated for patients with Chagas disease and, more recently, for patients with dilated CMP unrelated to Chagas disease (see Fig. 27-1). In fact, the epicardial approach may be required in about 30% of patients with nonischemic CMP and VT.15 Percutaneous epicardial ablation of VT can be guided by activation, entrainment, and pace mapping (see Chap. 27).
Ablation
For patients with mappable VTs, focal ablation targets the critical isthmus of the reentrant circuit as defined by activation, entrainment, and pace mapping techniques. For unmappable VTs, linear ablation lesions are guided by substrate mapping and pace mapping in the scar border zone. Linear lesions are created in regions that cross the border zone and intersect the best pace map site, which approximates the exit site of the VT circuit, similar to strategies described for unmappable post-MI VT (see Chap. 22).
An irrigated-tip ablation catheter is preferred for ablation because it can create larger lesions and also can be used for radiofrequency (RF) energy delivery within the coronary venous system if needed. The power setting is adjusted to an impedance drop of 10 Ω starting with 30 to 35 W. If RF energy is applied during VT, it is applied for 30 seconds and, if VT does not terminate, the catheter is moved to an alternate site. If VT terminates during the energy application, the application is continued for a total of 120 milliseconds. In the setting of nontolerated VTs, RF energy is delivered for 60 to 120 milliseconds at each site to create ablation lines as guided by substrate mapping.15
The endpoint of the procedure is noninducibility of all the VTs for which an appropriate target site can be identified. Induction of multiple VTs is not uncommon in patients with dilated CMP. Whether targeting all inducible VTs is superior to targeting only the VTs that have been clinically documented is still unclear.15
There are only a few single-center reports on acute outcome of VT ablation in the setting of nonischemic CMP, with short- to intermediate-term follow-up in relatively small numbers of patients. Acute success in eliminating inducible VT has varied from 56% to 74% with VT recurrence of 42% to 75% with endocardial ablation. Satisfactory control of VTs previously refractory to medical treatment can be achieved in a majority of patients (60% to 70%) with continuing antiarrhythmic medications, if tolerated. Outcome appears to be somewhat improved with epicardial ablation, but long-term follow-up in a large cohort of patients is lacking.13,15
The success of endocardial ablation of VTs associated with nonischemic CMP is lower than that observed for post-MI VTs. Reentry circuits deep to the endocardium and in the epicardium appear to be a likely explanation. Combined endocardial and epicardial mapping approaches are likely to improve the success of ablation.7
Less than 5% of patients have major complications, such as cardiac tamponade, thromboembolic events, or death. Electromechanical dissociation after multiple VT inductions can occur in patients with very low ejection fractions. Limiting the number of inductions of VT may help to prevent this life-threatening complication.15
Sarcoid Cardiomyopathy
Pathophysiology
The inflammatory process in cardiac sarcoidosis often is initiated in the myocardium, creating lesions (granulomas) that then extend to the epicardium, endocardium, or both. Inflammation and fibrosis participate in ventricular arrhythmogenesis, one of the hallmarks of cardiac sarcoidosis. Surviving muscle bundles within scar tissue most likely form the substrate for reentry.16
Multiple, monomorphic VTs, which are common in cardiac sarcoidosis, are predominantly due to a scar-related, reentrant mechanism. Low-amplitude and fragmented potentials are recorded both in the endocardial and epicardial regions of both ventricles. The presence of isolated potentials during sinus rhythm at most effective ablation sites suggests that a process similar to post-MI VT is responsible for VTs in patients with cardiac sarcoidosis. The most common site of VT circuit is the peritricuspid area, which is consistent with the predominance of basal involvement of the right ventricular septum in cardiac sarcoidosis. The disease process can be located intramurally and may be reachable by neither the endocardial nor the epicardial approach.13,16
Clinical Considerations
Determining cardiac involvement in patients with sarcoidosis is difficult because granulomas may be present without clinical dysfunction. Patients with cardiac sarcoidosis can present with congestive heart failure, atrioventricular block, supraventricular arrhythmia, and/or ventricular tachyarrhythmia. Importantly, cardiac involvement of sarcoidosis is associated with a mortality rate greater than 40% at 5 years, and many of the deaths are caused by ventricular tachyarrhythmias. Approximately 50% of patients with cardiac sarcoidosis require treatment for ventricular arrhythmias. These patients usually have multiple monomorphic VTs, with either left or right bundle branch block morphology. Arrhythmias can be refractory to a combination of steroids and antiarrhythmic drugs in almost half of the patients. ICD implantation is the mainstay therapy in these patients. Nonetheless, RF catheter ablation as an adjunct therapy to ICD can be effective in eliminating VT or in markedly reducing the VT burden, with success rates in a recently reported registry ranging from 25% to 70%, depending on the location of the reentrant circuit.13,16
EP testing can potentially provide prognostic information in asymptomatic patients with cardiac sarcoidosis. VT inducibility may help to identify those at risk for ventricular arrhythmia. A negative EP study appears to predict a benign course within the first several years after diagnosis. More studies are needed to guide prophylactic ICD therapy in this population.17
For cardiac sarcoidosis, cardiac biopsy is one of the few ways to confirm the diagnosis, although cardiac MR imaging with delayed enhancement can sometimes detect the granular cells, which resemble clumps of sand or salt grains, and help to regionalize the disease process and indicate whether scarring is located intramurally, epicardially, endocardially, or transmurally.18 About one-third of the patients with cardiac sarcoidosis have detectable abnormalities visible in an echocardiogram.16
Chagas Cardiomyopathy
Pathophysiology
Chagas disease is an endemic disease in Latin America caused by a unicellular parasite, Trypanosoma cruzi. Almost 18 million people are infected and almost 25% of them will develop chronic myocardial disease in the following years or decades.19
Chronic Chagas heart disease, the most serious manifestation of Chagas disease, is an inflammatory form of dilated CMP. The panmyocarditis of Chagas heart disease progressively involves the various cardiac tissues and results in extensive cardiac fibrosis. When the extent of myocardial damage is severe, the disease manifests as myocardial dysfunction that may be segmental, typically a ventricular aneurysm, or global, resembling a dilated CMP.19,20
Myocardial damage can occur in various areas of both ventricles, but the inferolateral segment of the LV is the most commonly involved site, with frequently observed wall motion abnormalities. Apical, septal, and apical inferior aneurysms have also been described. The classic lesion of Chagas disease is a localized aneurysm of the LV apex, with relatively normal surrounding wall motion. This results in a narrow neck when visualized by echocardiography or ventriculography; when present, this can usually distinguish an aneurysm of Chagas heart disease from one due to coronary artery disease. The aneurysms and segmental abnormalities are thought to result from localized destruction of extracellular matrix collagen along with myocyte loss, which leads to focal weakening of the ventricular wall. The apical location is particularly vulnerable because of the nature of the collagen structure at this location, normal apical thinning, and a relatively increased wall stress, which would promote the gradual development of aneurysmal dilation of the weakened segment. Regional dyssynergy caused by segmental conduction abnormalities could also contribute to aneurysm formation.19,21
Histological examination reveals focal and diffuse myocardial fibrosis, predominantly in the subepicardium, interspersed with viable but often damaged myocardial fibers. VT can arise from various regions in both ventricles, but LV inferolateral scar is the main source of sustained VT reentrant circuits. In theses areas, endocardial mapping frequently shows fragmented and late potentials during sinus rhythm as well as continuous or diastolic activity during VT. Histological analysis of those segments has shown focal and diffuse fibrosis that is predominantly subepicardial with nonuniform anisotropy of the surviving fibers. Epicardial VT reentrant circuits occur frequently in Chagas CMP; approximately 70% of VTs are epicardial in origin.19,20
Clinical Considerations
Cardiac abnormalities can be detected in all phases or forms of Chagas disease. The natural history and the type of cardiac involvement can vary widely in patients with Chagas disease. Patients can present with a wide variety of clinical manifestations; the most important of these are ventricular arrhythmias, sudden death, congestive heart failure, thromboembolism, stroke, and heart block.20,21
VT in Chagas disease can have heterogeneous presentations. SCD, usually due to VF and VT, is the most common cause of death, occurring more often than in other types of dilated CMP, with an incidence ranging from 51% to 65%. Frequently, the arrhythmic episodes are clustered in short periods, causing electrical storms (“chagasic storm”). Ventricular ectopy is remarkably frequent in all stages of the disease, even when there is no other evidence of cardiac involvement. Ectopy is dense and temporally unvarying, with patients often having tens of thousands of ectopic beats per day. In one report, 14% of patients presented with aborted sudden death, and sustained VT or sudden death occurred subsequently in 39% of patients with LV aneurysm or dysfunction. Nonsustained VT has been found by ambulatory monitoring in 10% of patients with mild wall motion abnormalities, in 56% of those with severe wall motion abnormalities or aneurysms without heart failure, and in 87% of those with advanced congestive heart failure.19,20
Because of the high incidence of thromboembolic phenomena, oral anticoagulants are recommended for patients with atrial fibrillation, previous embolism, and apical aneurysm with thrombus, even in the absence of controlled clinical trials demonstrating their efficacy.20,21
1. Hamilton R.M., Azevedo E.R. Sudden cardiac death in dilated cardiomyopathies. Pacing Clin Electrophysiol. 2009;32(Suppl 2):S32-S40.
2. Kleber A.G., Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431-488.
3. Boussy T., Paparella G., de Asmundis .C., et al. Genetic basis of ventricular arrhythmias. Heart Fail Clin. 2010;6:249-266.
4. Aliot E.M., Stevenson W.G., Mendral-Garrote J.M., et al. EHRA/HRS Expert Consensus on Catheter Ablation of Ventricular Arrhythmias: developed in a partnership with the European Heart Rhythm Association (EHRA), a Registered Branch of the European Society of Cardiology (ESC), and the Heart Rhythm Society (HRS); in collaboration with the American College of Cardiology (ACC) and the American Heart Association (AHA). Heart Rhythm. 2009;6:886-933.
5. Yokokawa M., Tada H., Koyama K., et al. The characteristics and distribution of the scar tissue predict ventricular tachycardia in patients with advanced heart failure. Pacing Clin Electrophysiol. 2009;32:314-322.
6. Yokokawa M., Tada H., Koyama K., et al. Nontransmural scar detected by magnetic resonance imaging and origin of ventricular tachycardia in structural heart disease. Pacing Clin Electrophysiol. 2009;32(Suppl 1):S52-S56.
7. Nakahara S., Tung R., Ramirez R.J., et al. Characterization of the arrhythmogenic substrate in ischemic and nonischemic cardiomyopathy implications for catheter ablation of hemodynamically unstable ventricular tachycardia. J Am Coll Cardiol. 2010;55:2355-2365.
8. Kuhne M., Abrams G., Sarrazin J.F., et al. Isolated potentials and pace-mapping as guides for ablation of ventricular tachycardia in various types of nonischemic cardiomyopathy. J Cardiovasc Electrophysiol. 2010;21:1017-1023.
9. Vest R.N.III, Gold M.R. Risk stratification of ventricular arrhythmias in patients with systolic heart failure. Curr Opin Cardiol. 2010;25:268-275.
10. Das M.K., Zipes D.P. Fragmented QRS: a predictor of mortality and sudden cardiac death. Heart Rhythm. 2009;6(Suppl 3):S8-S14.
11. Das M.K., Maskoun W., Shen C., et al. Fragmented QRS on twelve-lead electrocardiogram predicts arrhythmic events in patients with ischemic and nonischemic cardiomyopathy. Heart Rhythm. 2010;7:74-80.
12. Myerburg R.J., Reddy V., Castellanos A. Indications for implantable cardioverter-defibrillators based on evidence and judgment. J Am Coll Cardiol. 2009;54:747-763.
13. Natale A., Raviele A., Al-Ahmad A., et al. Venice Chart International Consensus document on ventricular tachycardia/ventricular fibrillation ablation. J Cardiovasc Electrophysiol. 2010;21:339-379.
14. Josephson M.E. Catheter and surgical ablation in the therapy of arrhythmias. In: Josephson M.E., editor. Clinical cardiac electrophysiology. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2008:746-888.
15. Bogun F., Morady F. Ablation of ventricular tachycardia in patients with nonischemic cardiomyopathy. J Cardiovasc Electrophysiol. 2008;19:1227-1230.
16. Jefic D., Joel B., Good E., et al. Role of radiofrequency catheter ablation of ventricular tachycardia in cardiac sarcoidosis: report from a multicenter registry. Heart Rhythm. 2009;6:189-195.
17. Mehta D., Mori N., Goldbarg S.H., et al. Primary prevention of sudden cardiac death in silent cardiac sarcoidosis: role of programmed ventricular stimulation. Circ Arrhythm Electrophysiol. 2011;4:43-48.
18. Cheong B.Y., Muthupillai R., Nemeth M., et al. The utility of delayed-enhancement magnetic resonance imaging for identifying nonischemic myocardial fibrosis in asymptomatic patients with biopsy-proven systemic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009;26:39-46.
19. Rassi A.Jr., Rassi A., Marin-Neto J.A. Chagas disease. Lancet. 2010;375:1388-1402.
20. Rassi A.Jr., Rassi A., Rassi S.G. Predictors of mortality in chronic Chagas disease: a systematic review of observational studies. Circulation. 2007;115:1101-1108.
21. Bern C., Montgomery S.P., Herwaldt B.L., et al. Evaluation and treatment of Chagas disease in the United States: a systematic review. JAMA. 2007;298:2171-2181.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 25 Ventricular Tachycardia in Nonischemic Dilated Cardiomyopathy
Pathophysiology
The diagnosis of nonischemic dilated CMP is established by the absence of significant (>75% stenosis) coronary artery disease and prior myocardial infarction (MI). Nonischemic dilated CMP is not a single disease entity. Valvular heart disease, hypertension, sarcoidosis, amyloidosis, Chagas disease, alcohol abuse, infections, and pregnancy, among others, need to be considered as possible etiologies. An underlying etiology for adult dilated CMP is found in only 50% of patients. The remaining 50% are considered idiopathic. Idiopathic dilated CMP is characterized by an increase in myocardial mass and a reduction in ventricular wall thickness. The heart assumes a globular shape, and there is a pronounced ventricular chamber dilation and atrial enlargement.1
To date, 33 genes have been linked to nonsyndromic familial dilated CMP. Notably, the frequencies of dilated CMP mutations in any one gene are low (<1% to 8%), and a genetic cause is identified in only 30% to 35% of familial dilated CMP cases. Although usually nonsyndromic, dilated CMP can be included in syndromic disease involving various organ systems, most commonly skeletal muscle disease (muscular dystrophy).2
As compared with sporadic cases of idiopathic dilated CMP, familial CMP patients are younger and tend to have higher left ventricular ejection fraction (LVEF) and more significant myocardial fibrosis. In patients with idiopathic dilated CMP, the proposed diagnostic criteria for the familial form of the disease are the existence of two or more affected family members, or of one first-degree relative with a documented history of unexplained sudden death before 35 years of age. In most cases, proof of a genetic cause of a CMP has a limited impact on the treatment of the index patient, but can have important implications in regard to family screening and genetic counseling. Dilated CMP in patients who do not have a known family history may also have a genetic basis.3
In contrast to ischemic heart disease, the electrophysiological (EP) substrate for sustained monomorphic ventricular tachycardia (VT) in patients with nonischemic dilated CMP is not clearly defined. Although bundle branch reentry (BBR) VT is identified as the VT mechanism in a significant percentage of patients with monomorphic VT in the setting of nonischemic CMP, the majority (80%) of VTs appear to originate from the myocardium and are due to scar-related reentry rather than BBR.4 BBR VT is discussed separately in Chapter 26.
Myocardial fibrosis, myocyte disarray, and membrane abnormalities are important factors in the substrate causing VT in patients with dilated CMP. Sustained VT is associated with more extensive myocardial fibrosis and nonuniform anisotropy involving both the endocardium and epicardium, compared with patients without sustained reentry. The reentry circuits are typically associated with regions of low-voltage electrograms, consistent with scar. Catheter mapping studies of patients with nonischemic CMP point to reentry around scar deep in the myocardium, near the ventricular base and in the perivalvular region, as the underlying mechanism for VT. Studies of explanted hearts with dilated nonischemic CMP have found inexcitable fibrosis creating regions of conduction block and surviving myocardium providing the substrate for potential reentry circuits. Slow conduction through muscle bundles separated by interstitial fibrosis can cause a zigzag path and promote reentry. Furthermore, patients with nonischemic CMP and predominance of scar distribution involving 26% to 75% of wall thickness (as quantified by magnetic resonance [MR] imaging) are more likely to have inducible VT. Delayed-enhancement MR imaging typically reveals nontransmural scar areas often distributed in the basal portion of the ventricular free wall or basal to midportion of the septum. Sustained VTs are observed more frequently in patients having a greater volume of hyperenhanced areas and greater number of hyperenhanced segments, and nontransmural scar tissue was present at the VT circuit exit site in the majority of patients.5,6
The scar and fibrosis resulting from nonischemic etiologies are distinctly different from post-MI scar; hence, the reentrant circuit may have different anatomical and functional properties that affect propagation. Compared with post-MI VT, the scar tends to be smaller and less confluent, the total number of the transmural scar segments is significantly smaller, and with less endocardial involvement in nonischemic CMP.4 Whereas ischemia produces a predictable wavefront of necrosis progressing from subendocardium to epicardium (and scar areas larger endocardially than epicardially), usually confined to a specific coronary vascular territory, scars in nonischemic CMP have been shown to have a predilection for the midmyocardium and epicardium. In contrast to the dense scar with isolated surviving myocardial bundles, scar in nonischemic CMP is patchy and may have fewer fixed boundaries and protected channels or isthmuses, which can alter the extent of local conduction slowing.7
Clinical Considerations
Epidemiology
Ventricular arrhythmias, both symptomatic and asymptomatic, are common in patients with nonischemic dilated CMP. Nonsustained VT can be observed in 30% to 50% of patients, but its incidence decreases significantly after optimization of medical treatment.8 However, syncope and SCD are infrequent initial manifestations of the disease. The incidence of SCD is highest among patients with indicators of more advanced cardiac disease who are also at highest risk of all-cause mortality. Although VT, ventricular fibrillation (VF), or both are considered the most common mechanism of SCD, bradycardia, pulmonary embolism, electromechanical dissociation, and other causes account for up to 50% of SCDs in patients with advanced heart failure.
Risk Stratification
LVEF has remained the most studied and the most powerful predictor and is the primary method currently used in clinical decisions for the prevention of SCD in patients with heart failure. Depressed LVEF is also a powerful predictor of cardiac mortality. On the basis of the results of large clinical trials, in clinical practice an LVEF of 35% or less has become the primary criterion used for prophylactic ICD placement. The use of LVEF as the predominant risk stratifier has serious limitations, however, because LVEF lacks sensitivity for prediction of SCD. Even a low LVEF (<20%) may not have high positive predictive value for SCD. Clinical factors such as functional class, history of heart failure, nonsustained VT, age, LV conduction abnormalities, inducible sustained VT, and atrial fibrillation influence arrhythmic death and total mortality risk and, consequently, potentially influence the prognostic value of a depressed LVEF. Therefore, patients with an LVEF greater than 30% and other risk factors may have a higher mortality and a higher risk of SCD than those with an LVEF less than 30% but no other risk factors.9
PVCs and nonsustained VT correlate with the severity of cardiac disease and occur in the majority of patients with severe LV dysfunction. This limits the usefulness of ventricular arrhythmias as risk stratifiers as they would be expected to be sensitive but not specific. Additionally, the presence and characteristics (frequency, length, and rate) of nonsustained VT do not appear to predict increased risk of subsequent life-threatening ventricular arrhythmias in patients with severe LV impairment receiving optimal medical treatment. Nevertheless, it has been suggested that the presence of nonsustained VT may be more specific in the individual with better LV systolic function. Nonsustained VT significantly increases the risk of malignant ventricular arrhythmias in the subgroup with an LVEF greater than 35%. In these patients, even without worsening LV systolic function and symptoms, survival free from malignant ventricular arrhythmias is similar to that of patients with an LVEF less than 35% with or without nonsustained VT.8
Cardiac MR can be used to evaluate the presence and magnitude of nontransmural scar tissue. Patients with nonischemic CMP and sustained VT typically have a greater volume and number of hyperenhanced (scar) areas compared with those without sustained VT.5
Patients typically have wide QRS complexes during the baseline rhythm, often with left bundle branch block or a nonspecific intraventricular conduction defect. Prolonged QRS duration has been associated with increased mortality in heart failure patients, but association with SCD has not been proven. Recently, fragmentation of the QRS complex on the 12-lead surface electrocardiogram (ECG) (filter range, 0.15 to 100 Hz; AC filter, 60 Hz, 25 mm/sec, 10 mm/mV) has been found to potentially predict increased risk of appropriate ICD therapies as well as a higher combined endpoint of ICD therapy and mortality in nonischemic CMP patients who received an ICD for primary and secondary prevention. The usefulness of this parameter needs further evaluation.10 During VT, QRS complexes are typically very wide and fragmented; most patients have multiple QRS morphologies of VT (Fig. 25-1).11
Prolongation of the QT interval, QT dispersion, and QT variability have had mixed predictive results with limited clinical applicability at present. Furthermore, studies evaluating the association of abnormal heart rate turbulence and reduced heart rate variability and SCD in heart failure patients have had conflicting results.9
[/not-level-membership-for-cardiovascular-category]
