Chapter 29 Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia
Pathophysiology
Arrhythmogenic right ventricular cardiomyopathy-dysplasia (ARVD) is an inherited primary disease of the myocardium characterized by ventricular arrhythmias and structural abnormalities of the right ventricle (RV). The hallmark pathological findings are progressive myocyte loss and fibrofatty (fibrous and adipose) tissue replacement, with a predilection for the RV, but the left ventricle (LV) and septum also can be affected. Fibrofatty replacement of the myocardium produces “islands” of scar that can lead to reentrant VTs, and these patients have an increased risk of sudden cardiac death (SCD), mostly secondary to ventricular tachycardia (VT).1
Molecular Genetics
The autosomal dominant nonsyndromic ARVD-1 to ARVD-12 as well as the two rare recessive forms (Naxos disease and Carvajal syndrome) have been mapped to 12 genetic loci, with mutations in eight gene loci identified (Table 29-1). Although some identified gene mutations such as RYR2 may be phenocopies of ARVD, a common theme of desmosomal protein mutations has been emerging. In fact, five of the eight gene loci encode desmosomal proteins (desmoplakin [DSP], plakophilin-2 [PKP2], desmoglein-2 [DSG2], desmocollin-2 [DSC2], junctional plakoglobin [JUP]) and have been found in association with the ARVD phenotype in up to 50% of cases. Among the known genes, mutations in PKP2 appear to be the most common causes of ARVD, accounting for approximately 20% of cases. Mutations in DSG2 and DSP each account for approximately 10% to 15% of cases. Therefore, ARVD is currently considered, at least in a subset, a disease of the cardiac desmosome.2,3 The majority of these mutations are insertion/deletion or nonsense mutations, which are expected to cause premature termination of the encoded proteins.1
TABLE 29-1 Known Genetic Loci for Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia
| LOCUS NAME | CAUSATIVE GENE | MODE OF INHERITANCE |
|---|---|---|
| ARVD-1 | Transforming growth factor-β3 | Autosomal dominant |
| ARVD-2 | Cardiac ryanodine receptor (RYR2) | Autosomal dominant |
| ARVD-3 | Unknown | Autosomal dominant |
| ARVD-4 | Unknown | Autosomal dominant |
| ARVD-5 | Transmembrane protein-43 | Autosomal dominant |
| ARVD-6 | Unknown | Autosomal dominant |
| ARVD-7 | Unknown | Autosomal dominant |
| ARVD-8 | Desmoplakin (DSP) | Autosomal dominant |
| ARVD-9 | Plakophilin-2 (PKP2) | Autosomal dominant |
| ARVD-10 | Desmoglein-2 | Autosomal dominant |
| ARVD-11 | Desmocollin-2 | Autosomal dominant |
| ARVD-12 | Plakoglobin (JUP) | Autosomal dominant |
| Naxos disease | Plakoglobin (JUP) | Autosomal recessive |
| Carvajal syndrome | Desmoplakin (DSP) | Autosomal recessive |
Mutations in genes encoding nondesmosomal proteins also can potentially cause ARVD. Alterations of the 5′ and 3′ regulatory elements of TGFB3, encoding transforming growth factor-β3, have each been reported. More recently, a mutation in TMEM43, encoding a transmembrane protein with ties to an adipogenic transcription factor, was reported as the cause for ARVD-5, a subtype of ARVD with prominent LV involvement. Mutations in RYR2, encoding the cardiac ryanodine receptor (the major calcium release channel of the sarcoplasmic reticulum in cardiomyocytes, also implicated in familial catecholaminergic polymorphic VT), result in a form of arrhythmogenic cardiomyopathy (ARVD-2) characterized by exercise-induced polymorphic VT that does not appear to have a reentrant mechanism, occurring in the absence of significant structural abnormalities. Patients do not develop characteristic features of ARVD on the 12-lead electrocardiogram (ECG) or signal-averaged ECG, and global RV function remains unaffected. ARVD-2 shows a closer resemblance to familial catecholaminergic polymorphic VT in both etiology and phenotype; its inclusion under the umbrella term of ARVD remains controversial.2–4
Pathogenesis
Cardiac desmosomes are specialized, multiprotein complexes in the intercalated disks that anchor intermediate filaments to the cytoplasmic membrane in adjacent cardiomyocytes, thereby forming a three-dimensional (3-D) scaffolding and providing mechanical strength and cohesion of cardiomyocytes in the beating heart (Fig. 29-1). Cardiac desmosomes are also responsible for regulating transcription of genes involved in adipogenesis and apoptosis as well as maintaining proper electrical conductivity through regulation of gap junctions and calcium homeostasis. Cardiac desmosomes are composed of three groups of proteins: the cadherin family, the Armadillo family, and the plakin family. The cadherin family is composed of three desmocollins and three desmogleins, which are primarily responsible for anchoring the structure to the membrane. The Armadillo family is composed of plakoglobin and three plakophilins, which form the core structure and possess signaling capabilities. The plakin family is composed of DSP, envoplakin, periplakin, plectin, and pinin, which are responsible for the attachment of the desmosomes to intermediary filaments.1,5
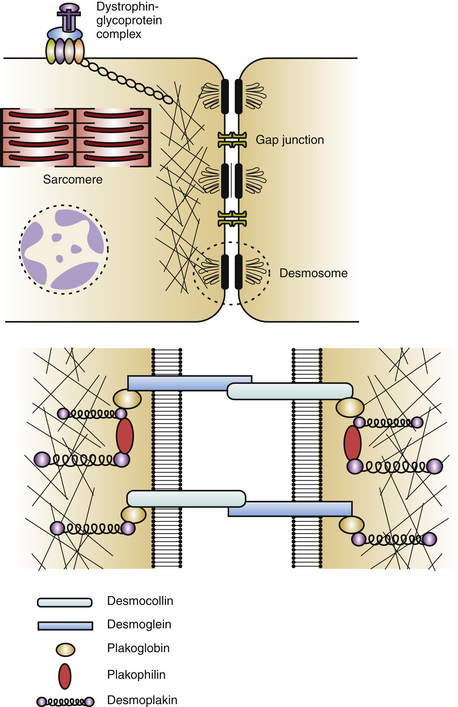
The mechanisms by which the affected desmosomes cause myocyte apoptosis, fibrogenesis, adipogenesis, and slow ventricular conduction, thus leading to impaired RV function and increased arrhythmogenicity, remain to be determined. Mutations in desmosomal genes alter the number or integrity of desmosomes and thereby lead to impaired mechanical coupling and failure of cell-to-cell adhesion structures during exposure to physical strain, resulting in cardiomyocyte detachment and degeneration, with subsequent inflammation and replacement by fibrofatty tissue. Fibrofatty replacement is a nonspecific repair process also observed in the muscular dystrophies. The architecture of thin surviving myocardial bundles within the fibrofatty tissue creates lengthened conduction pathways, conduction slowing at pivotal points, and conduction block. All factors contribute to activation delay and can create an electrophysiological (EP) substrate for reentry and VT. Ventricular dysfunction can ensue in the later stages owing to progressive myocyte detachment and death.6
Additionally, mutations in desmosomal proteins can impair expression of interacting proteins at the intercalated disk (e.g., gap junction or ion channel proteins), giving rise to impairment of intercellular conductance and promoting ventricular arrhythmogenesis, even in the absence of fibrofatty tissue replacement. Impaired desmosomal structure and function also can affect other cell-to-cell contact structures in the myocardium. In particular, connexin-43 remodeling in the gap junctions, which contributes to delayed conduction and ventricular arrhythmogenesis, is often observed in ARVD patients, and these potential electrical conduction abnormalities may favor arrhythmic events that characterize the disease and predispose patients to high risk of SCD.2–4
Pathology
Regardless of the mechanism, the patchy replacement of the RV myocardium by fibrofatty tissue provides a substrate for reentrant ventricular arrhythmias. The most striking morphological feature of the disease is the diffuse or segmental loss of RV myocytes, with replacement by fibrofatty tissue and thinning of the RV wall. Patchy inflammatory infiltrates can be present in areas of myocardial damage. Fibrofatty replacement usually begins in the subepicardium or midmural layers and progresses to the subendocardium. Only the endocardium and myocardium of the trabeculae may be spared. The sites of involvement can be localized and in early disease are often confined to the so-called “triangle of dysplasia”—namely, the RV outflow tract (RVOT), RV apex, and inferolateral wall near the tricuspid valve. RV aneurysms (Fig. 29-2), and segmental RV hypokinesia are typical. Diffuse myocardial involvement leads to global RV dilation. However, the fibrofatty pattern of ARVD is limited not only to the RV; the disease also can migrate to the LV free wall (commonly observed in advanced disease), with a predilection for the posteroseptal and posterolateral areas, with relative sparing of the septum.2–4
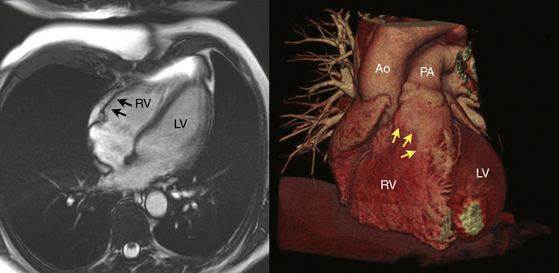
Despite the widespread impression that ARVD is universally a progressive disease process, a recent report found that the extent of the endocardial scar as measured by bipolar voltage mapping in patients with ARVD and VT remained relatively stable, despite progressive RV dilation.7
Clinical Considerations
Clinical Presentation
Four clinicopathological stages of ARVD can be considered: the early “concealed” phase, followed by the “overt electrical disorder,” the “phase of RV failure,” and finally, the phase of “biventricular failure.” Although ARVD is a genetically transmitted disease, it is associated with a long asymptomatic lead time and individuals in their teens may not have any characteristics of ARVD clinically or on screening tests. Early ARVD is often asymptomatic (the “concealed” phase), occasionally associated with minor ventricular arrhythmias and subtle structural changes. Nonetheless, these patients are still at risk of SCD, especially during intense physical exertion. With disease progression, the “overt electrical disorder” typically causes symptomatic ventricular arrhythmia and more obvious morphological abnormalities detectable by imaging. Further disease extension results in RV dilation and dysfunction (the “phase of RV failure”), precipitating symptoms and signs of right heart failure. Unless SCD occurs, progressive impairment of cardiac function can result in biventricular heart failure late in the evolution of ARVD, usually within 4 to 8 years after typical development of complete right bundle branch block (RBBB). End-stage disease is often indistinguishable from dilated cardiomyopathy and manifests with congestive heart failure, atrial fibrillation, and an increased incidence of thromboembolic events. Overall, judging the accurate position of the patient on the time scale of the spectrum can be difficult, and some patients may remain stable in the same phase of the disease for several decades.8
ARVD is one of the most arrhythmogenic human heart diseases known. Electrical instability is present at the very early stages of ARVD. Ventricular arrhythmias range from isolated ectopy to sustained VT or ventricular fibrillation (VF).8 Approximately 50% of patients with ARVD present with symptomatic ventricular arrhythmias, most commonly sustained and nonsustained VT, manifested by palpitations, dizziness, and/or syncope. The frequency of ventricular arrhythmias in ARVD varies with the severity of the disease, ranging from 23% in patients with mild disease to almost 100% in patients with severe disease. Ventricular arrhythmias characteristically occur during exercise; up to 50% to 60% of patients with ARVD show monomorphic VT during exercise testing.
Initial Evaluation
Electroanatomical voltage mapping, which seems to be an effective technique to detect RV low-voltage regions reflecting fibrofatty myocardial atrophy in patients with ARVD, has been shown to improve the diagnostic accuracy of myocardial biopsy by reducing the sampling errors. Endomyocardial biopsies are obtained from low-voltage areas, preferably from the border zone, in order to minimize the risk of perforation.9
Additionally, immunohistochemical analysis of conventional biopsy samples to detect a change in the distribution of desmosomal proteins can also improve the sensitivity and specificity of this diagnostic tool. Reduced immunoreactive signal levels of plakoglobin at intercalated disks were found to be a consistent feature in patients with ARVD but not seen in other forms of myocardial disease.10
Recognition of the problems in diagnosing ARVD and the fact that there is no “gold standard” or single test that is diagnostic of ARVD led to the formation of a task force in 1994 that proposed major and minor criteria to aid in the diagnosis, based on family history as well as structural, histological, functional, arrhythmic, and ECG abnormalities (Table 29-2).11 The diagnosis of ARVD is based on the presence of two major criteria, one major plus two minor criteria, or four minor criteria. It is important to recognize, however, that these criteria were defined retrospectively from aggregated series of referrals to tertiary care institutions dominated by the overt or severe end of the disease spectrum (i.e., symptomatic index cases and SCD victims) and have never been prospectively validated; therefore, their applicability is limited in situations where the prior probability of ARVD is different from the initial derivation set of patients. Consequently, the original task force criteria are highly specific but lacked sensitivity for early and familial disease. Furthermore, this diagnostic approach does not specify the preferred order of imaging techniques to examine and score RV morphology and function.5,11
TABLE 29-2 Original and Revised Task Force Diagnostic Criteria for Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia
| ORIGINAL TASK FORCE CRITERIA | REVISED TASK FORCE CRITERIA | |
|---|---|---|
| I. Global or Regional Dysfunction and Structural Alterations* | ||
| Major | ||
By MRI:
By RV angiography:
By MRI:
• Late potentials by SAECG in at least one of three parameters in the absence of a QRS duration ≥ 110 msec on the standard ECG
• Filtered QRS duration (fQRS) ≥ 114 msec
• Duration of terminal QRS < 40 µV (low-amplitude signal duration) ≥ 38 msec
• Root-mean-square voltage of terminal 40 msec ≥ 20 µV
• Terminal activation duration of QRS ≥ 55 msec measured from the nadir of the S wave to the end of the QRS, including R′, in V1, V2, or V3, in the absence of complete right bundle branch block
• ARVC/D confirmed in a first-degree relative who meets current task force criteria
• ARVC/D confirmed pathologically at autopsy or surgery in a first-degree relative
• Identification of a pathogenic mutation† categorized as associated or probably associated with ARVC/D in the patient under evaluation
• History of ARVC/D in a first-degree relative in whom it is not possible or practical to determine whether the family member meets current task force criteria
• Premature sudden death (<35 yr of age) due to suspected ARVC/D in a first-degree relative
• ARVC/D confirmed pathologically or by current task force criteria in second-degree relative
Diagnostic terminology for original criteria: This diagnosis is fulfilled by the presence of two major, or one major plus two minor, criteria or by four minor criteria from different groups. Diagnostic terminology for revised criteria: definite diagnosis: two major or one major and two minor criteria, or four minor from different categories; borderline: one major and one minor or three minor criteria from different categories; possible: one major or two minor criteria from different categories.
2-D = two-dimensional; ARVC/D = arrhythmogenic right ventricular cardiomyopathy/dysplasia; aVF = augmented voltage unipolar left foot lead; aVL = augmented voltage unipolar left arm lead; BSA = body surface area; LV = left ventricle; MRI = magnetic resonance imaging; PLAX = parasternal long-axis view; PSAX = parasternal short-axis view; RVOT = right ventricular outflow tract; RV = right ventricle; SAECG = signal-averaged ECG.
* Hypokinesis is not included in this or subsequent definitions of RV regional wall motion abnormalities for the proposed modified criteria.
† A pathogenic mutation is a DNA alteration associated with ARVC/D that alters or is expected to alter the encoded protein, is unobserved or rare in a large non-ARVC/D control population, and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree.
Adapted with permission from Marcus FI, McKenna WJ, Sherrill D, et al: Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria, Circulation 121:1533-1541, 2010.
A recent study of a large number (108) of newly diagnosed patients suspected of having ARVD found that a combination of diagnostic imaging tests is needed to evaluate the presence of RV structural, functional, and electrical abnormalities. ECG, echocardiography, RV angiography, signal-averaged ECG (SAECG), and Holter monitoring provide optimal clinical evaluation of patients suspected of having ARVD. The diagnostic performance of MR imaging and RV biopsy seemed to be inferior in comparison with the five tests recommended.12 Based on these findings, the original task force criteria have been recently modified to incorporate the emerging diagnostic modalities and advances in the genetics of ARVD to improve diagnostic sensitivity while maintaining diagnostic specificity. In this modification of the task force criteria, quantitative criteria were proposed and abnormalities were defined on the basis of comparison with normal subject data.11 Scoring by major and minor criteria was maintained, structural abnormalities were quantified, and task force criteria highly specific for ARVD were upgraded to major. Furthermore, new criteria were added: terminal activation duration of QRS equal to or greater than 55 milliseconds, VT with left bundle branch block (LBBB) morphology and superior axis, and genetic criteria.13
A recent study found that the new task force criteria are increasing the diagnostic yield of ARVD when applied to patients with suspected disease.13 When performing and analyzing imaging studies, it is important to recognize, because ARVD is a rare disease, the importance of expert interpretation of the complex-shaped RV and the need for quantitation of RV structure and function as well as specific protocols for optimal evaluation. Referral to centers with expertise in this field should be considered.
Molecular genetic analysis can potentially facilitate timely diagnosis, guiding interpretation of borderline investigations, and enabling cascade screening of relatives. Unfortunately, sequence analysis of the known desmosomal ARVD-related genes only identifies a responsible mutation in approximately 50% of ARVD probands. Nevertheless, recent studies support the use of genetic testing as a new diagnostic tool in ARVD and also suggest a prognostic impact, as the severity of the disease appears different according to the underlying gene or the presence of multiple mutations.8,14 In a substantial number of probands, more than one disease-causing mutation can be found (mostly in different genes).15 Therefore, it is recommended that all desmosome genes be tested simultaneously in the proband. Whenever a pathogenic mutation is identified, it becomes possible to establish a presymptomatic diagnosis of the disease among family members and to provide them with genetic counseling to monitor the development of the disease and to assess the risk of transmitting the disease to offspring.16 Importantly, genetic testing is not recommended for patients with only a single minor criterion according to the 2010 task force criteria.17
Risk Stratification
Factors identifying patients with ARVD who are at risk for SCD have not yet been defined in large prospective studies focusing on survival. Nonetheless, several markers of increased risk for life-threatening ventricular arrhythmias have been described, including (1) previous history of cardiac arrest or a history of VT with hemodynamic compromise; (2) history of unexplained syncope (when VT or VF has not been excluded as the cause of syncope); (3) extensive RV disease; (4) LV involvement; (5) family history of cardiac arrest in first-degree relatives; (6) genotypes of ARVD associated with a high risk for SCD (e.g., patients with ARVD2, who can develop polymorphic VT and juvenile SCD associated with sympathetic stimulation); (7) increased QRS dispersion (maximal measured QRS duration minus minimal measured QRS duration is 40 milliseconds or more); (8) S wave upstroke equal to or greater than 0 milliseconds on the 12-lead ECG; (9) patients with Naxos disease; (10) induction of VT during EP testing; (11) detection of nonsustained VT on noninvasive monitoring; (12) male gender; and (13) young age at presentation (<5 years).18
Principles of Management
Pharmacological Therapy
Previous studies, which determined selection of antiarrhythmic therapy based on suppression of VT in the EP laboratory, found that sotalol was more effective than beta blockers and amiodarone in patients with inducible VT and in those with noninducible VT. Thus, sotalol was considered as a first-line antiarrhythmic agent in ARVD both for primary and secondary prevention. However, this was questioned in a recent study of a cohort of rigorously characterized ARVD population treated with empirically prescribed antiarrhythmic agents. In this study, beta blockers were neither protective nor harmful, sotalol was not effective, and amiodarone, although only received by a relatively small number of patients, showed superior efficacy in preventing sustained VT and implantable cardioverter-defibrillator (ICD) therapies.19
Therefore, among asymptomatic patients and those with mild disease, beta blockers may still be a reasonable recommendation to reduce the possibility of adrenergically induced arrhythmia. Patients with well-tolerated and non–life-threatening ventricular arrhythmias are at relatively low risk for SCD and can be treated with antiarrhythmic drugs, guided either noninvasively with ambulatory monitoring or with EP testing.20 Recent data argue against the empirical use of sotalol therapy, especially when permitting additional episodes of VT/VF is highly undesirable. It appears that, as in other settings, amiodarone is the most effective empirical antiarrhythmic drug therapy in patients with ARVD.19
When the disease has progressed to right or biventricular failure, treatment consists of the current therapy for heart failure, including diuretics, beta blockers, and angiotensin-converting enzyme inhibitors. Although angiotensin-converting enzyme inhibitors are well known for slowing progression in other cardiomyopathies, they have not been proven to be helpful in ARVD. Individuals with reduced RV ejection fraction with dyskinetic portions of the RV may benefit from long-term anticoagulation with warfarin to prevent thrombus formation and subsequent pulmonary embolism. For intractable RV failure, cardiac transplantation can be the only remaining alternative.20
Implantable Cardioverter-Defibrillator
An ICD is the most effective prevention against SCD. For secondary prevention, ICD implantation is recommended in patients with prior cardiac arrest and those with poorly tolerated arrhythmias that are not completely suppressed by antiarrhythmic drug therapy. During a mean follow-up of 39 months, nearly one-half of these patients had an appropriate ICD therapy, 16% had an inappropriate therapy, and 14% had a device-related complication.5,21
ICD implantation is also recommended for primary prevention in patients who are considered to be at high risk for SCD (see above). It is important to recognize, however, that there is not yet clear consensus on the specific risk factors that identify those patients with ARVD in whom the probability of SCD is sufficiently high to warrant an ICD for primary prevention.18
Importantly, ICD implantation in ARVD patients is associated with several risks. Areas of the RV myocardium in patients with ARVD may be thin and noncontractile and can be penetrated during placement of the RV leads, with subsequent cardiac tamponade. Also, the fibrofatty nature of the RV can lead to difficulty in attaining a location with acceptable R wave amplitude and pacing threshold. Marginal parameters may prevent adequate sensing of arrhythmias, resulting in improper ICD function or failure. A separate pace-sense lead may be necessary, positioned in the RV septum or outflow tract.20
Catheter Ablation
Catheter ablation of monomorphic VT has a 60% to 90% success rate in patients with ARVD. However, given the chance of long-term recurrence, ablation is not considered curative. Therefore, catheter ablation of VT has been considered mainly in patients with recurrent VT causing frequent ICD shocks and refractory to antiarrhythmic medications and those with VT storm or incessant VT refractory to antiarrhythmic medications. Nonetheless, with the recent advances in mapping and ablation techniques and the improved safety and success of the ablation procedures, as well as the lack of highly effective antiarrhythmic drug therapy, some investigators recommend catheter ablation as a first-line approach in patients presenting with recurrent monomorphic VT.8,22 ICD implantation is recommended as well, because of the unpredictability of recurrences.
Patients with limited endocardial substrate, those with late VT termination with RF delivery, and certainly those with persistent VT despite aggressive endocardial ablation should be considered for an epicardial approach.23
Participation in Sports
Because of the association between exercise and the induction of ventricular tachyarrhythmias, a diagnosis of ARVD is generally considered incompatible with competitive sports and moderate- to high-intensity-level recreational activities. Furthermore, any activity, competitive or not, that causes symptoms of palpitations, presyncope, or syncope should be avoided.20
Family Screening
First-degree relatives should be screened clinically with 12-lead ECG, echocardiography, and cardiac MR imaging. Given the low penetrance observed in most families, screening should be extended throughout the kindred to at least one generation beyond the last affected individual. Asymptomatic family members with a normal comprehensive evaluation are less likely to have inherited the gene defect, but should undergo follow-up at regular intervals (every 2 to 3 years) until definitive diagnostic tools are available.5 When the causative mutation in the index patient is identified, genetic testing is recommended in family members. A negative genetic test result for the familial mutation would obviate the need for repeated follow-up examinations.
Electrocardiographic Features
ECG during Sinus Rhythm
Approximately 40% to 50% of patients with ARVD have a normal ECG at presentation. However, by 6 years, almost all patients have one or more of several findings on ECG during normal sinus rhythm (NSR), including epsilon wave, T wave inversion, QRS duration equal to or greater than 110 milliseconds in leads V1 through V3, and RBBB. Significant differences exist in most ECG features among ARVD patients according to the degree of RV involvement, and they are more prevalent in diffuse ARVD than in the localized form of the disease.6,8
The hallmark feature of ARVD, the epsilon wave, is a marker of delayed activation of the RV free wall and outflow tract and is considered a major diagnostic criterion for ARVD. The epsilon wave has the appearance of a distinct wave just beyond the QRS, in the ST segment, in the right precordial leads, particularly in lead V1, and represents low-amplitude potentials caused by delayed activation of some portion of the RV (Fig. 29-3). Although the epsilon wave has been considered highly specific for ARVD, ECG markers that reflect ventricular conduction delay in ARVD (including the epsilon wave) are frequently observed in subjects with a spontaneous or drug-induced type 1 ECG pattern of Brugada syndrome.24 Additionally, this criterion is of limited sensitivity, observed in only 10% to 37% of ARVD patients when evaluated by standard ECG. The use of highly amplified (20 mV) precordial leads and modified limb leads can increase the detection of epsilon potentials to 75% but is rarely utilized.6,8
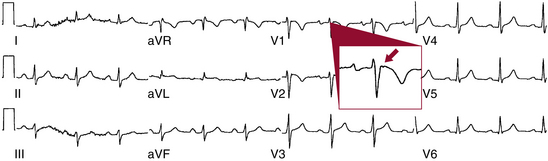
Other markers of delayed activation of the RV include prolongation of the QRS duration to at least 110 milliseconds in leads V1 through V3, and a ratio of the sum of the QRS duration in leads (V1 + V2 + V3)/(V4 + V5 + V6) ≥ 1.2 (sensitivity of 35% to 98%). In fact, a QRS duration greater than 110 milliseconds in lead V1 has a sensitivity of 26% to 75% and a specificity of 100% in patients suspected of having ARVD based on historical features. Additionally, incomplete or complete RBBB can be observed in 18% and 15% of patients, respectively. Epicardial mapping suggests that these patterns are usually caused by parietal block, rather than by disease of the bundle branch itself. In the presence of an RBBB pattern, selective prolongation of the QRS duration in leads V1 to V3 compared with lead V6 (>25 milliseconds, parietal block) is an important hallmark of ARVD (sensitivity of 52% to 64%).6,8,11,12 Furthermore, T wave inversion in leads V1 through V3 in the absence of RBBB is considered a minor diagnostic criterion for ARVD and its prevalence in ARVD has been reported as 55% to 94% in different series. The extent of right precordial T wave inversion relates to the degree of RV involvement. Increased QT dispersion (i.e., inter-lead variability of the QT interval) has been observed in ARVD.
Prolongation of the upstroke of the S wave (≥55 milliseconds from the nadir of the S wave to the isoelectric line) in leads V1 through V3 in the absence of RBBB, which presumably represents altered depolarization, was found in the original report to be a very sensitive ECG criterion for ARVD (present in all cases with diffuse ARVD and in 90% in the localized form of the disease). However, the sensitivity of this criterion was less than 60% in several other reports. This measurement has the highest diagnostic usefulness for differentiating the localized form of ARVD from idiopathic RVOT VT, correlates with the degree of RV involvement, and is an independent predictor of VT induction. It is recognized that a prolonged S wave upstroke directly relates to QRS width in the right precordial leads; nonetheless, it is superior to localized QRS prolongation in distinguishing the mild form of ARVD from idiopathic RVOT VT.6,11,12
Another ECG marker of ARVD described recently is the “terminal activation delay.” This parameter is measured from the nadir of the S wave to the end of all depolarization deflections, thereby including not only the S wave upstroke but also both late fractionated signals and epsilon waves, to the completion of the QRS complex. A prolonged terminal activation delay (≥55 milliseconds) was observed in 71% of ARVD patients and only 4% of controls.6,25
QRS fragmentation in standard ECG leads, defined as deflections at the beginning of the QRS complex, on top of the R wave or in the nadir of the S wave, has a sensitivity of 85% for the diagnosis of ARVD.26
The SAECG is a highly amplified and signal-processed ECG that can detect microvolt-level electrical potentials in the terminal QRS complex, known as late potentials. Late potentials, which reflect the slow conduction in the ventricular myocardium and electrical potentials that extend beyond the activation time of normal myocardium, typically arise from scarred myocardium, an anatomical substrate potentially responsible for reentrant ventricular arrhythmias. The SAECG and its three components (filtered QRS duration, low-amplitude signal duration, and root-mean-square voltage of the last 40 milliseconds of the QRS) are highly associated with the diagnosis of ARVD. Late potentials and fragmented electrical activity can be detected by signal-averaged ECG in 50% to 80% of patients with ARVD. Although there are no guidelines for abnormal SAECG in patients with ARVD, it is common practice to categorize the SAECG as abnormal if two or more of these parameters are abnormal. In a recent report, the sensitivity of using SAECG for diagnosis of ARVD was increased from 47% using two of the three criteria to 69% by using any one of the three criteria, while maintaining a high specificity of 90% to 95%. Abnormal SAECG was strongly associated with dilated RV volumes and decreased RV ejection fraction detected by cardiac MR imaging. SAECG abnormalities did not vary with clinical presentation or reliably predict spontaneous or inducible VT, and had limited correlation with ECG findings.11,24,27 Additionally, SAECG abnormalities appear to correlate with the presence of low-voltage areas selectively in the RVOT as observed during electroanatomical voltage mapping, whereas surface ECG abnormalities are associated with a more diffuse RV involvement.28 The usefulness of SAECG for predicting inducible VT in ARVD remains uncertain.24
ECG during Ventricular Tachycardia
Ventricular ectopy in ARVD usually arises from the RV and therefore has an LBBB pattern similar to that seen in idiopathic RVOT VT. The ECG morphology of the VT is predominantly LBBB, but RBBB can be observed and does not exclude an RV origin (Fig. 29-4). The QRS axis during VT is typically between –90 and +110 degrees; an extreme rightward direction of the QRS axis is uncommon.29 No correlation has been observed between any specific ECG morphology and a particular activation pattern, although all RBBB morphology VTs exhibit a peritricuspid circuit.
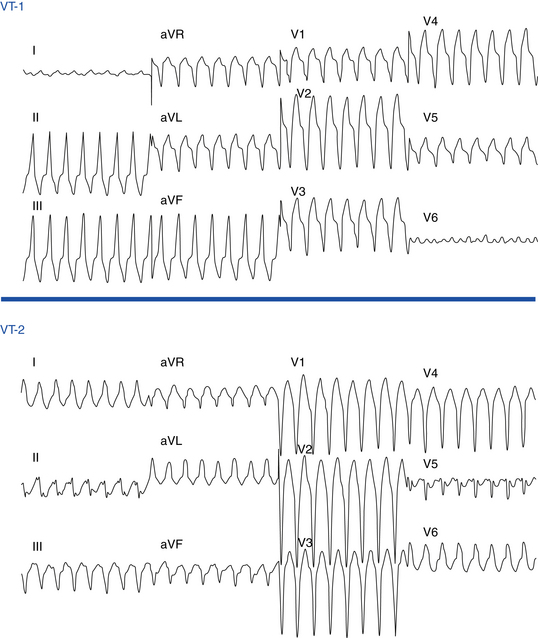
Additionally, patients with ARVD frequently demonstrate the presence of more than one VT morphology, either spontaneously (64%) or during an EP study (88%).6
Electrophysiological Features
Tachycardia Features
Reentry in areas of abnormal myocardium is the most likely mechanism of VT in ARVD. Most reentrant circuits cluster around the tricuspid annulus and the RVOT. Multiple morphologies of inducible VT are common. A variety of different reentry circuit sites can be identified with entrainment mapping. A single region can give rise to multiple VT morphologies. Previous studies showed mean numbers of different VT morphologies per patient ranging from 1.8 to 3.8.6
Exclusion of Other Arrhythmia Mechanisms
In clinical practice, the disease that most frequently mimics ARVD is idiopathic RVOT VT. Although RVOT VT is associated with a benign prognosis with no familial basis, it can be extremely difficult to distinguish from the concealed phase of ARVD, in which typical ECG and imaging abnormalities are absent.
The resting surface ECG in NSR is typically normal in patients with idiopathic VT, with no epsilon wave, QRS widening or fragmentation, or other markers of delayed activation of the RV typically observed in ARVD patients. However, the ECG is also normal in 40% to 50% of patients with ARVD at presentation, but in almost no patient after 6 years.30
Electroanatomical voltage mapping appears to be an effective way of distinguishing early or concealed ARVD from idiopathic VT by detecting RV electroanatomical scars that correlate with the histopathological features pathognomonic of the disease.31
Mapping
VTs in ARVD share many features observed in post-MI VTs. First, the tachycardias typically are monomorphic and have a macroreentrant mechanism. Second, the circuits are composed of zones of abnormal conduction, characterized by low-amplitude abnormal electrograms, with identifiable exit regions to the surrounding myocardium. Third, outer loops, which can be broad portions of the reentry circuit in communication with the surrounding myocardium, have also been observed. Fourth, the same types of targets for ablation previously identified in post-MI VT (critical isthmus) can also be useful for targeting VT in ARVD. Therefore, mapping techniques for ARVD-related VT are similar to those used for post-MI VT (see Chap. 22).8
Activation Mapping
Most reentrant circuits cluster around the inferolateral tricuspid annulus, the RVOT, and the RV apex, which are the typical areas of fibrous and fatty infiltration in ARVD. Hence, these areas are targeted first by activation mapping. Because the VT circuit is composed of zones of abnormal conduction, characterized by low-amplitude abnormal electrograms, with identifiable exit regions to the surrounding myocardium, endocardial activation mapping during VT typically detects abnormal fragmented or split low-voltage electrograms, with diastolic potentials (Fig. 29-5).8
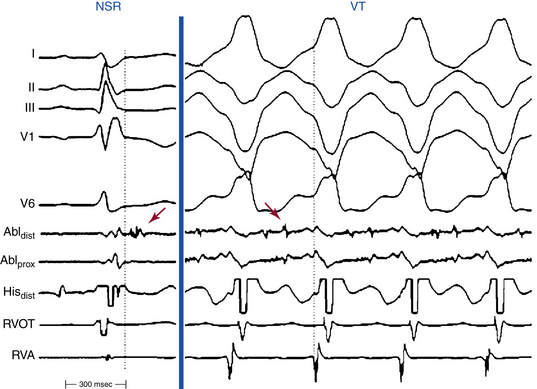
Detailed mapping will usually reveal more than one site of presystolic activity. It is therefore essential to demonstrate that the presystolic site recorded is in fact the earliest site. A normal presystolic bipolar electrogram (amplitude, >3 mV; duration, <70 milliseconds) should prompt further search for earlier activity. If, after very detailed mapping, the earliest recorded site is not at least 50 milliseconds presystolic, this suggests that either the map is inadequate (most common) or the VT arises deeper than the subendocardium, in the midmyocardium, or even incorporating the subepicardium. VTs in patients with ARVD exhibiting a focal activation pattern have also been described; however, an epicardial reentrant circuit with a defined endocardial exit may explain the focal activation pattern of the RV endocardium.8,29
An isolated mid-diastolic potential is defined as a low-amplitude, high-frequency diastolic potential separated from the preceding and subsequent ventricular electrograms by an isoelectric segment. It is likely that isolated mid-diastolic potentials that cannot be dissociated from the VT are generated in segments of the zone of slow conduction or common pathway (isthmus), which are integral components of the reentry circuit. It is important to recognize that not all presystolic activity recorded during VT is related to the mechanism; it could represent delayed activity not related to the tachycardia mechanism, dead-end pathways, or even an artifact. Therefore, it is important to confirm that presystolic activity is related to the reentrant circuit. Demonstration of a fixed relationship of the diastolic electrograms to the subsequent VT QRS, despite spontaneous or induced oscillations of the tachycardia CL, helps exclude the possibility that those electrograms reflect late activation from unrelated dead-end pathways.8
Additionally, 3-D electroanatomical mapping can help in the precise description of VT reentrant circuits, sequence of ventricular activation during the VT and rapid visualization of the activation wavefront, and identification of slow-conducting pathways and appropriate sites for entrainment mapping. Local activation times are assigned according to the onset of the bipolar electrogram registered at the tip of the mapping catheter and are color-coded. These systems also help in navigation of the ablation catheter, planning of ablation lines, and maintaining a log of sites of interest (e.g., sites with favorable entrainment or pace mapping findings), which can then be revisited with precision. Additionally, voltage (scar) mapping is a helpful feature of some of the electroanatomical mapping systems (see later).8
Entrainment Mapping
Entrainment mapping is used to characterize reentry circuits in ARVD, identify critical isthmuses, and guide ablation. Pacing is performed during VT at a CL 10 to 30 milliseconds shorter than the VT CL, at RV sites identified during activation mapping, demonstrating continuous activity or isolated mid-diastolic potentials. For stable VTs, the following three criteria are used to define the critical isthmus and predict the success of RF application in termination of the VT: (1) entrainment with concealed fusion; (2) PPI equal to the tachycardia CL (within 20 to 30 milliseconds); (3) and S-QRS interval equal to the local electrogram-to-QRS interval (±20 milliseconds). Other predictors of successful ablation sites include S-QRS interval–to–VT CL ratio less than 0.7, long S-QRS interval, and alteration of the tachycardia CL and/or termination by subthreshold or nonpropagated stimuli. However, because of the diffuse nature of the disease in ARVD, isthmuses may sometimes be wide and good entrainment maps can be obtained from a rather large area. In this case, linear lesions across the diastolic pathway may be necessary.8
Pace Mapping
Pace mapping can also be used in conjunction with substrate mapping when other mapping techniques are not feasible, so that it can provide information on where ablation can be directed. The VT circuit’s exit site is approximated by the site of pace mapping that generates QRS complexes similar to those of VT. From that point, evaluation of the S-QRS interval (measured to the onset of the earliest QRS on the 12-lead ECG) can help trace the course of the VT isthmus. Sites along the VT isthmus are associated with good pace maps but with progressively longer S-QRS intervals.8
Electroanatomical Substrate Mapping
Electroanatomical substrate mapping provides a helpful tool in reconstructing the VT circuit in patients with ARVD and hemodynamically stable VTs, in conjunction with activation and entrainment mapping techniques.29 Additionally, in a subset of ARVD patients, conventional mapping during VT can be limited by changing multiple morphologies, nonsustainability, or hemodynamic instability. Substrate-based voltage mapping during sinus rhythm can be used in these cases to identify regions of scar and abnormal myocardium and guide, in conjunction with pace mapping, ablation of linear lesions to connect or encircle the abnormal regions.22
As noted, in ARVD, the normal myocardium is replaced by fatty and then fibrous tissue, causing thinning and scarring, predominantly in the RV. Studies have found that regions of abnormal voltage suggestive of scar were observed in all ARVD patients. The endocardial bipolar voltage abnormalities tend to be perivalvular and, although affecting predominantly the RV free wall, involve some aspect of the septum in most (76%) patients. This pattern is consistent with pathological studies demonstrating a predilection for scar to occur in these particular areas in ARVD. Whether an extensive perivalvular fibrotic process is specific for patients who present with sustained monomorphic VT in the setting of ARVD is yet to be determined.22,29 In contrast to the post-MI pattern of dense scar surrounded by a border zone, the regions of abnormal myocardium in ARVD are commonly patchy and inhomogeneous, occurring in anatomically disparate areas. As a consequence, low-voltage identification in ARVD is less useful as a marker for ablation because it is very widespread. Nonetheless, evaluating the existence of different voltage areas within the 0.5-mV scar (i.e., the voltage limits are reset to 0.3 to 0.5 mV as the upper limit and to 0.1 mV as the lower, representing dense scar) can potentially help visualize channels of slow conduction within scar tissue.8 The critical isthmus of each of the reentrant circuit typically is bounded by an area of low-voltage scar on one side and an anatomical barrier (tricuspid annulus) on the other side, or by two parallel lines of double potentials.29
A recent report found that identifying ventricular electrograms with an isolated delayed component during sinus rhythm (defined as a distinct electrogram after the QRS separated [by ≥40 milliseconds] by an isoelectric interval or very low-amplitude signal of <0.1 mV) can be used to define the substrate for the VT circuit and, when combined with other mapping maneuvers, can help guide catheter ablation in ARVD patients with noninducible or unmappable VTs.32
Importantly, the disease in ARVD has been reported to begin epicardially and progress to the endocardium; therefore, endocardial fibrosis, as reflected by the voltage map findings, can be a manifestation of more extensive disease involvement and can be anticipated to be the more common substrate for uniform sustained VT (Fig. 29-6).23
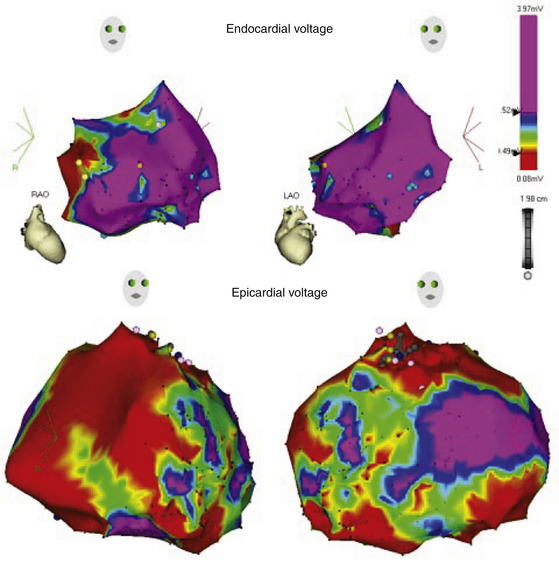
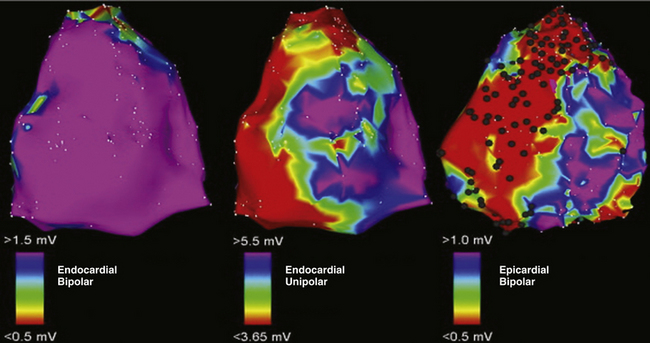
Endocardial unipolar voltage mapping (with a cutoff of 5.5 mV) can potentially provide a more global assessment of myocardial voltage and more closely approximate the degree and location of epicardial bipolar voltage abnormalities that serve as a substrate for VT in ARVD patients with no or limited endocardial bipolar voltage abnormalities. Therefore, endocardial unipolar voltage mapping may help in decision making related to proceeding to epicardial substrate mapping and ablation (Fig. 29-7).33
Noncontact Mapping
Once chamber geometry has been delineated, VT is induced and mapping can begin. The data acquisition process is performed automatically by the system, and all data for the entire chamber are acquired simultaneously. The system then reconstructs more than 3000 unipolar electrograms simultaneously and superimposes them onto the virtual endocardium, producing color-coded isopotential maps to depict graphically regions that are depolarized. Activation can be tracked on the isopotential map throughout the tachycardia cycle and wavefront propagation can be displayed as a user-controlled 3-D “movie.” The color range represents voltage or timing of onset. Sites of early (presystolic) endocardial activity, which are likely adjacent to reentry circuit exits, are usually identifiable; in some cases, isthmuses can be identified. Diastolic depolarization is defined as activity on the isopotential map that can be continuously tracked back in time from VT exit sites, defined on the map as synchronous with the QRS onset. Diastolic activity and exit sites are then marked on the virtual endocardium, and a mapping catheter is navigated to them by the locator technology. Ablation lesions can be tagged, facilitating performing linear ablation devoid of gaps across the tachycardia critical isthmus.
Substrate mapping based on scar or diseased tissue, which has been recently introduced to the noncontact mapping technology, can be particularly helpful when the VT is not inducible during the procedure. Dynamic substrate mapping allows the creation of voltage maps from a single cardiac cycle and provides the ability to identify low-voltage areas, as well as fixed and functional block, on the virtual endocardium through noncontact methodology.34
Percutaneous Epicardial Mapping
A recent report found that, in patients with ARVD and VT who failed endocardial ablation, low-voltage areas consistent with scar were more extensive on the epicardium than the endocardium and uniformly included wide, split, and late electrograms consistent with abnormal conduction. The origin of VT defined by activation/entrainment mapping and suggested by the detailed substrate mapping observations and targeted for ablation was also frequently noted beyond the endocardially defined scar. Successful epicardial ablation sites were often opposite endocardial sites with normal voltage or where ablation had been ineffective.23
Therefore, patients with limited endocardial substrate, those with late VT termination with endocardial RF delivery, and certainly those with persistent VT despite aggressive endocardial ablation should be considered for an epicardial approach. Additionally, surface ECG criteria identifying a QS complex in lead V2 or leads III and aVF during VT can be helpful in identifying patients who should be considered for an epicardial approach with the initial procedure.23
The approach to epicardial mapping is essentially the same as for endocardial ablation, including activation mapping, entrainment mapping, substrate mapping, and pace mapping, and is discussed in detail in Chapter 27.
Ablation
Target of Ablation
For stable VTs, reentry circuit isthmuses are the target of ablation and are defined by activation and entrainment mapping as sites with continuous activity or isolated mid-diastolic potentials, entrainment with concealed fusion, PPI equal to the VT CL, and S-QRS interval less than 70% of the VT CL. Alteration of the tachycardia CL or termination of VT by subthreshold or nonpropagated stimuli, and repeated VT termination occurring with catheter manipulation or pacing at the site, also suggest an isthmus location (Fig. 29-8).29
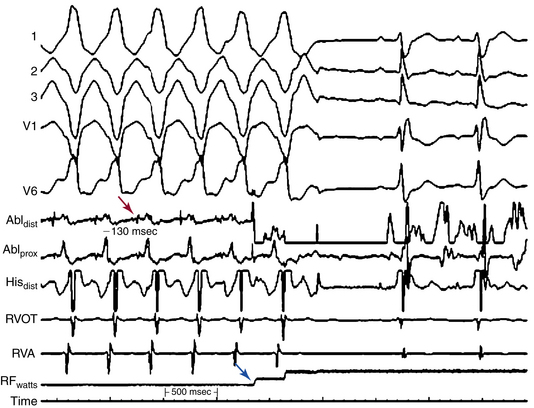
For hemodynamically unstable VTs, substrate mapping during sinus rhythm followed by brief inductions of VT (with the mapping catheter at the optimal site, as identified by substrate and pace mapping) can allow assessment of the relationship of the abnormal electrograms to the VT circuit. This can also allow entrainment maneuvers to be performed at those sites to help distinguish sites critical to the VT circuit from bystander sites. VT is then quickly terminated before significant hemodynamic compromise ensues. This approach can be facilitated by hemodynamic support with the use of intravenous vasopressors, intraaortic balloon pump, or both. This approach, however, requires being able to induce the same VT reproducibly. The use of noncontact mapping can be of value in these cases.
When the critical isthmuses of the reentrant circuit(s) can be identified, linear ablation lesions are delivered to sever those isthmuses. For unmappable VTs (due to poor inducibility or changing VT morphology), RF ablation can be performed as linear lesions targeting potential exit sites and isthmuses of VT circuits based on the location of the best pace map, location of valvular anatomical boundaries, and the substrate defined by the voltage mapping. Typically, linear lesions are created over target regions by sequential point lesions designed to: (1) connect scar or abnormal myocardium to a valve continuity; (2) connect scar or abnormal myocardium to another scar; (3) extend from the most abnormal endocardium (<0.5 mV) to normal myocardium (>1.5 mV); or (4) encircle the scar or abnormal region, depending on the scar location and size.22,32
Ablation Technique
Ablation may be performed with a 4-mm-tip (maximal target temperature of 60°C and maximal power of 50 W), an 8-mm-tip (maximal temperature of 65°C and maximal power of 75 W), or an irrigated-tip catheter (maximal temperature of 40-45°C and maximal power of 50 W). Irrigated RF ablation catheters are preferred, because power delivery can be limited in the trabeculated and annular regions where cooling from circulating blood flow is low. However, careful attention to power titration and monitoring impedance is necessary to reduce the risk of perforation when ablating in the free wall of the RV.35 Ablation is usually maintained for 60 to 120 seconds at the site where the tachycardia is terminated (in ablation during stable VT) or at each point along the linear ablation.8
Outcome
Ablation of VT in ARVD remains a clinical challenge and should be viewed as a potential palliative procedure in selected patients. Although VT ablation has been reported to successfully eliminate inducible VT acutely in 41% to 88% of patients, long-term success is low, and during average follow-ups of 11 to 24 months, VT recurs in 11% to 83% of patients, even following extensive or repeated ablation attempts.22,29,36 Recurrences usually manifest as nonsustained VT, a slower monomorphic sustained VT, or a new monomorphic VT.8,35
Multiple morphologies, hemodynamic instability, or noninducibility can limit the success of VT ablation. Additionally, ARVD can create a large number of potential circuits; an empirical approach may not eliminate all the possible pathways. Achieving adequate energy delivery can also be a problem, especially in regions difficult to access by catheter—in particular, abnormal regions along the tricuspid annulus, which can require the use of 8-mm-tip or irrigation-tip ablation catheters to achieve effective lesions underneath the tricuspid valve. It must be emphasized that caution must be taken when using irrigated-tip ablation in patients with very thin ventricular walls, although thick fibrous tissue is present in many areas. The risk of creating deeper lesions in thin walls of the RV body or apex potentially increases the risk of perforation. Although the magnitude of this risk is not well known, it seems to be infrequent.8
Furthermore, recent studies have shown a high prevalence of epicardial isthmuses in ARVD. Epicardial mapping and ablation have yielded a significant improvement in ablation efficacy; thus, it is reasonable to begin mapping endocardially, but be prepared to move to epicardial mapping following pericardial puncture if endocardial mapping and ablation do not quickly yield good results. In a recent report, percutaneous epicardial mapping and ablation were performed in 13 consecutive patients with ARVD after failed endocardial VT ablation. Twenty-seven distinct VTs were targeted for ablation from the epicardium; during 18 ± 13 months of follow-up, 77% of patients were free of VT.23
The discrepancy between the good acute results and the unfavorable long-term outcome may be explained by the progressive nature of ARVD, which predisposes to the occurrence of new and malignant arrhythmogenic substrates over time. This hypothesis, however, has been questioned by a recent study that demonstrated the absence of progression of the area of the endocardial scar as measured by voltage mapping in patients with ARVD and VT, despite demonstrating progressive RV dilation in these patients.7
1. Lombardi R., Marian A.J. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr Opin Cardiol. 2010;25:222-228.
2. Otten E., Asimaki A., Maass A., et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010;7:1058-1064.
3. Hamilton R.M., Fidler L. Right ventricular cardiomyopathy in the young: an emerging challenge. Heart Rhythm. 2009;6:571-575.
4. Maass K. Arrhythmogenic right ventricular cardiomyopathy and desmin: another gene fits the shoe. Heart Rhythm. 2010;7:1065-1066.
5. Boussy T., Paparella G., de Asmundis C., et al. Genetic basis of ventricular arrhythmias. Heart Fail Clin. 2010;6:249-266.
6. Cox M.G., Nelen M.R., Wilde A.A., et al. Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: toward improvement of diagnostic ECG criteria. J Cardiovasc Electrophysiol. 2008;19:775-781.
7. Riley M.P., Zado E., Bala R., et al. Lack of uniform progression of endocardial scar in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy and ventricular tachycardia. Circ Arrhythm Electrophysiol. 2010;3:332-338.
8. Arbelo E., Josephson M.E. Ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2010;21:473-486.
9. Avella A., d’Amati G., Pappalardo A., et al. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 2008;19:1127-1134.
10. Asimaki A., Tandri H., Huang H., et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075-1084.
11. Marcus F.I., McKenna W.J., Sherrill D., et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533-1541.
12. Marcus F.I., Zareba W., Calkins H., et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984-992.
13. Cox M.G., van der Smagt J.J., Noorman M., et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 2010;3:126-133.
14. Fressart V., Duthoit G., Donal E., et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861-868.
15. Bauce B., Nava A., Beffagna G., et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7:22-29.
16. Zipes D.P., Camm A.J., Borggrefe M., et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol. 2006;48:e247-e346.
17. Ackerman M.J., Priori S.G., Willems S., et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 2011;8:1308-1339.
18. Epstein A.E., DiMarco J.P., Ellenbogen K.A., et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: executive summary. Heart Rhythm. 2008;5:934-955.
19. Marcus G.M., Glidden D.V., Polonsky B., et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. J Am Coll Cardiol. 2009;54:609-615.
20. Kies P., Bootsma M., Bax J., et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 2006;3:225-234.
21. Hodgkinson K.A., Parfrey P.S., Bassett A.S., et al. The impact of implantable cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5),. J Am Coll Cardiol. 2005;45:400-408.
22. Verma A., Kilicaslan F., Schweikert R.A., et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation. 2005;111:3209-3216.
23. Garcia F.C., Bazan V., Zado E.S., et al. Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2009;120:366-375.
24. Letsas K.P., Efremidis M., Weber R., et al. Epsilon-like waves and ventricular conduction abnormalities in subjects with type 1 ECG pattern of Brugada syndrome. Heart Rhythm. 2011;8:874-878.
25. Cox M.G., van der Smagt J.J., Wilde A.A., et al. New ECG criteria in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2009;2:524-530.
26. Peters S., Trummel M., Koehler B. QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Heart Rhythm. 2008;5:1417-1421.
27. Folino A.F., Bauce B., Frigo G., Nava A. Long-term follow-up of the signal-averaged ECG in arrhythmogenic right ventricular cardiomyopathy: correlation with arrhythmic events and echocardiographic findings. Europace. 2006;8:423-429.
28. Santangeli P., Pieroni M., Dello R.A., et al. Noninvasive diagnosis of electroanatomic abnormalities in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2010;3:632-638.
29. Miljoen H., State S., De Chillou C.C., et al. Electroanatomic mapping characteristics of ventricular tachycardia in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Europace. 2005;7:516-524.
30. Shimizu W. Arrhythmias originating from the right ventricular outflow tract: how to distinguish “malignant” from “benign”? Heart Rhythm. 2009;6:1507-1511.
31. Corrado D., Basso C., Leoni L., et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J Am Coll Cardiol. 2008;51:731-739.
32. Nogami A., Sugiyasu A., Tada H., et al. Changes in the isolated delayed component as an endpoint of catheter ablation in arrhythmogenic right ventricular cardiomyopathy: predictor for long-term success. J Cardiovasc Electrophysiol. 2008;19:681-688.
33. Polin G.M., Haqqani H., Tzou W., et al. Endocardial unipolar voltage mapping to identify epicardial substrate in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2011;8:76-83.
34. Yao Y., Zhang S., He D.S., et al. Radiofrequency ablation of the ventricular tachycardia with arrhythmogenic right ventricular cardiomyopathy using non-contact mapping. Pacing Clin Electrophysiol. 2007;30:526-533.
35. Aliot E.M., Stevenson W.G., Mendral-Garrote J.M., et al. EHRA/HRS expert consensus on catheter ablation of ventricular arrhythmias: developed in a partnership with the European Heart Rhythm Association (EHRA), a Registered Branch of the European Society of Cardiology (ESC), and the Heart Rhythm Society (HRS); in collaboration with the American College of Cardiology (ACC) and the American Heart Association (AHA). Heart Rhythm. 2009;6:886-933.
36. Dalal D., Jain R., Tandri H., et al. Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:432-440.