Vasopressin, Diabetes Insipidus, and The Syndrome of Inappropriate Antidiuretic Hormone Secretion
Vasopressin
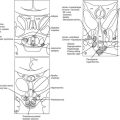
Arginine vasopressin (AVP) is a polypeptide that contains nine amino acids, with a disulfide bridge between cysteine residues, and considerable structural homology with oxytocin, which also is secreted from the posterior pituitary (Fig. 14-1).1 Vasopressin is synthesized predominantly in the magnocellular neurons in the paraventricular (PVN) and supraoptic (SON) nuclei of the anterior hypothalamus (Fig. 14-2). The cell bodies of the vasopressinergic neurons in the PVN and SON have axonal projections that connect to the posterior pituitary gland.
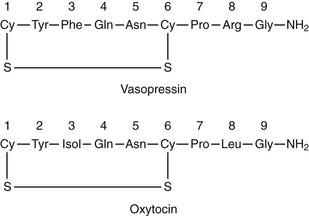
FIGURE 14-1 Amino acid structure of vasopressin and oxytocin. Note that only positions 3 and 8 have different amino acids.
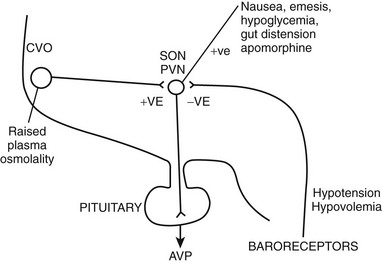
FIGURE 14-2 Factors governing arginine vasopressin (AVP) secretion. CVO, Circumventricular organ (site of osmoreceptors); PVN, paraventricular nuclei; SON, supraoptic nuclei.
In addition to magnocellular neurons, which project from the SON and the PVN to the posterior pituitary, parvocellular vasopressinergic neurons are present in the PVN and the suprachiasmatic nucleus.2 These smaller neurons project via the median eminence to the portal vascular system of the anterior pituitary while secreting vasopressin and corticotropin-releasing factor (CRF); they stimulate secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. Some parvocellular vasopressinergic neurons also project from the PVN to the forebrain, brain stem, and spinal cord. Vasopressin is released into the central nervous system, where it acts as a neurotransmitter and/or a neuromodulator.3
The gene that encodes the AVP precursor is expressed mainly in the hypothalamus but also in other tissues, including adrenal glands, gonads, cerebellum, and pituicytes in the posterior pituitary. It is structurally closely related to the gene that codes for the oxytocin (OT) precursor, which is expressed in different populations of hypothalamic magnocellular neurons. The vasopressin and oxytocin genes are closely linked in a tail-to-tail orientation on chromosome 20 with an intergenic region of 12 kb.4 Significant posttranslational processing is needed to form the mature hormone. The posttranslational product prepro-AVP-NPII is the precursor from which AVP is derived.4 The precursor consists of a signal peptide, AVP, neurophysin II (NPII; 95 amino acids), and a glycopeptide, copeptin (39 amino acids).
Neurophysin II is cleaved from prepro-AVP-NPII but remains complexed together with AVP before it is secreted into the bloodstream by exocytosis. Vasopressinergic neurons open voltage-gated calcium channels in nerve terminal membranes. This transient calcium ion influx results in fusion of neurosecretory granules with the nerve terminal membrane, as well as release of AVP and NPII in equimolar amounts into the circulation.5 Copeptin, although of unknown biological significance, may be a stable indirect marker of AVP release.
Magnocellular neurons in the supraoptic and paraventricular nuclei synthesize vasopressin within their cell bodies. The hormone is carried within neurosecretory granules down the predominantly unmyelinated axons of the magnocellular neurons, in the supraoptic-hypophyseal tract, at a speed of 2 mm per hour. The precursor undergoes successive cleavage by basic endopeptidases.6 These axons terminate in the posterior pituitary, which is rich in fenestrated capillaries.
Cellular Action of Vasopressin
Vasopressin binds to seven transmembrane, G-protein–coupled receptors on the plasma membrane of target cells. Three subtypes of vasopressin receptor exist (V1-V3) with distinct distribution, actions, and signal transduction.7,8
The V1 Receptor
Binding of AVP to V1 receptors at physiologic plasma concentrations has been shown to exert a weak pressor effect.8a The vasocontriction seen with vasopressin is most notable in the splanchnic, renal, and hepatic artery beds9–11 and has given rise to the successful use of the hormone in the treatment of bleeding esophageal varicies. At higher plasma concentrations, for instance, in response to significant hypotension or hypovolemia, vasopressin has been shown to have a significant pressor effect.12 A highly selective V1 vasopressin analogue has been shown to have similar effects in patients with sepsis.13 Rebound hypotension on withdrawal of treatment coupled with concerns regarding reduced cardiac output, pulmonary hypertension, hyponatremia, and bowel ischemia, has limited the widespread use of vasopressin in the setting of septic shock.
The V2 Receptor
When AVP binds to the V2 receptors on the basolateral membrane of the cells of the collecting ducts, it triggers an increase in cytoplasmic cyclic adenosine monophosphate (cAMP), which activates protein kinase (Fig. 14-3). Subsequently, movement of stored aquaporin-2 to the luminal membrane of the cell occurs. Aquaporin-2 forms tetramers in the cell membrane, with each monomer acting as a water channel14; this allows passage of tubular water into the cell. Free water reabsorption from the distal nephron and thus urine concentration are directly influenced by serum vasopressin levels. This relationship was established prior to the discovery of aquaporins. The AVP/V2 receptor interaction also simultaneously causes an increase in expression of the aquaporin-2 gene, with increased generation of mRNA for aquaporin-2.
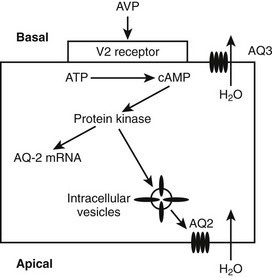
The V3 Receptor
The V3 receptor is found on corticotropic cells in the anterior pituitary. In isolation, AVP is a weak ACTH secretagogue, but when it acts synergistically with CRF, it causes significant secretion of ACTH, which is physiologically important. Parvocellular neurons, which coexpress AVP and CRF, project via the median eminence and terminate in the hypophyseal portal bed, which provides circulation to the anterior pituitary. AVP and CRF expression by these parvocellular neurons is subject to negative feedback control by glucocorticoids.15–17 AVP has no secretagogue properties with respect to the other anterior pituitary hormones.
Regulation of Vasopressin Release
Osmoregulation of Vasopressin Release
Plasma osmolality in healthy man varies by only 1% to 2% in physiologic conditions where there is free access to water. This precise regulation of plasma osmolality is maintained by the homeostatic process of osmoregulation.18
Changes in plasma osmolality are detected by specialized osmoreceptor cells in the anterior hypothalamus (see Fig. 14-2). Fenestrations in the blood-brain barrier cause the magnocellular cells of the circumventricular organs—the subfornical organ and the organum vasculosum lamina terminalis—to be bathed in plasma. Changes in plasma osmolality cause these hypothalamic nuclei to depolarize, sending neural signals, via the nucleus medianus, to the supraoptic and paraventricular nuclei. Elevation in plasma osmolality causes depolarization of these nuclei, leading to increased synthesis of AVP and secretion of AVP from the posterior pituitary. The osmoreceptor cells are solute specific, in that they respond vigorously to changes in plasma sodium concentration but less so to variations in blood urea.19 They respond to mannitol but are completely insensitive to changes in blood glucose concentration in experimental situations.
A linear relationship has been noted between plasma osmolality and plasma AVP concentrations throughout the physiologic range of plasma tonicity.20–22 If plasma concentrations are lowered by excessive ingestion of hypotonic fluid to below 280 to 285 mOsm/kg, secretion of AVP is suppressed, and plasma concentrations of the hormone are undetectable on radioimmunoassay measurement. In the absence of AVP-mediated water reabsorption from the kidney, hypotonic polyuria develops.22 The increase in free water clearance allows plasma osmolality to rise into the normal range. If plasma osmolality rises, owing to dehydration, plasma concentrations of AVP increase in proportion to the rise in plasma osmolality. The action of AVP in concentrating the urine and allowing reabsorption of water restores plasma osmolality to normal. This homeostatic process occurs continuously to maintain plasma osmolality within a narrow reference range. The relationship between plasma AVP concentration and urine osmolality is shown in Fig. 14-4.
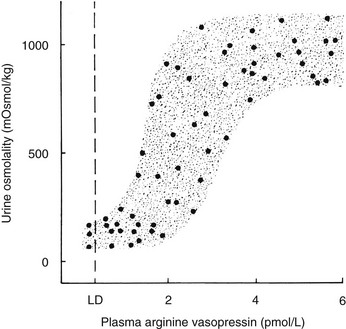
FIGURE 14-4 Relation between plasma arginine vasopressin (AVP) (pAVP) and urine osmolality. Data were obtained during water loading and fluid restriction in a group of healthy adults. Maximal urine concentration is achieved by pAVP values of 3 to 4 pmol/L. LD, Limit of detection of the AVP assay, 0.3 pmol/L.
The development of sensitive radioimmunoassays has allowed the nature of this relationship to be studied experimentally. When intravenous infusion of hypertonic sodium chloride solution is used to increase plasma osmolality in healthy volunteers, plasma AVP can be shown to increase in a linear fashion. Application of linear regression analysis to the data shows that a direct correlation defines the relationship between plasma osmolality and plasma AVP concentration (Fig. 14-5). The qualitative relationship is expressed by the following equation:
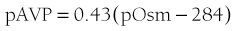
where pAVP indicates plasma AVP concentration, and pOsm indicates plasma osmolality.23
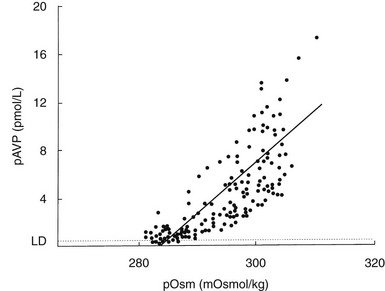
FIGURE 14-5 Relation between plasma osmolality (pOsm) and plasma arginine vasopressin (AVP) (pAVP). Increases in pAVP in response to hypertonicity induced by infusion of 855 mmol/L saline in a group of healthy adults. The mean regression line (dashed line) is defined by the following equation: pAVP = 0.43 (pOsm 284); r = 0.96; P < .001. LD, Limit of detection of the AVP assay, 0.3 pmol/L.
This mathematical formula describes two important physiologic characteristics of the relationship between plasma osmolality and plasma AVP that are clinically relevant. The first of these is the osmotic threshold for AVP release, which is defined by the abscissal intercept of the regression line. At a mean plasma osmolality of 284 mOsm/kg, plasma AVP is secreted, with increments in secretion as plasma osmolality rises. This represents the “set point,” or the osmotic threshold for AVP release, which is identical in man to the osmotic threshold for the onset of thirst.23 This is an important physiologic concept, as hypotonic polyuria develops when plasma osmolality is suppressed by water ingestion to concentrations below this “set point.” Therefore, AVP detectable in the plasma by radioimmunoassay at plasma osmolalities below this is, by definition, “inappropriately” elevated. Measurements of plasma AVP with ultrasensitive cytochemical methods at plasma osmolalities below the physiologic threshold have led to suggestions that secretion cannot be completely suppressed by hypotonicity,24 although the very low concentrations demonstrated experimentally could equally represent incomplete clearance of pre-secreted AVP.
The slope of the regression line, which represents the change in plasma AVP concentration per unit change in plasma osmolality, is the second important characteristic of the formula. This defines the sensitivity of the osmoreceptor-AVP releasing unit. Steeper slopes indicate a larger rise in plasma AVP concentrations after osmotic stimulation. Shallow slopes indicate lower rates of AVP secretion in response to hyperosmolality; a pathologic example of this can be seen in partial hypothalamic diabetes insipidus.25
In a few rare cases, complete disconnection of AVP-secreting neurons from their osmoreceptors has been described. The pathologic abnormality is persistent low-grade AVP release, despite plasma osmolalities that may fall below the osmotic threshold.26 AVP secretion can be increased to above this “basal” rate by stimulation of osmoreceptors, but secretion is never switched off entirely. It is hypothesized that some cells exert an inhibitory effect on AVP release, and if these cells malfunction, AVP secretion is abnormally continuous.
The osmotic threshold and sensitivity of AVP release vary significantly between individuals, but repeat testing has shown good reproducibility of these parameters within individuals.27 Studies in twins have suggested that the characteristics of the osmoregulatory line may be genetically determined, given that they are similar in monozygous but not dizygous twins.28
Although physiologic control of AVP secretion and thirst is almost entirely osmotic, the switch-off of the two is nonosmotic and is triggered by the act of drinking. In studies in healthy men who have been rendered hyperosmolar by hypertonic saline infusion29 or by dehydration,30 drinking is associated with an immediate fall in plasma AVP and thirst, before any changes in plasma osmolality can be measured. The fall in plasma AVP is so rapid that it mimics the half-life of AVP,29 suggesting that a neuroendocrine reflex, stimulated by oropharyngeal distention, switches off AVP secretion.
The conventional relationship between plasma osmolality and plasma AVP concentration is altered in a number of physiologic and pathophysiologic situations. Pregnancy causes a lowering of the threshold for AVP secretion in both rats31 and humans,32 which is responsible for the fall in basal plasma sodium concentration during pregnancy. The sensitivity of the osmoregulatory line remains unchanged. The mechanism is unclear but may be related to increased plasma concentrations of human chorionic gonadotropin (hCG). In the luteal phase of the ovulatory cycle, a small but significant decrease in plasma osmolality also occurs as a result of downward resetting of the osmotic thresholds for thirst and AVP release.33
Normal physiologic aging is associated with complex changes in osmoregulation.34 Basal circulating AVP concentrations increase with age, and the AVP response to osmotic stimulation has been shown to be enhanced in comparison with that in younger subjects.35,36 Thirst, as measured by visual analogue scales, is attenuated, however, and measured water intake after osmotic stimulation intake is reduced.37 This makes elderly humans more vulnerable to hypernatremia; many elderly patients in long-term care institutions exhibit permanent hypernatremia, although cognitive impairment may contribute to reduced thirst in these individuals.38
Osmoregulation in diabetes mellitus seems normal,39 even in the presence of hypernatremia,40 with preserved solute specificity of both AVP release and thirst; hyperglycemia stimulates neither thirst nor AVP secretion. Chronic hyperglycemia renders the renal tubules resistant to the antidiuretic effects of AVP, however, as the result of failure of recruitment of aquaporin-2.41 Improved glycemic control reverses the state of partial nephrogenic diabetes insipidus.41 Survivors of HONK, however, show typical osmoregulatory changes of aging, with exaggerated AVP secretion and attenuated thirst in response to dehydration.42
Baroregulation of Vasopressin Release
Although small variations in plasma osmolality can have profound effects on AVP secretion, physiologic fluctuations in blood pressure have almost no effect on plasma AVP secretion. However, pathophysiologic falls in both blood volume and pressure can stimulate AVP secretion. Although small falls in arterial blood pressure (on the order of 10%) have been shown to slightly increase plasma AVP concentrations (see Fig. 14-5), larger falls of 20% to 30% are required to stimulate sufficient AVP to exert a compensatory pressor effect.43,44 This makes teleologic sense as falls in blood pressure of up to 10% are common during a daily cycle, and if AVP were stimulated by these small falls, humans would be in a permanent state of antidiuresis. The relationship between blood pressure and plasma AVP is shown in Fig. 14-6.
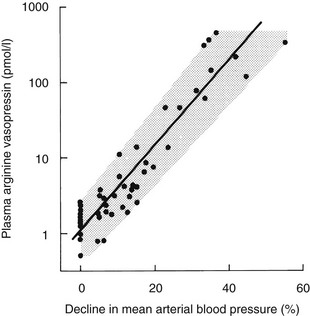
FIGURE 14-6 Relation of plasma arginine vasopressin (AVP) (pAVP) to the percentage decline in mean arterial blood pressure (MABP). Arterial blood pressure was reduced by infusing increasing doses of trimetaphan in healthy men. The regression line is defined by the following equation: log (pAVP) = 0.06 (MABP 0.67); r = 0.98; P < .001; N = 48.
The baroreceptors are situated in the cardiac atria, the carotid sinus, and the aortic arch. They exert a tonic inhibitory effect on AVP secretion in normal circumstances, but when they are unloaded during hypotension or hypovolemia, they can stimulate very high plasma concentrations of AVP via pathways that are separate from the osmoreceptors. The relation between blood pressure and AVP is exponential rather than linear and can be modified by neurohumoral influences such as atrial natriuretic peptides, which inhibit AVP responses, and norepinephrine, which augments baroregulatory AVP responses.45 Baroregulatory inputs also modify osmotic AVP secretion, in that hypovolemia augments the AVP response to hypernatremia.46,47
Other Mechanisms Regulating Vasopressin Release
In addition to osmotic and baroregulatory stimuli, AVP secretion occurs in response to nausea,48,49 manipulation of abdominal contents at surgery,50 and hypoglycemia.51 All of these secretagogues stimulate AVP secretion independently of the osmotic pathways.
Diabetes Insipidus
Diabetes insipidus is a clinical syndrome characterized by hypotonic polyuria due to failure to concentrate urine. Urine flow rates in excess of 40 mL/kg per 24 hours in adults, or more than 100 mL/kg per 24 hours in infants, are suggestive of diabetes insipidus. Three major categories of diabetes insipidus have been identified.52
In dipsogenic diabetes insipidus, excessive fluid intake reflects abnormal perception of thirst.
A full list of causes of diabetes insipidus is shown in Table 14-1.
Table 14-1

Hypothalamic Diabetes Insipidus
HDI (also known as neurogenic, central, or cranial DI) occurs when AVP secretion is insufficient to prevent the development of a hypotonic diuresis, such that the patient presents with polyuria and nocturia. The lesion in the vast majority of cases spares the thirst mechanism53; therefore, because thirst appreciation is intact, patients respond to the polyuria with appropriate polydipsia. The increase in fluid intake is nearly always sufficient to maintain normonatremia; hypernatremia in which fluid is freely available should always raise the suspicion of associated thirst abnormalities. The AVP deficiency may be complete, with no detectable plasma AVP, or partial, where AVP is detectable in the plasma at low concentrations inappropriate to the ambient plasma osmolality.
DI occurs in the immediate postoperative period in 18% to 30% of cases,54–57 with most cases presenting in the first 2 days following surgery. The most common natural history is spontaneous resolution over the next 2 to 5 days. The pathophysiology of acute transient DI is thought to be surgical contusion injury to the magnocellular neurons projecting from the supraoptic and paraventricular nuclei to the posterior pituitary. In some patients however, DI persists and AVP deficiency becomes permanent. Most neurosurgical series report a low incidence of 1% to 8% for permanent DI, following transsphenoidal surgery.54,56–58 Many published studies are retrospective, however, and use polyuria alone as the criteria for diagnosis of DI. We found that formal water deprivation testing identified permanent DI in 30% of patients who had transcranial surgery for pituitary adenoma and much higher figures of over 90% following craniopharyngioma surgery.55
The major factor influencing the risk of developing diabetes insipidus following pituitary surgery is tumor type. Craniopharyngioma is associated with preoperative DI, but in addition, surgical intervention is far more commonly associated with the development of DI than is surgery for any other intracranial tumor.55 Surgery for ACTH-secreting pituitary adenomas has been associated with a higher risk of developing DI in a number of series.54,56,58 Data from the literature are conflicting regarding the influence of tumor size on the risk of postoperative diabetes insipidus. Although pituitary adenomas rarely cause HDI, metastatic deposits in the hypothalamus, or more often the pituitary stalk, can result in HDI. Metastatic deposits usually have no endocrine effects, even when they are found in the stalk or the pituitary itself, but some, particularly from carcinoma of the breast or bronchus, produce HDI. Other tumors such as germinoma, pinealoma, and parasellar meningioma can cause HDI. It is interesting to note that although radiotherapy for nonpituitary cranial tumors commonly causes anterior pituitary dysfunction, it does not seem to cause DI.59
Lymphocytic infiltration of the neurohypophysis (infundibulo-neurohypophysitis), recognized by a thickened pituitary stalk and inflammatory infiltrates of T lymphocytes and plasma cells with eosinophils, is a well-described cause of HDI.60 HDI is present in up to 30% of adult patients with Langerhans’ cell histiocytosis (LCH).61
Idiopathic DI is diagnosed less often, as better imaging techniques have been introduced and awareness of autoimmune causes has increased.62,63 It has been estimated that one third of patients previously diagnosed to have idiopathic HDI may an autoimmune origin of the condition. The characteristics of autoimmune DI include the presence of circulating antibodies against AVP-secreting cells, young age of onset, and thickened pituitary stalk T1-weighted magnetic resonance imaging (MRI).64 Patients with autoimmune DI have an increased incidence of other organ-specific autoimmune endocrine disease, particularly thyroid disease.
Traumatic brain injury causes acute diabetes insipidus in 20% of cases,65 some of which result in a triple phase response similar to that seen after pituitary surgery. DI following brain injury is nearly always transient,66 however, and only 7% of long-term survivors of head injury have permanent DI when formally tested with water deprivation.67
Sheehan’s syndrome is now rare and is not usually associated with HDI, even when anterior hypopituitarism is widespread, but maximal urine-concentrating ability is impaired in some patients.68 HDI can occasionally occur in normal pregnancy as the result of increased activity of circulating vasopressinase, the placental aminopeptidase.69 Symptoms resolve post partum. The actions of vasopressinase can unmask partial HDI in patients with pituitary disease or can worsen symptoms in undiagnosed partial HDI. HDI in pregnancy must be differentiated from the transient NDI that is seen very occasionally.70
Familial HDI is extremely rare. Autosomal dominant HDI (adHDI) is associated with loss-of-function mutations in the AVP gene, resulting in the production of a folding-incompetent peptide precursor that accumulates in the secretory pathway apparatus of magnocellular neurons.71,72 This mutant precursor is responsible for an autophagocytic process that causes progressive damage and loss of VP neurons. A single mutant allele is sufficient for this process to occur. Most mutations affect exons 1 and 2 of the AVP gene.73–75 Familial HDI almost always presents in childhood.76
The Wolfram, or DIDMOAD, syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness) is a rare autosomal recessive, progressive neurodegenerative disorder characterized by the association of HDI with diabetes mellitus, optic atrophy, and bilateral sensorineural deafness, although other manifestations may include gonadal failure, renal outflow tract dilatation (secondary to reduced nerve fibers in the bladder wall), and progressive ataxia with brain stem atrophy.77 The syndrome is associated with premature death, usually due to ascending renal tract infection secondary to atonic bladder and hydronephrosis. Although DI is often reported to occur in only one third of cases, in one series where careful testing with AVP responses to hypertonic saline was performed, subnormal responses were seen in all patients.78 WS is associated with loss-of-function mutations in the WFSI gene on Ch.4p16.1, which encodes an 890 amino acid glycoprotein (wolframin) found in the endoplasmic reticulum. An additional locus for WS has been identified recently at Ch.4q22-24, suggesting that the syndrome may be genetically heterogeneous.79
Nephrogenic Diabetes Insipidus
Nephrogenic diabetes insipidus (NDI) occurs when renal resistance to the antidiuretic effects of VP allows hypotonic diuresis to develop. An outline of potential causes is given in Table 14-1. Lithium therapy is the most common cause of NDI in clinical practice, occurring in up to 30% of patients. Although polyuria usually disappears with cessation of treatment, a minority of patients develop interstitial nephritis, and permanent NDI occurs. Hypokalemia and hypercalcemia can produce acquired NDI, which is reversible with correction of the metabolic abnormality. Poorly controlled diabetes mellitus causes renal resistance to AVP, which seems to be due to failure to recruit aquaporin-2. The reversibility of the metabolic causes of NDI shows how the generation of aquaporins can be vulnerable to metabolic and pharmacologic insults.
Familial NDI is rare. X-linked recessive familial NDI (X-FNDI) is caused by inherited loss-of-function mutations in the V2-R. More than 70 different mutations have been described, affecting all aspects of receptor physiology, including expression, ligand binding, and G protein coupling. A vast majority cause complete loss of function and present in infancy, although some mutations are associated with partial loss of function.80 A minority of FNDI families have autosomal recessive (AR-FNDI), loss-of-function mutations of the gene encoding AQP2. Most mutations occur in the region of the gene that encodes the transmembrane domain of the water channel protein. Other kindreds have been described with an autosomal dominant NDI (AD-NDI) caused by loss-of-function mutations in AQP2 affecting the carboxyl-terminal intracellular domain of the protein. NDI is expressed in these kindreds because the protein product of the mutant allele forms mixed oligomers with the product of the wild-type allele, resulting in sequestration within the Golgi or mistargeting to the basolateral rather than the apical membrane in a dominant negative manner.81,82
Dipsogenic Diabetes Insipidus
Dipsogenic diabetes insipidus (DDI) occurs when excessive fluid intake lowers plasma osmolality to levels below the osmotic threshold for vasopressin secretion.83 In the absence of circulating AVP, hypotonic polyuria develops. Many patients seem to have no underlying illness, but careful osmoregulatory studies have identified a number of abnormalities of thirst regulation. Patients typically have a low osmotic threshold for thirst, which compels them to drink, even when plasma osmolality is lowered below the usual physiologic thirst threshold. In addition, they have an exaggerated thirst response to osmotic challenge and an inability to suppress thirst in response to drinking, so the volume of fluid drunk is in excess of that required for normal hydration.84 Some forms of DDI are associated with psychiatric disorders, and up to 20% of patients with chronic schizophrenia have DDI. If these patients are simultaneously prescribed drugs that can cause syndrome of inappropriate antidiuretic hormone secretion (SIADH), hyponatremia is inevitable and occasionally severe. Rarely, the problem may be associated with irritative structural abnormalities in the hypothalamus or posterior pituitary, such as hypothalamic sarcoidosis or craniopharyngioma, but usually brain imaging is normal.
Investigation of Diabetes Insipidus
In most patients, it is necessary to proceed to a water deprivation test.52 This is a two-step test of osmoregulatory function. The first step consists of an 8-hour period of dehydration. The normal physiologic response is to respond to the rise in plasma osmolality with secretion of AVP and concentration of urine to more than 750 mOsm/kg. Theoretically, patients with DDI have a normal osmoregulatory mechanism and should respond to dehydration with appropriate urine concentration. In the simplest use of this test, therefore, the key measurement is final urine osmolality, and the result answers the key question, “Does the patient have true DI, or not?” In patients who are being investigated for post-hypophysectomy HDI, only this step is needed to make the diagnosis.
In practice, the water deprivation test does not always produce straightforward results. Surreptitious water intake can give spurious results, so the test should be undertaken with strict supervision; results are best expressed in units and are optimal when attained by the clinician who has experience in performing the test. Partial forms of HDI and NDI cannot be differentiated from DDI.85 Furthermore, prolonged polyuria from any cause leads to partial resistance to the antidiuretic action of vasopressin (VP), caused by dilution of the renal medullary interstitium.86 This can mean that patients with severe polyuria due to dipsogenic DI may appear to have partial NDI. The accuracy of the test can be enhanced by the incorporation of AVP measurement with a reliable radioimmunoassay, if available. We incorporate measurement of thirst by visual analogue scale, which gives valuable information in the diagnosis of specific thirst problems.
The most accurate diagnosis of HDI is attained by direct measurement of plasma AVP concentration during intravenous infusion of hypertonic saline86,87 (Fig. 14-7). Patients with HDI are identified by undetectable or subnormal plasma AVP concentrations with respect to plasma osmolalities, whereas patients with DDI and NDI have plasma AVP responses in the normal range. DDI and NDI can be distinguished by relating plasma AVP concentration to urine osmolality at the end of the test. NDI is characterized by inappropriately high plasma AVP values for concomitant urine osmolality, whereas DDI patients show an appropriate relation. A visual analogue scale53 is equally useful in this form of osmotic stimulation.
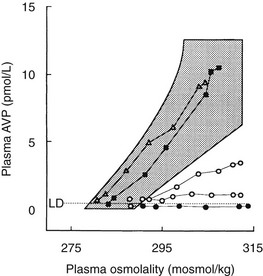
FIGURE 14-7 Dynamic tests of the arginine vasopressin (AVP) axis in patients with polyuria. Plasma AVP (pAVP) and osmolality (pOsm) responses to hypertonic (855 mmol/L) saline infusion. pAVP and urine osmolality (uOsm) responses to a period of fluid restriction in the same group of patients. Shaded areas, Range of the normal response. l and l, Patients with hypothalamic diabetes insipidus (HDI); n, patients with nephrogenic diabetes insipidus (NDI); s, patients with dipsogenic diabetes insipidus (DDI).
Because the concentrations of AVP in urine are higher than those in plasma, and thus are detectable by a wider range of available assays, measurement of urinary AVP concentration during osmotic stress has been proposed as an additional alternative to the measurement of plasma AVP in the diagnosis of HDI, but this method needs further evaluation.88
Once the diagnosis of HDI has been confirmed, imaging of the hypothalamic-pituitary region with MRI scanning is mandatory to exclude structural lesions. Sellar tumors in patients with HDI most often are craniopharyngiomas. A stalk mass has a wider differential diagnosis, including craniopharyngioma, germinoma, metastasis, lymphocytic hypophysitis, and stalk thickening in autoimmune HDI. Typical appearances may be found in sarcoid, hemochromatosis, and other inflammatory conditions. The posterior pituitary has as a characteristic bright signal on T1-weighted MRI, which is absent in most patients with HDI.89 The posterior lobe signal is also diminished in the elderly and in patients with anorexia nervosa, septic shock, and poorly controlled diabetes mellitus, and those on hemodialysis. It is thought that this may represent persistent hypersecretion and thus decreased VP-NPII complex content.
If the diagnosis is thought to be idiopathic DI, awareness of the possibility of an autoimmune origin is important. Routine measurement of thyroid function, vitamin B12, and autoantibodies is worthwhile, whereas tests for primary adrenal failure should be considered when clinically indicated. Serum angiotensin-converting enzyme concentration and chest films should be considered if sarcoid is suspected. It is important to repeat MRI imaging when initial films are normal, as some parasellar tumors such as germinomas can present with DI prior to the development of radiologic abnormalities. If stalk lesions are present, serum hCG and α-fetoprotein measurements may give a clue to the presence of germinoma, and early repeat MRI is important for assessing tumor growth. Biopsy should be considered early, as germinomas are best treated with chemotherapy and radiotherapy rather than surgical intervention. Dynamic tests of anterior pituitary function should be considered in cases in which structural pituitary lesions exist. Data in children suggest that growth hormone deficiency commonly accompanies autoimmune HDI,90 although anecdotal evidence suggests that this is not the case in adults.
Adipsic Diabetes Insipidus
Although diabetes insipidus is almost always associated with normal thirst and drinking appropriate to clinical needs, adipsic diabetes insipidus (ADI) is a rare but highly complex condition. It is seen most commonly after clipping of anterior communicating artery aneurysms, following subarachnoid hemorrhage91,92; the vascular supply to the osmoreceptors is derived from small arteries arising from the anterior communicating artery, and it is assumed that these vessels are damaged during aneurysm clipping, and that infarction of the circumventricular organs occurs where the osmoreceptors are sited. The osmoregulatory defect is almost always permanent, although anecdotal, unpublished reports have described recovery. Extensive hypothalamic surgery for craniopharyngioma,92 or, rarely, pituitary macroadenoma,93 have been reported to cause ADI.
In ADI secondary to clipping of the anterior communicating artery, a pure osmoreceptor defect is present. Osmotic stimulation with hypertonic saline causes no rise in plasma AVP or in thirst ratings, despite marked hyperosmolality. In contrast, nonosmotic baroregulated AVP secretion is completely normal, with secretion of AVP to produce plasma concentrations a hundred-fold higher than those needed for maximal urine concentration.94 The AVP synthesis/secretion unit of the SON/PVN/posterior pituitary is thus intact, but is not innervated by the osmoreceptors. In patients who undergo destructive surgery to the pituitary, all secretory function is lost.94
The diagnosis of ADI presents a management dilemma.95 Patients are unable to perceive changes in plasma tonicity via the usual thirst mechanisms and are vulnerable to marked swings in plasma sodium concentration. Severe hypernatremia is a particular hazard. Management requires regular desmopressin, fixed fluid intakes that may vary with climatic conditions, and regular review for measurement of plasma sodium. Recent data have also highlighted associated hypothalamic abnormalities that are often seen with ADI (Table 14-2), including hypothalamic obesity, seizure disorders, sleep apnea, and thermoregulatory disorders.92 Episodes of dehydration are often complicated by thrombotic complications, including pulmonary thromboembolism.92,96 Many patients die prematurely owing to postsurgical complications, electrolyte abnormalities, or sleep apnea.
The Syndrome of Inappropriate Antidiuretic Hormone
In 1956, in a presentation to the American Society for Clinical Investigation, Frederic Bartter and William Schwartz first described the syndrome of inappropriate antidiuretic hormone secretion (SIADH) as it occurred in two patients with lung carcinoma who had severe hyponatremia at presentation.97 By alternating fluid restriction with fluid loading, the investigators demonstrated a state of antidiuresis and hypothesized that their patients manifested a clinical syndrome of inappropriate antidiuresis secondary to excess circulating antidiuretic hormone. Once radioimmunoassays for the measurement of plasma AVP became available, their index clinic description and pathophysiologic hypothesis were substantiated by papers that reported elevated plasma vasopressin concentrations in the syndrome,98 which is now recognized to occur in a wide spectrum of disease states. However, differentiation of SIADH from other causes of hyponatremia remains important to the prescription of appropriate treatment regimens.
Causes of Siadh
SIADH is associated with a great number of illnesses and most often presents as a coincidental biochemical manifestation of the causative disease. The most common causes of SIADH are summarized in Table 14-3. The origin of SIADH is clinically divided into four categories: malignant, pulmonary, pharmacologic, and neurologic causes.
Table 14-3
Bartter and Schwartz first described SIADH in association with bronchogenic lung carcinoma. In small cell carcinoma of the lung, SIADH is relatively common, occasionally occurring as the presenting abnormality that prompts a search for the tumor.99 Most neoplasms have been reported to cause SIADH, and the association between malignant disease and SIADH is so strong that a patient presenting with SIADH, general malaise, and unexplained weight loss should be considered to have an underlying malignancy until proven otherwise.
SIADH is commonly associated with intracranial diseases, particularly traumatic brain injury,65–67 in which almost all cases resolve spontaneously with recovery from brain injury. More than 50% of patients with subarachnoid hemorrhage develop hyponatremia in the first week following the bleed, and 70% of these events are due to SIADH.100 SIADH also commonly occurs after hypophysectomy and after surgery for primary brain tumor.
Many drugs are important clinical causes of hyponatremia. The selective serotonin reuptake inhibitors (SSRIs) are thought to cause SIADH by direct stimulation of AVP secretion by serotonin.101 SIADH usually occurs in the first few weeks after SSRIs are introduced, particularly in elderly patients.102 It has been estimated that 12% of hospitalized patients on SSRI therapy develop SIADH.103 Psychotropic medications such as phenothiazines, haloperidol, and tricyclic antidepressants all cause SIADH. Many patients who have conditions treated with psychotropic drugs also have abnormal thirst; if these patients develop drug-induced SIADH, they can develop very significant hyponatremia. For instance, up to 20% of patients with chronic schizophrenia have psychogenic polydipsia, and excess fluid intake can precipitate severe hyponatremia. MDMA (“Ecstasy”) is thought to stimulate both AVP release and thirst as a result of hyperthermia, and this has been implicated in the development of cases of life-threatening hyponatremia.104
SIADH must be distinguished from exercise-associated hyponatremia (EAH),105 in which the electrolyte profile mimics SIADH. The pathophysiology is different, however. EAH occurs in athletes who ingest excessive hypotonic fluid during exercise, with resultant dilutional hyponatremia. Women, marathon runners racing for longer than 4 hours, and athletes of low body mass index are at greatest risk.106 Although some athletes with EAH fail to suppress AVP, EAH probably should be considered to be distinct from SIADH.
Pathophysiology of Siadh
SIADH occurs by definition when AVP secretion is not suppressed when plasma sodium concentration falls below the osmotic threshold for SIADH. However, Zerbe and colleagues were able to differentiate four different types of SIADH, depending on the pattern of AVP secretion 98 (Fig. 14-8).
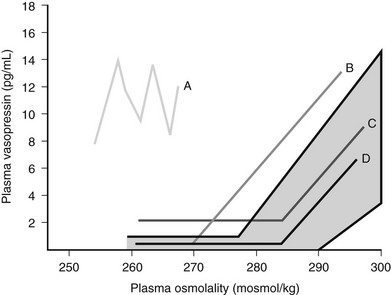
FIGURE 14-8 Summary of the four different patterns of arginine vasopressin (AVP) secretion in syndrome of inappropriate antidiuretic hormone secretion (SIADH).
Type A is the most common form of SIADH (40%). Characteristically, Type A patients exhibit excessive, random secretion of AVP, with loss of the close linear relationship between plasma osmolality and plasma AVP. Plasma AVP concentrations fluctuate widely in this variant, with no relationship to ambient plasma osmolality, but with consistent antidiuresis and inappropriate urine concentration despite hyponatremia. Type A is common in lung cancer; in vitro studies have demonstrated that some lung tumors synthesize AVP,107 and tumor tissue stains positive for AVP messenger RNA.108 However, Type A SIADH also occurs in non-neoplastic disease, for which ectopic AVP secretion could not be implicated. Plasma AVP concentrations in Type A SIADH are not suppressed by drinking,109 which makes patients vulnerable to the development of severe hyponatremia.
Type D provides a rare clinical picture of SIADH without detectable circulating AVP.110 It is postulated that a nephrogenic syndrome of inappropriate antidiuresis (NSIAD) may be responsible for the Type D picture of SIADH.111 Recent reports described two male infants in whom gain-of-function mutations in the V2 receptor led to a clinical picture of SIADH, with undetectable AVP levels. Both children were diagnosed in infancy. Identified mutations had different nucleotide substitution with different levels of V2 receptor activation.111
Studies in rat models of SIADH have demonstrated that the increase in water reabsorption is secondary to AVP-mediated expression of renal aquaporin-2,112 with a consequent increase in AQP protein excretion in the urine.113 Aquaporin-2 recruitment has been shown to be reversible through administration of a V2 receptor antagonist112,114 that provides the scientific rationale for the use of AVP antagonists in the treatment of this condition. Prolonged SIADH is associated with “escape” from antidiuresis, with downregulation of AQP2 mRNA and protein expression.
Although patients with SIADH have ambient plasma osmolalities that are below the physiologic osmotic threshold for thirst, they continue to drink apparently normal fluid volumes. In experiments in which patients with SIADH of mixed causes were infused intravenously with hypertonic sodium chloride solution, the osmotic threshold for thirst was lowered to a setting that was identical to that for AVP.109 Parallel lowering of the thresholds for thirst and AVP release ensured the maintenance of fluid intake, predisposing to persistent hyponatremia.
As fluid intake is maintained in the face of reduced free water clearance, patients with SIADH can develop severe hyponatremia. However, hyponatremia is often limited by “escape from antidiuresis.” This protective homeostatic mechanism occurs when the kidney begins to increase free water clearance despite inappropriate plasma AVP concentrations.115 Initial natriuresis is followed by an increase in urine flow116 with consequent water loss; this allows plasma sodium to stabilize and, occasionally, to rise. Although plasma sodium concentration does not usually rise into the normal physiologic range during escape from antidiuresis, the development of severe hyponatremia is prevented.
Experimental models of SIADH, in which hyponatremia was induced in Sprague-Dawley rats by the injection of subcutaneous desmopressin, showed that desmopressin administration alone did not lead to hyponatremia.117 Plasma volume expansion due to water loading, leading to increased renal perfusion pressure, was vital for initiation of escape from antidiuresis.118 A decrease in AQP2 protein expression and V2 receptor binding capacity119,120 has been postulated to cause the renal resistance to AVP observed during escape from antidiuresis.
In normal physiology, AVP has long-term effects on AQP2 via mRNA and protein expression, but it has short-term effects via the V2 receptor, leading to increased cyclic adenosine monophosphate (cAMP). It is likely that this short-term AVP action is also altered in escape, because reduced levels of cAMP in the collecting ducts of “escaped” rats have been demonstrated.121 This finding of reduced cAMP activity suggests both the short-term regulation of AQP activity by reduced vesicle “shuttling” and the long-term downregulation of AQP mRNA expression in the genesis of “escape from antidiuresis.”
Diagnosis of Siadh
Essential criteria for the diagnosis of SIADH are presented in Table 14-4. Two additional supplemental criteria—raised plasma vasopressin concentration and abnormal water load test—are rarely used in clinical practice. Water loading of a patient with SIADH runs the risk of symptomatic hyponatremia; therefore, this procedure should be restricted to centers with considerable experience in managing disorders of water balance. Plasma AVP concentrations are elevated in almost all causes of hyponatremia and therefore are rarely of diagnostic benefit in hyponatremic patients; lack of access to local radioimmunoassays dictates that the results of tests may take weeks to come back, which further diminishes their clinical value.
Table 14-4
Essential Criteria for the Diagnosis of SIADH
Decreased plasma osmolality of the ECF (pOsm <280 mOsm/kg H2O)
Inappropriate urinary concentration during hypo-osmolality (uOsm >100 mOsm/kg H2O, normal renal function)
Clinical euvolemia
Elevated urinary sodium excretion (>30 mmol/L) on normal salt and water intake
Absence of other potential causes of euvolemic hypo-osmolality, particularly hypothyroidism, hypocortisolism, and diuretic use
ECF, Extracellular fluid; SIADH, syndrome of inappropriate antidiuretic hormone secretion.
Differential Diagnosis of Siadh
ACTH deficiency causes hyponatremia with a biochemical profile identical to classical SIADH. Hyponatremia responds to glucocorticoid replacement.122 Fluid restriction, which constitutes first-line therapy for SIADH, is deleterious and worsens hypovolemia.123 Measurement of plasma cortisol therefore is essential, as clinical signs are not always sufficiently reliable to differentiate between glucocorticoid deficiency and SIADH.124 Glucocorticoid-deficient patients have impairment of free water clearance, causing a relative excess of body water to sodium.125,126 Glucocorticoids are thought to suppress AVP release, and elevation of plasma AVP concentrations has been reported in ACTH-deficient patients who develop hyponatremia.127
The differentiation of SIADH from glucocorticoid deficiency is particularly important in patients with neurosurgical conditions, who commonly present with hyponatremia, which is traditionally ascribed to SIADH.128 Recent evidence indicates that acute pituitary dysfunction occurs commonly following traumatic brain injury and subarachnoid hemorrhage, with ACTH deficiency reported in 16% of patients with acute65 and 12.7% of patients with chronic head injury129 and in 2.5% of patients after subarachnoid hemorrhage.130 Glucocorticoid deficiency may present with hyponatremia; coexistent hypoglycemia or hypotension suggests the diagnosis of hypopituitarism.131 We have seen acute ACTH deficiency severe enough to cause a biochemical picture, which mimics SIADH in traumatic brain injury, subarachnoid hemorrhage, and intracranial hemorrhage. Therefore, in patients presenting with SIADH against a background of sudden intracerebral catastrophe, we consider that the possibility of acute hypopituitarism should be considered in the differential diagnosis.
Hypothyroidism
Hyponatremia in hypothyroidism is rare, but life-threatening hyponatremia is occasionally reported.132,133 Hypothyroid patients have elevated AVP responses to subtle volume contraction, with increased recruitment of AQP2.134 However, the hyponatremia is probably multifactorial. Thyroxine has direct effects on sodium reabsorption from the tubule and on renal ability to excrete free water135,136; decreased glomerular filtration rate also occurs in hypothyroidism, which corrects with thyroid hormone replacement.137
Treatment for Siadh
Symptomatic cerebral irritation is the most important factor in determining that hyponatremia should be treated. However, the degree of hyponatremia is also important in that symptoms, morbidity, and mortality are all related to the degree of hyponatremia. Modest falls in plasma sodium to 130 mmol/L are unlikely to be symptomatic or to require any intervention. However, the likelihood of symptoms increases as the plasma sodium concentration falls.138 At plasma sodium concentrations below 120 mmol/L, morbidity and mortality are more common. Two other factors increase the likelihood of symptoms. The first of these is the coexistence of other structural or biochemical abnormalities; the likelihood of seizures, for instance, is increased at any plasma sodium concentration in the presence of intracerebral lesions such as tumor or hemorrhage. In addition, cerebral irritation due to hyponatremia is increased if the patient is hypoxic, hypotensive, or dehydrated.
The second important factor is the rate of development of hyponatremia, which is even more important than comorbidity in determining the likelihood and severity of cerebral symptoms. Patients with chronic hyponatremia (>48 hours’ duration) are much less likely to develop symptoms than those in whom the fall in plasma sodium concentration occurs rapidly (acute hyponatremia, <48 hours’ duration) and may have no symptoms at all despite marked hyponatremia. For instance, many patients can withstand chronic hyponatremia due to diuretics for long periods with no symptoms other than postural dizziness due to hypotension. These patients undergo cerebral adaptation by extruding solutes such as potassium and organic osmolytes into the plasma. This prevents osmotic movement of water into the brain, where it can cause cerebral edema. In contrast, in acute hyponatremia, for instance, in neurosurgical conditions, this time-dependent process does not occur with the subsequent development of brain edema and cerebral irritation.139 Rapid treatment therefore is more likely to be necessary when hyponatremia has developed quickly, as serious symptoms are more likely, and failure to treat can be fatal.138
Treatment for Chronic Hyponatremia
Modest hyponatremia of >130 mmol/L will rarely need treatment, but the frequency of symptoms and adverse effects, such as seizures, increases with plasma sodium concentrations below this. After careful exclusion of treatable underlying conditions such as glucocorticoid deficiency, water restriction is the treatment of choice. Fluid restriction of 1 L per day is occasionally sufficient to correct hyponatremia,140 with a slow, steady rise in plasma sodium. If the patient has been polydipsic, however, fluid restriction can cause a more rapid rise in plasma sodium concentration. Fluid restriction is difficult to maintain in patients with cognitive difficulties, in those with a history of polydipsia, and in patients receiving intravenous medications. In addition, in more severe hyponatremia, the degree of fluid restriction necessary to correct hypo-osmolality may be too difficult to maintain.
The development of specific V2 receptor antagonists offers a specific antidote to AVP-mediated hyponatremia.141 These agents act as “aquaretics,” which increase free water excretion without a natriuresis and offer a therapeutic effect regardless of the cause of raised AVP.
Selective V2 antagonists (e.g., tolvaptan, lixivaptan) and combined V1a/V2 antagonists (e.g., conivaptan) may be used.142 Pure V2 antagonism may be suitable for patients with SIADH but may lead to a rise in AVP levels. Few clinical data are available for this group of patients. V2 antagonists appear to be more effective in SIADH than in cirrhosis.143
References
1. Schally, AV. Hormones of the neurohypophysis. In: Lock W, Schally AV, eds. The Hypothalamus and Pituitary in Health and Disease. Springfield, IL: Charles C Thomas; 1972:154–171.
2. Zimmerman, EA, Nilaver, G, Hou-ya, A, et al. Vasopressinergic and oxytocinergic pathways in the central nervous system. Fed Proc. 1984;43:91–96.
3. Sofroniew, MV. Projections from vasopressin, oxytocin and neurophysin neurones to neural targets in the rat and human. J Histochem Cytochem. 1980;28:475–478.
4. Mohr, E, Schmitz, E, Richter, D. A single rat genomic cDNA fragment encodes both the oxytocin and vasopressin genes separated by 11 kilobases and orientated in opposite transcriptional directions. Biochemie. 1988;70:649–654.
5. Dutton, A, Dyball, REJ. Phasic firing enhances vasopressin release from the rat neurohypophysis. J Physiol. 1979;290:433–440.
6. Russell, JT, Brownstein, MJ, Gainer, H. Biosynthesis of vasopressin, oxytocin and neurophysins: isolation and characterization of two common precursors (propressophysin and prooxyphysin). Endocrinology. 1980;107:1880–1891.
7. Zing, HH. Vasopressin and oxytocin receptors. Ballieres Clin Endocrinol Metab. 1996;10:75–96.
8. Thibonnier, M, Conarty, DM, Preston, JA, et al. Molecular pharmacology of human vasopressin receptors. Adv Exp Med Biol. 1988;449:251–276.
8a. Montani, JP, Liard, JF, Schoun, J, et al. Hemodynamic effects of exogenous and endogenous vasopressin at low plasma concentrations in conscious dogs. Circ Res. 1980;47:346–355.
9. Altura, BM, Altura, BT. Actions of vasopressin, oxytocin, and synthethic analogs on vascular smooth muscle. Fed Proc. 1984;43:80–86.
10. Liard, J-F. Acute hemodynamic effects of antidiuretic agents. In: Comley AW, Liard J-F, Ausiello DA, eds. Vasopressin: Cellular and Integrative Functions. New York: Raven Press; 1988:461–466.
11. Aisenbrey, GA, Handelman, WA, Arnold, P, et al. Vascular effects of arginine vasopressin during fluid deprivation in the rat. J Clin Invest. 1981;67:961–968.
12. Dunser, MW, Mayr, AJ, Ulmer, H, et al. Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003 May 13;107(18):2313–2319.
13. Morelli, A, Rocco, M, Conti, G, et al. Effects of terlipressin on systemic and regional haemodynamics in catecholamine-treated patients. Intensive Care Med. 2004 Apr;30(4):597–604.
14. Gonen, T, Walz, T. The structure of aquaporins. Q Rev Biophys. 2006;39(4):361–396.
15. Du Pasquier, D, Dreifuss, JJ, Dubois-Dauphin, M, et al. Binding sites for vasopressin in the human pituitary are associated with corticotrophs and may differ from other known vasopressin receptors. J Neuroendocrinol. 1991;3:237–247.
16. Sawchenko, PE, Swanson, LW, Vale, WW. Co-expression of corticotrophin releasing factor and vasopressin immunoreactivity in parvocellular neurosecretory neurons of the adrenalectomized rat. Proc Natl Acad Sci U S A. 1984;81:1877–1883.
17. Baldino, F, Davis, LG. Glucocorticoid regulation of vasopressin messenger RNA. In: Uhl GR, ed. In Situ Hybridization in the Brain. New York: Plenum Press; 1986:97.
18. Robertson, GL, Aycitema, P, Zerbe, RL. Neurogenic disorders of osmoregulation. Am J Med. 1982 Feb;72(2):339–353.
19. Zerbe, RL, Robertson, GL. Osmoregulation of thirst and vasopressin secretion in human subjects: effect of various solutes. Am J Physiol. 1983;244:E607–E614.
20. Robertson, GL, Shelton, RL, Athar, S. The osmoregulation of vasopressin. Kidney Int. 1976;10:25–37.
21. Hammer, M, Ladefoged, J, Olgaard, K. Relationship between plasma osmolality and plasma vasopressin in human subjects. Am J Physiol. 1980;238:E313–E317.
22. Baylis, PH, Thompson, CJ. Osmoregulation of vasopressin secretion and thirst in health and disease. Clin Endocrinol. 1988;29:549–576.
23. Thompson, CJ, Bland, J, Burd, J, et al. The osmotic thresholds for thirst and vasopressin release are similar in a healthy man. Clin Sci. 1986;71:651–656.
24. Baylis, PH, Pippard, C, Gill, GV, et al. Development of a cytochemical assay for plasma vasopressin: application to studies on water loading normal man. Clin Endocrinol. 1986;24:383–392.
25. Baylis, PH, Gill, GV. Investigation of polyuria. Clin Endocrinol Metab. 1984;13:295–310.
26. Robertson, GL. Physiology of ADH secretion. Kidney Int. 1987;32(Suppl 21):S20–S26.
27. Thompson, CJ, Selby, P, Baylis, PH. Reproducibility of osmotic and non-osmotic tests of vasopressin secretion in man. Am J Physiol. 1991;60:R533–R539.
28. Zerbe, RL. Genetic factors in normal and abnormal regulation of vasopressin secretion. In: Schrier RW, ed. Vasopressin. New York: Raven Press; 1985:213–220.
29. Thompson, CJ, Burd, JM, Baylis, PH. Acute suppression of plasma vasopressin and thirst after drinking in hypernatraemic man. Am J Physiol. 1986;252:R1138–R1142.
30. Seckl, JR, Williams, TD, Lightman, SL. Oral hypertonic saline causes transient fall in vasopressin in humans. Am J Physiol. 1986;251:R214–R217.
31. Durr, JA, Stamoutsos, BA, Lindheimer, MD. Osmoregulation during pregnancy in the rat: evidence for resetting of the threshold for vasopressin secretion during gestation. J Clin Invest. 1981;68:337–346.
32. Davison, JM, Gilmore, EA, Durr, J, et al. Altered threshold for vasopressin secretion and thirst in human pregnancy. Am J Physiol. 1984;246:F105–F109.
33. Spruce, BA, Baylis, PH, Burd, J, et al. Variation in osmoregulation of arginine vasopressin during the human menstrual cycle. Clin Endocrinol. 1985;22:37–42.
34. Miller, M. Fluid and electrolyte homeostasis in the elderly: physiological changes of ageing and clinical consequences. Baillieres Clin Endocrinol Metab. 1997;11:367–387.
35. Johnson, AG, Crawford, GA, Kelly, D. Arginine vasopressin and osmolality in the elderly. J Am Geriatr Soc. 1994;42:399–404.
36. Helderman, JH, Vestal, RE, Rowe, JW, et al. The response of arginine vasopressin to intravenous alcohol and hypertonic saline in man: The impact of ageing. J Gerontol. 1978;33:39–47.
37. Phillips, PA, Rolls, BJ, Ledingham, JGG. Reduced thirst after water deprivation in healthy elderly men. N Engl J Med. 1984;311:753–759.
38. Crowe, MJ, Forsling, ML, Rolls, BJ. Altered water excretion in healthy elderly men. Age Ageing. 1987;16:285–293.
39. Thompson, CJ, David, SN, Butler, PC, et al. Osmoregulation of thirst and vasopressin secretion in insulin dependent diabetes mellitus. Clin Sci. 1988;74:599–606.
40. Thompson, CJ, Davis, SN, Baylis, PH. Effects of blood glucose concentration on osmoregulation in insulin dependent diabetes mellitus. Am J Physiol. 1989;256:R597–R604.
41. McKenna, K, Morris, AD, Newton, RW, et al. Renal resistance to vasopressin in poorly controlled insulin dependent diabetes. Am J Physiol. 2000;279:E155–E160.
42. McKenna, K, Morris, AD, Azam, H, et al. Exaggerated vasopressin secretion and attenuated osmoregulated thirst in human survivors of hyperosmolar coma. Diabetologia. 1999 May;42(5):534–538.
43. Johnson, JA, Zehr, JE, Moore, WW. Effects of separate and concurrent osmotic and volume stimuli on plasma ADH in sheep. Am J Physiol. 1970;218:1273–1280.
44. Baylis, PH. Posterior pituitary function in health and disease. Clin Endocrinol Metab. 1983;12:747–770.
45. Goetz, KL, Zhu, JL, Leadley, RJ, et al. Hemodynamic and hormonal influences on the secretion of vasopressin. In: Jard S, Jamison R, eds. Vasopressin. Montrouge, France: John Libbey; 1991:279–286.
46. Dunn, FL, Brennan, TJ, Nelson, AE, et al. The role of blood osmolality and volume in regulating vasopressin secretion in the rat. J Clin Invest. 1973;52:3212–3219.
47. Goldsmith, SR, Dodge, D, Cowley, AW. Nonosmotic influences on osmotic stimulation of vasopressin in humans. Am J Physiol. 1987;252:H85–H88.
48. Rowe, JW, Shelton, RL, Helderman, JH, et al. Influence of the emetic reflex on vasopressin release in man. Kidney Int. 1979;16:729–735.
49. Verbalis, JG, Richardson, DW, Stricker, EM. Vasopressin release in response to nausea-producing agents and cholecystokinin in monkeys. Am J Physiol. 1987;252:R749–R753.
50. Ukei, M, Moran, WH, Zimmerman, B. The role of visceral afferent pathways on vasopressin secretion and urinary excretory patterns during surgical stress. Ann Surg. 1968;168:16–28.
51. Baylis, PH, Robertson, GL. Arginine vasopressin response to insulin induced hypoglycemia in man. J Clin Endocrinol Metab. 1981;53:935–940.
52. Thompson, CJ. Polyuric states in man. In: Baylis PH, ed. Water and salt homeostasis in health and disease. London: Bailliere Tindall; 1989:473–497.
53. Thompson, CJ, Baylis, PH. Thirst in diabetes insipidus: clinical relevance of quantitative assessment. Q J Med. 1987;56:853–862.
54. Hensen, J, Henig, A, Fahlbusch, R, et al. Prevalence, predictors and patterns of postoperative polyuria and hyponatraemia in the immediate course after transsphenoidal surgery for pituitary adenomas. Clin Endocrinol. 1999;50:431–439.
55. Smith, D, Finucane, F, Phillips, J, et al. Abnormal regulation of thirst and vasopressin secretion following surgery for craniopharyngioma. Clin Endocrinol. 2004;61:273–279.
56. Nemergut, EC, Zuo, Z, Jane, JA, Jr., et al. Predictors of diabetes insipidus after transsphenoidal surgery: a review of 881 patients. J Neurosurg. 2005;103:403–454.
57. Adams, JR, Blevins, LS, Jr., Allen, GS, et al. Disorders of water metabolism following transsphenoidal pituitary surgery: a single institution’s experience. Pituitary. 2006;9:93–99.
58. Sudhakar, N, Ray, A, Vafidis, JA. Complications after trans-sphenoidal surgery: our experience and a review of the Literature. Br J Neurosurg. 2004 October;18(5):507–512.
59. Agha, A, Sherlock, M, Brennan, S, et al. Hypothalamic-pituitary dysfunction following irradiation of non-pituitary brain tumours in adults. J Clin Endocrinol Metab. 2005 Dec;90(12):6355–6360.
60. Imura, H, Nakao, K, Shimatsu, A. Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med. 1993;329:683–689.
61. Arico, M, Girschikofsky, M, Genereau, T. Langerhans cell histiocytosis in adults: report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39:2341–2348.
62. Blotner, H. Primary or idiopathic diabetes insipidus: a system disease. Metabolism. 1958;7:191–206.
63. Baylis, PH. Understanding the cause of idiopathic cranial diabetes insipidus: a step forward. Clin Endocrinol. 1994;40:171–172.
64. Pivoello, R, De Bellis, A, Faggiano, A, et al. Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin-secreting cells and clinical, immunological, and radiological features in a large cohort of patients with central diabetes insipidus of known and unknown etiology. J Clin Endocrinol Metab. 2003;88:1629–1636.
65. Agha, A, Rogers, B, Mylotte, D, et al. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin Endocrinol. 2004;60:584–591.
66. Agha, A, Sherlock, M, Phillips, J, et al. The natural history of post-traumatic neurohypophysial dysfunction. Eur J Endocrinol. 2005;152:371–377.
67. Agha, A, Thornton, E, O’Kelly, P, et al. Posterior pituitary dysfunction following traumatic brain injury. J Clin Endocrinol Metab. 2004;89:5987–5992.
68. Jialal, I, Desai, K, Rajput, MC. An assessment of posterior pituitary function in patients with Sheehan’s syndrome. Clin Endocrinol. 1987;27:91–95.
69. Baylis, PH, Thompson, CJ, Burd, JM, et al. Recurrent pregnancy-induced polyuria and thirst due to hypothalamic diabetes insipidus: An investigation into possible mechanisms responsible for polyuria. Clin Endocrinol. 1986;24:459–466.
70. Barron, WM, Cohen, LH, Ulland, LA. Transient vasopressin-resistant diabetes insipidus of pregnancy. N Engl J Med. 1984;310:442–444.
71. Ito, M, Jameson, JL, Ito, M. Molecular basis of autosomal dominant neurohypophyseal diabetes insipidus: cellular toxicity caused by the accumulation of mutant vasopressin precursors within the endoplasmic reticulum. J Clin Invest. 1997;99:2897–2905.
72. Eubank, S, Nguyen, TL, Deeb, R. Effects of diabetes insipidus mutations on neurophysin folding and function. J Biol Chem. 2001;276:29671–29680.
73. Davies, J, Murphy, D. Autophagy in hypothalamic neurones of rats expressing a familial neurohypophyseal diabetes insipidus transgene. J Neuroendocrinol. 2002;14:629–637.
74. Rittig, S, Sigaard, C, Ozata, M. Autosomal dominant neurohypophyseal diabetes insipidus due to substitution of histidine for tyrosine-2 in the vasopressin moiety of the hormone precursor. J Clin Endocrinol Metab. 2002;87:3351–3355.
75. Christenson, JH, Sigaard, C, Corydon, TJ. Impaired trafficking of mutated AVP prohormone in cells expressing rare disease genes causing autosomal dominant familial neurohypophyseal diabetes insipidus. Clin Endocrinol. 2004;60:125–136.
76. Elias, PCL, Ellias, LLK, Torres, N. Progressive decline of vasopressin secretion in familial autosomal dominant neurohypophyseal diabetes insipidus presenting a novel mutation in the vasopressin-neurophysin II gene. Clin Endocrinol. 2003;59:511–518.
77. Barrett, TG, Bundey, SE. Wolfram (DIDMOAD) syndrome. J Med Genet. 1997;34:838–841.
78. Thompson, CJ, Charlton, J, Walford, S, et al. Vasopressin secretion in the DIDMOAD (Wolfram) syndrome. Q J Med. 1989;71:333–345.
79. Cryns, K, Sivakumaran, TA, Van den Ouweland, JMW. Mutational spectrum of the WFS1 gene in Wolfram syndrome, non-syndromic hearing impairment, diabetes mellitus and psychiatric disease. Hum Mutat. 2003;22:275–287.
80. Barbieris, C, Mouillac, B, Durroux, T. Structural basis of vasopressin/oxytocin receptor function. J Endocrinol. 1998;156:223–229.
81. Mulders, SM, Bichet, DG, Rijss, JPL. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J Clin Invest. 1998;102:57–66.
82. Asai, T, Kuwahara, M, Kurihara, H. Pathogenesis of nephrogenic diabetes insipidus by aquaporin-2 C-terminus mutations. Kidney Int. 2003;64:2–10.
83. McKenna, K, Thompson, C. Osmoregulation in clinical disorders of thirst and thirst appreciation. Clin Endocrinol. 1998;49:139–152.
84. Thompson, CJ, Edwards, CRW, Baylis, PH. Osmotic and non-osmotic regulation of thirst and vasopressin secretion in patients with compulsive water drinking. Clin Endocrinol. 1991;35:221–228.
85. Zerbe, RL, Robertson, GL. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N Engl J Med. 1981;305:1539–1546.
86. Robertson, GL, Diagnosis of diabetes insipidus. Diabetes Insipidus in Man: Frontiers of Hormone Research. Czernichow, P, Robinson, AG, eds. Diabetes Insipidus in Man: Frontiers of Hormone Research, vol 13. Basel: S. Karger, 1985:176–189.
87. Baylis, PH, Robertson, GL. Vasopressin response to hypertonic saline infusion to assess posterior pituitary function. J R Soc Med. 1980;73:255–260.
88. Diedrich, S, Eckmanns, T, Exner, P. Differential diagnosis of polyuric/polydipsic syndromes with the aid of urinary vasopressin measurement in adults. Clin Endocrinol. 2001;54:665–671.
89. Fujisawa, I. Magnetic resonance imaging of hypothalamic-neurohypophyseal system. J Neuroendocrinol. 2004;16:297–302.
90. Ghirardello, S, Garrè, ML, Rossi, A, et al. The diagnosis of children with central diabetes insipidus. J Pediatr Endocrinol Metab. 2007 Mar;20(3):359–375.
91. McIver, B, Connacher, A, Whittle, I, et al. Adipsic hypothalamic diabetes insipidus after clipping of anterior communicating artery aneurysm. BMJ. 1991;303(6815):1465–1467.
92. Crowley, RK, Sherlock, M, Agha, A, et al. Clinical insights into adipsic diabetes insipidus: a large case series. Clin Endocrinol. 2007;66:475–482.
93. Sherlock, M, Agha, A, Crowley, R, et al. Adipsic diabetes after surgery for macroprolactinoma. Pituitary. 2006;9:59–64.
94. Smith, D, McKenna, K, Moore, K, et al. Baroregulation of vasopressin release in adipsic diabetes insipidus. J Clin Endocrinol Metab. 2002;87(10):4564–4568.
95. Ball, SG, Vaidja, B, Baylis, PH. Hypothalamic adipsic syndrome: diagnosis and management. Clin Endocrinol. 1997;47(4):405–409.
96. Bergada, I, Aversa, L, Heinrich, JJ. Peripheral venous thrombosis in children and adolescents with adipsic hypernatraemia secondary to hypothalamic tumours. Horm Res. 2004;61(3):108–110.
97. Schwartz, WB, Bennett, W, Curelop, S, et al. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. Am J Med. 1957;23(4):529–542.
98. Zerbe, R, Stropes, L, Robertson, G. Vasopressin function in the syndrome of inappropriate antidiuresis. Annu Rev Med. 1980;31:315–327.
99. Seute, T, Leffers, P, ten Velde, GP, et al. Neurologic disorders in 432 consecutive patients with small cell lung carcinoma. Cancer. 2004;100(4):801–806.
100. Sherlock, M, O’Sullivan, E, Agha, A, et al. The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage. Clin Endocrinol (Oxf). 2006 Mar;64(3):250–254.
101. Matsumoto, H. Hyponatremia associated with selective serotonin reuptake inhibitors. Intern Med. 2005;44(3):173–174.
102. Miehle, K, Paschke, R, Koch, CA. Citalopram therapy as a risk factor for symptomatic hyponatremia caused by the syndrome of inappropriate secretion of antidiuretic hormone (SIADH): a case report. Pharmacopsychiatry. 2005;38(4):181–182.
103. Bouman, WP, Pinner, G, Johnson, H. Incidence of selective serotonin reuptake inhibitor induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone secretion in the elderly. Int J Geriatr Psychiatry. 1998;13:123–125.
104. Budisavljevic, MN, Stewart, L, Sahn, SA. Hyponatremia associated with 3,4-methylenedioxymethylamphetamine (“Ecstasy”) abuse. Am J Med Sci. 2003;326(2):89–93.
105. Hew-Butler, T, Almond, C, Ayus, JC, et al. Consensus Statement of the 1st International Exercise-Associated Hyponatraemia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med. 2005;15(4):208–213.
106. Almond, CSD, Shin, AY, Fortescue, EB. Hyponatraemia among runners in the Boston Marathon. N Engl J Med. 2005;352(15):1550–1556.
107. George, JM, Capen, CC, Philips, AS. Biosynthesis of vasopressin in vitro and ultrastructure of a bronchogenic cancer. J Clin Invest. 1972;51:141–148.
108. Gross, AJ, Steinberg, SM, Reilly, JG. Atrial natriuretic factor and arginine vasopressin production in tumor cell lines from patients with lung cancer and their relationship to serum sodium. Cancer Res. 1993 Jan 1;53(1):67–74.
109. Smith, D, Moore, K, Tormey, W, et al. Downward resetting of the osmotic threshold for thirst in patients with SIADH. Am J Physiol Endocrinol Metab. 2004;287(5):E1019–E1023.
110. Baylis, PH. The syndrome of inappropriate antidiuretic hormone secretion. Int J Biochem Cell Biol. 2003;35(11):1495–1499.
111. Feldman, BJ, Rosenthal, SM, Vargas, GA. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005;352(18):1884–1890.
112. Ishikawa, S, Saito, T, Kasono, K. Pathological role of aquaporin-2 in impaired water excretion and hyponatremia. J Neuroendocrinol. 2004;16(4):293–296.
113. Saito, T, Higashiyama, M, Nagasaka, S, et al. Role of aquaporin-2 gene expression in hyponatremic rats with chronic vasopressin-induced antidiuresis. Kidney Int. 2001;60(4):1266–1276.
114. Ishikawa, SE, Schrier, RW. Pathophysiological roles of arginine vasopressin and aquaporin-2 in impaired water excretion. Clin Endocrinol. 2003;58(1):1–17.
115. Verbalis, JG, Murase, T, Ecelbarger, CA, et al. Studies of renal aquaporin-2 expression during renal escape from vasopressin-induced antidiuresis. Adv Exp Med Biol. 1998;449:395–406.
116. Levinsky, NG, Davidson, DG, Berliner, RW. Changes in urine concentration during prolonged administration of vasopressin and water. Am J Physiol. 1959;196(2):451–456.
117. Verbalis, JG. Escape from vasopressin-induced antidiuresis: insights at the molecular level. News Physiol Sci. 1999;14:221.
118. Verbalis, JG. Escape from antidiuresis: a good story. Kidney Int. 2001;60(4):1608–1610.
119. Ecelbarger, CA, Nielsen, S, Olson, BR. Role of renal aquaporins in escape from vasopressin-induced antidiuresis in rat. J Clin Invest. 1997;99(8):1852–1863.
120. Tian, Y, Sandberg, K, Murase, T, et al. Vasopressin V2 receptor binding is down-regulated during renal escape from vasopressin-induced antidiuresis. Endocrinology. 2000;141(1):307–314.
121. Ecelbarger, CA, Chou, CL, Lee, AJ, et al. Escape from vasopressin-induced antidiuresis: role of vasopressin resistance of the collecting duct. Am J Physiol. 1998;274(6 Pt 2):F1161–F1166.
122. Diederich, S, Franzen, NF, Bahr, V, et al. Severe hyponatremia due to hypopituitarism with adrenal insufficiency: report on 28 cases. Eur J Endocrinol. 2003;148(6):609–617.
123. Chen, YC, Cadnapaphornchai, MA, Summer, SN. Molecular mechanisms of impaired urinary concentrating ability in glucocorticoid-deficient rats. J Am Soc Nephrol. 2005;16(10):2864–2871.
124. Olchovsky, D, Ezra, D, Vered, I, et al. Symptomatic hyponatremia as a presenting sign of hypothalamic-pituitary disease: a syndrome of inappropriate secretion of antidiuretic hormone (SIADH)-like glucocorticosteroid responsive condition. J Endocrinol Invest. 2005;28(2):151–156.
125. Saito, T, Ishikawa, SE, Ando, F, et al. Vasopressin-dependent upregulation of aquaporin-2 gene expression in glucocorticoid-deficient rats. Am J Physiol Renal Physiol. 2000;279(3):F502–F508.
126. Okada, K, Nomura, M, Furusuyo, N, et al. Amelioration of extrapontine myelinolysis and reversible parkinsonism in a patient with asymptomatic hypopituitarism. Intern Med. 2005;44(7):739–742.
127. Oelkers, W. Hyponatraemia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med. 1989;321:492–496.
128. Fox, J, Falik, JL, Shalboub, RJ. Neurosurgical hyponatraemia: the role of inappropriate antidiuresis. J Neurosurg. 1971;34:506–514.
129. Agha, A, Rogers, B, Sherlock, M, et al. Anterior pituitary dysfunction in survivors of traumatic brain injury. J Clin Endocrinol Metab. 2004;89(10):4929–4936.
130. Aimaretti, G, Ambrosio, MR, Di Somma, C, et al. Traumatic brain injury and subarachnoid haemorrhage are conditions at high risk for hypopituitarism: screening study at 3 months after the brain injury. Clin Endocrinol (Oxf). 2004;61(3):320–326.
131. Agha, A, Sherlock, M, Thompson, CJ. Post-traumatic hyponatraemia due to acute hypopituitarism. QJM. 2005;98(6):463–464.
132. Sari, R, Sevinc, A. Life-threatening hyponatremia due to cessation of L-thyroxine. J Natl Med Assoc. 2003;95(10):991–994.
133. Taskapan, C, Sahin, I, Taskapan, H, et al. Possible malignant neuroleptic syndrome that is associated with hypothyroidism. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(5):745–748.
134. Chen, YC, Cadnapaphornchai, MA, Yang, J, et al. Nonosmotic release of vasopressin and renal aquaporins in impaired urinary dilution in hypothyroidism. Am J Physiol Renal Physiol. 2005;289(4):F672–F678.
135. Hanna, FW, Scanlon, MF. Hyponatraemia, hypothyroidism and role of arginine-vasopressin. Lancet. 1997;350:755–756.
136. Schmitz, PH, de Meijer, PH, Meinders, AE. Hyponatremia due to hypothyroidism: a pure renal mechanism. Neth J Med. 2001;58(3):143–149.
137. Montenegro, J, Gonzalez, O, Saracho, R, et al. Changes in renal function in primary hypothyroidism. Am J Kidney Dis. 1996;27(2):195–198.
138. Adrogue, HJ. Consequences of inadequate management of hyponatremia. Am J Nephrol. 2005;25(3):240–249.
139. Verbalis, JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471–503.
140. Yeates, KE, Singer, M, Morton, AR. Salt and water: a simple approach to hyponatremia. CMAJ. 2004;170(3):365–369.
141. Verbalis, JG. Vasopressin V2 receptor antagonists. J Mol Endocrinol. 2002;29(1):1–9.
142. Serradeil-Le Gal, C, Wagnon, J, Valette, G, et al. Nonpeptide vasopressin receptor antagonists: development of selective and orally active V1a, V2 and V1b receptor ligands. Prog Brain Res. 2002;139:197–210.
143. Palm, C, Reimann, D, Gross, P. The role of V2 vasopressin antagonists in hyponatremia. Cardiovasc Res. 2001;51(3):403–408.