[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 5
Hypothalamic Syndromes
The hypothalamus houses multiple nuclei along with afferent and efferent nerve fibers that connect the hypothalamus to the various portions of the brain and brainstem. It is divided into four regions: from anterior to posterior, the preoptic, supraoptic, tuberal, and mamillary regions; and three zones: laterally from the third ventricle, the periventricular, medial, and lateral1–4 zones (Table 5-1, Figs. 5-1 and 5-2).
Table 5-1
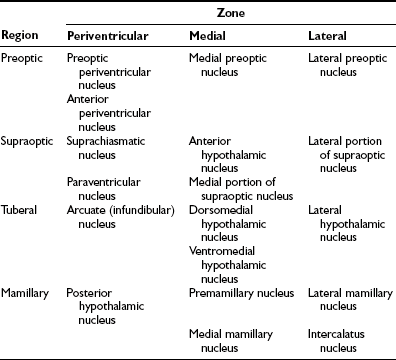
From Braunstein GD: The hypothalamus. In Melmed S (ed): The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.
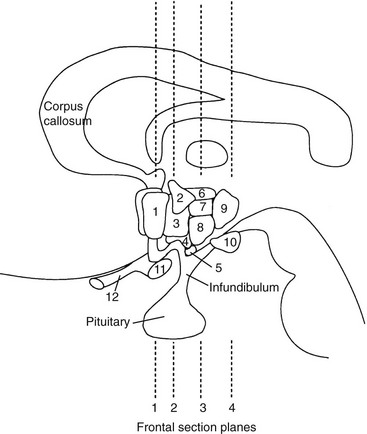
FIGURE 5-1 Schematic representation of lateral brain section demonstrating hypothalamic nuclei. Dashed lines represent the frontal (coronal) section planes illustrated in Figures 5-2 and 5-3. 1, preoptic nucleus; 2, paraventricular nucleus; 3, anterior hypothalamic areas; 4, supraoptic nucleus; 5, arcuate nucleus; 6, dorsal hypothalamic area; 7, dorsomedial nucleus; 8, ventromedial nucleus; 9, posterior hypothalamic area; 10, mamillary body; 11, optic chiasm; 12, optic nerve. (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
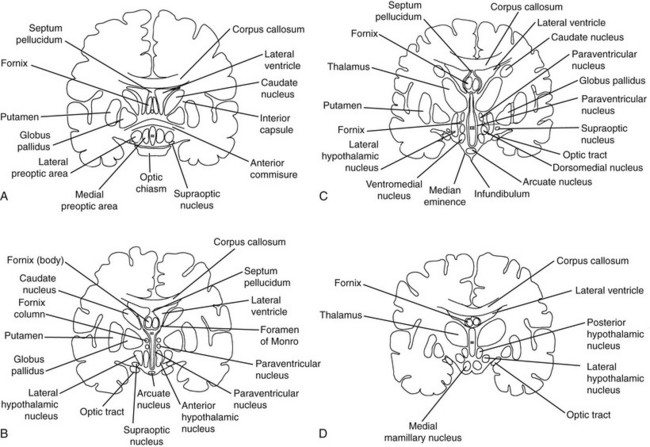
FIGURE 5-2 Frontal (coronal) sections of the hypothalamic regions. A, Preoptic region (frontal section plane 1 in Fig. 5-1). B, Supraoptic region (frontal section plane 2 in Fig. 5-1). C, Tuberal region (frontal section plane 3 in Fig. 5-1). D, Mamillary region (frontal section plane 4 in Fig. 5-1). (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
The hypothalamus is responsible for many of the body’s homeostatic mechanisms, including water metabolism, temperature regulation, appetite control, the sleep/wake cycle, circadian rhythms, and control of the sympathetic and parasympathetic nervous systems. In addition, this area has activity in regard to emotional expression, behavior, and memory. Finally, the hypothalamus is essential to the neuroendocrine control of anterior pituitary function. Table 5-2 lists the various functions of the hypothalamus, the hypothalamic nuclei or hypothalamic regions that have been identified as being responsible for these functions, and the disorders that result from either destructive or stimulatory lesions in or around the nuclei or region.1,4–25
Table 5-2
Hypothalamic Functions, the Nuclei or Regions Involved with the Specific Functions, and the Disorders Resulting from Stimulatory or Destructive Lesions in the Regions

SIADH, Syndrome of inappropriate secretion of antidiuretic hormone.
Hypothalamic Disorders: Pathophysiologic Principles
First, the small overall size of the hypothalamus and the close association of the nuclei and nerve tracts mean that a variety of different pathologic processes may give rise to the same signs and symptoms of neurologic and hypothalamic dysfunction.4 The spectrum of disorders that can affect the hypothalamus is shown in Table 5-3. Tumors, infiltrative disorders, and infections, among other conditions, frequently give rise to headaches, neuro-ophthalmologic disorders, pyramidal tract or sensory nerve dysfunction, extrapyramidal cerebellar signs, and recurrent vomiting.9,10 Other common manifestations include gonadal dysfunction (either hypogonadism or precocious puberty), diabetes insipidus, somnolence, dysthermia, and evidence of a caloric imbalance (either with hyperphagia and obesity or anorexia with emaciation).9,10
Table 5-3
Causes of Hypothalamic Dysfunction
Congenital
Tumors
Infiltrative
Immunologic
Nutritional, Metabolic
Degenerative
Infectious
Vascular
Trauma
Functional
Other
DIDMOAD, Diabetes insipidus, diabetes mellitus, optic atrophy, deafness.
Modified from Braunstein GD: The hypothalamus. In Melmed S (ed): The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.
Fourth, most lesions that result in chronic hypothalamic syndromes involve more than one nucleus. As can be seen in Table 5-2, most of the hypothalamic functions are controlled by more than one nucleus, and this redundancy allows some degree of compensation should one nucleus be affected. In addition, most of the nuclei are paired, and destruction of a single nucleus may not be sufficient to result in a clinical syndrome. Thus lesions that affect the basal tuberal region of the hypothalamus (pituitary adenomas with suprasellar extension, optic gliomas, and craniopharyngiomas), are multiple (granulomatous disorders, metastatic tumors, infiltrative disease), or cause enlargement of the third ventricle (aqueductal stenosis, colloid cysts, pinealomas, germ cell tumors, midbrain gliomas) will more likely result in clinical hypothalamic dysfunction than will disorders affecting the more lateral portions of the hypothalamus.
Manifestations of Hypothalamic Disease
Central Diabetes Insipidus
Complete or partial central diabetes insipidus results from (1) destruction of the antidiuretic hormone (ADH)-producing magnocellular neurons in the supraoptic and paraventricular nuclei or (2) interruption of the transport of ADH through their axons, which terminate in the pituitary stalk and posterior pituitary. Diabetes insipidus is relatively common in patients with chronic hypothalamic disorders, being found in approximately 35% of such patients.9,10 It also is frequently found in patients with acute insults to the hypothalamus or pituitary stalk, as is seen in vascular accidents and neurosurgical trauma. Obesity and hypogonadism frequently are present in patients with diabetes insipidus due to tumors or infiltrative disorders (Fig. 5-3).
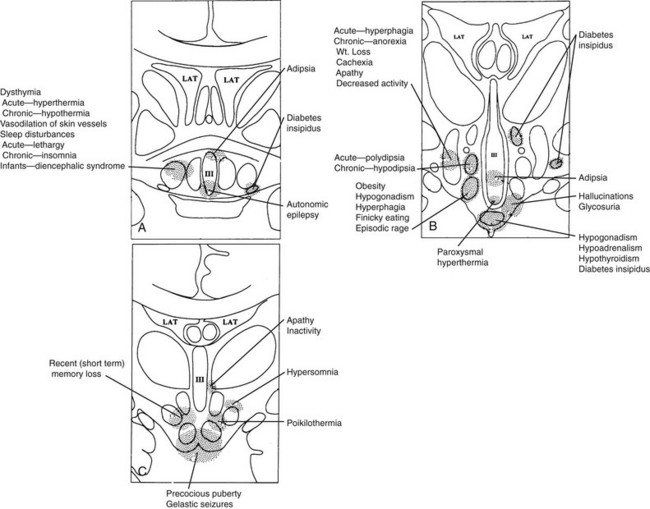
FIGURE 5-3 Clinical findings associated with hypothalamic lesions located at various anatomic sites. Clinicopathologic correlation based on multiple studies.15–25A, Corresponds to region depicted in Figure 5-2A. B, Corresponds to region depicted in Figure 5-2C. C, Corresponds to section depicted in Figure 5-2D. (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
The majority of patients with diabetes insipidus have idiopathic or familial diabetes insipidus associated with gliosis of the supraoptic and paraventricular nuclei.26 Approximately one third of patients with idiopathic diabetes insipidus have detectable anti-ADH–producing cell antibodies, suggesting an autoimmune cause.27 Autosomal-recessive, X-linked-recessive, and autosomal-dominant forms of familial diabetes insipidus have been described. In the more common autosomal-dominant form, nucleotide deletions or substitutions in the ADH gene on chromosome 20 have been identified.28 The DIDMOAD syndrome (Wolfram’s syndrome) represents a rare autosomal-recessive form of central diabetes insipidus (DI) associated with type 1 diabetes mellitus (DM), optic atrophy (OA), bilateral sensorineural deafness (D), and occasionally ataxia and autonomic neurogenic bladder.29 Diabetes insipidus is a frequent manifestation of suprasellar and pineal germinomas, sarcoidosis, lymphocytic infundibuloneurohypophysitis, and the chronic disseminated form of Langerhans’ cell histiocytosis.30–37
Adipsic or Essential Hypernatremia
Adipsic hypernatremia occurs when the osmoreceptors that are present in the anterior medial and anterior lateral preoptic regions are damaged. Affected patients have an impaired thirst mechanism, which results in insufficient fluid intake despite the hypernatremia. Although most of the affected patients have partial diabetes insipidus, their extracellular fluid volume remains normal, and they are not dehydrated. Therefore, they exhibit chronic elevations of serum sodium but normal blood pressure, pulse rate, serum creatinine, and creatinine clearance and can release ADH and concentrate their urine during fluid deprivation. When serum sodium concentrations are less than 160 mmol/L, few symptoms are present. However, between 160 and 180 mmol/L, patients may have fatigue, weakness, lethargy, muscle tenderness, cramps, anorexia, depression, and irritability; at 180 mmol/L, stupor and coma may be present. Close to half of these patients have hypothalamic obesity, and almost three fourths demonstrate some degree of anterior pituitary hormone deficiency.6,9,10,13,38,39
Essential hypernatremia has been described with a variety of lesions, including craniopharyngiomas, suprasellar germinomas, optic nerve gliomas, pineal tumors, Langerhans’ cell histiocytosis, sarcoidosis, trauma, hydrocephalus, cysts, inflammatory conditions, ruptured aneurysms of the anterior communicating artery, and toluene exposure.38–40 The Hayek-Peake syndrome is the association of essential hypernatremia with hypodipsia, obesity, lethargy, increased perspiration, central hypoventilation, hyperprolactinemia, hypothyroidism, and hyperlipidemia without an identifiable structural hypothalamic defect.41
Syndrome of Inappropriate Secretion of Antidiuretic Hormone
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) is characterized by serum hyponatremia and hypo-osmolarity, with an inappropriately elevated urine osmolarity, in a patient with normal renal, adrenal, and thyroid function and no clinical evidence of intravascular or extracellular fluid volume expansion. The clinical symptoms depend on the rate of decrease of serum sodium, as well as the absolute serum sodium concentration. At serum sodium levels greater than 120 mmol/L, symptoms are generally mild and nonspecific and include anorexia, nausea, headache, weakness, and lethargy. At less than 120 mmol/L, these symptoms are accompanied by nausea, vomiting, and mental confusion; at very low levels, by seizure and coma.42 The syndrome is found with a variety of intracranial abnormalities, including head trauma, intracranial bleeding, meningitis, encephalitis, neurosurgery, hydrocephalus, acute intermittent porphyria, craniopharyngiomas, germinomas, and pinealomas.6,13,30 An idiopathic form has been described in young women who exhibit menstrual irregularities, have enlarged lateral ventricles, and have SIADH cyclically. No structural defect has been described in these patients.4
Reset Osmostat
The osmoreceptors, located in the circumventricular organs of the lamina terminalis, may become “reset” in some patients with tuberculosis, malnutrition, quadriplegia, and psychosis. The osmoreceptors activate release of ADH at a lower serum osmolality than normal and appropriately decrease ADH release when the serum osmolality falls further.42
Cerebral Salt Wasting
Hyponatremia and the other manifestations of SIADH also co-occur in the syndrome of cerebral salt wasting, which is primarily seen in postoperative neurosurgical patients treated for subarachnoid bleeding, intracranial aneurysms, or following head injury. In contrast to SIADH in which patients are euvolemic or have an expansion of their effective arterial blood volume, patients with cerebral salt wasting are hypovolemic due to renal salt loss. The salt wasting and hypovolemia are felt to be the result of disruption of the normal sympathetic nervous system input into the kidneys and/or relapse of a natriuretic factor from the brain.43
Dysthermia
The warm receptors present in the preoptic anterior hypothalamus are stimulated by an increase in the temperature of the blood. Together with signals from peripheral warm receptors that respond to an increase in external temperature, the afferent signals travel through the median forebrain bundle to the lateral portion of the posterior hypothalamus, which leads to vasodilation and sweating to dissipate heat. Conversely, stimulation of the preoptic anterior hypothalamic cold receptors through a decrease in temperature of the blood, or stimulation of the peripheral cold receptors through a decrease in ambient temperature, results in medial neurons in the posterior hypothalamus activating heat production through muscular shivering and heat conservation through vasoconstriction.5,6
Acute injury to the anterior hypothalamic and preoptic areas may result in a rapid temperature elevation (as high as 41°C) associated with tachycardia and unconsciousness from failure of the heat-dissipating mechanisms to function while heat production continues. Chronic hyperthermia may be secondary to lesions in the tuberoinfundibular region. In contrast to patients with elevated temperature due to the inflammation of infections, these patients generally do not experience malaise and paradoxically may have peripheral vasoconstriction.1,5,6,9,11
Wolff and colleagues44 described a syndrome of hyperthermia associated with shaking chills, fever, hypertension, vomiting, and peripheral vasoconstriction that occurred cyclically at 3-week intervals, without a pathologic lesion in the hypothalamus being found. Similar paroxysms of hyperthermia have been noted in other patients without the cyclicity, and together these episodes may represent a variant of diencephalic epilepsy.6,44,45
Between 0.02% and 2.4% of patients receiving neuroleptic drugs develop the neuroleptic malignant syndrome (NMS), which is characterized by hyperthermia to 38°C or higher; severe extrapyramidal signs, including “lead-pipe” muscle rigidity and tremor; signs of autonomic nervous system dysfunction such as pallor, tachycardia, arrhythmias, blood pressure lability, and diaphoresis; and changes in mental status, including mutism, delirium, and coma.46 All antipsychotic medications have been reported to cause NMS, and most evidence suggests that disruption of dopamine neurotransmission by neuroleptic-induced dopamine receptor blockade is the major pathophysiologic abnormality in susceptible individuals. Indeed, the greater the potency of the neuroleptic in regard to its dopamine D2-receptor antagonism activity, the greater the frequency of NMS occurrence.46 NMS is successfully treated with a variety of dopamine agonists. Injury to the preoptic medial and tuberal nuclei has been demonstrated at autopsy, as has a depletion of hypothalamic norepinephrine concentrations.47 The syndrome generally begins within 2 weeks of initiating the neuroleptic and evolves over a 24- to 72-hour period. The most common complication is rhabdomyolysis, which may result in myoglobinuria and acute renal failure. The mortality of this syndrome is currently less than 10%, which reflects increased recognition of the disorder and initiation of prompt therapy.46
The serotonin syndrome is closely related to NMS and presents with the triad of altered mental status (somnolence, confusion, agitation, seizures, and coma), autonomic instability (fever, diaphoresis, tachycardia, hypo- or hyperthermia, mydriasis), and abnormal neuromuscular activity (myoclonus, rigidity, hyperreflexia). Any drug or combination of drugs which elevates the concentration of serotonin in the central nervous system can cause the syndrome. These include selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants, monamine oxidase inhibitors, cocaine, and amphetamines. These drugs either directly or indirectly activate thermogenesis via the hypothalamus.46
Hypothermia
Large, destructive lesions of the anterior or posterior hypothalamus may result in inability to generate heat through vasoconstriction and muscular shivering. This occurs in 10% to 15% of patients with a variety of hypothalamic lesions, especially neoplasms, infiltrative disorders, and infections.9,10,13 It also has been noted in patients with Parkinson’s disease and Wernicke’s encephalopathy, which are associated with lesions in the posterior hypothalamus and mamillary bodies, respectively.48,49
Diencephalic autonomic epilepsy refers to episodic or paroxysmal hypothermia during which the body temperature decreases to 32°C or less over minutes to days, along with evidence of autonomic nervous system dysfunction, including flushing, sweating, hypotension, bradycardia, salivation, lacrimation, pupillary dilation, Cheyne-Stokes respiration, nausea, vomiting, asterixis, ataxia, and obtundation.6,15,50–53 Electroencephalographic (EEG) abnormalities occur during the episodes. Autopsy studies have shown gliosis and loss of the arcuate nucleus and premamillary area in some patients, whereas others have been found to have tumors involving the floor and lower portion of the third ventricle.15,50 The corpus callosum has been found to be absent in approximately half of patients with episodic hypothermia; these individuals may also exhibit diabetes insipidus, reset osmostat, growth hormone deficiency, hypogonadism, or precocious puberty (Shapiro’s syndrome).54
Poikilothermia
When both the heat-loss and heat-conserving homeostatic mechanisms are impaired, wide fluctuations of body temperature might take place without the patient’s experiencing thermal discomfort. This condition, known as poikilothermia, is found with both anterior and posterior hypothalamic destruction, as well as in patients with large lesions that may involve the posterior hypothalamus and rostral mesencephalon.6,9,10 Rarely, patients with Wernicke’s encephalopathy may experience poikilothermia.6
Disorders of Appetite Control and Caloric Balance
Approximately 25% of patients with structural hypothalamic lesions exhibit hyperphagia and obesity.9,10 Usually, the patients have lesions involving a large portion of the hypothalamus, although bilateral destruction of only the ventromedial nucleus may lead to hypothalamic obesity.1,9,10,17,18,22,25,55 The majority of patients harbor a neoplasm, especially craniopharyngioma, with a minority having inflammatory or granulomatous processes, a history of trauma, or infiltrative disorders.55 Common clinical findings in these patients include headaches, visual abnormalities, hypogonadism, diabetes insipidus, and somnolence. Less commonly, behavioral abnormalities, such as antisocial behavior or sham rage, and seizures may be present.55 Hypothalamic obesity also is found with defects of the leptin and leptin receptor genes, the melanocortin 4 receptor gene, and the proopiomelanocortin gene.56–59
Diencephalic Syndrome of Infancy
Infants harboring a low-grade hypothalamic or optic nerve glioma, or rarely ependymomas, gangliogliomas, or dysgerminomas that destroy the ventromedial nuclei, may develop an unusual syndrome at approximately age 1 to 2 years, in which they begin to lose weight and subcutaneous fat, while maintaining an apparently good food intake and normal growth. They exhibit hyperactivity and a cheerful affect and often demonstrate nystagmus, pallor, vomiting, tremor, and optic atrophy.60–62 Endocrine evaluation is generally normal or may show nonspecific abnormalities that include elevated growth hormone levels.63 If the patients live beyond age 2 years, they begin to gain weight and become obese. Their euphoria and cheerful affect disappear and are replaced by rage and irritability. Somnolence and precocious puberty also may be present.5,60–63 A similar syndrome rarely has been described in adults with tumors involving the optic chiasm or anterior hypothalamic region.64
Hypothalamic Cachexia in Adults
In patients with destructive lesions of the lateral hypothalamus, rapid weight loss, decreased activity, hypophagia, muscle wasting, cachexia, and death may ensue, usually due to a neoplasm.9,10,17,18,21 Malignant multiple sclerosis also may cause the lateral hypothalamic syndrome.17,21
Anorexia Nervosa
Anorexia nervosa is a common disorder, usually seen in young women beginning before the age of 25 years. Although it is not associated with a structural hypothalamic defect, functional hypothalamic abnormalities are present. These patients may exercise excessively, induce vomiting, and have amenorrhea with a prepubertal pattern of gonadotropin release.65 Elevations of basal serum growth hormone, ghrelin, and peptide YY are found, with reduction of insulin-like growth factor 1 (IGF-1) and leptin concentrations. Patients may demonstrate abnormalities in hypothalamic-pituitary-adrenal activity, with elevated plasma cortisol concentrations, decreased adrenocorticotropic hormone (ACTH) levels, and an attenuated ACTH response to corticotropin-releasing hormone (CRH). Low concentrations of thyroxine (T4) and triiodothyronine (T3) are found, with elevated reverse T3 and a thyroid-stimulating hormone (TSH) response to thyrotropin-releasing hormone (TRH) that is either normal or demonstrates a delayed peak consistent with hypothalamic hypothyroidism.66,67 Additionally, these patients may have hyperprolactinemia with galactorrhea, evidence of thermal dysregulation, and a partial diabetes insipidus.68 The neuroendocrine and functional hypothalamic abnormalities remit when the patients regain their weight.
Diencephalic Glycosuria
Acute injuries to the tuberoinfundibular region from basal skull fractures, intracranial hemorrhage, or neurosurgical intervention around the third ventricle may lead to transient hyperglycemia and glycosuria.1,69 Although many of the “stress hormones” with glucose contraregulatory activity are elevated in these patients, they do not appear to be responsible for the glucose abnormality.
Sleep/Wake Cycle and Circadian Abnormalities
Approximately 10% of patients with hypothalamic disease will first be seen with somnolence, and this condition is found in 30% of such patients at some time during the course of their illness.9,10 Somnolence is commonly seen in lesions involving the posterior hypothalamus, often in association with hypothermia.1,6,70 Approximately 40% of patients with hypersomnolence also have hypothalamic obesity.55 Most patients with these manifestations have neoplasms, especially craniopharyngiomas, epithelial pineal tumors, and suprasellar germinomas.31,71 Encephalitis and Wernicke’s nutritional encephalopathy are other causes of hypothalamic hypersomnia.1,5,6 As previously noted, acute hypothalamic injury may lead to a transient coma. Narcolepsy, which is characterized by sudden episodes of sleep that last minutes to hours, may in some instances have a hypothalamic cause. The syndrome has been found in patients with third ventricular tumors, with multiple sclerosis, after head injuries, and with encephalitis.1 Deficiency of the hypothalamic orexin, hypocretin-1, have been found in the cerebrospinal fluid (CSF) of patients with narcolepsy, and a loss of hypocretin neurons occurs in the lateral hypothalamus in affected patients.72,73
Patients with lesions of the anterior and preoptic hypothalamic nuclei may exhibit hyperactivity and insomnia or, more commonly, alterations in the sleep/wake cycle, with daytime sleepiness and nighttime hyperactivity.6,23,74 This is characteristically seen in patients with cystic craniopharyngiomas. Anterior tuberal lesions also may lead to alterations in the sleep/wake cycle, as well as an akinetic mutism type of syndrome in which the patient appears awake but does not respond to verbal stimuli and demonstrates little spontaneous movement.74
The suprachiasmatic nuclei are responsible for the maintenance of many of our circadian rhythms, and lesions involving this region will alter the sleep/wake cycle, temperature control, and cognitive function.72,75
Abnormalities of Emotional Expression or Behavior
Sham rage reactions with emotional lability; marked agitation; and aggressive, destructive behavior are found in patients with lesions involving the ventromedial nuclei.6,23,70 Activation of the sympathetic nervous system is present during the episodes. In contrast, apathy, somnolence, and hypoactivity, as well as vocal and auditory unresponsiveness and akinetic mutism, have been found in patients with destruction of the mamillary bodies or lesions in the medial posterior hypothalamus.1,6
Hypersexual behavior is seen in individuals with lesions involving the caudal hypothalamus.76 The Kleine-Levin syndrome is believed to represent a functional abnormality of the hypothalamus. It generally affects adolescent boys, who have recurrent episodes of somnolence with periodic arousal that is associated with irritability, abnormal speech, forgetfulness, food gorging, and masturbation and other sexual activity. The episodes may occur at 3- to 6-month intervals and generally last 5 to 7 days. The disorder usually remits spontaneously in late adolescence or early adulthood.77
Gelastic or laughing seizures are a form of diencephalic epilepsy due to lesions involving the floor of the third ventricle and mamillary area, especially hamartomas of the tuber cinereum.78 The affected child does not lose consciousness but stops his or her activity and begins to laugh or giggle or make bubbling noises, associated with a grimace from tightening of the facial muscles.79,80 EEG abnormalities are present during the seizure.
Disordered Control of Anterior Pituitary Function
Precocious Puberty: Isosexual pubertal development in girls younger than 8 years or boys younger than 9 years represents sexual precocity, which most often is due to premature activation of the hypothalamic-pituitary-gonadal axis. The majority of girls have no discernible lesion and are therefore classified as having idiopathic central precocious puberty, whereas only 10% of boys have idiopathic precocious puberty.81 In the latter, close to half have hypothalamic hamartomas, and a third have other benign or malignant neoplasms that are located in the posterior hypothalamus or near the mamillary bodies.81 The spectrum of pathologic conditions that can cause central precocious puberty are listed in Table 5-4.4,81–89 Some of these lesions may bring about early activation of the hypothalamic-pituitary-gonadal axis through increased intracranial pressure or irritation of the basal hypothalamus. Hypothalamic hamartomas involving the tuber cinereum are often associated with precocious puberty through premature activation of the normal hypothalamic gonadotropin-releasing hormone (GnRH) secretory mechanisms or through direct secretion of GnRH by the hamartoma, because GnRH has been located immunohistochemically within hamartomatous neurons.90,91 In addition to pressure effects, germ cell tumors may result in precocious puberty through the secretion of human chorionic gonadotropin (hCG), which may stimulate the child’s gonads to secrete sex steroid hormones, bringing about precocious sexual development. Finally, premature activation of the normal hypothalamic-pituitary-gonadal axis has been described in some patients who have had incomplete sexual precocity from congenital adrenal hyperplasia or polyostotic fibrous dysplasia (McCune-Albright) syndrome, in which the hypothalamus is exposed to elevated sex steroid hormone levels at an early age. Premature activation of the normal hypothalamic-pituitary-gonadal axis has also occurred in patients with primary hypothyroidism, who also may exhibit galactorrhea with elevated prolactin levels (Van Wyk-Grumbach syndrome), in which the mechanism for the premature activation is unknown but usually ceases with correction of the hypothyroidism.89
Table 5-4
Causes of Central Precocious Puberty
Idiopathic
Congenital Abnormalities
Hypothalamic hamartoma
Arachnoid cyst
Myelomeningocele
Aqueductal stenosis with hydrocephalus
Tuberous sclerosis
Congenital optic nerve hypoplasia
Congenital adrenal hyperplasia
McCune-Albright syndrome
Septo-optic dysplasia
Neoplasms
Optic nerve glioma
Hypothalamic glioma
Neurofibroma
Astrocytoma
Ependymoma
Infundibuloma
Pinealoma
Neuroblastoma
Germinoma
Craniopharyngioma
Inflammatory Conditions
Tuberculosis
Sarcoidosis
Meningoencephalitis
Subdural Hematoma
Primary Hypothyroidism
From Braunstein GD: The hypothalamus. In Melmed S (ed): The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.
Acromegaly: Acromegaly due to the ectopic secretion of GHRH is rare. In most instances, the source of the ectopic GHRH is a bronchial carcinoid, islet cell neoplasm, adrenal tumor, or lung carcinoma.92,93 However, acromegaly has also been found in patients with hypothalamic hamartomas, gangliocytomas, gliomas, and choristomas.92–95 Some of these tumors have been shown to contain GHRH, and they presumably secrete the releasing factor, which in turn stimulates the somatotrophs to hypersecrete growth hormone.
Cushing’s Disease: Several lines of evidence suggest that Cushing’s disease has a hypothalamic component to its pathophysiology.96 First, it has been noted that the onset of the disease often follows an emotionally stressful event.97 Because depression may be associated with pseudo-Cushing’s syndrome with hypersecretion of glucocorticoids, it is conceivable that chronic corticotroph stimulation by hypothalamic CRH could lead to the development of a corticotroph adenoma and Cushing’s disease and may account for the recurrence of Cushing’s disease after apparently successful removal of an ACTH-secreting corticotroph adenoma.98,99 Second, most patients with Cushing’s disease, when given exogenous glucocorticoids in sufficient quantities, are able to suppress their ACTH secretion, a fact that has been known for some time and forms the basis for the high-dose portion of the dexamethasone suppression test.96 Presumably, this phenomenon reflects an increased set point for negative feedback of glucocorticoids at the hypothalamic level. Third, some patients with Cushing’s disease exhibit a reduction in ACTH and cortisol secretion and amelioration of symptoms after the administration of cyproheptadine, bromocriptine, or sodium valproate, which may work through the hypothalamus.100–103 Nevertheless, most if not all corticotroph adenomas are of clonal origin rather than polyclonal, as would be anticipated if CRH hypersecretion were responsible for the pituitary abnormality.104 Hypothalamic factors such as CRH may promote clonal expansion of corticotroph cells that have become intrinsically abnormal.105 An unusual cause of pituitary-dependent Cushing’s disease is the secretion of CRH by an intracranial neoplasm, as was demonstrated with an intrasellar gangliocytoma.106
Hyperprolactinemia: Because prolactin secretion by the lactotrophs is under hypothalamic dopamine inhibitory control, it is not surprising that patients with a variety of hypothalamic disorders may exhibit hyperprolactinemia. This is seen in 79% of patients with suprasellar germinomas, 36% of patients with craniopharyngiomas, and 14% of patients with pineal germinomas.32 Most patients have a prolactin concentration less than 70 ng/mL, and galactorrhea is infrequently seen, probably because of the coexistence of hypogonadism.107 Nevertheless, amenorrhea and galactorrhea may be present in women, and erectile dysfunction in men.
Idiopathic hyperprolactinemia in patients without any demonstrable structural abnormality in the pituitary or hypothalamus is presumably due to a hypothalamic dopamine deficiency. The prolactin secretory dynamics of these patients in response to various stimulatory and inhibitory agents is similar to that seen in patients with prolactin-secreting pituitary adenomas. Indeed, when followed for a long period, some patients with idiopathic hyperprolactinemia will eventually be found to have a prolactin-secreting pituitary microadenoma. Additional evidence supporting a hypothalamic cause for the pituitary adenoma is the finding of lactotroph hyperplasia in some patients who have had documented adenomas, as well as the recurrence of prolactin-secreting pituitary adenoma after successful removal of a microadenoma and an interval of normal prolactin-secretory dynamics.108,109
Hypofunction Syndromes
Hypothalamic Hypogonadism: Kallmann’s syndrome (olfactory-genital dysplasia) is the most common form of congenital isolated gonadotropin deficiency and can occur sporadically or in a familial setting as an X-linked, autosomal-dominant, or autosomal-recessive trait with incomplete penetrance.110 The X-linked disorder, which accounts for 10% to 15% of sporadic cases and 30% to 60% of familial cases, is due to a defect in the KAL-1 gene whose product, anosmin-1, normally directs the migration of GnRH neurons from the olfactory placode to the hypothalamus. This results in a deficiency or absence of GnRH-secreting neurons in the hypothalamus, as well as agenesis or hypoplasia of the olfactory bulb, the latter defect being responsible for the hyposmia or anosmia seen in this syndrome. Boys are affected more commonly than girls and often exhibit cryptorchidism and microphallus at birth, reflecting the lack of fetal gonadotropins, which stimulate testosterone secretion from the fetal testes. At the time of expected puberty, there is a failure of gonadotropins to increase, of testicular enlargement, and secondary sexual characteristic development. After a single bolus injection of GnRH, little or no increase in gonadotropin levels is seen. However, if GnRH is given in a pulsatile fashion every 90 minutes, an increase in luteinizing hormone (LH) and follicle-stimulating hormone (FSH) will occur, reflecting the fact that the gonadotrophs are normal but understimulated in this syndrome. Pulsatile GnRH therapy may result in full virilization. Other components of this syndrome include color blindness, nerve deafness, cleft palate, exostosis, and renal abnormalities.110 An autosomal-dominant form of anosmic and normosmic congenital hypogonadotropic hypogonadism is due to mutations of the fibroblast growth factor-1 receptor and accounts for 7% to 10% of patients with hypogonadotropic hypogonadism.111 Another 5% of patients have a loss-of-function mutation of the GnRH receptor.110
Hypogonadotropic hypogonadism has been found with leptin and leptin-receptor gene mutations, as well as GPR54 and DAX1 mutations.110 Congenital gonadotropin deficiency also is seen as a manifestation of panhypopituitarism (which may be on a hypothalamic basis), as well as with several complex hypothalamic disorders, including the Prader-Willi, Bardet-Biedl, and Laurence-Moon syndromes.
Hypogonadism is a relatively common manifestation of hypothalamic tumors and infiltrative disease, especially those that involve the floor of the third ventricle and median eminence. Obesity, diabetes insipidus, and neuro-ophthalmologic abnormalities often accompany the hypogonadism.9,10
Growth Hormone Deficiency: A variety of congenital structural defects involving the hypothalamus, such as anencephaly, holoprosencephaly, encephalocele, and septo-optic dysplasia, may result in growth hormone deficiency, either alone or with other anterior pituitary hormone deficiencies.4 Monotropic growth hormone deficiency may occur sporadically or on a familial basis because of a deficient production or secretion of GHRH. Such patients will demonstrate an increase in growth hormone secretion after multiple injections of GHRH. Growth hormone deficiency also occurs as a manifestation of panhypopituitarism and is the hormone that is most frequently absent in these patients. As in patients with isolated growth hormone deficiency, those with panhypopituitarism generally have a hypothalamic basis for the abnormality, with deficiencies of multiple hypothalamic-releasing hormones.112
At birth, patients with congenital growth hormone deficiency have a normal length and weight but may exhibit microphallus. During the first year, growth retardation is seen, with a delay in both height and bone ages. Hypoglycemia due to loss of the glucose contraregulation effect of growth hormone may be found. During childhood, an increase in subcutaneous fat along with proportional short stature is noted. Even in the presence of normal gonadotrophs, puberty is often delayed in these patients. Treatment with growth hormone increases linear growth, reduces subcutaneous fat and glucose intolerance, and stimulates pubertal progression.112
Growth hormone deficiency is generally the earliest endocrine manifestation of a hypothalamic tumor or infiltrative process and results in growth retardation. Even in patients with structural hypothalamic disease who have no clinical evidence of growth retardation, provocative testing reveals a high frequency of inadequate growth hormone secretion.110,113
Hypothalamic Hypoadrenalism: Congenital or acquired isolated ACTH deficiency is quite rare. However, ACTH deficiency does commonly occur in association with other anterior pituitary hormone deficiencies due to craniopharyngiomas, suprasellar germinomas, and septo-optic dysplasia.31,82,83,113–116 Clinical manifestations include nausea, vomiting, hypotension, and hypoglycemia, without the hyperpigmentation and electrolyte abnormalities from aldosterone deficiency seen in primary adrenocortical insufficiency.
Hypothalamic Hypothyroidism: Isolated TSH deficiency also is quite rare. However, TSH deficiency is found in approximately one third of the patients with craniopharyngiomas, suprasellar germinomas, and in patients with septo-optic dysplasia.30,31,82,83,113,115 Clinically, patients may exhibit dry skin, puffiness, pallor, lethargy, bradycardia, hypothermia, and weight gain, with evidence of an atrophic thyroid gland. Serum free-T4 levels are low, and the serum TSH may be low or slightly elevated, the latter reflecting abnormal glycosylation of the TSH molecule, which results in decreased biologic activity.117 After an injection of TRH, a delayed and prolonged increase in TSH is seen in patients with hypothalamic hypothyroidism.30,31,77
Specific Hypothalamic Disorders
Prader-Willi syndrome, first described in 1956, occurs in approximately 1 in 25,000 live births.118,119 The major clinical manifestations include infantile hypotonia; feeding problems; failure to thrive; rapid weight gain occurring between ages 1 and 6 years; a characteristic dysmorphic facial appearance with a narrow bitemporal diameter, almond-shaped eyes, palpebral fissures, down-turned mouth; developmental delay and mental retardation. Hypogonadism may be present at birth, with cryptorchidism, scrotal hypoplasia, and a small penis in boys; poor development of the labia minora and clitoris in girls; and delayed onset of puberty associated with low sex steroid hormones, low gonadotropins, and blunting of the gonadotropin response to GnRH. In addition, these patients have short stature (associated with growth hormone deficiency) and behavioral problems that appear during childhood and are characterized by temper tantrums, aggressive behavior, and obsessive-compulsiveness. One of the major characteristics of these patients is marked, indiscriminate hyperphagia and central obesity. These patients will exhibit abnormal food-seeking behavior, often eating discarded or spoiled food or pet food. Sleep disturbances and abnormalities in temperature control and heat generation also suggest a hypothalamic cause.118–121 The only anatomic abnormality found in these patients is a decrease in the size of the paraventricular nuclei and oxytocin-producing neurons.72
Prader-Willi syndrome is a disorder of genetic (genomic) imprinting, in most cases due to a microdeletion of the paternally contributed chromosome 15q11-q13.119,120 Because only the paternal genes are normally expressed in this region, a mutation of the paternal gene results in absence of expression. A minority of patients have maternal uniparental disomy (both members of the chromosome pair inherited from the same parent). An even rarer cause is a translocation involving chromosome 15.119,120
Bardet-Biedl and Related Syndromes
The Bardet-Biedl syndrome represents an autosomal-recessive disorder characterized by retinal pigmentary dystrophy (retinitis pigmentosa), mental retardation, central obesity, polydactyly, a variety of renal abnormalities, and hypogonadotropic hypogonadism.122 Those with the Laurence-Moon syndrome also exhibit retinal pigmentary dystrophy, mental retardation, and hypogonadotropic hypogonadism. These patients also have progressive spastic paraparesis and distal muscle weakness but do not exhibit polydactyly.123 The Biemond syndrome is another autosomal-recessive condition with mental retardation, polydactyly or brachydactyly, obesity, and hypogonadotropic hypogonadism. Retinal pigmentary dystrophy does not occur in this condition; rather, these patients have iris coloboma. The autosomal-recessive Alström syndrome is associated with atypical retinal pigmentary dystrophy, obesity, nerve deafness, diabetes mellitus, and acanthosis nigricans. Affected patients have hypogonadism due to primary gonadal failure rather than hypothalamic dysfunction.123,124 The overlapping features of these different syndromes raise the possibility that they are due to a similar genetic abnormality.
Septo-Optic Pituitary Dysplasia
The anatomic features of septo-optic pituitary dysplasia, a midline developmental abnormality, are an absence of the septum pellucidum, agenesis of the corpus callosum, unilateral or bilateral hypoplasia of the optic nerves, and absence of the supraoptic and paraventricular nuclei, with posterior pituitary hypoplasia.82,83,114,115,125–127 The nonendocrine manifestations of this disease include visual abnormalities, mental retardation, nystagmus, seizures, and various forms of cerebral palsy.82 Approximately two thirds of affected patients have short stature associated with growth hormone deficiency; approximately 40% have ACTH deficiency; 20% TSH deficiency; and one fourth exhibit gonadotropin deficiency. Close to one fourth of the patients exhibit diabetes insipidus, and approximately 20% have hyperprolactinemia.82,83,114,115,125–127 This disorder is caused by recessive mutation in the homeobox gene on chromosome 3, HESX1.128
Hyperphagic Short Stature (Psychosocial Dwarfism)
Hyperphagic short stature, a rare syndrome which has its onset before age 2 years, occurs in some children exposed to a disturbed parent-child home environment. The clinical manifestations include short stature and delayed bone age associated with abnormal growth-hormone response to provocative tests; low body weight despite an enormous appetite that is associated with gorging, pica, food hoarding, vomiting, and production of foul-smelling stools; polydipsia; bizarre behavior; emotional or mental retardation; and a protuberant abdomen. In addition to the abnormal growth hormone responses, these patients may have an inadequate ACTH response to provocative testing, although thyroid function and urine-concentrating ability are normal. The clinical findings are reversible and disappear when the children are placed in a nurturing environment.129,130
Pseudocyesis
An extreme example of a functional hypothalamic disorder is seen in women who develop a conversion reaction in which they think they are pregnant but in fact are not. Amenorrhea, morning nausea, breast enlargement and engorgement, and abdominal distension due to retained colonic gas are present in these women. Hyperprolactinemia is found, and some women exhibit galactorrhea.131,132 Elevated levels of LH may account for the persistent corpus luteum activity that is seen in this syndrome.133 When these women are informed of the diagnosis, the clinical manifestations rapidly disappear.
Hypothalamic Hamartoma
These benign hyperplastic malformations contain ganglion cells, myelinated nerve fibers, and glial matrix, and are generally located between the tuber cinereum and mamillary bodies.79,134 Most patients exhibit onset of clinical symptoms before age 2 years, with the major endocrine abnormality being isosexual precocious puberty. Other common manifestations include gelastic seizures, emotional lability, hyperactivity, and neurodevelopmental delay.78,80,89,90,134–138 During late childhood or adolescence, obesity develops in many of these patients. The precocious puberty in these patients responds to long-acting GnRH agonists that down-regulate GnRH receptors.89 Neurosurgical removal of the hamartomas is indicated if signs of increased intracranial pressure, progressive growth, intractable seizures, or neurologic deterioration are present.90
The Pallister-Hall syndrome consists of hypothalamic hamartomas, panhypopituitarism, polydactyly, imperforate anus, and multiple craniofacial and limb abnormalities.139–142 This may occur sporadically or be transmitted as an autosomal-dominant trait associated with a mutation of the GLP3 gene on chromosome 7p13.143
Suprasellar Arachnoid Cyst
Suprasellar arachnoid cyst, an uncommon developmental anomaly of the arachnoid membrane, leads to a CSF-filled cyst that obstructs CSF flow through the foramen of Monro, leading to hydrocephalus and increased intracranial pressure. Thus headache, vomiting, lethargy, and increased head size are commonly found in these patients. The cysts also may compress the brainstem, optic nerve, and optic chiasm, leading to spasticity, ataxia, tremor, decreased visual acuity, and visual field defects. Endocrine abnormalities include growth hormone and ACTH deficiency, as well as precocious puberty.144 Surgical decompression or percutaneous ventriculocystostomy is used to drain the cyst and reduce intracranial pressure.145
Infiltrative Disorders
Sarcoidosis may involve the basal hypothalamus and floor of the third ventricle and lead to diminished visual acuity, visual field abnormalities, diabetes insipidus, thermal dysregulation, somnolence, personality changes, obesity, and hypothalamic hypopituitarism. Most patients with hypothalamic sarcoidosis also have involvement outside of the central nervous system.34,35,37,146–147
The chronic disseminated form of Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) is classically composed of the triad of membranous bone lesions, exophthalmos, and diabetes insipidus. Growth retardation, hyperprolactinemia, hypogonadism, hypodipsia or adipsia, sleep disturbances, hyperphagia with obesity, temperature dysregulation, and behavioral abnormalities also may be found in these patients.33,148,149
Hypothalamic Dysfunction After Brain Irradiation
Both whole-brain irradiation and localized radiotherapy for brain or head or neck neoplasms are associated with delayed onset of hypothalamic dysfunction, most often manifested by progressive loss of growth hormone secretion and hyperprolactinemia. ACTH and gonadotropin deficiency are also found, as are changes in personality and abnormalities in thirst, sleep/wake cycle, and appetite regulation. Children are more susceptible to hypothalamic damage than are adults, and the incidence of hypothalamic abnormality increases with increasing radiation dose and decreasing intervals over which the radiation is administered.150,151
Traumatic Brain Injury
Posttraumatic hypopituitarism with multiple deficiencies of anterior pituitary hormones and elevation of prolactin due to damage to the hypothalamus, pituitary stalk, the hypophyseal artery or the pituitary is a relatively common occurrence following head trauma. Following the acute phase, anterior pituitary function may recover.152,153
References
1. Boshes, B, Syndromes of the diencephalons: The hypothalamus and the hypophysis. Localization in Clinical Neurology: Handbook of Clinical Neurology. Vinken, PJ, Bruyn, GW, eds. Localization in Clinical Neurology: Handbook of Clinical Neurology, Vol 2. Amsterdam: North-Holland, 1969:432–468.
2. Kirgis, HD, Locke, W. Anatomy and embryology. In: Locke W, Schally AV, eds. The Hypothalamus and Pituitary in Health and Disease. Springfield, IL: Charles C Thomas; 1972:3–21.
3. Bruesch, SR. Anatomy of the human hypothalamus. In: Givens JR, Kitabchi AE, Robertson JT, eds. The Hypothalamus. St Louis: Mosby-Year Book; 1984:1–16.
4. Braunstein, GD. The hypothalamus. In: Melmed S, ed. The Pituitary. 2nd ed. Cambridge, MA: Blackwell Scientific; 2002:317–348.
5. Carmel, PW. Surgical syndromes of the hypothalamus. Clin Neurosurg. 1980;27:133–159.
6. Plum, F, Van Uitert, R. Nonendocrine diseases and disorders of the hypothalamus. In: Reichlin S, Baldessarini RJ, Martin JB, eds. The Hypothalamus. New York: Raven Press; 1978:415–473.
7. Sano, K, Mayanagi, Y, Sekino, H, et al. Results of stimulation and destruction of the posterior hypothalamus in man. J Neurosurg. 1970;33:689–707.
8. Garnica, AD, Netzloff, ML, Rosenbloom, AL. Clinical manifestations of hypothalamic tumors. Ann Clin Lab Sci. 1980;10:474–485.
9. Bauer, HG. Endocrine and other clinical manifestations of hypothalamic disease: A survey of 60 cases, with autopsies. J Clin Endocrinol Metab. 1954;14:13–31.
10. Bauer, HG. Endocrine and metabolic conditions related to pathology in the hypothalamus: A review. J Nerv Ment Dis. 1959;128:323–338.
11. Thompson, HJ, Tkacs, NC, Saatman, KE, et al. Hypothermia following traumatic brain injury: A critical evaluation. Neurobiol Dis. 2003;12:163–173.
12. Dott, NM. Surgical aspects of the hypothalamus. In: Le Gros Clark WE, Beattie J, Riddoch G, et al, eds. The Hypothalamus: Morphological, Functional, Clinical and Surgical Aspects. London: Oliver & Boyd; 1938:131–185.
13. Frohman, LA. Clinical aspects of hypothalamic disease. In: Motta M, ed. The Endocrine Functions of the Brain. New York: Raven Press; 1980:419–446.
14. Riddoch, G. Clinical aspects of hypothalamic derangement. In: Le Gros Clark WE, Beattie J, Riddoch G, et al, eds. The Hypothalamus: Morphological, Functional, Clinical and Surgical Aspects. London: Oliver & Boyd; 1938:101–130.
15. McLean, AJ. Autonomic epilepsy. Arch Neurol. 1934;32:189–197.
16. Rothballer, AB, Dugger, GS. Hypothalamic tumor: Correlation between symptomatology, regional anatomy, and neurosecretion. Neurology. 1955;5:160–177.
17. White, LE, Hain, RF. Anorexia in association with a destructive lesion of the hypothalamus. Arch Pathol Lab Med. 1959;68:275–281.
18. Reeves, AG, Plum, F. Hyperphagia, rage, and dementia accompanying a ventromedial hypothalamic neoplasm. Arch Neurol. 1969;20:616–624.
19. Fox, RH, Davies, TW, Marsh, FP, et al. Hypothermia in a young man with an anterior hypothalamic lesion. Lancet. 1970;2:185–188.
20. Lewin, K, Mattingly, D, Millis, RR. Anorexia nervosa associated with hypothalamic tumour. Br Med J. 1972;2:629–630.
21. Kamalian, N, Keesey, RE, Zurhein, GM. Lateral hypothalamic demyelination and cachexia in a case of “malignant” multiple sclerosis. Neurology. 1975;25:25–30.
22. Celesia, GG, Archer, CR, Chung, HD. Hyperphagia and obesity: Relationship to medial hypothalamic lesions. JAMA. 1981;246:151–153.
23. Haugh, RM, Markesbery, WR. Hypothalamic astrocytomas: Syndrome of hyperphagia, obesity, and disturbances of behavior and endocrine and autonomic function. Arch Neurol. 1983;40:560–563.
24. Schwartz, WJ, Busis, NA, Hedley-Whyte, ET. A discrete lesion of ventral hypothalamus and optic chiasm that disturbed the daily temperature rhythm. J Neurol. 1986;233:1–4.
25. Pinkney, J, Wilding, J, Williams, G, et al. Hypothalamic obesity in humans: what do we know and what can be done? Obes Rev. 2002;3:27–34.
26. Bergeron, C, Kovacs, K, Ezrin, C, et al. Hereditary diabetes insipidus: An immunohistochemical study of the hypothalamus and pituitary gland. Acta Neuropathol. 1991;81:345–348.
27. Maghnie, M, Ghirardello, S, De Bellis, A, et al. Idiopathic central diabetes insipidus in children and young adults is commonly associated with vasopressin-cell antibodies and markers of autoimmunity. Clin Endocrinol (Oxf). 2006;65:470–478.
28. McLeod, JF, Kouvacs, L, Gaskill, MB, et al. Familial neurohypophyseal diabetes insipidus associated with a signal peptide mutation. J Clin Endocrinol Metab. 77, 1997. [599A–599G].
29. Minton, JA, Ranibow, LA, Ricketts, C, et al. Wolfram syndrome. Rev Endocr Metab Disord. 2003;4:53–59.
30. Verbalis, JG. Management of disorders of water metabolism in patients with pituitary tumors. Pituitary. 2002;22:119–132.
31. Buchfelder, M, Fahlbusch, R, Walther, M, et al. Endocrine disturbances in suprasellar germinomas. Acta Endocrinol. 1989;120:337–342.
32. Jennings, MT, Gelman, R, Hochberg, F. Intracranial germ-cell tumors: Natural history and pathogenesis. J Neurosurg. 1985;63:155–167.
33. Kaltsas, GA, Powles, TB, Evanson, J, et al. Hypothalamo-pituitary abnormalities in adult patients with Langerhans cell histiocytosis: Clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab. 2000;85:1370–1376.
34. Delaney, P. Neurologic manifestations in sarcoidosis: Review of the literature, with a report of 23 cases. Ann Intern Med. 1977;87:336–345.
35. Stuart, CA, Neelon, FA, Lebovitz, HE. Hypothalamic insufficiency: The cause of hypopituitarism in sarcoidosis. Ann Intern Med. 1978;88:589–594.
36. Jawadi, MH, Hanson, TJ, Schemmel, JE, et al. Hypothalamic sarcoidosis and hypopituitarism. Horm Res. 1980;12:1–9.
37. Vesely, DL, Maldonado, A, Levey, GS. Partial hypopituitarism and possible hypothalamic involvement in sarcoidosis. Am J Med. 1977;62:425–431.
38. Ouma, JR, Farrell, VJR. Lymphocytic infundibulo-neurohypophysitis with hypothalamic and optic pathway involvement: Report of a case and review of the literature. Surg Neurol. 2002;57:49–54.
39. McKenna, K, Thompson, C. Osmoregulation in clinical disorders of thirst appreciation. Clin Endocrinol. 1998;49:139–152.
40. Teelucksingh, S, Steer, CR, Thompson, CJ, et al. Hypothalamic syndrome and central sleep apnoea associated with toluene exposure. Q J Med. 1991;78:185–190.
41. Hayek, A, Peake, GT. Hypothalamic adipsia without demonstrable structural lesion. Pediatrics. 1982;70:275–278.
42. Ellison, DH, Berl, T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072.
43. Palmer, BF. Hyponatremia in patients with central nervous system disease: SIADH versus CSW. Trends Endocrinol Metab. 2003;14:182–187.
44. Wolff, SM, Adler, RC, Buskirk, ER, et al. A syndrome of periodic hypothalamic discharge. Am J Med. 1964;36:956–967.
45. Martin, JB, Reichlin, S. Clinical Neuroendocrinology. Philadelphia: FA Davis; 1987.
46. Rusyniak, DE, Sprague, JE. Hyperthermic syndromes induced by toxins. Clin Lab Med. 2006;26:165–184.
47. Horn, E, Lach, B, Lapierre, Y, et al. Hypothalamic pathology in the neuroleptic malignant syndrome. Am J Psychiatry. 1988;145:617–620.
48. Sandyk, R, Iacono, RP, Bamford, CR. The hypothalamus in Parkinson disease. Ital J Neurol Sci. 1987;8:227–234.
49. Haak, HR, van Hilten, JJ, Roos, RAC, et al. Functional hypothalamic derangement in a case of Wernicke’s encephalopathy. Neth J Med. 1990;36:291–296.
50. Penfield, W. Diencephalic autonomic epilepsy. Arch Neurol Psychiatry. 1929;22:358–369.
51. Fox, RH, Wilkins, DC, Bell, JA, et al. Spontaneous periodic hypothermia: Diencephalic epilepsy. Br Med J. 1973;2:693–695.
52. Mooradian, AD, Morley, GK, McGeachie, R, et al. Spontaneous periodic hypothermia. Neurology. 1984;34:79–82.
53. Flynn, MD, Sandeman, DD, Mawson, DM, et al. Cyclical hypothermia: Successful treatment with ephedrine. J R Soc Med. 1991;84:752–753.
54. Kloos, RT. Spontaneous periodic hypothermia. Medicine (Baltimore). 1995;74:268–280.
55. Bray, GA, Gallagher, TF, Jr. Manifestations of hypothalamic obesity in man: A comprehensive investigation of eight patients and a review of the literature. Medicine (Baltimore). 1975;54:301–330.
56. Montague, CT, Farooqi, IS, Whitehead, JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908.
57. Clement, K, Vaisse, C, Lahlou, N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401.
58. Yeo, GS, Farooqi, IS, Aminian, S, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112.
59. Krude, H, Biebermann, H, Luck, W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–157.
60. Russell, A. A diencephalic syndrome of emaciation in infancy and childhood. Arch Dis Child. 1951;26:274.
61. Burr, IM, Slonim, AE, Danish, RK, et al. Diencephalic syndrome revisited. J Pediatr. 1976;88:439–444.
62. Poussaint, TY, Barnes, PD, Nichols, K, et al. Diencephalic syndrome: clinical features and imaging findings. Am J Neuroradiol. 1997;18:1499–1505.
63. Fleishchman, A, Brue, C, Poussaint, TY, et al. Diencephalic syndrome: a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics. 2005;115:e742–e748.
64. Miyoshi, Y, Yunoki, M, Yano, A, et al. Diencephalic syndrome of emaciation in an adult associated with a third ventricle intrinsic craniopharyngioma: Case report. Neurosurgery. 2003;52:224–227.
65. Rome, ES. Eating disorders. Obstet Gynecol Clin North Am. 2003;30:353–377.
66. Muñoz, MT, Argente, J. Anorexia nervosa in female adolescents: Endocrine and bone mineral density disturbances. Eur J Endocrinol. 2002;147:275–286.
67. Misra, M, Miller, KK, Tsai, P, et al. Elevated peptide YY levels in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab. 2006;91:1027–1033.
68. Mecklenberg, RS, Loriaux, DL, Thompson, RH, et al. Hypothalamic dysfunction in patients with anorexia. Medicine (Baltimore). 1974;53:147–159.
69. Clark, LG. The hypothalamus in man. In: Le Gros Clark WE, Beattie J, et al, eds. The Hypothalamus: Morphological, Functional, Clinical and Surgical Aspects. London: Oliver & Boyd; 1938:59–68.
70. Carpenter, MB, Sutin, J. Human Neuroanatomy. Baltimore: Williams & Wilkins; 1983.
71. Locke, W, Schally, AV. The Hypothalamus and Pituitary in Health and Disease. Springfield, IL: Charles C Thomas; 1972.
72. Overeem, S, van Vliet, JA, Lammers, GJ, et al. The hypothalamus in episodic brain disorders. Lancet. 2002;1:437–444.
73. Nishino, S. Clinical and neurobiological aspects of narcolepsy. Sleep Med. 2007;8:373–399.
74. Martin, JB, Reichlin, S. Clinical Neuroendocrinology. Philadelphia: FA Davis; 1987.
75. Cohen, RA, Albers, HE. Disruption of human circadian and cognitive regulation following a discrete hypothalamic lesion: A case study. Neurology. 1991;41:726–729.
76. Fenzi, F, Simonati, A, Crosato, F, et al. Clinical features of Kleine-Levin syndrome with localized encephalitis. Neuropediatrics. 1993;24:292–295.
77. Arnulf, I, Seitzer, JM, File, J, et al. Kleine-Levin syndrome: a systemic review of 186 cases in the literature. Brain. 2005;128:2763–2776.
78. Breningstall, GN. Gelastic seizures, precocious puberty, and hypothalamic hamartoma. Neurology. 1985;35:1180–1183.
79. Sharma, RR. Hamartoma of the hypothalamus and tuber cinereum: A brief review of the literature. J Postgrad Med. 1987;33:1–13.
80. Harvey, AS, Freeman, JL. Epilepsy in hypothalamic hamartoma: clinical and EEG features. Semin Pediatr Neurol. 2007;14:60–64.
81. Shankar, RR, Pescovitz, OH. Precocious puberty. Adv Endocrinol Metab. 1995;6:55–89.
82. Margalith, D, Jan, JE, McCormick, AQ, et al. Clinical spectrum of congenital optic nerve hypoplasia: Review of 51 patients. Dev Med Child Neurol. 1984;26:311–322.
83. Margalith, D, Tze, WJ, Jan, JE. Congenital optic nerve hypoplasia with hypothalamic-pituitary dysplasia. Am J Dis Child. 1985;139:361–366.
84. Gross, RE. Neoplasms producing endocrine disturbances in childhood. Am J Dis Child. 1940;59:579–628.
85. Laue, L, Comite, F, Hench, K, et al. Precocious puberty associated with neurofibromatosis and optic gliomas. Am J Dis Child. 1985;139:1097–1100.
86. Gillett, GR, Symon, L. Hypothalamic glioma. Surg Neurol. 1987;28:291–300.
87. Banna, M. Pathology and clinical manifestations. In: Hankinson J, Banna M, eds. Pituitary and Parapituitary Tumours. London: Saunders; 1976:13–58.
88. Weinberger, LM, Grant, FC. Precocious puberty and tumors of the hypothalamus. Arch Intern Med. 1941;67:762–792.
89. Chemaitilly, W, Trivin, C, Adan, L, et al. Central precocious puberty: clinical and laboratory features. Clin Endocrinol (Oxf). 2001;54:289–294.
90. Rosenfeld, JV, Harvey, AS, Wrennal, J, et al. Transcallosal resection of hypothalamic hamartomas, with control of seizures, in children with gelastic epilepsy. Neurosurgery. 2001;48:108–118.
91. Hochman, HI, Judge, DM, Reichlin, S. Precocious puberty and hypothalamic hamartoma. Pediatrics. 1981;67:236–244.
92. Losa, M, von Werder, K. Pathophysiology and clinical aspects of the ectopic GH-releasing hormone syndrome. Clin Endocrinol. 1997;47:123–135.
93. Saeger, W, Puchner, MJA, Ludecke, DK. Combined sellar gangliocytomas and pituitary adenoma in acromegaly or Cushing’s disease. Virchows Arch. 1994;425:93–99.
94. Asa, SL, Bilbao, JM, Kovacks, K, et al. Hypothalamic neuronal hamartoma associated with pituitary growth hormone cell adenoma and acromegaly. Acta Neuropathol. 1980;52:231–234.
95. Asa, SL, Scheithauer, BW, Bilbao, JM, et al. A case for hypothalamic acromegaly: A clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone releasing factor. J Clin Endocrinol Metab. 1984;58:796–803.
96. Biller, BMK. Pathogenesis of pituitary Cushing’s syndrome: Pituitary versus hypothalamic. Endocrinol Clin North Am. 1994;23:547–554.
97. Gifford, S, Gunderson, JG. Cushing’s disease as a psychosomatic disorder: A selective review of the clinical and experimental literature and a report of ten cases. Perspect Biol Med. 1970;13:169–221.
98. Bigos, ST, Somma, M, Rasio, E, et al. Cushing’s disease: Management by transsphenoidal pituitary microsurgery. J Clin Endocrinol Metab. 1980;50:348–354.
99. Lamberts, SW, Stefanko, SZ, DeLang, SE, et al. Failure of clinical remission after transsphenoidal removal of a microadenoma in a patient with Cushing’s disease: Multiple hyperplastic and adenomatous cell nests in surrounding pituitary tissues. J Clin Endocrinol Metab. 1980;50:793–795.
100. Krieger, DT, Amorosa, L, Linick, F. Cyproheptadine-induced remission of Cushing’s disease. N Engl J Med. 1975;293:893–896.
101. Lankford, HU, Tucker, HS, Blackard, WG. A cyproheptadine-reversible defect in ACTH control persisting after removal of the pituitary tumor in Cushing’s disease. N Engl J Med. 1981;305:1244–1248.
102. Cavagnini, F, Invitti, C, Polli, EE. Sodium valproate in Cushing’s disease. Lancet. 1984;2:162–163.
103. Lamberts, SWJ, Klijn, JG, deQuijada, M, et al. The mechanism of the suppressive action of bromocriptine on adrenocorticotropin secretion in patients with Cushing’s disease and Nelson’s syndrome. J Clin Endocrinol Metab. 1980;51:307–311.
104. Biller, BMK, Alexander, JM, Zervas, NT, et al. Clonal origins of adrenocorticotropin-secreting pituitary tissue in Cushing’s disease. J Clin Endocrinol Metab. 1992;75:1303–1309.
105. Faglia, G, Spada, A. The role of hypothalamus in pituitary neoplasia. Clin Endocrinol. 1995;9:225–242.
106. Asa, SL, Kovacs, K, Tindall, GT, et al. Cushing’s disease associated with an intrasellar gangliocytoma producing corticotrophin-releasing factor. Ann Intern Med. 1984;101:789–793.
107. Kapcala, LP, Molitch, ME, Post, KD, et al. Galactorrhea, oligo/amenorrhea, and hyperprolactinemia in patients with craniopharyngiomas. J Clin Endocrinol Metab. 1980;51:798–800.
108. McKeel DW Jr, Fowler M, Jacobs LS: The high prevalence of prolactin cell hyperplasia in the human adenohypophysis. In Proceedings of the Endocrine Society 60th Annual Meeting, Miami Beach, 1978, abstract 353.
109. Feigenbaum, SL, Downey, DE, Wilson, CB, et al. Transsphenoidal pituitary resection for preoperative diagnosis of prolactin-secreting pituitary adenoma in women: Long-term follow-up. J Clin Endocrinol Metab. 1996;81:1711–1719.
110. Layman, LC. Hypogonadotropic hypogonadism. Endocrinol Metab Clin North Am. 2007;36:283–296.
111. Gonzalez-Martinez, D, Kim, SH, Hu, Y, et al. Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism. J Neurosci. 2004;24:10384–10392.
112. Dattani, M, Preece, M. Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet. 2004;363:1977–1987.
113. Fahlbusch, R, Muller, OA, Werder, KV. Functional endocrinological disturbances in parasellar processes. Acta Neurochir. 1979;28(Suppl):456–460.
114. Arslanian, SA, Rothfus, WE, Foley, TP, Jr., et al. Hormonal, metabolic, and neuroradiologic abnormalities associated with septo-optic dysplasia. Acta Endocrinol. 1984;107:282–288.
115. Willnow, S, Kiess, W, Butenandt, O, et al. Endocrine disorders in septo-optic dysplasia (De Morsier syndrome): Evaluation and follow up of 18 patients. Eur J Pediatr. 1996;155:179–184.
116. Korsgaard, O, Lindholm, J, Rasmussen, P. Endocrine function in patients with suprasellar and hypothalamic tumours. Acta Endocrinol. 1976;83:1–8.
117. Beck-Peccoz, P, Amr, S, Menezes-Ferreira, M, et al. Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism. N Engl J Med. 1985;312:1085–1090.
118. State, MW, Dykens, EM. Genetics of childhood disorders: XV. Prader-Willi syndrome: genes, brain, and behavior. J Am Acad Child Adolesc Psychiatry. 2000;39:797–800.
119. Cassidy, SB. Prader-Willi syndrome. J Med Genet. 1997;34:917–923.
120. Hoybye, C, Hilding, A, Jacobsson, H, et al. Metabolic profile and body composition in adults with Prader-Willi syndrome and severe obesity. J Clin Endocrinol Metab. 2002;87:3590–3597.
121. Goldstone, AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab. 2004;15:12–20.
122. Beales, PL, Elcioglu, N, Woolf, AS, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:437–446.
123. Beales, PL, Warner, AM, Hitman, GA, et al. Bardet-Biedl syndrome: A molecular and phenotypic study of 18 families. J Med Gent. 1997;34:922–928.
124. Charles, SJ, Moore, AT, Yates, JAW, et al. Alström’s syndrome: Further evidence of autosomal recessive inheritance and endocrinological dysfunction. J Med Genet. 1990;27:590–592.
125. Birkebaek, NH, Patel, L, Wright, NB, et al. Endocrine status in patients with optic nerve hypoplasia: Relationship to midline central nervous system abnormalities and appearance of the hypothalamic-pituitary axis on magnetic resonance imaging. J Clin Endocrinol Metab. 2003;88:5281–5286.
126. Roessmann, U, Velasco, ME, Small, EJ, et al. Neuropathology of “septo-optic dysplasia” (de Morsier syndrome) with immunohistochemical studies of the hypothalamus and pituitary gland. J Neuropathol Exp Neurol. 1987;46:597–608.
127. Yukizane, S, Kimura, Y, Yamashita, Y, et al. Growth hormone deficiency of hypothalamic origin in septo-optic dysplasia. Eur J Pediatr. 1990;150:30–33.
128. Polizzi, A, Pavone, P, Iannetti, P, et al. Septo-optic dysplasia complex: a heterogeneous malformation syndrome. Pediatr Neurol. 2006;34:66–71.
129. Skuse, D, Albanese, A, Stanhope, R, et al. A new stress-related syndrome of growth failure and hyperphagia in children, associated with reversibility of growth-hormone insufficiency. Lancet. 1996;348:353–358.
130. Gilmour, J, Skuse, D, Pembrey, M. Hyperphagic short stature and Prader-Willi syndrome: A comparison of behavioural phenotypes, genotypes and indices of stress. Br J Psychiatry. 2001;179:129–137.
131. Zuber, T, Kelly, J. Pseudocyesis. Am Fam Physician. 1984;30:131–134.
132. Bray, MA, Muneyyirci-Delale, A, Kofinas, GD, et al. Circadian, ultradian, and episodic gonadotropin and prolactin secretion in human pseudocyesis. Acta Endocrinol. 1991;124:501–509.
133. Yen, SSC, Rebar, RW, Quesenberry, W. Pituitary function in pseudocyesis. J Clin Endocrinol Metab. 1976;43:132–136.
134. List, CF, Dowman, CE, Bagchi, BS, et al. Posterior hypothalamic hamartomas and gangliogliomas causing precocious puberty. Neurology. 1958;8:164–174.
135. Diebler, C, Ponsot, G. Hamartomas of the tuber cinereum. Neuroradiology. 1983;25:93–101.
136. Comite, F, Psescovitz, OH, Rieth, KG. Luteinizing hormone-releasing hormone analog treatment of boys with hypothalamic hamartoma and true precocious puberty. J Clin Endocrinol Metab. 1984;59:888–892.
137. Nguyen, D, Singh, S, Zaatreh, M, et al. Hypothalamic hamartomas: seven cases and review of the literature. Epilepsy Behav. 2003;4:246–258.
138. Arita, K, Kurisu, K, Kiura, Y, et al. Hypothalamic hamartoma. Neurol Med Chir (Tokyo). 2005;45:221–231.
139. Hall, JG, Pallister, PD, Clarren, SK, et al. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly: A new syndrome? Part I: Clinical, causal, and pathogenetic considerations. Am J Med Genet. 1980;7:47–74.
140. Clarren, SK, Alvord, EC, Jr., Hall, JG. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly: A new syndrome? Part II: Neuropathological considerations. Am J Med Genet. 1980;7:75–83.
141. Biesecker, LG, Abbott, M, Allen, J, et al. Report from the Workshop on Pallister-Hall Syndrome and Related Phenotypes. Am J Med Genet. 1996;65:76–81.
142. Biesecker, LG, Graham, JM, Jr. Pallister-Hall syndrome. J Med Genet. 1996;33:585–589.
143. Boudreau, EA, Liow, K, Frattali, CM, et al. Hypothalamic hamartomas and seizures: distinct natural history of isolated and Pallister-Hall syndrome cases. Epilepsia. 2005;46:42–47.
144. Pradilla, G, Jallo, G. Arachnoid cysts: case series and review of the literature. Neurosurg Focus. 2007;22:E7.
145. Kirollos, RW, Javadpour, M, May, P, et al. Endoscopic treatment of suprasellar and third ventricle-related arachnoid cysts. Childs Nerv Syst. 2001;17:713–718.
146. Bihan, H, Christozova, V, Dumas, JL, et al. Sarcoidosis: clinical, hormonal, and magnetic resonance imaging (MRI) manifestations of hypothalamic-pituitary disease in 9 patients and review of the literature. Medicine (Baltimore). 2007;86:259–268.
147. Porter, N, Beynon, HL, Randeva, HS. Endocrine and reproductive manifestations of sarcoidosis. QJM. 2003;96:553–561.
148. Makras, P, Alexandraki, KI, Chrousos, GP, et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab. 2007;18:252–257.
149. Amato, MC, Elias, LL, Elias, J, et al. Endocrine disorders in pediatric-onset Langerhans Cell Histiocytosis. Horm Metab Res. 2006;38:746–751.
150. Rutter, MM, Rose, SR. Long-term endocrine sequelae of childhood cancer. Curr Opin Pediatr. 2007;19:480–487.
151. Gurney, JG, Kadan-Lottick, NS, Packer, RJ, et al. Endocrine and cardiovascular late effects among adult survivors of childhood brain tumors: Childhood Cancer Survivor Study. Cancer. 2003;97:663–673.
152. Agha, A, Thompson, CJ. Anterior pituitary dysfunction following traumatic brain injury (TBI). Clin Endocrinol (Oxf). 2006;64:481–488.
153. Schneider, HJ, Kreitschmann-Andermahr, I, Ghigo, E, et al. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: a systematic review. JAMA. 2007;298:1429–1438.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 5
Hypothalamic Syndromes
The hypothalamus houses multiple nuclei along with afferent and efferent nerve fibers that connect the hypothalamus to the various portions of the brain and brainstem. It is divided into four regions: from anterior to posterior, the preoptic, supraoptic, tuberal, and mamillary regions; and three zones: laterally from the third ventricle, the periventricular, medial, and lateral1–4 zones (Table 5-1, Figs. 5-1 and 5-2).
Table 5-1
From Braunstein GD: The hypothalamus. In Melmed S (ed): The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.
FIGURE 5-1 Schematic representation of lateral brain section demonstrating hypothalamic nuclei. Dashed lines represent the frontal (coronal) section planes illustrated in Figures 5-2 and 5-3. 1, preoptic nucleus; 2, paraventricular nucleus; 3, anterior hypothalamic areas; 4, supraoptic nucleus; 5, arcuate nucleus; 6, dorsal hypothalamic area; 7, dorsomedial nucleus; 8, ventromedial nucleus; 9, posterior hypothalamic area; 10, mamillary body; 11, optic chiasm; 12, optic nerve. (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
FIGURE 5-2 Frontal (coronal) sections of the hypothalamic regions. A, Preoptic region (frontal section plane 1 in Fig. 5-1). B, Supraoptic region (frontal section plane 2 in Fig. 5-1). C, Tuberal region (frontal section plane 3 in Fig. 5-1). D, Mamillary region (frontal section plane 4 in Fig. 5-1). (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
The hypothalamus is responsible for many of the body’s homeostatic mechanisms, including water metabolism, temperature regulation, appetite control, the sleep/wake cycle, circadian rhythms, and control of the sympathetic and parasympathetic nervous systems. In addition, this area has activity in regard to emotional expression, behavior, and memory. Finally, the hypothalamus is essential to the neuroendocrine control of anterior pituitary function. Table 5-2 lists the various functions of the hypothalamus, the hypothalamic nuclei or hypothalamic regions that have been identified as being responsible for these functions, and the disorders that result from either destructive or stimulatory lesions in or around the nuclei or region.1,4–25
Table 5-2
Hypothalamic Functions, the Nuclei or Regions Involved with the Specific Functions, and the Disorders Resulting from Stimulatory or Destructive Lesions in the Regions
SIADH, Syndrome of inappropriate secretion of antidiuretic hormone.
Hypothalamic Disorders: Pathophysiologic Principles
First, the small overall size of the hypothalamus and the close association of the nuclei and nerve tracts mean that a variety of different pathologic processes may give rise to the same signs and symptoms of neurologic and hypothalamic dysfunction.4 The spectrum of disorders that can affect the hypothalamus is shown in Table 5-3. Tumors, infiltrative disorders, and infections, among other conditions, frequently give rise to headaches, neuro-ophthalmologic disorders, pyramidal tract or sensory nerve dysfunction, extrapyramidal cerebellar signs, and recurrent vomiting.9,10 Other common manifestations include gonadal dysfunction (either hypogonadism or precocious puberty), diabetes insipidus, somnolence, dysthermia, and evidence of a caloric imbalance (either with hyperphagia and obesity or anorexia with emaciation).9,10
Table 5-3
Causes of Hypothalamic Dysfunction
Congenital
Tumors
Infiltrative
Immunologic
Nutritional, Metabolic
Degenerative
Infectious
Vascular
Trauma
Functional
Other
DIDMOAD, Diabetes insipidus, diabetes mellitus, optic atrophy, deafness.
Modified from Braunstein GD: The hypothalamus. In Melmed S (ed): The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.
Fourth, most lesions that result in chronic hypothalamic syndromes involve more than one nucleus. As can be seen in Table 5-2, most of the hypothalamic functions are controlled by more than one nucleus, and this redundancy allows some degree of compensation should one nucleus be affected. In addition, most of the nuclei are paired, and destruction of a single nucleus may not be sufficient to result in a clinical syndrome. Thus lesions that affect the basal tuberal region of the hypothalamus (pituitary adenomas with suprasellar extension, optic gliomas, and craniopharyngiomas), are multiple (granulomatous disorders, metastatic tumors, infiltrative disease), or cause enlargement of the third ventricle (aqueductal stenosis, colloid cysts, pinealomas, germ cell tumors, midbrain gliomas) will more likely result in clinical hypothalamic dysfunction than will disorders affecting the more lateral portions of the hypothalamus.
Manifestations of Hypothalamic Disease
Central Diabetes Insipidus
Complete or partial central diabetes insipidus results from (1) destruction of the antidiuretic hormone (ADH)-producing magnocellular neurons in the supraoptic and paraventricular nuclei or (2) interruption of the transport of ADH through their axons, which terminate in the pituitary stalk and posterior pituitary. Diabetes insipidus is relatively common in patients with chronic hypothalamic disorders, being found in approximately 35% of such patients.9,10 It also is frequently found in patients with acute insults to the hypothalamus or pituitary stalk, as is seen in vascular accidents and neurosurgical trauma. Obesity and hypogonadism frequently are present in patients with diabetes insipidus due to tumors or infiltrative disorders (Fig. 5-3).
FIGURE 5-3 Clinical findings associated with hypothalamic lesions located at various anatomic sites. Clinicopathologic correlation based on multiple studies.15–25A, Corresponds to region depicted in Figure 5-2A. B, Corresponds to region depicted in Figure 5-2C. C, Corresponds to section depicted in Figure 5-2D. (From Braunstein GD: The hypothalamus. In Melmed S [ed]: The Pituitary, 2nd ed. Cambridge, MA: Blackwell Scientific, 2002, pp 317–348.)
The majority of patients with diabetes insipidus have idiopathic or familial diabetes insipidus associated with gliosis of the supraoptic and paraventricular nuclei.26 Approximately one third of patients with idiopathic diabetes insipidus have detectable anti-ADH–producing cell antibodies, suggesting an autoimmune cause.27 Autosomal-recessive, X-linked-recessive, and autosomal-dominant forms of familial diabetes insipidus have been described. In the more common autosomal-dominant form, nucleotide deletions or substitutions in the ADH gene on chromosome 20 have been identified.28 The DIDMOAD syndrome (Wolfram’s syndrome) represents a rare autosomal-recessive form of central diabetes insipidus (DI) associated with type 1 diabetes mellitus (DM), optic atrophy (OA), bilateral sensorineural deafness (D), and occasionally ataxia and autonomic neurogenic bladder.29 Diabetes insipidus is a frequent manifestation of suprasellar and pineal germinomas, sarcoidosis, lymphocytic infundibuloneurohypophysitis, and the chronic disseminated form of Langerhans’ cell histiocytosis.30–37
Adipsic or Essential Hypernatremia
Adipsic hypernatremia occurs when the osmoreceptors that are present in the anterior medial and anterior lateral preoptic regions are damaged. Affected patients have an impaired thirst mechanism, which results in insufficient fluid intake despite the hypernatremia. Although most of the affected patients have partial diabetes insipidus, their extracellular fluid volume remains normal, and they are not dehydrated. Therefore, they exhibit chronic elevations of serum sodium but normal blood pressure, pulse rate, serum creatinine, and creatinine clearance and can release ADH and concentrate their urine during fluid deprivation. When serum sodium concentrations are less than 160 mmol/L, few symptoms are present. However, between 160 and 180 mmol/L, patients may have fatigue, weakness, lethargy, muscle tenderness, cramps, anorexia, depression, and irritability; at 180 mmol/L, stupor and coma may be present. Close to half of these patients have hypothalamic obesity, and almost three fourths demonstrate some degree of anterior pituitary hormone deficiency.6,9,10,13,38,39
Essential hypernatremia has been described with a variety of lesions, including craniopharyngiomas, suprasellar germinomas, optic nerve gliomas, pineal tumors, Langerhans’ cell histiocytosis, sarcoidosis, trauma, hydrocephalus, cysts, inflammatory conditions, ruptured aneurysms of the anterior communicating artery, and toluene exposure.38–40 The Hayek-Peake syndrome is the association of essential hypernatremia with hypodipsia, obesity, lethargy, increased perspiration, central hypoventilation, hyperprolactinemia, hypothyroidism, and hyperlipidemia without an identifiable structural hypothalamic defect.41
Syndrome of Inappropriate Secretion of Antidiuretic Hormone
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) is characterized by serum hyponatremia and hypo-osmolarity, with an inappropriately elevated urine osmolarity, in a patient with normal renal, adrenal, and thyroid function and no clinical evidence of intravascular or extracellular fluid volume expansion. The clinical symptoms depend on the rate of decrease of serum sodium, as well as the absolute serum sodium concentration. At serum sodium levels greater than 120 mmol/L, symptoms are generally mild and nonspecific and include anorexia, nausea, headache, weakness, and lethargy. At less than 120 mmol/L, these symptoms are accompanied by nausea, vomiting, and mental confusion; at very low levels, by seizure and coma.42 The syndrome is found with a variety of intracranial abnormalities, including head trauma, intracranial bleeding, meningitis, encephalitis, neurosurgery, hydrocephalus, acute intermittent porphyria, craniopharyngiomas, germinomas, and pinealomas.6,13,30 An idiopathic form has been described in young women who exhibit menstrual irregularities, have enlarged lateral ventricles, and have SIADH cyclically. No structural defect has been described in these patients.4
Reset Osmostat
The osmoreceptors, located in the circumventricular organs of the lamina terminalis, may become “reset” in some patients with tuberculosis, malnutrition, quadriplegia, and psychosis. The osmoreceptors activate release of ADH at a lower serum osmolality than normal and appropriately decrease ADH release when the serum osmolality falls further.42
Cerebral Salt Wasting
Hyponatremia and the other manifestations of SIADH also co-occur in the syndrome of cerebral salt wasting, which is primarily seen in postoperative neurosurgical patients treated for subarachnoid bleeding, intracranial aneurysms, or following head injury. In contrast to SIADH in which patients are euvolemic or have an expansion of their effective arterial blood volume, patients with cerebral salt wasting are hypovolemic due to renal salt loss. The salt wasting and hypovolemia are felt to be the result of disruption of the normal sympathetic nervous system input into the kidneys and/or relapse of a natriuretic factor from the brain.43
Dysthermia
The warm receptors present in the preoptic anterior hypothalamus are stimulated by an increase in the temperature of the blood. Together with signals from peripheral warm receptors that respond to an increase in external temperature, the afferent signals travel through the median forebrain bundle to the lateral portion of the posterior hypothalamus, which leads to vasodilation and sweating to dissipate heat. Conversely, stimulation of the preoptic anterior hypothalamic cold receptors through a decrease in temperature of the blood, or stimulation of the peripheral cold receptors through a decrease in ambient temperature, results in medial neurons in the posterior hypothalamus activating heat production through muscular shivering and heat conservation through vasoconstriction.5,6
Acute injury to the anterior hypothalamic and preoptic areas may result in a rapid temperature elevation (as high as 41°C) associated with tachycardia and unconsciousness from failure of the heat-dissipating mechanisms to function while heat production continues. Chronic hyperthermia may be secondary to lesions in the tuberoinfundibular region. In contrast to patients with elevated temperature due to the inflammation of infections, these patients generally do not experience malaise and paradoxically may have peripheral vasoconstriction.1,5,6,9,11
Wolff and colleagues44 described a syndrome of hyperthermia associated with shaking chills, fever, hypertension, vomiting, and peripheral vasoconstriction that occurred cyclically at 3-week intervals, without a pathologic lesion in the hypothalamus being found. Similar paroxysms of hyperthermia have been noted in other patients without the cyclicity, and together these episodes may represent a variant of diencephalic epilepsy.6,44,45
Between 0.02% and 2.4% of patients receiving neuroleptic drugs develop the neuroleptic malignant syndrome (NMS), which is characterized by hyperthermia to 38°C or higher; severe extrapyramidal signs, including “lead-pipe” muscle rigidity and tremor; signs of autonomic nervous system dysfunction such as pallor, tachycardia, arrhythmias, blood pressure lability, and diaphoresis; and changes in mental status, including mutism, delirium, and coma.46 All antipsychotic medications have been reported to cause NMS, and most evidence suggests that disruption of dopamine neurotransmission by neuroleptic-induced dopamine receptor blockade is the major pathophysiologic abnormality in susceptible individuals. Indeed, the greater the potency of the neuroleptic in regard to its dopamine D2-receptor antagonism activity, the greater the frequency of NMS occurrence.46 NMS is successfully treated with a variety of dopamine agonists. Injury to the preoptic medial and tuberal nuclei has been demonstrated at autopsy, as has a depletion of hypothalamic norepinephrine concentrations.47 The syndrome generally begins within 2 weeks of initiating the neuroleptic and evolves over a 24- to 72-hour period. The most common complication is rhabdomyolysis, which may result in myoglobinuria and acute renal failure. The mortality of this syndrome is currently less than 10%, which reflects increased recognition of the disorder and initiation of prompt therapy.46
The serotonin syndrome is closely related to NMS and presents with the triad of altered mental status (somnolence, confusion, agitation, seizures, and coma), autonomic instability (fever, diaphoresis, tachycardia, hypo- or hyperthermia, mydriasis), and abnormal neuromuscular activity (myoclonus, rigidity, hyperreflexia). Any drug or combination of drugs which elevates the concentration of serotonin in the central nervous system can cause the syndrome. These include selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants, monamine oxidase inhibitors, cocaine, and amphetamines. These drugs either directly or indirectly activate thermogenesis via the hypothalamus.46
Hypothermia
Large, destructive lesions of the anterior or posterior hypothalamus may result in inability to generate heat through vasoconstriction and muscular shivering. This occurs in 10% to 15% of patients with a variety of hypothalamic lesions, especially neoplasms, infiltrative disorders, and infections.9,10,13 It also has been noted in patients with Parkinson’s disease and Wernicke’s encephalopathy, which are associated with lesions in the posterior hypothalamus and mamillary bodies, respectively.48,49
Diencephalic autonomic epilepsy refers to episodic or paroxysmal hypothermia during which the body temperature decreases to 32°C or less over minutes to days, along with evidence of autonomic nervous system dysfunction, including flushing, sweating, hypotension, bradycardia, salivation, lacrimation, pupillary dilation, Cheyne-Stokes respiration, nausea, vomiting, asterixis, ataxia, and obtundation.6,15,50–53 Electroencephalographic (EEG) abnormalities occur during the episodes. Autopsy studies have shown gliosis and loss of the arcuate nucleus and premamillary area in some patients, whereas others have been found to have tumors involving the floor and lower portion of the third ventricle.15,50 The corpus callosum has been found to be absent in approximately half of patients with episodic hypothermia; these individuals may also exhibit diabetes insipidus, reset osmostat, growth hormone deficiency, hypogonadism, or precocious puberty (Shapiro’s syndrome).54
Poikilothermia
When both the heat-loss and heat-conserving homeostatic mechanisms are impaired, wide fluctuations of body temperature might take place without the patient’s experiencing thermal discomfort. This condition, known as poikilothermia, is found with both anterior and posterior hypothalamic destruction, as well as in patients with large lesions that may involve the posterior hypothalamus and rostral mesencephalon.6,9,10 Rarely, patients with Wernicke’s encephalopathy may experience poikilothermia.6
Disorders of Appetite Control and Caloric Balance
Approximately 25% of patients with structural hypothalamic lesions exhibit hyperphagia and obesity.9,10 Usually, the patients have lesions involving a large portion of the hypothalamus, although bilateral destruction of only the ventromedial nucleus may lead to hypothalamic obesity.1,9,10,17,18,22,25,55 The majority of patients harbor a neoplasm, especially craniopharyngioma, with a minority having inflammatory or granulomatous processes, a history of trauma, or infiltrative disorders.55 Common clinical findings in these patients include headaches, visual abnormalities, hypogonadism, diabetes insipidus, and somnolence. Less commonly, behavioral abnormalities, such as antisocial behavior or sham rage, and seizures may be present.55 Hypothalamic obesity also is found with defects of the leptin and leptin receptor genes, the melanocortin 4 receptor gene, and the proopiomelanocortin gene.56–59
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
