[level-membership-for-basic-science-category]
Chapter 24 Vasodilators and Nitric Oxide Synthase
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| CHF | Congestive heart failure |
| NO | Nitric oxide |
| PDE | Phosphodiesterase |
Therapeutic Overview
A summary of the uses of these compounds is provided in the Therapeutic Overview Box.
| Therapeutic Overview | |
|---|---|
| Clinical Problem | Goal of Drug Intervention |
| Hypertension | Decrease blood pressure |
| Congestive heart failure | Increase cardiac output and decrease O2 consumption |
| Coronary artery insufficiency | Increase effective flow through coronary arteries and decrease O2 consumption by the heart |
| Peripheral vascular disease | Increase blood flow to the ischemic area |
| Hemostasis | Slow bleeding into surgical field |
| Impotence | Increased erectile function |
Mechanisms of Action
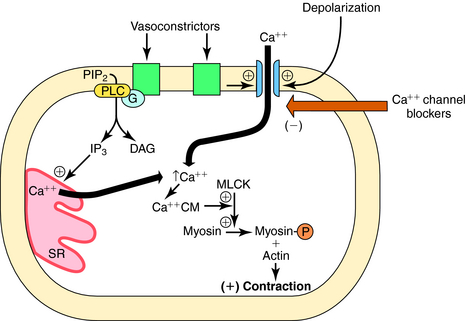
Vasodilators act at different sites in the cascade of events that couple excitation of vascular smooth muscle to contraction (Table 24-1). Thus, to understand the mechanisms of action of these agents and their uses, it is critical to be familiar with the processes involved in the contraction of smooth muscle cells.
Vascular Smooth Muscle Cell Contraction and Relaxation
Smooth muscle contraction is ultimately regulated by intracellular Ca++ concentrations. Excitation-contraction coupling occurs by several mechanisms. Depolarization of vascular smooth muscle cell membranes allows Ca++ entry through voltage-gated channels. When these channels open, Ca++ flows into the cell down its concentration gradient (Fig. 24-1). Activation of receptors for certain vasoconstrictor substances can also open Ca++ channels. In addition to elevating intracellular Ca++ by opening channels, receptor activation can also increase intracellular Ca++ by activating phospholipase C, which hydrolyzes phosphatidylinositol 4,5-bisphosphate to diacylglycerol and inositol 1,4,5-trisphosphate, both of which contribute to contraction (see Chapters 1 and Chapters 9). Inositol trisphosphate releases Ca++ from intracellular stores, whereas diacylglycerol activates protein kinase C, an enzyme that phosphorylates several substrates involved in the contractile response. When Ca++ enters the smooth muscle cell, it combines with calmodulin, and the Ca++-calmodulin complex activates myosin light-chain kinase, which in turn phosphorylates the myosin light chain, promoting the interaction of myosin and actin and cross-bridge formation, leading to contraction. Because Ca++-channel antagonists block or limit the entry of Ca++ through voltage-gated channels, these drugs dilate blood vessels that have some endogenous degree of vasoconstrictor tone, or limit vasoconstriction caused by endogenous or exogenous vasoactive stimulants (see Chapter 20).
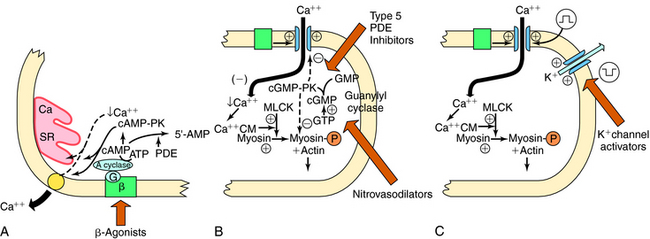
Increases in cyclic adenosine monophosphate (cAMP) also lead to smooth muscle relaxation. Increased cAMP activates cAMP-dependent protein kinase A, which phosphorylates several proteins, leading to decreased intracellular Ca++ as a consequence of reduced influx, enhanced uptake into the sarcoplasmic reticulum, and/or enhanced extrusion through the cell membrane (Fig. 24-2, A). Myosin light-chain kinase may also be phosphorylated, leading to enzyme inactivation and inhibition of contraction. Because adrenergic β receptor agonists such as isoproterenol activate adenylyl cyclase and increase cAMP, these agents lead to relaxation of vascular smooth muscle. Similarly, drugs that inhibit phosphodiesterases (PDEs), which metabolize cAMP and cyclic guanosine monophosphate (cGMP), promote smooth muscle relaxation. Drugs such as papaverine may act by this mechanism.
 -hyperpolarization,
-hyperpolarization,  -depolarization.
-depolarization.Nitrovasodilators are organic nitrates that provide a source of nitric oxide (NO), which activates a soluble guanylyl cyclase in vascular smooth muscle, causing an increase in intracellular cGMP, which activates a cGMP-dependent protein kinase (see Fig. 24-2, B). This kinase leads to the phosphorylation of proteins, which results in smooth muscle relaxation. Although the cellular mechanisms involved are not entirely clear, they may include decreased entry of Ca++ through membrane channels, inhibition of phosphatidylinositol hydrolysis, stimulation of Ca++ pumps to extrude or sequester Ca++, and decreased sensitivity of contractile proteins to Ca++.
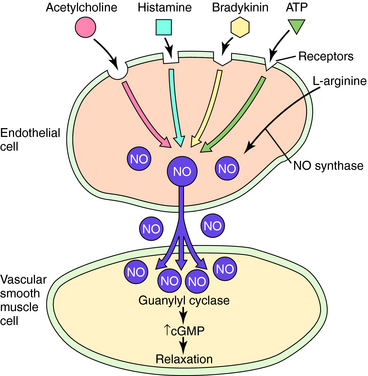
NO may be the final common mediator for several vascular smooth muscle relaxants. In addition to nitrovasodilators, which may form NO or a related molecule, some endogenous agents that cause vasodilation do so in whole or in part by releasing NO from endothelial cells. Included among these are bradykinin, histamine, adenosine triphosphate, adenosine diphosphate, substance P, and acetylcholine (Fig. 24-3). Because the endothelium is an important structure for communicating between the blood and the vascular media, it has the potential to be an important target for vasodilator therapy.
Agents such as minoxidil cause vasodilation by activating K+ channels in vascular smooth muscle. The increased K+ conductance results in hyperpolarization of the cell membrane and relaxation (see Fig. 24-2, C). The hyperpolarizing effect also counteracts stimulants that act by depolarization, promoting Ca++ entry.
Phosphodiesterase Type 5 Inhibitors
Sildenafil, tadalafil, and vardenafil are selective inhibitors of cGMP-specific PDE type 5, which is found in high concentrations in the penile corpus cavernosum and is responsible for the degradation of cGMP. NO release during sexual stimulation activates guanylyl cyclase, resulting in increased levels of cGMP, which causes smooth muscle relaxation in the corpus cavernosum, allowing the inflow of blood. The PDE5 inhibitors prevent the catabolism of cGMP, thereby prolonging its actions (see Fig. 24-2, B). These drugs have no direct relaxant effect on isolated human corpus cavernosum and, at recommended doses, have no effect in the absence of sexual stimulation. Rather, they cause specific vasodilation in the presence of appropriate sexual stimulation and have become increasingly popular for treating erectile dysfunction in men.
Although hydralazine was one of the first antihypertensive drugs, its mechanism of action has not been definitively elucidated. It is a vasodilator with effects on arterioles and not venules. Hydralazine has been shown to decrease intracellular Ca++ concentrations, and recent evidence suggests that this action may be due to its ability to inhibit the inositol 1,4,5-trisphosphate-induced release of Ca++ from the sarcoplasmic reticulum in vascular smooth muscle cells (see Fig. 24-1). Additional sites of action have also been proposed (see Table 24-1).
Pharmacokinetics
Selected pharmacokinetic parameter values for the nitrovasodilators are summarized in Table 24-2. Organic nitrates are almost completely absorbed from the gastrointestinal tract and fairly completely absorbed from the buccal mucosa. After sublingual administration, peak plasma concentrations are achieved in 1 to 2 minutes. Absorption is much slower with topical ointments and transdermal patches, and plasma concentrations attained with transdermal preparations are lower and more variable than those obtained with ointments. The nitrates are metabolized in the liver by glutathione nitrate reductase (e.g., nitroglycerin is rapidly converted to inorganic nitrite and to denitrated metabolites). Isosorbide dinitrate is also metabolized by hepatic glutathione reductase and converted to inactive products and to an active metabolite, 5-isosorbide mononitrate, which may account for its longer duration of antianginal activity.
| Drug | Route of Administration | Remarks |
|---|---|---|
| Nitroglycerin | Sublingual | Onset 2-4 min, duration 30-60 min depending on patient activity, minimal first-pass effect, all organic nitrates metabolized by liver |
| Oral | Onset 10-20 min, duration 2-3 hrs, significant first-pass effect | |
| IV | Immediate onset, used to maintain stable blood concentration | |
| Transdermal | Discs or patches: slower onset, 10-18 hrs variable duration; ointment less variable, duration 20-24 hrs, for nocturnal angina | |
| Aerosol | Rapid onset, difficult to control | |
| Isosorbide dinitrate* | Sublingual | Similar in onset to nitroglycerin, longer duration (2-4 hrs) |
| Oral | Onset 10-20 min, duration 4-8 hrs | |
| Erythrityl tetranitrate | Sublingual | Onset 3-5 min, duration 1-2 hrs |
| Pentaerythritol tetranitrate | Oral | Onset 15-30 min, duration 4-8 hrs |
IV; intravenous
* Active metabolite; oral preparations: onset varies with dose, and duration depends on extent of first-pass metabolism.
Relationship of Mechanisms of Action to Clinical Response
Nitrovasodilators and Angina Pectoris
The goal of therapy in coronary artery disease is to reduce pain and increase the patient’s exercise tolerance. This can be accomplished by administration of organic nitrates, the prototype of which is nitroglycerin. Organic nitrates are the mainstay of antianginal therapy, used effectively for this purpose for approximately 100 years.
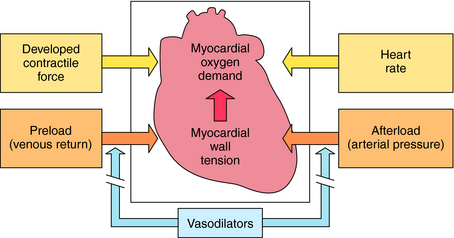
The pharmacological properties of the organic nitrates that make them useful depend on the underlying cause of the angina. If pain is associated with atherosclerosis, the chief benefit arises from actions on the peripheral circulation and not on coronary vessels. Nitrates produce vasodilation of the venous vasculature. Dilation of venous capacitance vessels diminishes venous return to the heart, reducing ventricular volume and pressure. This decreases ventricular wall tension, a major contributor to the O2 demands of the heart (Fig. 24-4). Thus, by decreasing preload on the heart, O2 needs diminish, and demand is consistent with supply.
Other Drugs for Angina Pectoris
Additional drugs used for the treatment of angina pectoris are adrenergic β receptor antagonists and Ca++ channel-blocking drugs (see Chapters 11 and 20). The beneficial effect of β receptor blockers in angina is their ability to decrease O2 demands of the heart. These drugs decrease heart rate and ventricular contractile force (see Chapter 11). Heart rate and contractile force, together with ventricular wall tension, are major determinants of myocardial O2 demand (see Fig. 24-4). In addition, chronic therapy with β-blockers reduces blood pressure. Thus these drugs also decrease afterload. Unlike organic nitrates, β-blockers are not used to terminate an acute attack of angina pectoris but rather to increase exercise tolerance of the patient and to reduce the frequency of anginal attacks.
Vasodilators and Congestive Heart Failure
Recent studies indicate that vasodilator therapy is extremely effective in treatment of CHF. Drugs used more frequently for treating CHF are those that increase the force of cardiac contraction (see Chapter 23) or minimize Na+ and H2O retention (see Chapter 21). Cardiac glycosides affect only two of several determinants of cardiac function (e.g., contractility and rate).
Patients who are refractory to cardiac glycosides frequently do well if treated with vasodilators. Among the direct-acting vasodilators used to treat CHF are the nitrates, hydralazine, minoxidil, and sodium nitroprusside. Angiotensin-converting enzyme (ACE) inhibitors such as captopril enalapril, and lisinopril (see Chapter 20 and 23) are also of proven effectiveness, as is the adrenergic α1 receptor antagonist prazosin. Long-term treatment with hydralazine alone is only minimally effective in treatment of CHF, but the combination of hydralazine with a nitrate, isosorbide dinitrate, effectively decreases mortality. Minoxidil is also generally not very effective when used alone.
Pharmacovigilance: Side-Effects, Clinical Problems, and Toxicity
Another concern is that by decreasing peripheral vascular resistance, vasodilators cause reflex activation of the sympathetic nervous system (see Chapter 11). Enhanced sympathetic activity can lead to unwanted cardiac effects. The release of renin from juxtaglomerular cells is also enhanced by reflex sympathetic nerve stimulation caused by vasodilators. To counteract this action, adrenergic β receptor antagonists are frequently administered in conjunction with direct-acting vasodilators.
Major side effects of vasodilators are summarized in the Clinical Problems Box.
Phosphodiesterase Type 5 Inhibitors
Type 5 PDE inhibitors can cause abnormalities in color vision, although this is most common with sildenafil.
Inhibition of PDE5 in other tissues, such as esophageal smooth muscle, can result in a reduced tone of the esophageal sphincter and increased gastroesophageal reflux, as well as dyspepsia. In addition, as a consequence of PDE type 5 inhibition in the brain, these compounds have been reported to result in emotional, neurological, and psychological side effects. They also have the potential to cause hypotension.
New Horizons
This compound is released in response to physiological challenges, such as hypoxia or stress, or by endogenous hormones, such as angiotensin. Endothelin-1 initially dilates smooth muscle but subsequently produces an intense, long-lasting vasoconstriction. Two types of endothelin receptors have been characterized (ETA and ETB). Current evidence indicates that endothelin does not act as a circulating hormone but rather as an autocrine or paracrine substance. Abnormally high levels may play a pathogenic role in some forms of vasospasm. Antagonists of endothelin receptors have been developed, and their therapeutic potential is being investigated.
Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271-324.
Vanhoutte PM. Endothelial control of vasomotor function: From health to coronary disease. Circ J. 2003;67:572-575.
Webb RC. Smooth muscle contraction and relaxation. Adv Physiol Educ. 2003;27:201-206.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 24 Vasodilators and Nitric Oxide Synthase
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| CHF | Congestive heart failure |
| NO | Nitric oxide |
| PDE | Phosphodiesterase |
Therapeutic Overview
A summary of the uses of these compounds is provided in the Therapeutic Overview Box.
| Therapeutic Overview | |
|---|---|
| Clinical Problem | Goal of Drug Intervention |
| Hypertension | Decrease blood pressure |
| Congestive heart failure | Increase cardiac output and decrease O2 consumption |
| Coronary artery insufficiency | Increase effective flow through coronary arteries and decrease O2 consumption by the heart |
| Peripheral vascular disease | Increase blood flow to the ischemic area |
| Hemostasis | Slow bleeding into surgical field |
| Impotence | Increased erectile function |
Mechanisms of Action
Vasodilators act at different sites in the cascade of events that couple excitation of vascular smooth muscle to contraction (Table 24-1). Thus, to understand the mechanisms of action of these agents and their uses, it is critical to be familiar with the processes involved in the contraction of smooth muscle cells.
Vascular Smooth Muscle Cell Contraction and Relaxation
Smooth muscle contraction is ultimately regulated by intracellular Ca++ concentrations. Excitation-contraction coupling occurs by several mechanisms. Depolarization of vascular smooth muscle cell membranes allows Ca++ entry through voltage-gated channels. When these channels open, Ca++ flows into the cell down its concentration gradient (Fig. 24-1). Activation of receptors for certain vasoconstrictor substances can also open Ca++ channels. In addition to elevating intracellular Ca++ by opening channels, receptor activation can also increase intracellular Ca++ by activating phospholipase C, which hydrolyzes phosphatidylinositol 4,5-bisphosphate to diacylglycerol and inositol 1,4,5-trisphosphate, both of which contribute to contraction (see Chapters 1 and Chapters 9). Inositol trisphosphate releases Ca++ from intracellular stores, whereas diacylglycerol activates protein kinase C, an enzyme that phosphorylates several substrates involved in the contractile response. When Ca++ enters the smooth muscle cell, it combines with calmodulin, and the Ca++-calmodulin complex activates myosin light-chain kinase, which in turn phosphorylates the myosin light chain, promoting the interaction of myosin and actin and cross-bridge formation, leading to contraction. Because Ca++-channel antagonists block or limit the entry of Ca++ through voltage-gated channels, these drugs dilate blood vessels that have some endogenous degree of vasoconstrictor tone, or limit vasoconstriction caused by endogenous or exogenous vasoactive stimulants (see Chapter 20).
Increases in cyclic adenosine monophosphate (cAMP) also lead to smooth muscle relaxation. Increased cAMP activates cAMP-dependent protein kinase A, which phosphorylates several proteins, leading to decreased intracellular Ca++ as a consequence of reduced influx, enhanced uptake into the sarcoplasmic reticulum, and/or enhanced extrusion through the cell membrane (Fig. 24-2, A). Myosin light-chain kinase may also be phosphorylated, leading to enzyme inactivation and inhibition of contraction. Because adrenergic β receptor agonists such as isoproterenol activate adenylyl cyclase and increase cAMP, these agents lead to relaxation of vascular smooth muscle. Similarly, drugs that inhibit phosphodiesterases (PDEs), which metabolize cAMP and cyclic guanosine monophosphate (cGMP), promote smooth muscle relaxation. Drugs such as papaverine may act by this mechanism.
Nitrovasodilators are organic nitrates that provide a source of nitric oxide (NO), which activates a soluble guanylyl cyclase in vascular smooth muscle, causing an increase in intracellular cGMP, which activates a cGMP-dependent protein kinase (see Fig. 24-2, B). This kinase leads to the phosphorylation of proteins, which results in smooth muscle relaxation. Although the cellular mechanisms involved are not entirely clear, they may include decreased entry of Ca++ through membrane channels, inhibition of phosphatidylinositol hydrolysis, stimulation of Ca++ pumps to extrude or sequester Ca++, and decreased sensitivity of contractile proteins to Ca++.
NO may be the final common mediator for several vascular smooth muscle relaxants. In addition to nitrovasodilators, which may form NO or a related molecule, some endogenous agents that cause vasodilation do so in whole or in part by releasing NO from endothelial cells. Included among these are bradykinin, histamine, adenosine triphosphate, adenosine diphosphate, substance P, and acetylcholine (Fig. 24-3). Because the endothelium is an important structure for communicating between the blood and the vascular media, it has the potential to be an important target for vasodilator therapy.
Agents such as minoxidil cause vasodilation by activating K+ channels in vascular smooth muscle. The increased K+ conductance results in hyperpolarization of the cell membrane and relaxation (see Fig. 24-2, C). The hyperpolarizing effect also counteracts stimulants that act by depolarization, promoting Ca++ entry.
Phosphodiesterase Type 5 Inhibitors
Sildenafil, tadalafil, and vardenafil are selective inhibitors of cGMP-specific PDE type 5, which is found in high concentrations in the penile corpus cavernosum and is responsible for the degradation of cGMP. NO release during sexual stimulation activates guanylyl cyclase, resulting in increased levels of cGMP, which causes smooth muscle relaxation in the corpus cavernosum, allowing the inflow of blood. The PDE5 inhibitors prevent the catabolism of cGMP, thereby prolonging its actions (see Fig. 24-2, B
[/not-level-membership-for-basic-science-category]
