Chapter 55 Adjuvant Antineoplastic Drugs
| Abbreviations | |
|---|---|
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| ATP | Adenosine triphosphate |
| EGFR | Epidermal growth factor receptor |
| HPV | Human papilloma virus |
| IFN-α | Interferon alpha |
| IL-2 | Interleukin-2 |
| IV | Intravenous |
| mRNA | Messenger ribonucleic acid |
| NK | Natural killer (cell) |
| PDGFR | Platelet-derived growth factor receptor |
| VEGFR | Vascular endothelial growth factor receptor |
Therapeutic Overview
Immunotherapy, vaccines, drugs affecting angiogenesis, specific tumor growth factor receptor antagonists, and other forms of biological therapies are becoming more common in the treatment of neoplastic disease. Traditional chemotherapy, as discussed in Chapter 54, is aimed at destruction of rapidly dividing tumor cells. However, newer approaches involve drugs that are aimed at more specific cellular targets on the cancer cells with reduced effects on normal cells. Thus these agents typically have fewer side effects than the traditional chemotherapeutic agents. In addition to being administered alone for the treatment of certain cancers, many of these compounds are also used in combination with traditional chemotherapeutic regimens.
Mechanisms of Action
| Therapeutic Overview |
|---|
| Targets |
| The immune system |
| Growth factors and their receptors |
| Intracellular signaling molecules |
| Angiogenesis |
| Agents |
| Hormones and antihormones |
| Cytokines |
| Monoclonal antibodies |
| Vaccines |
| Small molecules |
nucleus and interact with specific hormone-responsive elements on chromatin to induce synthesis of specific messenger ribonucleic acids (mRNAs). Translation of these mRNA species leads to formation of new proteins that alter physiological or biochemical reactions in a beneficial manner. Most of the proteins involved, however, have not yet been identified and characterized.
Antihormonal drug treatment strategies for breast cancer include:
Approximately 70% of all postmenopausal patients whose breast tumors show the presence of estrogen receptors respond favorably to antiestrogen therapy, as opposed to only approximately 10% of those whose tumors do not express receptors. Tamoxifen is the main antiestrogen used clinically, and it acts by binding to estrogen receptors and blocking estrogen-dependent transcription of cells in the G1 phase. By blocking the binding of estrogens, tamoxifen (see Chapter 40) may decrease estrogen stimulation of the production of transforming growth factor-α and secretion of associated proteins. Toremifene, closely related to tamoxifen, is a newer estrogen receptor antagonist that is also used to treat estrogen receptor-positive breast cancer, or when estrogen sensitivity is unknown. It is indicated in postmenopausal women whose cancer has metastasized. Fulvestrant has also been recently approved for the treatment of estrogen receptor-positive breast cancer in postmenopausal women. However, in contrast to tamoxifen and toremifene, instead of blocking estrogen receptors, fulvestrant destroys them.
Although postmenopausal women have very low levels of estrogen, small amounts are produced through the conversion of androstenedione, secreted by the adrenal glands, to estrogen via an aromatase enzyme (see Fig 38-1). Even these low levels can stimulate estrogen-sensitive breast cancer. The aromatase inhibitors letrozole, anastrazole, and exemestane effectively block the production of estrogen from the adrenals and are alternatives to the estrogen receptor antagonists in postmenopausal women. These drugs do not affect ovarian secretion of estrogen and for this reason are not used in the treatment of breast cancer in premenopausal women.
Harnessing intrinsic biological systems for treatment of disease is an appealing concept, because theoretically it could be highly targeted and of limited toxicity. “Biological therapy” attempts to use our native host defense system and its humoral and cellular components as weapons to fight cancer (see Chapter 6). Many of these weapons are intended to stimulate the immune system for destruction of malignant cells. The immune system involves the action of many different cell types acting in concert, particularly lymphocytes, which can be classified as B, T, or null cells. These cells can secrete proteins, including antibodies, which possess unique affinity for their conjugate antigens and impart specificity to the immune system. They also secrete cytokines, which have wide-ranging cellular influences. Use of these systems may introduce remarkable therapeutic target specificity at the risk of induction of autoimmunity and unique toxicities. Agents currently in use include cytokines, monoclonal antibodies, and vaccines.
Rituximab is a chimeric immunoglobulin G1-κ monoclonal antibody raised against the CD20 antigen, constructed with a murine light and heavy-chain variable region and a human constant region sequence. CD20 is a transmembrane protein expressed by most B cells at various stages of development and by malignant B lymphocytes. It is found on more than 90% of B cell non-Hodgkin’s lymphomas, but importantly, is not expressed by hematopoietic stem cells or other normal tissues. Rituximab binds to B lymphocytes after intravenous (IV) administration; therefore serum concentrations vary inversely with tumor burden. Complement-dependent cytotoxicity and ADCC are both mechanisms through which this agent may cause target cell lysis. Rituximab is used in the treatment of indolent low-grade non-Hodgkin’s lymphoma, where it may be used as monotherapy, and in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy as treatment for CD20-positive diffuse large B-cell non-Hodgkin’s lymphoma. Rituximab is a component of a combination regimen using ibritumomab tiuxetan and is being evaluated in the treatment of chronic lymphocytic leukemia, thrombocytopenic purpura, and Waldenström’s macroglobulinemia.
Growth Factor Receptors: Anti-EGFR Therapy
The EGFR is present on a variety of solid tumors including non-small-cell lung cancer, head and neck cancer, and malignant gliomas. EGFR expression correlates with poor clinical outcome and resistance to cytotoxic agents. The EGFR consists of an extracellular ligand-binding domain, a hydrophobic transmembrane domain, and an intracellular domain with tyrosine kinase activity. Upon stimulation by ligand, the EGFR dimerizes, which initiates an intracellular pro-survival signaling cascade, resulting in increased cell proliferation, metastasis, and decreased apoptosis (Fig. 55-1). The EGFR pathway can be inhibited by either blocking the extracellular domain with monoclonal antibodies or by small molecule tyrosine kinase inhibitors that block adenosine triphosphate (ATP)-binding and inhibit kinase activity.
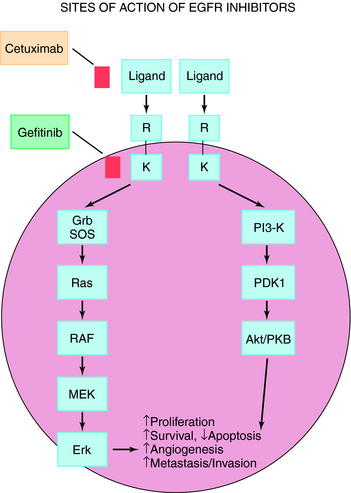
Trastuzumab is a recombinant deoxyribonucleic acid-derived humanized monoclonal antibody that selectively binds with high affinity to the extracellular domain of the human EGFR2 protein HER2. The HER2 (or C-ERBB2) protooncogene encodes a transmembrane receptor protein structurally related to the EGFR. Trastuzumab inhibits the proliferation of human tumor cells that overexpress HER2 and is approved for treatment of patients with metastatic breast cancer. As a single agent, it is indicated for treatment of patients with metastatic breast cancer whose tumors overexpress the HER2 protein and who have received one or more chemotherapy regimens for metastatic disease. It is also indicated for administration in combination with paclitaxel for treatment of patients with metastatic breast cancer who are chemotherapy naive and whose tumors overexpress the HER2 protein. Cetuximab is the first monoclonal antibody directed against the EGFR to be approved for treatment of colorectal cancer, either alone or in combination with irinotecan. Cetuximab is a genetically engineered version of a mouse antibody that contains both human and mouse components targeting EGFRs.
Pharmacokinetics
The pharmacokinetics of the drugs described in this chapter are summarized in Table 55-1. Of major consideration when administering these drugs is the fact that many of them are extensively metabolized by cytochrome P450 enzymes; thus the potential for serious drug interactions exists. Patients receiving these drugs typically receive other medications as well, including chemotherapeutic drugs, anti-infectives, anti-emetics, and drugs to stimulate the bone marrow.
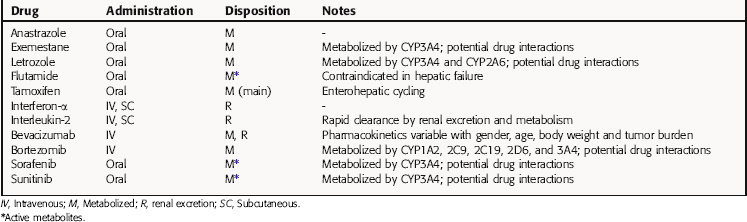
It is important to note that the pharmacokinetics of bevacizumab vary with age, gender, body weight, and tumor burden. Therefore the dosing of this agent must be individualized for each patient. Dosage adjustments may be required for any of these drugs in the presence of hepatic or renal failure, and therefore careful monitoring of the patient’s status is required.
Relationship of Mechanisms of Action to Clinical Response
Most of these agents are used in multiple-drug protocols in which the cytolytic effects of the different agents interact in a complex manner. The clinical use of combination chemotherapy is discussed in Chapter 53.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Tamoxifen can induce changes in the endometrium leading to endometrial hyperplasia, endometriosis, or
endometrial cancer. The incidence of these alterations is lower in premenopausal as compared with postmenopausal women; however, continued surveillance during therapy is required.
Aromatase inhibitors, by virtue of blocking the production of even low levels of estrogen that are produced in postmenopausal women, lead to the development of osteoporosis. The use of bone density-supportive drugs such as the bisphosphonates (see Chapter 44) is often required to diminish this effect. Additional adverse effects include bone and joint pain and elevated serum cholesterol levels.
New Horizons
normal cells. Many of these agents will continue to be adjuvant therapy administered in conjunction with, or after, traditional cytoxic chemotherapy.
The development of the HPV vaccine is an example of an antigen-based vaccine to stimulate immunity against a specific virus. Tumor cell vaccines are yet another approach currently under development. The production of these vaccines is patient-specific and involves removal of tumor cells from the patient and treating the cells in vitro with radiation so that they cannot form new tumors. Specific tumor antigens are identified that will be recognized by the patient’s immune system, amplified either chemically or through gene amplification, and then injected back into the patient. The goal is to stimulate the patient’s immune response to the foreign tumor cells. Newer modifications of this type of patient-specific vaccine are to fuse the tumor cells to dendritic cells, again with the objective of stimulating an immune response.
Anonymous. Chemotherapy for esophageal, gastric and colorectal cancers. Treat Guidel Med Lett. 2006;4:55-60.
Anonymous. A human papillomavirus vaccine. Med Lett. 2006;48:65-66.
Anonymous. Two new drugs for renal cell carcinoma. Med Lett. 2007;49:18-19.
Carlson et al. 2006 Carlson RW, Brown E, Burstein HJ, et al. NCCN taskforce report: Adjuvant Therapy for Breast Cancer. J Nat Comp Cancer Network. 2006;4:S1-S26.