Chapter 36 Drugs to Control Pain
| Abbreviations | |
|---|---|
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| GI | Gastrointestinal |
| 5-HT | Serotonin |
| IV | Intravenous |
| MAO | Monoamine oxidase |
| NAPQI | N-acetyl-p-benzoquinoneimine |
| NE | Norepinephrine |
| NSAID | Nonsteroidal antiinflammatory drug |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| TX | Thromboxane |
Therapeutic Overview
The third group of analgesics includes compounds that do not relieve pain from tissue damage (i.e., nociceptive pain) but can provide relief in specific pain syndromes such as neuropathic pain, gout, migraine headache, and fibromyalgia. Neuropathic pain results from changes in sensory neurons that render them hyperactive, even in the absence of nociceptive stimuli. It is often a chronic condition that is impervious to standard analgesic drugs. However, neuropathic pain is ameliorated by tricyclic antidepressants and compounds used to treat seizure disorders, drugs that are not thought of as primary analgesic agents.
| Therapeutic Overview |
|---|
| Opioids |
| Relief of most types of moderate to severe visceral or somatic pain |
| Symptomatic treatment of acute diarrhea |
| Cough suppression |
| Treatment of opiate addiction and alcoholism |
| Anesthetic adjunct |
| Overdose can be reversed by opioid receptor antagonists |
| NSAIDs and Acetaminophen |
| Relief of mild to moderate somatic pain including headache, toothache, myalgia, and arthralgia |
| Reduce fever |
| Prophylaxis of myocardial infarction and stroke |
| NSAIDs only |
| Relief in inflammatory disorders including rheumatoid arthritis, osteoarthritis, gout, and ankylosing spondylitis |
Mechanisms of Action
Sensations of pain are modulated by both ascending and descending pathways in the central nervous system (CNS). Noxious or nociceptive stimuli activate highly developed endings on primary afferent neurons, termed nociceptors (pain receptors). These stimuli give rise to action potentials that are transmitted along afferent neurons into the dorsal horn of the spinal cord. A-delta (Aδ) fibers are small, myelinated, rapidly conducting afferent neurons that terminate in lamina I of the spinal cord. They have a relatively high threshold for activation by mechanical and thermal stimuli and mediate sharp and localized pain, often termed somatic pain. C-fibers are even smaller unmyelinated afferent neurons and hence are slower conducting. They are polymodal and are activated by mechanical, thermal, or chemical stimuli. They terminate in lamina II of the spinal cord (substantia gelatinosa) and mediate dull, diffuse, aching, or burning pain sometimes called visceral pain (see Chapter 13, Fig. 13-5). Aδ and C-fibers release excitatory amino acids in the dorsal horn; C-fibers, which are stimulated by bradykinin and prostaglandins (PGs) released from local damaged cells, release substance P and other neuropeptides. These neurotransmitters activate secondary neurons that form the ascending spinothalamic pathway, which projects to supraspinal nuclei in the thalamus and then to the limbic system and cerebral cortex (Fig. 36-1, A).
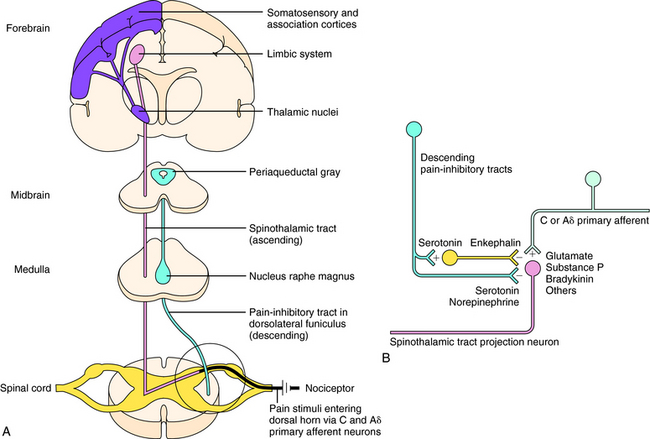
Descending pain-inhibitory systems originate in the periaqueductal gray region of the midbrain and from several nuclei of the rostroventral medulla oblongata and project downward to the dorsal horn. These descending systems release norepinephrine (NE), 5-HT, and other neurotransmitters and thereby inhibit the activity of the ascending pain pathways, either through direct synaptic contacts or indirectly by activating inhibitory interneurons. These pathways are illustrated in Figure 36-1, B.
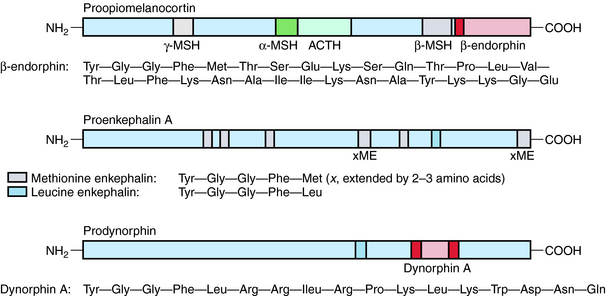
Three major families of opioid peptides have been identified: the enkephalins, endorphins, and dynorphins. They are derived from precursor molecules encoded by separate genes—proenkephalin, proopiomelanocortin, and prodynorphin, respectively. Although found primarily in the CNS, some opioid peptides, notably the enkephalins, also exist in peripheral tissues such as nerve plexuses of the gastrointestinal (GI) tract and the adrenal medulla (Table 36-1).
| Opioid Family | Precursor | Distribution |
|---|---|---|
| Enkephalins | Proenkephalin | Widely throughout the CNS, especially in interneurons, including those associated with pain pathways and emotional behavior; also found in some peripheral tissues |
| Endorphins | Proopiomelanocortin | β-Endorphin in hypothalamus, nucleus tractus solitarius, and anterior lobe of the pituitary where it is co-released with adrenocorticotrophin in response to stress |
| Dynorphins | Prodynorphin | Dynorphin A (1-17) in the magnocellular cells of the hypothalamus and posterior lobe of the pituitary gland where it co-localizes with vasopressin; shorter-chain dynorphins distributed widely in the CNS, some associated with pain pathways, especially in the spinal cord |
β-Endorphin and longer-chain dynorphins, such as dynorphin A (1-17), have a more limited distribution in the CNS and may not influence pain processing directly. Rather, they may have hormonal roles in responses to stress and fluid homeostasis, respectively. The structures of the three classical families of opioid peptides are shown in Figure 36-2.
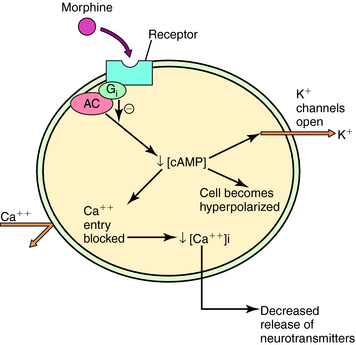
Three major opioid receptors have been identified by pharmacological means and molecular cloning and are designated μ, κ, and δ (Table 36-2). They are also referred to by either their International Union of Pharmacology nomenclature (OP3, OP2, and OP1, respectively) or their molecular biological nomenclature (MOP, KOP, and DOP, respectively). All three receptors belong to the superfamily of G-protein-coupled receptors with the characteristic seven transmembrane-spanning regions (see Chapter 1). Activation of these receptors decreases synthesis of cyclic adenosine monophosphate, increases K+ conductance, and decreases Ca++ conductance, effects illustrated in Figure 36-3. Because changes in K+ and Ca++ conductances inhibit neuronal activity, activation of any of the three opioid receptors usually results in decreased neuronal transmission.
TABLE 36–2 Opioid Receptors and their Ligands
| Receptor | Endogenous Ligand | Drug Ligands |
|---|---|---|
| μ Receptor (OP3/MOP) | Enkephalins | Morphine |
| β-Endorphin | Buprenorphine | |
| Endomorphins (?) | Methadone | |
| Meperidine | ||
| Fentanyl | ||
| κ Receptor (OP2/KOP) | Dynorphins | Butorphanol |
| Pentazocine | ||
| δ Receptor (OP1/DOP) | Enkephalins | None to date |
| β-Endorphin |
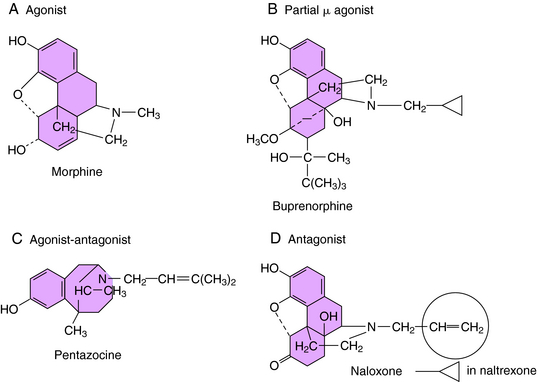
The selectivity of opioid receptors for endogenous and drug ligands is shown in Table 36-2. The anatomical distribution of opioid receptors is consistent with the actions of the opioids—that is, they are found prominently among structures of the ascending and descending pain-modulatory pathways. All clinically important effects of morphine and morphine-like drugs are mediated by μ receptors. Some of the effects of mixed-action opioids, including analgesia at the level of the spinal cord, sedation, and the dysphoria that occurs at high doses, are mediated by κ receptors. With the exception of the opioid antagonists, there are currently no therapeutic agents that interact with μ receptors in a clinically meaningful way. In cases in which opioids can be resolved into optical isomers, the levorotatory isomer usually has a considerably higher affinity for opioid receptors than does its dextrorotatory counterpart. The structures of morphine and representative agonist/antagonist compounds are shown in Figure 36-4.
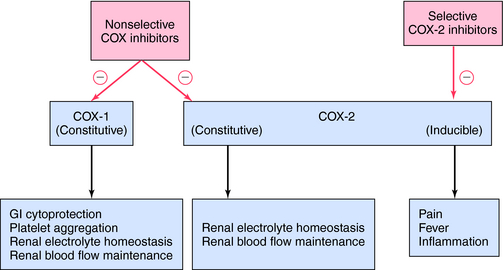
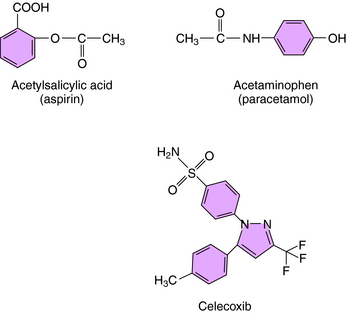
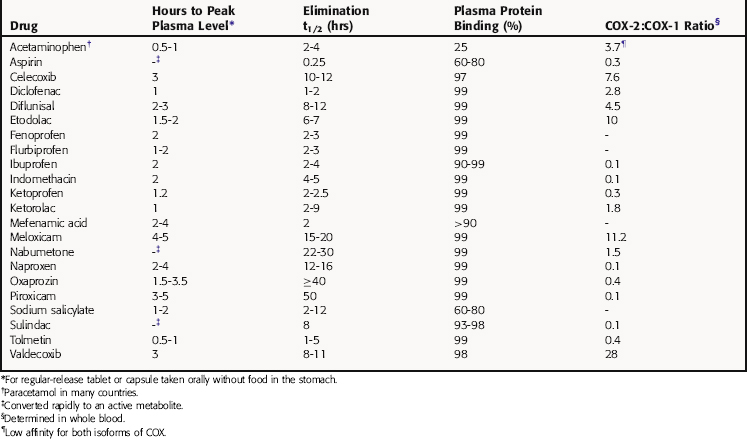
The mechanism of action, all of the therapeutic effects, and many of the side effects of the NSAIDs are due to inhibition of cyclooxygenase (COX), an enzyme involved in the metabolism of the eicosanoids. The eicosanoids are derivatives of arachidonic acid and include the leukotrienes synthesized by the action of 5-lipoxygenases, and the PGs and thromboxanes (TXs) synthesized by the action of the COXs (see Chapter 15, Fig. 15-1). Two distinct COX enzymes have been identified. COX-1 is constitutively expressed and is involved in “housekeeping tasks” in cells. COX-2 occurs constitutively in some tissues but is largely inducible, and induction results in a marked increase in the rate of synthesis and release of COX products, particularly the PGs. Aspirin acetylates both COX enzymes, inhibiting their activity irreversibly, whereas other nonselective NSAIDs inhibit the COX enzymes reversibly. The COX-2 inhibitors are 8- to 35-fold more selective for COX-2 relative to COX-1 and inhibit COX-2 irreversibly in a time-dependent manner. The functional consequences of inhibition of COX-2 relative to COX-1 are depicted in Figure 36-5. Structures of aspirin, acetaminophen, and the COX-2 inhibitor celecoxib are shown in Figure 36-6.
Drugs for Specific Pain Syndromes
Neuropathic pain is the result of injury to peripheral sensory nerves and is different from the nociceptive pain caused by tissue damage, in which sensory nerves are activated by chemical mediators of pain. There are many causes of nerve injury, including physical trauma, metabolic and autoimmune disorders, viral infection, chemotoxicity, and chronic inflammation. Nearly half of all diabetic patients experience peripheral neuropathies eventually. Neuropathic pain states often are associated with hyperalgesia (increased sensitivity to normally painful stimuli) and allodynia (pain caused by stimuli that are not normally painful; e.g., touch).
In neuropathic pain, primary afferent neurons are hyperactive, discharging spontaneously (in the absence of an identifiable noxious stimulus), and there is a cascade of changes to neurons in dorsal root ganglia and in the dorsal horn of the spinal cord. These changes present multiple targets for pharmacological intervention, among which are increases in the expression and activity of Na+ channels. Several drugs introduced to treat seizure disorders have been shown to be effective in the treatment of neuropathic pain. These agents include carbamazepine and lamotrigine, which inhibit voltage-dependent Na+ channels, and pregabalin, which binds to an auxiliary subunit of voltage-gated Ca++ channels to decrease the release of several neurotransmitters (see Chapter 34). The tricyclic antidepressants (see Chapter 30) have also been shown to be effective for this condition.
Drugs from several pharmacological classes are used to treat or prevent gout. The NSAIDs and the corticosteroids (see Chapter 39) attenuate inflammatory responses to urate crystals and the associated pain. Colchicine also reduces the inflammatory response, but through a different mechanism. Colchicine binds to tubulin in leukocytes, causing microtubules to disaggregate. This affects the structure of the cells, inhibiting their migration into the inflamed area and reducing phagocytic activity.
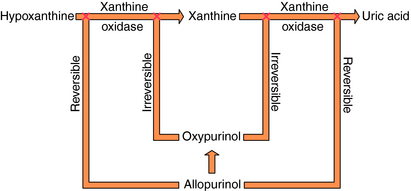
Specific drugs are used to correct the underlying hyperuricemia in gout. Allopurinol, a structural analog of the purine hypoxanthine, inhibits the enzyme xanthine oxidase (Fig. 36-7), blocking the metabolism of hypoxanthine and xanthine to uric acid and lowering blood urate concentrations. Normally, approximately 90% of filtered urate is resorbed and only 10% is excreted. The uricosuric agents probenecid and sulfinpyrazone increase urate excretion by competing with uric acid for the renal tubular acid transporter so less urate is resorbed.
The NSAIDs also bring relief from migraine episodes in many patients. They are presumed to attenuate the neurogenically induced inflammatory response through inhibition of COX-2. Other drugs are also used as preventive therapy, including tricyclic antidepressants, especially amitriptyline (see Chapter 30), and the β adrenergic receptor blockers propranolol and timolol (see Chapter 11).
Pharmacokinetics
Aspirin (acetylsalicylic acid) has a low pKa and is well absorbed from the acidic environment of the stomach and duodenum, the part of the GI tract that accounts for much of the absorption of the NSAIDs. Aspirin has a plasma half-life of only 15 minutes because it undergoes rapid hydrolysis to salicylic acid, which has therapeutic effects similar to those of the parent drug. The half-life of salicylic acid ranges from 2 to 3 hours at doses used to treat pain and fever to as high as 12 hours at doses sometimes used to treat inflammatory disorders. Approximately 75% is conjugated with glycine in the liver to form the inactive salicyluric acid, which is excreted by the kidneys, along with glucuronide conjugates and 10% free salicylic acid. At alkaline pH, up to 30% of a dose may be excreted as free salicylic acid, which is why sodium bicarbonate is administered to alkalinize the urine in treating toxic concentrations. The limited hepatic pool of glycine and glucuronide available for conjugation results in elimination of salicylate by first-order kinetics at low doses and by zero-order kinetics at higher doses. This accounts for the increasing half-life with increasing dose.
Acetaminophen (paracetamol in many countries), like the NSAIDs, is a weak acid that is almost completely absorbed from the GI tract and the rectum. Peak plasma concentration is achieved within 1 hour, and distribution is relatively uniform throughout the body. Acetaminophen is converted almost completely to inactive metabolites in the liver. A small proportion is oxidatively metabolized via cytochrome P450 enzymes to N-acetyl-p-benzoquinoneimine (NAPQI), which is conjugated with glutathione and excreted. NAPQI is highly reactive with and binds covalently to sulfhydryl groups. If the glutathione content of the liver is depressed by disease or fasting, or is depleted by high concentrations of the intermediate metabolite, NAPQI interacts with sulfhydryl-containing hepatocellular proteins, which can lead to hepatic necrosis. Glutathione-depleting concentrations of NAPQI occur after acute overdose with acetaminophen and can also occur after high doses (>4gm/day) in patients taking drugs that induce cytochrome P450s.
The pharmacokinetic parameters for the NSAIDs and acetaminophen are shown in Table 36-4.
Drugs for Specific Pain Syndromes
Neuropathic Pain and Fibromyalgia
The pharmacokinetics of the anticonvulsants and antidepressants used for neuropathic pain and fibromyalgia are presented in Chapter 34 and Chapter 30, respectively.
Allopurinol is oxidized to oxypurinol (alloxanthine), which, like the parent compound, is an inhibitor of xanthine oxidase (see Fig. 36-7) and is largely excreted by the kidneys. Inhibition of xanthine oxidase by oxypurinol is irreversible and accounts for most of the therapeutic effects of allopurinol.
Probenecid inhibits the renal tubular secretion of weak acids and can elevate plasma concentrations of weakly acidic drugs taken concomitantly—for example, many NSAIDs. This is used to advantage in situations in which it is necessary to maintain high plasma levels of cephalosporins, penicillin, and other β-lactam antibiotics (see Chapter 46).
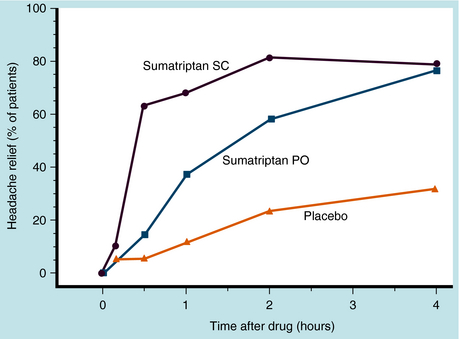
Sumatriptan is administered orally, subcutaneously, or intranasally. Oral bioavailability is 15%, and peak plasma concentrations are reached in 1.5 to 2 hours. In contrast, subcutaneous administration results in 97% bioavailability and peak plasma concentrations in 10 to 20 minutes. Protein binding is low, and the elimination half-life is 2 to 2.5 hours. Sumatriptan is metabolized in the liver by MAO-A. After subcutaneous administration, approximately 60% of a dose is excreted renally (20% unchanged) and the rest by the biliary-fecal route. Relief from pain of severe migraine begins within 10 minutes of injection, and half of patients experience relief within 30 minutes. The onset of action is slower when given orally, with peak relief in more than half of patients occurring within 2 hours (Fig. 36-8).
The newer triptans have better oral bioavailability, ranging from 40% (zolmitriptan) to 60% to 75% (almotriptan, naratriptan) and are taken only by this route. They are metabolized 25% to 50% in the liver by MAO-A (except naratriptan, which is metabolized by microsomal enzymes), and metabolites and unchanged drug are excreted in urine and bile. Among the available drugs, only zolmitriptan has an important active metabolite; it is more potent than the parent compound and probably contributes to the therapeutic effect. Elimination half-lives range from 2 to 6 hours.
Relationship of Mechanisms of Action to Clinical Response
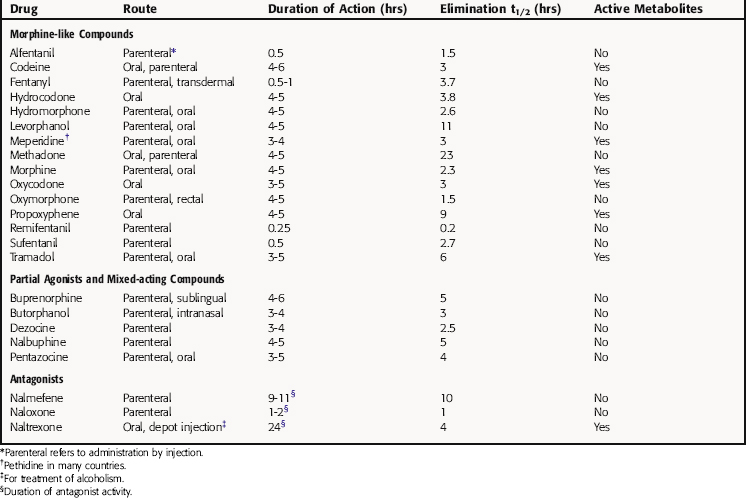
In general, all morphine-like drugs are equally effective in alleviating pain except for codeine, propoxyphene, and tramadol, which are less effective. A particular drug is often chosen based on its speed of onset, duration of action, and oral bioavailability. Fentanyl and its derivatives, with rapid onsets and short durations of action, are used almost exclusively IV in anesthesiology to manage pain during and immediately after surgery (see Chapter 35). Virtually all opioids exert their analgesic effects through μ opioid receptors in brain and spinal cord. Tramadol is an exception, being a racemic mixture with the ‘d or +’ isomer binding to μ opioid receptors and inhibiting neuronal 5-HT reuptake, and the ‘l or -’ isomer inhibiting NE reuptake and stimulating α2 adrenergic receptors.
Opioid analgesics are most effective in the management of dull, diffuse, continuous pain, with adequate doses relieving even sharp, localized, intermittent pain. A standard dose produces satisfactory relief in approximately 90% of patients with mild to moderate postoperative pain and in 65% to 70% of patients with moderate to severe postoperative pain. The degree of relief may decline after several days or more of frequent administration, as tolerance develops. Within limits, tolerance is overcome by increasing the dose and restoring the analgesic response. The pain relief conferred by opioids is often accompanied by drowsiness, mental clouding, and an elevated mood (i.e., euphoria). Although the euphoria is associated with a potential for abuse, in patients with pain it is more likely to be a secondary consequence of pain relief.
The pure opioid antagonists bind with high affinity and selectivity to opioid receptors but lack intrinsic activity and do not activate the receptors. The prototype naloxone (see Fig. 36-4) is used clinically, chiefly to reverse the respiratory depressant effect of opioid agonists. Administered IV, naloxone rapidly restores normal respiration and reverses virtually all effects of the agonist. If naloxone is administered before or along with an agonist, the effects of the agonist are blocked. Naloxone must be readministered periodically because of its short half-life. Its affinity for μ receptors is significantly greater than its affinity for κ or δ receptors. Therefore higher doses are required to reverse the effects of mixed-action opioids (mediated at κ receptors).
Naloxone and other antagonists can precipitate a full-blown withdrawal syndrome in people who are physically dependent on an opioid (see Chapter 37). If there is a possibility of physical dependence in an overdosed patient, it is recommended that he or she be started on a low dose of naloxone. The dose can be increased gradually to reverse respiratory depression and restore consciousness while minimizing withdrawal intensity.
Naltrexone has the same pharmacological characteristics as naloxone but has a longer duration of action and superior oral bioavailability. High doses taken orally produce prolonged blockade of opioid receptors, an effect used to advantage in therapy of some abusers. Naltrexone is also approved for the treatment of alcoholism by blocking the role of endogenous opioids in alcohol craving (see Chapter 32).
Aspirin, the prototype NSAID, remains one of the most commonly used and effective agents for treating headache and mild to moderate pain arising from muscles, tendons, joints, and soma. A dose of 650 to 1000 mg produces acceptable relief in 60% to 80% of patients. Aspirin is far less effective in providing relief of severe pain and pain from visceral organs, which usually require an opioid. However, unlike the opioids, aspirin and other NSAIDs do not cause analgesic tolerance or physical dependence and are not abused. Some of the major differences between aspirin and morphine, the prototype opioid analgesic, are listed in Table 36-5.
TABLE 36–5 Comparison of Analgesic Effects of Morphine and Aspirin
| Parameter | Morphine | Aspirin |
|---|---|---|
| Type of pain relieved | Visceral, somatic | Somatic |
| Intensity of pain relieved | Moderate to severe | Mild to moderate |
| Site of action | CNS | Local |
| Tolerance development | Yes | No |
| Physical dependence development | Yes | No |
| Abuse potential | High | None |
Drugs for Specific Pain Syndromes
Neuropathic pain syndromes are caused by many factors, some yet to be identified, and are difficult to manage. They are largely unresponsive to NSAIDs and only occasionally respond to opioid analgesics. Opioids can be used in responsive patients, although the side effects associated with chronic administration, including tolerance, must be addressed. However, neuropathic pain often can be alleviated by drugs ineffective in treating acute nociceptive pain. Notable among these are anticonvulsant drugs and tricyclic antidepressants.
Carbamazepine, lamotrigine, gabapentin, and pregabalin are currently the anticonvulsant drugs used most often to treat neuropathic pain (see Chapter 34). Carbamazepine has well-documented efficacy in the treatment of trigeminal neuralgia and may be beneficial in several other neuropathic pain syndromes including diabetic neuropathy. Lamotrigine is also of benefit in trigeminal neuralgia, while gabapentin is indicated for post-herpetic neuropathy and appears to reduce pain associated with a variety of syndromes, including phantom-limb pain, Guillain-Barré syndrome, and diabetic neuropathy. Pregabalin appears to have a profile similar to gabapentin and has also been found to be of benefit for the pain associated with sciatica. Because gabapentin seems to improve measures of mood and quality of life and has a relatively favorable profile of side effects, it is becoming the antiepileptic drug of choice for treating a range of neuropathies.
Clinical studies have demonstrated the efficacy of tricyclic antidepressants for pain relief in diabetic and post-herpetic neuropathies and in several other syndromes. Presumably, the drugs act via descending pain-inhibitory pathways, which contain noradrenergic and serotonergic neurons (see Fig. 36-1). Indeed, tricyclic antidepressants that inhibit reuptake of both NE and 5-HT such as amitriptyline, and perhaps the newer combination agents (see Chapter 30), may be more effective than drugs that selectively block reuptake of only one neurotransmitter. The analgesic effect of these agents is likely independent of their antidepressant effect, because onset of pain relief occurs more rapidly and at lower doses.
NSAIDs also provide symptomatic relief from the inflammation and pain of acute gout. Indomethacin is used most often, although ibuprofen, naproxen, sulindac, and piroxicam are also effective. Corticosteroids are efficacious antiinflammatory agents (see Chapter 39), and when used appropriately for short-term treatment of acute gout, are a safe alternative.
Individuals exhibiting frequent episodes or those who cannot take vasoconstrictors or are refractory to acute treatment should receive prophylactic treatment to prevent headaches. For predictable and limited attacks of migraine, such as those that may occur during menstruation, a brief course of NSAIDs, an ergot alkaloid, or a triptan is recommended beginning several days before the anticipated onset. For continuous prophylaxis, several groups of agents are effective including the β adrenergic receptor antagonists propranolol and timolol, the anticonvulsants valproic acid and topiramate, the tricyclic antidepressants including amitriptyline and nortriptyline, and the Ca++-channel blocker verapamil. Drugs used to abort migraine are not typically used prophylactically, owing largely to unacceptable side effects with prolonged administration.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Depression of respiration is the most serious side effect of opioid analgesics and is the principal cause of death from overdose. Opioids decrease the sensitivity of chemoreceptors in the brainstem to carbon dioxide, a normal stimulus of ventilatory reflexes. The result is a blunting of the ventilatory response to increases in the PCO2 in blood and cerebrospinal fluid (see Fig. 35-9). At equally effective analgesic doses, most opioids, including partial agonists and mixed-acting drugs, produce a similar degree of respiratory depression, as indexed by elevation of blood PCO2. Depression of respiration increases with increasing dose, but partial agonists and mixed-action opioids produce proportionately smaller changes in respiration than do morphine-like drugs. The respiratory depressant effect of opioids is at least additive, if not super-additive, with that produced by other CNS depressants such as general anesthetics, sedative-hypnotics, and alcohol. Tolerance tends to parallel analgesic tolerance but does not protect against the respiratory depressant effect of non-opioid drugs.
The cardiovascular system is relatively unaffected by opioid analgesics. High IV doses of morphine and some related drugs cause a decrease in peripheral resistance and a decline in blood pressure. These effects are rarely clinically significant in supine patients. Some opioids cause a vagally mediated reflex bradycardia, which can be blocked with atropine. Morphine, meperidine, and several other opioids can release histamine from mast cells in peripheral tissues upon IV administration, resulting in transient vasodilation, hypotension, and itching (see Chapter 14). Histamine release is one of the few effects of opioid drugs not mediated by opioid receptors and prevented by naloxone. Pentazocine and butorphanol have mild sympathomimetic effects, causing minor increases in heart rate and blood pressure.
Tolerance develops to most effects of the opioids. With repeated drug administration, larger doses are necessary to produce the original response. Tolerance develops rapidly to emetic effects; more gradually to analgesic, endocrine, and respiratory depressant effects; and virtually not at all to constipating and miotic effects. A point may be reached in highly tolerant patients where further dose increases no longer achieve pain relief. Tolerance also develops to the effects of mixed-action opioids, but at a slower rate.
Continuous exposure to an opioid analgesic results in development of physical dependence, a state in which the body has adapted to the presence of the drug and requires it for normal function. When administration is terminated or an antagonist is administered, withdrawal symptoms occur as discussed in Chapter 37.
The adverse GI side effects are due to two distinct actions. The first, a physical interaction between the drug and the gastric mucosa, can be reduced by taking the medication with meals and with adequate amounts of fluids to facilitate complete dissolution of tablets. The second is due to COX-1 inhibition and the resulting loss of cytoprotective eicosanoids (see Chapter 15). Because they largely spare COX-1, selective COX-2 inhibitors produce a lower incidence of significant upper GI complications than do nonselective NSAIDs.
Acute overdose with aspirin or acetaminophen results in effects not seen at therapeutic doses. The syndrome produced by each drug is unique. Among the events in acute aspirin intoxication are ventilatory changes, metabolic acidosis, nausea, vomiting, hyperthermia, and stupor (Table 36-6). Treatment depends upon the severity of intoxication, which can be determined by measuring plasma salicylate levels, and is largely supportive: gastric lavage, alkalinizing the urine, cooling the body, and IV fluids containing HCO3− and glucose. Acute aspirin overdose is a leading cause of accidental poisoning in children.
TABLE 36–6 Relationship Between Blood Salicylate Level and Therapeutic and Toxic Effects
| Blood Salicylate Level (μg/mL) | Effect | Consequence |
|---|---|---|
| 50-100 | Analgesia, antipyresis | |
| 150-300 | Antiinflammatory | |
| 200-350 | Salicylism | Tinnitus, dizziness, nausea |
| ≤350 | Hyperventilation | Respiratory alkalosis |
| 450-800 | Disrupted carbohydrate metabolism, sweating, vomiting, uncoupled oxidative phosphorylation, depressed respiration, increasing acidosis and body temperature | Metabolic acidosis, dehydration, hyperthermia, respiratory acidosis, delirium, convulsions, coma |