Chapter 83 Vasodilators and antihypertensives
Vasodilators are a generic group of drugs that are primarily used in the intensive care unit (ICU) for the management of acute hypertensive states and emergencies. In addition, they have an important role in the management of hypertension and cardiac failure.1
PHYSIOLOGY
Blood pressure is controlled by a complex physiological neurohormonal system involving all components of the cardiovascular system.2,3 Traditionally, clinical practice has focused on the arterial circulation as the major regulator of systemic pressure. The importance of venous circulation in determining mean arterial pressure and cardiac output is discussed in Chapter 82.
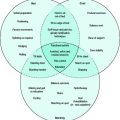
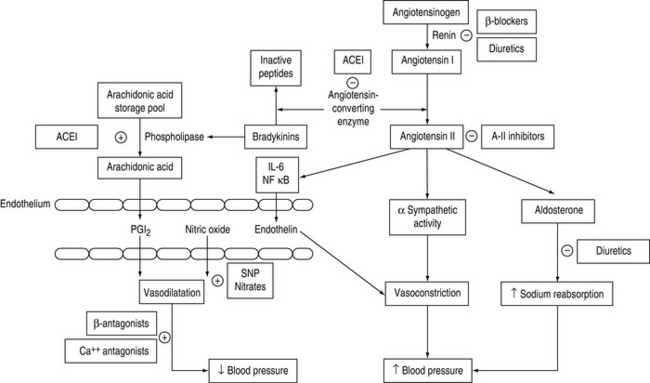
The role of the peripheral vasculature, including both arteriolar and venous systems, in the regulation of blood pressure may be conceptually regarded as a balance between vasodilatation and vasoconstriction3 (Figure 83.1).
CALCIUM FLUX
The concentration of intracellular ionised calcium is the primary determinant of vascular smooth muscle tone: increases lead to smooth muscle contraction, decreases cause relaxation. Control of calcium influx and efflux is determined by adrenergic receptor occupation and changes in membrane potential, mediated through voltage gated channels (see Chapter 82, Figure 82.2).
ENDOTHELIAL SYSTEM
The endothelium has a central role in blood pressure homeostasis by secreting substances such as nitric oxide, prostacyclin and endothelin.3 These substances are continuously released by the endothelium and are integral in regional autoregulation.4
ADRENERGIC SYSTEM
The sympathetic nervous system is integrally involved with all of the above systems, regulating vascular tone at central, ganglionic and local neural levels. Adrenergic stimulation of β-receptors is associated with vasodilatation; α-receptor stimulation results in vasoconstriction. The vascular effects of the catecholamines and vasopressors are discussed in Chapter 82.
Adrenergic stimulation is the predominant system in regulating venous tone.5 This is due to endothelial differences in veins resulting in less production of nitric oxide and reduced responsiveness to angiotensin II.
PATHOPHYSIOLOGY
Hypertensive states develop as a result of impaired or abnormal homeostatic processes, causing an imbalance between vasoconstrictive and vasodilatory effects.
CALCIUM ANTAGONISTS
Calcium antagonists have numerous effects on the cardiovascular system, influencing heart rate conduction, myocardial contractility and vasomotor tone. Entry of calcium through voltage-gated calcium channels is a major determinant of arteriolar, but not venous, tone.6
Magnesium is a physiological calcium antagonist, and is used therapeutically as magnesium sulphate.
NIFEDIPINE
Nifedipine is frequently used to treat angina pectoris, especially that due to coronary artery vasospasm. Peripheral vasodilatation results in decreased systemic blood pressure, often associated with sympathetic stimulation resulting in increased cardiac output and heart rate that may counter the negative inotropic, chronotropic and dromotropic effects of nifedipine. Nevertheless, nifedipine may be associated with profound hypotension in patients with ventricular dysfunction, aortic stenosis and/or concomitant β-blockade. For this reason, the use of sublingual nifedipine as a method of treating hypertensive emergencies is no longer recommended.7
Nifedipine and related drugs may cause diuretic-resistant peripheral oedema that is due to redistribution of extracellular fluid rather than sodium and water retention.
NIMODIPINE
It may be used to attenuate cerebral arterial vasospasm following aneurysmal subarachnoid haemorrhage. Improved outcomes have been demonstrated in patients with Grade 1 and 2 subarachnoid haemorrhage.8 Systemic hypotension may result from peripheral vasodilatation that may compromise cerebral blood flow in susceptible patients. Similarly, cerebral vasodilatation may increase intracranial pressure in patients with reduced intracranial elastance.
It may be given by intravenous infusion or enterally with equal effect.
AMLODOPINE
Amlodipine is an oral preparation that has a similar pharmacodynamic profile to nifedipine. In addition to arteriolar vasodilatory and cardiac effects, amlodipine has been shown to exert specific anti-inflammatory effects in hypertension, diabetic nephropathy and in modulating high-density lipoprotein (HDL) in patients with hypercholesterolaemia.9 These effects have seen amlodipine increasingly being used for treatment of hypertension in high-risk patients, and may have a role in stable critically ill patients with associated comorbidities.
VERAPAMIL
Verapamil is not as active as nifedipine in its effects on smooth muscle and therefore causes less pronounced decrease in systemic blood pressure and reflex sympathetic activity. It has a limited role as a vasodilator.10
MAGNESIUM SULPHATE
Magnesium regulates intracellular calcium and potassium levels by activation of membrane pumps and competition with calcium for transmembrane channels. Physiological effects are widespread, affecting cardiovascular, central and peripheral nervous systems and the musculoskeletal junction.11
Consequently, it has an established role in the treatment of pre-eclampsia and eclampsia,12 perioperative management of phaeochromocytoma13 and treatment of autonomic dysfunction in tetanus.14
DIRECT-ACTING VASODILATORS
These drugs act directly on vascular smooth muscle and exert their effects predominantly by increasing the concentration of endothelial nitric oxide. These drugs are also known as nitrovasodilators.15
SODIUM NITROPRUSSIDE
Sodium nitroprusside produces direct venous and arterial vasodilatation, resulting in a prompt decrease in systemic blood pressure. The effect on cardiac output is variable. Decreases in right atrial pressure reflect pooling of blood in the venous system, which may decrease cardiac output. This may result in reflex tachycardia that may oppose the overall reduction in blood pressure. In patients with left ventricular failure, the effect on cardiac output will depend on initial left ventricular end-diastolic pressure. Sodium nitroprusside has unpredictable effects on calculated systemic vascular resistance. Homeostaticmechanisms in preserving cardiac output may explain tachyphylaxis to prolonged infusions.
The prolonged use of large doses of sodium nitroprusside may be associated with toxicity related to the production and cyanide and, to a lesser extent, methaemoglobin.16
ISOSORBIDE DINITRATE
Isosorbide dinitrate is the most commonly administered oral nitrate for the prophylaxis of angina pectoris. It has a physiological effect that lasts up to 6 hours in doses of 60–120 mg. The mechanism of action is the same as glyceryl trinitrate. Hypotension may follow acute administration, but tolerance to this develops with chronic therapy.17
HYDRALAZINE
Following intravenous administration, hydralazine has a rapid onset of action, usually within 5–10 minutes. It may also be administered intramuscularly or orally. The drug is partially metabolised by acetylation, for which there is marked inter-individual variability (35% population are slow acetylators). Whilst this does not have much clinical significance regarding the antihypertensive effects, it is important with respect to toxicity.17
DIAZOXIDE
Diazoxide is chemically related to the thiazide diuretics and is a potent, non-selective, direct-acting vasodilator. The mechanism of action is unclear, but it is a predominantly arteriolar vasodilator.18 Diazoxide is administered intravenously or intramuscularly. It has a rapid onset (3–5 minutes) and prolonged duration of action (1–2 hours), often with precipitous reductions in blood pressure. Diazoxide has similar cardiovascular effects to hydralazine and is associated with significant reflex sympathetic stimulation, resulting in increased cardiac output and heart rate.
It is associated with metabolic side-effects such as hyperglycaemia and sodium and water retention.
α-ADRENERGIC ANTAGONISTS
Several groups of compounds act as α-adrenergic blockers with variable affinity for populations of α-receptors. Physiology and pathophysiology may influence the responsiveness of the drug receptor–effector relationship. Receptor pathobiology is discussed in Chapter 82. Consequently, there may be marked inter- and intra-individual variability in the patient’s response to these drugs.
PHENTOLAMINE
Phentolamine is a non-selective, competitive antagonist at α1– and α2-receptors. At low doses, phentolamine causes prejunctional inhibition of noradrenaline release (via α2-receptor inhibition). At higher doses, more complete α-receptor blockade is achieved, with enhancement of effects of β-agonists due to increased local concentration of noradrenaline produced by α2-blockade (see Chapter 82, Figure 82.3a).
PHENOXYBENZAMINE
Prolonged use is associated with increased β-adrenergic effects, predominantly increased heart rate, for which combination therapy with β-blockade is used. Phenoxybenzamine is primarily used in the management of phaeochromocytoma, either preoperatively or long term in inoperable patients. It may also be used to control autonomic hyperreflexia in patients with spinal cord transection.19
PRAZOSIN
It is administered orally and usually used for essential or renovascular (hyper-reninaemia) hypertension. It is frequently used in combination with β-blockers and diuretics, particularly in patients with renal dysfunction.
CARVEDILOL
It is administered orally; no intravenous preparation is available.
Recent studies have demonstrated slowing of progression of congestive cardiac failure and improved mortality, particularly when used in conjunction with ACE inhibitors in patients with mild to moderate cardiac failure.20,21 It may also be used in patients who cannot be treated with ACE inhibitors.
SYMPATHOMIMETICS
The peripheral vascular effects of β-adrenergic agonists such as adrenaline (epinephrine), noradrenaline, dopamine and the synthetic catecholamines dobutamine and isoprenaline are discussed in Chapter 82.
ANGIOTENSIN-CONVERTING ENZYME INHIBITORS
Angiotensin converting enzyme (ACE) inhibition has become a cornerstone in the management of patients with hypertension, cardiac failure and ischaemic heart disease.22,23 These drugs act by non-selective, competitive, irreversible inhibition to the angiotensin I binding site.
CARDIOVASCULAR EFFECTS
ACE inhibition is associated with improved myocardial performance following acute myocardial infarction due to left ventricular remodelling and improvement in neurohumoral activation. These drugs have been shown to improve survival following myocardial infarction in patients with left ventricular dysfunction.24
RENOVASCULAR EFFECTS
As a rule in intensive care patients, ACE inhibitors are started in suitable patients once renal function has stabilised and the patient no longer requires inotropic support.
SIDE-EFFECTS AND TOXICITY
In addition to renal dysfunction, ACE inhibitors may be associated with a number of side-effects. The most common of these is cough, which is due to the increased production of kinins.25
Severe angioneurotic oedema causing upper-airway obstruction may occur with all ACE inhibitors, although this is less common with enalapril and lisinopril. This is due to increased activation of bradykinins. ACE inhibitors are contraindicated in patients with a history of hereditary or idiopathic angioneurotic oedema.26
ANGIOTENSIN RECEPTOR BLOCKERS
These are a newer class of antihypertensive drugs that cause irreversible, selective blockade of angiotensin II at AT1 receptors.27,28
The selective blockade of angiotensin II offers several possible advantages over ACE inhibitors. These drugs are long acting and may be given once daily; onset of action is slow, thereby avoiding first-dose hypotension; side-effects such as cough and angioneurotic oedema are less common.29
CENTRALLY ACTING AGENTS
These agents modulate adrenergic stimulation at central nervous system and spinal cord level.
OTHER ANTIHYPERTENSIVE AGENTS
β-ADRENERGIC ANTAGONISTS
β-Blockers have been used for the treatment of hypertension for over 30 years and have an increasingly important role in the management of cardiac failure.30–32
In addition to decreasing heart rate and contractility, β-blockers have other neurohumoral effects that affect vascular tone. These relate to inhibition of renin release from juxtaglomerular cells (Figure 83.1) and prejunctional inhibition of noradrenaline that result in reduction in vascular tone and blood pressure. A central effect of β-blockers has also been proposed.
The mode of action has been described in terms of selectivity to blockade of β-adrenergic receptors – β1 and/or β2. While this is an appropriate pharmacological distinction, the clinical activity of these drugs is not as predictable due to mixed populations of β1– and β2-receptors in most organs and variable receptor responsiveness in physiological and pathophysiological conditions. Consequently,there is marked inter-individual variability in the response to these drugs. In high enough doses, whether intentionally or due to toxicity, all β-blockers will cause generalised antagonism with resultant therapeutic and toxic effects.
DRUG SELECTION
The clinical use of vasodilators in intensive care is different from their use in ambulatory patients. In the critically ill patient, these drugs are primarily used to control acute rises in mean arterial pressure associated with sympathetic stimulation, or as specific treatment of hypertensive emergencies.33
The selection of drug to treat hypertensive states will depend on the predominant cause of hypertension and the mechanism of action in the homeostatic pathway outlined in Figure 83.1.
There are no large studies investigating optimum therapy in patients presenting with hypertensive emergencies. These conditions occur in a heterogenous group of patients and drug selection is essentially determined by the underlying pathophysiology, personal preference and experience.33
MONITORING
The principles of haemodynamic monitoring in patients receiving vasoactive drugs is outlined in Chapter 82.
DOSAGES AND DRUG ADMINISTRATION
Infusion lines should be free of injection portals and clearly marked with identifying labels.
Concentrations of infusions should be standardised in accordance with individual unit protocols. Suggested infusion concentrations and common drug doses are shown in Table 83.1.
Table 83.1 Dose and infusion concentrations of commonly used vasodilators and antihypertensives in intensive care
| Agent | Infusion/Dose | Caution |
|---|---|---|
| Sodium nitroprusside | 50 mg/250 ml 5% Dextrose; range 3–40 ml/hour | Cyanide toxicity (> total dose 0.5 mg/kg per 24 hours) |
| Photodegradation | ||
| Raised intracranial pressure | ||
| Rebound hypotension | ||
| Shunt and oxygen desaturation | ||
| Glyceryl trinitrate | 30 mg/100 ml 5% Dextrose; range 2–25 ml/hour | Drug binding to polyvinylchloride |
| Tachyphylaxis | ||
| Raised intracranial pressure | ||
| Hydralazine | 10–20 mg i.v. bolus | Tachycardia |
| 20–40 mg 6–8-hourly | Myocardial ischaemia | |
| Diazoxide | 50–100 mg i.v. boluses | Precipitous hypotension |
| 15–30 mg/min infusion | Hyperglycaemia | |
| Trimetaphan | 1–4 mg/min infusion | Mydriasis, ileus |
| Bradycardia | ||
| Phentolamine | 1–10 mg i.v. boluses | Tachycardia |
| 5–30 mg/hour infusion | ||
| Phenoxybenzamine | Oral: 10 mg/day until postural hypotension | Idiosyncratic hypotension |
| i.v.: 1 mg/kg per day | ||
| Prazosin | 2–10 mg/day, 8-hourly | |
| Nifedipine | 5–10 mg oral/sublingual | Precipitous hypotension |
| Amlodipine | 5–10 mg oral b.d. | Caution in renal impairment |
| Captopril | 6.25–50 mg orally, 8-hourly | Caution in renovascular hypertension and renal failure |
| Acute hypertension: 6.5–25 mg sublingually p.r.n. | Pregnancy | |
| Angioneurotic oedema | ||
| Enalapril | 5–20 mg 8-hourly | |
| Enalaprilat | 0.625–5 mg bolus | Caution in renal failure and hypovolaemia |
| Losartan | 25–100 mg/day | Caution in renal failure |
| Clonidine | 25 μg to 150 mg i.v. bolus | Acute, perioperative centrally mediated hypertension |
| May cause rebound hypertension with chronic use | ||
| Atenolol | 1–10 mg i.v. boluses | Caution in poor left ventricular function, asthma |
| 25–100 mg oral b.d. | Hyperkalaemia | |
| Potentiated in renal failure | ||
| Metoprolol | As for atenolol, safe in renal failure | |
| Esmolol | Loading dose 0.5 mg/kg | |
| 10–40 mg/hour infusion | ||
| Labetalol | 20–80 mg i.v. boluses | |
| 0.5–4 mg/min infusion | ||
| Magnesium sulphate | 40–60 mg/kg loading (or 6 g) | Maintain serum magnesium > 1.5–2 mmol/l |
| 2–4 g/hour infusion |
SPECIFIC SITUATIONS
ACUTE HYPERTENSION
The majority of instances of acute hypertension in the ICU will respond to simple measures addressing the above.34
HYPERTENSIVE ENCEPHALOPATHY
Hypertensive encephalopathy is defined as an acute organic brain syndrome occurring as a result of failure of cerebrovascular autoregulation. There may be differences in the degree of hypertension that cause encephalopathy. It may present as confusion, visual disturbances, blindness, seizures or stroke. If not adequately treated, hypertensive encephalopathy may result in intracerebral haemorrhage, coma or death.35
The aim of drug therapy in these patients is to reduce blood pressure in a controlled, predictable and safe way. Acutely, short-acting, titratable parenteral drugs are suitable in emergency situations. Sodium nitroprusside can be used safely in most circumstances. Although sodium nitroprusside may increase intracranial pressure, associated reductions in mean arterial pressure offset this effect. Phentolamine is equally effective. Esmolol may be useful as an adjunctive agent.36
Patients with hypertensive emergencies are frequently hypovolaemic due to excessive sympathetic stimulation. In the absence of left ventricular failure, judicious fluid replacement may reduce blood pressure and improve renal function, thereby minimising precipitous hypotension that may result following administration of some drugs. Diuretics are generally avoided in these conditions unless there is evidence of left ventricular failure.36
ACUTE STROKE
Acute stroke syndromes frequently occur in the setting of severe hypertension. The reduction of mean arterial pressure must be balanced by the maintenance of adequate cerebral perfusion pressure and cerebral blood flow. Ischaemic or infarcted brain is vulnerable to critical reductions in cerebral blood flow, while excessive mean arterial pressure may increase the risk of cerebral haemorrhage.37
AORTIC DISSECTION
Aortic dissection is the most dramatic and most rapidly fatal complication of severe hypertension. Blood pressure should be decreased as rapidly as possible to normal or slightly hypotensive levels. Titrations are usually made to achieve systolic blood pressures of 100–110 mmHg or mean arterial pressure of 55–65 mmHg. This will depend on the patient’s premorbid blood pressure and the accuracy of blood pressure measurement. It is important to maintain blood pressure at levels compatible with adequate cerebral and renal perfusion.38
ACUTE MYOCARDIAL ISCHAEMIA
ACE inhibitors may be used in the acute situation and may be required for longer term treatment.
PHAEOCHROMOCYTOMA
Tumours of the adrenal medulla secrete catecholamines that result in initial paroxysmal, then sustained, severe hypertension. They may present to the ICU as a hypertensive emergency or perioperatively for surgical ablation.39
Acute hypertensive crises associated with phaeochromocytoma are managed with incremental doses orinfusions of phentolamine. Untreated patients may be significantly hypovolaemic and may require judicious volume replacement. β-Blockers should not be used in the acute stage as these will potentiate unopposed α-adrenergic stimulation.
Phenoxybenzamine forms the mainstay of treatment and preparation for surgery. This is commenced in 20–30 mg increments and continued until blood pressure is controlled. Excessive β-adrenergic effects are treated with β-blockers after sufficient α-blockade with phenoxybenzamine.19
Magnesium sulphate is useful in the perioperative management of phaeochromocytoma. It is given by infusion at 2–4 g/hour.13
RENAL FAILURE
Patients in the recovery phase of acute renal failure are usually hypertensive. This is a normal physiological response and should not be treated unless there is associated myocardial or cerebral ischaemia.40
PRE-ECLAMPSIA AND ECLAMPSIA
In addition to delivery of the baby and placenta, parenteral magnesium sulphate is the treatment of choice to prevent the evolution of pre-eclampsia to eclampsia (seizures and deteriorating encephalopathy12). Other parenteral drugs that have been used for many years for pregnancy-induced hypertensive states include hydralazine, phentolamine, diazoxide and labetalol.41
ACE inhibitors and angiotensin receptor blockers are contraindicated in pregnancy. This is discussed in Chapter 55.
1 Erdmann E. The management of heart failure – an overview. Basic Res Cardiol. 2000;95(Suppl 1):I3-17.
2 Cohn JN. Left ventricle and arteries: structure, function, hormones, and disease. Hypertension. 2001;37:346-349.
3 Spieker LE, Flammer AJ, Luscher TF. The vascular endothelium in hypertension. Handb Exp Pharmacol. 2006;176:249-283.
4 Egan K, FitzGerald GA. Eicosanoids and the vascular endothelium. Handb Exp Pharmacol. 2006;176:189-211.
5 Magder S, De Varennes B. Clinical death and the measurement of stressed vascular volume. Crit Care Med. 1998;26:1061-1064.
6 Schulman IH, Zachariah M, Raij L. Calcium channel blockers, endothelial dysfunction, and combination therapy. Aging Clin Exp Res. 2005;17:40-45.
7 Varon J, Marik PE. The diagnosis and management of hypertensive crises. Chest. 2000;118:214-227.
8 Rinkel GJ, Feigin VL, Algra A, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2005:CD000277.
9 Zanchetti A, Julius S, Kjeldsen S, et al. Outcomes in subgroups of hypertensive patients treated with regimens based on valsartan and amlodipine: an analysis of findings from the VALUE trial. J Hypertens. 2006;24:2163-2168.
10 De Cicco M, Macor F, Robieux I, et al. Pharmacokinetic and pharmacodynamic effects of high-dose continuous intravenous verapamil infusion: clinical experience in the intensive care unit. Crit Care Med. 1999;27:332-339.
11 Saris NE, Mervaala E, Karppanen H, et al. Magnesium. An update on physiological, clinical and analytical aspects. Clin Chim Acta. 2000;294:1-26.
12 Altman D, Carroli G, Duley L, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet. 2002;359:1877-1890.
13 James MF, Cronje L. Pheochromocytoma crisis: the use of magnesium sulfate. Anesth Analg. 2004;99:680-686.
14 Thwaites CL, Yen LM, Loan HT, et al. Magnesium sulphate for treatment of severe tetanus: a randomised controlled trial. Lancet. 2006;368:1436-1443.
15 Vassalle C, Domenici C, Lubrano V, et al. Interaction between nitric oxide and cyclooxygenase pathways in endothelial cells. J Vasc Res. 2003;40:491-499.
16 Alaniz C, Watts B. Monitoring cyanide toxicity in patients receiving nitroprusside therapy. Ann Pharmacother. 2005;39:388-389.
17 Ferdinand KC. Isosorbide dinitrate and hydralazine hydrochloride: a review of efficacy and safety. Expert Rev Cardiovasc Ther. 2005;3:993-1001.
18 Frank G. Diazoxide and trimethaphan used? Chest. 2001;119:316.
19 Prys-Roberts C. Phaeochromocytoma – recent progress in its management. Br J Anaesth. 2000;85:44-57.
20 Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651-1658.
21 Kopecky SL. Effect of beta blockers, particularly carvedilol, on reducing the risk of events after acute myocardial infarction. Am J Cardiol. 2006;98:1115-1159.
22 Stone PH. Review: ACE inhibitors reduce mortality and cardiovascular endpoints in stable coronary artery disease. ACP J Club. 2006;145:32.
23 Remuzzi G, Ruggenenti P. Overview of randomised trials of ACE inhibitors. Lancet. 2006;368:555-556.
24 Nickenig G, Ostergren J, Struijker-Boudier H. Clinical evidence for the cardiovascular benefits of angiotensin receptor blockers. J Renin Angiotensin Aldosterone Syst. 2006;7(Suppl 1):S1-7.
25 Dicpinigaitis PV. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest. 2006;129:S169-173.
26 Beltrami L, Zingale LC, Carugo S, et al. Angiotensin-converting enzyme inhibitor-related angioedema: how to deal with it. Expert Opin Drug Saf. 2006;5:643-649.
27 See S. Angiotensin II receptor blockers for the treatment of hypertension. Expert Opin Pharmacother. 2001;2:1795-1804.
28 Bhatia V, Bhatia R, Mathew B. Angiotensin receptor blockers in congestive heart failure: evidence, concerns, and controversies. Cardiol Rev. 2005;13:297-303.
29 Cooper ME, Webb RL, de Gasparo M. Angiotensin receptor blockers and the kidney: possible advantages over ACE inhibition? Cardiovasc Drug Rev. 2001;19:75-86.
30 Krum H. Guidelines for management of patients with chronic heart failure in Australia. Med J Aust. 2001;174:459-466.
31 Packer M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001;110(Suppl 7A):S81-94.
32 Gheorghiade M, Eichhorn EJ. Practical aspects of using beta-adrenergic blockade in systolic heart failure. Am J Med. 2001;110(Suppl 7A):S68-73.
33 Moser M, Izzo JLJr, Bisognano J. Hypertensive emergencies. J Clin Hypertens (Greenwich). 2006;8:275-281.
34 Slama M, Modeliar SS. Hypertension in the intensive care unit. Curr Opin Cardiol. 2006;21:279-287.
35 Mabie WC. Management of acute severe hypertension and encephalopathy. Clin Obstet Gynecol. 1999;42:519-531.
36 Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet. 2000;356:411-417.
37 Sokol SI, Kapoor JR, Foody JM. Blood pressure reduction in the primary and secondary prevention of stroke. Curr Vasc Pharmacol. 2006;4:155-160.
38 Ahmad F, Cheshire N, Hamady M. Acute aortic syndrome: pathology and therapeutic strategies. Postgrad Med J. 2006;82:305-312.
39 Graham GW, Unger BP, Coursin DB. Perioperative management of selected endocrine disorders. Int Anesthesiol Clin. 2000;38:31-67.
40 Palmer BF. Impaired renal autoregulation: implications for the genesis of hypertension and hypertension-induced renal injury. Am J Med Sci. 2001;321:388-400.
41 Watson D. The detection, investigation and management of hypertension in pregnancy. Aust N Z J Obstet Gynaecol. 2000;40:361.