[level-membership-for-pathology-category]9
Vascular disease and infarcts
In the setting of rapidly evolving neurologic deficits, stroke is not synonymous with brain infarct, since the types of cerebrovascular disease that usually result in a stroke may involve infarction or intracranial hemorrhage (see Chapter 10). Infarct, a localized area of ischemic brain injury, should also be differentiated from global hypoxic-ischemic brain injury (see Chapter 8). Stroke has been defined by the WHO as ‘rapidly developing clinical signs of a focal (or global) disturbance of cerebral function, lasting more than 24 hours or leading to death and with no apparent cause other than that of vascular origin’. Stroke ranks as the second most common single cause of death in the developed world.
To place these concepts in a more practical context, brain infarcts can be caused by:
 Large vessel (or macrovascular) arterial disease.
Large vessel (or macrovascular) arterial disease.
 Small vessel (or microvascular) arterial disease (arteries <400–500 μm in diameter).
Small vessel (or microvascular) arterial disease (arteries <400–500 μm in diameter).


 STROKE TERMINOLOGY
STROKE TERMINOLOGY Infarction describes rapidly developing focal clinical signs of CNS (and/or retinal) dysfunction due to ischemia, and either radiologic evidence of CNS (and/or retinal) ischemia in a defined vascular distribution, or symptoms and signs of CNS dysfunction persisting for at least 24 hours, with exclusion of non-ischemic etiologies.
Infarction describes rapidly developing focal clinical signs of CNS (and/or retinal) dysfunction due to ischemia, and either radiologic evidence of CNS (and/or retinal) ischemia in a defined vascular distribution, or symptoms and signs of CNS dysfunction persisting for at least 24 hours, with exclusion of non-ischemic etiologies.










 GENETICS OF ISCHEMIC CEREBROVASCULAR DISEASE
GENETICS OF ISCHEMIC CEREBROVASCULAR DISEASE
Brain infarcts occur as one of many systemic manifestations in various hereditary disorders.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke (MELAS)



 SCD is an autosomal recessive hemoglobinopathy characterized by chronic hemolytic anemia and ‘sickle-shaped’ erythrocytes.
SCD is an autosomal recessive hemoglobinopathy characterized by chronic hemolytic anemia and ‘sickle-shaped’ erythrocytes.
 SCD results from point mutation in the hemoglobin β-chain.
SCD results from point mutation in the hemoglobin β-chain.
 Number of strokes/100 patient-years increases from 0.13 (<2years of age) to 1.28 (>50years of age).
Number of strokes/100 patient-years increases from 0.13 (<2years of age) to 1.28 (>50years of age).
 At least 10% of SCD patients have a stroke during their lifetimes – 75% ischemic, 25% hemorrhagic.
At least 10% of SCD patients have a stroke during their lifetimes – 75% ischemic, 25% hemorrhagic.




















 FACTORS IMPORTANT IN ISCHEMIC BRAIN INJURY
FACTORS IMPORTANT IN ISCHEMIC BRAIN INJURY





 RISK FACTORS FOR ATHEROSCLEROSIS
RISK FACTORS FOR ATHEROSCLEROSIS







 ATHEROSCLEROSIS AND STROKE
ATHEROSCLEROSIS AND STROKE






 EPIDEMIOLOGIC ASPECTS OF STROKE
EPIDEMIOLOGIC ASPECTS OF STROKE







LARGE ARTERIAL DISEASE
MACROSCOPIC APPEARANCES
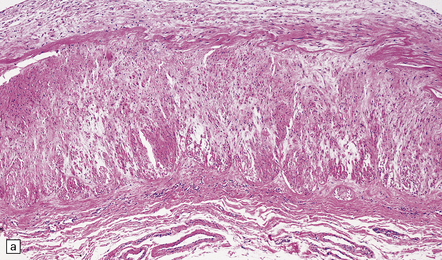
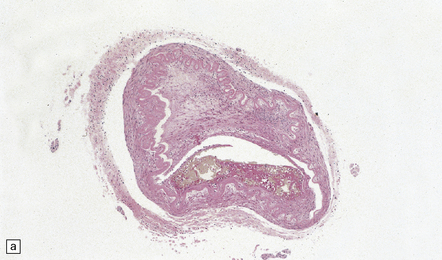
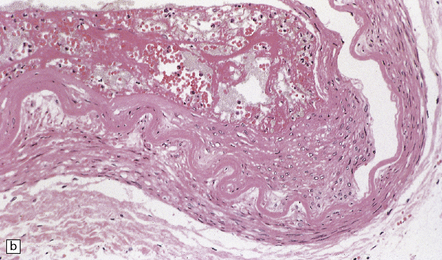
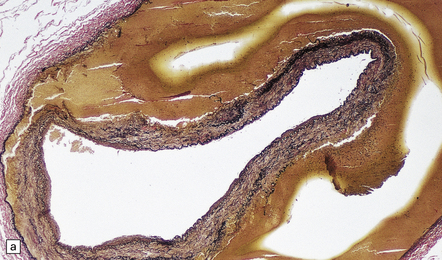
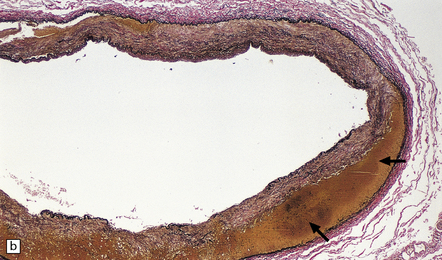
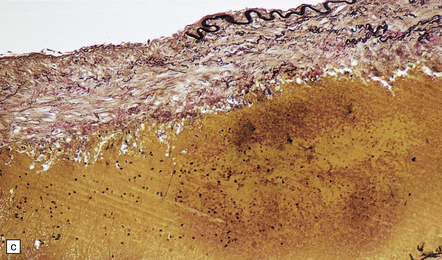
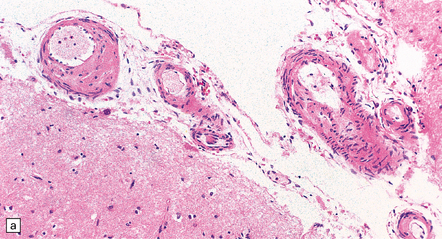
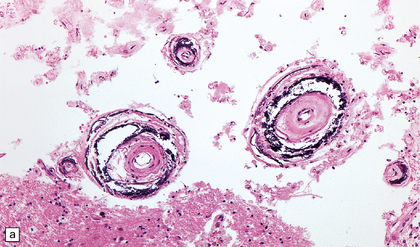
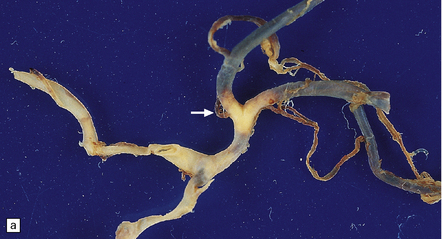
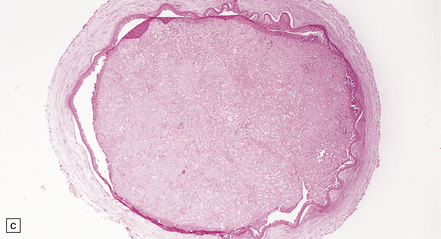
The severity of atherosclerosis can vary significantly in different arteries (e.g. severe basilar artery involvement may accompany less prominent MCA involvement). The degree and extent of aortic or coronary atherosclerosis do not predict its severity in the intracranial basal cerebral vasculature, i.e. compartments of the circle of Willis. Atheroma is often most severe at the origins of the vertebral arteries and carotid bifurcation (Figs 9.1, 9.2). Intracranial atherosclerosis is most severe in major branches of the circle of Willis and vertebrobasilar system (Fig. 9.3). Atheroma in distal arterial branches is more common in Asian and African-American subjects. The extent and topography of atherosclerosis in the basal vessels are often best documented by removing the circle of Willis from the fixed brain (Fig. 9.3b). In such a specimen from a subject with severe atherosclerosis, decalcification prior to histologic examination is recommended. Carotid endarterectomy specimens from individuals with TIA or threatened ischemic stroke are often submitted for histologic examination; extent and severity of atheroma within plaques, as well as the presence of plaque ulceration, necrosis and thrombus adherent to intima must be assessed (Fig. 9.2).
9.1 Atheroma of carotid artery.
Sections of common carotid (right) and internal carotid arteries removed at necropsy from a patient with severe atherosclerotic cerebrovascular disease and systemic atherosclerosis. Note severe stenosis of the lumina and eccentric intimal thickening of the internal carotid artery with thrombosis.
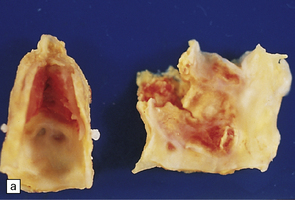
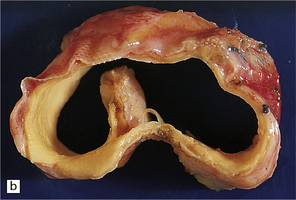
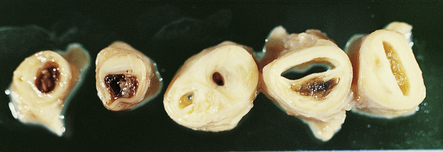
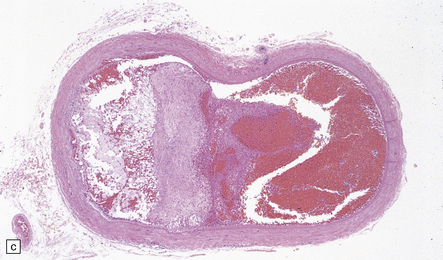
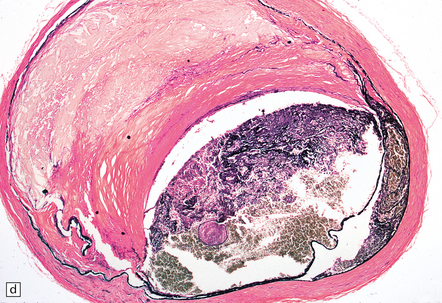
9.2 Carotid endarterectomy specimens from two patients.
(a) A specimen cut open to reveal grumous fractured eggshell-like atheromatous material and superimposed mural thrombus. (b) Another specimen cut in cross section showing atheroma at the carotid bifurcation. (Courtesy of Dr Sophia Apple, Department of Pathology and Laboratory Medicine, UCLA Medical Center.)
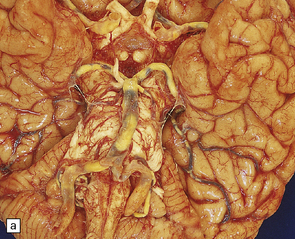
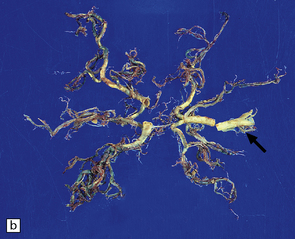
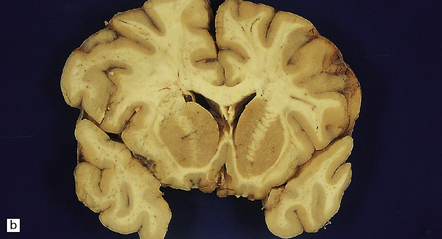
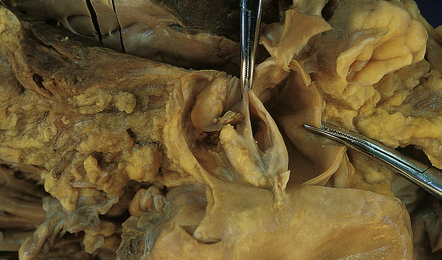
9.3 Severe atherosclerosis of the circle of Willis and its major branches.
(a) Patchy yellow discoloration of the arterial branches indicating underlying atheroma. (b) Severe atheroma involving the basal arteries of an elderly patient with ischemic-vascular dementia. Arrow indicates basilar artery. Note especially severe atherosclerosis of the basilar, posterior cerebral and middle cerebral arteries. However, patchy atheroma extends even into distal branches. (Courtesy of Dr Ivan Klement.)
MICROSCOPIC APPEARANCES
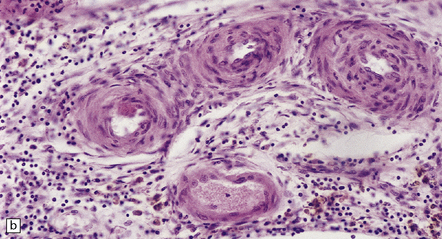
Histopathologic features of atheroma are best highlighted with stains that differentiate elastica, fibrous tissue, and smooth muscle (e.g. elastica van Gieson). Immunohistochemistry (IHC) using primary antibodies to vascular smooth muscle actin, endothelium (Factor 8, lectins), and macrophages may be helpful. Fibromuscular intimal hyperplasia with an intact endothelium and variable narrowing of the vascular lumen is noted in ‘early’ and asymptomatic vascular lesions, and often discovered incidentally at necropsy (Fig. 9.4). Complicated plaques show cholesterol clefts and prominent lipid-/hemosiderin-laden macrophages and may be heavily calcified. There is usually significant narrowing of the arterial vessel lumen, sometimes in association with ulceration and overlying mural or occlusive thrombus (Fig. 9.5). Immunohistochemistry (IHC) using primary antibodies to smooth muscle actin often demonstrates a thick smooth muscle cell ‘cap’ over a lipid-rich subendothelial plaque. It is quite rare for even severely narrowed segments of the circle of Willis to show plaque ulceration, in contrast to the frequency of this phenomenon in ICA endarterectomy specimens. Severely atherosclerotic arterial segments, especially in the basilar artery may show ectasia or even a fusiform aneurysm.
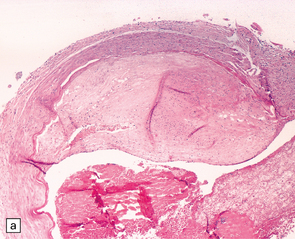
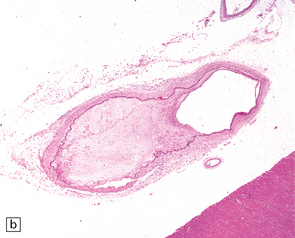
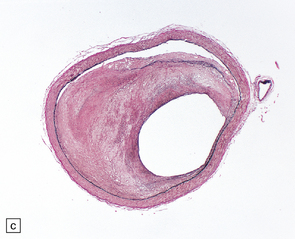
9.4 Atherosclerosis with fibromuscular intimal hyperplasia.
Eccentric atheroma is prominent in these arteries, but is largely composed of smooth muscle cells and ‘ground substance’ (glycosaminoglycans), though scattered cholesterol clefts are also present. Note the intact endothelium in all instances. The single elastic lamina (as is the case in intracerebral arteries) is also largely intact and best demonstrated in panels b and c. (a) Middle cerebral artery. (b) Meningeal artery, a branch of the posterior cerebral artery. (c) Section of posterior cerebral artery stained with Elastic van Gieson.
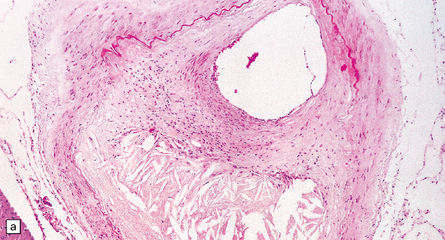
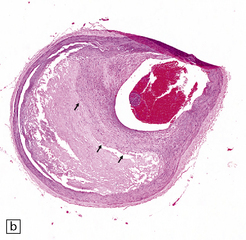
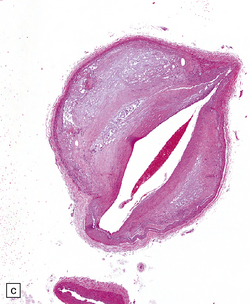
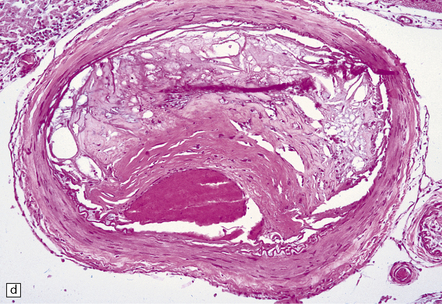
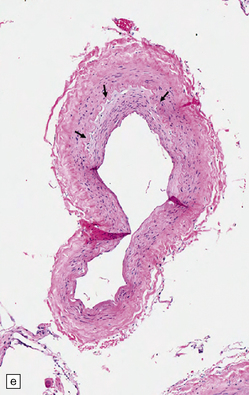
9.5 Complicated atherosclerosis.
(a) Prominent eccentric atheroma with smooth muscle cell hyperplasia overlying a region with abundant cholesterol clefts in an intracranial meningeal artery. The atheroma has produced significant stenosis. Note discontinuity in elastica. (b) Low magnification view of cross-section through a major branch of the circle of Willis, with severe stenosis of the lumen. Arrows indicate smooth muscle cell ‘cap’ adjacent to lumen and overlying grumous cholesterol material; no ulceration or mural thrombus is noted adjacent to the lumen. (c) Basal artery showing severe stenosis of the lumen by atheroma on two aspects of the vessel wall. (d) Small meningeal artery almost occluded by atheromatous material; only a small residual lumen remains. (e) Small meningeal artery (branch of ACA) with mild atheroma consisting almost exclusively of fibromuscular intimal hyperplasia; arrows indicate internal elastic lamina.
The distal circulation, both meningeal and parenchymal arteries, may show atherosclerotic changes or deposits of platelet-fibrin material (Fig. 9.6). Rarely, examination of an autopsy brain specimen from an individual with severe atherosclerosis may yield the finding of numerous atheroemboli within infarcted regions (Fig. 9.7).
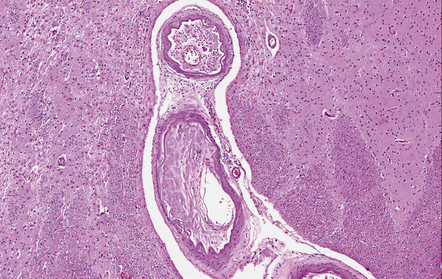
9.6 Intracerebral atherosclerosis.
Microatheroma almost occluding a tortuous artery in the basal ganglia of a patient with longstanding hypertension.
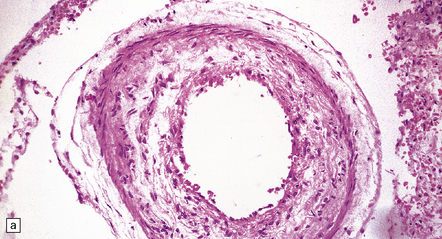
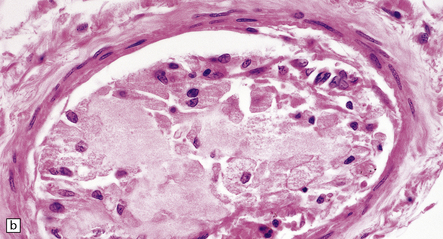
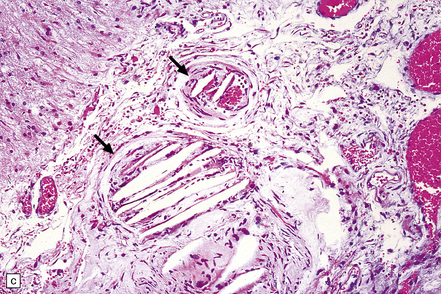
9.7 Cerebral atherosclerosis and complications of atheroma.
(a) Atherosclerosis in a small artery (external diameter approximately 500 μm) manifesting as fibromuscular intimal hyperplasia. (b) Intraluminal macrophages originating in embolic material from fragmented atheromatous plaques in a 58-year-old hypertensive woman with multiple cerebral and cerebellar infarcts in both watershed and large artery territories. (c) Cholesterol clefts within atheroemboli from fragmented atheromatous plaques occluding lumina of two meningeal arteries (arrows) within brain specimen of a patient with multiple cerebral infarcts. (d) Small intraparenchymal artery occluded by atheromatous material, including foreign body giant cell around cholesterol ‘clefts’. (e) Larger artery in another patient showing occlusive thrombus composed primarily of atheromatous material (‘atheroembolus’) within which prominent residual cholesterol clefts are seen. An ‘embolic’ origin of the atheromatous material is suggested by the relative absence of intrinsic atherosclerosis in the arterial wall.
 THE AUTOPSY OF A STROKE PATIENT
THE AUTOPSY OF A STROKE PATIENT
 Autopsy studies of stroke should not be used to infer epidemiologic data, since patients that come to necropsy are a highly selected group. However, autopsies are crucial in highlighting vascular mechanisms that are of importance in stroke pathogenesis.
Autopsy studies of stroke should not be used to infer epidemiologic data, since patients that come to necropsy are a highly selected group. However, autopsies are crucial in highlighting vascular mechanisms that are of importance in stroke pathogenesis.


Especially important for infarction/ischemic stroke
 Assess the patency of major neck arteries (carotid/vertebral) by injecting water at their origins and monitoring flow into the cranial cavity after brain removal.
Assess the patency of major neck arteries (carotid/vertebral) by injecting water at their origins and monitoring flow into the cranial cavity after brain removal.
• If any vessel is obstructed, the prosector should attempt to establish:
– site(s) of occlusion/severe stenosis
– cause(s) of occlusion (e.g. atheroma, embolus, dissection, other).
• Vessels (arteries) should be dissected until significant lesions are documented; this sometimes requires painstaking removal of the entire carotid or vertebral artery.
• Dissection of intraosseous portions of the vertebral arteries is facilitated by prior removal of the cervical vertebral bodies. This is achieved from an anterior approach by making a parasagittal saw cut through the medial aspect of the vertebral pedicles (a laterally placed cut will damage the arteries).
 Carefully examine the heart, looking particularly for:
Carefully examine the heart, looking particularly for:
• Endocarditis (infective or non-infective/marantic) – culture/histology of any valvular lesion possibly helpful.
• Any cause of a right-to-left shunt (e.g. septal defect, patent foramen ovale).
• Valvular abnormalities, especially in the left heart (e.g. myxomatous degeneration/prolapse of mitral valve, calcific aortic sclerosis with stenosis).
• Evidence of recent myocardial infarction ± mural thrombus or old infarction with ventricular aneurysm ± mural thrombus.
 Look for old/recent infarcts in other organs, which may suggest a source of embolism.
Look for old/recent infarcts in other organs, which may suggest a source of embolism.

Especially important for hemorrhage:
 In the medical history and general autopsy, seek evidence of disease that may be of etiologic significance, even though this may not be discovered in many individuals:
In the medical history and general autopsy, seek evidence of disease that may be of etiologic significance, even though this may not be discovered in many individuals:
• Hypertension – cardiomegaly with left ventricular hypertrophy, nephrosclerosis.
• Dementia/cognitive impairment – especially in elderly patients this may point to Aβ cerebral amyloid angiopathy (CAA), which is commonly associated with Alzheimer’s disease.
• Neoplasia – hemorrhage in association with metastasis or primary glioma, especially oligodendroglioma.
• History suggesting recreational drug abuse, e.g. amphetamines, heroin.
 When a hematoma is documented at the time of brain removal, the prosector must establish:
When a hematoma is documented at the time of brain removal, the prosector must establish:
• In which compartment it is present and (optimally) in which compartment it originated (e.g. subdural, subarachnoid, combined parenchymal/subarachnoid or intraventricular).
• Its approximate volume and effect(s) on surrounding brain (e.g. edema).
• By dissecting (prior to fixation) the circle of Willis and its major branches, whether a ruptured saccular aneurysm has caused any subarachnoid (or intraparenchymal) hematoma.

 For special studies regarding the evaluation of evacuated intracerebral blood clot, see Chapter 10.
For special studies regarding the evaluation of evacuated intracerebral blood clot, see Chapter 10.
 HYPERHOMOCYSTEINEMIA, ATHEROMA, AND BRAIN INFARCTS
HYPERHOMOCYSTEINEMIA, ATHEROMA, AND BRAIN INFARCTS
 Hyperhomocysteinemia occurs in a variety of inherited disorders, including homocystinuria (secondary to cystathione β-synthase deficiency) and several rare disorders of vitamin B12 or folate metabolism.
Hyperhomocysteinemia occurs in a variety of inherited disorders, including homocystinuria (secondary to cystathione β-synthase deficiency) and several rare disorders of vitamin B12 or folate metabolism.

 Several mechanisms are probably of importance. Homocysteine:
Several mechanisms are probably of importance. Homocysteine:

 FIBROMUSCULAR DYSPLASIA
FIBROMUSCULAR DYSPLASIA FMD shows female predominance.
FMD shows female predominance.
 Familial cases make up ~10% of total.
Familial cases make up ~10% of total.
 FMD usually becomes symptomatic in the 4th or 5th decade, but occurs over a very broad age range.
FMD usually becomes symptomatic in the 4th or 5th decade, but occurs over a very broad age range.
 Connective tissue disorders and α1-antitrypsin deficiency are associated clinical conditions.
Connective tissue disorders and α1-antitrypsin deficiency are associated clinical conditions.
 Ischemic (brain) infarcts may occur secondary to vascular stenosis/occlusion.
Ischemic (brain) infarcts may occur secondary to vascular stenosis/occlusion.
 As many as 20% of patients with carotid dissections show angiographic features of FMD.
As many as 20% of patients with carotid dissections show angiographic features of FMD.
 FMD is rarely associated with formation of aneurysms on intracranial or extracranial arteries.
FMD is rarely associated with formation of aneurysms on intracranial or extracranial arteries.
FIBROMUSCULAR DYSPLASIA (FMD)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Several pathologically distinct subtypes of FMD are described:
 Intimal (up to 10%): There is circumferential or eccentric deposition of collagen in the intima, with fragmented or duplicated internal elastic lamina. Angiography shows long smooth narrowing.
Intimal (up to 10%): There is circumferential or eccentric deposition of collagen in the intima, with fragmented or duplicated internal elastic lamina. Angiography shows long smooth narrowing.


Histologic findings that are characteristic include fibrosis, non-atherosclerotic smooth muscle cell hyperplasia or thinning, destruction of the internal elastic lamina, negligible inflammation, absence of macrophages, and generalized disorganization of arterial wall components (Fig. 9.8).
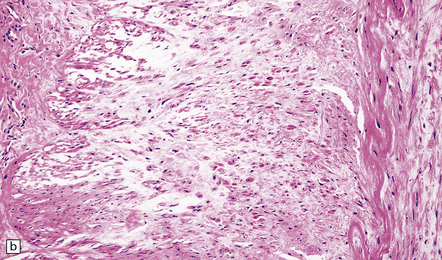
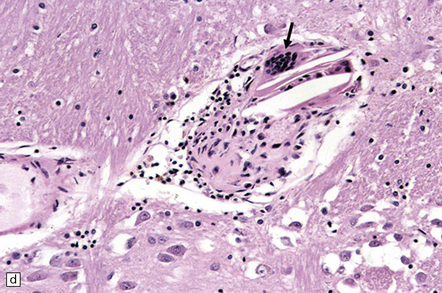
9.8 FMD of extracranial ICA associated with aneurysm formation.
(a) Tunica media showing disorganization and loss of smooth muscle cells. (b) Abnormal orientation and loss of smooth muscle cells. (c) Irregular or disrupted internal elastic lamina. (Courtesy of Drs M Miyauchi and S Shionoya.)
 MOYAMOYA DISEASE
MOYAMOYA DISEASE Annual incidence is approximately 1/1 million.
Annual incidence is approximately 1/1 million.
 Females are affected more commonly than males.
Females are affected more commonly than males.
 Moyamoya disease is most common in Asians and has peak incidences at ages 10–14years and 40–50years.
Moyamoya disease is most common in Asians and has peak incidences at ages 10–14years and 40–50years.

 An upper respiratory tract infection often precedes initial symptoms.
An upper respiratory tract infection often precedes initial symptoms.





 The prognosis is poor if both anterior and posterior branches of circle of Willis are involved.
The prognosis is poor if both anterior and posterior branches of circle of Willis are involved.
MOYAMOYA DISEASE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
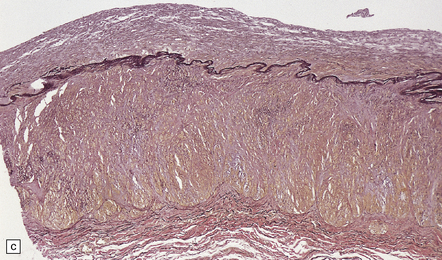
Arterial branches of the circle of Willis show thrombotic lesions in over 50% of patients. Those most commonly affected are the ICA, posterior communicating and posterior cerebral arteries. Severely stenotic non-complicated atherosclerosis, with intimal fibromuscular hyperplasia, but negligible lipid, cholesterol, inflammation, or disruption of the elastica is found (Fig. 9.9). Platelet-fibrin thrombi in various stages of organization are often seen at the intimal surface.
9.9 Moyamoya disease.
(a,b) Sections of internal carotid artery from a 15-year-old boy with Moyamoya disease. There is marked intimal thickening with platelet fibrin thrombi on the intimal surface. (c) Posterior cerebral artery from a 52-year-old woman with Moyamoya disease. The artery contains mural thrombus composed of red blood cells and fibrin. (Courtesy of Professor E Ikeda and Professor Y Hosoda, Tokyo, Japan.)
 INTRACRANIAL ARTERIAL DOLICHOECTASIA (IADE)
INTRACRANIAL ARTERIAL DOLICHOECTASIA (IADE)





 ETIOLOGIC FACTORS IN ARTERIAL DISSECTION
ETIOLOGIC FACTORS IN ARTERIAL DISSECTION







ARTERIAL DISSECTION
This is rare and tends to affect young and middle-aged adults. The dissection (Fig. 9.10) is usually spontaneous, but can be initiated by blunt trauma, often quite mild (e.g. neck injury in a motor vehicle accident or chiropractic manipulation of the neck). The dissection may involve extracranial or intracranial parts of the vertebral artery (more common in women) or carotid artery (more common in men). An intimal tear leads to a medial or subendothelial hematoma. The expanding hematoma may occlude the arterial lumen, usually producing infarction of CNS tissue, less commonly hemorrhage. Dissection of intracranial arteries, especially the vertebral artery, may rarely extend through the adventitia, producing subarachnoid hemorrhage. Dissection of intracranial arteries is likely to become more common as aggressive endovascular revascularization procedures (e.g. thromboembolectomy after ischemic stroke) become more widely utilized in clinical neurologic practice (Fig. 9.11).
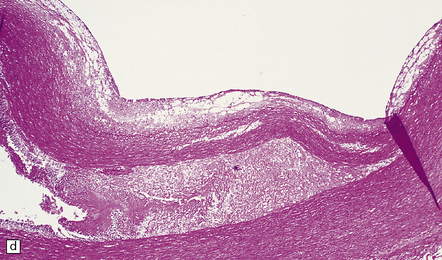
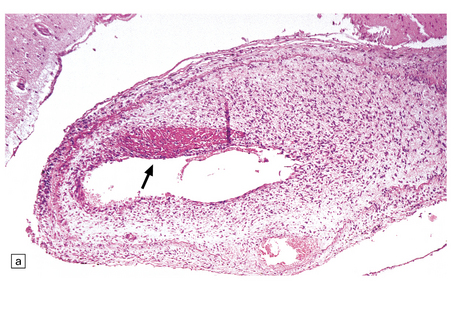
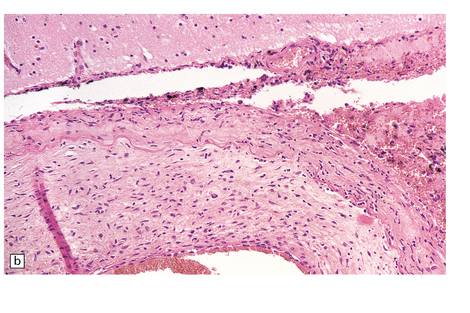
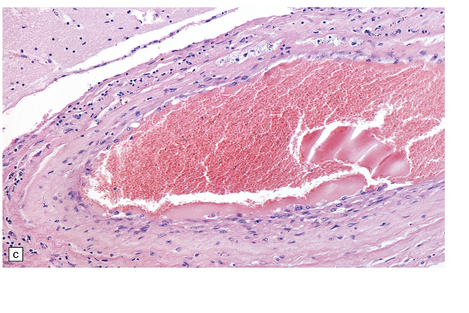
9.10 Vertebral artery dissection following chiropractic neck manipulation (a–c).
(a) Almost circumferential dissection of blood between internal and external elastic laminae. (b) A thin ‘strip’ of blood (arrows) between internal and external elastic laminae. (c) Breach of internal elastic lamina (possibly related to the entry point for the dissection). (Material studied by kind courtesy of Professor Michael A Farrell, Dublin, Ireland.) (d) Intracranial basilar artery dissection in another patient. A subintimal ‘wedge’ of blood extends into the media.
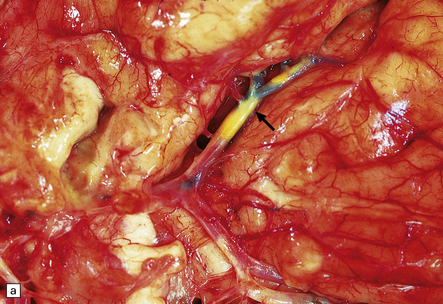
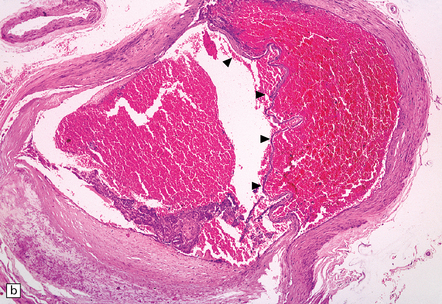
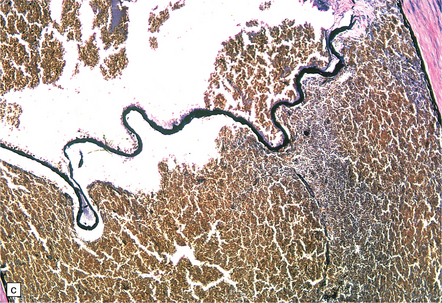
9.11 Dissection of right MCA following attempted thrombectomy using a fine catheter device.
(a) Fresh autopsy specimen showing severe atheromatous plaque in the intra-Sylvian segment of the right MCA (arrow). Note necrosis of adjacent temporal lobe. (b) A relatively non-atherosclerotic segment of the artery showing extensive sub-intimal dissection (arrowheads indicate the ‘raised’ intima, under which is abundant acute hemorrhage). (c) Magnified view showing the elastica overlying fresh blood. (d) A severely atherosclerotic segment of the MCA showing only a small amount of subintimal blood.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Subarachnoid hemorrhage or ischemic infarct with cerebral edema is evident macroscopically. Demonstration of the point of origin of the dissection usually necessitates complete examination of intracranial or extracranial portions of the affected carotid or vertebral artery. A subendothelial or intramural hematoma is found within the vessel wall (Fig. 9.10).
HUMAN IMMUNODEFICIENCY VIRUS (HIV)-ASSOCIATED STROKE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
HIV-associated ‘vasculopathy’ has been described as showing non-specific features, including intimal fibromuscular hyperplasia, fragmentation of the internal elastic lamina, and sometimes aneurysmal dilatation of vessel walls, a pathology consistent with ‘healed arteritis’ (Fig. 9.12). HIV-infected individuals (including those responsive to combined retroviral therapy) may be at risk for accelerated atherosclerosis, though factors responsible for this are unclear. Brain parenchymal arteriosclerotic change has also been observed in HIV-infected individuals that develop cerebral microinfarcts. HIV-1 has been demonstrated immunohistochemically in affected vessel walls (possibly within the cytoplasm of macrophages), but its pathogenic role in this location is unclear.
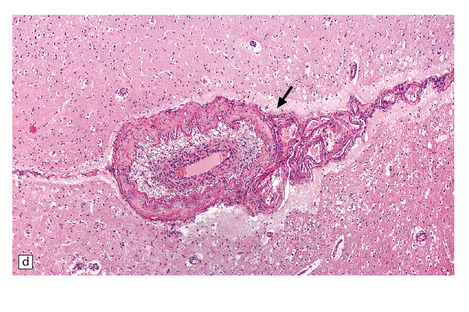
9.12 HIV-associated arteriopathy.
All illustrations are from the brain of a patient with AIDS, which had multifocal areas of necrosis and hemorrhage at necropsy. (a) Eccentric intimal fibromuscular hyperplasia with focal attenuation of the elastic lamina and a small organizing mural thrombus (arrow). (b) Another artery with a relatively intact elastic lamina and superimposed intimal hyperplasia, including some cells with macrophage morphology. (c,d) Two arteries showing intimal hyperplasia with variable numbers of foamy histiocytes in the ‘plaque’. The foam cell component dominates intimal thickening in the artery indicated by arrow in panel (d).
 ANTIPHOSPHOLIPID ANTIBODY (APLA) SYNDROME
ANTIPHOSPHOLIPID ANTIBODY (APLA) SYNDROME
 APLAs are associated with risk of thrombosis in both arterial and venous systems.
APLAs are associated with risk of thrombosis in both arterial and venous systems.




 Most SLE patients in whom thrombotic cerebrovascular disease occurs have high titers of APLAs.
Most SLE patients in whom thrombotic cerebrovascular disease occurs have high titers of APLAs.
 The mainstay of treatment is warfarin or other forms of anticoagulation.
The mainstay of treatment is warfarin or other forms of anticoagulation.
 Sneddon syndrome is the association of livedo reticularis and strokes with APLAs.
Sneddon syndrome is the association of livedo reticularis and strokes with APLAs.




ANGIITIS AND VASCULITIS AFFECTING LARGE ARTERIES


 GIANT CELL ARTERITIS
GIANT CELL ARTERITIS




GIANT CELL ARTERITIS (GCA)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
GCA is characterized by widespread granulomatous inflammation within arterial walls, sometimes causing cerebral infarction. Commonly affected vessels include the aorta and coronary arteries, cerebral arteries, and other head or neck arteries, including the central retinal artery (Fig. 9.13). Multinucleated giant cells are usually a prominent feature of the inflammatory infiltrate, and their cytoplasm may contain fragments of elastica. The polyclonal and polymorphous lymphocytic infiltrate seen within affected arterial walls includes mainly T-cells, with CD4 (helper) cells exceeding CD8 (suppressor) cells, thus implicating cellular immunity in the disease.
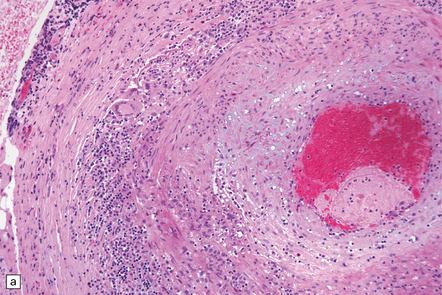
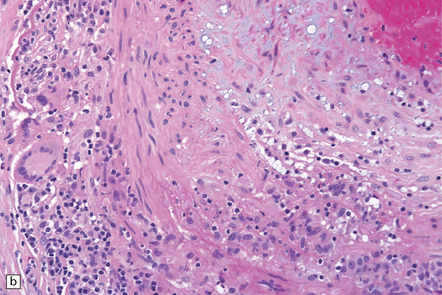
9.13 Giant cell arteritis (GCA).
(a) Temporal artery biopsy showing marked intimal hyperplasia, and transmural chronic inflammation, particularly prominent in the adventitia and including multinucleated giant cells. (b) Magnified view of the inflammatory infiltrate, including several multinucleated giant cells.
PATHOLOGIC FINDINGS AFTER ENDOVASCULAR INTERVENTIONS
The various procedures may result in idiosyncratic surgical or autopsy specimens:
 Thrombectomy specimens (extracted from occluded arteries) generally contain aggregates of platelets/fibrin, linear collections of leukocytes, and erythrocyte-rich regions, but cholesterol clefts or ‘crystals’ (indicating probable atheroemboli) are notably absent. Unexpected findings in such specimens have included mycotic emboli and fragments of atheroma with attached arterial intima. Early endothelialization may be seen within and over the thrombus.
Thrombectomy specimens (extracted from occluded arteries) generally contain aggregates of platelets/fibrin, linear collections of leukocytes, and erythrocyte-rich regions, but cholesterol clefts or ‘crystals’ (indicating probable atheroemboli) are notably absent. Unexpected findings in such specimens have included mycotic emboli and fragments of atheroma with attached arterial intima. Early endothelialization may be seen within and over the thrombus.
 Patients that come to autopsy after unsuccessful thromboembolectomy or post-treatment complications usually show bland/hemorrhagic infarcts, thrombi (residual or post-treatment) and subintimal dissection (Figs 9.14, 9.15).
Patients that come to autopsy after unsuccessful thromboembolectomy or post-treatment complications usually show bland/hemorrhagic infarcts, thrombi (residual or post-treatment) and subintimal dissection (Figs 9.14, 9.15).
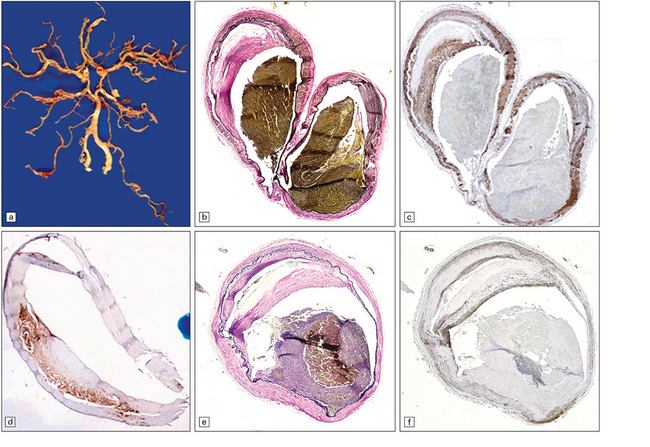
9.14 Endovascular intervention.
These images and those in Fig. 9.15 are from patients who underwent autopsy after thromboembolectomy using a mechanical retriever device. (a) Severely atherosclerotic circle of Willis dissected from base of brain. (b,c) Parallel sections from moderately atherosclerotic left MCA bifurcation. (c) Immunohistochemistry with anti-smooth muscle actin antibody to highlight smooth muscle cell hyperplasia. (d) section of basilar artery immunostained with anti-CD68 antibody to highlight dense accumulation of macrophages in the plaque. (e,f) Adjacent sections through left MCA. (f) Immunohistochemistry with anti-smooth muscle actin antibody.
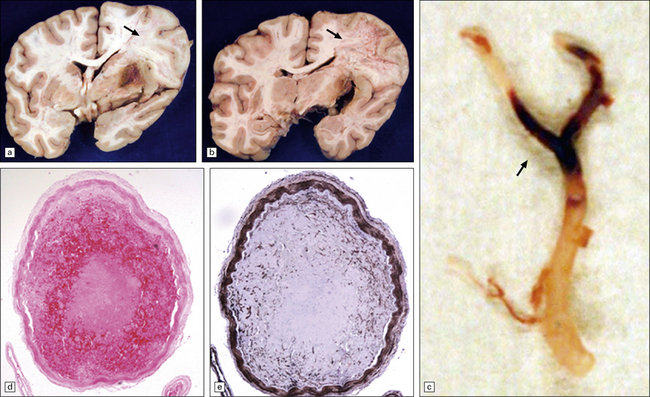
9.15 Endovascular intervention.
These images and those in Fig. 9.14 are from patients who underwent autopsy after thromboembolectomy using a mechanical retriever device. (a,b) Coronal slices of fixed brain from patient that expired 38 days after attempted thromboembolectomy. Note large right MCA territory infarct undergoing liquefactive necrosis (arrows). (c) ‘Saddle’ thromboembolus at right MCA bifurcation. (d,e) Representative cross-sections of right MCA showing a fairly well-organized thromboembolus in a relatively non-atherosclerotic artery. (e) Immunohistochemistry with anti-smooth muscle actin antibody

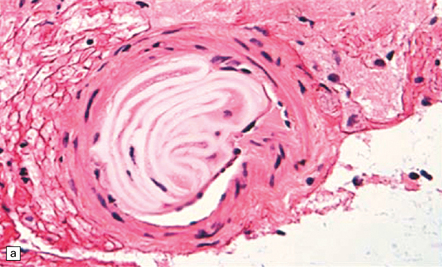
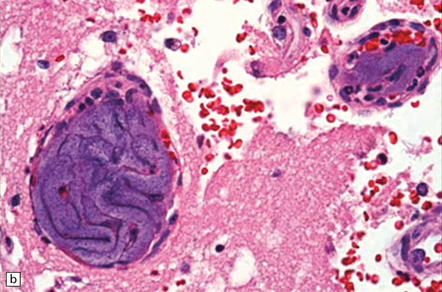
 Even subjects that have not undergone a therapeutic endovascular intervention, but have had intra-arterial procedures (e.g. catheter angiography) may have intravascular foreign materials (e.g. from catheter sheath coverings) that are associated with a brisk foreign body giant cell reaction and brain ischemia (Fig. 9.16). Such materials may rarely be seen in brain biopsies from encephalopathic patients with a past history of many angiographic/endovascular procedures.
Even subjects that have not undergone a therapeutic endovascular intervention, but have had intra-arterial procedures (e.g. catheter angiography) may have intravascular foreign materials (e.g. from catheter sheath coverings) that are associated with a brisk foreign body giant cell reaction and brain ischemia (Fig. 9.16). Such materials may rarely be seen in brain biopsies from encephalopathic patients with a past history of many angiographic/endovascular procedures.
SMALL VESSEL DISEASE (MICROANGIOPATHY)
 arteriosclerosis/arteriolosclerosis (Fig. 9.17)
arteriosclerosis/arteriolosclerosis (Fig. 9.17)
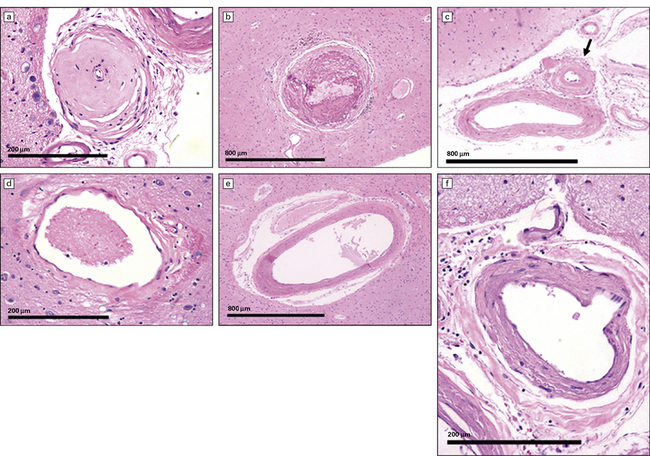
9.17 Arteriosclerosis – arteries/veins/capillaries of various sizes variably affected by disease.
(a,c) small arteries (arrow in c) affected by hyaline mural thickening. (b) Thromboembolus distending a medium-sized parenchymal artery. (d) Parenchymal vein showing slight collagenosis of its wall. (e,f) Small arteries showing moderate ‘onion-skin’ thickening of their walls.


 PRIMARY ANGIITIS (VASCULITIS) OF THE CNS (PACNS)
PRIMARY ANGIITIS (VASCULITIS) OF THE CNS (PACNS)
 Annual incidence is estimated at 2.4/million person-years.
Annual incidence is estimated at 2.4/million person-years.
 Most (~70%) patients are diagnosed by angiography; ~30% by CNS biopsy.
Most (~70%) patients are diagnosed by angiography; ~30% by CNS biopsy.
 Laboratory/hematologic/serologic results are usually normal.
Laboratory/hematologic/serologic results are usually normal.
 CSF is abnormal in 88% of patients – slightly increased WBC count ± protein concentration.
CSF is abnormal in 88% of patients – slightly increased WBC count ± protein concentration.

 Three histopathologies are found in biopsies, with relatively little overlap:
Three histopathologies are found in biopsies, with relatively little overlap:

PRIMARY ANGIITIS OF THE CNS (PACNS)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
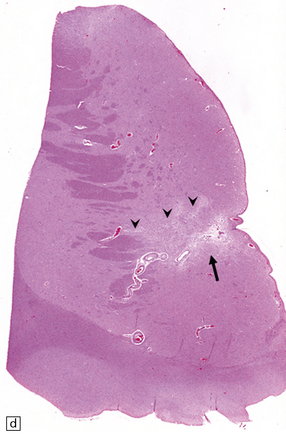
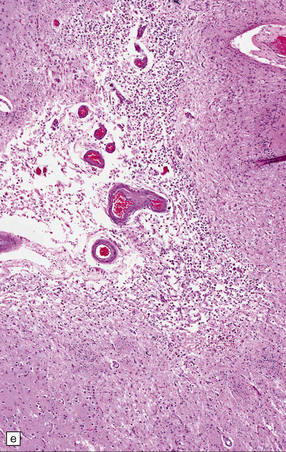
In approximately 20% of PACNS cases, the pathology may be similar to that of systemic giant cell (granulomatous) angiitis, comprising widespread granulomatous inflammation of leptomeningeal and parenchymal blood vessels and occasionally including branches of the circle of Willis (it is therefore not exclusively a microangiopathy). Giant cells may be present within granulomas, admixed with abundant lymphocytes and plasma cells, but conversely may be a negligible component of the pathology. Focal fibrinoid necrosis of affected vessel walls is common (Figs 9.18, 9.19). Rarely, PACNS can affect the spinal cord predominantly or exclusively.
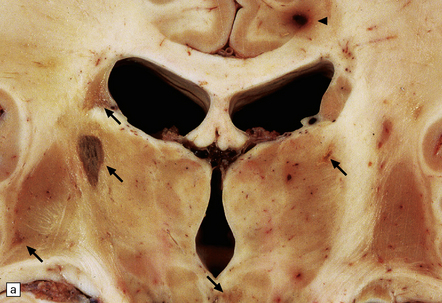
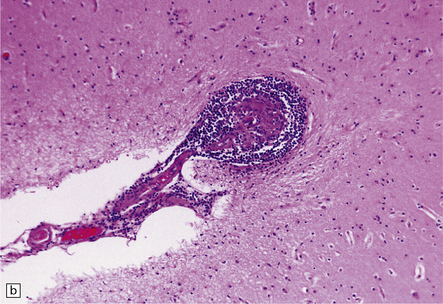
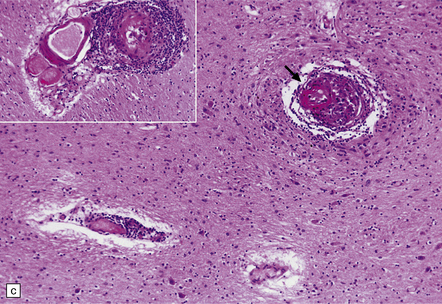
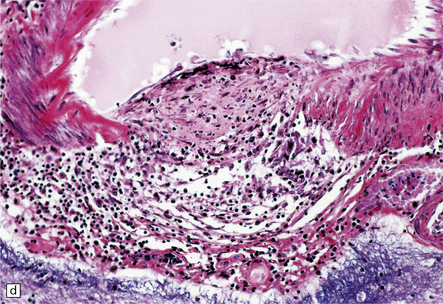
9.18 Primary angiitis of the CNS (PACNS).
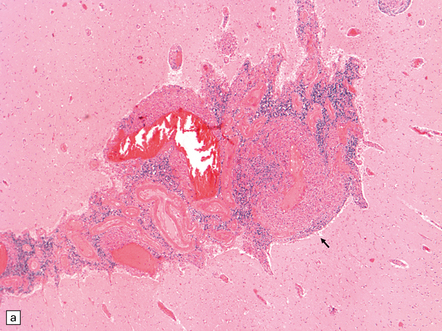
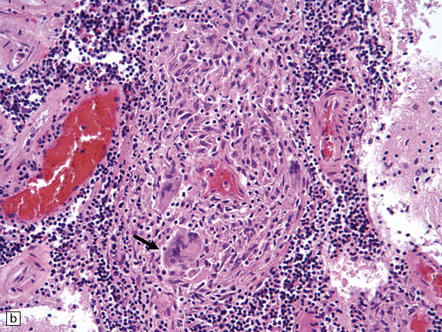
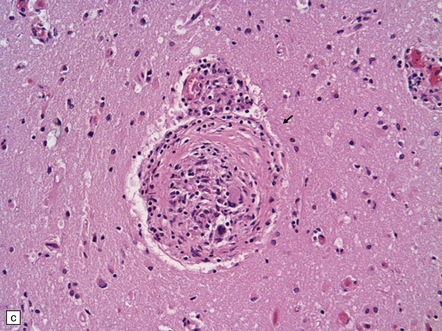
(a) Coronal slice through the basal ganglia and thalamus. Note the scattered ischemic lesions (arrows) and a focus of hemorrhage (arrowhead). (b) Focal granulomatous inflammation at the point of entry of a small leptomeningeal artery into the superficial cortex.(c) Angiitis involving small arteries in the white matter (the inset shows a blood vessel in the base of the pons). Note the discrete perivascular granulomas and focal fibrinoid necrosis (arrow). (d) An affected artery in the putamen. Note the segmental nature of the transmural inflammation, which has destroyed the internal elastic lamina as well as other vessel wall elements.
Severe granulomatous inflammation, presenting as angiitis, is occasionally associated with advanced CAA (Fig. 9.20). Some have designated this entity as amyloid-b-related angiitis (ABRA). Affected patients present and progress differently to those with AD-related CAA, usually showing a more rapid progression of cognitive impairment and a partial response to anti-inflammatory agents. The diagnostic lesson is that, when severe angiitis is encountered in the brain of an elderly individual, associated Aβ CAA should be suspected and appropriately investigated.
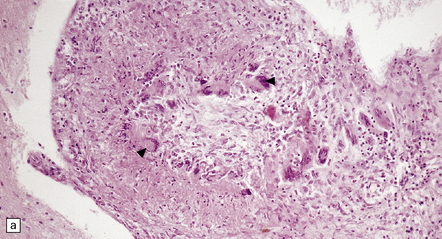
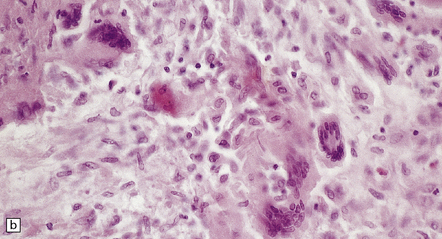
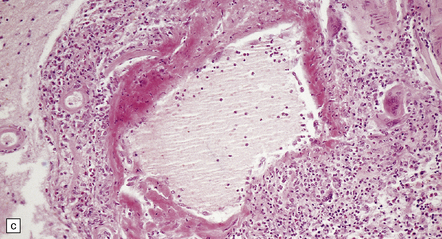
9.20 PACNS associated with severe β-amyloid angiopathy (CAA).
(a) Severe transmural inflammation with occlusion of the arterial lumen and numerous multinucleated giant cells (arrowheads) in vessel wall. (b) Detail of the giant cells shown in (a). (c) Fibrinoid necrosis of another artery from the same patient. (d) Aβ amyloid immunoreactivity (arrows) in walls of both meningeal and cortical arteries.
 MISCELLANEOUS VASCULITIDES AND VASCULITIS-LIKE CONDITIONS
MISCELLANEOUS VASCULITIDES AND VASCULITIS-LIKE CONDITIONS











ANGIITIS DUE TO MISCELLANEOUS VASCULITIDES
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 Polyarteritis nodosa is a multifactorial panarteritis that causes destruction of the elastica and affects small and medium-sized arteries. The acute phase is characterized by neutrophil infiltration of the vessel wall and fibrinoid necrosis. The chronic/healing phase is characterized by vascular fibrosis. End-organ damage results from thrombosis, ischemia and infarction.
Polyarteritis nodosa is a multifactorial panarteritis that causes destruction of the elastica and affects small and medium-sized arteries. The acute phase is characterized by neutrophil infiltration of the vessel wall and fibrinoid necrosis. The chronic/healing phase is characterized by vascular fibrosis. End-organ damage results from thrombosis, ischemia and infarction.


 Lymphomatoid granulomatosis typically involves the lungs, but other organs, including the CNS, can be affected. It is characterized by angiodestructive lesions of parenchymal arteries and arterioles, in which a highly polymorphous transmural infiltrate of atypical leukocytes is almost always combined with fibrinoid necrosis. The disease is now thought in most cases to be a form of diffuse large B cell lymphoma, usually driven by EBV infection and with a prominent and somewhat atypical T cell infiltrate (Figs 9.21, 9.22). Rare angiotropic lymphomas of T-cell or Ki-1 anaplastic large cell type have been reported.
Lymphomatoid granulomatosis typically involves the lungs, but other organs, including the CNS, can be affected. It is characterized by angiodestructive lesions of parenchymal arteries and arterioles, in which a highly polymorphous transmural infiltrate of atypical leukocytes is almost always combined with fibrinoid necrosis. The disease is now thought in most cases to be a form of diffuse large B cell lymphoma, usually driven by EBV infection and with a prominent and somewhat atypical T cell infiltrate (Figs 9.21, 9.22). Rare angiotropic lymphomas of T-cell or Ki-1 anaplastic large cell type have been reported.
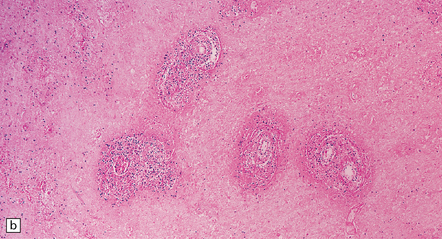
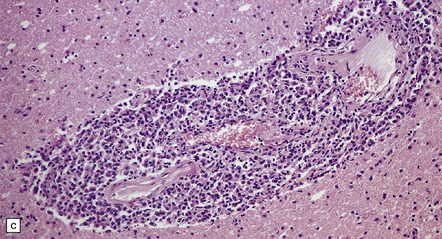
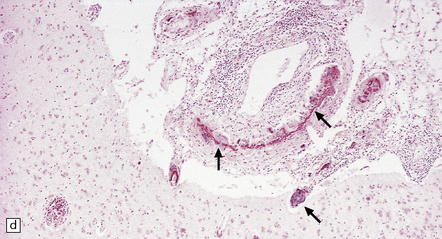
9.21 Diffuse large B cell lymphoma (pathology previously described as lymphomatoid granulomatosis) in a patient with AIDS.
(a) Section of brain showing a focus of necrosis adjacent to the left lateral ventricle. (b) Section from a necrotic region that includes small vessels with transmural and adventitial inflammatory infiltrates. (c) A non-necrotic focus showing a polymorphous inflammatory infiltrate, primarily in the vascular adventitia. (d) Severely inflamed vessel wall with patchy fibrinoid necrosis (arrows).
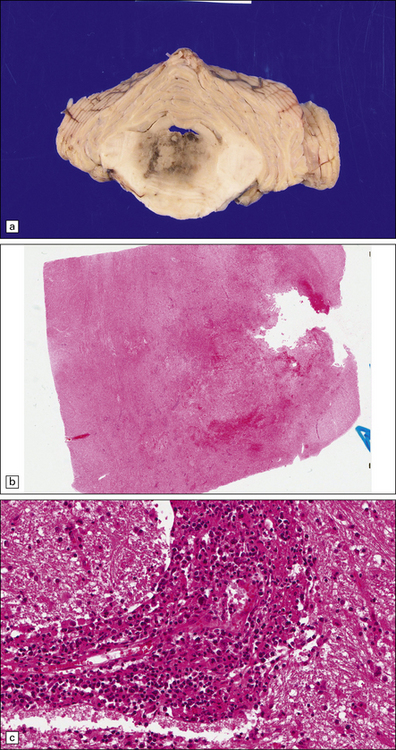
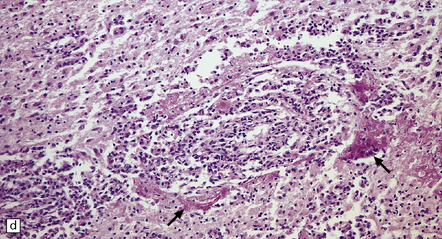
9.22 Diffuse large B cell lymphoma (pathology previously described as lymphomatoid granulomatosis).
This lesion presented in an elderly man on steroid therapy for autoimmune hepatitis that had progressed to cirrhosis. (a) Poorly demarcated hemorrhagic and necrotic mass in pons. (b,c) Histology showing extensive areas of hemorrhagic necrosis, thickened blood vessels, and a pronounced infiltrate of polymorphous moderately atypical lymphoid cells. (Numerous infiltrating cells were immunoreactive for Epstein–Barr virus antigen.)
MICROVASCULOPATHIES ASSOCIATED WITH DEMENTIA
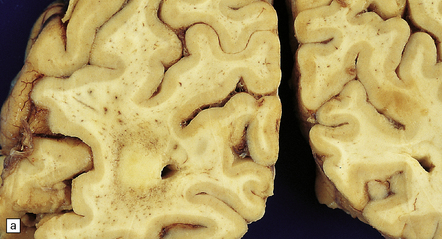
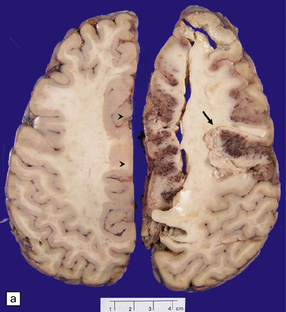
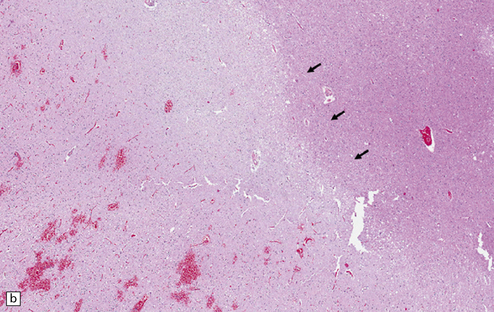
‘Pure’ ischemic-vascular dementia (IVD), with negligible Alzheimer disease pathology, is also known as multi-infarct dementia, is quite rare, and has varied neuropathologic substrates. These include large infarcts and a combination of cystic, lacunar and micro-infarcts throughout the brain, each associated with different forms of cerebrovascular disease (Fig. 9.23). Infarction may result from basal or cervical (carotid/vertebral) atherosclerosis and intraparenchymal microvascular disease. Hippocampal injury that manifests as small glial scars or mimics the hippocampal sclerosis seen in temporal lobe epilepsy may also be evident. ‘Sub-infarctive’ ischemic lesions, e.g. within subcortical white matter, may contribute to IVD, but their etiology is poorly characterized. Occasionally, white matter arteries show adventitial fibrosis and enlarged perivascular spaces, though surrounding brain tissue can be remarkably unaffected in terms of reactive change; mild proliferation of microglia and astrocytes may be one manifestation of such borderline ischemia.
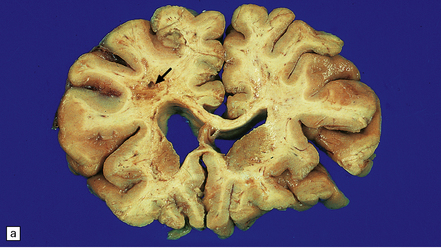
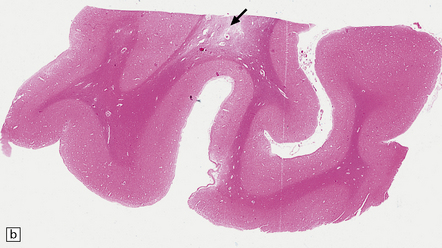
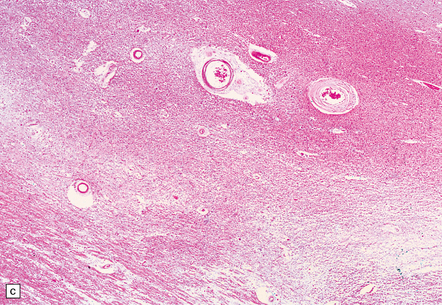
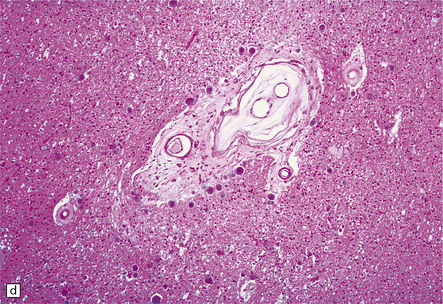
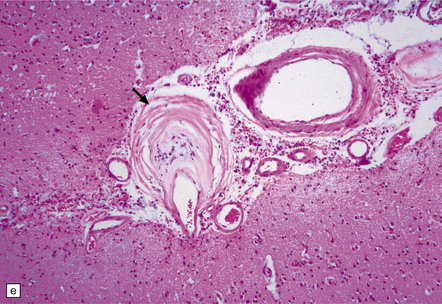
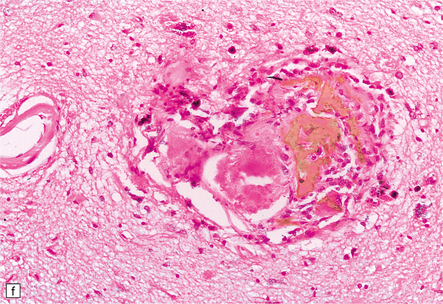
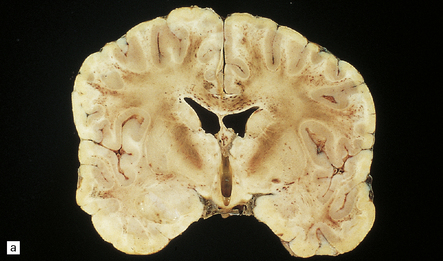
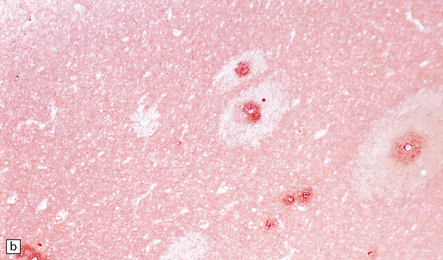
9.23 Ischemic-vascular/multi-infarct dementia.
(a) Coronal section through the brain of an elderly professor with a long history of neurologic deterioration. Note severe encephalomalacia of the white matter, asymmetric between the cerebral hemispheres, with a lacunar infarct (arrow) in the left centrum semiovale. Subsequent microscopic sections are all from this patient’s brain. (b) Whole mount section (right parietal region) showing leukomalacia (arrow), but preservation of the overlying cerebral cortex. (c) Transition between relatively preserved white matter (containing sclerotic arteries) at top of panel, and white matter with cystic microcavitation, pallor and astrocytic gliosis (bottom of panel). (d) Sclerotic white matter arteries (from superior temporal region) showing severe adventitial fibrosis. (e) Variably thickened meningeal arteries. One shows a Charcot–Bouchard type microaneurysm (arrow). (f) Parenchymal artery showing possible remnant of a Charcot–Bouchard microaneurysm, with fragments of an arterial wall filled and surrounded by blood breakdown products.
 BINSWANGER’S SUBCORTICAL ARTERIOSCLEROTIC LEUKOENCEPHALOPATHY
BINSWANGER’S SUBCORTICAL ARTERIOSCLEROTIC LEUKOENCEPHALOPATHY




MACROSCOPIC AND MICROSCOPIC APPEARANCES
There is diffuse, often poorly demarcated (unless lacunes are present) white matter cavitation and softening; well-defined foci of necrosis may be seen (Fig. 9.24). Microscopic sections confirm infarction and reactive change. Microvascular changes are usually those of arteriosclerosis/lipohyalinosis, with prominent ‘onion skin’ type thickening of affected arteries. Microscopy also reveals astrocytic gliosis, microglial activation and an inflammatory response (Fig. 9.24). Chronic lympho-histiocytic inflammation may sometimes be seen in thickened arterial walls.
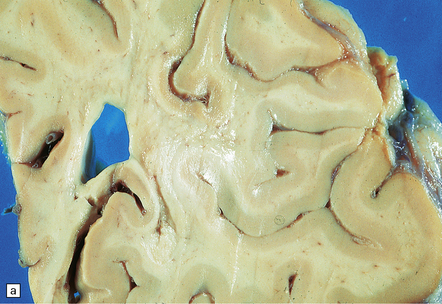
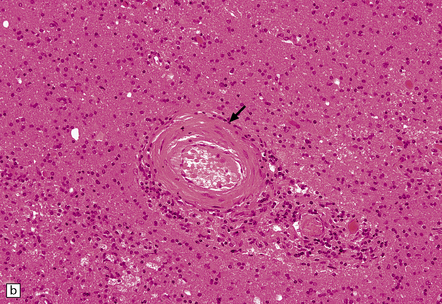
9.24 BSAL.
(a) Coronal section of fixed brain (parietal lobe) showing poorly delineated regions of leukomalacia, with an intact overlying cerebral cortex. (b) Representative histologic section from white matter showing a thickened artery (arrow) with an ‘onion skin’ type appearance. Note also scattered chronic inflammatory cells in the adventitia. Many other vessels throughout the cerebral white matter showed similar features. (c) Another representative section of white matter, immunostained with a primary antibody to a macrophage/microglial marker (CD68) showing a prominent microglial/macrophage infiltrate within white matter, accentuated in the walls of blood vessels (arrow).
 MISCELLANEOUS MICROANGIOPATHIES
MISCELLANEOUS MICROANGIOPATHIES
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)





















MACROSCOPIC AND MICROSCOPIC APPEARANCES
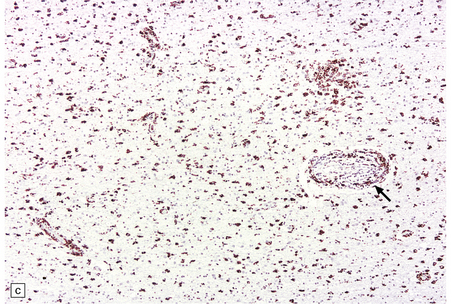
These include cortical and subcortical atrophy with myelin loss, astrocytosis and small foci of cystic encephalomalacia in the subcortical white matter (Fig. 9.25). Small arteries show fibrosis and replacement of smooth muscle cells in the media by eosinophilic, periodic acid-Schiff (PAS)-positive, Congo red-negative, granular material (Fig. 9.26). Ultrastructural studies reveal compact electron-dense GOM around remaining smooth muscle cells (Fig. 9.25).
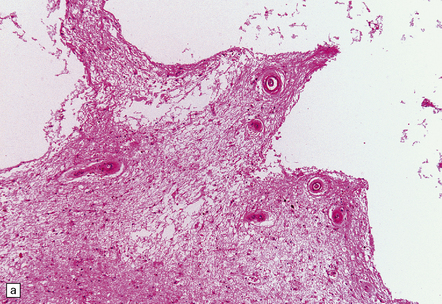
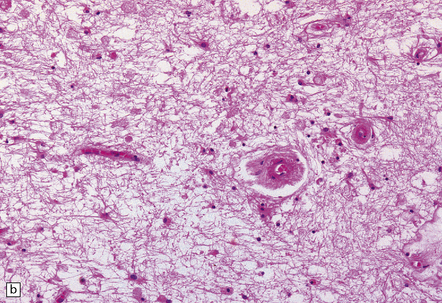
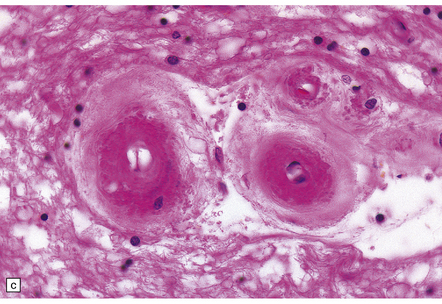
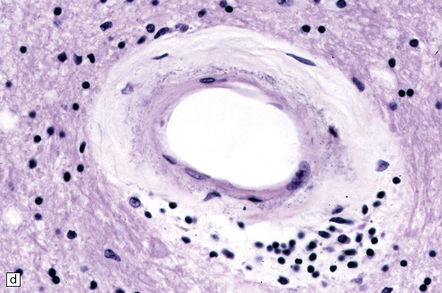
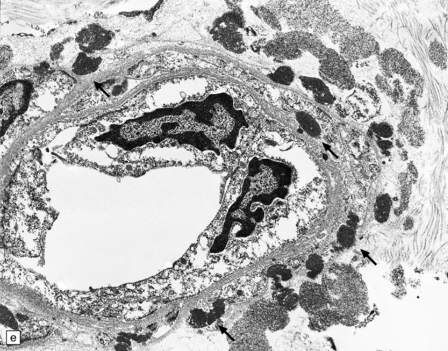
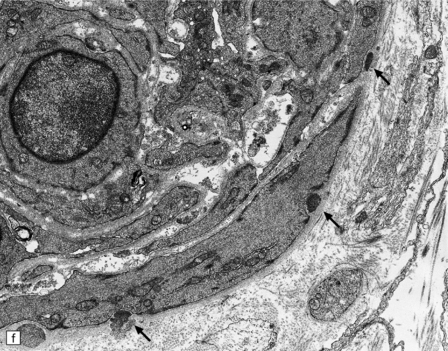
9.25 CADASIL.
(a) The deep cerebral white matter is rarefied and gliotic, and includes cavitated infarcts. (b) Higher magnification reveals a meshwork of astrocytes, and scattered macrophages. The tunica media of arteries and arterioles within the abnormal white matter has been largely replaced by granular, deeply eosinophilic material. (c) The granular material in the vessel walls is more obvious at high magnification. (d) Characteristic adventitial fibrosis, in which a few lymphocytes may be found, as well as punctate material immediately adjacent to media. (e) Electron microscopy of a leptomeningeal arteriole reveals numerous aggregates of granular osmiophilic material (arrows) adjacent to smooth muscle cells. (f) In most but not all patients with CADASIL, aggregates of granular osmiophilic material can also be detected in skin biopsies, adjacent to smooth muscle cells in the walls of dermal arterioles. The abnormal aggregates are typically most numerous on the abluminal aspect of the muscle cells, and tend to be situated in small indentations in the plasmalemma (arrows).
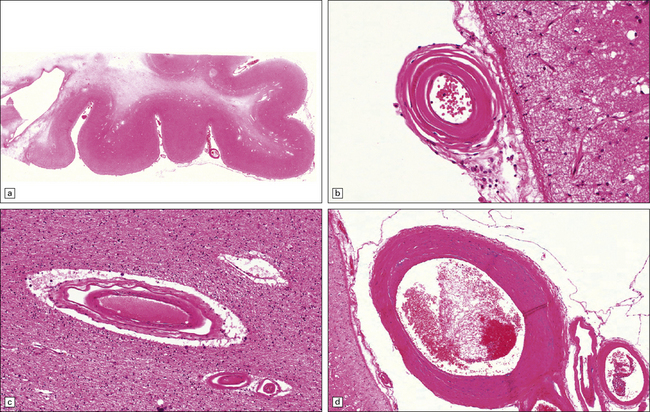
9.26 CARASIL.
(a) Diffuse pallor of subcortical white matter. (b–d) Meningeal and parenchymal arteries showing degrees of atherosclerotic and arteriosclerotic change. (Case studied by kind courtesy of Professors Takao Masaki, Shigeo Murayama, Fukutake and Hajime Miyata, Japan.)
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (MAEDA syndrome)
Clinically, there is presenile dementia (age of onset 29–50years), together with alopecia and spondylosis. Pathologically, there is an ischemic non-hypertensive microangiopathy, with white matter infarcts. The arterial changes resemble non-complicated atherosclerosis with intimal hyperplasia and affect slightly larger blood vessels than those involved in CADASIL (Table 9.1). Arteriolar sclerotic changes are also noted (Fig. 9.26).

Cerebroretinal vasculopathies and hereditary endotheliopathy with retinopathy nephropathy and stroke (HERNS)
These may be similar or identical entities. HERNS is a multi-system disorder that has an IVD-like component. Only one necropsy has been performed on a patient with HERNS. Cavitation was evident in the subcortical white matter, extending into the basal ganglia (Fig. 9.27). Other features were large areas of ischemic necrosis and small unusual angiomatoid formations surrounded by calcifications (Fig. 9.28).
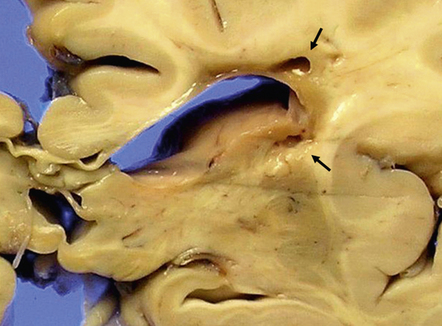
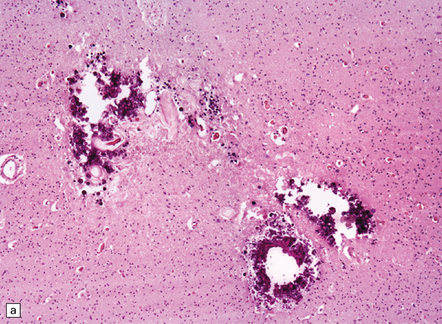
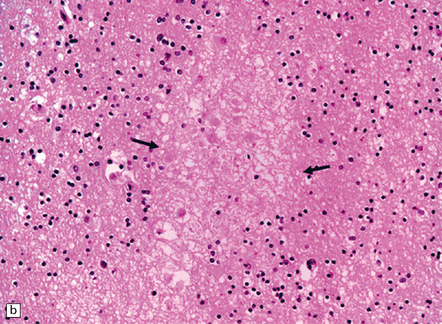
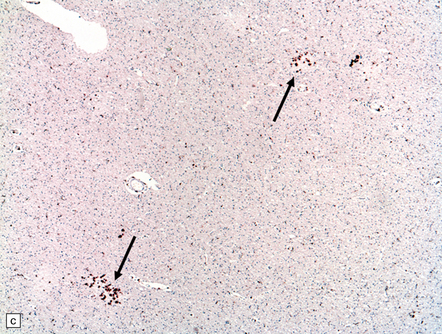
9.28 HERNS.
(a) Large calcified concretions associated with angiomatoid vascular formations. (b) Microinfarction (arrows), predominantly in subcortical white matter, identified by tissue rarefaction and neuroaxonal spheroids. (c) Small collections of CD68-immunopositive macrophages highlighting areas of infarction (arrows).
MISCELLANEOUS (SPORADIC) MICROANGIOPATHIES
Systemic lupus erythematosus is more commonly associated with brain infarcts than hemorrhages. Stroke may result from emboli associated with Libman–Sacks endocarditis or hypertension associated with renal disease. Microinfarcts and small hemorrhages may be found in affected brains, but true vasculitis is rarely observed. Hyaline change in the walls of thickened arteries is, however, sometimes associated with perivascular lymphocytes (Fig. 9.29).
9.29 Microangiopathy associated with SLE.
(a) Small blood vessels showing thickened hyalinized walls. This patient died of non-neurologic disease, but had scattered microinfarcts throughout the CNS. (b) Perivascular lymphocytes.
Thrombotic thrombocytopenic purpura preferentially involves gray matter. An interaction of platelets with platelet-aggregating factor or unusual multimers of factor VIII:von Willebrand factor may cause abnormal platelet agglutination resulting in microvascular thrombi and end-organ (including cerebral) ischemia (Fig. 9.30).
9.30 Microangiopathy of TTP.
Brain section from 39-year-old woman with TTP. Multiple cerebral regions of necrosis and hemorrhage were identified at necropsy. Microvessels contain microthrombi and have plump endothelium.
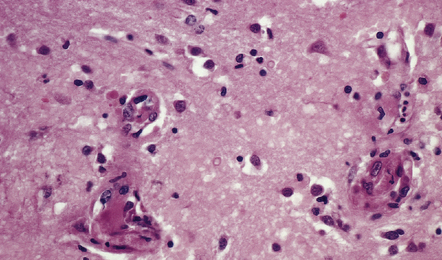
Siderocalcinosis and ferruginization are the encrustation of microvessels with calcium and iron respectively. Arteries and arterioles, particularly those in the basal ganglia and endplate region/dentate fascia of the hippocampus, are affected. The encrustation is accompanied by variable collagenous thickening of the intima and narrowing of the vascular lumen (Fig. 9.31). Capillaries may also be affected (Fig. 9.32). In a few elderly people, some of whom have hypoparathyroidism, many small and medium-sized blood vessels in the basal ganglia and cerebellum become calcified. This vascular change is also frequently noted in the CNS of patients with AIDS and can occur in mitochondrial encephalopathies.
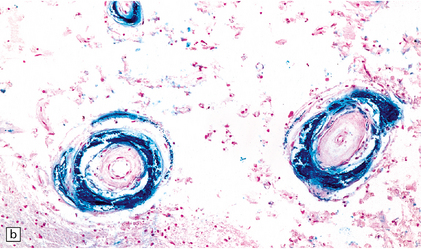
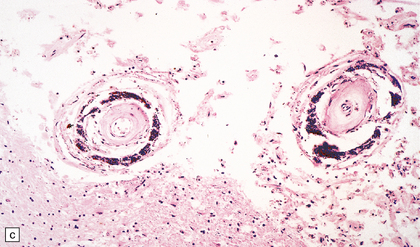
9.31 Siderocalcinosis and ferruginization of microvessels in the globus pallidus.
(a–c) Dense mineral deposits in the vascular media and adventitia. The intima is fibrotic, but the mineralization and intimal fibrosis are not usually associated with specific parenchymal abnormalities or necrosis. Focal ischemic change may, however, occur in severe cases. (b) Perls Prussian blue to demonstrate iron. (c) von Kossa’s technique to show calcium.
EMBOLIC DISEASES (STROKE)
An embolic infarct may occur when any solid/particulate material:



The resultant infarct is usually:
 clinically abrupt in onset due to sudden cessation of blood flow
clinically abrupt in onset due to sudden cessation of blood flow
 hemorrhagic, possibly because of dissolution of the embolus with re-establishment of blood flow into necrotic tissue or into viable tissue in which blood vessel walls have been rendered necrotic (Figs 9.33, 9.34).
hemorrhagic, possibly because of dissolution of the embolus with re-establishment of blood flow into necrotic tissue or into viable tissue in which blood vessel walls have been rendered necrotic (Figs 9.33, 9.34).
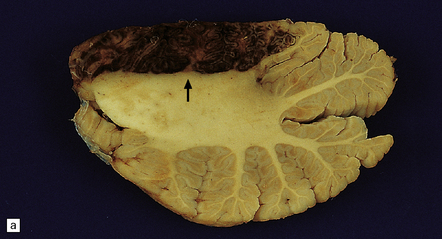
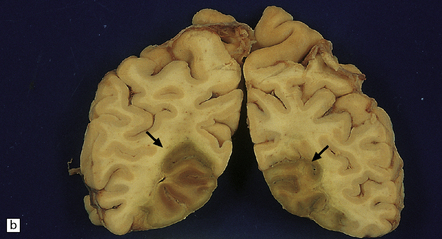
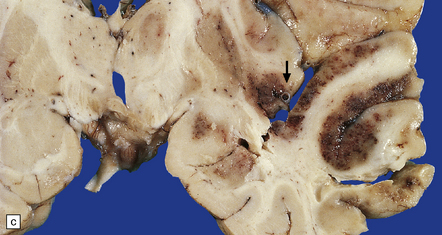
9.33 Hemorrhagic embolic infarction.
(a,b) Acute and organizing hemorrhagic infarcts, in the superior cerebellum (arrow) and inferomedial parieto-occipital region (arrows), respectively, in brain sections from two elderly patients with basilar artery atherosclerosis and distal artery-to-artery emboli of atheromatous material. (c) Recent hemorrhagic infarct involving the superior part of the temporal lobe, the insula, and inferior part of the frontal lobe and resulting from an embolus, which is visible in the MCA (arrow).
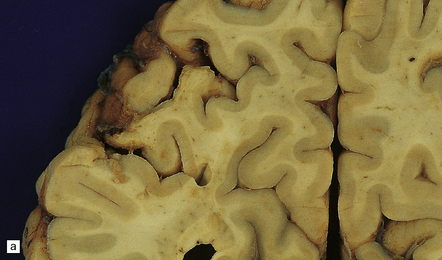
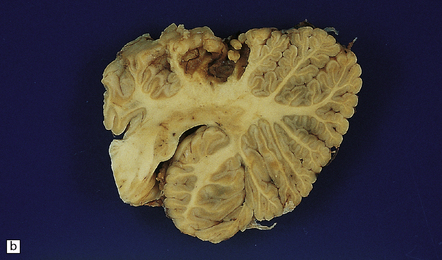
The differential diagnosis of embolic stroke includes:


 SOURCES OF BRAIN AND SPINAL CORD EMBOLI
SOURCES OF BRAIN AND SPINAL CORD EMBOLI
 Plaques of complicated (calcified and focally necrotic) atherosclerosis within the aorta and its branches serve as a nidus for material that can embolize distally to produce brain/spinal cord infarction. ‘Showers’ of atheroemboli passing from the aorta through radicular branches to the spinal cord may cause progressive myelopathy.
Plaques of complicated (calcified and focally necrotic) atherosclerosis within the aorta and its branches serve as a nidus for material that can embolize distally to produce brain/spinal cord infarction. ‘Showers’ of atheroemboli passing from the aorta through radicular branches to the spinal cord may cause progressive myelopathy.
Emboli associated with cardiac lesions are an important cause of ischemic stroke, especially in young people who are relatively free of atherosclerosis. Principal sources of cardiogenic emboli are:
 Thrombus originating in the non-contractile left atrium of a patient with atrial fibrillation.
Thrombus originating in the non-contractile left atrium of a patient with atrial fibrillation.

 Endocarditis, either infective (e.g. in association with a prosthetic valve or after rheumatic fever) or non-bacterial thrombotic (marantic) endocarditis; resultant emboli may be either septic (Fig. 9.35) or composed of platelets/fibrin (Fig. 9.36).
Endocarditis, either infective (e.g. in association with a prosthetic valve or after rheumatic fever) or non-bacterial thrombotic (marantic) endocarditis; resultant emboli may be either septic (Fig. 9.35) or composed of platelets/fibrin (Fig. 9.36).
9.35 Embolism due to (infective) bacterial endocarditis.
Section of MCA from a man with HIV who developed sepsis due to Streptococcus viridans and bacterial endocarditis of the mitral valve. Note the large thrombus surrounded by masses of inflammatory cells (arrows) and blood cells outside the internal elastic lamina. Had the patient survived, an infective (mycotic) aneurysm might have developed at this site.

 Fragments of valvular material that break away during cardiothoracic surgical or endovascular therapeutic procedures (Fig. 9.37, and see Fig. 9.40).
Fragments of valvular material that break away during cardiothoracic surgical or endovascular therapeutic procedures (Fig. 9.37, and see Fig. 9.40).
 Paradoxical embolus in association with a right-to-left shunt.
Paradoxical embolus in association with a right-to-left shunt.







 Iatrogenic emboli may result from a variety of materials introduced into the arterial circulation during vascular surgery, angiography, or increasingly common and aggressive endovascular therapeutic procedures. Cotton fiber is one of the most common of these and has a characteristic appearance when examined using polarized light (Fig. 9.40).
Iatrogenic emboli may result from a variety of materials introduced into the arterial circulation during vascular surgery, angiography, or increasingly common and aggressive endovascular therapeutic procedures. Cotton fiber is one of the most common of these and has a characteristic appearance when examined using polarized light (Fig. 9.40).




 CEREBRAL VENOUS (SINUS) THROMBOSIS (CVT)
CEREBRAL VENOUS (SINUS) THROMBOSIS (CVT)
 Women are more commonly affected than men, usually aged 20–40years.
Women are more commonly affected than men, usually aged 20–40years.


 Oral contraceptives are implicated in >50% of cases, prothrombotic conditions in 34% of cases.
Oral contraceptives are implicated in >50% of cases, prothrombotic conditions in 34% of cases.
 Superior sagittal and lateral sinuses are most commonly involved.
Superior sagittal and lateral sinuses are most commonly involved.



CEREBRAL VENOUS (SINUS) THROMBOSIS (CVT)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Macroscopic findings depend upon the interval between onset of CVT and death. The most consistent feature is severe and extensive hemorrhagic necrosis in the brain, often with evolution into parenchymal hematomas (Fig. 9.43). The hemorrhagic infarcts may be indistinguishable from those caused by emboli, except that they are not usually defined by the boundaries of a specific arterial territory. The thrombosed sinus is usually engorged (Fig. 9.44).
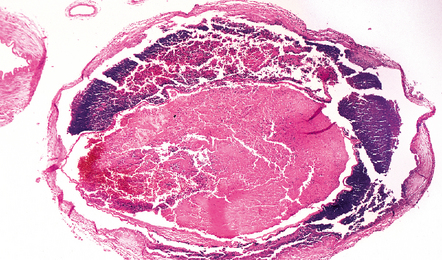
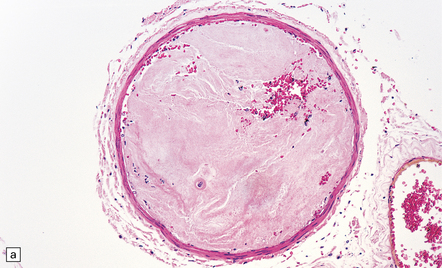
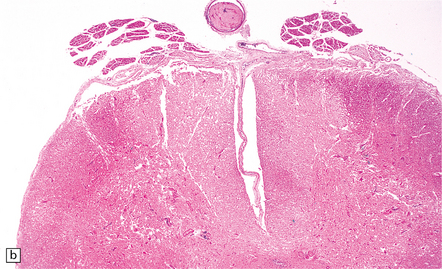
9.36 Cerebrovascular consequences of non-bacterial thrombotic (marantic) endocarditis.
(a) Solid yellow-white ‘saddle embolus’ (arrow) at the MCA bifurcation. (b) Large subacute left hemispheric infarct, with early midline shift, in a 44-year-old woman who had metastatic breast carcinoma complicated by NBTE. (c) Left MCA embolus producing infarct in ‘b’. (d) Mitral valve vegetation that gave rise to embolus. The MCA embolus shows a comparable degree of organization to that seen in the mitral valve vegetation itself.
9.38 Cerebral fat emboli.
(a) Multiple petechial hemorrhages in the cerebral white matter. (b) Frozen section of brain tissue stained for lipid. (Courtesy of Dr Cynthia T Welsh, Charleston, South Carolina.)
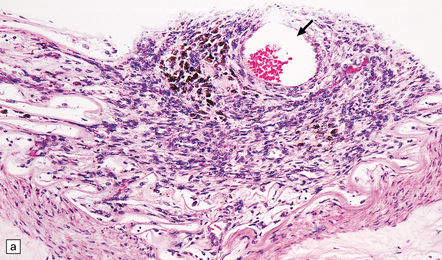
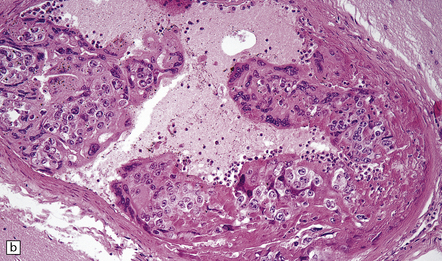
9.39 Neoplastic emboli in the brain.
(a) Neoplastic embolus in a patient who had disseminated sarcoma and an old cerebral infarct underlying the occluded vessel. Note the small recanalized lumen (arrow). (b) Embolic choriocarcinoma in the meningeal artery with focal invasion of the vessel wall and early aneurysm formation. The patient died of multiple cerebral hemorrhages.
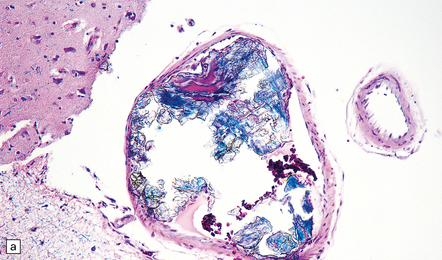
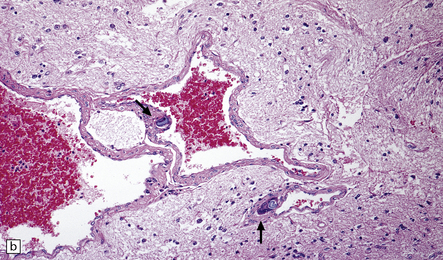
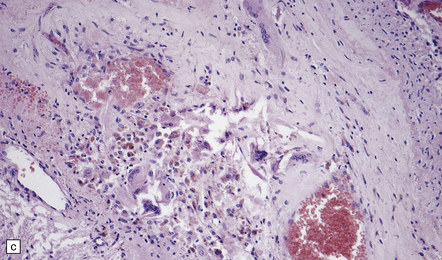
9.40 Intentional or accidental, iatrogenic emboli take many forms.
(a) Particulate material found in meningeal artery from a patient who underwent a complicated cardiothoracic surgical procedure. (Courtesy of Dr Virginia M Walley, Peterborough, Canada.) (b) Small basophilic particles surrounded by foreign body giant cells and embedded within the vessel walls (arrows) of an AVM that was not the target of therapeutic embolization. Such ‘foreign’ materials are particularly likely to lodge in AVMs because of their rapid and turbulent blood flow, which is associated with arteriovenous shunting. (c) Longstanding mesencephalic AVM in a patient who had undergone multiple therapeutic embolizations. A foreign body giant cell reaction is noted in the depths of the AVM. (d) Same section as in (c) viewed after polarization and demonstrating acellular refractile particles. These probably represent cotton fiber introduced during an interventional procedure or angiography.
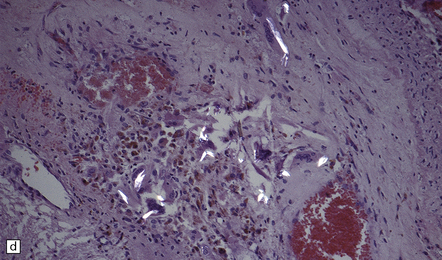
9.41 Embolism of material from intervertebral disc.
(a) Embolus of intervertebral disc material to the anterior spinal artery resulting in an extensive spinal cord infarct (b). (Courtesy of Professor Michael A Farrell, Dublin.)
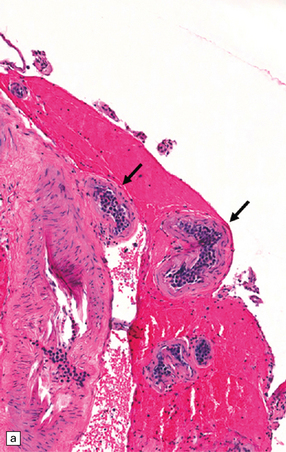
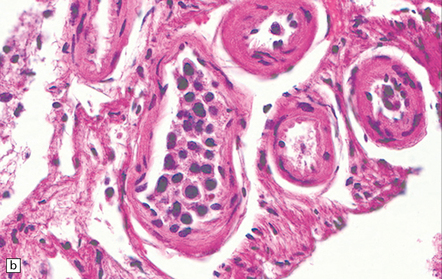
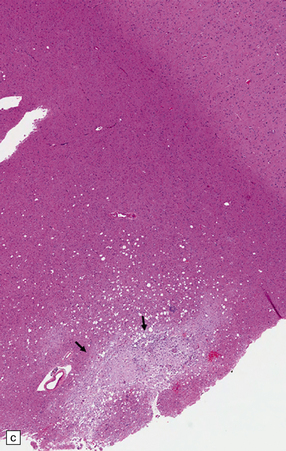
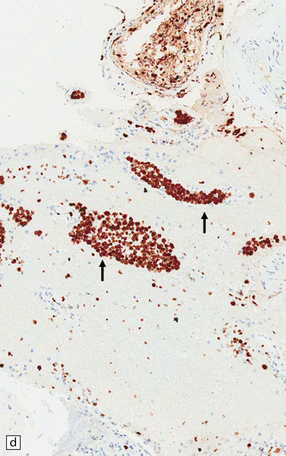
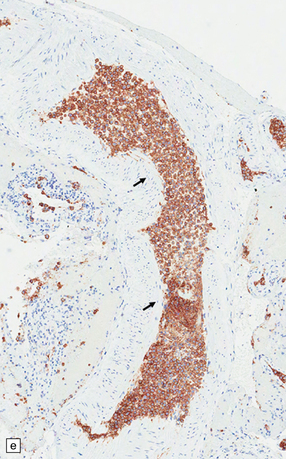
9.42 Disseminated intravascular (angiotropic) large B cell lymphoma.
(a) Many leptomeningeal arteries (arrows) occluded by atypical lymphoid cells. (b) Similar pathology at higher magnification. (c) Adjacent brain parenchyma with acute small infarct (arrows). Immunohistochemistry showed that most intraluminal cells (arrows) were immunoreactive for Ki-67 (d) and CD20 (e).
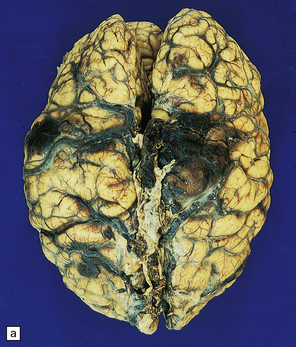
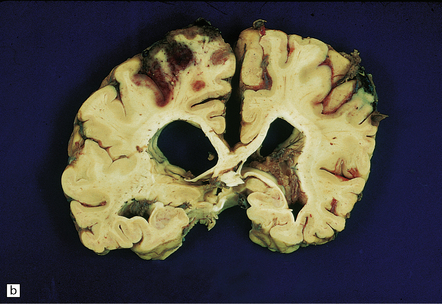
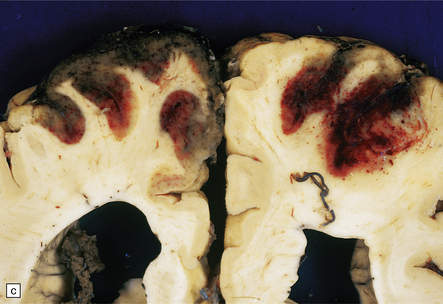
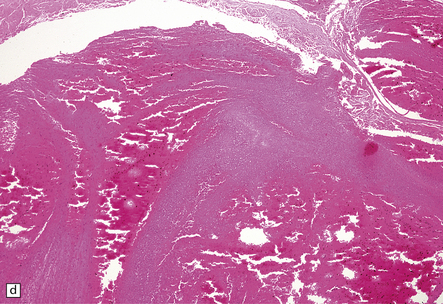
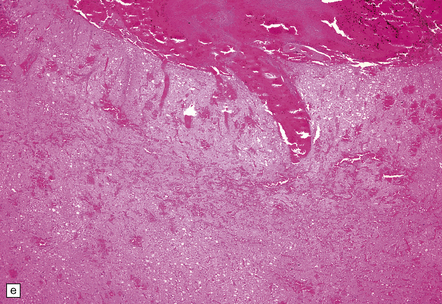
9.43 Venous sinus thrombosis.
This brain is from an elderly woman in a chronic care facility for severe dementia. (a) The cerebral convexities show thrombosed surface veins and irregular asymmetric regions of superficial hemorrhagic necrosis. (b,c) Coronal sections of fixed brain confirm the presence of variably hemorrhagic cortical and white matter necrosis. Enlarged lateral ventricles are related to the patient’s longstanding Alzheimer’s disease. Microscopically, dural vein contains recent thrombus (d), and nearby cortex shows extensive subacute hemorrhagic necrosis (e).
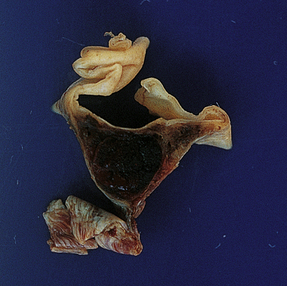
9.44 Superior sagittal sinus thrombosis.
Coronal section through the superior sagittal sinus showing recent thrombus.
Histologic sections show hemorrhagic necrosis (Fig. 9.43). If there is a septic component, microorganisms may be demonstrated with appropriate stains, and inflammatory cells are abundant in the thrombus.
 CORTICAL DYSFUNCTION DUE TO MAJOR CEREBRAL ARTERIAL TERRITORY INFARCTION
CORTICAL DYSFUNCTION DUE TO MAJOR CEREBRAL ARTERIAL TERRITORY INFARCTION
Clinical features of infarction in specific arterial territories:



 HEMATOLOGIC DISORDERS (INCLUDING INHERITED COAGULOPATHIES) INCREASING THE RISK OF DEVELOPING BRAIN INFARCTS
HEMATOLOGIC DISORDERS (INCLUDING INHERITED COAGULOPATHIES) INCREASING THE RISK OF DEVELOPING BRAIN INFARCTS Hematologic disorders cause 1–2% of arterial ischemic stroke and include myeloproliferative disorders, multiple myeloma, lymphoma, chronic lymphocytic leukemia, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, APL antibody syndrome, and factor V mutation associated with lupus anticoagulant.
Hematologic disorders cause 1–2% of arterial ischemic stroke and include myeloproliferative disorders, multiple myeloma, lymphoma, chronic lymphocytic leukemia, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, APL antibody syndrome, and factor V mutation associated with lupus anticoagulant.
 Principal inherited coagulation disorders linked to brain infarcts include:
Principal inherited coagulation disorders linked to brain infarcts include:
• Protein C deficiency. This increases the risk of arterial and venous brain infarction, especially if combined with oral contraceptive use or other prothrombotic conditions (e.g. protein S deficiency). The defective gene maps to chromosome 2q13–14.
• Factor V Leiden mutation (R506Q). This mutation prevents inactivation of factor V by protein C (also known as activated protein C resistance, APC-R). Clinically, there is a risk of developing systemic venous thromboses/thromboemboli. Rarely, factor V Leiden mutation is found in children with brain infarction.
• Protein S deficiency. This is an occasional finding in children or young adults with brain infarcts, and the defective gene maps to 3q11.
• Antithrombin III abnormalities. Deficient or defective antithrombin III is associated with venous thrombosis and is a rare cause of brain infarction.
• Carbohydrate-deficient glycoprotein synthase type I. This rare enzyme deficiency affects synthesis of many coagulation factors and their inhibitors, substantially increasing the risk of venous thromboses and brain infarcts.
CNS INFARCTION
MACROSCOPIC APPEARANCES
 Cystic – usually in the territory of a major artery (e.g. MCA, ACA, PCA) or one of its branches and >1 cm across its maximal dimension.
Cystic – usually in the territory of a major artery (e.g. MCA, ACA, PCA) or one of its branches and >1 cm across its maximal dimension.


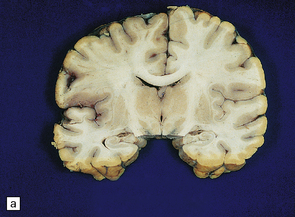
Acute infarction
Changes that develop within 8–36 hours include slight blurring of the gray/white matter interface, dusky discoloration of gray matter and slight softening of the tissue on palpation. These changes may be relatively easy to detect, especially if confined to the territory of a major cerebral artery (Fig. 9.45).
Subacute infarction
Infarction with an antemortem time frame of 2–4 days appears as distinct softening, with blurring of the gray/white matter junction and dusky gray discoloration of brain tissue. At this stage, the most telling abnormality is cerebral edema (Figs 9.46, 9.47).
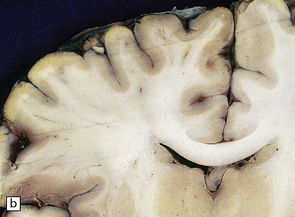
9.46 Acute infarction (a) Left MCA territory infarct, consistent in appearance with having occurred 2–4 days prior to death.
Early ‘cracking’ artefact outlines the region of necrosis (arrows), and there is early edema with shift of midline structures from left to right. (b) Axial cut of another brain at a comparable stage of ‘evolution’ at the time of death. The infarct involves a significant proportion of left MCA territory (arrows).
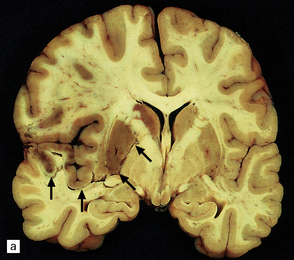
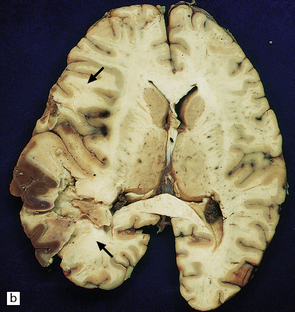
9.47 Subacute infarction.
Extensive left MCA territory infarct with necrosis (especially involving the basal ganglia) with massive left to right shift of midline structures, a major contributing factor to the patient’s death. Patient had experienced left internal carotid artery thrombosis 4–5 days prior to death.
Chronic infarction
 Stage I: As edema subsides antemortem, the region of infarction starts to undergo liquefactive necrosis, eventually leading to cavitation (Fig. 9.48).
Stage I: As edema subsides antemortem, the region of infarction starts to undergo liquefactive necrosis, eventually leading to cavitation (Fig. 9.48).
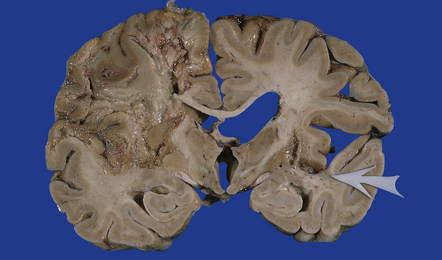
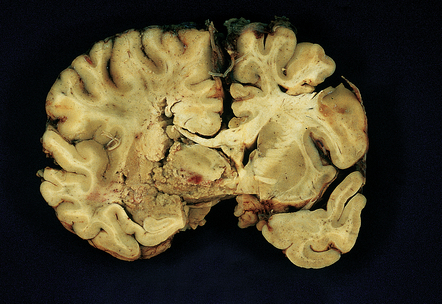
9.48 Subacute infarction.
Liquefactive necrosis in a left MCA territory infarct that occurred a few days before death. Much of the edema has subsided. The arrow in the right cerebral hemisphere indicates foci of cystic encephalomalacia (old infarction) in the right putamen and adjacent insular cortex. Bilateral thrombotic occlusions were noted in both internal carotid arteries.
 Stage II: An old infarct (older than several months) appears as a cystic cavity within the brain substance and is surrounded by atrophic brain tissue (Figs 9.49, 9.50). In some cases a well-defined cavity does not form; instead, there is a ‘moth-eaten’ appearance to the area of encephalomalacia.
Stage II: An old infarct (older than several months) appears as a cystic cavity within the brain substance and is surrounded by atrophic brain tissue (Figs 9.49, 9.50). In some cases a well-defined cavity does not form; instead, there is a ‘moth-eaten’ appearance to the area of encephalomalacia.
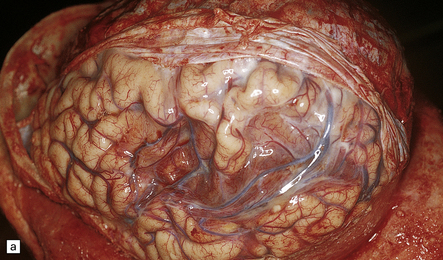
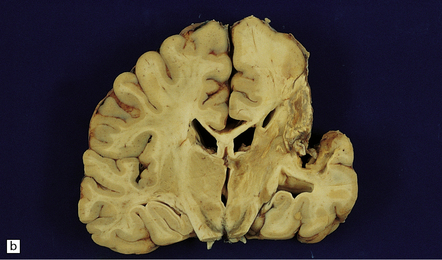
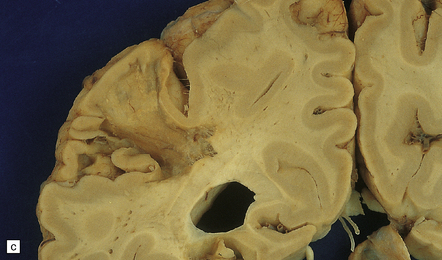
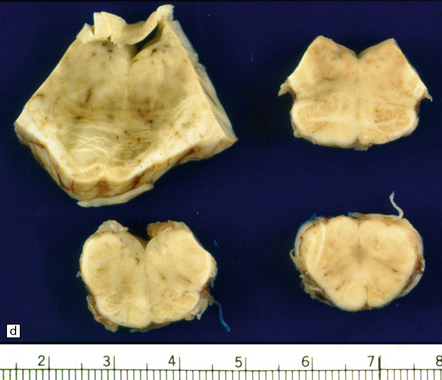
9.49 Old cystic MCA territory infarction (different patients)
(a) Collapse of brain substance (left fronto-parietal region) with apparent opacification and hypervascularity of overlying leptomeninges. (b) Surrounding reactive changes partly due to secondary infection of necrotic tissue, a distinctly unusual complication of cerebral infarction. (c) A predominantly subcortical infarct in the left parietal lobe. (d) Wallerian degeneration in brain stem of patient whose cerebral hemispheres are illustrated in panel (b). Note atrophy in right basis pontis and right pyramid, i.e. ipsilateral to the hemispheric infarct.
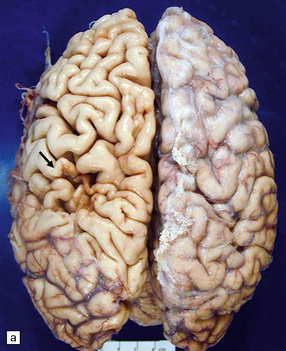
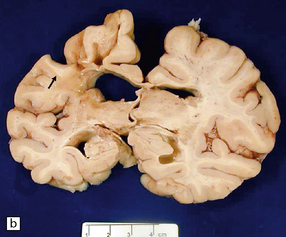
9.50 Old cystic infarction involving portion of left MCA territory.
(a) Cerebral convexities after removal of meninges. Arrow indicates an old left fronto-parietal infarct. Note orange-brown discoloration of the affected parenchyma, suggesting a hemorrhagic component to the infarct. (b) Coronal section from the same brain. The infarct has produced atrophy (arrow). Note compensatory enlargement of the left lateral ventricle.
MICROSCOPIC APPEARANCES
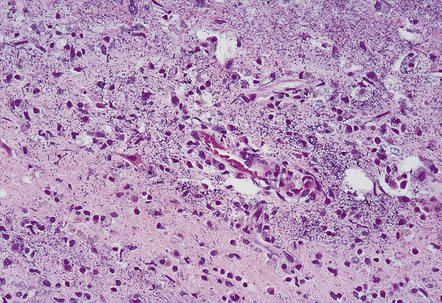
Acute infarction (approximately 1–4 days)
This results in neuronal eosinophilia with pyknosis, as in anoxic-ischemic encephalopathy (see Chapter 8), and is associated with vacuolation of the surrounding neuropil (Fig. 9.51).
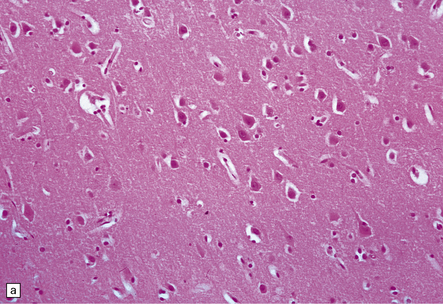
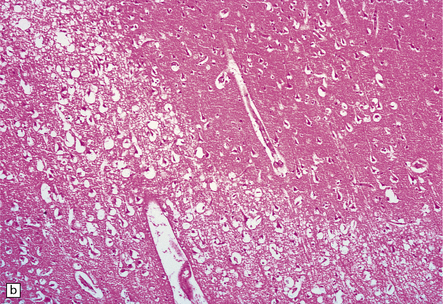
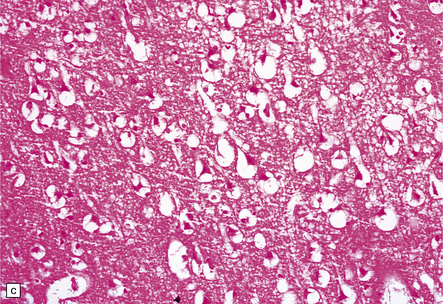
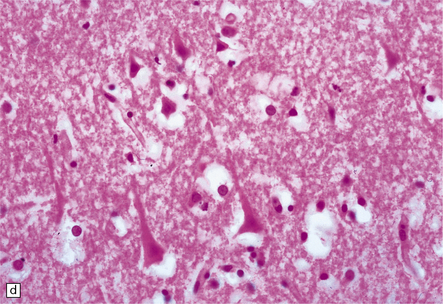
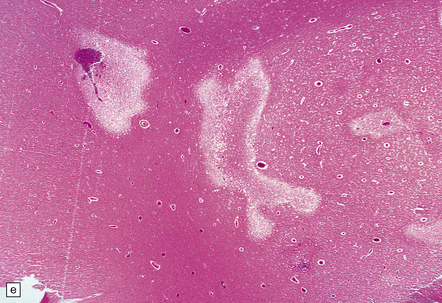
Subacute infarction (approximately 5–30 days)
Neuronal eosinophilia may persist at this stage. Variable degrees of neutrophil infiltration are evident, often adjacent to necrotic microvessels (‘diapedesis’). Early microglial activation and the subsequent formation of foamy macrophages are responses to the tissue damage; the macrophages include many transformed monocytes that have moved into the infarct from the circulation. Endothelial proliferation and neovascularization are found at 5–12 days, but are variable in extent and timing (Fig. 9.52).
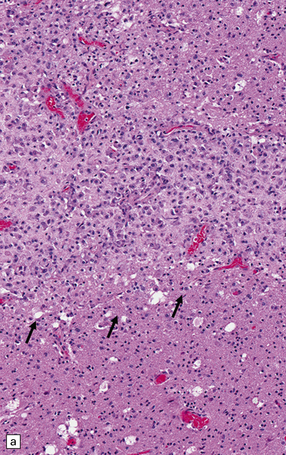
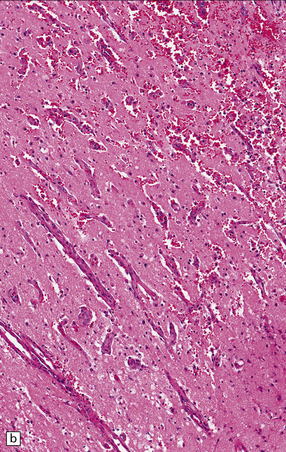
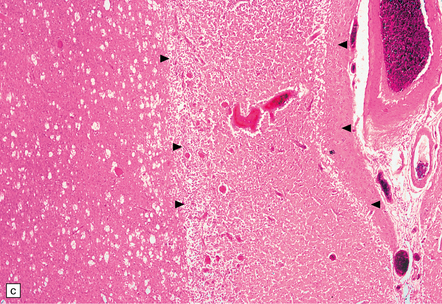
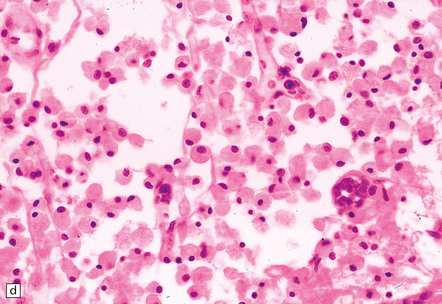
9.52 Histology of subacute to chronic cerebral infarction.
(a) Clear demarcation (arrows) between normal brain (at bottom) and old infarct of 1–2weeks duration (at top). Infarcted tissue is replaced by macrophages, and there is early neovascularization. (b) Infarct examined in brain sections approximately 7–10 days post-stroke. Note the prominent capillaries. (c) Infarct of several weeks duration (between arrowheads). Note dense macrophage infiltration that replaces much of the cortex. There is conspicuous sparing of the superficial cortex (layer I). Vacuolization deep to the infarct may represent edema. (d) Macrophage response. The macrophages that resorb necrotic brain tissue and that may persist for the life of the patient have a characteristic round shape, eccentric nucleus, and foamy or granular cytoplasm, which in some instances contains altered blood pigment, especially if the infarct had a significant hemorrhagic component.
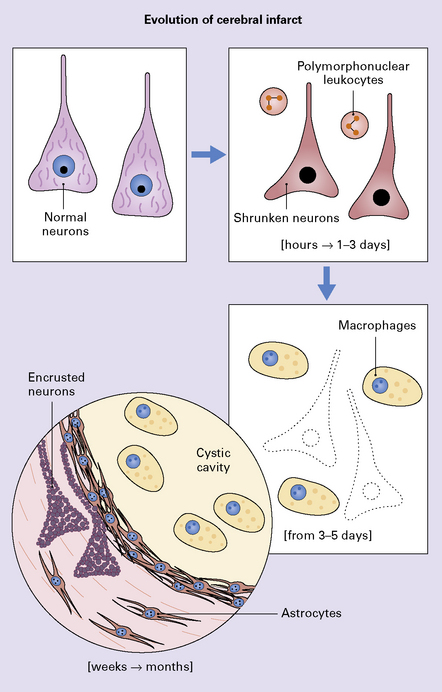
Chronic infarction (weeks to months or years)
Foamy macrophages, often containing hemosiderin (if the infarct had a significant hemorrhagic component), may persist over many months, often as long as the patient lives. Thin-walled blood vessels pass through the cystic cavity. Abundant reactive astrocytes (often with gemistocytic morphology) encircle the infarct. In large cortical infarcts, there is a characteristic pattern of relative preservation of the molecular layer (lamina I), which contains abundant reactive astrocytes (Fig. 9.53). Residual neurons and axons in and around the infarct may become encrusted with iron or calcium (Fig. 9.54). Very rarely, thrombosed vessels adjacent to the infarct are found. The cellular and tissue events in the evolution of an infarct are shown diagrammatically in Figure 9.55. Depending upon the size of the infarct, wallerian degeneration may be noted in the descending (corticospinal) tracts, especially if a cerebral infarct involves a large region of motor cortex or underlying white matter, including the internal capsule (Fig. 9.49).
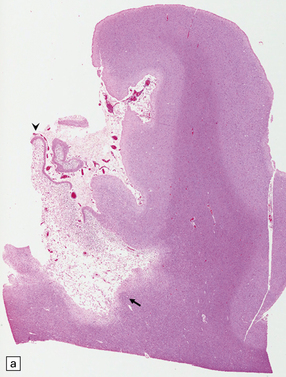
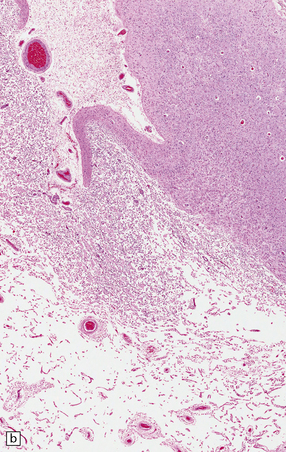
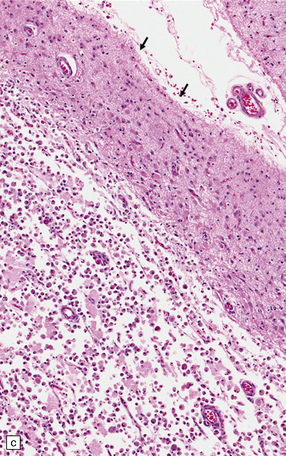
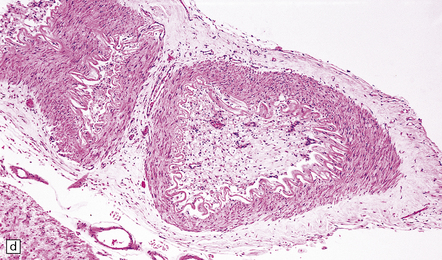
9.53 Histology of ‘old’ (remote) infarction.
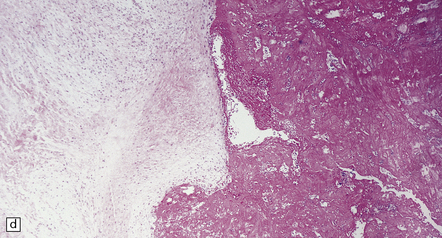
After a few weeks, the ‘age’ of an infarct becomes increasingly difficult to judge. (a) Old cystic infarct (arrow), well demarcated from surrounding intact cortex. Arrowhead indicates a thin rim of preserved subpial tissue. A meningeal artery near the arrowhead shows thrombosis. (b) Magnified view of part of the infarct showing a cystic cavity traversed by a few intact blood vessels and retaining large numbers of macrophages. (c) Evolving cystic cavity. With time, macrophages partly or completely resorb necrotic tissue, leaving a cystic cavity surrounded by a dense rim of reactive astrocytes. Arrows indicate the densely gliotic but preserved cortical layer I, with underlying macrophages. (d) A meningeal artery adjacent to the infarct containing a well-organized thrombus.
INFARCTS CAUSED BY (THROMBOEMBOLIC) OCCLUSION OF LARGE ARTERIES
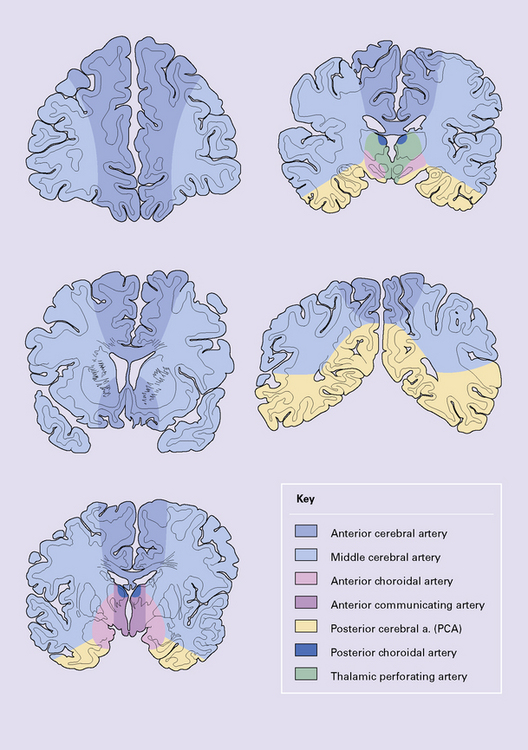
The territories supplied by the major cerebral arteries are illustrated in Figure 9.56. If a destructive lesion corresponds to one of these discrete regions, its etiology is likely to be ischemic, resulting from occlusion of a major cerebral artery (Fig. 9.57). If a destructive lesion appears between two major arterial territories, it is likely to represent a watershed/borderzone infarct (see Chapter 8). Infarcts resulting from thromboembolic occlusion of the ICA resemble (in size and distribution) those caused by MCA thrombosis, though the ACA territory may also be involved (Fig. 9.58). Anatomic territories supplied by branches of the vertebrobasilar system are more variable. An example of an extensive brainstem infarct, which is secondary to basilar artery thrombosis (Fig. 9.59), contrasts with a smaller well-delineated infarct resulting from occlusion of the vertebral artery (Fig. 9.60).
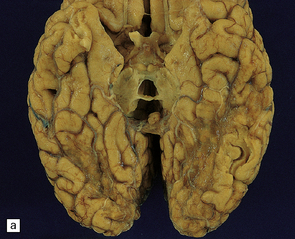
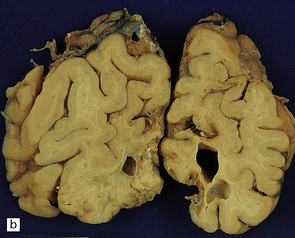
9.57 PCA territory infarction.
(a) View of the undersurface of the brain. (b,c) Coronal slices through the same specimen as in (a). Note that atherosclerosis (unusually) extends even into leptomeningeal arteries.
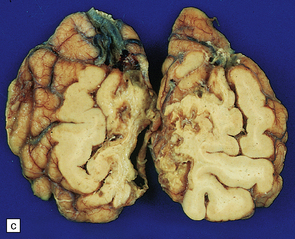
9.58 Hemorrhagic infarction.
Patient was a severe diabetic with unstable ICA atherosclerotic plaque. (a) Extensive acute hemorrhagic infarct involving both MCA (arrow) and ACA (arrowheads) territories. The pattern of petechial hemorrhages in the cortex is typical of hemorrhagic infarction. (b) Microscopic section of one of the infarcts. Arrows indicate demarcation between normal brain and pale infarct. Note numerous petechial hemorrhages are seen within the infarcted tissue.
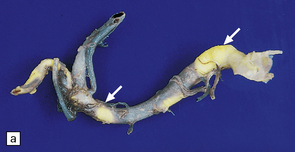
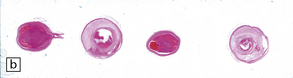
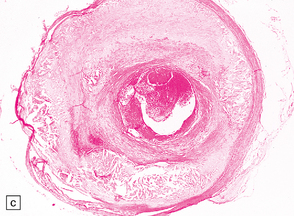
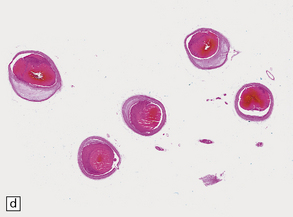
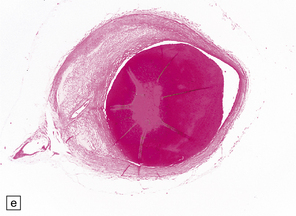
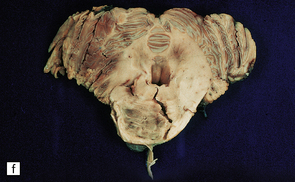
9.59 Basilar artery thrombosis with resultant brain stem infarct.
(a) Severe atheroma at the origin of the basilar artery (arrow at left – union of the vertebral arteries) and at its tip (arrow at right). (b–e) Sections from the basilar artery tip (b) and magnified view of a representative section (c) showing severe complicated atheroma with compromise of the lumen and superimposed thrombus formation. By contrast, the proximal basilar artery (d,e) shows moderate atheroma consisting of eccentric intimal thickening. Thrombus seen at this level (e) probably represents retrograde thrombus consequent to basilar tip occlusion. (f) Resultant necrosis throughout brain stem and cerebellum.
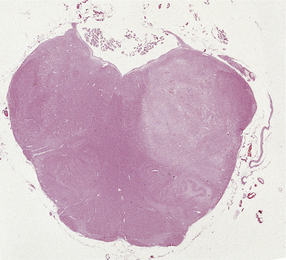
SPINAL CORD INFARCTION
Infarction of the spinal cord is much less frequent than infarction in the brain. Atherosclerosis of the spinal arteries occurs only rarely, and atherosclerotic plaques in the aorta tend not to occlude the ostia of arteries that supply the cord (Fig. 9.61).
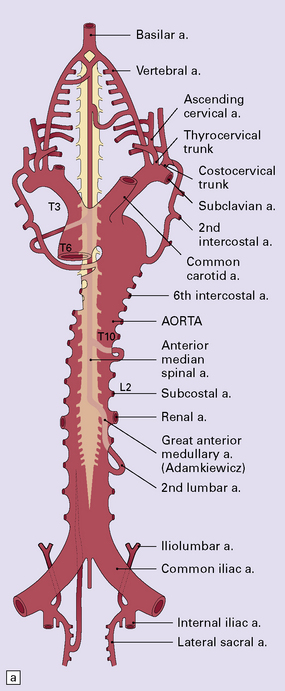
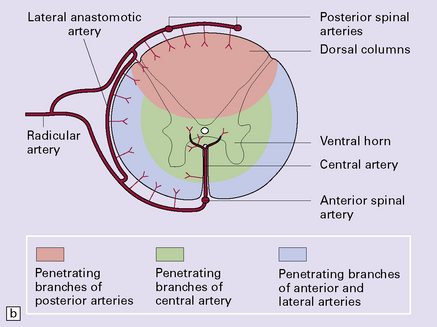
9.61 Blood supply of the spinal cord.
(a) The major arterial branches to the spinal cord. (b) The regions supplied by the arteries of the spinal cord.
Spinal cord infarction is caused by:

• material from complicated atherosclerotic plaques in the aorta (postmortem studies indicate that this may be asymptomatic, depending upon the regions of spinal cord affected)
• fibrocartilaginous material, usually following trauma to the back.
 Severe hypotension (e.g. during a complicated surgical procedure or secondary to trauma).
Severe hypotension (e.g. during a complicated surgical procedure or secondary to trauma).





MICROSCOPIC APPEARANCES
The histologic features of spinal cord infarcts appear and evolve identically to those of brain infarcts (see above). Infarcts in the distribution of the anterior spinal artery are most common and affect the anterior gray matter and adjacent white matter tracts (Fig. 9.62). Infarction resulting from hypotension preferentially affects the anterior zones of the cord and in mild cases may target anterior horn motor neurons, resulting in their loss and a limited astrocytic gliosis. The most vulnerable watershed zone is around the level of T4.
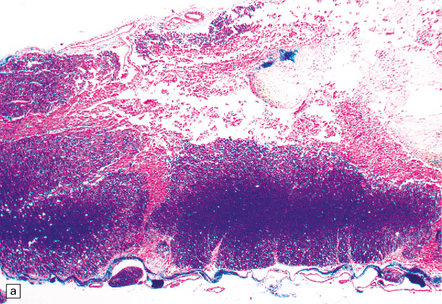
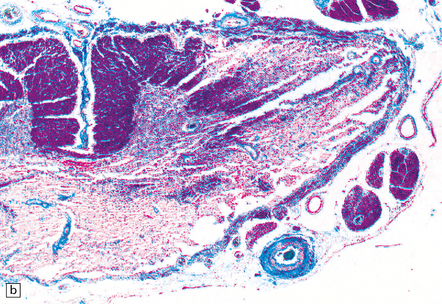
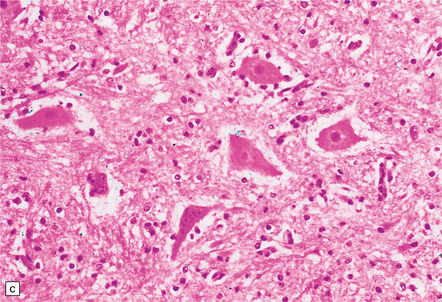
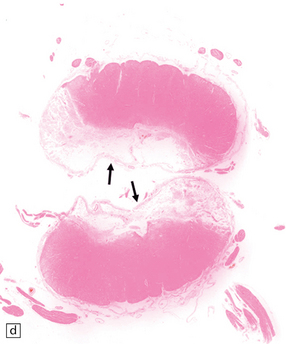
9.62 Spinal cord infarct.
(a) Section through part of the upper thoracic cord, in a patient with anterior spinal artery thrombosis (not shown in this section). There is preservation of most of the posterior columns and left lateral column, but extensive necrosis of the gray matter and right anterior and lateral columns (solochrome cyanin). (b) Section through part of the upper thoracic cord, in a patient with severe atheroma and infarction of the cord in the posterior spinal artery territory. Note the sparing of the anterior columns and most of the gray matter (solochrome cyanin). (c) Eosinophilic neurons in the ventral horn of the spinal cord following a severe episode of hypoxia–ischemia (‘hypoxic myelopathy’). (d) Sections of cervical spinal cord from a young woman who experienced thrombosis of the proximal anterior spinal artery, probably secondary to Factor V Leiden deficiency, which led to a prothrombotic state. She survived the spinal cord infarct by several days. At autopsy, there was extensive cystic cavitation of the anterior third of the cervical cord, including the anterior horns and portions of the anterolateral white matter tracts (arrows indicate anterior surface of each spinal cord section).
FOIX–ALAJOUANINE SYNDROME (ANGIODYSGENETIC MYELOMALACIA)
The surfaces of the caudal and less often rostral cord are covered by tortuous, relatively large caliber blood vessels. By histopathologic examination, these vessels have thickened fibrotic walls lacking an elastic lamina. The cord may show focal atrophy. Zones of necrotic cord are surrounded by small caliber thick-walled tortuous vessels, some of which may become mineralized (Fig. 9.63). Areas of necrosis contain lipid-laden macrophages and are surrounded by astrogliosis. Occasionally, perivascular lymphocytes are seen, though the appearances are not those of vasculitis.
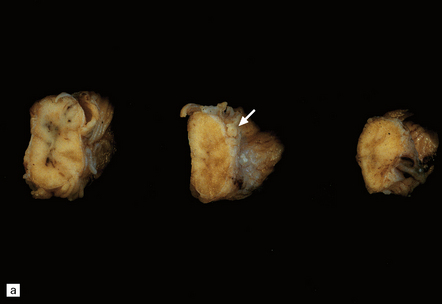
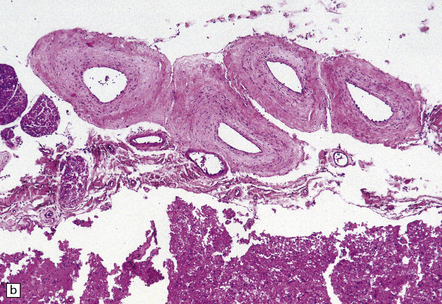
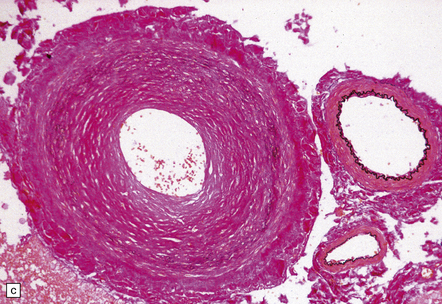
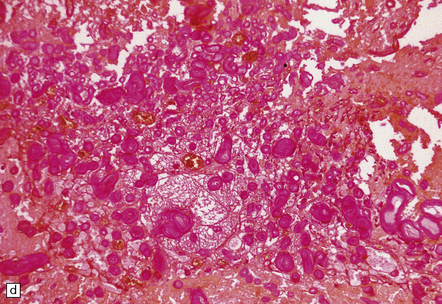
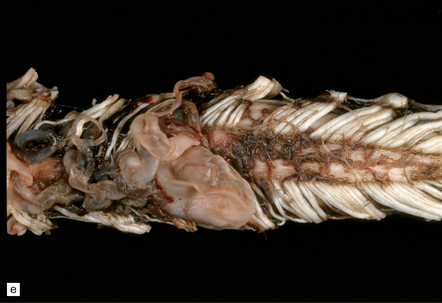
9.63 Foix–Alajouanine syndrome.
(a) Slices through the spinal cord, which is partly necrotic and shows patchy brown discoloration of the parenchyma. Note the large-caliber, thick-walled blood vessel (arrow) on the dorsal aspect of the cord. (b) A section through the tortuous, thick-walled, fistulous blood vessel on the dorsum of the cord. (c) The wall of the fistulous blood vessel consists largely of collagen, with no elastic lamina. Note for comparison an adjacent, normal posterior spinal artery. (d) Typical cluster of fibrotic, thick-walled, small-caliber blood vessels within the spinal parenchyma adjacent to a region of old infarction. The lumen of most of the vessels is completely obliterated by hyaline thickening of the wall. (e) Congenital spinal arteriovenous fistula – an incidental finding at necropsy. Note the tortuous, large-caliber but still thin-walled blood vessel on the dorsum of the cord. It is likely that this type of arteriovenous fistula leads eventually to the development of Foix–Alajouanine syndrome. (Courtesy of Dr Michael Ashworth, Royal Liverpool Children’s NHS Trust.)
WATERSHED OR BORDERZONE INFARCTS
These usually occur in elderly individuals with significant atherosclerosis that experience one or more episodes of prolonged hypotension and reduced CNS perfusion (e.g. intraoperatively, while under general anesthesia, or after cardiac arrest). Watershed infarcts are also a complication of markedly increased intracranial pressure, especially after head injury. The infarcts are usually wedge-shaped, with their base at the pial surface. Infarction may be symmetrical within the cerebral hemispheres and focally hemorrhagic (an appearance attributed to reperfusion of necrotic tissue) (Fig. 9.64).
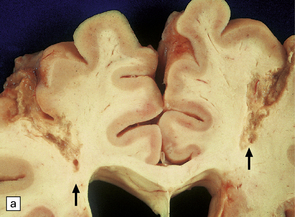
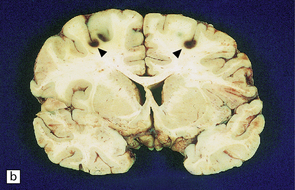
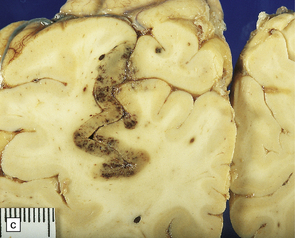
9.64 Watershed infarcts in coronal slices of brain from three patients.
(a) Asymmetric ACA/MCA watershed zone infarcts (arrows), with wedge-shaped areas of old necrosis extending from cortex into deep white matter. (b) Smaller more prominently hemorrhagic ACA/MCA watershed infarcts (arrowheads). (c) Section from parieto-occipital region showing a hemorrhagic ACA/MCA watershed infarct.
LACUNAR INFARCTS (LACUNES)
A lacunar infarct is small (≤1.0 cm across its maximal dimension) and often due to microvascular disease (Fig. 9.65), but it can also be embolic. Lacunar infarcts occur in regions where hypertensive microvascular disease is most common (basal ganglia, pons, internal capsule).
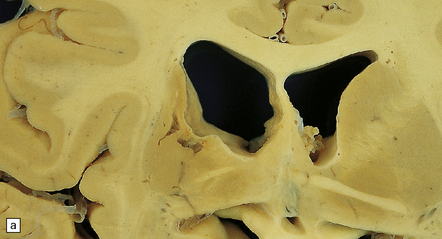
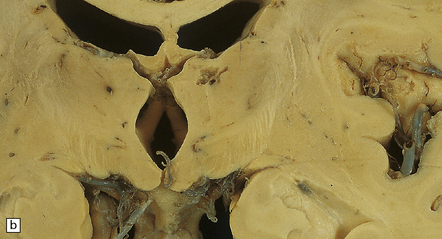
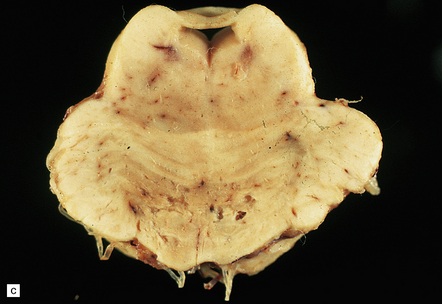
9.65 Lacunar infarcts in the basal ganglia.
(a) Lacunar infarcts in left caudate, putamen, and internal capsule. They are so extensive that there is compensatory enlargement of the overlying left lateral ventricle. (b) Lacunar infarcts in the right diencephalon and thalamus at the coronal level of the mamillary bodies. (c) Lacunar infarcts in the basis pontis imparting a ‘moth-eaten’ appearance to this structure. (d) Lacunar infarcts within the basal ganglia. The largest lacune in this section (arrow) has caused cavitation within the caudate nucleus, resulting in ‘pitting’ of the overlying ependymal surface. Arrowheads indicate small regions of rarefaction. Note microatheroma within small parenchymal arteries adjacent to the lacunes. (e) Higher magnification showing shared microscopic features between lacunes and infarcts. There are collections of macrophages within the lacunar cavity, which is usually surrounded by an irregular border containing reactive astrocytes.


REFERENCES
Balami, J.S., Chen, R-L, Grunwald, I.Q., et al. Neurological complications of acute ischemic stroke. Lancet Neurol.. 2011;10:357–371.
Barnett, H.J.M. Forty years of progress in stroke. Stroke.. 2010;41:1068–1072.
Batjer, H.H. Cerebrovascular disease. Philadelphia: Lippincott-Raven, 1997;1276.
Caplan, L.R. Stroke thrombolysis – growing pains. Mayo Clin Proc.. 1997;72:1090–1092.
Caplan, L.R., Arenillas, J., Cramer, S.C., et al. Stroke-related translational research. Arch Neurol.. 2011;68(9):1110–1123.
Carmichael, S.T. Cellular and molecular mechanisms of neural repair after stroke: Making waves. Ann Neurol.. 2006;59:735–742.
Demaerschalk, B.M., Hwang, H-M, Leung, G. US cost of ischemic stroke: A systematic literature review. Am J Manag Care.. 2010;16:525–533.
Fisher, M. Clinical atlas of cerebrovascular disorders. London: Mosby-Wolfe; 1994.
Hachinski, V., Norris, J.W. The acute stroke. Philadelphia: FA Davis; 1985.
Jickling, G.C., Sharp, FR. Blood biomarkers of ischemic stroke. Neurotherapeutics.. 2011;8:349–360.
Marsh, J.D., Keyrouz, S.G. Stroke prevention and treatment. J Am Coll Cardiol.. 2010;56:683–691.
Mohr, J.P., Wolf, P.A., Grotta, J.C., et al. Stroke. Pathophysiology, diagnosis, and management, 5th ed., Philadelphia: Elsevier Saunders, 2011.
Saver, J.L. Proposal for a universal definition of cerebral infarction. Stroke.. 2008;39:3110–3115.
Sierra, C., Coca, A., Schiffrin, E.L. Vascular mechanisms in the pathogenesis of stroke. Curr Hypertens Rep.. 2011;13:200–207.
Stehbens, W.E. Pathology of the cerebral blood vessels. St Louis: Mosby; 1972.
Vinters, H.V. Cerebrovascular disease – practical issues in surgical and autopsy pathology. Curr Top Pathol.. 2001;95:51–99.
Whisnant, J.P., Wiebers, D.O., O’Fallon, W.M., et al. A population-based model of risk factors for ischemic stroke: Rochester, Minnesota. Neurology.. 1996;47:1420–1428.
Atherosclerosis and atheroemboli
Adraktas, D.D., Brasic, N., Furtado, A.D., et al. Carotid atherosclerosis does not predict coronary, vertebral, or aortic atherosclerosis in patients with acute stroke symptoms. Stroke.. 2010;41:1604–1609.
Barnett, H.J.M., Gunton, R.W., Eliasziw, M., et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA.. 2000;283:1429–1436.
Fisher, C.M., Gore, I., Okabe, N., et al. Atherosclerosis of the carotid and vertebral arteries – extracranial and intracranial. J Neuropathol Exp Neurol.. 1965;24:455–476.
Masuda, J., Yutani, C., Ogata, J., et al. Atheromatous embolism in the brain: A clinicopathologic analysis of 15 autopsy cases. Neurology.. 1994;44:1231–1237.
Ogata, J., Masuda, J., Yutani, C., et al. Mechanisms of cerebral artery thrombosis: a histopathological analysis on eight necropsy cases. J Neurol Neurosurg Psychiatry.. 1994;57:17–21.
Ogata, J., Yamanishi, H., Ishibashi-Ueda, H. Review: Role of cerebral vessels in ischemic injury of the brain. Neuropathol Appl Neurobiol.. 2011;37:40–55.
Ogata, J., Yutani, C., Otsubo, R., et al. Heart and vessel pathology underlying brain infarction in 142 stroke patients. Ann Neurol.. 2008;63:770–781.
Resch, J.A., Okabe, N., Loewenson, R.B., et al. Pattern of vessel involvement in cerebral atherosclerosis. J Atheroscler Res.. 1969;9:239–250.
Ross, R. Atherosclerosis – an inflammatory disease. N Engl J Med.. 1999;340:115–126.
Torvik, A., Jorgensen, L. Thrombotic and embolic occlusions of the carotid arteries in an autopsy material. Part 1. Prevalence, location and associated diseases. J Neurol Sci.. 1964;1:24–39.
Torvik, A., Jorgensen, L. Thrombotic and embolic occlusions of the carotid arteries in an autopsy series. Part 2. Cerebral lesions and clinical course. J Neurol Sci. 1966;3:410–432.
Yasaka, M., Yamaguchi, T., Shichiri, M. Distribution of atherosclerosis and risk factors in atherothrombotic occlusion. Stroke.. 1993;24:206–211.
Complications of endovascular techniques
Almekhlafi, M.A., Hu, W.Y., Hill, M.D., et al. Calcification and endothelialization of thrombi in acute stroke. Ann Neurol.. 2008;64:344–348.
Marder, V.J., Chute, D.J., Starkman, S., et al. Analysis of thrombi retrieved from cerebral arteries of patients with acute ischemic stroke. Stroke.. 2006;37:2086–2093.
Mehta, R.I., Solis, O.E., et al. Hydrophilic polymer emboli: an under-recognized iatrogenic cause of ischemia and infarct. Mod Pathol.. 2010;23:921–930.
Yin, N.S., Benavides, S., Starkman, S., et al. Autopsy findings after intracranial thrombectomy for acute ischemic stroke: a clinicopathologic study of 5 patients. Stroke.. 2010;41:938–947.
Fibromuscular dysplasia and moyamoya disease
Dusick, J.R., Gonzalez, N.R., Martin, N.A. Clinical and angiographic outcomes from indirect revascularization surgery for Moyamoya disease in adults and children: A review of 63 procedures. Neurosurgery.. 2010;68:34–43.
Furie, D.M., Tien, R.D. Fibromuscular dysplasia of arteries of the head and neck: Imaging findings. AJR Am J Roentgenol.. 1994;162:1205–1209.
Gosalakkal, J.A. Moyamoya disease: A review. Neurol India.. 2002;50:6–10.
Ikeda, E., Hosoda, Y. Distribution of thrombotic lesions in the cerebral arteries in spontaneous occlusion of the circle of Willis: cerebrovascular Moyamoya disease. Clin Neuropathol.. 1993;12:44–48.
Masuda, J., Ogata, J., Yutani, C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in Moyamoya disease. Stroke.. 1993;24:1960–1967.
Schievink, W.I., Bjornsson, J., Piepgras, D.G. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke.. 1994;25:2492–2496.
Slavin, R.E., Saeki, K., Bhagavan, B., et al. Segmental arterial mediolysis: A precursor to fibromuscular dysplasia? Mod Pathol.. 1995;8:287–294.
Touze, E., Oppenheim, C., Trystram, D., et al. Fibromuscular dysplasia of cervical and intracranial arteries. Int J Stroke.. 2010;5:296–305.
Caplan, L.R., Zarins, C.K., Hemmati, M. Spontaneous dissection of the extracranial vertebral arteries. Stroke.. 1985;16:1030–1038.
Farrell, M.A., Gilbert, J.J., Kaufmann, J.C.E. Fatal intracranial arterial dissection: clinical pathological correlation. J Neurol Neurosurg Psychiatry.. 1985;48:111–121.
Ferro, P., Bonafe, A., Arrue, P., et al. Dissection of intracranial arteries – Ten cases. J Neuroradiol.. 1996;23:139–148.
Hart, R.G. Vertebral artery dissection. Neurology.. 1988;38:987–989.
Hart, R.G., Easton, J.D. Dissections. Stroke.. 1985;16:925–927.
Karacagil, S., Hardemark, H.G., Bergqvist, D. Spontaneous internal carotid artery dissection – Review. Int Angiol.. 1996;15:291–294.
Krings, T., Choi, I-S. The many faces of intracranial arterial dissections. Interventional Neuroradiology.. 2010;16:151–160.
Connor, M.D., Lammie, G.A., Bell, J.E., et al. Cerebral infarction in adult AIDS patients. Observations from the Edinburgh HIV autopsy cohort. Stroke. 2000;31:2117–2126.
Dobbs, M.R., Berger, J.R. Stroke in HIV infection and AIDS. Expert Rev Cardiovasc Ther.. 2009;7:1263–1271.
Dubrovsky, T., Curless, R., Scott, G., et al. Cerebral aneurysmal arteriopathy in childhood AIDS. Neurology.. 1998;51:560–565.
Smith, T.W., DeGirolami, U., Henin, D., et al. Human immunodeficiency virus (HIV) leukoencephalopathy and the microcirculation. J Neuropathol Exp Neurol.. 1990;49:357–370.
Vinters, H.V., Anders, K.H. Neuropathology of AIDS. Boca Raton: CRC Press Inc; 1990.
Anti-phospholipid syndromes and systemic (e.g. coagulation, hematologic) factors
Gray, J.M., Khamashta, M.A. Antiphospholipid syndrome: coming of age. Rheumatol Clin.. 2011;7:151–153.
Hademenos, G.J., Alberts, M.J., Awad, I., et al. Advances in the genetics of cerebrovascular disease and stroke. Neurology.. 2001;56:997–1008.
Kalaria, R.N., Kalimo, H. Introduction: Non-atherosclerotic cerebrovascular disorders. Brain Pathol.. 2002;12:337–342.
Munts, A.G., van Genderen, P.J.J., Dippel, D.W.J., et al. Coagulation disorders in young adults with acute cerebral ischaemia. J Neurol.. 1998;245:21–25.
Anders, K.H., Wang, Z.Z., Kornfeld, M., et al. Giant cell arteritis in association with cerebral amyloid angiopathy: Immunohistochemical and molecular studies. Hum Pathol.. 1997;28:1237–1246.
Ghanchi, F.D., Dutton, G.N. Current concepts in giant cell (temporal) arteritis. Surv Ophthalmol.. 1997;42:99–123.
Hajj-Ali, R.A., Calabrese, L.H. Central nervous system vasculitis. Curr Opin Rheumatol.. 2009;21:10–18.
Ho, M.G., Chai, W., Vinters, H.V., et al. Unilateral hemispheric primary angiitis of the central nervous system. J Neurol.. 2011;258:1714–1716.
Gordon, L.K., Levin, L.A. Visual loss in giant cell arteritis. JAMA.. 1998;280:385–386.
Hayreh, S.S., Podhajsky, P.A., Raman, R., et al. Giant cell arteritis: Validity and reliability of various diagnostic criteria. Am J Ophthalmol.. 1997;123:285–296.
Rhodes, R.H., Madelaire, N.C., Petrelli, M., et al. Primary angiitis and angiopathy of the central nervous system and their relationship to systemic giant cell arteritis. Arch Pathol Lab Med.. 1995;119:334–339.
Rigby, H., Easton, A., Bhan, V. Amyloid beta-related angiitis of the central nervous system: Report of 3 cases. Can J Neurol Sci.. 2011;38:626–630.
Salvarini, C., Brown, R.D., Jr., Calamia, K.T., et al. Primary central nervous system vasculitis: analysis of 101 patients. Ann Neurol.. 2007;62:442–451.
Scolding, N.J., Joseph, F., Kirby, P.A., et al. Aβ-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain. 2005;128:500–515.
Vinters, H.V. Inflammation complicates an ‘age-related’ cerebral microangiopathy. Can J Neurol Sci.. 2011;38:543–544.
Microvascular disease (including familial forms and those associated with dementia)
Brulin, P., Godfraind, C., Leteurtre, E., et al. Morphometric analysis of ultrastructural vascular changes in CADASIL: analysis of 50 skin biopsy specimens and pathogenic implications. Acta Neuropathol (Berl).. 2002;104:241–248.
Chabriat, H., Joutel, A., Dichgans, M., et al. CADASIL. Lancet Neurol.. 2009;8:643–653.
Fisher, C.M. Binswanger’s encephalopathy: a review. J Neurol.. 1989;236:65–79.
Greenberg, B.M. The neurologic manifestations of systemic lupus erythematosus. Neurologist.. 2009;15:115–121.
Hachinski, V., Iadecola, C., Petersen, R.C., et al. National Institute of Neurological Disorders and Stroke – Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke.. 2006;37:2220–2241.
Hara, K., Shiga, A., Fukutake, T., et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med.. 2009;360:1729–1739.
Jen, J., Cohen, A.H., Yue, Q., et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology.. 1997;49:1322–1330.
Kalimo, H., Miao, Q., Tikka, S., et al. CADASIL: the most common hereditary subcortical vascular dementia. Future Neurol.. 2008;3:683–704.
Lammie, G.A. Hypertensive cerebral small vessel disease and stroke. Brain Pathol.. 2002;12:358–370.
Lanfranconi, S., Markus, H.S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease. A systematic review. Stroke.. 2010;41:e513–e518.
Selnes, O.A., Vinters, H.V. Vascular cognitive impairment. Nat Clin Pract Neurol.. 2006;2:538–547.
Tanoi, Y., Okeda, R., Budka, H. Binswanger’s encephalopathy: serial sections and morphometry of the cerebral arteries. Acta Neuropathol (Berl).. 2000;100:347–355.
Verbeek M.M., de Waal R.M.W., Vinters H.V., eds. Cerebral amyloid angiopathy in Alzheimer’s disease and related disorders. Dordrecht: Kluwer Academic, 2000.
Vinters, H.V., Ellis, W.G., Zarow, C., et al. Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol.. 2000;59:931–945.
Biller, J., Challa, V.R., Toole, J.F., et al. Nonbacterial thrombotic endocarditis. A neurologic perspective of clinicopathologic correlations of 99 patients. Arch Neurol. 1982;39:95–98.
Hess, D.C., D’Cruz, I.A., Adams, R.J., et al. Coronary artery disease, myocardial infarction, and brain embolism. Neurol Clin.. 1993;11:399–417.
Kamenar, E., Burger, P.C. Cerebral fat embolism: A neuropathological study of a microembolic state. Stroke.. 1980;11:477–484.
Vinters, H.V., Jahan, R. Interactions between heart and brain. In: Silver M.D., Gotlieb A.I., Schoen F.J., eds. Cardiovascular pathology. 3 rd ed. New York: Churchill Livingstone; 2001:471–492.
DiNubile, M.J. Septic thrombosis of the cavernous sinuses. Arch Neurol.. 1988;45:567–572.
Einhaupl, K.M., Villringer, A., Meister, W., et al. Heparin treatment in sinus venous thrombosis. Lancet.. 1991;338:597–600.
Saposnik, G., Barinagarrementeria, F., Brown, R.D., Jr., et al. Diagnosis and management of cerebral venous thrombosis: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke.. 2011;42:1158–1192.
Infarcts (major vessel territory, watershed, lacunar, spinal cord, etc.)
Bogousslavsky, J., Barnett, H.J.M., Fox, A.J., et al. Atherosclerotic disease of the middle cerebral artery. Stroke.. 1986;17:1112–1120.
Castaigne, P., Lhermitte, F., Gautier, J.C., et al. Arterial occlusions in the vertebro-basilar system. A study of 44 patients with post-mortem data. Brain. 1973;96:133–154.
Cheshire, W.P., Santos, C.C., Massey, E.W., et al. Spinal cord infarction: Etiology and outcome. Neurology.. 1996;47:321–330.
Chuaqui, R., Tapia, J. Histologic assessment of the age of recent brain infarcts in man. J Neuropathol Exp Neurol.. 1993;52:481–489.
Fisher, C.M. Lacunes: Small, deep cerebral infarcts. Neurology.. 1965;15:774–784.
Gacs, G., Fox, A.J., Barnett, H.J.M., et al. Occurrence and mechanisms of occlusion of the anterior cerebral artery. Stroke.. 1983;14:952–959.
Gan, R., Sacco, R.L., Kargman, D.E., et al. Testing the validity of the lacunar hypothesis: The Northern Manhattan Stroke Study experience. Neurology.. 1997;48:1204–1211.
Helgason, C., Caplan, L.R., Goodwin, J., et al. Anterior choroidal artery-territory infarction. Report of cases and review. Arch Neurol.. 1986;43:681–686.
Kubik, C.S., Adams, R.D. Occlusion of the basilar artery – a clinical and pathological study. Brain.. 1946;69:73–121.
Torvik, A. The pathogenesis of watershed infarcts in the brain. Stroke.. 1984;15:221–223.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]9
Vascular disease and infarcts
In the setting of rapidly evolving neurologic deficits, stroke is not synonymous with brain infarct, since the types of cerebrovascular disease that usually result in a stroke may involve infarction or intracranial hemorrhage (see Chapter 10). Infarct, a localized area of ischemic brain injury, should also be differentiated from global hypoxic-ischemic brain injury (see Chapter 8). Stroke has been defined by the WHO as ‘rapidly developing clinical signs of a focal (or global) disturbance of cerebral function, lasting more than 24 hours or leading to death and with no apparent cause other than that of vascular origin’. Stroke ranks as the second most common single cause of death in the developed world.
To place these concepts in a more practical context, brain infarcts can be caused by:
Large vessel (or macrovascular) arterial disease.
Small vessel (or microvascular) arterial disease (arteries <400–500 μm in diameter).
Infarction describes rapidly developing focal clinical signs of CNS (and/or retinal) dysfunction due to ischemia, and either radiologic evidence of CNS (and/or retinal) ischemia in a defined vascular distribution, or symptoms and signs of CNS dysfunction persisting for at least 24 hours, with exclusion of non-ischemic etiologies.
GENETICS OF ISCHEMIC CEREBROVASCULAR DISEASE
Brain infarcts occur as one of many systemic manifestations in various hereditary disorders.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke (MELAS)
SCD is an autosomal recessive hemoglobinopathy characterized by chronic hemolytic anemia and ‘sickle-shaped’ erythrocytes.
SCD results from point mutation in the hemoglobin β-chain.
Number of strokes/100 patient-years increases from 0.13 (<2years of age) to 1.28 (>50years of age).
At least 10% of SCD patients have a stroke during their lifetimes – 75% ischemic, 25% hemorrhagic.
LARGE ARTERIAL DISEASE
MACROSCOPIC APPEARANCES
The severity of atherosclerosis can vary significantly in different arteries (e.g. severe basilar artery involvement may accompany less prominent MCA involvement). The degree and extent of aortic or coronary atherosclerosis do not predict its severity in the intracranial basal cerebral vasculature, i.e. compartments of the circle of Willis. Atheroma is often most severe at the origins of the vertebral arteries and carotid bifurcation (Figs 9.1, 9.2). Intracranial atherosclerosis is most severe in major branches of the circle of Willis and vertebrobasilar system (Fig. 9.3). Atheroma in distal arterial branches is more common in Asian and African-American subjects. The extent and topography of atherosclerosis in the basal vessels are often best documented by removing the circle of Willis from the fixed brain (Fig. 9.3b). In such a specimen from a subject with severe atherosclerosis, decalcification prior to histologic examination is recommended. Carotid endarterectomy specimens from individuals with TIA or threatened ischemic stroke are often submitted for histologic examination; extent and severity of atheroma within plaques, as well as the presence of plaque ulceration, necrosis and thrombus adherent to intima must be assessed (Fig. 9.2).
9.1 Atheroma of carotid artery.
Sections of common carotid (right) and internal carotid arteries removed at necropsy from a patient with severe atherosclerotic cerebrovascular disease and systemic atherosclerosis. Note severe stenosis of the lumina and eccentric intimal thickening of the internal carotid artery with thrombosis.
9.2 Carotid endarterectomy specimens from two patients.
(a) A specimen cut open to reveal grumous fractured eggshell-like atheromatous material and superimposed mural thrombus. (b) Another specimen cut in cross section showing atheroma at the carotid bifurcation. (Courtesy of Dr Sophia Apple, Department of Pathology and Laboratory Medicine, UCLA Medical Center.)
9.3 Severe atherosclerosis of the circle of Willis and its major branches.
(a) Patchy yellow discoloration of the arterial branches indicating underlying atheroma. (b) Severe atheroma involving the basal arteries of an elderly patient with ischemic-vascular dementia. Arrow indicates basilar artery. Note especially severe atherosclerosis of the basilar, posterior cerebral and middle cerebral arteries. However, patchy atheroma extends even into distal branches. (Courtesy of Dr Ivan Klement.)
MICROSCOPIC APPEARANCES
Histopathologic features of atheroma are best highlighted with stains that differentiate elastica, fibrous tissue, and smooth muscle (e.g. elastica van Gieson). Immunohistochemistry (IHC) using primary antibodies to vascular smooth muscle actin, endothelium (Factor 8, lectins), and macrophages may be helpful. Fibromuscular intimal hyperplasia with an intact endothelium and variable narrowing of the vascular lumen is noted in ‘early’ and asymptomatic vascular lesions, and often discovered incidentally at necropsy (Fig. 9.4). Complicated plaques show cholesterol clefts and prominent lipid-/hemosiderin-laden macrophages and may be heavily calcified. There is usually significant narrowing of the arterial vessel lumen, sometimes in association with ulceration and overlying mural or occlusive thrombus (Fig. 9.5). Immunohistochemistry (IHC) using primary antibodies to smooth muscle actin often demonstrates a thick smooth muscle cell ‘cap’ over a lipid-rich subendothelial plaque. It is quite rare for even severely narrowed segments of the circle of Willis to show plaque ulceration, in contrast to the frequency of this phenomenon in ICA endarterectomy specimens. Severely atherosclerotic arterial segments, especially in the basilar artery may show ectasia or even a fusiform aneurysm.
9.4 Atherosclerosis with fibromuscular intimal hyperplasia.
Eccentric atheroma is prominent in these arteries, but is largely composed of smooth muscle cells and ‘ground substance’ (glycosaminoglycans), though scattered cholesterol clefts are also present. Note the intact endothelium in all instances. The single elastic lamina (as is the case in intracerebral arteries) is also largely intact and best demonstrated in panels b and c. (a) Middle cerebral artery. (b) Meningeal artery, a branch of the posterior cerebral artery. (c) Section of posterior cerebral artery stained with Elastic van Gieson.
9.5 Complicated atherosclerosis.
(a) Prominent eccentric atheroma with smooth muscle cell hyperplasia overlying a region with abundant cholesterol clefts in an intracranial meningeal artery. The atheroma has produced significant stenosis. Note discontinuity in elastica. (b) Low magnification view of cross-section through a major branch of the circle of Willis, with severe stenosis of the lumen. Arrows indicate smooth muscle cell ‘cap’ adjacent to lumen and overlying grumous cholesterol material; no ulceration or mural thrombus is noted adjacent to the lumen. (c) Basal artery showing severe stenosis of the lumen by atheroma on two aspects of the vessel wall. (d) Small meningeal artery almost occluded by atheromatous material; only a small residual lumen remains. (e) Small meningeal artery (branch of ACA) with mild atheroma consisting almost exclusively of fibromuscular intimal hyperplasia; arrows indicate internal elastic lamina.
The distal circulation, both meningeal and parenchymal arteries, may show atherosclerotic changes or deposits of platelet-fibrin material (Fig. 9.6). Rarely, examination of an autopsy brain specimen from an individual with severe atherosclerosis may yield the finding of numerous atheroemboli within infarcted regions (Fig. 9.7).
9.6 Intracerebral atherosclerosis.
Microatheroma almost occluding a tortuous artery in the basal ganglia of a patient with longstanding hypertension.
9.7 Cerebral atherosclerosis and complications of atheroma.
(a) Atherosclerosis in a small artery (external diameter approximately 500 μm) manifesting as fibromuscular intimal hyperplasia. (b) Intraluminal macrophages originating in embolic material from fragmented atheromatous plaques in a 58-year-old hypertensive woman with multiple cerebral and cerebellar infarcts in both watershed and large artery territories. (c) Cholesterol clefts within atheroemboli from fragmented atheromatous plaques occluding lumina of two meningeal arteries (arrows) within brain specimen of a patient with multiple cerebral infarcts. (d) Small intraparenchymal artery occluded by atheromatous material, including foreign body giant cell around cholesterol ‘clefts’. (e) Larger artery in another patient showing occlusive thrombus composed primarily of atheromatous material (‘atheroembolus’) within which prominent residual cholesterol clefts are seen. An ‘embolic’ origin of the atheromatous material is suggested by the relative absence of intrinsic atherosclerosis in the arterial wall.
THE AUTOPSY OF A STROKE PATIENT
Autopsy studies of stroke should not be used to infer epidemiologic data, since patients that come to necropsy are a highly selected group. However, autopsies are crucial in highlighting vascular mechanisms that are of importance in stroke pathogenesis.
Especially important for infarction/ischemic stroke
Assess the patency of major neck arteries (carotid/vertebral) by injecting water at their origins and monitoring flow into the cranial cavity after brain removal.
• If any vessel is obstructed, the prosector should attempt to establish:
– site(s) of occlusion/severe stenosis
– cause(s) of occlusion (e.g. atheroma, embolus, dissection, other).
• Vessels (arteries) should be dissected until significant lesions are documented; this sometimes requires painstaking removal of the entire carotid or vertebral artery.
• Dissection of intraosseous portions of the vertebral arteries is facilitated by prior removal of the cervical vertebral bodies. This is achieved from an anterior approach by making a parasagittal saw cut through the medial aspect of the vertebral pedicles (a laterally placed cut will damage the arteries).
Carefully examine the heart, looking particularly for:
• Endocarditis (infective or non-infective/marantic) – culture/histology of any valvular lesion possibly helpful.
• Any cause of a right-to-left shunt (e.g. septal defect, patent foramen ovale).
• Valvular abnormalities, especially in the left heart (e.g. myxomatous degeneration/prolapse of mitral valve, calcific aortic sclerosis with stenosis).
• Evidence of recent myocardial infarction ± mural thrombus or old infarction with ventricular aneurysm ± mural thrombus.
Look for old/recent infarcts in other organs, which may suggest a source of embolism.
Especially important for hemorrhage:
In the medical history and general autopsy, seek evidence of disease that may be of etiologic significance, even though this may not be discovered in many individuals:
• Hypertension – cardiomegaly with left ventricular hypertrophy, nephrosclerosis.
• Dementia/cognitive impairment – especially in elderly patients this may point to Aβ cerebral amyloid angiopathy (CAA), which is commonly associated with Alzheimer’s disease.
• Neoplasia – hemorrhage in association with metastasis or primary glioma, especially oligodendroglioma.
• History suggesting recreational drug abuse, e.g. amphetamines, heroin.
When a hematoma is documented at the time of brain removal, the prosector must establish:
• In which compartment it is present and (optimally) in which compartment it originated (e.g. subdural, subarachnoid, combined parenchymal/subarachnoid or intraventricular).
• Its approximate volume and effect(s) on surrounding brain (e.g. edema).
• By dissecting (prior to fixation) the circle of Willis and its major branches, whether a ruptured saccular aneurysm has caused any subarachnoid (or intraparenchymal) hematoma.
For special studies regarding the evaluation of evacuated intracerebral blood clot, see Chapter 10.
HYPERHOMOCYSTEINEMIA, ATHEROMA, AND BRAIN INFARCTS
Hyperhomocysteinemia occurs in a variety of inherited disorders, including homocystinuria (secondary to cystathione β-synthase deficiency) and several rare disorders of vitamin B12 or folate metabolism.
Several mechanisms are probably of importance. Homocysteine:
FMD shows female predominance.
Familial cases make up ~10% of total.
FMD usually becomes symptomatic in the 4th or 5th decade, but occurs over a very broad age range.
Connective tissue disorders and α1-antitrypsin deficiency are associated clinical conditions.
Ischemic (brain) infarcts may occur secondary to vascular stenosis/occlusion.
As many as 20% of patients with carotid dissections show angiographic features of FMD.
FMD is rarely associated with formation of aneurysms on intracranial or extracranial arteries.
FIBROMUSCULAR DYSPLASIA (FMD)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Several pathologically distinct subtypes of FMD are described:
Intimal (up to 10%): There is circumferential or eccentric deposition of collagen in the intima, with fragmented or duplicated internal elastic lamina. Angiography shows long smooth narrowing.
Histologic findings that are characteristic include fibrosis, non-atherosclerotic smooth muscle cell hyperplasia or thinning, destruction of the internal elastic lamina, negligible inflammation, absence of macrophages, and generalized disorganization of arterial wall components (Fig. 9.8).
9.8 FMD of extracranial ICA associated with aneurysm formation.
(a) Tunica media showing disorganization and loss of smooth muscle cells. (b) Abnormal orientation and loss of smooth muscle cells. (c) Irregular or disrupted internal elastic lamina. (Courtesy of Drs M Miyauchi and S Shionoya.)
Annual incidence is approximately 1/1 million.
Females are affected more commonly than males.
Moyamoya disease is most common in Asians and has peak incidences at ages 10–14years and 40–50years.
An upper respiratory tract infection often precedes initial symptoms.
The prognosis is poor if both anterior and posterior branches of circle of Willis are involved.
MOYAMOYA DISEASE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Arterial branches of the circle of Willis show thrombotic lesions in over 50% of patients. Those most commonly affected are the ICA, posterior communicating and posterior cerebral arteries. Severely stenotic non-complicated atherosclerosis, with intimal fibromuscular hyperplasia, but negligible lipid, cholesterol, inflammation, or disruption of the elastica is found (Fig. 9.9). Platelet-fibrin thrombi in various stages of organization are often seen at the intimal surface.
9.9 Moyamoya disease.
(a,b) Sections of internal carotid artery from a 15-year-old boy with Moyamoya disease. There is marked intimal thickening with platelet fibrin thrombi on the intimal surface. (c) Posterior cerebral artery from a 52-year-old woman with Moyamoya disease. The artery contains mural thrombus composed of red blood cells and fibrin. (Courtesy of Professor E Ikeda and Professor Y Hosoda, Tokyo, Japan.)
ARTERIAL DISSECTION
This is rare and tends to affect young and middle-aged adults. The dissection (Fig. 9.10) is usually spontaneous, but can be initiated by blunt trauma, often quite mild (e.g. neck injury in a motor vehicle accident or chiropractic manipulation of the neck). The dissection may involve extracranial or intracranial parts of the vertebral artery (more common in women) or carotid artery (more common in men). An intimal tear leads to a medial or subendothelial hematoma. The expanding hematoma may occlude the arterial lumen, usually producing infarction of CNS tissue, less commonly hemorrhage. Dissection of intracranial arteries, especially the vertebral artery, may rarely extend through the adventitia, producing subarachnoid hemorrhage. Dissection of intracranial arteries is likely to become more common as aggressive endovascular revascularization procedures (e.g. thromboembolectomy after ischemic stroke) become more widely utilized in clinical neurologic practice (Fig. 9.11).
9.10 Vertebral artery dissection following chiropractic neck manipulation (a–c).
(a) Almost circumferential dissection of blood between internal and external elastic laminae. (b) A thin ‘strip’ of blood (arrows) between internal and external elastic laminae. (c) Breach of internal elastic lamina (possibly related to the entry point for the dissection). (Material studied by kind courtesy of Professor Michael A Farrell, Dublin, Ireland.) (d) Intracranial basilar artery dissection in another patient. A subintimal ‘wedge’ of blood extends into the media.
9.11 Dissection of right MCA following attempted thrombectomy using a fine catheter device.
(a) Fresh autopsy specimen showing severe atheromatous plaque in the intra-Sylvian segment of the right MCA (arrow). Note necrosis of adjacent temporal lobe. (b) A relatively non-atherosclerotic segment of the artery showing extensive sub-intimal dissection (arrowheads indicate the ‘raised’ intima, under which is abundant acute hemorrhage). (c) Magnified view showing the elastica overlying fresh blood. (d) A severely atherosclerotic segment of the MCA showing only a small amount of subintimal blood.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Subarachnoid hemorrhage or ischemic infarct with cerebral edema is evident macroscopically. Demonstration of the point of origin of the dissection usually necessitates complete examination of intracranial or extracranial portions of the affected carotid or vertebral artery. A subendothelial or intramural hematoma is found within the vessel wall (Fig. 9.10).
HUMAN IMMUNODEFICIENCY VIRUS (HIV)-ASSOCIATED STROKE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
HIV-associated ‘vasculopathy’ has been described as showing non-specific features, including intimal fibromuscular hyperplasia, fragmentation of the internal elastic lamina, and sometimes aneurysmal dilatation of vessel walls, a pathology consistent with ‘healed arteritis’ (Fig. 9.12). HIV-infected individuals (including those responsive to combined retroviral therapy) may be at risk for accelerated atherosclerosis, though factors responsible for this are unclear. Brain parenchymal arteriosclerotic change has also been observed in HIV-infected individuals that develop cerebral microinfarcts. HIV-1 has been demonstrated immunohistochemically in affected vessel walls (possibly within the cytoplasm of macrophages), but its pathogenic role in this location is unclear.
9.12 HIV-associated arteriopathy.
All illustrations are from the brain of a patient with AIDS, which had multifocal areas of necrosis and hemorrhage at necropsy. (a) Eccentric intimal fibromuscular hyperplasia with focal attenuation of the elastic lamina and a small organizing mural thrombus (arrow). (b) Another artery with a relatively intact elastic lamina and superimposed intimal hyperplasia, including some cells with macrophage morphology. (c,d) Two arteries showing intimal hyperplasia with variable numbers of foamy histiocytes in the ‘plaque’. The foam cell component dominates intimal thickening in the artery indicated by arrow in panel (d).
ANTIPHOSPHOLIPID ANTIBODY (APLA) SYNDROME
APLAs are associated with risk of thrombosis in both arterial and venous systems.
Most SLE patients in whom thrombotic cerebrovascular disease occurs have high titers of APLAs.
The mainstay of treatment is warfarin or other forms of anticoagulation.
[/not-level-membership-for-pathology-category]
