Chapter 73 Uveal Effusion Syndrome and Hypotony Maculopathy
Uveal effusion syndrome
Introduction
Spontaneous exudative detachment of the choroid and ciliary body was first reported by Schepens and Brockhurst in 19631; these authors used the term “uveal effusion” in their description of this disorder. Almost two decades later, Gass and Jallow in 1982 coined the term “idiopathic uveal effusion syndrome” to describe idiopathic serous detachment of the choroid, ciliary body, and retina.2 Uveal effusion syndrome is a rare ocular disorder that typically manifests itself in an otherwise healthy middle-aged male. In their original report, Schepens and Brockhurst described 17 patients, only one of whom was female. The diagnosis of uveal effusion syndrome is based on characteristic clinical findings and exclusion of other known causes of uveal effusion. Bilateral involvement is common and unilateral cases tend to occur in older males. In addition to the accumulation of serous fluid in the ciliary body and choroid, nonrhegmatogenous retinal detachment with marked shifting of the subretinal fluid are commonly observed in patients with uveal effusion syndrome. Retinal detachment often begins inferiorly, as in other causes of exudative retinal detachment. Other ocular findings include dilation of the episcleral blood vessels, blood in Schlemm’s canal, normal intraocular pressure, mild vitreous cells, leopard-spot retinal pigment epithelial alterations, elevation of the subretinal fluid protein levels, and elevation of the cerebrospinal fluid protein. Without treatment, a protracted clinical course with remissions and exacerbations over many months to years may cause significant visual decline and morbidity. Unlike other causes of ciliochoridal effusion, patients with idiopathic uveal effusion syndrome respond poorly to nonsurgical treatment, including corticosteroids or antimetabolites. Similarly, surgical treatment of nonrhegmatogenous retinal detachment in uveal effusion syndrome using conventional techniques, including scleral buckling or pars plana vitrectomy, fails to reattach the neurosensory retina secondary to persistent serous exudation. In most cases, successful reattachment of the nonrhegmatogenous retinal detachment requires a scleral-thinning procedure, including quadrantic partial-thickness sclerectomies and sclerostomies.
Pathophysiology of ciliochoroidal effusions
General mechanisms
Since idiopathic uveal effusion syndrome represents only a small percentage of ciliochoroidal effusions, it is important to discuss the general mechanisms of serous accumulation in the ciliary body and choroid. Most cases of ciliochoroidal effusion can be classified into one of the following pathophysiologic categories: (1) hydrodynamic; (2) inflammatory; (3) neoplastic; or (4) associated with abnormal sclera.3 Under physiologic conditions, a normal eye has equilibrium between the transmural hydrostatic pressure gradient, defined as the difference between the intravascular blood pressure and intraocular pressure, and the colloid osmotic pressure gradient of the choriocapillaris (Fig. 73.1). Albumin is the most abundant protein in the choroidal capillaries and is the primary driver of the colloid osmotic pressure. This pressure gradient draws fluid into blood vessels and maintains relative dehydration of the suprachoroidal space due to a low extravascular colloid concentration.4 Fenestrated capillaries of the choroid allow albumin to escape into the extravascular space. To maintain the colloid osmotic gradient, albumin leaves the choroid across the sclera and this transscleral protein flow is facilitated by intraocular pressure.5–7
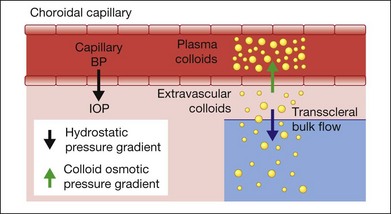
The fluid equilibrium across the layers of the choroid may be disturbed by several factors affecting one or more components of this intricate system.3 Ocular hypotony decreases the driving force for transscleral protein flow and increases the transmural hydrostatic pressure gradient. These changes then facilitate the accumulation of protein and fluid in the suprachoroidal space. Elevated uveal venous pressure increases the transmural hydrostatic pressure gradient and leads to increased fluid movement into the extravascular space. Vascular competence may be compromised by inflammation, which then increases capillary protein permeability and accumulation of protein in the extravascular space. This reduces the colloid osmotic pressure gradient and the absorption of extravascular fluid into the capillaries. Abnormal scleral composition or thickness may increase resistance to transscleral protein outflow and accumulation of protein-rich fluid in the suprachoroidal space. These alterations are more likely to affect the choroidal fluid dynamics when two or more are present simultaneously. Indeed, the creation of ciliochoroidal effusion in animal models requires experimental alteration of two or more pathophysiologic factors.8,9
Idiopathic and nanophthalmic uveal effusion
In patients with uveal effusion syndrome or the closely related condition of nanophthalmos, abnormal sclera, referred to here as scleropathy, is the most likely primary ocular anomaly affecting choroidal fluid dynamics. In nanophthalmos, scleropathy is congenital in origin and associated with other ocular abnormalities. Acquired scleropathy may be secondary to a systemic disorder, such as the accumulation of amyloid in systemic amyloidosis or mucopolysaccharide in Hunter syndrome.10,11 In uveal effusion syndrome, scleropathy appears to be secondary to the abnormal accumulation of glycosaminoglycan-like deposits and thickening of the sclera in the absence of any known systemic disorder.12–14 Ward et al. reported that electron microscopy of excised sclera showed increased glycosaminoglycan-like deposits between the scleral fibers.14 In a following report, Forrester and colleagues performed histochemical studies on scleras excised from patients with uveal effusion syndrome and showed deposition of the glycosaminoglycan proteodermatan sulfate and a smaller amount of proteochondroitin sulfate, indicating a primary defect in scleral proteodermatan metabolism and representing a form of ocular mucopolysaccharidosis.12 Histologic similarities between scleras isolated from eyes with uveal effusion syndrome and nanophthalmos were demonstrated by Uyama et al., who found disorganized scleral fibers and proteoglycan deposits in 6 nanophthalmic eyes and 11 nonnanophthalmic eyes with uveal effusion syndrome.13 As discussed above, abnormal scleral composition increases resistance to transscleral protein outflow, which in turn leads to the accumulation of protein in the extravascular space of the choroid and higher colloid osmotic pressure. This results in reduced movement of fluid from the suprachoroidal space into the choroidal capillaries and leads to serous ciliochoroidal effusion. In vitro experimental evidence is consistent with this model, as chondroitinase ABC digestion, which removes glycosaminoglycans, improves scleral transport in human cadaver eyes.15
Ciliochoroidal effusion and nonrhegmatogenous retinal detachment can be successfully treated in patients with uveal effusion syndrome by quadrantic partial-thickness sclerectomies.16,17 The disappearance of serous fluid after partial-thickness sclerectomies is consistent with the hypothesis that abnormally thickened sclera prevents outflow of protein and suggests that the removal of excess extravascular protein may be improved by reducing scleral thickness and resistance. Under normal conditions, eyes with congenital or acquired scleropathies may have enough redundancy in choroidal protein transport mechanisms to achieve physiologic fluid equilibrium and dehydration of the suprachoroid. However, when choroidal fluid dynamics are further stressed by additional pathologic factors, such as compression of the vortex veins, these compensatory mechanisms may no longer be sufficient to overcome the effect of increased colloids in the suprachoroidal space, which may then lead to increased extravascular fluid retention and ciliochoroidal detachment.
Vortex vein compression was first suggested by Schaffer in 1975 as a possible mechanism of uveal effusion in nanophthalmic eyes following glaucoma filtration surgery.18 Relative obstruction of venous outflow secondary to compressed vortex veins may cause congestion of the choriocapillaris and alter the transmural hydrostatic pressure gradient, favoring increased retention of fluid in the suprachoroidal space. Brockhurst reported successful reattachment of the retina and resolution of ciliochoroidal effusion in nanophthalmic eyes after surgical decompression of the vortex veins.19 Additional evidence for the role of increased intravascular pressure secondary to reduced ocular venous drainage in uveal effusion was provided by Casswell and colleagues, who reported resolution of retinal detachment after vortex vein decompression in patients with uveal effusion syndrome.20 As proposed by Gass, compression of the vortex veins by an abnormally thickened sclera may contribute to increased fluid retention and serous exudation in the choroid and ciliary body in uveal effusion syndrome.16
Clinical features
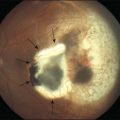
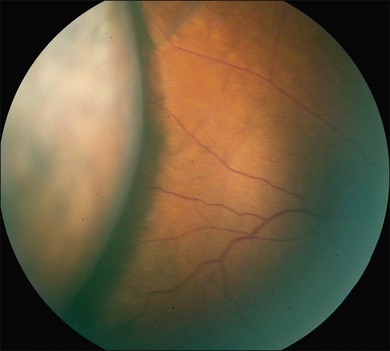
Ciliochoroidal detachments in uveal effusion syndrome are brown-orange, solid-appearing elevations with smooth, convex surfaces (Fig. 73.2). Transillumination of the globe confirms the serous nature of the exudation. Choroidal detachments do not undulate appreciably with ocular movements, and this helps to distinguish them from rhegmatogenous retinal detachments. In early or mild cases, the diagnosis is suggested when the ora serrata is visible without the use of scleral depression secondary to shallow elevation of the pars plana and peripheral choroid (Fig. 73.3). As the effusion progresses, annular or lobular choroidal detachment may be seen. The characteristic four-lobed configuration results from the attachment of the choroid to the sclera at the vortex vein ampullae. The fluid accumulation is always greater anteriorly, as the anterior connecting fibers attaching choroid to the sclera are long and tangentially oriented, unlike the posterior fibers that are short and run more directly from uvea to sclera.21
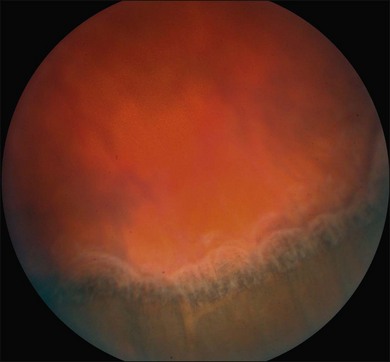
Long-standing uveal effusion causes decompensation of the retinal pigment epithelial fluid barrier, resulting in increased protein and fluid accumulation in the subretinal space and development of nonrhegmatogenous retinal detachment (Fig. 73.4). There is greater absorption of fluid from the subretinal space compared with protein outflow, which results in rising protein concentration and marked shifting of subretinal fluid with changes in head position. Progressive subretinal fluid accumulation may lead to total retinal detachment. Chronic serous effusion and subretinal fluid accumulation may result in diffuse depigmentation and multifocal hyperplasia of the retinal pigment epithelium, forming the characteristic clinical finding of leopard spots in the fundus (Figs 73.4 and 73.5).
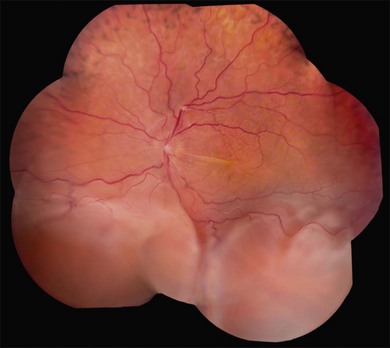
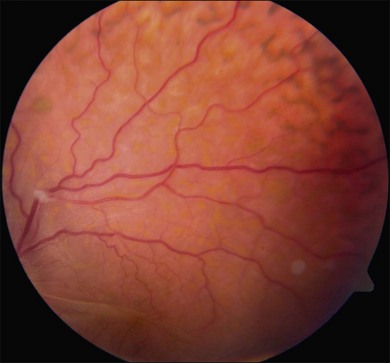
Anterior-segment examination in a patient with uveal effusion syndrome may reveal dilation of the episcleral blood vessels. Blood may be present in the Schlemm’s canal on gonioscopy. The anterior chamber is free of any signs of inflammation and the presence of anterior-chamber cell should increase suspicion of another ocular disorder with secondary uveal effusion. Mild vitreous cells may be present. Intraocular pressure is normal in patients with uveal effusion syndrome and hypotony would indicate the presence of an alternative etiology. Elevation of subretinal fluid protein levels has been documented.2,22,23 Although not commonly tested today, earlier studies of patients with uveal effusion syndrome demonstrated increased cerebrospinal fluid pressure and protein levels in some cases.1,2
Diagnostic studies
Ophthalmic ultrasound
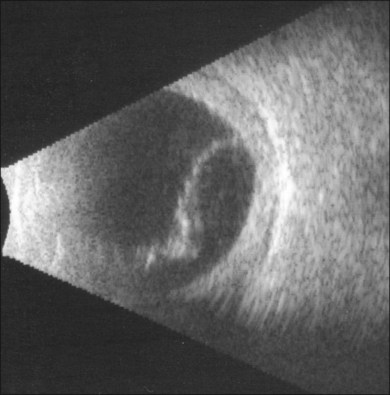
B-scan ultrasound examination typically shows a smooth, thick, dome-shaped membrane with little aftermovement.24 Ciliochoroidal effusion may be distinguished from retinal detachment by extension of the detachment anterior to the ora serrata. Highly bullous ciliochoroidal detachments may extend posteriorly and insert near the edge of the optic nerve. A-scan evaluation demonstrates a thick, 100% spike at tissue sensitivity, which at low sensitivity can often be seen to be double-peaked.24 In early presentations of uveal effusion syndrome, the only evidence for ciliochoroidal effusion may be subtle degrees of supraciliary effusion on high-frequency ultrasound biomicroscopy. Diffuse, high-reflective thickening of the posterior choroid may be seen (Fig. 73.6). Low-reflective choroidal thickening should raise the suspicion for an infiltrative process secondary to an inflammatory or neoplastic condition.24
Angiography and optical coherence tomography
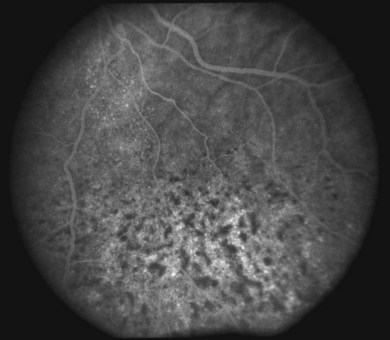
The diagnostic value of fluorescein and indocyanine green angiography and optical coherence tomography is limited in uveal effusion syndrome and serves mostly to exclude other etiologies. Angiography may demonstrate a leopard-skin appearance of hyperfluorescence and hypofluorescence (Fig. 73.7). The presence of focal fluorescein leaks and focal pigment epithelial detachments should increase the suspicion for idiopathic central serous chorioretinopathy as the underlying diagnosis. Multiple pinpoint leaks may indicate the presence of an inflammatory or neoplastic choroidal infiltration. Indocyanine green angiography shows diffuse granular choroidal hyperfluorescence in the early phase, indicating marked hyperpermeability of the choroidal vessels.13 This choroidal hyperfluorescence persists into the late-phase angiogram and becomes more diffuse, demonstrating increased accumulation of fluid in the choroid. Spectral-domain optical coherence tomography may show focal thickening of the retinal pigment epithelium layer corresponding to the areas of leopard spots.25
Differential diagnosis
Uveal effusion syndrome is a diagnosis of exclusion. The differential diagnosis may be categorized by primary pathogenic factors causing ciliochoroidal effusion (Table 73.1). As discussed above, serous effusions in the ciliary body and choroid generally require the simultaneous presence of multiple pathogenic factors.
Table 73.1 Differential diagnosis of ciliochoroidal effusions
| Scleropathies
Congenital Acquired Hydrodynamic factors Ocular hypotony Elevated uveal venous pressure Malignant hypertension Inflammatory factors Neoplastic conditions |
Congenital and acquired scleropathies
As discussed above, congenital or acquired scleropathy with abnormal thickening of the sclera significantly alters the choroidal fluid dynamics and may cause uveal effusion, especially in the presence of other pathogenic factors such as vortex vein compression with reduced venous outflow and elevated uveal venous pressure. Uveal effusion syndrome and nanophthalmos can be considered in the same spectrum of diseases with a congenital primary scleral abnormality. Secondary scleral abnormalities are seen in association with systemic disorders, including amyloidosis or mucopolysaccharidosis.10,11
Uveal effusion in patients with nanophthalmos is remarkably similar to that seen in the idiopathic uveal effusion syndrome. Nanophthalmos is a pure form of microphthalmia characterized by small eyes and extremely thick sclera with no other identifiable ocular or systemic abnormalities. The axial length is generally less than 20 mm and high hyperopia (> +7.00 D) is typical. Other findings include small corneal diameter, shallow anterior chamber, high lens-to-eye volume ratio, and strong predisposition to develop angle closure glaucoma.26 Uveal effusion and nonrhegmatogenous retinal detachment may occur spontaneously and are frequently induced by intraocular surgery in nanophthalmic eyes.26,27 The detection of nanophthalmos is important prior to any planned intraocular procedure since prophylactic use of sclerectomies may prevent significant postoperative complications secondary to uveal effusion.
Hydrodynamic effusions
Hydrodynamic factors associated with ciliochoroidal effusion include ocular hypotony, elevated uveal venous pressure, and malignant hypertension. Long-standing low intraocular pressure induces uveal effusion and may in turn be worsened by ciliary body detachment. The most common reason for hypotony is glaucoma filtering or drainage device surgery, especially in the early postoperative period. Resuturing the scleral flap, scleral patch grafting, autologous blood injection, and other methods have been described in the literature to reverse postoperative hypotony.28–30 Hypotony and uveal effusion complicating anterior-segment surgery are usually secondary to a wound leak or inadvertent filtering bleb. Other ocular disorders that may lead to hypotony and secondary ciliochoroidal effusion include cyclodialysis, penetrating ocular trauma, rhegmatogenous retinal detachment, and ciliary body dysfunction.
Elevated uveal venous pressure and increased transudation from choroidal vessels may be seen in the setting of carotid-cavernous sinus fistula or dural arteriovenous fistulas. When an intracranial fistula is suspected in a patient with uveal effusion, nonrhegmatogenous retinal detachment, and other associated findings, neuroimaging should be obtained. Patients with idiopathic prominent episcleral vessels or Sturge–Weber syndrome have elevated episcleral venous pressure and likely secondary increase in choriocapillaris pressure, which predisposes them to a higher risk of significant ciliochoroidal effusion during intraocular surgery when the intraocular pressure drops to zero.31 Compression of the vortex veins and reduction of ocular venous outflow may lead to uveal effusion after scleral buckling surgery.
Treatment of idiopathic uveal effusion syndrome
Scleral thinning procedures
Gass first described sclerectomy with sclerostomy for the treatment of uveal effusion syndrome in 1983.16 The idea of using this procedure in patients with uveal effusion syndrome arose after an unsuccessful attempt at vortex vein decompression in another patient. Gass operated on both eyes of a patient with thick sclera and uveal effusion and amputated or ruptured the vortex veins during attempted decompression. Despite the occurrence of suprachoroidal hemorrhage in both eyes, ciliochoroidal and nonrhegmatogenous detachment resolved in both eyes postoperatively. This observation led him to hypothesize that the excision of large scleral flaps at the sites of vortex veins was responsible for the successful outcome. To test this hypothesis, he performed quadrantic partial-thickness sclerectomies and sclerostomies without vortex vein decompression or drainage of subretinal fluid in both eyes of a patient with long-standing uveal effusion and bullous nonrhegmatogenous retinal detachment. Complete resolution of ciliochoroidal and subretinal fluid occurred 10–12 weeks after surgery.16 In 1990, Johnson and Gass expanded on this initial result in a retrospective study of 23 eyes of 20 patients with uveal effusion syndrome who underwent a scleral-thinning procedure without vortex vein decompression.17 The investigators observed complete resolution of subretinal and supraciliochoroidal fluid in 96% of eyes after one or two procedures. The mean time for resolution of uveal effusion and nonrhegmatogenous retinal detachment was 2.4 months. Other investigators reported similar results using scleral-thinning procedures for treatment of uveal effusion and nonrhegmatogenous retinal detachment associated with Hunter syndrome, nanophthalmos, and uveal effusion syndrome.11,13,20,32,33
The surgical procedure for scleral thinning involves the creation of 5 × 7 mm, one-half to two-thirds thickness sclerectomies in each quadrant, centered 1–2 mm anterior to the equator and placed outside the meridian of each vortex vein to avoid its intrascleral course (Fig. 73.8).16,17 The long axis of the sclerectomy is oriented circumferentially. An approximately 2-mm linear sclerostomy may be created in the center of each sclerectomy bed and enlarged with a 1–2-mm scleral punch. Similar scleral-thinning techniques have been reported by others treating nanophthalmic or idiopathic uveal effusion.13,34
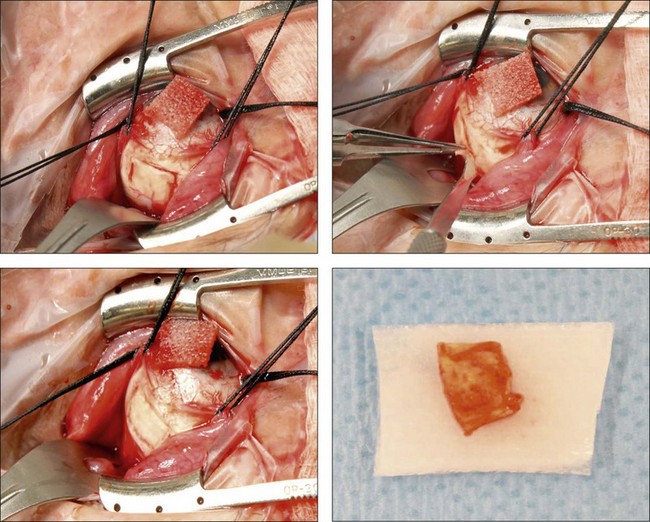
Pars plana vitrectomy
The extent of visual improvement after scleral-thinning procedures may be limited by photoreceptor and retinal pigment epithelial damage secondary to chronic retinal detachment. In the series reported by Johnson and Gass, visual acuity improved by two or more Snellen lines in 56% of eyes, remained stable in 35% of eyes, and worsened in 9% of eyes.17 Overall visual acuity was 20/400 or better in 96% of eyes and 20/40 or better in 35% of eyes. Among the 12 of 23 eyes whose final visual acuity was worse than 20/40, the primary vision-limiting factor was atrophic photoreceptor and retinal pigment epithelial damage due to chronic retinal detachment. The mean time for retinal reattachment following scleral-thinning surgery in this series was 2.4 months. To facilitate rapid retinal reattachment and prevent ongoing damage to photoreceptors and the retinal pigment epithelium, Schneiderman and Johnson performed pars plana vitrectomy and internal drainage of subretinal fluid at the time of quadrantic partial-thickness sclerectomies in a 73-year-old man with uveal effusion syndrome and total retinal detachment.35 This patient’s vision improved from hand motions to 20/70 1 year postoperatively. This combined approach of sclerectomies and pars plana vitrectomy for macula-off nonrhegmatogenous retinal detachment in uveal effusion syndrome allows immediate approximation of the photoreceptors and the retinal pigment epithelium and prevents further photoreceptor cell death. An alternative approach is external drainage of the subretinal fluid,22 which presents significant risk of subretinal hemorrhage, profound ocular hypotony, and difficulty accessing the posteriorly shifting subretinal fluid.
Vortex vein decompression
In 1980, Brockhurst reported successful use of scleral-thinning procedure with vortex vein decompression in the treatment of nanophthalmic ciliochoroidal effusion.19 He described the isolation of the intrascleral portion of the vortex vein and decompressing all four veins. Since scleral-thinning procedures alone are successful in the treatment of uveal effusion syndrome, vortex vein decompression for the treatment of ciliochoroidal effusion in uveal effusion syndrome or nanophthalmos is no longer performed by most vitreoretinal surgeons.
Hypotony maculopathy
Introduction
Loss of central vision may develop in patients with reduced intraocular pressure following trauma or ocular surgery. One of the causes of reduced vision in the setting of hypotony is significant folding of the choroid, neurosensory retina, and retinal pigment epithelium in the posterior pole, termed hypotony maculopathy by Gass in 1972.36–38 The initial description of fundus changes associated with hypotony dates back to the 1950s, when Dellaporta described patients with reduced vision following glaucoma procedures or perforating eye injuries.36 He noted that hypotony was associated with several fundus abnormalities, including optic nerve swelling, vascular tortuosity, and chorioretinal folds.36 The incidence of hypotony maculopathy significantly increased after the introduction of antimetabolites in glaucoma-filtering surgery.39–41 Younger age, myopia, and male gender are significant risk factors for the development of hypotony maculopathy after glaucoma procedures.40–42
Clinical features
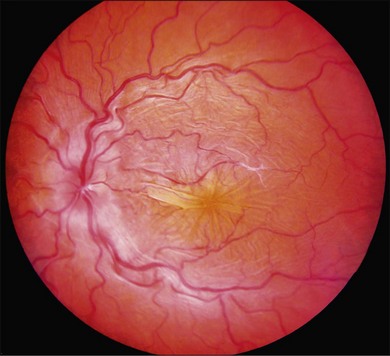
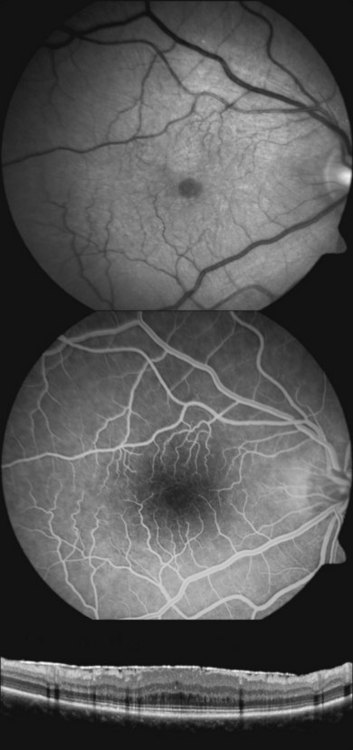
Posterior-segment examination in patients with hypotony maculopathy shows irregular folding of the neurosensory retina, retinal pigment epithelium, and choroid (Fig. 73.9). These folds are initially broad and indistinct and tend to radiate outward in a branching fashion from the optic disc temporally and appear concentric or irregular nasally. Around the center of the fovea, retinal folds may be arranged in a stellate pattern. The elevated crests of the folds appear yellow with dark narrow troughs in between. Reduction of the ocular anteroposterior diameter causes relative hyperopia. Marked optic disc edema may be present. Retinal vasculature is typically tortuous and may be engorged in some eyes. Anterior-segment examination may show a shallow anterior chamber if there is ciliochoroidal effusion. As the intraocular pressure returns to the normal range, the choroidal folds flatten and may disappear. If the hypotony was chronic, permanent retinal pigment epithelial changes may cause pigmented lines in the fundus.
Diagnosis
Fluorescein angiography
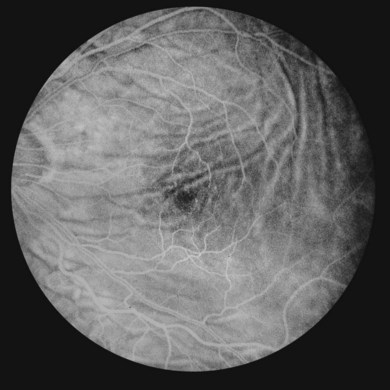
Fluorescein angiography is useful in demonstrating chorioretinal folds, especially in mild cases with a normal-appearing fundus examination. In the initial stages of hypotony, fluorescein angiography demonstrates an irregular increase in background choroidal fluorescence corresponding to the crest of the choroidal folds (Fig. 73.10). This produces hyperfluorescent streaks that may be seen in the early arterial phase. The hyperfluorescence of these streaks are multifactorial: (1) thinning of the retinal pigment epithelium on the crest of the folds; (2) pooling of the choroidal fluorescein under the crest; and (3) shorter course of the incident blue and reflected yellow-green light during angiography. The troughs of the folds are occupied by compressed retinal pigment epithelial cells, which reduce the transmission of the background choroidal fluorescence and cause hypofluorescence on angiography. Leakage of fluorescein may be seen in the capillaries of the optic nerve and there is typically no leakage in the retinal capillaries.43 In long-standing hypotony, angiography may show permanent alterations in the retinal pigment epithelium. The chorioretinal folds may be differentiated from the folds of the neurosensory retina, which do not alter the background fluorescence.
Ocular ultrasound
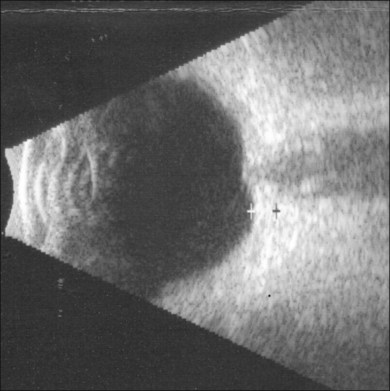
B-scan ultrasonography typically shows flattening and thickening of the sclera and choroid in the posterior pole44 (Fig. 73.11). Anterior-segment evaluation with ultrasound biomicroscopy may be useful for the identification of the underlying etiology of hypotony. The presence of a cyclodialysis cleft or anterior ciliary detachment may only be apparent on ultrasound biomicroscopy.45,46
Optical coherence tomography
In patients with low intraocular pressure and reduced visual acuity, optical coherence tomography imaging of the posterior pole can help to demonstrate subtle chorioretinal folds of the macula that may otherwise be difficult to detect on biomicroscopy47–49 (Fig. 73.12). High-resolution optical coherence tomography may detect chorioretinal folding in early hypotony, which may not show any changes in the background choroidal fluorescence during fluorescein angiography.48 However, the clinical significance of subtle chorioretinal folds detected only by optical coherence tomography in a patient with unchanged vision is unclear. Optical coherence tomography in a patient with symptomatic hypotony maculopathy and stereotypical biomicroscopic findings typically shows prominent chorioretinal folds (Fig. 73.12).

Pathogenesis
Hypotony
The reduction of intraocular pressure may be secondary to decreased aqueous production or increased aqueous outflow. In most cases, both a reduction in aqueous production and an increase in aqueous outflow are present in a hypotonous eye. Table 73.2 lists various etiologies of decreased intraocular pressure, some of which will be briefly discussed below.
| Postoperative
Ocular trauma Inflammatory Retinal detachment Ciliochoroidal detachment Ciliary body hypoperfusion Photocoagulation or cryoablation of the ciliary body Excessive reduction of intraocular pressure by pharmacologic agents Systemic |
Increased aqueous outflow
When the aqueous production is no longer sufficient to match the increased outflow, hypotony develops. Most cases of ocular hypotony are secondary to increased aqueous outflow. Under physiologic conditions, the aqueous leaves the eye primarily through the trabecular meshwork and Schlemm’s canal in a pressure-dependent manner, with much smaller amounts exiting through uveoscleral outflow. Several factors may increase aqueous outflow, including wound leak after anterior-segment surgery and excessive aqueous filtration after glaucoma filtering or drainage device surgery. After the introduction of antimetabolites in glaucoma-filtering procedures, the incidence of hypotony increased.39 Antimetabolites inhibit subconjunctival healing and scarring, which would otherwise limit the overfiltration rate. With the introduction of sutureless small-gauge vitrectomy technology, vitreoretinal surgeons encounter ocular hypotony with increasing frequency in their postoperative patients.50 Ocular trauma may be associated with scleral rupture and wound leak. Postoperative or traumatic cyclodialysis creates a free communication between the anterior chamber and the suprachoroidal space, increasing the uveoscleral outflow.
Mechanism of maculopathy
Chorioretinal folds form in the hypotonous eye due to scleral shrinkage and reduction of the inner surface area of the sclera. This loss of surface area then leads to folding of the inner portion of choroid and the outer retina.51–54 Maculopathy develops when the neurosensory retina is thrown into a series of stellate folds around the center of the fovea. This central stellate retinal wrinkling is caused by the thickened posterior scleral wall and the choroid displacing the normally thick retina surrounding the thin foveola.43 The folding of the neurosensory retina is the primary cause of vision loss in hypotony maculopathy. The risk of hypotony maculopathy is highest in young myopic patients, and this may indicate that the sclera is more vulnerable to swelling and contraction in young patients.
Mechanism of optic disc edema
Reduced intraocular pressure may lead to broad areas of choroidal swelling and retinal folding around the optic nerve and this may cause disc edema.43 Another proposed mechanism is the anterior bowing of the lamina cribrosa and constriction of axonal bundles in the lamina scleralis, causing reduced orthograde and retrograde axoplasmic transport and swelling of the optic nerve.55 Disc edema may be less impressive in eyes with pre-existing optic nerve disease and loss of axons.
Differential diagnosis
The finding of chorioretinal folds may be seen in any condition that reduces the inner surface area of the sclera secondary to thickening or shrinkage (Table 73.3).
Idiopathic chorioretinal folds
Horizontal macular chorioretinal folds as well as folds radiating from the optic nerve may be seen in young hyperopic patients on routine evaluation.52,56 These idiopathic chorioretinal folds are usually confined to the area around the macula and optic nerve, but may at times involve most of the posterior pole. Patients usually do not report any visual dysfunction. The appearance of the folds is usually symmetric in both eyes, but may occasionally be seen unilaterally. As the name indicates, the etiology of these chorioretinal folds is unknown and may be secondary to the shrinkage of the fibrous tunic of the eye after an inflammatory process during early years of life.43
Retrobulbar mass lesions
Any space-occupying mass in the orbit may cause scleral edema, choroidal congestion, and chorioretinal folds.57,58 A mass in the extraconal space may induce astigmatism, whereas intraconal location of a tumor may lead to acquired hyperopia.57 Both benign and malignant tumors have been reported to present with chorioretinal folds.
Scleral inflammation
Scleral inflammation with edema, thickening, and choroidal congestion may lead to the development of chorioretinal folds. Thyroid eye disease, idiopathic orbital inflammation, and other autoimmune or infectious uveitic diseases presenting with scleritis may have associated chorioretinal folds.43,59,60
Choroidal tumors
Malignant melanoma or metastatic lesions of the choroid may cause vascular congestion, choroidal edema, and scleral thickening, leading to chorioretinal folds surrounding the tumor.43
Choroidal neovascularization
Spontaneous or laser-induced contraction of subretinal pigment epithelial choroidal neovascular membrane may induce the formation of chorioretinal folds that radiate away from the membrane.61
Focal chorioretinal scars
Focal scars of the choroid and the retina may sometimes cause contraction and induce chorioretinal folds radiating towards the center of the scar.62
Optic nerve head disorders
Idiopathic intracranial hypertension and papilledema may be associated with chorioretinal folds. These folds are usually horizontal in the macula and converge on the nasal side of the swollen optic nerve.63,64 Chorioretinal folds have also been seen in patients with unexplained optic atrophy.65 In some patients with increased intracranial pressure, chorioretinal folds may present in the absence of any detectable papilledema and a lumbar puncture with an opening pressure may need to be performed to rule out idiopathic intracranial hypertension.66
Retinal folds
Retinal folds may be mistaken for chorioretinal folds in some eyes. Usually, folds of the neurosensory retina are narrow and seen radiating away from a focal inner retinal contraction caused by macular pucker or outer retinal contraction secondary to a chorioretinal scar (Fig. 73.13).43 Fluorescein angiography is helpful in differentiating retinal folds from chorioretinal folds, since folds of the neurosensory retina do not change the fluorescence pattern and are not seen on angiography.
Treatment
The effective treatment of hypotony maculopathy requires correcting the underlying ocular abnormality and restoring normal intraocular pressure. Since postoperative hypotony accounts for a significant portion of hypotony maculopathy patients, treatment is typically managed by an anterior-segment surgeon. Injection of viscoelastic or fluid into the anterior chamber to increase intraocular pressure produces short-lived effects. Wound leaks after anterior-segment surgery need to be addressed immediately with a bandage contact lens or suturing the wound to prevent further aqueous loss. Ocular inflammation requires treatment with a topical or oral corticosteroid. Posttraumatic cyclodialysis cleft may require laser or surgical closure to normalize intraocular pressure. Overfiltration after glaucoma surgery may require resuturing the scleral flap, donor scleral patch graft placement, or autologous blood injection.28–30,67
Chorioretinal folds may resolve following the restoration of normal intraocular pressure. In cases of chronic hypotony, the retinal pigment epithelium will show permanent changes, including hyperpigmentation at the troughs of the folds, causing irregularly dark, pigmented lines in the posterior pole detected by fluorescein angiography.43 The choroid and the sclera return to normal thickness and the tortuosity and engorgement of the retinal vessels disappear.
Early detection of hypotony maculopathy is important for implementing the necessary measures to restore normal intraocular pressure and recovery of vision. The prognosis for visual recovery depends on the duration of the hypotony and the chronicity of the chorioretinal folds. Prolonged folding of the choroid and the neurosensory retina may cause irreversible structural changes in the macula and poor visual recovery after normalization of the intraocular pressure. However, significant visual improvement may still occur in some patients after treatment of chronic hypotony.68 Duker and Schuman reported that pars plana vitrectomy and mechanical flattening of the posterior pole with intraoperative instillation of perfluorocarbon liquid significantly improved visual acuity in a patient who had minimal visual recovery after increasing the intraocular pressure with previous bleb revision.69 The efficacy of this approach has not been confirmed in larger patient cohorts.
1 Schepens CL, Brockhurst RJ. Uveal effusion. 1. Clinical picture. Arch Ophthalmol. 1963;70:189–201.
2 Gass JD, Jallow S. Idiopathic serous detachment of the choroid, ciliary body, and retina (uveal effusion syndrome). Ophthalmology. 1982;89:1018–1032.
3 Johnson MW. Uveal Effusion. In: Guyer DR, Yannuzzi LA, Chang S, et al. Retina–vitreous–macula. Philadelphia: W.B. Saunders; 1999:658–668.
4 Toris CB, Pederson JE, Tsuboi S, et al. Extravascular albumin concentration of the uvea. Invest Ophthalmol Vis Sci. 1990;31:43–53.
5 Brubaker RF, Pederson JE. Ciliochoroidal detachment. Surv Ophthalmol. 1983;27:281–289.
6 Inomata H, Bill A. Exit sites of uveoscleral flow of aqueous humor in cynomolgus monkey eyes. Exp Eye Res. 1977;25:113–118.
7 Alm A, Bill A. Ocular circulation. In: Moses RA, Hart WM, Jr. Adler’s physiology of the eye. St Louis: Mosby; 1987:199.
8 Aaberg TM, Maggiano JM. Choroidal edema associated with retinal detachment repair: experimental and clinical correlation. Mod Probl Ophthalmol. 1979;20:6–15.
9 Hawkins WR, Schepens CL. Choroidal detachment and retinal surgery. Am J Ophthalmol. 1966;62:813–819.
10 Liew SC, McCluskey PJ, Parker G, et al. Bilateral uveal effusion associated with scleral thickening due to amyloidosis. Arch Ophthalmol. 2000;118:1293–1295.
11 Vine AK. Uveal effusion in Hunter’s syndrome. Evidence that abnormal sclera is responsible for the uveal effusion syndrome. Retina. 1986;6:57–60.
12 Forrester JV, Lee WR, Kerr PR, et al. The uveal effusion syndrome and trans-scleral flow. Eye (Lond). 1990;4:354–365.
13 Uyama M, Takahashi K, Kozaki J, et al. Uveal effusion syndrome: clinical features, surgical treatment, histologic examination of the sclera, and pathophysiology. Ophthalmology. 2000;107:441–449.
14 Ward RC, Gragoudas ES, Pon DM, et al. Abnormal scleral findings in uveal effusion syndrome. Am J Ophthalmol. 1988;106:139–146.
15 Boubriak OA, Urban JP, Bron AJ. Differential effects of aging on transport properties of anterior and posterior human sclera. Exp Eye Res. 2003;76:701–713.
16 Gass JD. Uveal effusion syndrome. A new hypothesis concerning pathogenesis and technique of surgical treatment. Retina. 1983;3:159–163.
17 Johnson MW, Gass JD. Surgical management of the idiopathic uveal effusion syndrome. Ophthalmology. 1990;97:778–785.
18 Calhoun FP, Jr. The management of glaucoma in nanophthalmos. Trans Am Ophthalmol Soc. 1975;73:97–122.
19 Brockhurst RJ. Vortex vein decompression for nanophthalmic uveal effusion. Arch Ophthalmol. 1980;98:1987–1990.
20 Casswell AG, Gregor ZJ, Bird AC. The surgical management of uveal effusion syndrome. Eye (Lond). 1987;1:115–119.
21 Moses RA. Detachment of ciliary body – anatomical and physical considerations. Invest Ophthalmol. 1965;4:935–941.
22 Brockhurst RJ, Lam KW. Uveal effusion. II. Report of a case with analysis of subretinal fluid. Arch Ophthalmol. 1973;90:399–401.
23 Gass JD. Uveal effusion syndrome: a new hypothesis concerning pathogenesis and technique of surgical treatment. Trans Am Ophthalmol Soc. 1983;81:246–260.
24 Green RL, Byrne SF. Diagnostic ophthalmic ultrasound. In: Ryan SJ, ed. Retina. St. Louis: Mosby, 1989.
25 Okuda T, Higashide T, Wakabayashi Y, et al. Fundus autofluorescence and spectral-domain optical coherence tomography findings of leopard spots in nanophthalmic uveal effusion syndrome. Graefes Arch Clin Exp Ophthalmol. 2010;248:1199–1202.
26 Singh OS, Simmons RJ, Brockhurst RJ, et al. Nanophthalmos: a perspective on identification and therapy. Ophthalmology. 1982;89:1006–1012.
27 Brockhurst RJ. Nanophthalmos with uveal effusion. A new clinical entity. Arch Ophthalmol. 1975;93:1989–1999.
28 Cohen SM, Flynn HW, Jr., Palmberg PF, et al. Treatment of hypotony maculopathy after trabeculectomy. Ophthalmic Surg Lasers. 1995;26:435–441.
29 Haynes WL, Alward WL. Rapid visual recovery and long-term intraocular pressure control after donor scleral patch grafting for trabeculectomy-induced hypotony maculopathy. J Glaucoma. 1995;4:200–201.
30 Haynes WL, Alward WL. Combination of autologous blood injection and bleb compression sutures to treat hypotony maculopathy. J Glaucoma. 1999;8:384–387.
31 Bellows AR, Chylack LT, Jr., Epstein DL, et al. Choroidal effusion during glaucoma surgery in patients with prominent episcleral vessels. Arch Ophthalmol. 1979;97:493–497.
32 Allen KM, Meyers SM, Zegarra H. Nanophthalmic uveal effusion. Retina. 1988;8:145–147.
33 Good WV, Stern WH. Recurrent nanophthalmic uveal effusion syndrome following laser trabeculoplasty. Am J Ophthalmol. 1988;106:234–235.
34 Faulborn J, Kolli H. Sclerotomy in uveal effusion syndrome. Retina. 1999;19:504–507.
35 Schneiderman TE, Johnson MW. A new approach to the surgical management of idiopathic uveal effusion syndrome. Am J Ophthalmol. 1997;123:262–263.
36 Dellaporta A. Fundus changes in postoperative hypotony. Am J Ophthalmol. 1955;40:781–785.
37 Gass JDM. Hypotony maculopathy. In: Bellows JC, ed. Contemporary ophthalmology, honoring Sir Stewart Duke-Elder. Baltimore: Williams & Wilkins; 1972:343–366.
38 Hovanesian JA, Higginbotham EJ, Lichter PR, et al. Long-term visual outcome of ocular hypotension after thermosclerostomy. Am J Ophthalmol. 1993;115:603–607.
39 Costa VP, Wilson RP, Moster MR, et al. Hypotony maculopathy following the use of topical mitomycin C in glaucoma filtration surgery. Ophthalmic Surg. 1993;24:389–394.
40 Jampel HD, Pasquale LR, Dibernardo C. Hypotony maculopathy following trabeculectomy with mitomycin C. Arch Ophthalmol. 1992;110:1049–1050.
41 Stamper RL, McMenemy MG, Lieberman MF. Hypotonous maculopathy after trabeculectomy with subconjunctival 5-fluorouracil. Am J Ophthalmol. 1992;114:544–553.
42 Fannin LA, Schiffman JC, Budenz DL. Risk factors for hypotony maculopathy. Ophthalmology. 2003;110:1185–1191.
43 Gass JDM. Hypotony maculopathy. Stereoscopic atlas of macular diseases: diagnosis and treatment. St. Louis: Mosby; 1997. p. 294–5
44 Cappaert WE, Purnell EW, Frank KE. Use of B-sector scan ultrasound in the diagnosis of benign choroidal folds. Am J Ophthalmol. 1977;84:375–379.
45 Chan TK, Talbot JF, Rennie IG, et al. The application of ultrasonic biomicroscopy in the management of traumatic hypotony. Eye (Lond). 2000;14(Pt 5):805–807.
46 Roters S, Engels BF, Szurman P, et al. Typical ultrasound biomicroscopic findings seen in ocular hypotony. Ophthalmologica. 2002;216:90–95.
47 Budenz DL, Schwartz K, Gedde SJ. Occult hypotony maculopathy diagnosed with optical coherence tomography. Arch Ophthalmol. 2005;123:113–114.
48 Lima VC, Prata TS, Castro DP, et al. Macular changes detected by Fourier-domain optical coherence tomography in patients with hypotony without clinical maculopathy. Acta Ophthalmol (Copenh). 2011;89:e274–e277.
49 Martinez de la Casa JM, Garcia Feijoo J, Castillo Gomez A, et al. [Hypotony maculopathy diagnosed by optical coherence tomography.]. Arch Soc Esp Oftalmol. 2003;78:567–569.
50 Thompson JT. Advantages and limitations of small gauge vitrectomy. Surv Ophthalmol. 2011;56:162–172.
51 Cangemi FE, Trempe CL, Walsh JB. Choroidal folds. Am J Ophthalmol. 1978;86:380–387.
52 Leahey AB, Brucker AJ, Wyszynski RE, et al. Chorioretinal folds. A comparison of unilateral and bilateral cases. Arch Ophthalmol. 1993;111:357–359.
53 Newell FW. Choroidal folds. The seventh Harry Searls Gradle memorial lecture. Am J Ophthalmol. 1973;75:930–942.
54 Newell FW. Fundus changes in persistent and recurrent choroidal folds. Br J Ophthalmol. 1984;68:32–35.
55 Minckler DS, Bunt AH. Axoplasmic transport in ocular hypotony and papilledema in the monkey. Arch Ophthalmol. 1977;95:1430–1436.
56 Kalina RE, Mills RP. Acquired hyperopia with choroidal folds. Ophthalmology. 1980;87:44–50.
57 Friberg TR, Grove AS, Jr. Choroidal folds and refractive errors associated with orbital tumors. An analysis. Arch Ophthalmol. 1983;101:598–603.
58 Shields JA, Shields CL, Rashid RC. Clinicopathologic correlation of choroidal folds: secondary to massive cranioorbital hemangiopericytoma. Ophthal Plast Reconstr Surg. 1992;8:62–68.
59 Krist D, Wenkel H. Posterior scleritis associated with Borrelia burgdorferi (Lyme disease) infection. Ophthalmology. 2002;109:143–145.
60 Haruyama M, Yuzawa M, Kawamura A, et al. Indocyanine green angiographic findings of chorioretinal folds. Jpn J Ophthalmol. 2001;45:293–300.
61 Diskin J, Maguire AM, Margherio RR. Choroidal folds induced with diode endolaser. Arch Ophthalmol. 1992;110:754.
62 Johnson RN, Schatz H, McDonald HR. Photic maculopathy: early angiographic and ophthalmoscopic findings and late development of choroidal folds. Case report. Arch Ophthalmol. 1987;105:1633–1634.
63 Bird AC, Sanders MD. Choroidal folds in association with papilloedema. Br J Ophthalmol. 1973;57:89–97.
64 Lavinsky J, Lavinsky D, Lavinsky F, et al. Acquired choroidal folds: a sign of idiopathic intracranial hypertension. Graefes Arch Clin Exp Ophthalmol. 2007;245:883–888.
65 Sarraf D, Schwartz SD. Bilateral choroidal folds and optic neuropathy: a variant of the crowded disk syndrome? Ophthalmology. 2003;110:1047–1052.
66 Griebel SR, Kosmorsky GS. Choroidal folds associated with increased intracranial pressure. Am J Ophthalmol. 2000;129:513–516.
67 Schwartz GF, Robin AL, Wilson RP, et al. Resuturing the scleral flap leads to resolution of hypotony maculopathy. J Glaucoma. 1996;5:246–251.
68 Delgado MF, Daniels S, Pascal S, et al. Hypotony maculopathy: improvement of visual acuity after 7 years. Am J Ophthalmol. 2001;132:931–933.
69 Duker JS, Schuman JS. Successful surgical treatment of hypotony maculopathy following trabeculectomy with topical mitomycin C. Ophthalmic Surg. 1994;25:463–465.