Chapter 131 Tuberous Sclerosis and the Eye
Introduction
History, diagnosis, and genetic basis
Tuberous sclerosis complex (TSC) is a rare multisystem genetic disorder, characterized by hamartomatous tumors of the brain, skin, viscera, and eye. It is inherited in an autosomal dominant fashion,1 with a high degree of penetrance2 with variable phenotypic expression. The first publication of a color plate by Rayner of a patient with an apparent facial angiofibroma took place in 1835.3 In 1880, D.M. Bourneville, from a neuropathological study of a young patient with seizures, hemiplegia, mental subnormality, who also had renal tumors, coined the term tuberous sclerosis.4 However, the eponym Bourneville’s disease was commonly used to describe this condition until only recently. In 1908, Vogt5 first proposed epilepsy, mental retardation, and the skin lesions of adenoma sebaceum as a diagnostic triad. In 1920, Van der Hoeve6 first recognized retinal involvement in tuberous sclerosis. Ophthalmologists now recognize astrocytic hamartomas of both the retina and the optic nerve as common features of this condition.
The diagnostic criteria are based on the premise now that there are no truly pathognomonic features or clinical criteria for the diagnosis of tuberous sclerosis or tuberous sclerosis complex (TSC).5 Signs that were once regarded as sufficiently specific are now known to occur as isolated findings in normal individuals without TSC.7 In theory, the perfect disease classification scheme should have both a high sensitivity (low rate of missing the diagnosis of tuberous sclerosis) and a high specificity (low rate of incorrectly labeling a patient with tuberous sclerosis). The committee recommended the adoption of an ordinal classification of tuberous sclerosis: definite, probable, and suspect tuberous sclerosis.7 Patients can be classified into one of three categories based on the presence of clinical features of varying specificity (primary features: high specificity; secondary features: moderate specificity; tertiary features: lower degree of specificity).8 The revised criteria require either TSC-associated lesions in two or more organ systems or two dissimilar lesions in one organ system to confirm the diagnosis. The classic triad included seizures, mental retardation, and cutaneous angiofibromas, which, however, only occurs in 29% of cases.9 Mental retardation and seizures were also left out of the revised criteria by the 1998 expert consensus panel.10 The addition of DNA testing complements the clinical diagnosis and helps with genetic counseling, especially in concert with prenatal testing.11
Systemic manifestations
Neurological
Seizures
Children with tuberous sclerosis often have seizures that are referred to as “infantile spasms” or “salaam attacks.”7 These seizures are characterized by repetitive myoclonic spasms of the head, neck, and limbs. Originally described by West,12 an English GP, through the experience of his own son in 1841, the seizures last for seconds but may occur in bouts of 10–50. Hoyt13 described infantile spasms as “lightning fast nodding of the head, often with extension or flexion of the trunk and frequently also of the arms.” According to Pampliglione and Pugh,14 25% of children affected by “infantile spasms” develop other stigmata of tuberous sclerosis within 4 years of diagnosis. Infantile seizures, the most common feature of the disease, tend to evolve into grand mal seizures at a later time and are seen in 93% of patients.11,15 Presently, the treatment of epilepsy in TSC remains a major challenge, even with new anticonvulsant medications.16 Even after neurosurgery, seizures recur in approximately 30% of cases.17
Cognitive and behavioral disability
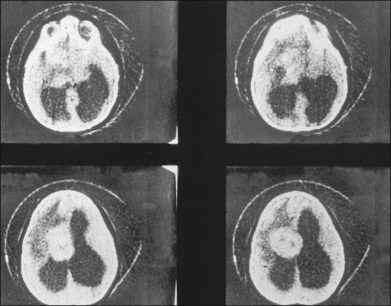
Children with tuberous sclerosis may have normal or above-average intelligence.15 However, Lagos and Gomez15 reported that 44 (62%) of 71 tuberous sclerosis patients were labeled as mentally retarded. Subsequent reports confirm that neurocognitive manifestations ranging from profound cognitive disabilities to mild learning impairment occur in around 45% of patients.18 While the majority of individuals with TSC have normal intelligence, they may still be prone to specific cognitive memory, attentional and executive skills deficits.19 It should be noted that early reports emphasizing the prevalence of mental retardation in tuberous sclerosis suffered from selection bias because they were based on patients who were institutionalized.11 In the Lagos and Gomez series,15 intracranial calcifications were more commonly seen in patients with normal intelligence. The cause of both seizures and mental retardation may be related to the presence of cortical tubers (Fig. 131.1) and subependymal hamartomas (Fig. 131.2). These tumor masses represent benign astrocytic hamartomas, which characteristically involve the basal ganglia, lateral and third ventricles, and the posterior fossa. Histopathologically, they consist of well-differentiated large astrocytes in an admixture of fibrillary astrocytes and calcospherules.20 They frequently undergo cystic degeneration and dystrophic calcification, accounting for their typical radiologic features (Fig. 131.3) and for the name tuberous sclerosis.21 Most significant variables associated with a poor cognitive outcome include a history of refractory seizures, TSC2 mutations, and cortical tubers in specific locations of the brain.22
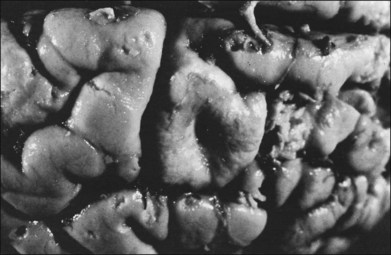
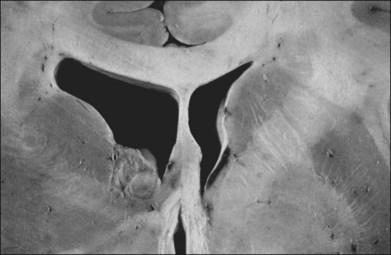
Brain tumors are associated with tuberous sclerosis. These well-circumscribed lesions are classified as subependymal giant-cell astrocytomas and should not be confused with the giant-cell variant of glioblastoma multiforme, a malignant astrocytoma.23
A case of giant cerebral aneurysm with visual loss as a result of optic nerve compression in a patient with tuberous sclerosis has been reported.24
Skin features
Facial angiofibromas formerly referred to as adenoma sebaceum, occur as a reddish-brown papular rash found characteristically in a “butterfly” distribution over the face. This rash is a pathognomonic hallmark of tuberous sclerosis and is very sensitive, occurring in over 85% of patients.15 Histopathologically, adenoma sebaceum consists of multiple smooth papules that are benign angiofibromas.25 The rash is generally not apparent at birth but appears in childhood, generally before the age of 9 years, and tends to intensify with time.20 It is most prominent in the nasolabial folds and over the malar areas.
The skin lesion formerly believed to be pathognomonic for tuberous sclerosis is the hypopigmented macule or patch (Fig. 131.4). These lesions are variable in shape, with only an occasional one justifying the term ash-leaf sign. Often present at birth, this is the first clinical sign of disease. Its occurrence is highly variable, being present in up to 75% of affected children.26 Pathologically, the ash-leaf lesion is an achromic nevus,27 as opposed to vitiligo, in which the melanocytes are actually missing. Ultraviolet light (classically a Wood’s lamp) may be used effectively in a darkened room to screen for the ash-leaf sign.28 The importance of this clinical sign in the workup of a child with infantile spasms of unknown cause cannot be overemphasized.26
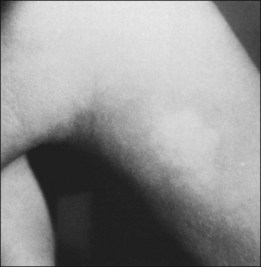
Fig. 131.4 Hypopigmented skin macule or patch (achromic nevus), sometimes referred to as an ash-leaf sign.
(Courtesy of P. McLeod.)
Shagreen patches represent areas of skin affected by fibromatous infiltration. This sign is seen in approximately 20% of cases, often in the lower back.29 This flesh-colored, leathery plaque on the lumbosacral area is highly characteristic of the tuberous sclerosis complex.28 Subungual fibromas may be seen on the hands and feet.15,28
Visceral features
Although Vogt’s classic triad of mental deficiency, epilepsy, and facial angiofibromas implies central nervous system and skin involvement, it is important to be cognizant of the possible presence of visceral tumors. Renal angiolipomas are hamartomas that occur singularly or in multiples, in unilateral or bilateral fashion. They are present in 80% of patients with tuberous sclerosis.20 They are benign tumors that are not known to metastasize to distant sites,30 although extension to the renal vein has been noted.31 Cardiac hamartomas also occur. They are rhabdomyomas that are single or multiple whitish nodules that characteristically protrude into the ventricles.20 Slowly, progressive subpleural cysts may form and can rupture, causing a spontaneous pneumothorax.32 Hamartomas of the liver, thyroid, pancreas, and testis have been reported.20
Skeletal features
The skeletal system is affected in 40% of cases.7 Sclerotic calcified areas are seen in the skull and spine. Skull radiographs may show opacities over the calvaria that represent intraosseous sclerotic areas of intracranial calcified astrocytic hamartomas. Computed tomography (CT) scans are particularly helpful in the investigation of tuberous sclerosis.33 Phalangeal cysts affect the hands and feet.7
Ocular manifestations
Retinal manifestations
The retinal phakomas of tuberous sclerosis are astrocytic hamartomas of the retina, seen in an estimated 53% of patients, according to Lagos and Gomez15 in their Mayo Clinic series of 71 cases. Shelton34 found evidence of retinal hamartomatous lesions in all seven patients examined. Solitary astrocytic hamartomas may be seen in otherwise healthy patients, but in patients with tuberous sclerosis they may be multifocal or bilateral. Although most frequently seen in the setting of tuberous sclerosis, these hamartomas may also be noted in patients with neurofibromatosis.35,36 Although astrocytic hamartomas of the retina are the principal ocular manifestation of the tuberous sclerosis complex, two patients have been reported in whom stromal depigmentation of the iris and atypical colobomata were correlated on histopathological examination with hamartomas of the iris pigment epithelium and ciliary body epithelium.37
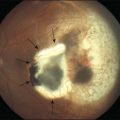
Typically, astrocytic hamartomas can be classified morphologically as one of two types: (1) large, whitish (calcified) nodular masses or (2) flat, translucent (noncalcified) smooth tumors.38 Nyboer et al.39 have described an intermediate type of retinal hamartoma having features of both types. These lesions typically occur at or near the optic disc. More peripheral lesions may occur, and these may be more easily confused with retinoblastoma. Although almost all astrocytic hamartomas of the retina in tuberous sclerosis are endophytic in nature, an exophytic case has been described (Figs 131.5, 131.6 illustrate a different case).31 This case consisted of the pathologic examination of an eye enucleated for neovascular glaucoma from a 10-year-old boy with tuberous sclerosis and seizures. The retina was totally detached and was associated with a large multinodular mulberry-like astrocytic tumor in the subretinal space.
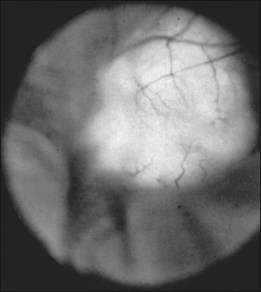
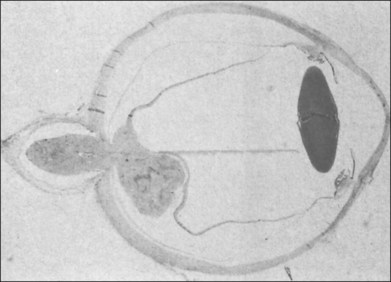
Fig. 131.6 Pathologic correlate (low-power photograph) of the eye containing the lesion illustrated in Figure 131.5.
(Courtesy of J. Augsburger.)
The exophytic tumors are astrocytic tumors in the subretinal space.40 The endophytic tumors are astrocytic hamartomas of the nerve fiber layer of the retina (Figs 131.7, 131.8).12 These retinal tumors are benign and non-neoplastic,20 generally showing minimal or slow evidence of growth,13 although progressive enlargement associated with the development of calcification has been reported.39 Retinal astrocytomas can, therefore, occasionally show progressive enlargement, retinal detachment, and vitreous seeding – findings that can “mislead the clinician towards a diagnosis of retinoblastoma.”41 The reverse, however, has also been reported, since spontaneous regression of retinal astrocytic hamartoma has been described.42
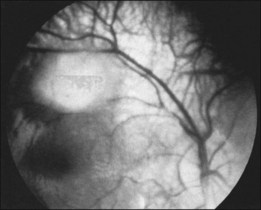
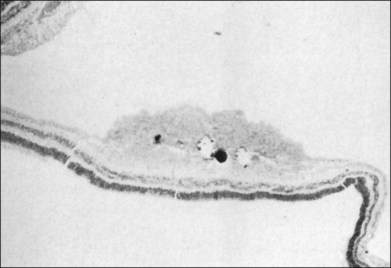
Fig. 131.8 Pathologic correlate of an endophytic astrocytic hamartoma of the retina.
(Courtesy of R. Bell.)
Most astrocytic (glial) hamartomas of the retina are composed of elongated fibrous astrocytes with small oval nuclei and cytoplasmic processes.36 Larger tumors, particularly those located on or near the optic nerve, may have large round and moderately pleomorphic astrocytes.43 Foci of calcification may be seen. Clinically, Gass35 has noted that these lesions may show varying degrees of tumor vascularization, which is more evident angiographically than ophthalmoscopically. Fluorescein angiography of retinal astrocytomas characteristically shows late-phase relative hyperfluorescence with leakage of dye into the vitreous cavity.35 Fluorescein angiography, however, has not been considered helpful in differentiating astrocytic hamartomas from solitary retinoblastoma.44
Ocular complications related to the presence of retinal hamartomas have been reported as rare complications of tuberous sclerosis. These include vitreous hemorrhage, retinal vascular abnormalities (including telangiectasia, neovascularization, and exudation), and vitreous seeding, which is characteristically described in association with optic nerve head or epipapillary astrocytic hamartomas. Vitreous hemorrhage may arise from the substance of retinal or optic nerve astrocytomas, according to a clinicopathologic report in which the vitreous hemorrhage was thought to be arising from the surface of an epipapillary astrocytic hamartoma.45 The hemorrhage was observed intraoperatively during pars plana vitrectomy surgery to remove old vitreous hemorrhage. Attempts to use endodiathermy to cauterize an apparent surface bleeding site exposed cystic spaces within the tumor that were filled with hemorrhagic material and actively bled. A myriad of small vascular channels were confirmed pathologically.36 Blood-filling intratumor cystic spaces in a large epipapillary tumor, resulting in vitreous hemorrhage, were first reported by Van der Hoeve in 1921.46 Atkinson et al.47 reported vitreous hemorrhage in two cases of optic disc astrocytomas. Koch and Walsh48 also reported vitreous hemorrhage in a case associated with increased intracranial pressure and papilledema. Vitreous hemorrhage may also arise from retinal neovascularization associated with a retinal astrocytoma.49 The angiogliomatous nature of retinal astrocytic hamartomas has been stressed in a pathologic report by Barsky and Wolter,38 giving rise to speculation regarding the possible angioblastic and astroblastic origin of these lesions. Retinal telangiectasis with surrounding intraretinal lipid exudation in the parafoveal region and serous retinal detachment affecting the macula have been reported to occur in association with multiple astrocytic hamartomas.40 A single case of a peripapillary lesion that hemorrhaged into the vitreous, increased in size, and underwent spontaneous necrosis has been reported.50
Optic nerve phakomas
As reported by Lagos and Gomez,15 retinal and optic nerve astrocytomas occur in 53% of cases. These lesions “protrude above or overlie the optic disc” (Fig. 131.9) and, according to Miller,51 “may initially have a grayish or grayish pink appearance, but later develop a glistening, yellow, mulberry appearance.” Optic nerve astrocytomas have long been known to be capable of vitreous seeding and vitreous hemorrhage, as noted earlier, since Van der Hoeve’s 1921 report.46,48 In 1984, vitreous seeding associated with an intense vitritis in a patient with tuberous sclerosis was reported by De Juan et al.52 These authors speculated that the development of a posterior vitreous detachment with associated traction at the optic disc may have resulted in vitreous seeding.
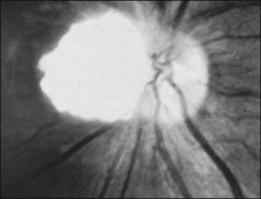
Ocular adnexal lesions
Angiofibromas (adenoma sebaceum) of the skin of the lids occur in tuberous sclerosis, giving rise to a typical salmon-colored lid.53 Isolated white eyelashes (poliosis) located among normally pigmented ones have been reported in a patient in whom the fundus of one eye was remarkable, not only for an astrocytic hamartoma of the retina, but also for a leaf-shaped area of hypopigmentation in the retinal periphery,54 similar in appearance to the hypopigmented skin lesions (ash-leaf spots). Hypopigmented iris spots have also been reported as an early sign of disease.55 Other isolated associations with tuberous sclerosis have been summarized by Williams and Taylor.11
Differential diagnosis
The differential diagnosis of astrocytic hamartomas of the retina and optic nerve depends to a great extent on whether the diagnosis of tuberous sclerosis can be established. This diagnosis is based on various primary, secondary, and tertiary features, most of which were initially put forth by Gomez56 and recently revised by the Diagnostic Criteria Committee of the National Tuberous Sclerosis Association.7 The major criteria are: facial angiofibromas (adenoma sebaceum); ungual fibromas; cortical tuber(s) (at necropsy); subependymal nodule(s) or giant-cell astrocytoma (histologically confirmed); multiple calcified subependymal nodules protruding into the ventricle (radiologic evidence), and multiple retinal astrocytes. Secondary features include: an affected first-degree relative; cardiac rhabdomyoma; other retinal hamartoma or achromic patch; cerebral tubers; noncalcified subependymal nodule; shagreen patch; forehead plaque; pulmonary lymphangiomatosis; renal angiomyolipoma, and renal cysts. Tertiary criteria include, but are not limited to, hypomelanotic macules, hamartoma of other organs, and infantile spasms. Definite tuberous sclerosis requires either one primary feature and two secondary features or one secondary feature plus two tertiary ones. Probable tuberous sclerosis requires either one secondary plus one tertiary feature or three tertiary features. Suspect tuberous sclerosis requires either one secondary feature or two tertiary features.7 Therefore, in the presence of a single retinal (or optic nerve) hamartoma, only one secondary criterion or two tertiary criteria are required to make the diagnosis of tuberous sclerosis. The first step for the ophthalmologist should be carefully to reexamine both eyes for signs of one or more retinal or optic nerve hamartomas. Retinal and optic nerve astrocytic hamartomas can also be present in patients with neurofibromatosis.35
If the diagnosis of tuberous sclerosis or neurofibromatosis cannot be clearly established, the appearance of the retinal or optic nerve hamartoma may provide clinical clues related to the associated systemic condition. Shields and Shields21 state that a tumor that consists of a mulberry-like calcification is pathognomonic of tuberous sclerosis. However, when the only retinal tumor seen is a small, translucent, and whitish noncalcified thickening of the nerve fiber layer, it may be difficult, if not impossible, to distinguish it from myelinated nerve fibers or retinoblastoma.21 This difficulty in differentiating between small astrocytic hamartomas of the retina and retinoblastoma has also been stressed by others.57 Gass44 has reported that fluorescein angiography, although helpful in outlining the rich capillary network in these tumors, is not helpful in differentiating astrocytoma from retinoblastoma. However, despite this potential source of major diagnostic error, case series that examine cases of possible retinoblastoma rarely include a single case of astrocytic hamartoma of the retina.13,58 In Howard and Ellsworth’s series of 500 cases of suspected retinoblastoma in 1965,58 not a single case of astrocytic hamartoma was mentioned, although there were 265 cases without retinoblastoma. Shields and Shields,21 who list astrocytic hamartoma in the differential diagnosis of retinoblastoma, confirm the rarity of a mistaken diagnosis in their own series. Of the 136 patients sent to their clinic with the diagnosis of possible retinoblastoma, only one patient had an astrocytic hamartoma. Shields et al.36 reported the use of fine-needle biopsy to help differentiate an astrocytic hamartoma from a retinoblastoma.
Other disorders that can mimic astrocytic hamartoma include myelinated nerve fibers and inflammatory lesions.7 Furthermore, given the angiomatous component of these lesions, Coats disease and other causes of retinal telangestasis36,49,50,57,59 should be included in the differential diagnosis. A more thorough list of clinical simulators of astrocytic hamartoma is provided in Box 131.1.
Box 131.1
Differential diagnosis of astrocytic hamartomas
• Other causes of retinal telangiectasis
• Toxocara canis, toxoplasmosis
• Other causes of choroiditis and scleritis
• Other causes of exudative retinal detachment with underlying mass lesion
• Optic disc astrocytic hamartoma
• Retinitis pigmentosa associated with optic disc excrescences (hyaline bodies) versus astrocytic hamartomas
• Optic disc glioma (variant of optic nerve glioma)
• Other primary optic disc tumor
• Other causes of unilateral optic disc swelling or papillitis
Optic disc hamartomas, after they have developed the yellow, glistening, mulberry appearance so characteristic of these lesions,51 may be confused with optic disc drusen. However, optic disc drusen lie within the disc, whereas astrocytic hamartomas protrude above it51 and obscure both the optic nerve and retinal blood vessels.60 Heckenlively61 reviewed the controversy of the origin of the “globular excrescences or hyaline bodies of the optic nerve head.” These hyaline bodies are suspected of being drusen based on the pathologic studies of Spencer,43,62 Puck et al.,63 and Novak and Foos.64 Heckenlively’s interpretation is that these studies do not put to rest the issues raised by Robertson,65 namely, that vitreal epipapillary excrescences in patients with retinitis pigmentosa represent astrocytic hamartomas.61 Optic disc glioma may also be confused with astrocytic hamartomas. Gliomas of the optic disc may initially appear with disc swelling or obscuration of the disc by a whitish, protuberant mass. These lesions can be easily confused with astrocytic hamartomas, especially in the case of a noncalcified hamartoma. Although more commonly described in neurofibromatosis, Shields and Shields21 note that optic disc glioma can also appear in patients with tuberous sclerosis. Other neoplastic lesions of the optic disc should be considered in the differential diagnosis, including capillary and cavernous hemangioma.51
Genetics
Tuberous sclerosis is a systemic disorder that is inherited in an autosomal dominant fashion in which mutations in the tumor suppressor genes TSC1 or TSC2 lead to the development of benign hamartomas throughout the body as described, with incomplete penetrance and variable expressivity. However, approximately 70% of cases are said to arise from new mutations.66 The prevalence of disease has been approximated at 1 in 15 000.67 Linkage studies have demonstrated the genetic heterogeneity of this condition, with two separate mutations accounting for the vast majority of cases.68–70 The two genes responsible for most cases of tuberous sclerosis are located on chromosome 9 at q34 (TSC1) and on chromosome 16 at p13 (TSC2).71 No phenotypic differences are noted in patients with the two different mutations, although mutations in TSC1 appear to be associated with a milder clinical phenotype.69,72 Tuberin, the product of TSC2, has partial homology to Rap 1-GTPase-activating protein, which supports the theory that tuberin plays a role in cell growth regulation.68 The product of TSC1 has been identified as hamartin.73 Both proteins are widely expressed in the brain and may form part of a cascade modulating cellular differentiation, tumor suppression and intracellular signaling.73
Genetic counseling is essential because patients with the tuberous sclerosis gene have a 50% chance of transmitting the gene to their offspring.68 Parents of patients with tuberous sclerosis should be made aware of the possibility of germline mosaicism. It is important that these individuals have a thorough systemic evaluation, which should include CT scanning of the head and renal ultrasonography to rule out tuberous sclerosis.68 If the results of these examinations are normal in the parents, the child likely suffers from a new mutation, and the subsequent risk of another child having tuberous sclerosis has been estimated at 1%.58
1 Fryer AE, Chalmers A, Connor JM, et al. Evidence that the gene for tuberous sclerosis is on chromosome 9. Lancet. 1987;1:659–661.
2 Napolioni V, Moavero R, Curatolo P. Recent advances in the neurobiology of tuberous sclerosis complex. Brain Dev. 2009;31:104–113.
3 Gomez M. History of tuberous sclerosis complex. Brain Dev. 1995;17:55–57.
4 Bourneville DM. Selerose tubereuse des circonvolutions cerebrales: idiotie et epilepsie hemiplegique. Arch Neurol (Paris). 1880;1:81–91.
5 Vogt H. Zur diagnostik der tuberosen sclerose. Z Erforschung Behandlung Jugendl Schwachsinns. 1908;2:1–16.
6 Van der Hoeve J. Eye symptoms in tuberous sclerosis of the brain. Trans Ophthalmol Soc UK. 1920;40:329–334.
7 Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643–649.
8 Rowley SA, O’Callaghan FJ, Osborne JP. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85:420–423.
9 Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis advances in diagnosis, genetics and management. J Am Acad Dermatol. 2007;57:189–202.
10 Rosser T Panigrahy A, McClintock W. The diverse clinical manifestations of tuberous sclerosis complex: a review. Semin Pediatr Neurol. 2006;13:27–36.
11 Williams R, Taylor D. Tuberous sclerosis. Surv Ophthalmol. 1985;30:143–153.
12 West WJ. On a peculiar form of infantile convulsion. Lancet. 1841;1:724–725.
13 Hoyt CS. The ocular findings in infantile spasms. Ophthalmology. 1979;86:1794–1802.
14 Pampliglione G, Pugh E. Infantile spasms and subsequent appearance of tuberous sclerosis syndrome. Lancet. 1975;2:1046.
15 Lagos JC, Gomez MR. Tuberous sclerosis: reappraisal of a clinical entity. Mayo Clin Proc. 1967;42:26–49.
16 Curatalo P, Bombardieri R, Cerminara C. Current Management for Epilepsy in tuberous sclerosis complex. Curr Opin Neurol. 2006;19:119–123.
17 Jansen FE, van Huffelen AC, Bourez-Swart M, et al. Consistent localization of interictal epileptiform activity on EEG’s of patients with tuberous sclerosis complex. Epilepsia. 2005;46:415–419.
18 Joinson C, O’Callaghan FJ, Osborne JP, et al. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33:335–344.
19 Prather P, DeVries PJ. Behavioral and neuropsychological aspects of tuberous sclerosis complex. J Child Neurol. 2004;19:666–674.
20 Font RL, Ferry AP. The phakomatoses. Int Ophthalmol Clin. 1972;12:1–50.
21 Shields JA, Shields CL. Diagnosis and management of intraocular tumors. St Louis: Mosby; 1983.
22 Raznahan A, Higgins NJ, deVries PJ, et al. Neuroanatomical correlates of memory deficits in tuberous sclerosis complex. Cereb Cortex. 2007;17:261–271.
23 Russel DS, Rubinstein LJ. Pathology of tumors of the central nervous system, 3rd ed. London: Edward Arnold; 1971.
24 Guttan M. Visual loss secondary to a giant aneurysm in a patient with tuberous sclerosis. Can J Neurol Sci. 1984;11:472–474.
25 Nickel WR, Reed WB. Tuberous sclerosis. Special reference to the microscopic alterations in the cutaneous hamartomas. Arch Dermatol. 1962;85:209–226.
26 Roth JC, Epstein CJ. Infantile spasms and hypopigmented macules. Arch Neurol. 1971;25:547–551.
27 Gold AP, Freeman JM. Depigmented nevi: the earliest sign of tuberous sclerosis. Pediatrics. 1965;35:1003–1005.
28 Monaghan HP, Krafchik BR, MacGregor DL, et al. Tuberous sclerosis complex in children. Am J Dis Child. 1981;135:912–917.
29 Callen JP. The skin, the eye, and systemic disease. Cutis. 1979;24:501–511.
30 Farrow GM, Harrison EG, Jr., Utz DC, et al. Renal angiomyolipoma: a clinicopathologic study of 32 cases. Cancer. 1968;22:564–570.
31 Charlot-Charles J, Jones GW. Renal angiomyolipoma associated with tuberous sclerosis: review of the literature. Urology. 1974;3:465–469.
32 Reed WB, Nickel WR, Campion G. Internal manifestations of tuberous sclerosis. Arch Dermatol. 1963;87:715–728.
33 Sanders MD, Gawler J. Computerized tomographic scanning (EMI scan) in neuro-ophthalmology. Trans. Ophthalmol Soc UK. 1975;95:237–245.
34 Shelton RW. The incidence of ocular lesions in tuberous sclerosis. Ann Ophthalmol. 1975:771–774.
35 Gass JDM. Stereoscopic atlas of macular diseases: diagnosis and treatment, 3rd ed. St Louis: Mosby; 1987.
36 Shields JA, Shields CL, Ehya H, et al. Atypical retinal astrocytic hamartoma diagnosed by fine-needle biopsy. Ophthalmology. 1996;103:949–952.
37 Eagle RC, Jr., Shields JA, Shields CL, et al. Hamartomas of the iris and ciliary epithelium in tuberous sclerosis complex. Arch Ophthalmol. 2000;118:711–715.
38 Barsky D, Wolter JR. The retinal lesions of tuberous sclerosis: an angiomatosis hamartoma. J Pediatr Ophthalmol. 1971;8:261–265.
39 Nyboer JH, Robertson DM, Gomez MR. Retinal lesions in tuberous sclerosis. Arch Ophthalmol. 1976;94:1277–1280.
40 Wolter JR, Reimer K, et al. Exophytic retinal astrocytoma in tuberous sclerosis. J Pediatr Ophthalmol. 1969;6:186–191.
41 Shields CL, Shields JA, Eagle RC, Jr., et al. Progressive enlargement of acquired retinal astrocytoma in 2 cases. Ophthalmology. 2004;111:363–368.
42 Kirati H, Bilgic S. Spontaneous regression of retinal astrocytic hamartoma in a patient with tuberous sclerosis. Am J Ophthalmol. 2002;133:715–716.
43 Spencer WH, ed. Ophthalmic pathology: an atlas and textbook, vol. 2. Philadelphia: WB Saunders, 1985.
44 Gass JDM. Fluorescein angiography: an aid in the differential diagnosis of intraocular tumors. Int Clin Ophthalmol. 1972;12:85.
45 Kroll AJ, Richer DP, Robb RM, et al. Vitreous hemorrhage complicating retinal astrocytic hamartoma. Surv Ophthalmol. 1981;26:31–38.
46 Van der Hoeve J. Augengeschwulste bei der tuberosen Hirnklerose (Bourneville). Albrecht von Graefes Arch Ophthalmol. 1921;105:880–898.
47 Atkinson A, Sanders MD, Wong V. Vitreous hemorrhage in tuberous sclerosis: report of two cases. Br J Ophthalmol. 1973;57:773–779.
48 Koch FLP, Walsh M. Syndrome of tuberous sclerosis: report of a case. Arch Ophthalmol. 1939;21:465–475.
49 Jost BF, Olk RJ. Atypical retinitis proliferans, retinal telangiectasis, and vitreous hemorrhage in a patient with tuberous sclerosis. Retina. 1986;6:534–536.
50 Coppeto JR, Lubin JR, Albert DM. Astrocytic hamartoma in tuberous sclerosis mimicking necrotizing retinochoroiditis. J Pediatr Ophthalmol Strabismus. 1982;19:306–313.
51 Miller NR. Walsh and Hoyt’s clinical neuro-ophthalmology, 4th ed. Baltimore: Williams and Wilkins; 1982.
52 De Juan E, Jr., Green WR, Gupta PK, et al. Vitreous seeding by retinal astrocytic hamartoma in a patient with tuberous sclerosis. Retina. 1984;4:100–102.
53 Zolli C, Rodrigues MM, Shannon GM. Unusual eyelid involvement in tuberous sclerosis. J Pediatr Ophthalmol. 1976;13:156–158.
54 Awan KJ. Leaf-shaped lesions of ocular fundus and white eyelashes in tuberous sclerosis. South Med J. 1982;75:227–278.
55 Gutman I. Hypopigmented iris spots: an early sign of tuberous sclerosis. Ophthalmology. 1982;89:1155–1159.
56 Gomez MR. Tuberous sclerosis. New York: Raven Press; 1979.
57 Cleasby GW, Fung WE, Shelker WB. Astrocytoma of the retina. Am J Ophthalmol. 1967;64:465–469.
58 Howard GM, Ellsworth RM. Differential diagnosis of retinoblastoma. I. Relative frequency of lesions which simulate retinoblastoma. Am J Ophthalmol. 1965;60:610–618.
59 Panzo GJ, Meyers SM, Gutman FA, et al. Spontaneous regression of parafoveal exudates and serous retinal detachment in a patient with tuberous sclerosis and retinal astrocytoma. Retina. 1984;4:242–245.
60 Hogan MJ, Zimmerman LE. Ophthalmic pathology. Philadelphia: WB Saunders; 1962.
61 Heckenlively JR. Retinitis pigmentosa. Philadelphia: Lippincott; 1988.
62 Spencer WH. Drusen of the optic disc and aberrant axoplasmic transport. Ophthalmology. 1978;85:21–38.
63 Puck A, Tso MOM, Fishman GA. Drusen of the optic nerve associated with retinitis pigmento. Arch Ophthalmol. 1985;103:231–234.
64 Novak RL, Foos RY. Drusen of the optic disk in retinitis pigmentosa. Am J Ophthalmol. 1987;103:44–47.
65 Robertson DM. Hamartomas of the optic disc with retinitis pigmentosa. Am J Ophthalmol. 1972;74:526–531.
66 O’Callaghan F. Tuberous sclerosis complex. Pediatr Child Health. 2007;18:30–36.
67 Hunt A, Lindenbaum RH. Tuberous sclerosis: a new estimate of prevalence within the Oxford region. J Med Genet. 1984;21:272–277.
68 MacDonald IM, Bech-Hansen T, Britten WA, Jr., et al. The phakomatoses: recent advances in genetics. Can J Ophthalmol. 1997;32:6–11.
69 Povey S, Burley MW, Attwood J, et al. Two loci for tuberous sclerosis: one on 9q34 and one on 16p13. Ann Hum Genet. 1994;58:107–127.
70 Sampson JR, Janssen JAJ, Sandkujji LA, et al. Linkage investigation of three putative tuberous sclerosis determining loci on chromosome 9q, 11q, and 12q. J Med Genet. 1992;29:861–866.
71 European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315.
72 Dabora SL, et al. Am J Human Genet. 2001;68:1305–1315.
73 Crino PB, Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology. 1999;53:1384–1390.