[level-membership-for-basic-science-category]
Chapter 28 Treatment of Parkinson’s and Alzheimer’s Diseases
| Abbreviations | |
|---|---|
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| BBB | Blood-brain barrier |
| ChE | Cholinesterase |
| COMT | Catechol-O-methyl transferase |
| DA | Dopamine |
| l-DOPA | 3,4-Dihydroxy-phenylalanine |
| MAO | Monoamine oxidase |
| NMDA | N-methyl-d-aspartate |
Therapeutic Overview
While several neurotransmitter systems deteriorate in Alzheimer’s disease, one of the first and most pronounced reductions occurs within the acetylcholine (ACh) containing projections from the nucleus basalis in the basal forebrain to the cerebral cortex. The septo-hippocampal cholinergic pathway is similarly affected, whereas the
| Therapeutic Overview |
|---|
| Parkinson’s Disease |
| Pathology |
| Degeneration of nigrostriatal DA neurons |
| Presence of Lewy bodies in surviving neurons |
| Treatment |
| l-DOPA |
| MAO inhibitors |
| COMT inhibitors |
| DA agonists |
| Muscarinic receptor antagonists |
| NMDA receptor antagonists |
| Alzheimer’s Disease |
| Pathology |
| Degeneration of basal forebrain cholinergic neurons |
| Presence of amyloid plaques and neurofibrillary tangles |
| Neuron and synapse loss in cerebral cortex and hippocampus |
| Treatment |
| AChE inhibitors |
| NMDA receptor antagonists |
intrinsic striatal cholinergic system remains largely intact. Muscarinic cholinergic receptors in the cerebral cortex and hippocampus remain more or less intact, but nicotinic cholinergic receptors decline.
Patients with both Parkinson’s and Alzheimer’s diseases may manifest neuropsychiatric disturbances as a result of their primary disease, including psychoses, depression, anxiety, and agitation. These can be treated with the atypical antipsychotics, antidepressants, and anxiolytic compounds discussed in Chapters 29 to Chapters 31. A summary of the treatment of Parkinson’s and Alzheimer’s diseases is provided in the Therapeutic Overview Box.
Mechanisms of Action
Current strategies for the treatment of Parkinson’s disease are directed at increasing dopaminergic activity in the striatum to compensate for the loss of nigrostriatal DA neurons (see Fig. 27-8). The major drugs used include compounds that increase the synthesis and decrease the catabolism of DA or directly stimulate DA receptors; secondary compounds block muscarinic cholinergic receptors, enhance DA release, and perhaps antagonize NMDA receptors.
l-DOPA was introduced for the treatment of Parkinson’s disease in 1970. It is the precursor of DA and crosses the BBB, whereas DA does not. l-DOPA increases DA synthesis but does not stop progression of the disease. When given alone, only 1% to 3% of an administered dose of l-DOPA reaches the brain; the rest is metabolized peripherally as shown in Figure 28-1. To prevent its peripheral metabolism and increase its availability to the brain, l-DOPA is administered with carbidopa, an aromatic l-amino acid decarboxylase inhibitor that does not cross the BBB. Carbidopa does not have any therapeutic benefit when used alone but increases the amount of l-DOPA available to the brain. However, as a consequence of peripheral inhibition of aromatic l-amino acid decarboxylase, more precursor is metabolized by plasma catechol-O-methyltransferase (COMT) producing 3-o-methyldopa. To overcome this, the COMT inhibitors tolcapone or entacapone are used in combination with l-DOPA/carbidopa. These compounds prolong the plasma t1/2 of l-DOPA, increasing the time the drug is available to cross the BBB. They also prevent the buildup of 3-o-methyldopa, which competitively inhibits l-DOPA transport across the BBB. Entacapone does not cross the BBB, and its actions are limited to the periphery. However, tolcapone does cross the BBB and also prevents the formation of 3-o-methyldopa in brain and the catabolism of DA (Fig. 28-1).
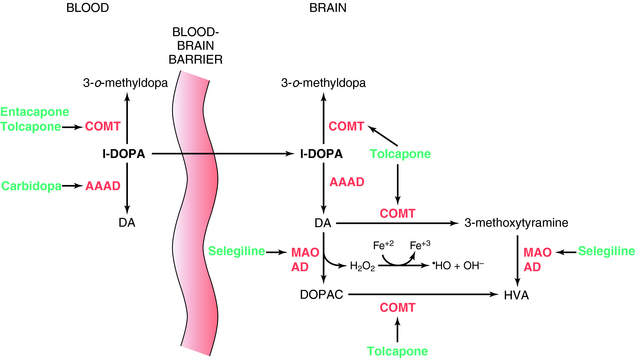
Another approach to increase brain DA levels involves the use of the monoamine oxidase type B (MAO-B) irreversible inhibitors selegiline or rasagiline to inhibit the catabolism of DA in the brain (see Fig. 28-1). In addition, by inhibiting the catabolism of DA to DOPAC, these compounds decrease production of the byproduct hydrogen peroxide, limiting the possible formation of free radicals that form when the peroxide reacts with ferrous iron.
Current strategies for the treatment of Alzheimer’s disease are directed primarily at increasing cholinergic activity to compensate for the loss of basal forebrain cholinergic neurons (see Fig. 27-9). Most available compounds are AChE inhibitors, including donepezil, galantamine, and rivastigmine. All of these drugs cross the BBB and are reversible AChE inhibitors. Donepezil and galantamine are specific for AChE, whereas rivastigmine inhibits both AChE and butyrylcholinesterase (ChE). As a consequence of AChE inhibition, ACh released from remaining cholinergic terminals is not rapidly hydrolyzed, leading to prolonged cholinergic receptor activation. In addition, galantamine stimulates presynaptic nicotinic cholinergic receptors in the brain through an allosteric mechanism, enhancing ACh release.
Memantine is the first low-affinity NMDA receptor channel blocker approved to treat Alzheimer’s disease. Memantine is a derivative of amantadine (see above) and binds to the open state of the glutamate NMDA receptor to block ion flux through this channel (see Chapter 1). NMDA receptors are critical to learning and memory and neural plasticity in the brain and serve an integrating function by remaining closed until a sufficient dendritic depolarization occurs to overcome blockade of the channel by Mg++. Once open, Na+ and Ca++ enter the cell, with Ca++ activating multiple signaling cascades. Excessive opening of the channel can be associated with excitotoxicity in neurons. Although this may contribute to neurodegeneration in Alzheimer’s disease, there is no evidence yet that memantine protects neurons or modifies the course of the disease. It is hypothesized that the low-affinity antagonism of NMDA receptors prevents tonic activation while still permitting opening of the channel during periods of elevated activity critical for memory formation.
Pharmacokinetics
l-DOPA is always administered with carbidopa to increase plasma levels and the t1/2 of l-DOPA and to ensure sufficient l-DOPA is available to cross the BBB. l-DOPA/carbidopa is rapidly absorbed by the gastrointestinal tract but competes with dietary protein for both intestinal absorption and transport across the BBB. l-DOPA/carbidopa is available in an immediate-release form, which has a t1/2 of only 60 to 90 minutes. Controlled-release formulations are available to minimize the number of daily doses required and prolong therapeutic plasma concentrations. A rapidly dissolving formulation of l-DOPA/carbidopa that dissolves on the tongue is available for Parkinson’s patients who have difficulty swallowing. This preparation dissolves immediately and releases the active drugs within 30 minutes, with other pharmacokinetic parameters similar to the oral preparations.
Selected pharmacokinetic parameters for drugs used for Parkinson’s disease are listed in Table 28-1.
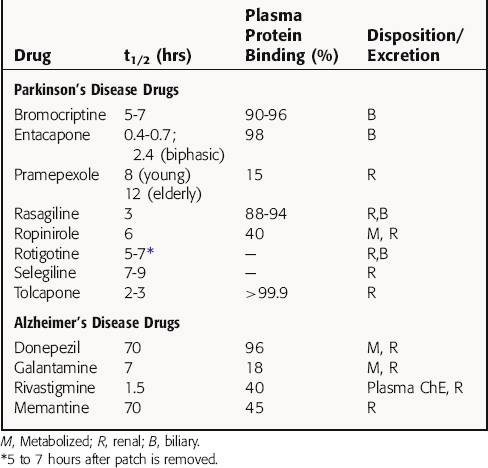
All drugs used to treat Alzheimer’s disease have good oral bioavailability but differ widely in their pharmacokinetic profiles (Table 28-1). Donepezil, which has a very long t1/2, is highly bound to plasma proteins and is metabolized in the liver by CYP2D6 and CYP3A4. Its primary metabolite is equally effective as the parent compound in blocking AChE activity. Galantamine, which is available in immediate-release and extended-release formulations, is also metabolized by CYP2D6 and CYP3A4, but its metabolites are inactive. More than 50% of a dose of donepezil is excreted unchanged, and its hepatic metabolism does not appear to lead to drug interactions or limitations in special populations. In contrast, galantamine is metabolized by CYP2D6 and CYP3A4, and inhibitors of both of these metabolic pathways increase its bioavailability. In addition, poor metabolizers (7% of the population with a genetic variation with decreased CYP2D6 activity) exhibit a significant reduction in clearance of galantamine.
Relationship of Mechanisms of Action to Clinical Response
The DA receptor agonists are effective as monotherapy early in the disease or as an adjunct to l-DOPA in later stages. These compounds are not as efficacious as l-DOPA and have a lower propensity to cause dyskinesias or motor fluctuations. As monotherapy, these compounds are effective for 3 to 5 years, at which time l-DOPA/carbidopa must be initiated. As adjunctive therapy in advanced disease, DA receptor agonists contribute to clinical improvement and allow a reduction in the dose of l-DOPA required. Although the ergot derivatives bromocriptine and pergolide were used for many years, the use of bromocriptine has declined in favor of the non-ergot compounds, and pergolide was withdrawn from the united States market because of its association with cardiac-valve regurgitation.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Central nervous system effects include depression, anxiety, agitation, insomnia, hallucinations, and confusion, particularly in the elderly, and may be attributed to enhanced mesolimbic and mesocortical dopaminergic activity (see Fig. 27-8). The tricyclic antidepressants or selective serotonin reuptake inhibitors (see Chapter 30) may be used for depression, but the latter may cause worsening of motor symptoms. The atypical antipsychotics clozapine and quetiapine are beneficial for psychotic reactions and do not exacerbate the motor symptoms like the typical antipsychotics (see Chapter 29).
Selegiline and rasagiline can cause nausea and orthostatic hypotension. At doses recommended for Parkinson’s disease, which inhibit MAO-B but not MAO-A, selegiline is unlikely to induce a tyramine interaction (see Chapters 11 and Chapters 30); similar data for rasagiline are unavailable. Selegiline may cause rare toxic interactions with fluoxetine and meperidine and can increase the adverse effects of l-DOPA, particularly dyskinesias and psychoses in the elderly. Rasagiline has been reported to increase the incidence of melanoma, and because rasagiline is metabolized by CYP1A2, plasma concentrations may increase in the presence of CYP1A2 inhibitors such as ciprofloxacin and fluvoxamine. MAO inhibitors must be used with caution in patients taking any drug enhancing serotonergic activity, including the antidepressants, dextromethorphan, and tryptophan (see Chapter 30). Combinations of these compounds could induce “serotonin syndrome,” a serious condition characterized by confusion, agitation, rigidity, shivering, autonomic instability, myoclonus, coma, nausea, diarrhea, diaphoresis, flushing, and even death.
DA receptor agonists cause side effects similar to those with l-DOPA, including nausea and postural hypotension. These compounds cause more central nervous system-related effects than l-DOPA, including hallucinations, confusion, cognitive dysfunction, and sleepiness.
Several studies have reported that patients maintained on DA receptor agonists develop increased impulsivity and exhibit pathological gambling, perhaps reflecting stimulation of the midbrain dopaminergic ventral tegmental-nucleus accumbens pathway thought to mediate addictive behaviors (see Chapter 27).
The muscarinic receptor antagonists all cause typical anticholinergic effects as discussed extensively in Chapter 10. Because Parkinson’s disease is predominantly an age-related disorder, and older individuals show increased vulnerability to other dysfunctions including dementia and glaucoma, anticholinergics must be used with caution in the elderly, because these drugs impair memory, exacerbate glaucoma, and may cause urinary retention.
All AChE inhibitors currently available are relatively free of serious side effects. As expected, these compounds have a high incidence of peripheral cholinergic effects such as nausea, vomiting, anorexia, and diarrhea. Rivastigmine is associated with a greater incidence of these effects than the other drugs; it is uncertain if inhibition of ChE activity is responsible. Fortunately, many of these effects demonstrate tolerance, and gradual dosage escalation permits many patients to tolerate their full therapeutic doses. Adverse events with memantine were low in clinical trials, with none exceeding twice the placebo values in 5% or more of patients. High doses can produce dissociative anesthetic type effects similar to ketamine, including confusion, hallucination, hypnosis, and stupor (see Chapter 35).
Common side effects associated with drugs used for Parkinson’s and Alzheimer’s diseases are listed in the Clinical Problems Box.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 28 Treatment of Parkinson’s and Alzheimer’s Diseases
| Abbreviations | |
|---|---|
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| BBB | Blood-brain barrier |
| ChE | Cholinesterase |
| COMT | Catechol-O-methyl transferase |
| DA | Dopamine |
| l-DOPA | 3,4-Dihydroxy-phenylalanine |
| MAO | Monoamine oxidase |
| NMDA | N-methyl-d-aspartate |
Therapeutic Overview
While several neurotransmitter systems deteriorate in Alzheimer’s disease, one of the first and most pronounced reductions occurs within the acetylcholine (ACh) containing projections from the nucleus basalis in the basal forebrain to the cerebral cortex. The septo-hippocampal cholinergic pathway is similarly affected, whereas the
| Therapeutic Overview |
|---|
| Parkinson’s Disease |
| Pathology |
| Degeneration of nigrostriatal DA neurons |
| Presence of Lewy bodies in surviving neurons |
| Treatment |
| l-DOPA |
| MAO inhibitors |
| COMT inhibitors |
| DA agonists |
| Muscarinic receptor antagonists |
| NMDA receptor antagonists |
| Alzheimer’s Disease |
| Pathology |
| Degeneration of basal forebrain cholinergic neurons |
| Presence of amyloid plaques and neurofibrillary tangles |
| Neuron and synapse loss in cerebral cortex and hippocampus |
| Treatment |
| AChE inhibitors |
| NMDA receptor antagonists |
intrinsic striatal cholinergic system remains largely intact. Muscarinic cholinergic receptors in the cerebral cortex and hippocampus remain more or less intact, but nicotinic cholinergic receptors decline.
Patients with both Parkinson’s and Alzheimer’s diseases may manifest neuropsychiatric disturbances as a result of their primary disease, including psychoses, depression, anxiety, and agitation. These can be treated with the atypical antipsychotics, antidepressants, and anxiolytic compounds discussed in Chapters 29 to Chapters 31. A summary of the treatment of Parkinson’s and Alzheimer’s diseases is provided in the Therapeutic Overview Box.
Mechanisms of Action
Current strategies for the treatment of Parkinson’s disease are directed at increasing dopaminergic activity in the striatum to compensate for the loss of nigrostriatal DA neurons (see Fig. 27-8). The major drugs used include compounds that increase the synthesis and decrease the catabolism of DA or directly stimulate DA receptors; secondary compounds block muscarinic cholinergic receptors, enhance DA release, and perhaps antagonize NMDA receptors.
l-DOPA was introduced for the treatment of Parkinson’s disease in 1970. It is the precursor of DA and crosses the BBB, whereas DA does not. l-DOPA increases DA synthesis but does not stop progression of the disease. When given alone, only 1% to 3% of an administered dose of l-DOPA reaches the brain; the rest is metabolized peripherally as shown in Figure 28-1. To prevent its peripheral metabolism and increase its availability to the brain, l-DOPA is administered with carbidopa, an aromatic l-amino acid decarboxylase inhibitor that does not cross the BBB. Carbidopa does not have any therapeutic benefit when used alone but increases the amount of l-DOPA available to the brain. However, as a consequence of peripheral inhibition of aromatic l-amino acid decarboxylase, more precursor is metabolized by plasma catechol-O-methyltransferase (COMT) producing 3-o-methyldopa. To overcome this, the COMT inhibitors tolcapone or entacapone are used in combination with l-DOPA/carbidopa. These compounds prolong the plasma t1/2 of l-DOPA, increasing the time the drug is available to cross the BBB. They also prevent the buildup of 3-o-methyldopa, which competitively inhibits l-DOPA transport across the BBB. Entacapone does not cross the BBB, and its actions are limited to the periphery. However, tolcapone does cross the BBB and also prevents the formation of 3-o-methyldopa in brain and the catabolism of DA (Fig. 28-1).
Another approach to increase brain DA levels involves the use of the monoamine oxidase type B (MAO-B) irreversible inhibitors selegiline or rasagiline to inhibit the catabolism of DA in the brain (see Fig. 28-1). In addition, by inhibiting the catabolism of DA to DOPAC, these compounds decrease production of the byproduct hydrogen peroxide, limiting the possible formation of free radicals that form when the peroxide reacts with ferrous iron.
