[level-membership-for-basic-science-category]
Chapter 30 Treatment of Affective Disorders
| Abbreviations | |
|---|---|
| DA | Dopamine |
| 5-HT | Serotonin |
| MAO | Monoamine oxidase |
| MAOI | Monoamine oxidase inhibitor |
| NE | Norepinephrine |
| SNRI | Serotonin/norepinephrine reuptake inhibitor |
| SSRI | Serotonin selective reuptake inhibitor |
| TCA | Tricyclic antidepressant |
Therapeutic Overview
changes characterized by severe highs and gut-wrenching lows, which may worsen to a psychotic state. In addition, depression is often associated with comorbid anxiety disorders.
Although the molecular and cellular etiology of depression remains unknown, it is generally accepted that depression involves impaired monoaminergic neurotransmission, leading to alterations in the expression of specific genes. This is supported by studies demonstrating that antidepressants increase the expression of the transcription factor cyclic adenosine monophosphate response element-binding protein (CREB) and brain-derived neurotrophic factor (BNDF), both of which are critical for maintaining normal cell structure in limbic regions of the brain that are targets for monoaminergic projections. In addition, postmortem and imaging studies have demonstrated neuronal loss and shrinkage in the prefrontal cortex and hippocampus in depressed patients, some of which could be reversed by antidepressants.
The treatment of bipolar disorder has changed over the past decade. Lithium has been the mainstay of treatment for many years, particularly for control of the manic phase. However, the anticonvulsants lamotrigine, valproic acid, and carbamazepine have been frequently used as well (see Chapter 34), especially in cases in which the bipolar disorder was characterized by rapid cycling. Recently, the atypical antipsychotic drugs aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone were approved as monotherapy for bipolar disorder (see Chapter 29). Antidepressants may also be warranted to treat the depressive phase of the illness.
The pharmacology of the antipsychotics is discussed in Chapter 29 and that of the anticonvulsants in Chapter 34. Therapeutic actions related to the antidepressants and lithium are summarized in the Therapeutic Overview Box.
Mechanisms of Action
| Therapeutic Overview |
|---|
| Antidepressants |
| Prolong the action of biogenic amines at the synapse by inhibiting amine reuptake, increasing amine release or decreasing amine catabolism |
| Enhance neurogenesis and dendritic branching in the adult hippocampus |
| Lithium |
| Interferes with receptor-activated phosphatidylinositol turnover; blocks the conversion of inositol phosphate to free inositol |
| Antagonizes 5-HT1A and 5-HT1B autoreceptors, alleviating feedback inhibition of 5-HT release |
| Enhances glutamate reuptake system, clearing glutamate from the synapse |
was the first agent demonstrated to have antidepressant efficacy. The TCAs have a three-ring structure with a side chain containing a tertiary or secondary amine attached to the central ring, resembling the phenothiazine antipsychotics (Fig. 30-1). The tertiary amines include imipramine, amitriptyline, clomipramine, and doxepin; the secondary amines include desipramine and nortriptyline.
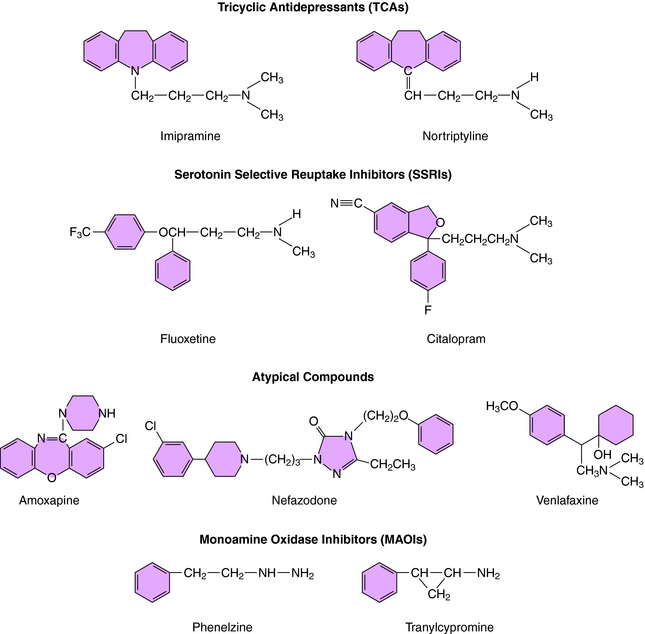
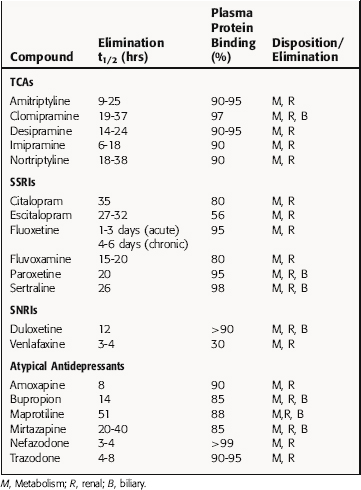
The TCAs block the reuptake of NE, 5-HT, or both into noradrenergic and/or serotonergic nerve terminals, respectively, by specific interactions with plasma membrane transporters (Fig. 30-2). As a consequence of this inhibition, the actions of NE and 5-HT released from these neurons are not rapidly terminated, resulting in a prolonged stimulation of NE receptors, 5-HT receptors, or both. The TCAs do not affect the reuptake of DA by dopaminergic nerve terminals, and their selectivity for NE versus 5-HT transporters differs among the different compounds (Table 30-1).
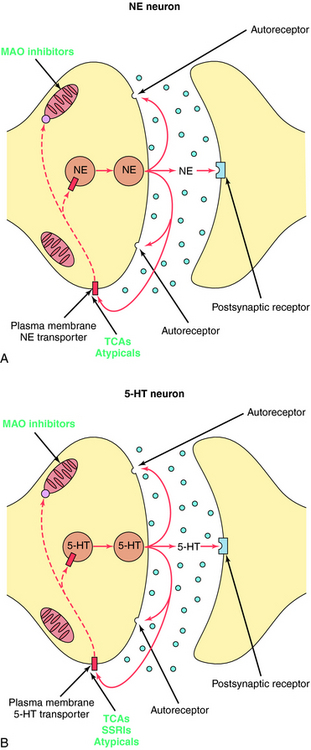
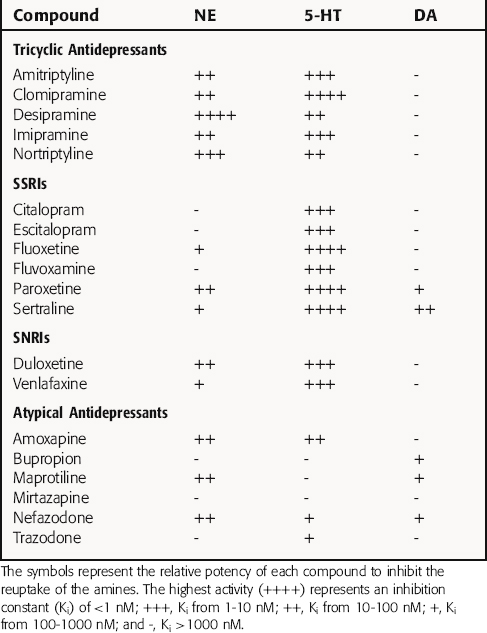
The SSRIs and SNRIs also inhibit the reuptake of biogenic amines, and as their name implies, the SSRIs have the highest affinity for 5-HT transporters, whereas the SNRIs have high affinity for 5-HT transporters and moderate affinity for NE transporters. It is important to note, however, that specificity and selectivity are always dose-related such that the SSRIs sertraline and paroxetine inhibit both NE and DA reuptake at the upper end of their dose ranges (see Table 30-1). Similarly, it is also important to keep in mind that the classification of newly developed compounds is based on their affinity for specific transporters, whereas that of the TCAs is based on chemical structure. Thus, although clomipramine is classified chemically as a TCA, its ability and selectivity to inhibit 5-HT and NE reuptake matches that of the SSRI paroxetine. Likewise, the selectivity of the TCAs imipramine and amitriptyline resemble that of the SNRI duloxetine. Thus, at times, classification schemes may be misleading.
As mentioned, the atypical compounds are a very heterogeneous group of drugs. Among these, maprotiline and nefazodone are relatively selective inhibitors of NE reuptake (see Table 30-1). Trazodone is a weak inhibitor of 5-HT reuptake, bupropion weakly inhibits DA reuptake, and mirtazapine appears devoid of activity at any reuptake transporter.
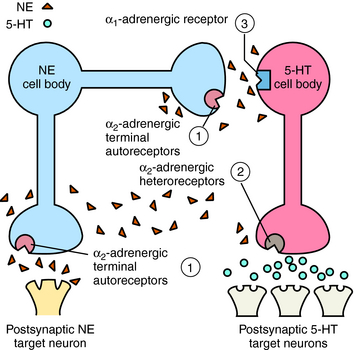
Mirtazapine also blocks α2 adrenergic receptors on noradrenergic and serotonergic nerve terminals and on noradrenergic dendrites (Fig. 30-3). Stimulation of α2 autoreceptors on noradrenergic neurons decreases NE release, whereas stimulation of α2 heteroreceptors on serotonergic neurons inhibits 5-HT release. In addition, stimulation of α1 adrenergic receptors on serotonergic cell bodies and dendrites increases their firing rate. Thus mirtazapine, by inhibiting α2 autoreceptors, enhances noradrenergic cell firing and the release of NE, which activates α1 adrenergic receptors to increase 5-HT release while concurrently blocking α2 heteroreceptors, further facilitating the release of 5-HT.
The MAOIs used for the treatment of depression are phenelzine and tranylcypromine (see Fig. 30-1) and the recently approved selegiline transdermal patch. Phenelzine and selegiline are irreversible MAO inhibitors, and tranylcypromine is a long-lasting MAO inhibitor. At the doses used for depression, all these compounds are nonselective and inhibit both MAO-A and MAO-B. These enzymes are distinct gene products with MAO-A present in human placenta, intestinal mucosa, liver, and brain—responsible for the catabolism of 5-HT, NE, and tyramine; and MAO-B present in human platelets, liver, and brain—responsible predominantly for the catabolism of DA and tyramine. These enzymes are located in the outer membrane of mitochondria and function to maintain low cytoplasmic concentrations of the monoamines, facilitating inward-directed transporter activity (i.e., monoamine reuptake). MAO inhibition causes an increase in monoamine concentrations in the cytosol of the nerve terminal. All the effects of the MAOIs have been attributed to enhanced aminergic activity resulting from enzyme inhibition.
Research with selective MAOIs has shown that inhibition of MAO-A is necessary for antidepressant activity. Thus, although selegiline is a selective inhibitor of MAO-B at low doses and is used at these doses for the treatment of Parkinson’s disease (see Chapter 28), at higher doses selectivity is lost, and selegiline inhibits both MAO-A and MAO-B and has antidepressant activity.
Although lithium has been the standard prophylactic agent for the treatment of bipolar disorder for decades, its cellular mechanisms of action remain unclear. Currently, three actions of lithium have been postulated to mediate its clinical efficacy. The first is interference with receptor-activated phosphoinositide turnover (see Chapter 1). Lithium blocks the hydrolysis of inositol phosphate to free inositol, thereby reducing free inositol concentrations and depleting further formation of phosphatidylinositol in the cell membrane. Hence, the effects of agonists working through this signaling system will be blunted. Lithium has also been shown to inhibit 5-HT1A and 5-HT1B autoreceptors on serotonergic dendrites and nerve terminals, thereby preventing feedback inhibition of 5-HT release. Last, sustained lithium exposure enhances glutamate reuptake by glutamatergic neurons, thereby decreasing the time glutamate is present at glutamatergic synapses and dampening the ability of glutamate to stimulate its receptors. Clearly, additional studies are needed to elucidate the actions of lithium that underlie its unique efficacy in bipolar disorder.
Pharmacokinetics
Pharmacokinetic parameters of representative antidepressants are presented in Table 30-2. In general, antidepressants are readily absorbed, primarily in the small intestine, and undergo significant first-pass hepatic metabolism. Peak plasma concentrations are achieved within hours after ingestion, with steady-state concentrations achieved after 4 to 7 days at a fixed dose.
Because phenelzine and selegiline inhibit MAO irreversibly and tranylcypromine inhibits it persistently but noncovalently, the biological effects of these compounds outlast their physical presence in the body; that is, a loss of enzyme activity persists after the drugs are metabolized and eliminated. New enzyme must be synthesized for MAO activity to return to normal, a process that takes several weeks.
Relationship of Mechanisms of Action to Clinical Response
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Amine Reuptake Inhibitors and Atypical Compounds
Antihistaminergic, Anticholinergic, and Antiadrenergic Effects
Many of the most common side effects of the TCA and atypical drugs result from their antagonist actions at H1 histaminergic, muscarinic cholinergic, and α1 adrenergic receptors, leading to marked sedative effects, atropine-like effects including dry mouth, constipation, and cycloplegia, and prazosin-like effects such as orthostatic hypotension, respectively (Table 30-3). As a group, these drugs are more potent at blocking H1 than muscarinic and α1 adrenergic receptors.
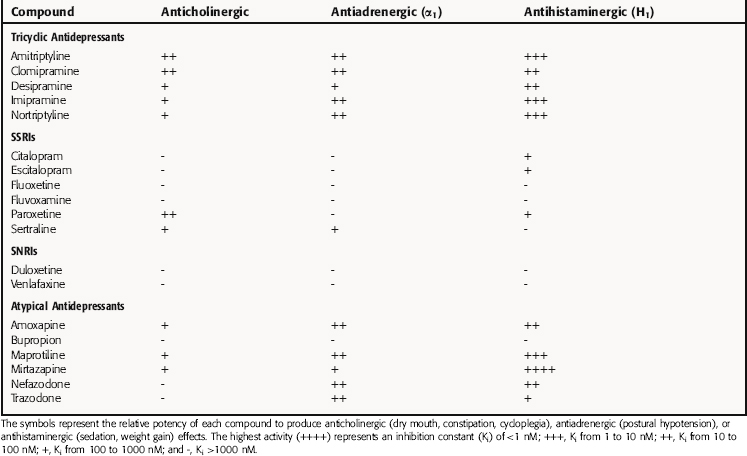
The TCAs have fairly marked anticholinergic activity at clinical doses. Although the atypical compounds are somewhat less potent than TCAs in blocking muscarinic cholinergic receptors, amoxapine, maprotiline, and mirtazapine do have anticholinergic effects. The SSRI paroxetine also inhibits muscarinic receptors and produces anticholinergic activity, albeit less than that of the TCAs, reflecting a dose-related difference. Other SSRIs, the SNRIs, and other atypical antidepressants are very weak antagonists at muscarinic cholinergic receptors and do not cause anticholinergic side effects.
Central Nervous System Effects
TCA-induced toxicity of the central nervous system can produce delirium, especially in the elderly, which is easily recognizable. Such delirium is usually preceded by what appears to be a worsening of depression. This may lead to administration of increased doses of the TCA, with further worsening of the delirium. In most cases the antimuscarinic activity of the TCAs is the driving force behind the delirium. If the patient is hospitalized, physostigmine can be used to reverse the delirium. The incidence of insomnia, nervousness, restlessness, and anxiety appears to be relatively high in patients taking fluoxetine. It has an activating effect that can be anxiogenic in some patients and should be started at a lower dose in those with an anxiety component to their illness. Venlafaxine has side effects similar to those of SSRIs. Bupropion can cause nervousness and insomnia, as well as tremors and palpitations, and has more of a stimulant than a sedative effect; therefore it should not be administered in the evening. Headaches commonly occur in patients on SSRIs and venlafaxine.
The SSRIs and SNRIs have the potential to lead to serious consequences if combined with other compounds that increase brain levels of 5-HT or stimulate 5-HT receptors. Among these are the MAOIs and other antidepressants, as well as meperidine and dextromethorphan, which are potent inhibitors of 5-HT reuptake, and tryptophan, which can enhance 5-HT synthesis. This interaction can lead to a condition known as serotonin syndrome. This syndrome is characterized by alterations in autonomic function (fever, chills, and diarrhea), cognition and behavior (agitation, excitement, hypomania), and motor systems (myoclonus, tremor, motor weakness, ataxia, hyper-reflexia), and may often resemble neuroleptic malignant syndrome (see Chapter 29). Currently it is believed that activation of 5-HT receptors in the brainstem and spinal cord may mediate these effects. The incidence of the disorder is not known, but as the use of SSRIs increases, it may become more prevalent. Thus a heightened awareness is required for prevention, recognition, and prompt treatment. This involves discontinuation of the suspected drugs, administration of 5-HT antagonists such as cyproheptadine or methysergide, administration of the skeletal muscle relaxant dantrolene, and other supportive measures. The syndrome usually resolves within 24 hours but can be fatal.
Nefazodone is associated with hepatic failure and received a black box warning from the United States Food and Drug Administration. Amoxapine produces many of the same side effects as antipsychotic agents, including dystonic reactions, tardive dyskinesia, and neuroleptic malignant syndrome (see Chapter 29).
Poisoning accounts for approximately 20% of all suicides, and TCAs are the most commonly used drugs in such cases. TCA overdose can produce coma, seizures, hypertension, and cardiac abnormalities, with death resulting primarily from cardiac arrest. A lethal dose of a TCA may be as low as 1 g, which is roughly 4 or 5 days of medication. In general, the SSRIs, SNRIs, and the atypical compounds (except amoxapine and maprotiline) are much safer in overdoses than the TCAs. However, as noted earlier, there have been reports of cardiac conduction problems associated with citalopram overdose.
The interaction between the MAOIs and the other antidepressants may produce serotonin syndrome, as discussed. In addition, because MAOIs lead to increased intracellular stores of NE within adrenergic nerve terminals, these compounds can enhance the action of indirect-acting sympathomimetics that stimulate the release of NE from these sites. Of major importance is the potential for the MAOIs to induce a hypertensive reaction after the ingestion of tyramine-containing compounds, an action known as the tyramine or cheese effect. Tyramine, which is an indirect-acting sympathomimetic, is normally metabolized by MAO within the gastrointestinal tract after ingestion. When MAO activity in the gastrointestinal tract is inhibited, such as occurs after the oral administration of MAOIs, tyramine is not metabolized and enters the circulation, where it can release stored NE from sympathetic nerve endings. Because the amount of NE in the adrenergic nerve ending is increased as a consequence of MAO inhibition, the result is a massive increase in NE released into the synapse, with a resultant hypertensive crisis (see Chapter 11). Therefore patients taking MAOIs are maintained on a tyramine-restricted diet. This may not be a problem with the use of the selegiline transdermal system, because sufficient MAO activity in the gastrointestinal tract remains intact after transdermal drug administration. Although hypertensive crises are associated with tyramine ingestion during MAOI treatment, in all actuality, hypotension is a much more common side effect of MAOI treatment.
Subclinical hypothyroidism can develop in patients taking lithium. Although obvious hypothyroidism is rare, a benign, diffuse, nontender thyroid enlargement (goiter), indicative of compromised thyroid function, occurs in some patients. This results from the ability of lithium to interfere with the iodination of tyrosine and, consequently, the synthesis of thyroxine (see Chapter 42).
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 30 Treatment of Affective Disorders
| Abbreviations | |
|---|---|
| DA | Dopamine |
| 5-HT | Serotonin |
| MAO | Monoamine oxidase |
| MAOI | Monoamine oxidase inhibitor |
| NE | Norepinephrine |
| SNRI | Serotonin/norepinephrine reuptake inhibitor |
| SSRI | Serotonin selective reuptake inhibitor |
| TCA | Tricyclic antidepressant |
Therapeutic Overview
changes characterized by severe highs and gut-wrenching lows, which may worsen to a psychotic state. In addition, depression is often associated with comorbid anxiety disorders.
Although the molecular and cellular etiology of depression remains unknown, it is generally accepted that depression involves impaired monoaminergic neurotransmission, leading to alterations in the expression of specific genes. This is supported by studies demonstrating that antidepressants increase the expression of the transcription factor cyclic adenosine monophosphate response element-binding protein (CREB) and brain-derived neurotrophic factor (BNDF), both of which are critical for maintaining normal cell structure in limbic regions of the brain that are targets for monoaminergic projections. In addition, postmortem and imaging studies have demonstrated neuronal loss and shrinkage in the prefrontal cortex and hippocampus in depressed patients, some of which could be reversed by antidepressants.
The treatment of bipolar disorder has changed over the past decade. Lithium has been the mainstay of treatment for many years, particularly for control of the manic phase. However, the anticonvulsants lamotrigine, valproic acid, and carbamazepine have been frequently used as well (see Chapter 34), especially in cases in which the bipolar disorder was characterized by rapid cycling. Recently, the atypical antipsychotic drugs aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone were approved as monotherapy for bipolar disorder (see Chapter 29). Antidepressants may also be warranted to treat the depressive phase of the illness.
The pharmacology of the antipsychotics is discussed in Chapter 29 and that of the anticonvulsants in Chapter 34. Therapeutic actions related to the antidepressants and lithium are summarized in the Therapeutic Overview Box.
Mechanisms of Action
| Therapeutic Overview |
|---|
| Antidepressants |
| Prolong the action of biogenic amines at the synapse by inhibiting amine reuptake, increasing amine release or decreasing amine catabolism |
| Enhance neurogenesis and dendritic branching in the adult hippocampus |
| Lithium |
| Interferes with receptor-activated phosphatidylinositol turnover; blocks the conversion of inositol phosphate to free inositol |
| Antagonizes 5-HT1A and 5-HT1B autoreceptors, alleviating feedback inhibition of 5-HT release |
| Enhances glutamate reuptake system, clearing glutamate from the synapse |
was the first agent demonstrated to have antidepressant efficacy. The TCAs have a three-ring structure with a side chain containing a tertiary or secondary amine attached to the central ring, resembling the phenothiazine antipsychotics (Fig. 30-1). The tertiary amines include imipramine, amitriptyline, clomipramine, and doxepin; the secondary amines include desipramine and nortriptyline.
The TCAs block the reuptake of NE, 5-HT, or both into noradrenergic and/or serotonergic nerve terminals, respectively, by specific interactions with plasma membrane transporters (Fig. 30-2). As a consequence of this inhibition, the actions of NE and 5-HT released from these neurons are not rapidly terminated, resulting in a prolonged stimulation of NE receptors, 5-HT receptors, or both. The TCAs do not affect the reuptake of DA by dopaminergic nerve terminals, and their selectivity for NE versus 5-HT transporters differs among the different compounds (Table 30-1).
The SSRIs and SNRIs also inhibit the reuptake of biogenic amines, and as their name implies, the SSRIs have the highest affinity for 5-HT transporters, whereas the SNRIs have high affinity for 5-HT transporters and moderate affinity for NE transporters. It is important to note, however, that specificity and selectivity are always dose-related such that the SSRIs sertraline and paroxetine inhibit both NE and DA reuptake at the upper end of their dose ranges (see Table 30-1). Similarly, it is also important to keep in mind that the classification of newly developed compounds is based on their affinity for specific transporters, whereas that of the TCAs is based on chemical structure. Thus, although clomipramine is classified chemically as a TCA, its ability and selectivity to inhibit 5-HT and NE reuptake matches that of the SSRI paroxetine. Likewise, the selectivity of the TCAs imipramine and amitriptyline resemble that of the SNRI duloxetine. Thus, at times, classification schemes may be misleading.
As mentioned, the atypical compounds are a very heterogeneous group of drugs. Among these, maprotiline and nefazodone are relatively selective inhibitors of NE reuptake (see Table 30-1). Trazodone is a weak inhibitor of 5-HT reuptake, bupropion weakly inhibits DA reuptake, and mirtazapine appears devoid of activity at any reuptake transporter.
Mirtazapine also blocks α2 adrenergic receptors on noradrenergic and serotonergic nerve terminals and on noradrenergic dendrites (Fig. 30-3). Stimulation of α2 autoreceptors on noradrenergic neurons decreases NE release, whereas stimulation of α2 heteroreceptors on serotonergic neurons inhibits 5-HT release. In addition, stimulation of α1 adrenergic receptors on serotonergic cell bodies and dendrites increases their firing rate. Thus mirtazapine, by inhibiting α2 autoreceptors, enhances noradrenergic cell firing and the release of NE, which activates α1 adrenergic receptors to increase 5-HT release while concurrently blocking α2 heteroreceptors, further facilitating the release of 5-HT.
The MAOIs used for the treatment of depression are phenelzine and tranylcypromine (see Fig. 30-1) and the recently approved selegiline transdermal patch. Phenelzine and selegiline are irreversible MAO inhibitors, and tranylcypromine is a long-lasting MAO inhibitor. At the doses used for depression, all these compounds are nonselective and inhibit both MAO-A and MAO-B. These enzymes are distinct gene products with MAO-A present in human placenta, intestinal mucosa, liver, and brain—responsible for the catabolism of 5-HT, NE, and tyramine; and MAO-B present in human platelets, liver, and brain—responsible predominantly for the catabolism of DA and tyramine. These enzymes are located in the outer membrane of mitochondria and function to maintain low cytoplasmic concentrations of the monoamines, facilitating inward-directed transporter activity (i.e., monoamine reuptake). MAO inhibition causes an increase in monoamine concentrations in the cytosol of the nerve terminal. All the effects of the MAOIs have been attributed to enhanced aminergic activity resulting from enzyme inhibition.
Research with selective MAOIs has shown that inhibition of MAO-A is necessary for antidepressant activity. Thus, although selegiline is a selective inhibitor of MAO-B at low doses and is used at these doses for the treatment of Parkinson’s disease (see Chapter 28), at higher doses selectivity is lost, and selegiline inhibits both MAO-A and MAO-B and has antidepressant activity.
[/not-level-membership-for-basic-science-category]
