31 Transfusion Medicine and Coagulation Disorders
Coagulation and bleeding assume particular importance when surgery is performed on the heart and great vessels using extracorporeal circulation. This chapter provides an understanding of the depth and breadth of hemostasis relating to cardiac procedures, beginning with coagulation pathophysiology. The pharmacology of heparin and protamine follows. This background is then applied to treatment of the bleeding patient. Coagulation monitoring is covered in Chapter 17, and fluid and blood management is further discussed in Chapter 30.
Overview of hemostasis
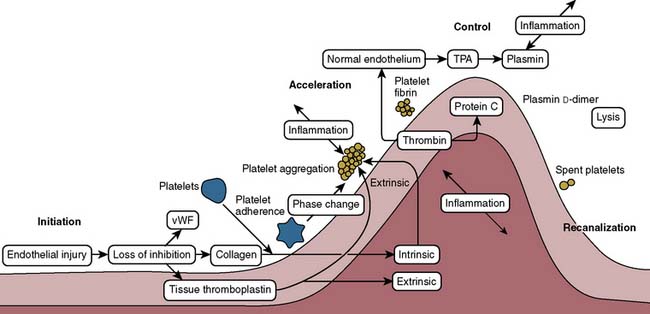
Proper hemostasis requires the participation of innumerable biologic elements (Box 31-1). This section groups them into four topics to facilitate understanding: coagulation factors, platelet function, the endothelium, and fibrinolysis. The reader must realize this is for simplicity of learning, and that in biology, the activation creates many reactions and control mechanisms, all interacting simultaneously. The interaction of the platelets, endothelial cells, and proteins to either activate or deactivate coagulation is a highly buffered and controlled process. It is perhaps easiest to think of coagulation as a wave of biologic activity occurring at the site of tissue injury (Figure 31-1).1 Although there are subcomponents to coagulation itself, the injury/control leading to hemostasis is a four-part event: initiation, acceleration, control, and lysis (recanalization/fibrinolysis). The initiation phase begins with tissue damage, which really is begun with endothelial cell destruction or dysfunction. This initiation phase leads to binding of platelets, as well as protein activations; both happen nearly simultaneously, and each has feedbacks into the other. Platelets adhere, creating an activation or acceleration phase that gathers many cells to the site of injury. From that adhesion a large number of events of cellular/protein messaging cascade. As the activation phase ramps up into an explosive set of reactions, counter-reactions are spun off, leading to control proteins damping the reactions. It is easiest, conceptually, to think of these control mechanisms as analogous to a nuclear reactor. The activation phase would continue to grow and overcome the whole organism unless control rods were inserted (e.g., thrombomodulin, proteins C and S, and tissue plasminogen activator [t-PA]) to stop the spread of the reaction. The surrounding normal endothelium acts quite differently from the disturbed (ischemic) endothelium. Eventually, the control reactions overpower the acceleration reactions and lysis comes into play. The diagram in Figure 31-1 shows lysis as relatively quick, but it can take 24 hours to days to have its full effects. A key concept is that hemostasis is part of a larger body system: inflammation. Most, if not all, of the protein reactions of coagulation control have importance in signaling inflammation leading to other healing mechanisms. Entire books have been written merely to examine these fascinating interactions. It is no wonder that cardiopulmonary bypass (CPB) has such profound inflammatory effects when it is considered that each of the activated coagulation proteins and cell lines then feeds into upregulation of inflammation.
During cardiac surgery, the endothelium (locally and systemically) is disturbed.2 The coronary arteries are made either partially or fully ischemic for periods with cardioplegia (perfused CPB being relatively rare today). Little known is that high concentrations of potassium are particularly insulting to endothelial cells. Ischemia/reperfusion injury is, therefore, the norm for every cardiac surgical case using CPB.3 Systemic ischemia and reperfusion occur throughout every capillary bed because microair, thrombus, and fat emboli are by-products of CPB4 (see Chapters 28 to 30 and 36). It is quite possible that transfusion of red blood cell (RBC) products, through blocking of the microcirculation, actually may contribute to ischemia/reperfusion injury. Transfusion has been shown to evoke endothelial cell hyper-reactivity mediated through cell membrane microparticles.4
 Protein Coagulation Activations
Protein Coagulation Activations
Coagulation Pathways
The coagulation factors participate in a series of activating and feedback inhibition reactions, ending with the formation of an insoluble clot.5 A clot is the sum total of platelet-to-platelet interactions, leading to the formation of a platelet plug (initial stoppage of bleeding). The cross-linking of platelets to each other by way of the final insoluble fibrin leads to a stable clot. Clot is not simply the activation of proteins leading to more protein deposition. Clinicians have been shaped in their thinking about coagulation by the historic way that coagulation proteins were discovered and the resulting coagulation tests. It is that teaching of the coagulation cascade, with resultant monitoring technology, that has led to some transfusion behaviors. The way coagulation has been classically taught (protein cascades) is not the way coagulation proceeds biologically.
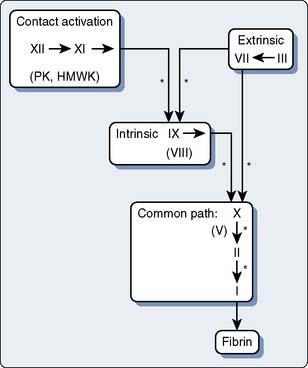
With few exceptions, the coagulation factors are glycoproteins (GPs) synthesized in the liver, which circulate as inactive molecules termed zymogens. Factor activation proceeds sequentially, each factor serving as substrate in an enzymatic reaction catalyzed by the previous factor in the sequence. Hence this classically has been termed a cascade or waterfall sequence. Cleavage of a polypeptide fragment changes an inactive zymogen to an active enzyme. The active form is termed a serine protease because the active site for its protein-splitting activity is a serine amino acid residue. Many reactions require the presence of calcium ion (Ca2+) and a phospholipid surface (platelet phosphatidylserine). The phospholipids occur most often either on the surface of an activated platelet or endothelial cell and occasionally on the surface of white cells. So anchored, their proximity to one another permits reaction rates profoundly accelerated (up to 300,000-fold) from those measured when the enzymes remain in solution. The factors form four interrelated arbitrary groups (Figure 31-2): the contact activation, intrinsic, extrinsic, and common pathways. They were so labeled historically by the human need for order. In biology, they are all highly interactive, occur simultaneously on the surface of cells, and have feedback loops with cross-reactions.
Contact Activation
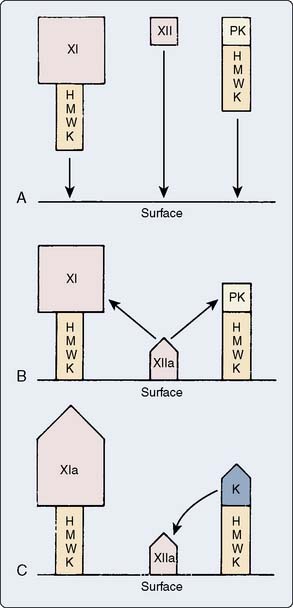
Factor XII, high-molecular-weight kininogen (HMWK), prekallikrein (PK), and factor XI form the contact or surface activation group. The in vivo events that activate factor XII remain unconfirmed. Clinicians do know that ex vivo contact with ionically charged surfaces will activate factor XII. Because factor XII autoactivates by undergoing a shape change in the presence of a negative charge, in vitro coagulation tests use glass, silica, kaolin, and other compounds with negative surface charge (see Chapter 17). The glycocalyx of endothelial cells has a repelling charge for coagulation proteins. One potential in vivo mechanism for factor XII activation is disruption of the endothelial cell layer, which exposes the underlying negatively charged collagen matrix. Activated platelets also provide negative charges on their membrane surfaces. HMWK anchors the other surface activation molecules, PK and factor XI, to damaged endothelium or activated platelets. Factor XIIa cleaves both factor XI, to form factor XIa, and PK, to form kallikrein. Figure 31-3 depicts the events of surface activation.
Intrinsic System
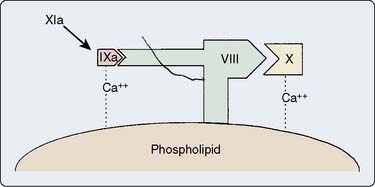
Intrinsic activation forms factor XIa from the products of surface activation. Factor XIa splits factor IX to form factor IXa, with Ca2+ required for this process. Then factor IXa activates factor X with help from Ca2+, a phospholipid surface (platelet-phosphatidylserine), and a GP cofactor, factor VIIIa. Figure 31-4 displays a stylized version of factor X activation. The phospholipids and GP cofactors are on the surface of platelets.
Extrinsic System
Activation of factor X can proceed independently of factor XII by substances classically thought to be extrinsic to the vasculature. This is of historic interest because today it is known that the expression of tissue factor is actually a highly regulated event in endothelial cells. Any number of endothelial cell insults can lead to the production of tissue factor by the endothelial cell.3–10 At rest, the endothelial cell is quite antithrombotic. However, with ischemia, reperfusion, sepsis, or cytokines (particularly interleukin-6), the endothelial cell will stimulate its production of intracellular nuclear factor-κB and send messages for the production of messenger RNA for tissue factor production.6 This can happen quickly and the resting endothelial cell can turn out large amounts of tissue factor. It is widely held today that the activation of tissue factor is what drives many of the abnormalities of coagulation after cardiac surgery, rather than contact activation.7,8 In some tissues, cells outside the vasculature contain large amounts of tissue factor. They are released when cells are damaged/ruptured. Thromboplastin, also known as tissue factor, released from tissues into the vasculature, acts as a cofactor for initial activation of factor X by factor VII. Factors VII and X then activate one another with the help of platelet phospholipid and Ca2+, thus rapidly generating factor Xa. (Factor VIIa also activates factor IX, thus linking the extrinsic and intrinsic paths.)
Thromboplastin straddles the extravascular cell membrane, with its extracellular portion available to bind factor VIIa. Cytokines (particularly tumor necrosis factor-α and interleukin-6) and endotoxins can stimulate its expression on endothelium.9,10 It anchors factor VIIa to the cell surface, thus facilitating activation of factor X. The amount of available factor Va also seems to be quite important for the adequate functioning of the normal coagulation cascades.
Common Pathway
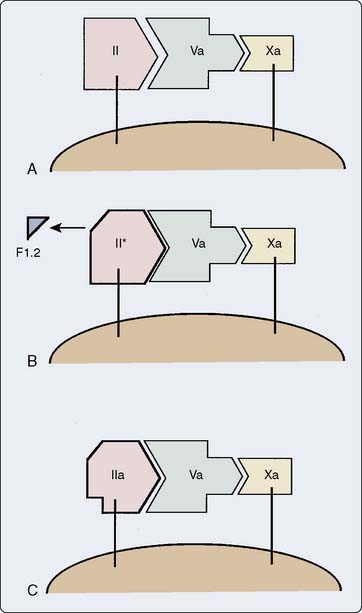
Figure 31-5 depicts the steps involved in formation of thrombin from its precursor, prothrombin. The by-product, fragment F1.2, serves as a plasma marker of prothrombin activation. An alternative scheme generates a different species, meizothrombin, involved more specifically in activation of coagulation inhibitors.11
Thrombin cleaves the fibrinogen molecule to form soluble fibrin monomer and polypeptide fragments termed fibrinopeptides A and B. Fibrin monomers associate to form a soluble fibrin matrix. Factor XIII, activated by thrombin, cross-links these fibrin strands to form an insoluble clot. Patients with lower levels of factor XIII have been found to have more bleeding after cardiac surgery.11,12
Modulators of the Coagulation Pathway
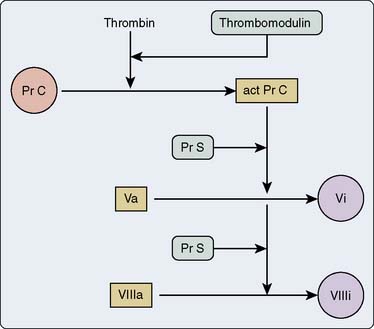
Thrombin, the most important coagulation modulator, exerts a pervasive influence throughout the coagulation factor pathways. It activates factors V, VIII, and XIII; cleaves fibrinogen to fibrin; stimulates platelet recruitment, chemotaxis of leukocytes and monocytes; releases t-PA, prostacyclin, and nitric oxide from endothelial cells; releases interleukin-1 from macrophages; and with thrombomodulin, activates protein C, a substance that then inactivates factors Va and VIIIa.12 Note the negative feedback aspect of this last action (Figure 31-6). Coagulation function truly centers around the effects of thrombin. The platelets, tissue factor, and contact activation all are interactive and are activated by a rent in the surface of the endothelium or through the loss of endothelial coagulation control. Platelets adhere to a site of injury and, in turn, are activated, leading to sequestration of other platelets. It is the interaction of all of those factors together that eventually creates a critical mass. Once enough platelets are interacting together, with their attached surface concomitant serine protease reactions, then a thrombin burst is created. Only when enough thrombin activation has been encountered in a critical time point is a threshold exceeded, and the reactions become massive and much larger than the sum of the whole. It is thought that the concentration and ability of platelets to react fully affect the ability to have a critical thrombin burst. CPB may affect the ability to get that full thrombin burst because of its effects on platelet number, platelet-to-platelet interactions, and the decreased amounts of protein substrates.
The many serine proteases that compose the coagulation pathways are balanced by serine protease inhibitors, termed serpins.13 This biologic yin and yang leads to an excellent buffering capacity. It is only when the platelet-driven thrombin burst so overwhelms the body’s localized anticoagulation or inhibitors that clot proceeds forward. Serpins include α1-antitrypsin, α2-macroglobulin, heparin cofactor II, α2-antiplasmin, antithrombin (AT; also termed antithrombin III [AT III]), and others.
An important note is that activated AT III is active only against free thrombin (fibrin-bound thrombin cannot be seen by AT III).14 Prothrombin circulates in the plasma but is not affected by heparin-AT III complexes; it is only thrombin, and thrombin does not circulate freely. Most thrombin in its active form is either bound to GP binding sites of platelets or in fibrin matrices. When blood is put into a test tube and clot begins to form (such as in an activated coagulation time [ACT]), 96% of thrombin production is yet to come. Most thrombin generation is on the surface of platelets and on clot-held fibrinogen. Platelets, through their GP binding sites and phospholipid folds, protect activated thrombin from attack by AT III. Therefore, the biologic role of AT III is to create an anticoagulant surface on endothelial cells. It is not present biologically to sit and wait for a dose of heparin before CPB.
CPB dilutes AT III substantially, and the further consumption of AT III during CPB (thrombin generation) leads, in some patients, to profoundly low levels of this important inhibitor.15 Research work adding AT III back to the CPB circuit has shown promise in that by doing so there is better preservation of serine protease proteins and platelets. Unfortunately, no pharmaceutical company has decided to pursue this line of research to the point that it would be either commonplace or economically feasible to add large amounts of AT III to CPB and, therefore, avoid consumptive coagulopathies. Congenital AT III deficiency can lead to in utero fetal destruction if the fetus is homozygous for the abnormal AT III. However, patients who are heterozygous for AT III abnormalities have about 40% to 60% of normal AT III activity. They have a particularly high risk for deep vein thrombosis. Low AT III levels have been described during extracorporeal membrane oxygenation, and the addition of AT III to the extracorporeal membrane oxygenation circuit has been effective in improving outcome and decreasing bleeding in some circumstances.16 It is not known how useful it would be in all patients on CPB because only small trials have been performed, but these have been encouraging.17,18 Both human AT III concentrate (harvested from multiple plasma donors and pasteurized) and a pharmaceutically engineered, goat milk–produced AT III (slightly different structure than human AT III) are commercially available.
Heparin cofactor II also inhibits thrombin, once it is activated. Although large doses of heparin activate heparin cofactor II, dermatans on endothelial cell surfaces activate it far more effectively, suggesting dermatans as alternative drugs to heparin.19 Dermatan sulfates are not available for use today in the United States.
Another serpin, protein C, degrades factors Va and VIIIa. Like other vitamin K–dependent factors, it requires Ca2+ to bind to phospholipid. Its cofactor, termed protein S, also exhibits vitamin K dependence. Genetic variants of protein C are less active and lead to increased risk for deep vein thrombosis and pulmonary embolism. When endothelial cells release thrombomodulin, thrombin then accelerates by 20,000-fold its activation of protein C20 (see Figure 31-6). Activated protein C also promotes fibrinolysis through a feedback loop to the endothelial cells to release t-PA.21
Regulation of the extrinsic limb of the coagulation pathway occurs via tissue factor pathway inhibitor (TFPI), a glycosylated protein that associates with lipoproteins in plasma.22 TFPI is not a serpin. It impairs the catalytic properties of the factor VIIa-tissue factor complex on factor X activation. Both vascular endothelium and platelets appear to produce TFPI.23,24 Heparin releases TFPI from endothelium, increasing TFPI plasma concentrations by as much as sixfold, which should be viewed as a biologic indicator of how poor heparin is as an anticoagulant. TFPI is not tested for in routine coagulation testing. It may be that some individuals with certain types of TFPI or who have very large amounts of circulating TFPI could be at risk for severe adverse bleeding after cardiac surgery.25–30 This area is just beginning to be examined today both in terms of whether TFPI is responsible for abnormal bleeding and whether its genetic variants have abnormal bleeding or thrombosis.
von Willebrand factor (vWF), a massive molecule composed of disulfide-linked glycosylated peptides, associates with factor VIII in plasma, protecting it from proteolytic enzymes. It circulates in the plasma in its coiled inactive form.31 Disruption of the endothelium either allows for binding of vWF from the plasma or allows for expression of vWF from tissue and from endothelial cells. Once bound, vWF uncoils to its full length and exposes a hitherto cryptic domain in the molecule. This A-1 domain has a very high affinity for platelet GPs. Initially, vWF attaches to the glycoprotein Iα (GPIα) platelet receptor, which slows the platelet forward movements against the shear forces of blood flow. Shear forces are activators of platelets. As the platelet’s forward movement along the endothelial brush border is slowed (because of vWF attachment), shear forces actually increase; thus, the binding of vWF to GP1 acts to provide a feedback loop for individual platelets, further activating them. The activation of vWF and its attachment to the platelet are not enough to bind the platelet to the endothelium, but it creates a membrane signal that allows for early shape change and expression of other GPs, GPIb and GPIIb/IIIa. Then, secondary GPIb binding connects to other vWF nearby, binding the platelet and beginning the activation sequence. It bridges normal platelets to damaged subendothelium by attaching to the GPIb platelet receptor. An ensuing platelet shape change then releases thromboxane, β-thromboglobulin, and serotonin, and exposes GPIIb/IIIa, which binds fibrinogen.
Deficiency States
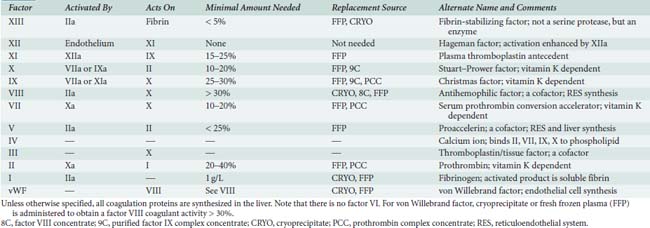
Decreased amounts of coagulation proteins may be inherited or acquired. Deficiencies of each part of the coagulation pathway are considered in turn. Table 31-1 summarizes the coagulation factors, their activation sequences, and vehicles for factor replacement when deficient.
TABLE 31-1 The Coagulation Pathway Proteins, Minimal Amounts Needed for Surgery, and Replacement Sources
Contact Activation
Although decreased amounts of factor XII, PK, and HMWK can occur, these defects do not have clinical sequelae. The autosomal dominant deficiency of factor XI is very rare. However, its incidence among Ashkenazi Jews is as high as 0.1% to 0.3%.32 Most of these patients require factor replacement with fresh-frozen plasma (FFP) for surgery. Spontaneous bleeding does not occur, but increased bleeding after a surgical event or trauma is possible. Factor XI concentrations do not directly correlate with bleeding after trauma or surgery, suggesting that factor XI deficiency can be easily overcome by activation of platelets, factor IX, and other signaling mechanisms. An FFP dose of 10 mL/kg will yield target concentrations of 20% activity, and it is often given for this rare deficiency.
Intrinsic
Hemophilia occurs worldwide, with a prevalence of 1 in 10,000. Hemophilia A, which constitutes about 80% to 85% of cases, originates from decreased activity of factor VIII. Because platelet function remains normal, minor cuts and abrasions do not bleed excessively. Joint and muscle hemorrhages ensue from minor trauma or, seemingly, spontaneously. Airway issues include epistaxis and obstruction from bleeding into the tongue. The bleeding time and prothrombin time (PT) remain normal, whereas the activated partial thromboplastin time (aPTT) is prolonged.33 Desmopressin, a synthetic analog of vasopressin, will increase factor VIII activity by releasing vWF from endothelial cells, except in patients with severe hemophilia A who have too little functional factor VIII available for vWF.34 Major surgery requires replenishment of factor VIII functional activity to greater than 80% of normal with FFP, cryoprecipitate, or factor VIII concentrate.35 Factor VIII concentrate is the preferred method today. After surgery, factor VIII concentrations should be maintained greater than 30% for 2 weeks with repeat doses. Current plasma-derived concentrates are solvent detergent and heat-treated to remove lipid-coated viruses (human immunodeficiency virus [HIV], hepatitis B, human T-lymphotropic virus [HTLV]. However, this was not historically the case, and in Europe during the early parts of the HIV/acquired immune deficiency syndrome (AIDS) crisis, most individuals with hemophilia contracted HIV/AIDS from contaminated products. A recombinant product also is available but costs about three times that of the plasma-derived one.
Factor IX deficiency manifests as hemophilia B, constituting 15% to 20% of all hemophilia cases. Patients present with symptoms identical to those with hemophilia A. No study has demonstrated a salutary effect of desmopressin here. Prothrombin complex (factor IX) concentrates will replenish levels, but consumptive coagulopathy remains a possible complication, stemming from the presence of activated coagulation factors, principally factor VIIa, in the preparation.36 Purified factor IX concentrate, a plasma-derived, solvent detergent and heat-treated product, currently constitutes the replacement vehicle of choice for patients with hemophilia B.37 Recombinant pure factor IX concentrate will be available, but at considerable expense (factor VIIa is now available as well). Consultation with an experienced hematologist aids in the care of patients with hemophilia undergoing surgery.
Common Pathway
Deficiency of either factor V or factor X, both extremely rare autosomal recessive disorders, increases both the PT and PTT. The bleeding time is normal in factor X deficiency but prolonged in one third of patients with factor V deficiency. The bleeding time prolongation arises from the role of factor V in platelet function.38 Prothrombin complex concentrate or FFP supplies prothrombin, factor V, and factor X. Numerous inherited abnormalities (polymorphisms) of prothrombin and fibrinogen occur, with varying characteristics. Cryoprecipitate, which contains 250 mg fibrinogen and 100 units factor VIII per 10-mL bag, as well as vWF and factor XIII, treats inherited or acquired disorders of fibrinogen. Many of these polymorphisms are associated with hypercoagulability and, perhaps, accelerated atherosclerosis rather than bleeding.
Warfarin
Administration of this vitamin K antagonist affects plasma levels of factors II, VII, IX, and X, as well as proteins C and S.39 Protein C has the shortest half-life, followed by factors VII (6 hours), IX (24 hours), X (2 days), and II (3 days).36 Substantial PT prolongation and some PTT prolongation accompany warfarin therapy. For immediate restoration of clotting function, FFP is given. Otherwise, parenteral vitamin K or cessation of warfarin (Coumadin) suffices. Clinicians should be extremely careful in administering these compounds if a patient is suspected of having heparin-induced thrombocytopenia (HIT). Treatment with commercially available factor VIIa restores PT to normal and appears to stop bleeding when warfarin therapy has not had time to be reversed. The use of factor VIIa for this intervention before surgery is both effective and, perhaps, worthwhile as it avoids the use of FFP. The time scale of factor VIIa effectiveness may not be as long as if one normalized circulating levels through FFP administration. It makes sense, therefore, that if the PT is prolonged again approximately 8 to 12 hours after a dose of factor VIIa, then redosage of the drug be given.
Inherited Thrombotic Disorders
A number of genetic abnormalities lead to thrombosis. The most prevalent (2% to 5%) in European-derived populations is factor V Leiden, in which a point mutation at residue 1691 on factor V renders it resistant to inactivation by activated protein C.40 Venous thromboembolism risk increases 7-fold in heterozygotes and 80-fold in homozygotes, but episodes are less severe than in other thrombotic disorders. Pregnancy and oral contraceptives greatly exacerbate the thrombotic tendency.41
Protein C or S deficiencies, if homozygous, present at birth as neonatal purpura fulminans. Protein C deficiency heterozygotes demonstrate 40% to 60% protein C activity and present with venous thrombosis beginning in adolescence. The role of reduced concentrations of protein S in causing thrombosis has come into question.41 Together, deficiencies of AT, protein C, and protein S account for 10% to 15% of inherited thrombosis.42
Homocysteinemia is the mild heterozygous state of cystathione β-synthetase deficiency, known as homocystinuria in its more serious homozygous form. Increased plasma concentrations of homocysteine induce endothelial cell tissue factor activity, stimulate factor V activation, and impair protein C activation, all of which contribute to thrombosis. Folic acid and vitamins B6 and B12 reduce homocysteine plasma concentrations.43

Platelet Function
Most clinicians think first of the coagulation proteins when considering hemostasis. Although no one element of the many that participate in hemostasis assumes dominance, platelets may be the most complex.44 Without platelets, there is no coagulation and no hemostasis, so it could be argued that they are most important. Without the proteins, there is hemostasis, but it lasts only about 10 to 15 minutes as the platelet plug is inherently unstable and breaks apart under the shear stress of the vasculature. Platelets provide phospholipid for coagulation factor reactions; contain their own microskeletal system and release coagulation factors; secrete active substances affecting themselves, other platelets, the endothelium, and other coagulation factors; and alter shape (through active actin-myosin contraction) to expose membrane GPs essential to hemostasis. Their cell signaling is highly regulated, is present in other cell lines (RBCs, leukocytes, and endothelial cells), and has been intensively studied. Platelets have perhaps as many as 30 to 50 different types of cell receptors, with many ways of these being activated and inhibited.
The initial response to vascular injury is formation of a platelet plug. Good hemostatic response depends on proper functioning of platelet adhesion, activation, and aggregation (Figure 31-7). This section first discusses these aspects and then follows with the effects of platelet disorders and platelet-inhibiting pharmaceuticals. Clinicians talk about platelet dysfunction, which is largely overarching and grossly too general a term. The complexity that is platelet function really needs careful study.
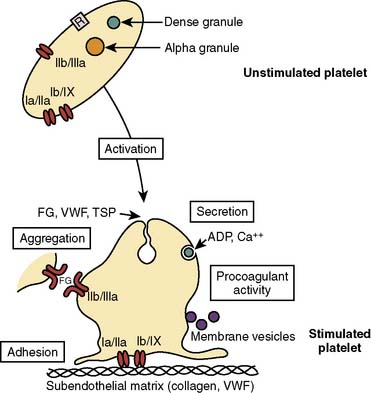
Figure 31-7 Platelet function in hemostasis.
(From George J, Shattil SJ: The clinical importance of acquired abnormalities of platelet function. N Engl J Med 324:27, 1991.)
Platelet Adhesion
Capillary blood exhibits laminar flow, which maximizes the likelihood of interaction of platelets with the vessel wall. Red cells and white cells stream near the center of the vessels and marginate platelets. However, turbulence causes reactions in endothelium that lead to the secretion of vWF, adhesive molecules, and tissue factor. Shear stress is high as fast-moving platelets interact with the endothelium. When the vascular endothelium becomes denuded or injured, the platelet has the opportunity to contact vWF, which is bound to the exposed collagen of the subendothelium. A platelet membrane component, GPIb, attaches to vWF, thus anchoring the platelet to the vessel wall. Independently, platelet membrane GPIa and GPIIa and IX may attach directly to exposed collagen, furthering the adhesion stage.44–46
After activation (see later), additional adhesive mechanisms come into play. Release of selectin GPs from α-granules allows their membrane expression, thus promoting platelet-leukocyte adhesion. This interaction ultimately may allow expression of tissue factor on monocytes, thus amplifying coagulation.47
The integrin GPs form diverse types of membrane receptors from combinations of 20 α and 8 β subunits.48 One such combination is GPIIb/IIIa, a platelet membrane component that initially participates in platelet adhesion. Platelet activation causes a conformational change in GPIIb/IIIa, which results in its aggregator activity.
Platelet adhesion begins rapidly—within 1 minute of endothelial injury—and completely covers exposed subendothelium within 20 minutes.45 It begins with decreased platelet velocity when GPIb/IX and vWF mediate adhesion, followed by platelet activation, GPIIb/IIIa conformational change, then vWF binding and platelet arrest on the endothelium at these vWF ligand sites.44,46
Platelet Activation and Aggregation
Agonists induce a graded platelet shape change (the amount based on the relative amount of stimulation), increase platelet intracellular Ca2+ concentration, and stimulate platelet G protein. In addition, serotonin and thromboxane A2 are potent vasoconstrictors (particularly in the pulmonary vasculature).49 The presence of sufficient agonist material results in platelet aggregation. Aggregation occurs when the integrin proteins (mostly fibrinogen) released from α granules form molecular bridges between the GPIIb/IIIa receptors of adjacent platelets (the final common platelet pathway).
Platelet Disorders
Dysfunctional vWF produces von Willebrand disease (vWD), an autosomal dominant disorder of variable expressivity.34 With an incidence of 1.4 to 5 cases per 1000 population, vWD is the most common inherited coagulopathy. Patients present with mucocutaneous hemorrhages rather than hemarthroses. Common symptoms include epistaxis, ecchymoses, and excessive bleeding after trauma, with surgery, or during menses. Because vWF concentrations vary greatly with time, symptoms have variable expressivity. Desmopressin reverses the prolonged bleeding time in patients with mild vWD.50 As with hemophilia A, severe cases of vWD do not benefit from desmopressin therapy. In one rare class of vWD (type IIB, 3% to 5% of vWD), desmopressin aggregates platelets, inducing thrombocytopenia and worsening rather than helping hemostasis. Table 31-2 summarizes features of the more common types of vWD. When blood products are needed, cryoprecipitate constitutes the replacement vehicle of choice in vWD, although recent factor VIII concentrates retain vWF activity and have been used successfully during cardiac surgery.51
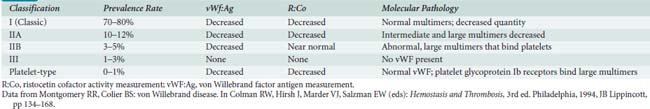
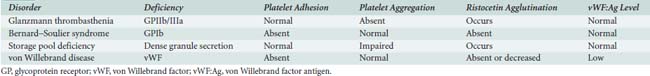
The addition of agonist (ADP or collagen) to platelets allows measurement of platelet aggregation in vitro. In Glanzmann thrombasthenia, the GPIIb/IIIa receptor is absent, preventing aggregation. However, ristocetin, a cationic antibiotic similar to vancomycin, can agglutinate platelets directly via GPIb receptors and vWF. Absence of the GPIb receptor, Bernard–Soulier syndrome, prevents adhesion and agglutination with ristocetin, but aggregation to ADP is normal, because the GPIIb/IIIa receptor is intact. Patients with vWD also exhibit impaired platelet adhesion and normal aggregation. Decreased amounts of vWF antigen distinguish it from the Bernard–Soulier syndrome. In platelet storage pool deficiency, impairment of dense granule secretion yields no ADP on adhesion. In vitro addition of collagen will not aggregate platelets because of absence of ADP release, whereas added ADP will initiate some aggregation. Table 31-3 summarizes these diagnostic findings. Uremia impairs the secretory and aggregating functions of platelets, resulting in an increased bleeding time. However, the most common effect of renal dysfunction is hypercoagulability. It is only with severe uremia that the platelets are poisoned. It is, therefore, a common misconception in the operating room that a patient with mild-to-moderate renal failure will be at increased risk for bleeding. The utilization of thromboelastography (TEG) can help in deciding whether the extent of renal failure is causing hypocoagulability. The cause and clinical significance remain poorly defined (see Chapter 17).
Prostaglandins and Aspirin
Endothelial cell cyclooxygenase synthesizes prostacyclin, which inhibits aggregation and dilates vessels. Platelet cyclooxygenase forms thromboxane A2, a potent aggregating agent and vasoconstrictor. Aspirin irreversibly acetylates cyclooxygenase, rendering it inactive. Low doses of aspirin, 80 to 100 mg, easily overcome the finite amount of cyclooxygenase available in the nucleus-free platelets. However, endothelial cells can synthesize new cyclooxygenase. Thus, with low doses of aspirin, prostacyclin synthesis continues whereas thromboxane synthesis ceases, decreasing platelet activation and aggregation. High doses of aspirin inhibit the enzyme at both cyclooxygenase sites.49
Reversible platelet aggregation is blocked by aspirin, as the platelet cyclooxygenase is inhibited. However, the more powerful agonists that yield the calcium release response can still aggregate and activate platelets, because cyclooxygenase is not required for those pathways (Figure 31-8).
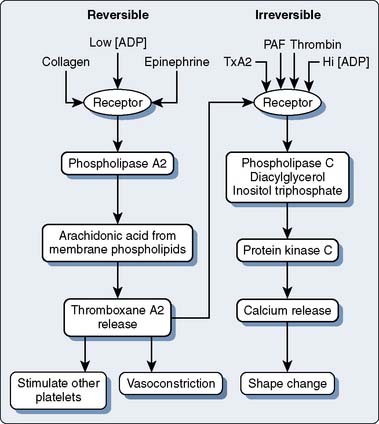
In many centers, a majority of the patients presenting for coronary artery bypass grafting (CABG) will have received aspirin within 7 days of surgery in hopes of preventing coronary thrombosis.52 Aspirin is a drug for which an increased risk for bleeding often has been demonstrated.53 Although most early research studies stated that aspirin leads to increased bleeding, since the mid-1990s, that early impression has not been confirmed. Today, it probably is more likely that, in some patients, a mild-to-moderate increased risk for bleeding is possible.
Unfortunately, most of the early studies of aspirin were not blinded, and it may be that the pervading belief that aspirin caused increased bleeding was actually self-fulfilling. Since the early 1990s, there have been a number of therapeutic changes including use of lower doses and greater use of antifibrinolytics. These changes alone may have decreased the overall risk for bleeding from aspirin. Follow-up prospective studies have, thus, yielded varied results. In the largest cohort of 772 men, aspirin increased bleeding after CABG by 33%,54 and a group of 101 aspirin-taking patients bled 25% to 56% more than a control group.55 In another study, the need to explore the mediastinum for excessive bleeding nearly doubled (factor of 1.82) in patients taking aspirin.56 However, many other studies have not shown increased bleeding with aspirin.57–62 Although a single aspirin can irreversibly inhibit platelet cyclooxygenase for the life of the platelet, aspirin-related bleeding after surgery usually requires more extensive exposure to the drug.
Drug-Induced Platelet Abnormalities
Many other agents inhibit platelet function. β-Lactam antibiotics coat the platelet membrane, whereas the cephalosporins are rather profound but short-term platelet inhibitors.63 Many cardiac surgeons may not realize that their standard drug regimen for antibiotics may be far more of a bleeding risk than aspirin. Hundreds of drugs can inhibit platelet function. Calcium channel blockers, nitrates, and β-blockers are ones commonly used in cardiac surgery. Nitrates are effective antiplatelet agents, and that may be part of why they are of such benefit in angina, not just for their vasorelaxing effect on large blood vessels. Nonsteroidal anti-inflammatory drugs reversibly inhibit both endothelial cell and platelet cyclooxygenase. In addition, anecdotal reports of platelet inhibition, without clear confirmatory studies, exist for many pharmaceuticals including dextran, and for innumerable foods (e.g., onion, garlic, alcohol) and spices (e.g., ginger, tumeric, cloves).24
Rofecoxib (Vioxx), a cyclooxygenase 2 (COX-2) inhibitor, was withdrawn from the U.S. market because of its cardiovascular risk profile (a small increase in associated acute myocardial infarctions).64 This COX-2 inhibitor has the highest selectivity for COX-2 versus COX-1 and thus leads to an imbalance between thromboxane A2 and prostacyclin production. The other COX-2 inhibitors are currently also undergoing cardiovascular investigation.
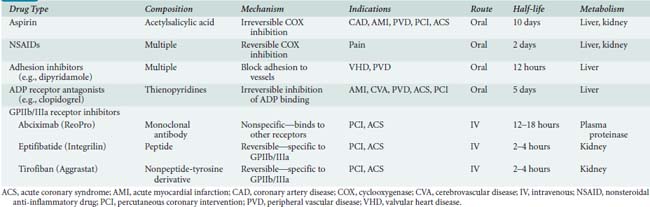
In addition to the partial inhibitory effects of aspirin and the other drugs mentioned earlier, new therapies that inhibit platelet function in a more specific manner have been developed. These drugs include platelet adhesion inhibitor agents, platelet-ADP-receptor antagonists, and GPIIb/IIIa receptor inhibitors (Table 31-4).65
Adenosine Diphosphate (ADP) Receptor Antagonists
Clopidogrel (Plavix), prasugrel (Effient), and ticlopidine (Ticlid) are thienopyridine derivatives that inhibit the ADP receptor pathway to platelet activation. They have a slow onset of action because they must be converted to active drugs, and their potent effects last the lifetime of the platelets affected (5 to 10 days). Clopidogrel and prasugrel are the preferred drugs. They are administered orally once daily to inhibit platelet function and are quite effective in decreasing myocardial infarctions after percutaneous coronary interventions (see Chapter 3). The combination of aspirin and clopidogrel has led to increased bleeding but is sometimes used in an effort to keep vessels and stents open. The TEG and now the RoTEM (a modified TEG) with ADP or other additives can be used to determine the degree of inhibition caused by these drugs (see Chapter 17). Other new tests are coming onto the market to allow testing for relative platelet inhibition caused by thienopyridines. Some of these are modifications of platelet flow cytometry or automated platelet aggregometers. Verify Now (Accumetrics, San Diego, CA) and the PFA-100 (Siemens USA, Deerfield, IL) have been used in dosing clopidogrel for cardiology procedures. These new platelet function tests are now finding their way into hospitals, and some are starting to use them before or after surgery.66–72 In the past, aprotinin was able to partially reverse the effects of these drug; however, it is currently not available. Today, treatment for bleeding caused by platelet dysfunction after cardiac surgery involves using factor VIIa, as well as repeated platelet transfusions. Although one report notes that in addition to factor VIIa, the investigators found it useful to infuse fibrinogen (available in Europe as a purified product) and factor XIII.73
Glycoprotein IIb/IIIa Receptor Inhibitors
GPIIb/IIIa receptor inhibitors are the most potent (> 90% platelet inhibition) and important platelet inhibitors because they act at the final common pathway of platelet aggregation with fibrinogen, no matter which agonist began the process. All of the drugs mentioned earlier work at earlier phases of activation of platelet function. These drugs are all administered by intravenous infusion, and they do not work orally. The GPIIb/IIIa inhibitors often are used in patients taking aspirin because they do not block thromboxane A2 production. The dose of heparin usually is reduced when used with these drugs (i.e., percutaneous coronary intervention to avoid bleeding at the vascular puncture sites). Platelet activity can be monitored to determine the extent of blockade. Excessive bleeding requires allowing the short-acting drugs to wear off, while possibly administering platelets to patients receiving the long-acting drug abciximab (see Table 31-4). Most studies have found increased bleeding in patients receiving these drugs who required emergency CABG.

Vascular Endothelium
Nitric oxide vasodilates blood vessels and inhibits platelets. Its mechanism involves activation of guanylate cyclase with eventual uptake of calcium into intracellular storage sites. Prostacyclin (prostaglandin I2) possesses powerful vasodilator and antiplatelet properties. Endothelium-derived prostacyclin opposes the vasoconstrictor effects of platelet-produced thromboxane A2. Prostacyclin also inhibits platelet aggregation, disaggregates clumped platelets, and at high concentrations, inhibits platelet adhesion. Prostacyclin increases intracellular concentrations of cyclic adenosine monophosphate, which inhibits aggregation. Thromboxane acts in an opposite manner. The mechanism of prostacyclin action is stimulation of adenylyl cyclase, leading to reduced intracellular calcium concentrations. Some vascular beds (e.g., lung) and atherosclerotic vessels secrete thromboxane, endothelins, and angiotensin, all vasoconstrictors, as well as prostacyclin. Activation of platelets releases endoperoxides and arachidonate. These substances, used by nearby damaged endothelial cells, provide substrate for prostacyclin production.5

Fibrinolysis
Extrinsic Fibrinolysis
Endothelial cells synthesize and release t-PA. Both t-PA and a related substance, urokinase plasminogen activator, are serine proteases that split plasminogen to form plasmin. The activity of t-PA magnifies on binding to fibrin. In this manner, also, plasmin formation remains localized to sites of clot formation. Epinephrine, bradykinin, thrombin, and factor Xa cause endothelium to release t-PA, as do venous occlusion and CPB.74 Fibrinolysis during and after CPB is discussed later.
Clinical Applications
Figure 31-9 illustrates the fibrinolytic pathway, with activators and inhibitors. Streptokinase, acetylated streptokinase plasminogen activator complex, and t-PA find application in the lysis of thrombi associated with myocardial infarction. These intravenous agents “dissolve” clots that form on atheromatous plaque. Clinically significant bleeding may result from administration of any of these exogenous activators or streptokinase.75
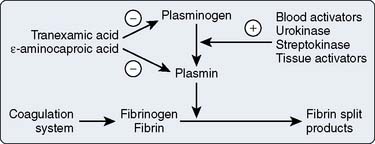
Figure 31-9 The fibrinolytic pathway.
(From Horrow JC, Hlavacek J, Strong MD, et al: Prophylactic tranexamic acid decreases bleeding after cardiac operations. J Thorac Cardiovasc Surg 99:70, 1990.)
Fibrinolysis also accompanies CPB. This undesirable breakdown of clot after surgery may contribute to postoperative hemorrhage and the need to administer allogeneic blood products. Regardless of how they are formed, the breakdown products of fibrin intercalate into sheets of normally forming fibrin monomers, thus preventing cross-linking. In this way, extensive fibrinolysis exerts an antihemostatic action. Factor XIII is an underappreciated coagulation protein. It circulates and, when activated, cross-links fibrin strands and protects fibrin from the lytic actions of plasmin. It has been known for some time that low levels of factor XIII are associated with increased hemorrhage after CPB. Factor XIII levels are reduced by hemodilution, but it also appears that there is active destruction in some patients with CPB. Several new studies have begun testing adding factor XIII to patients and assessing bleeding.76–78 The problem, however, is that currently there is no good way to assess factor XIII levels. Clearly, factor XIII will be quite expensive, and using it for most patients may well be ill advised.
Heparin
In 1916, in the course of experiments to determine whether the phospholipid component of cephalin caused clotting, a second-year medical student, Jay McLean, instead discovered a substance derived from liver that prolonged coagulation.79 His mentor, William Howell, named this substance heparin. Heparin has been used almost exclusively as the anticoagulant for CPB for more than 50 years.

Pharmacology
Chemical Structure
In the 1920s, Howell’s group isolated heparin and identified it as a carbohydrate containing glucuronic acid residues. In the 1930s, Jorpes demonstrated a hexosamine component to heparin (glucosamine, in particular) that is present in a ratio of 1:1 with glucuronic acid. Of greater importance, he discovered that heparin contains many sulfate groups—two per uronic acid residue—making it one of the strongest acids found in living things. In the 1950s, Jorpes’ group identified the sulfate groups at the N-position on glucosamine, where solely acetyl groups previously were thought to reside. In the 1960s, they corrected identification of the uronic acid as l-iduronic acid, an epimer of d-glucuronic acid, and refined its structural detail.80
The N-sulfated-d-glucosamine and l-iduronic acid residues of heparin alternate in copolymer fashion to form chains of varying length (Figure 31-10). As a linear anionic polyelectrolyte, the negative charges being supplied by sulfate groups, heparin demonstrates a wide spectrum of activity with enzymes, hormones, biogenic amines, and plasma proteins. A pentasaccharide segment binds to AT.81 Heparin is a heterogenous compound; the carbohydrates vary in both length and side-chain composition, yielding a range of molecular weights from 5000 to 30,000, with most chains between 12,000 and 19,000.82 Today, the standard heparin is called unfractionated heparin (UFH).
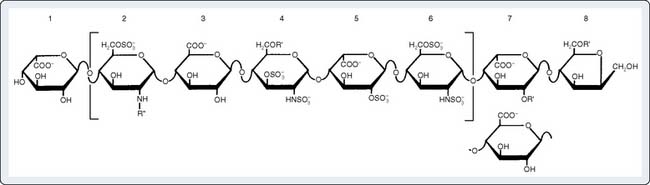
Heparin versus Heparan
Heparan, a glycosaminoglycan found in the connective tissue and the coating of the endothelial surfaces of nearly all species, can be distinguished from heparin by the following characteristics: (1) a predominance of glucuronic acid over iduronic acid; and (2) N-acetylation, rather than N-sulfation, of more than 20% of glucosamine residues. Bound to cellular proteins, heparan resides inside cells, on cell surfaces, and in the extracellular matrix.80–84
Source and Biologic Role
Heparin is found mostly in the lungs, intestines, and liver of mammals, with skin, lymph nodes, and thymus providing less plentiful sources.84 Abundance of heparin in tissues rich in mast cells suggests these as the source of the compound. Its presence in tissues with environmental contact suggests a biologic role relating to immune function. Heparin may assist white blood cell movements in the interstitium after an immunologic response has been triggered. Mollusks have no coagulation system yet possess heparin, arguing against a biologic role in hemostasis. It is clear that heparin, per se, was never intended biologically to be circulating in large dosages throughout the vascular tree.
Most commercial preparations of heparin now use pig intestine, 40,000 pounds of which yield 5 kg heparin.80 Prevention of postoperative thrombosis constituted the initial clinical use of heparin in 1935 by Best, Jaques, and colleagues in Toronto, and by Crafoord in Stockholm.80
Potency
Heparin potency is determined by comparing the test specimen against a known standard’s ability to prolong coagulation.85 Current United States Pharmacopeia (USP) and British Pharmacopoeia (BP) assays use a PT-like method on pooled sheep’s plasma obtained from slaughterhouses. The plasma commonly is contaminated with tissue extracts or other hemostatically active substances. The European Pharmacopoeia’s method, an aPTT on fresh sheep’s plasma, is superior to that of the USP.86 Modern research assays use human FFP, an aPTT-like method, and require linear log versus log plots of standard and test samples.85
UFH dose should not be specified by weight (milligrams) because of the diversity of anticoagulant activity expected from so heterogenous a compound. Unfortunately, because of the flawed USP assay, even units of activity often do not reflect clinical effects. As originally defined, 1 unit heparin prolongs the clotting of cat’s blood for only 24 hours at 0°C.87 Milligram usage, introduced in 1937 as a Swedish standard, was superseded by the first international standard, in which 130 units of activity corresponded to 1 mg. The fourth international standard uses a porcine mucosal preparation.85
One USP unit of heparin activity is the quantity that prevents 1.0 mL of citrated sheep’s plasma from clotting for 1 hour after addition of calcium.88 Units cannot be cross-compared among heparins of different sources, such as mucosal versus lung, or low-molecular-weight heparin (LMWH) versus UFH, or even lot to lot, because the assay used may or may not reflect actual differences in biologic activity. None of these measures has anything to do with the effect of a unit on anticoagulation effect for human cardiac surgery.

Pharmacokinetics and Pharmacodynamics
The heterogeneity of UFH molecules produces variability in the relation of dose administered to plasma level of drug. In addition, the relation of plasma level to biologic effect varies with the test system. A three-compartment model describes heparin kinetics in healthy humans: rapid initial disappearance, saturable clearance observed in the lower dose range, and exponential first-order decay at greater doses89,90 (Figure 31-11). The rapid initial disappearance may arise from endothelial cell uptake.91,92 The reticuloendothelial system, with its endoglycosidases and endosulfatases, and uptake into monocytes, may represent the saturable phase of heparin kinetics. Finally, renal clearance via active tubular secretion of heparin, much of it desulfated, explains heparin’s exponential clearance.
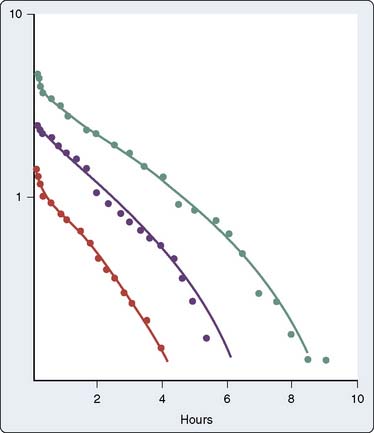
Male sex and cigarette smoking are associated with more rapid heparin clearance.93 The resistance of patients with deep vein thrombosis or pulmonary embolism to heparin therapy may be caused by the release from thrombi of PF4, a known heparin antagonist.94,95 Chronic renal failure prolongs elimination of high, but not low, heparin doses.93 Chronic liver disease does not change elimination.96
Loading doses for CPB (200 to 400 U/kg) are substantially greater than those used to treat venous thrombosis (70 to 150 U/kg). Plasma heparin levels, determined fluorometrically, vary widely (2 to 4 units/mL) after doses of heparin administered to patients about to undergo CPB.97 The ACT response to these doses of heparin displays even greater dispersion. Gravlee et al98 identified thrombocytosis and advanced age as causing a decreased ACT response to administered heparin. This effect may arise from alterations in pharmacokinetics, pharmacodynamics, or both. Interpatient variability in heparin response (pharmacodynamics) does affect the clotting time99,100; however, the clinical response to heparin administered to various patients is more consistent than suggested by in vitro measurements.
Actions and Interactions
Heparin exerts its anticoagulant activity via AT III, one of the many circulating serine protein inhibitors (serpins), which counter the effects of circulating proteases.101 The major inhibitor of thrombin and factors IXa and Xa is AT III; that of the contact activation factors XIIa and XIa is α1-proteinase inhibitor; kallikrein inhibition arises mostly from C1 inhibitor. AT activity is greatly decreased at a site of vascular damage, underscoring its primary role as a scavenger for clotting enzymes that escape into the general circulation.
AT inhibits serine proteases even without heparin. The extent to which heparin accelerates AT inhibition depends on the substrate enzyme; UFH accelerates the formation of the thrombin-AT complex by 2000-fold, but accelerates formation of the factor Xa-AT complex by only 1200-fold102 (Table 31-5). In contrast, LMWH fragments preferentially inhibit factor Xa. Enzyme inhibition proceeds by formation of a ternary complex consisting of heparin, AT, and the proteinase to be inhibited (e.g., thrombin, factor Xa). For UFH, inhibition of thrombin occurs only on simultaneous binding to both AT and thrombin. This condition requires a heparin fragment of at least 18 residues.101,103 A pentasaccharide sequence binds to AT (see Figure 31-10). LMWHs, consisting of chains 8 to 16 units long, preferentially inhibit factor Xa. In this case, the heparin fragment activates AT, which then sequentially inactivates factor Xa; heparin and factor Xa do not directly interact (Figure 31-12).104,105
TABLE 31-5 Some Coagulation Factor Inhibitors and the Effects of Heparin
| Factor | Major Inhibitor | Acceleration of Antithrombin Activity by Heparin |
|---|---|---|
| Kallikrein | C1 inhibitor | — |
| XIIa | α1-Proteinase inhibitor | — |
| Xia | α1-Proteinase inhibitor | 40-fold |
| Xa | Antithrombin | 1,200-fold |
| IXa | Antithrombin | 10,000-fold |
| IIa | Antithrombin | 2,000-fold |
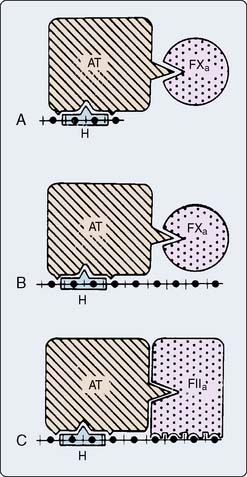
Several investigators have demonstrated continued formation of fibrinopeptides A106,107 and B108 (Figure 31-13), as well as prothrombin fragment F1.2 and thrombin-AT complexes,109 despite clearly acceptable anticoagulation for CPB by many criteria. These substances indicate thrombin activity. The clinical significance of this ongoing thrombin activity has had limited study. The ACT must be more prolonged to prevent fibrin formation during cardiac surgery compared with during extracorporeal circulation without surgery because surgery itself incites coagulation. UFH in conjunction with AT appears to work in plasma only on free thrombin. When considering what is known today about thrombin burst and thrombin activity, heparin appears to be relatively inefficient because there is not much free thrombin. Thrombin is held on the surface of activated platelets at various GP binding sites including the GPIIb/IIIa site. Most thrombin is fibrin bound, and heparin-AT complexes do not bind at all to this thrombin unless the level of heparin is pushed far above what is used routinely for CPB. The idea behind using heparin for CPB is that by creating a large circulating concentration of activated AT, whenever a thrombin molecule is produced, an available AT molecule will be there to immediately bind to it before it can have any further activating effect. Clearly, that is unrealistic with the knowledge that thrombin exerts its main activity by binding to the surface of platelets.
Bovine versus Porcine Preparations
Bovine lung heparin contains greater amounts of iduronic acid and sulfoamino groups than pork mucosal heparin. Because endothelial endoglycosidases degrade heparin at sulfoamino groups, elimination of beef lung heparin proceeds more quickly than that of pork mucosal heparin. Its AT III affinity and anticoagulant activity are less than those of porcine heparin. Either preparation can establish suitable anticoagulation for CPB. Beef lung heparin may be more amenable to protamine neutralization than the pork mucosal preparation because it exerts less anti–factor Xa activity.110 HIT is less common with pork heparin (see later). This information may be of historic value only because bovine heparin is no longer available in the United States.

Heparin Resistance
In the course of continuous infusions of UFH to treat venous thrombosis, some patients experience development of tachyphylaxis, requiring increasing amounts of heparin to maintain the laboratory measurement of anticoagulation, the aPTT, at its designated therapeutic level. In some reports, up to 22% of patients do not adequately respond to heparin and are termed heparin resistant.111–114 To most practitioners, that number seems high, but the definition of what constitutes heparin resistance is highly variable from institution to institution. Likewise, patients receiving UFH infusions exhibit a much diminished ACT response to full anticoagulating doses of UFH for CPB (200 to 400 U/kg). With widespread use of heparin infusions to treat myocardial ischemia and infarction, heparin resistance or, more appropriately, “altered heparin responsiveness” has become more problematic during cardiac surgery (Box 31-2).115,116
Mechanism
Although several observations suggest that decreased levels of AT III mediate heparin resistance, these observations lack sufficient evidence to establish this relation.116 In one study of 500 CABG patients, 21% demonstrated heparin resistance, and 65% of these responded to added AT III; this translates into 35% being nonresponders.112 First, a patient with congenital AT III deficiency displayed heparin resistance117; this hardly proves that all heparin resistance stems from AT III deficiency. Second, of six patients with venous thrombosis, three receiving heparin infusions displayed a 25% shorter half-life for AT III compared with the three untreated patients.118 Accelerated AT III consumption could have resulted from the thrombotic process rather than the heparin infusion. Third, plasma levels of AT III decreased by 17% to 33% during heparin administration by intravenous or subcutaneous routes.119–124 It is possible that this measurement resulted merely from formation of heparin-AT complexes. Perhaps accelerated elimination of AT III arises from some modification of the protein during or after its interaction with heparin.123 Also, it has been postulated that excesses of platelet activity, releases of PF4, could neutralize heparin in these patients. That has yet to be studied, as has a great deal to do with hypercoagulable states in cardiac surgery.125
Hemodilution accompanying CPB decreases AT levels to about half of normal levels.97 There are, however, outlier patients who have profoundly low AT levels. It is possible to see AT III levels as low as 20% of normal, and these levels correspond to levels seen in septic shock and diffuse intravascular coagulation.15 However, supplemental AT may not prolong the ACT, which means that the heparin available has been bound to sufficient or available AT. The only way that the ACT would be prolonged is if there is excess heparin beyond available AT. Reports of heparin resistance for CPB ascribe its occurrence variously to the use of autotransfusion,124 previous heparin therapy,126–129 infection,130,131 and ventricular aneurysm with thrombus.128,130 The differential diagnosis also includes hypereosinophilia, oral contraceptive therapy, consumptive coagulopathy, thrombocytosis, and congenital AT deficiency.128 Heparin resistance also might occur in patients with subclinical thrombotic processes releasing PF4.
The individual anticoagulant response to heparin varies tremendously.132 Some presumed cases of heparin resistance may represent nothing more than this normal variation. Regardless of cause, measurement of each individual’s anticoagulant response to heparin therapy for CPB is warranted.133 Heparin resistance helps focus the debate regarding whether anticoagulation monitoring should measure heparin concentrations or heparin effect; the goal of anticoagulation is not to achieve heparin presence in plasma but to inhibit the action of thrombin on fibrinogen, platelets, and endothelial cells (see Chapter 17). Therefore, the effect of heparin usually is measured.
Treatment
Most commonly, additional heparin prolongs the ACT sufficiently for the conduct of CPB. Amounts up to 800 U/kg may be necessary to obtain an ACT of 400 to 480 seconds or longer. Although administration of FFP, which contains AT,134 should correct AT depletion (Figure 31-14) and suitably prolong the ACT,135 such exposure to transfusion-borne infectious diseases should be avoided whenever possible. Today, the risks of blood transfusion have shifted away from blood-borne viral transmission, with transfusion-related acute lung injury being noted as the greatest mortality-associated event with transfusion. FFP and platelet transfusions carry the greatest risk for transfusion-related acute lung injury.136,137 Although transfusion-related acute lung injury has not been studied in relation to use of FFP for heparin resistance, it makes great sense to use one of the AT products. This modality is reserved for the rare refractory case. Rather than administer FFP, centers normally accepting only ACTs of 480 seconds or longer for CPB might consider accepting 400 seconds or less, or administering AT III concentrate.135,138
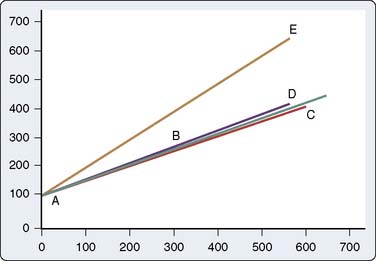
AT concentrate specifically addresses AT deficiency.15–18,134 Two products are available for utilization. One is a recombinant DNA engineered product made from goat’s milk and the other is a purified human plasma harvest derivative. There are currently no head-to-head studies to recommend one over the other at this time. The literature supports success in treating heparin resistance during cardiac surgery.116,139 A multicenter study on the efficacy of using a recombinant human antithrombin in heparin-resistant patients undergoing CPB was published.140 The patients received 75 U/kg recombinant human AT, which was effective in restoring heparin responsiveness in most patients. However, some patients still required FFP, and the patients bled more than did a control group after surgery.

Heparin Rebound
Several hours after protamine neutralization for cardiac surgery, some patients experience development of clinical bleeding associated with prolongation of coagulation times. This phenomenon is often attributed to reappearance of circulating heparin. Theories accounting for “heparin rebound” include late release of heparin sequestered in tissues, delayed return of heparin to the circulation from the extracellular space via lymphatics, clearance of an unrecognized endogenous heparin antagonist, and more rapid clearance of protamine in relation to heparin.141,142 Studies demonstrating uptake of heparin into endothelial cells suggest that these cells may slowly release the drug into the circulation once plasma levels decline with protamine neutralization.92 It is questionable how much heparin rebound contributes to actual bleeding. This phenomenon may be caused by TFPI release from the surface of endothelial cells or other causes of bleeding.
Incidence and Timing
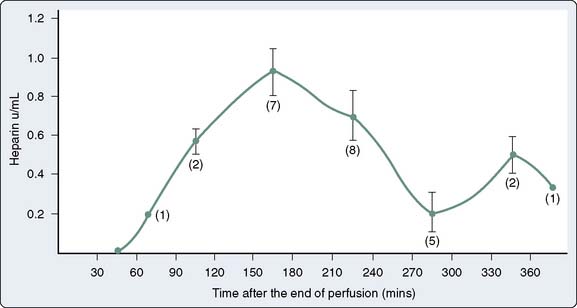
Although initial reports placed the incidence of heparin rebound after cardiac surgery at about 50%, modifications in the timing and amount of protamine administration decreased the incidence.143,144 Heparin rebound can occur as soon as 1 hour after protamine neutralization.127 When present, prolonged coagulation times or more direct evidence of circulating heparin may persist for 6 hours or longer.97,144–147
Treatment and Prevention
Although still debated by a few, most clinicians accept heparin rebound as a real phenomenon. However, clinical bleeding does not always accompany heparin rebound. When it does, administration of supplemental protamine will neutralize the remaining heparin (Box 31-3). Can the initial protamine dose be adjusted to prevent heparin rebound? Available studies yield conflicting results. All six patients who received protamine based on the estimated amount of remaining heparin developed heparin rebound, compared with none of six who received protamine based on the total administered dose of heparin.144 However, 42% of patients receiving large, fixed doses of both drugs experienced heparin rebound and increased bleeding compared with patients whose doses were titrated to clotting assays145 (Figure 31-15). Likewise, patients receiving smaller doses of protamine bled less than those receiving protamine doses based on the total amount of heparin administered.147 In contrast, a fourth study recommended that the ratio of protamine given to heparin remaining be as much as 1.6 mg/100 units.97
Although larger initial doses of protamine may decrease the likelihood of heparin rebound, two potential complications of protamine overdosage must be considered: adverse cardiovascular sequelae of protamine administration and the anticoagulant effects of protamine itself. Although protamine is an in vitro anticoagulant, doses up to 6 mg/kg in volunteers, in the absence of heparin, do not prolong the clotting time.148 However, doses four times in excess of a neutralizing dose doubled the ACT in dogs.149 The dose after CPB that causes anticoagulation in patients remains unknown. Clinical studies comparing fixed-ratio protamine doses with protocols that gauge protamine dose to remaining heparin activity and protamine drug lot potency demonstrated decreased doses of protamine, decreased chest tube drainage after surgery, and fewer transfusions.150,151

Heparin Effects Other Than Anticoagulation
UFH was never biologically intended to circulate freely in plasma.152,153 As such, it has a number of underappreciated and untoward effects. All too often the effects of CPB have been asserted as causing a coagulopathy; however, the effect of heparin contributing to this has not been widely studied. This is because there has not been an alternative anticoagulant to compare with heparin until now. In the future, there may be better anticoagulants to use during cardiac surgery (see later).
Heparin binds AT III during CPB, and through ongoing thrombin generation, the AT III levels are decreased over time, as well as via hemodilution. Thus, the AT III levels may become quite low, in the range seen during disseminated intravascular coagulopathy, septic shock, and eclampsia.15,154–156 AT III is not able to be constitutively increased by production. Therefore, the level at the beginning of a CPB case is all that is available. The liver will manufacture more AT III, but it may take 1 to 3 days to return to normal after cessation of CPB. In at least one study in which AT III was repleted to normal, the levels of coagulation proteins were much improved after CPB in patients who received exogenous AT III.15,134
UFH chelates calcium.157 When a large bolus dose of heparin is given, there is a slow and steady decline in blood pressure, probably because of decreased vascular resistance and decreased preload. Both arterial and venous vessels are dilated by the decrease in the calcium level. The heparin is given while patients are being prepared for CPB, and there are numerous mechanical events (i.e., catheters being inserted into the right atrium and vena cava and arrhythmias) that can be blamed for the hypotension, rather than the heparin itself.
Heparin, even in very small doses, partially and reversibly activates platelets and forces them to express many, if not all, of their GP binding sites.158–160 This fact alone makes clinicians wonder if there is not a better anticoagulant that might not do this. Heparan, the endothelial analog of heparin, does not break off freely into the circulation; and if it does, it is immediately neutralized by platelets through expression of PF4 and adsorption. Thus, it makes sense that platelets seeing loose heparin would suspect a site of tissue injury nearby and, therefore, an evolutionary advantage would be created by making these cells react and get ready to create a thrombus. Every coagulation activation is also an inflammatory signal. The fact that platelets take this reactive step means that they are now primed to either become highly reactive or contribute to the inflammatory events, or have their receptors targeted by other subsequent events. After CPB, platelets have many of their membrane GPs either destroyed or competitively occupied by a number of products of inflammation produced during CPB. The expression of binding sites in response to heparin, therefore, is important and probably has profound implications.
Heparin causes the competitive release of some heparan from endothelial cells and the release of TFPI.25,26 Endothelial cells all over the body change from being anticoagulant producing to rapidly producing tissue factor. What role large doses of heparin have in this whole-body event is hard to define, but it is known that heparin also causes the release of single-chain urokinase from endothelial cells.161 The amount of fibrinolysis caused by heparin infusion is not as great as the release of t-PA, which probably is mediated by cytokines as a result of the inflammatory reactions. When released from mast cells, heparin promotes leukocyte chemotaxis and movement through the interstitium.152,153 However, it is unclear whether heparin upregulates or decreases white cell activations.153
Heparin is important for a number of angiogenesis and repair activities of tissue,153 and these effects may have something to do with its antineoplastic effect.153 Heparin also affects lipid, sodium and potassium, and acid-base metabolism. These effects are not usually seen acutely but come into play when patients have been on heparin infusions for days in the ICU.
The immunologic effects of heparin are profound. The next section discusses HIT, but recent work shows that 30% to 50% of cardiac surgery patients have heparin antibodies present in their blood by the time of hospital discharge.162 The clinical implications of these prevalent antibodies remain unknown and are the subject of investigation.

Heparin-Induced Thrombocytopenia
Heparin normally binds to platelet membranes at GPIb and other sites, and aggregates normal platelets by releasing ADP.162,163 A moderate, reversible HIT, now termed type I, has been known for half a century.164 The fact that heparin actually triggers an acute decline in platelet count should be considered a biologic event, because heparin, even in trace amounts, triggers the expression of many different platelet GPs. This has been termed “activation of platelets,” but it is not total activation. Heparin’s prolongation of the bleeding time probably is related to activation of the platelets, as well as heparin binding to the GPIb surface. It may be that a number of platelets adhere to endothelial cells simply because of their expression of these GP binding sites. Margination, particularly within the pulmonary vasculature, may be an event of HIT type I.
Mechanism
These patients with HITT demonstrate a heparin-dependent antibody, usually IgG, although others are described, which aggregates platelets in the presence of heparin.161,162 During heparin therapy, measured antibody titers remain low because of antibody binding to platelets. Titers rise after heparin therapy ceases; but paradoxically, antibody may be undetectable a few months later.165 Two other features are unexpected: First, the antibody does not aggregate platelets in the presence of excess heparin; and second, not all re-exposed patients experience development of thrombocytopenia.166–168
The platelet surface contains complexes of heparin and PF4. Affected patients have an antibody to this complex. Antibody binding activates platelets via their FcγII receptors and activates endothelium169–171 (Figure 31-16). The activation of the platelet surface triggers a secondary thrombin release. Platelets can attach to each other creating what is known as a white-clot syndrome; but if secondary thrombin generation is created through antibody activation of the platelets, then a fibrin clot can be the result. In the absence of heparin, the heparin-PF4 antigen cannot form. However, there seems to be some sort of continuum between idiopathic thrombocytopenia and HIT.
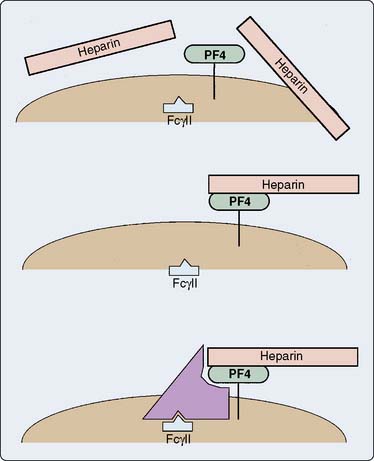
Figure 31-16 Presumed mechanism of the interaction among heparin, platelets, and antibody in heparin-induced thrombocytopenia.
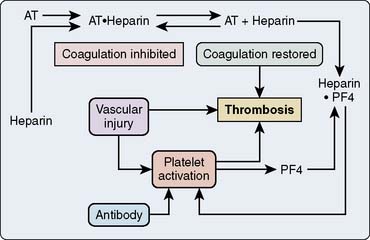
In the absence of an endothelial defect, the only responses to the antibody-antigen interaction are platelet consumption and thrombocytopenia. Atheroma rupture, endovascular interventions such as balloon angioplasty, vascular surgery, and other procedures that disrupt endothelium can provide a nidus for platelet adhesion and subsequent activation. PF4, released with platelet activation, binds to heparin locally, thus not only removing the inhibition of coagulation but also generating additional antigenic material (Figure 31-17). Clumps of aggregated platelets thrombose vessels, resulting in organ and limb infarction. Amputation, death, or both often occur with established HIT with thrombosis (HITT). The presence of heparin-PF4 antibodies recently has been associated with other adverse effects. It appears that if a patient undergoes cardiac surgery with positive antibodies, the risk for mortality or myocardial infarction, or both, may at least double.
Incidence and Diagnosis
Estimates of the true incidence of HIT are confounded by different diagnostic thresholds for platelet count, varying efforts to detect other causes, and incomplete reports.172,173 After 7 days of therapy with UFH, probably 1% of patients experience development of HIT; after 14 days of therapy, the prevalence rate is 3%.171 Using a platelet count of 100,000/mm3, multiple reports comprising more than 1200 patients revealed an overall incidence rate of HIT of 5.5% with bovine heparin and 1.0% with porcine heparin.161 Other recent research has found the preoperative incidence rate of enzyme-linked immunosorbent assay (ELISA)–positive patients to be between 6.5% to 10%. This means that antibodies are present, and that may not mean that thrombocytopenia is occurring. Of great interest is that many more patients develop positive tests for ELISA antibodies by days 7 to 30 after cardiac surgery. Somewhere between 25% and 50% of patients develop these antibodies.162 In a study of patients after cardiac surgery wherein all patients were screened for HIT antibodies, the group also looked for platelet counts well less than the patient’s baseline174; 21 of 153 patients (14%) tested positive for antibodies by heparin induced platelet activation (HIPA) testing. Those patients with a low platelet count and a high HIT antibody titer after surgery were at very high risk for mortality (59%). Therefore, a decline in platelet count or persistence of a low platelet count after cardiac surgery should be considered HIT until proved otherwise and taken very seriously. Anticoagulation with alternative nontriggering anticoagulants is imperative.174 Warfarin should be avoided until such time as the platelet count is recovered and vitamin K therapy can be instituted; this is because the use of warfarin compounds will decrease protein C, which has been known to trigger an HITT crisis.
In the bivalirudin trials, wherein patients thought to have HIT were given an alternative anticoagulant, the presence of antibodies after surgery was associated with adverse events. Particularly worrisome were the presence of a low platelet count or a blunted return toward a normal platelet count after surgery.175–177
Some particular lots of heparin may be more likely to cause HIT than others.178 HIT can occur not only during therapeutic heparin administration but with low prophylactic doses, although the incidence is dose related. Even heparin flush solution or heparin-bonded intravascular catheters can incite HIT.179–182 Cases of platelet-to-platelet adhesion creating a “white clot” in otherwise normal patients have been observed in the oxygenator and the reservoir of CPB machines. The fact that such events have been reported even when all other tests appeared normal signals the unpredictable nature of the heparin-PF4 antibody, as well as the biologic activity of UFH.
Although HIT usually begins 3 to 15 days (median, 10 days) after heparin infusions commence, it can occur within hours in a patient previously exposed to heparin. Platelet count steadily decreases to a nadir between 20,000 and 150,000/mm3. Absolute thrombocytopenia is not necessary; only a significant decrease in platelet count matters, as witnessed by patients with thrombocytosis who experience development of thrombosis with normal platelet counts after prolonged exposure to heparin. Occasionally, thrombocytopenia resolves spontaneously despite continuation of heparin infusion.183
Clinical diagnosis of HIT requires a new decrease in platelet count during heparin infusion. Laboratory confirmation is obtained from several available tests. In the serotonin release assay, patient plasma, donor platelets, and heparin are combined. The donor platelets contain radiolabeled serotonin, which is released when donor platelets are activated by the antigen-antibody complex. Measurement of serotonin release during platelet aggregation at both low and high heparin concentrations provides excellent sensitivity and specificity.166
A second assay measures more traditional markers of platelet degranulation in a mixture of heparin, patient plasma, and donor platelets.184 The most specific test is an ELISA for antibodies to the heparin-PF4 complex.160,161,185,186

Heparin-Induced Thrombocytopenia with Thrombosis
The incidence rate of HITT is 1.7% with bovine heparin and 0.3% with porcine heparin; thus, thrombosis accompanies more than one in five cases of HIT.161,162 It is clear that the longer patients are on heparin, the more likely it is that they will develop antibody; and with the knowledge that today close to 50% of cardiac patients develop antibodies, it is possible that a significant number of long-term or early mortalities might be because of undiagnosed HITT.162 In several studies in the catheterization laboratory, it has been shown that if HITT antibodies are present before the performance of angioplasty, the mortality and combined morbidity are greatly increased, perhaps double or more.187,188 One study has been conducted in almost 500 patients undergoing CABG surgery looking for the presence of antibodies and outcome. The incidence rate of antibody-positive patients was approximately 15%, and their length of stay in the hospital and mortality were more than doubled. Occasional rare situations in which the CPB circuit suddenly clots or when there is early graft thrombosis or whole-body clotting may all be variants of HITT, but none of these cases can be readily studied because they are so rare.187 If such an occurrence does happen, HITT should be in the differential diagnosis. The occurrence of thrombosis at first seems paradoxic. However, HITT has as its hallmark a huge thrombin burst that can occur all over the body. With such massive thrombin generation, the triggering of thrombosis is natural. Thrombosis may then activate the fibrinolytic system to produce a picture of consumptive coagulopathy.179
Warkentin and others189 have stressed the use of the 4Ts system (Thrombocytopenia, Timing, Thrombosis, and oTher [lack of other reasons]) to increase the index of suspicion with regard to HIT. The use of this scoring system is quite effective, but after cardiac surgery, the decline in platelet count is expected for at least 24 hours. If platelet count stays down after that time, then a high index of suspicion should be for HIT. A study of critically ill patients (noncardiac) using the 4Ts showed an incidence rate of 4.1% for HIT. Low scoring appears to be reliable, whereas high and intermediate scoring showed patients with antibody-positive tests.

Treatment and Prevention
In the absence of surgery, bleeding from thrombocytopenia with HIT is rare. In contrast with other drug-induced thrombocytopenia, in which severe thrombocytopenia commonly occurs, more moderate platelet count nadirs characterize HIT. Platelet transfusions are not indicated and may incite or worsen thrombosis. Heparin infusions must be discontinued, and an alternative anticoagulant should be instituted. LMWHs can be tested in the laboratory using serotonin release before patient administration. Although thrombosis may be treated with fibrinolytic therapy,190 surgery often is indicated. No heparin should be given for vascular surgery. Monitoring catheters should be purged of heparin flush, and heparin-bonded catheters should not be placed. Antiplatelet agents, such as aspirin, ticlopidine, or dipyridamole, which block adhesion and activation and, thus, PF4 release, provide ancillary help (see Table 31-4).
The patient presenting for cardiac surgery who has sustained HIT in the past presents a therapeutic dilemma. Antibodies may have regressed; if so, a negative serotonin release assay using the heparin planned for surgery will predict that transient exposure during surgery will be harmless. However, no heparin should be given at catheterization or in flush solutions after surgery.162,163
Patients with HIT who require urgent surgery may receive heparin once platelet activation has been blocked with aspirin and dipyridamole156–193 or, in the past, the prostacyclin analog iloprost.194–197 Unfortunately, iloprost is no longer available. The problem with this strategy is obtaining sufficient blockade of platelet activity. In the future, there may be ultra-short-acting platelet surface blocking agents available, and these would then seem perfect to create “platelet anesthesia.”
Another alternative, delaying surgery to wait for antibodies to regress, may fail because of the variable offset of antibody presence and the unpredictable nature of platelet response to heparin rechallenge. Plasmapheresis may successfully eliminate antibodies and allow benign heparin administration.198 Finally, methods of instituting anticoagulation without heparin may be chosen (see later). Of these, the alternative thrombin antagonists pose the greatest risk for uncontrolled bleeding, whereas LMWHs and heparinoids afford the greatest chance of success.199,200
LMWH heparin, as an alternative to UFH, has been used for urgent surgery.201–203 Although LMWHs also can induce thrombocytopenia,204–206 by displaying different antigenic determinants, they may prove acceptable alternatives for patients who experience development of HIT from UFH. Table 31-6 summarizes the therapeutic options available for urgent cardiac surgery in patients with HIT. For additional information on HIT, see reviews by Warkentin and Greinacher,162 Godal,163 and Chong.207
TABLE 31-6 Therapeutic Options for Anticoagulation for Bypass in Patients with Heparin-Induced Thrombocytopenia
No agent is currently indicated for anticoagulation in cardiopulmonary bypass.
RGD, receptor glycoprotein derived.

New Modes of Anticoagulation
The hemostatic goal during CPB is complete inhibition of the coagulation system. Unfortunately, even large doses of heparin do not provide this, as evidenced by formation of fibrinopeptides during surgery.107–109 Despite being far from the ideal anticoagulant, heparin still performs better than its alternatives. Heparans, dermatans, and other glycosaminoglycans with minimal antihemostatic properties may replace heparin in the future. Current substitutes for heparin include ancrod, a proteinase obtained from snake venom that destroys fibrinogen; heparin fragments, which provide less thrombin inhibition than the parent, unfractionated molecule; direct factor Xa inhibitors; and direct thrombin inhibitors (Box 31-4).
Ancrod
Ancrod abnormally cleaves fibrinogen, resulting in its rapid clearance by the reticuloendothelial system. Thrombin, thus, has no substrate on which to act. Proper patient preparation for CPB (plasma fibrinogen, 0.4 to 0.8 g/L) requires more than 12 hours. Figure 31-18 demonstrates the extent of fibrinogen depletion (from normal value of 1.5 to 4.5 g/L) and repletion for cardiac surgery using ancrod.208 Replenishment of fibrinogen via hepatic synthesis is slow; cryoprecipitate or FFP administration, or both, will speed restoration of coagulation. Patients anticoagulated in this fashion bleed more and require more cryoprecipitate and FFP compared with heparin-anticoagulated patients.208 One case was done with ancrod in which the coagulation activation was carefully studied.209 Unbridled thrombin production led to massive platelet activation with a secondary precipitous decline in platelet count. No clot formed, but a gray slime of platelets adhered to the wall of the oxygenator and the reservoir. The platelet count declined to less than 1000/mm3. However, transfusion with platelet concentrates and cryoprecipitate reestablished normal coagulation, and the patient had less than 500 mL of blood loss for the first 24 hours. No neurologic deficits occurred. However, ancrod is not commercially available in the United States.209
Low-Molecular-Weight Heparins
Figure 31-19 displays the effect of polysaccharide chain length on the inhibition of thrombin and factor Xa. Note that thrombin inhibition requires chains longer than 18 saccharide units and aPTT activity follows anti–factor IIa activity more closely than it does anti–factor Xa activity.103 Thrombin must bind to a portion of the heparin chain for AT to inhibit it. In contrast, factor Xa inhibition by AT does not require interaction of factor Xa with the heparin molecule. Only about 1% to 2% of standard heparin consists of low-molecular-weight (molecular weight, 6000 to 7000) fragments.93 Short polysaccharide chains of heparin can be synthesized, or extracted from standard heparin, but both processes cost much more than depolymerization of standard heparin utilizing nitric acid, peroxides, or the enzyme heparinase. Preparations of LMWH formed by these methods include Fraxiparine, dalteparin, and enoxaparin.
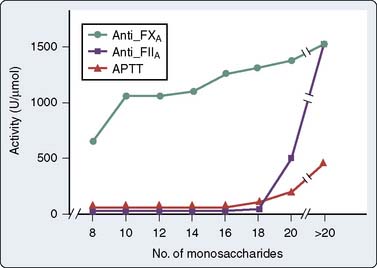
Figure 31-19 Heparin activity as a function of chain length.
(From Holmer E, Soderberg K, Bergqvist D, Lindahl U: Heparin and its low-molecular-weight derivatives. Anticoagulant and antithrombotic properties. Haemostasis 16[Suppl 2]:1, 1986.)
Standard heparin can inhibit thrombus formation in vivo (antithrombotic activity) and prolong in vitro clotting tests (anticoagulant activity). It also leads to clinical bleeding (antihemostatic activity). Table 31-7 highlights these definitions. LMWHs may dissociate these activities, displaying greater antithrombotic activity and less antihemostatic activity.93 Unfortunately, coagulation tests sensitive to thrombin inhibition and insensitive to inhibition of factor Xa, namely, the aPTT and ACT (see Figure 31-19), do not adequately monitor the antithrombotic effects of LMWHs. For additional information regarding LMWHs, there are several good reviews.210–212
* Properties of an ideal agent.
LMWHs have undergone clinical trials for antithrombosis after orthopedic procedures and for prevention of deep vein thrombosis.213–215 The traditional LMWHs, such as enoxaparin, have not been used for CPB cases because of a limited ability of protamine to neutralize them.216,217
Heparinoids
Danaparoid (Orgaran), a mixture of LMWHs and dermatans, can provide anticoagulation for CPB, but lack of readily available monitoring and sure neutralization limit its application to cases in which UFH clearly is contraindicated.217,218 Dermatans alone might prove suitable, but much more investigative work must first occur.7,218 Case reports of these agents have shown severe bleeding and, in some cases, death from hemorrhage after cardiac surgery. It appears at this point that dermatan sulfate will not be used for cardiac surgery, and danaparoid was removed from the U.S. market.
Fondaparinux (Arixtra) is a synthetic pentasaccharide identical to that in heparin. It is a primary factor Xa inhibitor that requires AT III, which does not affect thrombin, platelets, or fibrinolytic activity. It is being used for prophylaxis of deep vein thrombosis after surgery, but it does not alter routine coagulation tests (requires a factor Xa assay).219 Since 2008, there have been some reports of patients with HIT not responding successfully to therapy with fondaparinux.220,221 Supratherapeutic dosages for fondaparinux were required to inhibit binding of antibodies to platelets. Danaparoid in very low concentrations increased PF4 antibodies; however, in therapeutic concentrations, it decreased production of antibodies.221 There is no direct antidote for either of these agents, but patients may respond to factor VIIa (see later).
Direct Thrombin Inhibitors
Hirudin depends on renal excretion; renal failure prolongs its elimination half-life of 0.6 to 2.0 hours. Although there are no known direct neutralizing agents for these drugs, administration of prothrombin complex may partially restore coagulation by enhancing thrombin generation. Clinical trials of hirudin compounds have yielded mixed results. It has been used for patients with HITT, but the longer half-life of approximately 90 minutes means that many of these patients bleed after cardiac surgery.222 Hirudin is highly antigenic and will lead to immune complexes being created to itself in about 40% of patients. If it is used a second time, the overall incidence rate of anaphylaxis may be as high as 10% of all patients who have received it before.222 Currently, it is not recommended to turn to hirudin as a primary agent to perform CPB even if a patient has HIT antibodies.
New direct thrombin inhibitors are now available (Figure 31-20). These include argatroban and bivalirudin. Argatroban is a derivative of l-arginine, is a relatively small molecule, and functions as a univalent direct thrombin inhibitor.223,224 It binds at the active cleavage site of thrombin and stops thrombin’s action on serine proteases. It is completely hepatically cleared and has a reported half-life of 45 to 55 minutes with prolongation when liver function is depressed or liver blood flow is decreased. One case report examined argatroban’s half-life after CPB. It was found to be prolonged to 514 minutes in a patient undergoing heart transplantation.225 This led to the use of massive transfusion (55 units of RBCs, 42 units of FFP, 40 units of cryoprecipitate, 40 units of platelets, as well as 3 doses of factor VIIa).
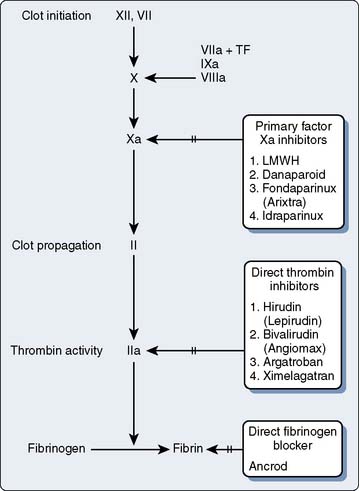
There is no reversal agent for argatroban, although factor VIIa has been given to increase thrombin generation. It has been U.S. Food and Drug Administration (FDA)–approved for anticoagulation in the face of HITT, but there has not been, to date, a large-scale, prospective, randomized trial for cardiac surgery or any type of comparison with heparin/protamine. Some case reports do exist of successful usage of argatroban in patients with HITT both on- and off-pump with acceptable amounts of postoperative bleeding.226–230 The dosing for off-pump cases has been reported to be about 2 to 3 μg/kg/min, with a goal of an ACT longer than 200 seconds. For on-pump cases, the dose is at least doubled (5 to 10 μg/kg/min), with an effort to achieve an ACT of 300 to 400 seconds.229 There is no safe and effective dose that has been clinically studied, so any use of this drug for cardiac surgery is off-label and based on experience, as well as case reports. Successful case completions have been noted without undue excess bleeding. However, as noted earlier in the transplant case, some very large bleeds have been encountered. Some cases of thrombosis also have occurred.231–236 It has been more commonly used in the ICU for patients with hypercoagulable syndromes and HITT.237,238 Argatroban is a viable direct thrombin inhibitor for use in the cardiac catheterization laboratory. It is far more likely that anesthesiologists will encounter the drug either in the ICU being used for patients with HIT or in patients who have come from the catheterization laboratory directly to the operating rooms.
Bivalirudin is a bivalent synthetic 20-amino acid peptide based on the structure of hirudin (previously called Hirulog).239–248 Pharmacologists have taken the active amino acids at either end of the hirudin molecule and biosynthesized them. One active site competitively binds to the fibrinogen-binding site of thrombin; and the other end of the molecule, the amino-terminal sequence, binds to the active serine cleavage site of thrombin. The two sequences of amino acids are connected together by a tetraglycine spacer. This fully manufactured molecule is highly specific for thrombin and has the unique property that it binds to both clot-bound and free thrombin. Heparin binds only to free plasma thrombin. Bivalirudin has a shorter half-life than argatroban and hirudin; the t1/2 is approximately 20 to 25 minutes (with normal renal function and not on CPB). One of the most unique features of bivalirudin is that its binding to thrombin is reversible and the molecule itself is cleaved by thrombin.
Like the other direct thrombin inhibitors, it also has no reversal agent analogous to protamine; so when it is used, it must wear off. Bivalirudin undergoes destruction by the molecule to which it binds and deactivates, thrombin; it is destroyed by thrombin (proteolytic cleavage). The more thrombin activation that is present (i.e., the less bivalirudin that is present), the shorter is the half-life. Only about 20% of the molecular activity is eliminated by renal clearance.240 In mild- to-moderate renal failure, the effect on bivalirudin clearance is thought to be small, but bleeding in patients with renal failure has been noted.
Several clinical trials of bivalirudin for cardiology procedures or cardiac surgery have been completed and published.240–248 Two pivotal trials aiming for FDA approval of bivalirudin for cardiac surgery with known/suspected HIT were conducted several years ago.175,176 In trials comparing bivalirudin with either heparin/protamine alone or heparin plus the use of a GP IIb/IIIa inhibitor for percutaneous interventions, bivalirudin was found to have at least equal or better safety and less bleeding than either of the other therapies. When compared with heparin/protamine alone in percutaneous coronary intervention, bivalirudin was found to be superior, not just in bleeding, but also in terms of morbidity and mortality (as a combined end point).247 In a trial of 100 off-pump routine CABG patients without suspected HIT, patients were randomized to receive either bivalirudin or heparin/protamine, and bleeding and outcome were equal between the groups.240 These patients underwent recatheterization at 3 months, and it was found that the bivalirudin patients had overall better flow down their grafts than did the patients who had received heparin/protamine. That finding is consistent with the work noted in the HIT section earlier wherein the presence of antibodies bodes poorly for postoperative complications. It also suggests that heparin-protamine in the face of ischemia and reperfusion may lead to adverse thrombotic outcomes with or without heparin-PF4 antibodies. This trial, in conjunction with subsequent trials, raises questions with regard to heparin-protamine and suggests that for off-pump CABG, a more routine usage of bivalirudin might produce improved outcomes.
A phase I/II safety trial of bivalirudin in 30 on-pump CABG patients has also shown good safety, but no comparison was conducted to look at advantages against heparin/protamine. When used, the doses for CPB have been a 0.50- to 0.75-mg/kg bolus followed by an infusion at 1.75 to 2.5 mg/kg/hr titrated to the ACT (target, 2.5 times baseline). The CPB system also was primed with 50 mg, and no stasis can be allowed in the CPB circuit because of metabolism of bivalirudin during CPB. The infusion is stopped about 15 to 30 minutes before CPB is discontinued, and patients bleed for up to 4 to 6 hours. OPCAB cases have used similar doses to ACT targets of 350 to 450 seconds.240 There certainly are some tricks to using bivalirudin for cardiac cases. The drug itself is broken down by thrombin, and thrombin is produced by CPB, as well as through tissue destruction. Any blood left alone without a continuous infusion of bivalirudin will, because of its generation of thrombin, overcome the anticoagulation of bivalirudin in time. Therefore, it is expected that stagnant blood in the mediastinal or the chest cavities, or both, will clot. This is alarming to the first-time user of bivalirudin and completely different from what is seen in cases with heparin anticoagulation. Also, the use of mediastinal suction during bypass is not recommended because the mediastinum is a source of a great deal of thrombin activity. Suctioning that back into the CPB reservoir has led to clots being present in a hard-shell reservoir wherein there is stasis or incomplete mixing of bivalirudin. Once the patient is separated from CPB, it is important to make a decision regarding whether the patient is likely to need to return to bypass. The bypass system, if left stagnant, will have ongoing production of thrombin. Over time, that thrombin will overcome the bivalirudin present in the plasma. Therefore, within 10 minutes of separation from CPB, it is wise to decide to either drain the blood from the pump, process it through a cell-saver machine, or reestablish flow and have a slow infusion of bivalirudin into the pump. The reestablishment of flow can be easily accomplished by reattaching the ends of the venous and arterial cannulae. If it is necessary to reestablish CPB, the system should be maintained warm and either a bolus (25 to 50 mg) of bivalirudin should be put into the pump or the infusion that had been running to the patient should be switched to the pump.241 Furthermore, some surgeons have suggested that in areas of stasis, such as in an internal mammary artery, it is important to flush the artery every 10 to 15 minutes to allow for new bivalirudin to be perfused, or clot could build up in the “dead end” if it is clamped. The other option is to not completely clamp off the internal mammary artery until just before it is to be anastomosed.
There has been some confusion regarding how best to monitor anticoagulation with bivalirudin for cardiac surgery. Originally, Koster et al 222 utilized the ecarin clotting time (ECT) to follow circulating levels of bivalirudin. The ECT has a straight-line dose-response relation in the critical range between 300 and 550 seconds. The ACT has a less specific and more variable relation. With circulating bivalirudin concentrations of 10 to 15 μg/mL, the ECT will be 400 to 450 seconds. It is known that clot will not be able to be generated with a bivalirudin concentration greater than 3 to 5 μg/mL, so a level of 10 to 12 will be well greater than a therapeutic threshold to ensure that fibrin formation does not occur on bypass. The dose-responsiveness of bivalirudin is highly predictable. There is no secondary reaction necessary such as with AT III and UFH. Therefore, when bivalirudin is given, there is an absolute amount of AT available. Debates among researchers have gone forward as to whether ACT or ECT monitoring is even necessary at all. The consensus is that ACT will work (the ECT is no longer commercially available). The other reason for using an ACT is that during CPB, if a drug pump malfunctions or the infusion is somehow disconnected, it is important to know that earlier rather than later. If the ACT begins to elevate to more than 500 seconds, then the team really does not know whether to back off on the bivalirudin infusion, stop it altogether, or attribute the effect to some other ACT-prolonging situation such as hemodilution or hypothermia. It is known that hypothermia retards the production of thrombin, but no studies have been done of bivalirudin half-lives in the face of mild-to-moderate hypothermia.
The two trials of bivalirudin in the face of known or suspected HIT antibodies did show effectiveness and safety.176,177 The CABG Hit On and Off-pump Safety and Efficacy (CHOOSE) and Evaluation of patients during coronary artery bypass Operation: Linking UTilization of bivalirudin to Improved Outcomes and New anticoagulant strategies (EVOLUTION) trials were performed as parts of a program to get bivalirudin approved for patients undergoing cardiac surgery with known or suspected HIT. EVOLUTION (ON and OFF) trials randomized patients to receive either heparin-protamine or bivalirudin as the primary anticoagulant regimen for either on- or off-pump CABG surgery. In EVOLUTION-OFF, 157 patients were scheduled for OPCAB at 21 centers. The dosing of bivalirudin was 0.75 mg/kg as a bolus and 1.75 mg/kg/hr while the grafts were being prepared and anastomosed. Heparin was dosed to reach an ACT target of 300 seconds and reversed with protamine. There were no differences in death, myocardial infarction, or need for repeat revascularization. However, there was a significant reduction in strokes seen with the use of bivalirudin. Bleeding was about the same with both groups. In the EVOLUTION-On trial, 150 patients underwent a number of cardiac procedures at the 21 sites. There were, again, no major differences in procedural success (death, MI, need for redo) in the heparin versus bivalirudin groups. There was a statistically, but clinically insignificant, difference (78 mL) in blood loss at 2 hours and not at 24 hours with bivalirudin.
The CHOOSE trials undertook to use bivalirudin in patients with known or suspected HIT. In the CHOOSE-On trial, 50 patients were enrolled. Immediate success of surgery was achieved in 94% of patients (treated with bivalirudin); at 30 days, success was 86%, and at 12 weeks that had decreased to 82%. Unfortunately, only historic controls could be examined to compare, and conjecture comes into play as to what might happen in such a high-risk group with known/suspected HIT. Bleeding and transfusion were greater in patients who received bivalirudin. In a single German center with a large experience using bivalirudin, 40 patients had heparin antibodies.242 These investigators noted that their procedural success rate was 99.4%; however, they did have an increased use of transfusion in those patients who received bivalirudin.
In the face of HITT syndrome, case reports continue to show effectiveness and utility of bivalirudin.244,245,248 This is an off-label use of the drug because it has not been FDA approved. In animal studies, bivalirudin does not activate platelets, and those animals that have received the drug for CPB have a better platelet count at the end of surgery. Other inflammatory mediators such as cytokines also may be decreased with bivalirudin administration as compared with heparin/protamine administration. Heparin, even in small doses, activates platelets to express their binding sites, whereas bivalirudin seems to leave the platelets quiescent. It also has no cross-reactivity with any immunoglobulin that is present to heparin/PF4 and does not produce an immune response of its own, which can be seen with hirudin.
The latest direct thrombin inhibitors being developed are for long-term oral use. Ximelagatran was in active phase III clinical trials, but they have been stopped. This is a prodrug that is rapidly biotransformed into the active drug Melagatran. The drug produces a predictable anticoagulant response, and no laboratory monitoring is required. This drug may be able to replace warfarin, which is a difficult drug to manage. Unfortunately, the FDA turned down ximelagatran in 2004 because of increased ALT levels in 6% of patients. It is approved in Europe for short-term use, and the FDA will reevaluate it after further clinical trials.249–252

Nonthrombogenic Surface
Considered the “Holy Grail” of extracorporeal circulation, an artificial surface that does not incite thrombus formation remains undiscovered. The endothelial surface, which the artificial one should mimic, performs a host of biochemical functions related to antithrombosis. Two that appear crucial are (1) secretion of substances that inhibit both platelet activation and aggregation and (2) surface and matrix adsorption of heparin and heparan, which may locally potentiate AT.253
Heparin may be immobilized onto surfaces by ionic bonding onto cationic surfactants. Unfortunately, both heparin and surfactant leach off the coated surface on exposure to blood. Covalent binding or surface grafting of heparin provides a more stable preparation. A properly heparin-bonded surface should bind AT sufficiently to prevent fibrinogen-induced platelet adhesion. Heparin-coated CPB tubing reduces the need for systemic heparin but has not been sufficiently effective to replace systemic anticoagulation.254–256 Other approaches include increasing surface hydrophilicity with polyethylene oxide and sequestration of the thrombogenic surface by endothelialization, albumin activation, or phospholipid mimicking.257 New materials might permit cardiac surgery with minimal doses of heparin or even without it (see Chapter 29).