22 Uncommon Cardiac Diseases
Cardiac tumors
Cardiac tumors increasingly are diagnosed before autopsy because of advancements in imaging, especially metastatic tumors of the heart and pericardium that account for a majority of cardiac tumors. Data pooled from 22 large autopsy series show the prevalence rate of adult primary cardiac tumors is 0.02% but causes considerable morbidity and mortality.1 Primary cardiac tumors in adults are usually benign, with only 20% to 25% malignant.2–5 Diagnosis can be elusive because these tumors may be associated with nonspecific symptoms mimicking other disease entities. Two-dimensional (2D) echocardiography (echo) modalities and magnetic resonance imaging (MRI) have allowed earlier, more frequent, and more complete assessment of cardiac tumors.6,7 The advent of three-dimensional (3D) echo increases the confidence of the diagnosis of a cardiac mass by giving more detailed characterization of mass size, composition, location, and relation to other structures.8
Primary cardiac tumors may originate from any cardiac tissue. Myxoma is the most common cardiac neoplasm, accounting for nearly 50% of tumors in adults (Table 22-1). Less frequently observed benign tumors that may require surgery include rhabdomyoma, fibroma, papillary fibroelastoma, lipoma, and angioma. Malignant primary cardiac tumors include sarcomas that are 95% of these tumors, followed by lymphoma. Sarcomas include angiosarcoma, rhabdomyosarcoma, and acquired immunodeficiency syndrome (AIDS)–related sarcomas.9 Rhabdomyosarcoma represents 20% of malignant neoplasms. Excision of these tumors seldom is curative, in part because of the delay in diagnosis caused by the nonspecific nature of the clinical symptoms. Surgery, radiation therapy, and chemotherapy may slow a tumor’s encroachment on intracavitary spaces or relieve obstruction.
| Neoplasms | Incidence Rate in Adults (%) |
|---|---|
| Myxoma | 45 |
| Lipoma | 20 |
| Papillary fibroelastoma | 15 |
| Angioma | 5 |
| Fibroma | 3 |
| Hemangioma | 5 |
| Rhabdomyoma | 1 |
| Teratoma | < 1 |
From Shapiro LM: Cardiac tumors: Diagnosis and management. Heart 85:218, 2001.
The incidence rate of metastatic cardiac tumors has increased from 0.2% to 10% as a result of improved survival.10 Metastatic cardiac tumors are much more common than primary cardiac tumors. Adenocarcinomas of the lung and breast, lymphomas that are commonly associated with AIDS or transplant immunosuppression, and melanoma are the most frequent metastatic cardiac tumors.11 Melanoma has a special tendency for metastasis to the heart and pericardium. However, metastasis of these tumors rarely is limited to the heart. The onset of arrhythmias or congestive heart failure (CHF) in patients with carcinomas suggests invasion of the myocardium by metastasis, but more than 90% of metastatic lesions to the heart are clinically silent.4
The most effective treatment of primary tumors generally is surgical resection, with 2% operative mortality. This is based on a recent retrospective study of 323 consecutive patients who underwent surgical resection of primary cardiac tumors over a period of 48 years in one institution.12 Recurrence rate in these tumors varied between 3% and 13%, but appeared to be related to a biologic propensity rather than surgical technique, as was previously believed. The overall rate of tumor embolization was 25% compared with previous reports of 12% to 45%. Embolic complications were seen more often in patients with minimal or no symptoms than those with large tumors associated with hemodynamic changes. Papillary fibroelastoma and aortic valve tumors are most commonly preceded by an embolic event. Even patients who are diagnosed by embolic events benefit greatly from surgical resection with excellent short- and long-term survival that is comparable with a cohort of patients with tumors undergoing surgery for other reasons.7 Orthotopic cardiac transplantation has been recommended for unresectable tumors,13 but the benefit is indeterminate. Although more infrequent, the surgical risk and outcome for malignant compared with benign tumor resection are usually much worse,2 especially in younger patients.12

Myxoma
Often a diagnostic challenge, myxoma, a benign, solitary neoplasm slowly proliferating, microscopically resembles an organized clot, which often obscures its identity as a primary cardiac tumor. The pedunculated mass is believed to arise from undifferentiated cells in the fossa ovalis and adjoining endocardium projecting into the left atrium (LA) and right atrium (RA) 75% and 20% of the time, respectively. However, myxomas appear in other locations of the heart, even occupying more than one chamber.14 The undifferentiated cells of a myxoma develop along a variety of cell lines, accounting for the multiple presentations and pathologies observed.15 Besides a variable amount of stroma, myxomas include hemorrhage, hemosiderin, thrombus, and calcium (Figure 22-1). Myxomas predominate in the 30- to 60-year-old age range, but any age group may be affected. More than 75% of the affected patients are women.16 Although most cases occur sporadically, 7% to 10% of atrial myxomas will occur in a familial pattern with an autosomal dominant transmission pattern.17
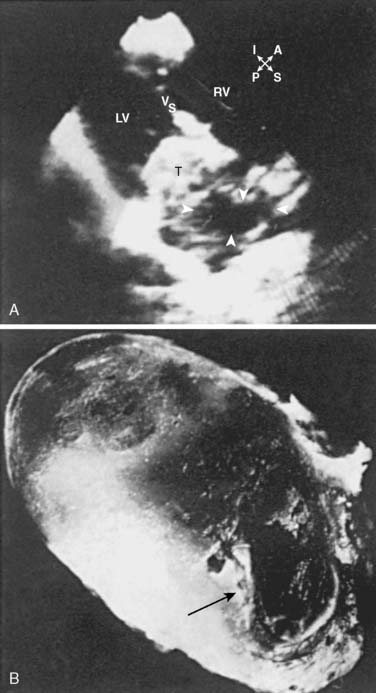
Rarely discovered by incidental echo examination, myxomas may manifest a variety of symptoms. The classic triad includes embolism, intracardiac obstruction, and constitutional symptoms. Approximately 80%14 of individuals will present with one component of the triad. Up to 10% may be asymptomatic even with mitral myxomas that arise from both atrial and ventricular sides of the anterior mitral leaflet.18 The most common initial symptom, dyspnea on exertion,16 reflects mitral valve obstruction usually present with left atrial myxomas (Figure 22-2). Because of the pedunculated nature of some myxomas, temporary obstruction of blood flow may cause hemolysis, hypotension, syncope, or sudden death. Other symptoms of mitral obstruction similar to mitral stenosis such as hemoptysis, systemic embolization, fever, and weight loss also may occur. If the tumor is obstructing the mitral valve, a “tumor plop” may be heard after the second heart sound on chest auscultation. The persistence of sinus rhythm in the presence of such symptoms may help distinguish atrial myxoma from mitral stenosis. Severe pulmonary artery hypertension (PAH) without significant mitral valve involvement suggests recurrent pulmonary emboli known to occur with a myxoma in the RA or right ventricle (RV). Occasionally, right-sided tumors may appear as cyanotic congenital heart lesions because of intracardiac shunting.19
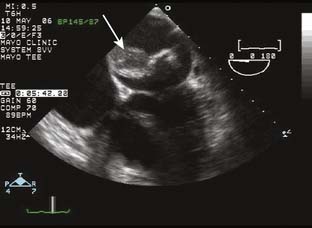
Recurrent fragmentation and embolization of the gelatinous-like tumor usually appear with systemic manifestations and are characteristic of myxoma. It is the smaller myxoma in the LA that does not create hemodynamic complications, exists for years undiagnosed, and is more likely to cause embolization.7 Cerebral aneurysms often exist in patients with recurrent systemic embolization from intracardiac myxoma probably secondary to damage by the systemic tumor emboli. The kidneys also are more susceptible to damage from myxoma emboli. Constitutional symptoms such as malaise, fever, and weight loss occur in about one third of patients, reflecting a possible autoimmune component but also delaying the diagnosis. Differential diagnosis includes endocarditis, connective tissue disorders, and malignancies. In general, the anatomic type of myxoma portends the clinical presentation. The solid, ovoid tumors are more often associated with CHF, whereas papillary myxomas present with cerebral embolization.20 Tumor size does not correlate with symptoms.16
Findings on a chest roentgenogram of a myxoma may be absent in one third of patients. Calcification on the chest roentgenogram is more diagnostic of right atrial myxoma, but rarely presents in left atrial myxoma. Before the availability of echo, angiography was used to identify all myxomas, but currently is probably only useful to determine coronary anatomy if considered necessary.2 Computerized tomography (CT) and MRI can help delineate the extent of the tumor and its relations to surrounding cardiac and thoracic structures.21 MRI is especially valuable in the diagnosis of myxoma when masses are equivocal or suboptimal by echo, or if the tumor is atypical in presentation.14 Difficulty may arise in differentiating thrombus from myxoma because both are so heterogenous.
Transthoracic echocardiography (TTE) is excellent for identifying intracavitary tumors because it is noninvasive, identifies tumor type, and permits complete visualization of each cardiac chamber (see Figure 22-1).22 It is the predominant imaging modality for screening.2 Transesophageal echocardiography (TEE) increases the diagnostic potential, as the nature of the tumor according to location, dimensions, number of masses, and echogenic pattern is better identified.23 Specifically, it yields morphologic detail in the evaluation of cardiac tumors, including points of tumor attachment and degree of mobility.
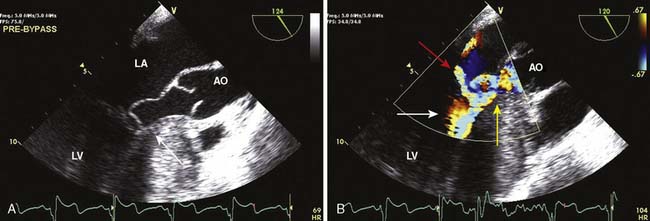
Myxomas have a typical echo appearance and often are irregular in shape with protruding fronds of tissue. There may be areas of calcification, and the echogenicity of the mass may not be homogenous.24 The presence of a large mass in the LA with an attachment to the interatrial septum is highly suggestive of myxoma. However, it must be emphasized that echo cannot provide a tissue diagnosis. Rarely, thrombus can be attached to the atrial septum.25 The degree of obstruction to ventricular filling caused by the tumor may be evaluated with Doppler echo (Figure 22-3). Qualitatively, color-Doppler imaging will show aliasing and flow acceleration through the atrioventricular valve when obstruction is present. Continuous-wave Doppler imaging is able to quantify the gradient between the atrium and ventricle. Before surgery, the goal of echo is to determine the site of tumor attachment, the absence of multiple masses, and to ensure that the tumor is not attached to the valve leaflets. If this cannot be accomplished with TTE, a TEE examination should be performed to aid in planning the surgical approach.
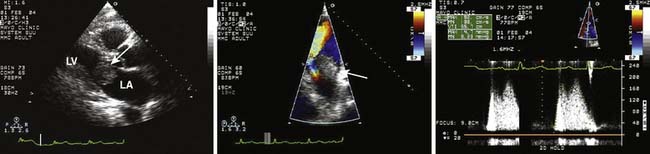
Currently, evaluation of cardiac tumors is a Class II indication for intraoperative TEE (supported by weaker evidence and expert opinion).26 When the primary reason for cardiac surgery is removal of an intracardiac mass, an intraoperative TEE evaluation should take place before the surgical incision to ensure that the mass is still present and has not embolized out of the heart (or dissolved as in the case of intracardiac thrombus). In the case of myxoma, an intraoperative examination in the presence of the surgeon can aid in finalizing the surgical plan. After tumor removal, the goal of TEE is to ensure that all visible mass was removed and there was no damage to adjacent structures. Specifically, in the case of a myxoma attached to the atrial septum, it is important to ensure that there is no interatrial shunting after removal. If the tumor was attached to, or near, a valve apparatus, the examiner must determine that the valve is competent after tumor removal.
The first surgical resection of an atrial myxoma was performed in 1954. Subsequently, surgical resection has been recommended even if the myxoma is discovered incidentally, mainly because the risk for embolization to the central nervous system may be 30% to 40%. Generally, the time interval between onset of symptoms and surgical resection is about 4 months, but surgery has been delayed for 10 years.16 Surgery is associated with a mortality rate of 0% to 7%.1,7 More importantly, it recently was documented for the first time that the long-term survival of an individual who underwent resection of myxoma was no different from an age- and sex-matched population.12
Anesthetic Considerations
Tumor location has a strong influence on anesthetic management. Left atrial myxomas most likely will cause mitral valve obstruction, often in conjunction with PAH and pulmonary venous hypertension. Anesthetic management will closely resemble a patient with mitral stenosis. In contrast, right atrial myxomas may produce signs of right-sided heart failure corresponding to tricuspid valve obstruction. Positioning of the patient for surgery must be carefully performed to detect severe restriction of venous return that may often be followed quickly with profound hypotension and arrhythmias. A large tumor increases the likelihood of hemodynamic instability, whereas a small tumor is associated with increased risk for embolization.7 Perioperative arrhythmias, especially atrial fibrillation or flutter, may arise in 25% of these patients, requiring immediate treatment. Hemodynamic instability with low cardiac output (CO) and arrhythmias are common.
Median sternotomy is recommended for resection of atrial myxoma, although anterior thoracotomy may be used in some benign tumors,2 as well as minimally invasive techniques. Femoral cannulation for initiation of cardiopulmonary bypass (CPB) may minimize the risk for dislodgment or fragmentation of the tumor. Subsequently, a venous cannula can be placed high in the superior vena cava because a biatrial approach to an atrial septal tumor is necessary. The most preferred surgical approach is the single atrial approach.12 Moderate systemic hypothermia, deep topical cooling, and cardioplegic arrest are often used, whereas circulatory arrest is reserved for malignant tumors with significant extension. The heart should not eject during CPB, to minimize systemic embolization of tumor fragments. Electrically induced ventricular fibrillation has been used to prevent ejection of blood after initiation of CPB. Wide excision of the septal base of the myxoma with Dacron or pericardial patching of the resulting defect is the preferred operation.27 Mitral valve replacement may be necessary in large tumors, ventricular side tumors, or tumors with other manifestations besides a propensity to embolize.18 Less extensive operations risk a greater incidence of tumor recurrence because of incomplete tumor excision or a second tumor originating in susceptible atrial tissue. The recurrence rate after complete excision of a sporadic cardiac myxoma is less than 5%.5 After surgery, the most common complication is a 25% incidence rate of transient arrhythmias, mostly supraventricular in nature.

Other Benign Tumors
Papillomas (papillary fibroelastoma) are rare tumors but are the third most common primary cardiac tumor (after myxoma and fibroma).5 Initially thought to be incidental findings during autopsies, today most are discovered in living patients.28 Mostly singular (90%), 1 to 4 cm in size, highly papillary, pedunculated, and avascular, papillomas are covered by a single layer of endothelium containing fine elastic fibrils in a hyaline stroma. Macroscopically, they resemble sea anemones. They originate most commonly from valvular endocardium,29 usually involving the ventricular surface of the aortic valve or the atrial surface of the mitral valve, but infrequently rendering the involved valve incompetent. They account for 75% of all primary cardiac valvular tumors.12,30 Adults between the ages of 40 and 80 primarily are affected, with the mean age at the time of detection of 60 years.5 Many patients are asymptomatic, so it is not surprising that 47% of these tumors are discovered incidentally during echo, catheterization, or even cardiac surgery. Echocardiographically, fibroelastomas have a typical appearance (Figure 22-4). They usually are small (mean size, 12 × 9 mm) and their motion is independent from that of the attached valve leaflet.31 They may appear similar to vegetations seen in endocarditis, or they may be confused with Lambl’s excrescences, which tend to be more nodular in appearance.
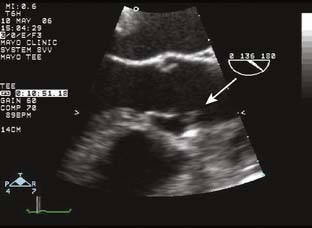
Currently, many tumors are found in a search to find the cause of embolic symptoms. These symptoms are most common when the aortic valve is involved. Previously believed to be harmless, postmortem studies have shown a high incidence of embolization to the cerebral and coronary circulations.4 Not surprisingly, symptoms often are related to stroke or transient ischemic attack and myocardial infarction (MI). Although embolization may be a tumor, it may be a thrombus because tumors are excellent sites for thrombus formation.5 It is important that the diagnosis is made because acute valvular dysfunction and even sudden death may occur.29 Surgical resection is curative but may require valvular repair or replacement in one third of cases.28 Recurrence is rare.
The incidence of cardiac lipomas4 is similar to papillary fibroelastoma. They occur as intracavitary tumors, intramyocardial masses, and pericardial tumors. Histologically, they usually consist of encapsulated groups of mature fat cells. Patients with these tumors often are asymptomatic, but if they occur in an intracavitary location, they may resemble myxomas. An intramyocardial location may provoke arrhythmias and conduction abnormalities. Pericardial lipomas are associated with tamponade and cardiac compression. These tumors tend to enlarge over time, and when symptoms appear, surgical excision is required.
Rhabdomyomas are primarily childhood tumors (majority before 1 year of age) located intramyocardially, and arise from all areas of the heart and at multiple locations of the heart. Tuberous sclerosis often is associated with these tumors.32 They represent 45% of all benign tumors of childhood, but only 1% of benign adult primary cardiac tumors.4 As pedunculated masses, rhabdomyomas usually originate in the ventricle, leading to inflow and outflow ventricular obstruction, and on the atrioventricular valves.5 Symptoms are caused by the obstruction of blood flow and arrhythmias, but life-threatening complications are rare. Although surgical resection may be necessary in 25% of cases, these tumors most often resolve spontaneously. Because of their space-occupying properties, rhabdomyomas may mimic other congenital defects such as left hypoplastic heart syndrome.
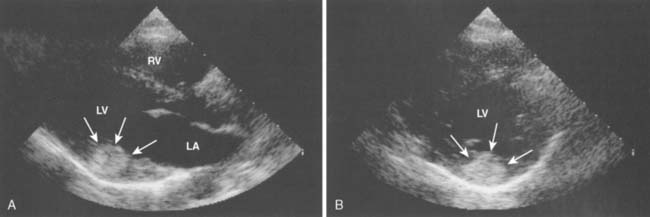
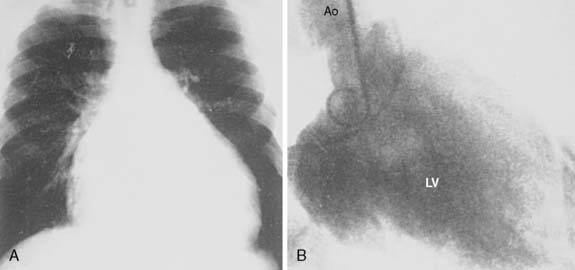
Fibromas are connective tissue tumors found primarily in children, making up 15% of pediatric benign primary cardiac tumors.4 They are usually solitary, located in the wall of the heart, and often involve the apical or septal areas of the left ventricle (LV) (Figure 22-5). Echocardiographically, fibromas tend to be well demarcated from the myocardium by calcifications (Figure 22-6).33 These tumors frequently interfere with the conduction pathways. Ventricular arrhythmias are common, and ventricular tachycardia with sudden death is not infrequent. The tumor may occlude or displace the coronary arteries. Patients may present with CHF or angina. Surgical resection is favored even for asymptomatic patients in view of the risk for fatal ventricular arrhythmias.34 Cardiac transplantation has been advocated for septal tumors that are unresectable; however, a subtotal resection achieves excellent late survival.
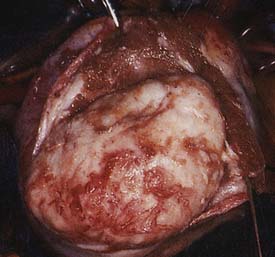
Figure 22-5 Dissection of ventricular fibroma (Patient 12; patient’s head is at top).
(From Cho JM, Danielson GK, Puga FJ, et al: Surgical resection of ventricular cardiac fibromas: Early and late results, Ann Thorac Surg 76:1933, 2003, by permission.)
Two vascular tumors, angiomas and pheochromocytomas of the heart (paraganglioma), are rare. Angiomas are found in the interventricular septum, and their vascular nature makes surgery difficult. Primary cardiac pheochromocytomas are located along the atrioventricular groove near the epicardial arteries.35 These tumors arise from neuroendocrine cells. Although cardiac pheochromocytomas became known based on symptoms related to catecholamine secretion, the symptoms are less dramatic than the corresponding adrenal tumors because norepinephrine is not converted to epinephrine in the cardiac tumors. Cardiac pheochromocytomas may go undiagnosed for years while undergoing exploratory laparotomy and multiple diagnostic tests before a cardiac location is considered. If these tumors are nonsecretors of catecholamines, often they are not detected until superior vena cava obstruction, pericardial effusion, or tamponade.36 Fifty percent of these tumors are hereditary, such as in neurofibromatosis or Hippel-Lindau disease. Although surgical excision is curative, total excision may be difficult. Severe intraoperative bleeding is known to occur with these tumors.35 Furthermore, clinicians should be prepared to treat these tumors as catecholamine-secreting tumors that may result in hemodynamic derangements during anesthesia with uncontrolled hypertension, pulmonary edema, and MI.36 Current perioperative management for these tumors has been reviewed.37

Malignant Tumors
Approximately 25% of primary cardiac tumors are malignant,5 and 95% of these are sarcomas. They are found infiltrating the RA and causing cavitary obstruction, but may have variable clinical presentations based on the location, causing diagnosis to be elusive. They usually occur between the ages of 30 and 50, preceded by vague symptoms such as dyspnea, but rapidly progressing to death. Angiosarcomas, the most common sarcoma,4 are rapidly spreading vascular tumors that arise most often from the RA, appearing near the inferior vena cava with extension to the mediastinum. They occur most commonly in adults and male individuals.5 Presenting symptoms include chest pain and dyspnea, progressive CHF, and bloody pericardial effusion.9 There are two clinicopathologic forms of the tumor. The first type deposits small tumor in the pericardium or epicardium and is associated with skin lesions or risk factors for Kaposi sarcoma. The second involves large tumor in the RA. Treatment is palliative as the response to chemotherapy and radiation is poor. Resection may be possible, but survival is less than 2 years. Rhabdomyosarcomas are aggressive tumors that have cellular elements that resemble striated muscle. These tumors occur in both sexes equally. They may originate in any chamber of the heart, but in contrast with angiosarcomas, they rarely become diffusely involved with the pericardium. They are bulky and invasive, growing rapidly. Surgical resection is possible, but distant metastasis reduces the chances of success. Chemotherapy and radiation are ineffective.5
Echo tends to be less helpful in the management of these patients than it is in patients with benign tumors. It may reveal physiologic complications of the tumor such as cavitary obliteration or valvular regurgitation, and it is helpful in finding associated pericardial effusions and tamponade physiology. However, these tumors have complex anatomy, and the perimeters of the tumor, as well as their involvement in valve apparatuses and coronary anatomy, can be difficult to determine even with TEE. In addition, echo does not image adjacent cardiac structures such as the lung and mediastinum in detail. When echo is combined with other imaging modalities such as CT and MRI, the clinician may obtain the anatomic information needed (from the CT or MRI), as well as the physiologic consequences (from echo).24
Other primary malignant tumors of the heart include malignant fibrous histiocytoma, fibrosarcomas, osteosarcoma, leiomyosarcoma (rarest malignant cardiac tumor), undifferentiated sarcoma, neurogenic sarcoma, and lymphomas. Primary cardiac lymphoma is defined as a non-Hodgkin lymphoma and accounts for about 1% of primary cardiac tumors.38 The prevalence of these tumors has been increasing in part because of AIDS and early detection from imaging advancements such as echo. Lymphomas are generally large masses with extensive infiltration into adjacent areas of the heart from the point of tumor origin. These commonly are located in the RA and RV. These primary tumors are rare, representing less than 2% of all cardiac tumors.39 Treatment involves a combination of chemotherapy and radiation, occasionally extending survival up to 5 years, but the median survival is 1 year. Malignant fibrous histiocytoma, in contrast with other sarcomas, generally is found in the LA. Despite resection, metastasis is common, as well as local recurrence. Surgical excision may be useful to alleviate symptoms but ultimately does not influence the poor survival.40
In general, malignant primary cardiac tumors may require a combination of surgery, radiation, and chemotherapy simply to limit cavitary obstruction to blood flow because of rapid growth and metastasis. Local recurrence is more likely to cause death than metastasis.41 More aggressive approaches for malignant tumors with extensive local disease before metastasis include autotransplantation.3 The heart is removed from the chest cavity and inverted to provide better exposure. Its value is still indeterminate, but no intraoperative deaths have occurred. Although still controversial, orthotopic heart transplantation may be considered for unresectable tumors that involve only the heart, but survival is not extended beyond 1 to 2 years.2 The rate of intraoperative death with malignant tumor resection is seven times that of benign resection, and there is twice the morbidity rate.
Most cardiac tumors are rarely associated with airway problems.42 Rather, the large pericardial effusions create significant hemodynamic instability and deterioration. Manipulation of these tumors also may exacerbate hemodynamics. If a large pericardial effusion is present before induction of anesthesia, it should be drained. Beyond the standard monitoring used for a cardiac surgical patient, the overall physical status and risk for embolization must be evaluated. Femoral arterial and central venous monitoring are recommended for patients with right atrial tumors. Sudden right-sided tumor embolization may be recognized based on an increased end-tidal carbon dioxide gradient. Left-sided embolization is difficult to assess during anesthesia.

Tumors with Systemic Cardiac Manifestations
Carcinoid tumors are metastasizing neuroendocrine tumors that arise primarily from the small bowel, occurring in 1 to 2 per 100,000 people in the population.43 On diagnosis, 20% to 30% of individuals with carcinoid tumors present with carcinoid syndrome. It is characterized by episodic vasomotor symptoms, bronchospasm, hypotension, diarrhea, and right-sided heart disease attributed to release of serotonin, histamine, bradykinins, and prostaglandins often in response to manipulation or pharmacologic stimulation. Manifestations of carcinoid syndrome occur primarily in patients with liver metastasis and impair the ability of the liver to inactivate large amounts of vasoactive substances.
Initially described in 1952,43 aspects of carcinoid heart disease may occur in 20% to 50% of patients with carcinoid syndrome.44,45 Carcinoid heart disease may be the initial feature of metastatic carcinoid disease in 20% of patients. The prognosis has improved substantially since the early 1980s for individuals with malignant carcinoid tumors and carcinoid heart disease, but it still causes considerable morbidity and mortality. The median life expectancy is 5.9 years without carcinoid heart disease, but declines to 2.6 years if it is present.45 Circulating serotonin levels had been found to be more than twice as high in persons with carcinoid syndrome who develop carcinoid heart disease,46 but this is no longer true because most patients receive somatostatin analogs.45
Carcinoid heart disease characteristically involves tricuspid regurgitation and pulmonic stenosis and regurgitation resulting in severe right-heart failure (Figure 22-7). Tumor growth in the liver permits large amounts of tumor products to reach the RV without the benefit of first-pass metabolism. Carcinoid plaques composed of myofibroblasts, collagen, and myxoid matrix47 are deposited primarily on the tricuspid and pulmonary valves, bringing about immobility and thickening of the valve leaflets, causing the distinctive valvular changes (Figure 22-8). After surgery, 80% of tricuspid valves are observed to be incompetent, with only 20% stenotic, whereas the affected pulmonary valves tend to be equally divided between incompetence and stenosis.47 The exact mechanism that causes valve injury is unknown, but high levels of serotonin sometimes are found in those patients with carcinoid heart disease.43 Less than 10% of those with carcinoid heart disease have left-heart involvement, possibly because of inactivation of serotonin in the lungs,48 but it may exist with the presence of a bronchial carcinoid or an interatrial shunt.
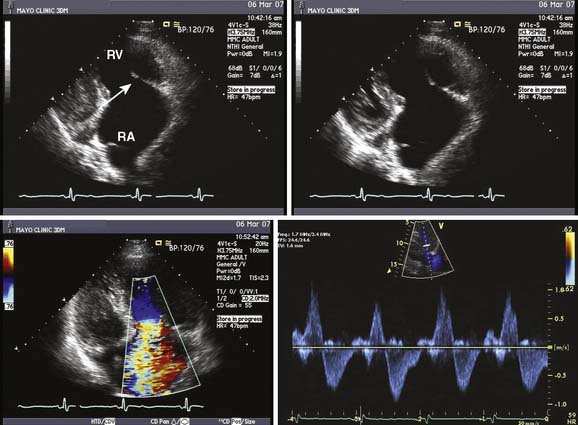
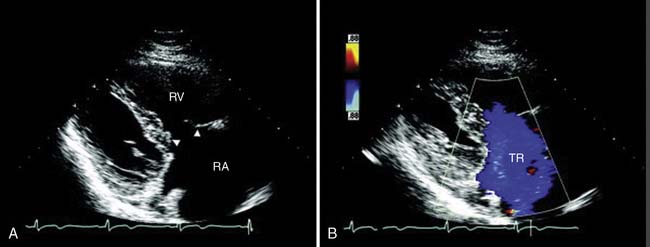
Echo features of carcinoid heart disease, particularly of the tricuspid valve, are practically diagnostic of the underlying disease process. The leaflets are thickened and retracted. The appearance of the tricuspid leaflets is often as if the leaflets were curled under (see Figure 22-7). The thickening and retraction result in a large coaptation defect and severe valvular regurgitation. The pulmonic valve may be difficult to image with TTE, but the midesophageal TEE view at 70 to 90 degrees often shows the valve well. With severe tricuspid regurgitation, the RV will dilate and abnormalities of ventricular septal motion may be noted. The thickness of the ventricular wall is usually normal. Doppler imaging of the hepatic veins will show systolic flow reversals consistent with severe tricuspid regurgitation. A careful search for a patent foramen ovale (PFO) should be undertaken because this has implications for left-sided valvular involvement.
Without treatment, median survival with carcinoid heart disease is 11 months.43 A large percentage of patients with carcinoid heart disease are asymptomatic because the disease is mild. Early detection can affect prognosis as progression of cardiac disease, especially to right ventricular failure, increases mortality.44 Treatment of the tumor and the malignant carcinoid syndrome does not result in regression of carcinoid heart disease.45 Surgery to replace both tricuspid and pulmonary valve with either bioprosthetic or mechanical valve is the only viable therapeutic option.49 The decision regarding mechanical or bioprosthetic valve(s) depends on individual risks and concerns.43,50 The optimal timing to operate is uncertain, but consideration should be given when signs of right ventricular failure appear. Even after surgery, right ventricular dysfunction will persist. Perioperative mortality rate is less than 10%.48
Anesthetic Considerations
Patients who have carcinoid heart disease and require cardiac surgery pose an anesthetic challenge.51–54 A carcinoid crisis with vasoactive mediator release is a life-threatening event that can be provoked by stress, physical stimulation, anesthesia, catecholamines, long-acting opioids (meperidine, morphine), or histamine-releasing muscle relaxants (atracurium). Preoperative control of carcinoid activity is a critical aspect of perioperative management made considerably easier with the administration of octreotide, a synthetic analog of somatostatin that inhibits the vasoactive compounds that produce carcinoid syndrome. It reduces the occurrence of symptoms in more than 70% of patients. The longer half-life of octreotide than somatostatin allows subcutaneous injection of 150 μg three times daily to control symptoms. After surgery, intravenous octreotide (50 to 100 μg/hr) should be started 2 hours before surgery and continued for 48 hours after surgery. Additional intermittent intravenous doses of 50 to 200 μg are given to stop severe hypotension and prevent further carcinoid symptoms.55 Anesthesiologists should be prepared to give large doses of octreotide (300 μg/hr) for stabilization during the intraoperative and postoperative periods. Atrial fibrillation is present in 18% of carcinoid patients, possibly because of the right-sided failure and chamber enlargement, as well as the proarrhythmic effects of seratonin.56 Severe hyperglycemia may occur with octreotide because of its inhibition of insulin secretion, especially in combination with steroids, so glucose monitoring is recommended.50
Preoperative medication to reduce anxiety is strongly suggested for these patients.51 Individuals with more active carcinoid disease experience greater reductions in systolic blood pressure with induction of anesthesia. Etomidate has been recommended for induction instead of thiopental (histamine-releasing) with short-acting opioids (fentanyl and sufentanil) and nonhistamine-releasing muscle relaxants.50 Benzodiazepines are especially valuable for preoperative anxiety, as well as part of anesthetic maintenance. The standard low-to-moderate dose of fentanyl or sufentanil anesthetic with isoflurane is recommended with a nonhistamine-producing muscle relaxant.57
Sudden intraoperative hypotension should be regarded as carcinoid crisis and intravenous octreotide administered until hemodynamic stability returns. Careful attention should be paid to physiologic parameters such as airway pressures as early warning signs of impending carcinoid crisis and treated before onset of severe hypotension. Previously, certain catecholamines (epinephrine, norepinephrine, dopamine, and isoproterenol) were considered to provoke mediator release in carcinoid syndrome, but a recent retrospective study of 100 consecutive patients who underwent cardiac surgery with carcinoid heart disease did not show a significant increase in intraoperative octreotide need or mortality with the use of vasoactive medications.54 The majority of patients in this study received vasopressor agents such as epinephrine, dopamine, and calcium. Because nearly 75% of these patients for cardiac surgery will be in New York Heart Association (NYHA) Class III and require multiple valve replacement, vasoactive medication should be administered according to hemodynamic indices. The safety of vasoactive medications in these patients with octreotide use has been confirmed in a subsequent study.57 Low CO from right ventricular failure and hypotension from vasoplegia are common in the postbypass period, so identifying carcinoid crisis in such circumstances is problematic.
The use of an antifibrinolytic is routine in many centers to reduce blood loss and transfusion requirements associated with CPB and cardiac surgery.58 Because patients with carcinoid heart disease often require surgery involving multiple valves in association with liver metastasis, coagulopathy and excessive hemorrhage after CPB are likely and considered one of the two major complications associated with surgery.55,57 Weingarten et al54 reported significant reductions in red blood cell (RBC) and non-RBC transfusions in patients undergoing cardiac surgery for carcinoid heart disease who received aprotinin compared with those who did not. Aprotinin use was not associated with a reduction in mortality. Aprotinin has the dual properties of antifibrinolysis and anti-inflammation compared with other popular synthetic antifibrinolytic agents (tranexamic acid and aminocaproic acid), but it is no longer available for administration. Synthetic antifibrinolytics appear safe for use in carcinoid heart disease.57
Cardiomyopathy
Previously, the World Health Organization (WHO)/International Society of Cardiology defined cardiomyopathy as heart muscle disease of unknown cause, unlike heart muscle disease attributed to a specific cause or associated with a disease process. With more knowledge concerning pathogenesis and causative factors, the difference between cardiomyopathy and specific heart disease has become less distinct. Previously, cardiomyopathy was classified as dilated, hypertrophic, and restrictive. Over time, each classification has become a recognized clinical condition. In 1995, the WHO/International Society of Cardiology redefined the cardiomyopathies according to dominant pathophysiology or, if possible, by “etiologic/pathogenetic factors.”59 Cardiomyopathies are now defined as “diseases of the myocardium associated with cardiac dysfunction.” The original cardiomyopathies classified as dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), and hypertrophic cardiomyopathy (HCM) were preserved, and arrhythmogenic right ventricular cardiomyopathy (ARVC) was added.
The annual incidence of cardiomyopathy in adults is 8.7 cases per 100,000 person-years.60 Underlying causes of cardiomyopathies are different but carry some prognostic significance.61 General characteristics of all four cardiomyopathies are displayed in Table 22-2. Reviews of the clinical, pathophysiologic, and therapeutic aspects of these cardiomyopathies are available.62–64 This section focuses on cardiomyopathy in relation to cardiac surgery, but it is relevant for the larger number of patients with a cardiomyopathy who undergo noncardiac surgery.
All the cardiomyopathies maintain some relation to genetic transmission. The genetics of these cardiomyopathies will not be addressed in detail, but this does not reflect a lack of importance. Genetic testing is advancing rapidly, with the ability to identify disease-causing mutations in those family members at risk but asymptomatic. The result is heightened clinical surveillance and, possibly, earlier intervention and prevention of the sequelae of the disease. More information is available in this review.65

Dilated Cardiomyopathy
Formerly referred to as congestive cardiomyopathy or idiopathic cardiomyopathy, DCM is by far the most common of the four cardiomyopathies in adults (60%). The term idiopathic has become less applicable to cardiomyopathies as developments in molecular biology and genetics have provided better insight into the pathogenesis of DCM. In the United States alone, nearly 550,000 individuals are newly diagnosed with CHF, whereas close to 4.6 million receive treatment for it. Even with current management, survival for adults at 1 and 5 years after diagnosis is 76% and 35%, respectively, representing a major health care concern.66 Interestingly, diagnosis of DCM in asymptomatic patients occurs in 30% of patients with extended quality of life and survival if medical treatment is initiated.67 This would suggest a less-advanced disease process and being more amenable to treatment. Nonischemic causes comprise 25% to 35% of adult patients with left ventricular dysfunction and CHF, of which DCM is a major component.63
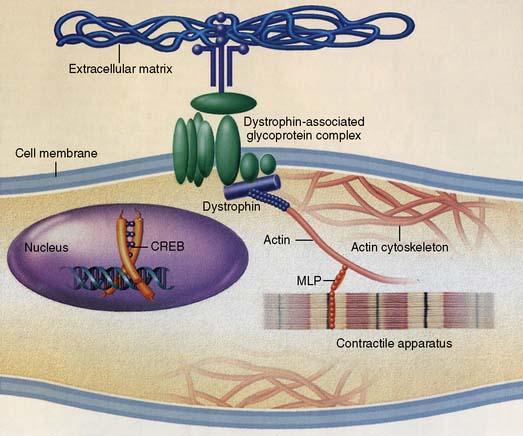
DCM has diverse causes, such as viral, inflammatory, toxic, or familial/genetic. It is associated with many cardiac and systemic disorders that influence the prognosis. Approximately 1230 patients with cardiomyopathy were evaluated and grouped according to a specific cause.61Table 22-3 shows that 50% of patients had common causes for DCM, but 50% were characterized as idiopathic. Currently, there is a greater appreciation for the role of genetic and familial factors in the cause of DCM (Figure 22-9).64,68,69 Between 10% and 35% of cases of DCM are familial with an autosomal dominant expression.63 Incomplete penetrance may account for differences in disease severity and progression in familial DCM despite sharing identical mutations.64 Similarities in clinical course that evoke a common set of molecular and cellular pathways may lead to better therapy in the future despite the many different causes of DCM.
TABLE 22-3 Common Clinicopathologic Diagnoses in 1230 Patients with Initially Unexplained Cardiomyopathy
| Diagnosis | No. of Patients (%) |
|---|---|
| Idiopathic dilated cardiomyopathy | 616 (50) |
| Myocarditis | 111 (9) |
| Ischemic cardiomyopathy | 91 (7) |
| Infiltrative disease | 59 (5) |
| Peripartum cardiomyopathy | 51 (4) |
| Hypertension | 49 (4) |
| Human immunodeficiency virus infection | 45 (4) |
| Connective tissue disease | 39 (3) |
| Substance abuse | 37 (3) |
| Doxorubicin related | 15 (1) |
| Other causes | 111 (10) |
From Wu LA, Lapeyre AC, Cooper LT: Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc 76: 1030–1038, 2001.
DCM is characterized morphologically by enlargement of right and left ventricular cavities with hypertrophied muscle fibers without an appropriate increase in the ventricular septal or free wall thickness, giving an almost spherical shape to the heart. These hearts are two to three times larger than normal.64 The valve leaflets may be normal, yet dilation of the heart has been associated with a regurgitant lesion secondary to displacement of the papillary muscle. Histologic changes are nonspecific and not associated with positive immunohistochemical, ultrastructural, or microbiologic tests. Microscopically, instead of large losses of myocardium, there is patchy and diffuse loss of tissue with interstitial fibrosis and scarring, uncharacteristic of ischemic myocardium.64 Degenerative changes are responsible for bundle branch block on the electrocardiogram (ECG).
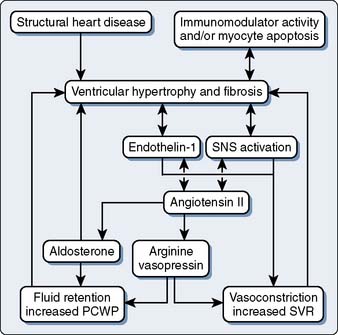
With DCM, there is more impairment of systolic function even though diastolic function is affected. As contractile function diminishes, stroke volume initially is maintained by augmentation of end-diastolic volume. Despite a severely decreased ejection fraction, stroke volume may be almost normal. Eventually, increased wall stress caused by marked left ventricular dilation and normal or thin left ventricular wall thickness occurs.63 Increasing left atrial size may indicate worsening diastolic dysfunction in these patients, contributing significantly to functional mitral regurgitation.70 It is important to expand the diagnosis of mitral regurgitation with echo beyond the left ventricular geometry and mitral orifice because contractility and dyssynchrony are essential for the correct diagnosis. Dilation, combined with valvular regurgitation, compromises the metabolic capabilities of heart muscle and produces overt circulatory failure. Compensatory mechanisms may allow symptoms of myocardial dysfunction to go unnoticed for an extended period. However, the onset of mitral regurgitation signals a poor prognosis as the ventricular function progressively worsens without intervention. The importance of corresponding neurohumoral influences, such as the renin-angiotensin system, on this pathologic process recently was appreciated as a major factor in the appearance of typical signs and symptoms of CHF and formation of therapeutic options (Figure 22-10).71,72 Additional evidence of cytokine activity and endothelium dysfunction provide a complex interplay of forces leading to circulatory failure. The possibility of blocking this neurohumoral response of CHF pharmacologically has not been fully realized, as early trials have been disappointing.73
Although DCM occurs in children, its presentation is generally in the fourth and fifth decades of life.64 The clinical picture of DCM typically includes signs and symptoms of CHF often corresponding to months of fatigue, weakness, and reduced exercise tolerance before diagnosis.62 One-third of individuals report chest pain.63 However, the first indication of DCM may be a stroke, arrhythmia, or even sudden death. Increasingly, individuals are presenting for routine medical screening to be informed of cardiomegaly on a routine chest roentgenogram. Symptoms may appear insidiously over a period of years or evolve rapidly after an unrelated illness. Physical signs of DCM, depending on the disease’s progression, include pulsus alternans, jugular venous distention, murmurs of atrioventricular valvular regurgitation, tachycardia, and gallop heart sounds.
A chest roentgenogram demonstrates variable degrees of cardiomegaly and pulmonary venous congestion (Figure 22-11). An ECG may be surprisingly normal or depict low QRS voltage, abnormal axis, nonspecific ST-segment abnormalities, left ventricular hypertrophy, conduction defects, and evidence of atrial enlargement. Atrial fibrillation is common, and about one fourth of patients have nonsustained ventricular tachycardia.62 Although the LV is affected, the RV may be spared in some patients, and this finding has been associated with improved survival.74 Coronary catheterization usually reveals normal coronary vessels. Coronary angiography also will distinguish between ischemic and idiopathic DCM, a finding that has therapeutic and prognostic implications. An endomyocardial biopsy rarely is valuable to identify the cause of DCM but may be useful to rule out other pathologies with similar presentation to DCM.64 The biopsy has no prognostic value or correlation with ventricular function.
Echo is extremely useful in the ambulatory management of patients with DCM. The characteristic 2D findings are a dilated LV with globally decreased systolic function. Indeed, all markers of systolic function (ejection fraction, fractional shortening, stroke volume, and CO) are uniformly decreased.33 Other associated findings may include a dilated mitral annulus with incomplete mitral leaflet coaptation, dilated atria, right ventricular enlargement, and thrombus in the left ventricular apex (Figure 22-12). In some instances, regional wall motion abnormalities will be present. Color Doppler imaging is useful in assessing the presence or absence of valvular regurgitation. Pulsed-wave and continuous-wave Doppler are used to quantify CO and evaluate filling pressures and pulmonary artery pressures. Well-compensated patients with DCM may have only mild impairment of diastolic function. As the disease progresses and patients become less well-compensated, the left ventricular diastolic filling pattern changes to that of restricted filling. Although systolic function may not change in these patients, the increased filling pressures associated with restrictive left ventricular filling will often worsen their CHF symptomatology.
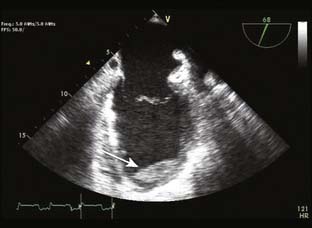
Figure 22-12 Transesophageal echocardiogram, midesophageal, two-chamber view in a patient with dilated cardiomyopathy.
Management of acute decompensated CHF continues to evolve, but the onset of overt CHF is a poor prognostic indicator for persons with DCM.71 However, compared with ischemic CHF, patients with nonischemic DCM show greater improvement in symptoms, left ventricular function, and remodeling during more contemporary therapy than in the past.75 Treatment revolves around management of symptoms and progression of DCM, whereas other measures are designed to prevent complications such as pulmonary thromboembolism and arrhythmias. The mainstay of therapy for DCM is vasodilators combined with digoxin and diuretics. All patients receive angiotensin-converting enzyme inhibitors to reduce symptoms, improve exercise tolerance, and reduce cardiovascular mortality without a direct myocardial effect.62,76,77Perhaps more important than the hemodynamic effects, angiotensin-converting enzyme inhibitors suppress ventricular remodeling and endothelial dysfunction, accounting for the improvement in mortality noted with this medication in DCM.78 Other afterload-reducing agents, such as selective phosphodiesterase-3 inhibitors like milrinone, may improve quality of life but do not affect mortality, and thus are rarely administered in chronic situations. More recently, spironolactone has assumed a greater role in treatment as mortality rate was reduced by 30% from all causes in patients receiving standard angiotensin-converting enzyme inhibitors for DCM with the addition of spironolactone in a large, double-blind, randomized trial.79 Although aldosterone increases sodium retention and reduces potassium loss, it also has been shown to cause myocardial and vascular fibrosis, impair baroreceptor function, and prevent catecholamine reuptake by the myocardium.
Until recently, β-blockers were contraindicated in DCM. In 1982, Bristow et al80 found decreased catecholamine sensitivity and β-receptor density in the failing human myocardium, leading to loss of contractility. The association between excess sympathetic activity and the failing heart has been aptly demonstrated.81 Recently, the dobutamine stress test has been shown to identify changes that reflect increased sympathetic stimulation in the ventricle of asymptomatic to mildly symptomatic patients with DCM.82 This finding may aid in the initiation of β-blockers in patients with normal resting parameters. The use of β-blockers in DCM has not only provided symptomatic improvement, but also substantial reductions in sudden and progressive death with NYHA Class II and III heart failure.83 This is especially significant because almost 50% of deaths are sudden.84
High-grade ventricular arrhythmias are common with DCM. Approximately 12% of all patients with DCM die suddenly,85 but overall prediction of sudden death in an individual with DCM is poor.84 Electrophysiologic (EP) testing has a poor negative predictive value that limits its usefulness. The best predictor of sudden death remains the degree of left ventricular dysfunction. Patients who have sustained ventricular tachycardia or out-of-hospital ventricular fibrillation are at increased risk for sudden death, but more than 70% of patients with DCM have nonsustained ventricular tachycardia during ambulatory monitoring.77 Furthermore, the prognostic significance of ventricular arrhythmias and response to prophylactic antiarrhythmia therapy in patients with DCM are not well established.
Antiarrhythmic medications are hazardous in patients with poor ventricular function because of their negative inotropic and sometimes proarrhythmic properties. Antiarrhythmic therapy may only be considered in DCM if inducible ventricular tachycardia or symptomatic arrhythmias are present. Class I agents are not indicated because they clearly have demonstrated increased mortality in patients with advanced CHF.62 Amiodarone is the preferred antiarrhythmic agent in DCM because its negative inotropic effect is less than other antiarrhythmic medications84 and its proarrhythmic potential is lowest.86 Counter to most trials of antiarrhythmic prophylaxis, results of a recent large multicenter trial of antiarrhythmic therapy in patients with CHF86 demonstrated that amiodarone was associated with a significantly lower mortality rate of 38% compared with 62% without therapy in persons with a higher resting heart rate. Not withstanding these results, implantable defibrillators reduce the risk for sudden death, as well as reducing mortality.84,85 Recent evidence has indicated that with previous cardiac arrest or sustained ventricular tachycardia, more benefit was gained from an implantable defibrillator.87 This was based on a 27% reduction in the relative risk for death attributed to a 50% reduction in arrhythmia-related mortality compared with treatment with amiodarone (see Chapters 4,10, and 25).
Other treatments for DCM include digoxin, which has been reaffirmed as clinically beneficial in two large trials of adults.74 The risk for thromboembolic complications is significant in DCM for adults. Patients with moderate ventricular dilation and moderate-to-severe systolic dysfunction have intracavitary stasis and a decreased ejection of blood, so they are likely to receive anticoagulants if any history of stroke, atrial fibrillation, or evidence of intracardiac thrombus exists.
Patients who are resistant to pharmacologic therapy for CHF have received dual-chamber pacing, cardiomyoplasty, left ventricular assist devices, cardiac surgery (nontransplantation) and transplantation in recent years. Cardiac resynchronization therapy with dual ventricular pacing improves NYHA functional class and ejection fraction 6 months after implantation.88 Placement of implantable left ventricular assist devices has enabled end-stage patients to reach transplantation or become destination therapy for those in whom transplantation is not an option.89 If mitral regurgitation develops in patients with DCM, mitral valve repair or replacement is recommended. The surgery in this high-risk population is safe and improves NYHA classification and survival.90 Transplantation can substantially prolong lives, with current survival at 15 years of 50% if younger than 55 years of age91; however, limited organ availability and drug-related morbidity for those with end-stage DCM looks to future improvements in assist devices and new surgical procedures to provide the best opportunity for increased survival. An example is a new surgical procedure called surgical ventricular restoration that may improve symptoms and cardiac status. It is performed with an arrested heart during CPB. Coronary artery bypass grafting (CABG) is first performed followed by ventriculotomy to insert the mannequin to reshape the ventricle (see Chapters 18 and 27).92
Anesthetic Considerations
Individuals with DCM are extremely sensitive to cardiodepressant anesthetic drugs. Intravenously administered anesthetic agents such as fentanyl (30 μg/kg) provide excellent anesthesia and hemodynamics in patients with ejection fractions less than 30%93 but contribute to prolonged respiratory depression delaying extubation. Shorter-acting narcotics such as remifentanil may be unsuitable for patients with poor left ventricular function undergoing cardiac surgery because of high incidences of bradycardia and severe hypotension.94 A comparison of remifentanil-sevoflurane with fentanyl-etomidate-isoflurane found significantly greater reduction in mean arterial pressure and greater incidence of bradycardia with the remifentanil anesthetic.95 Also, etomidate has been shown to have little effect on the contractility of the cardiac muscle in patients undergoing cardiac transplantation.96 Ketamine has been recommended for induction in critically ill patients97,98 because of its cardiovascular actions attributed mainly to a sympathomimetic effect from the central nervous system. Ketamine is a positive inotrope in the isolated rat papillary muscle and, more importantly, in a model of cardiomyopathic hamsters, did not display a negative inotropic effect.99 This makes ketamine (less than 0.5 mg/kg) an excellent choice to use in combination with fentanyl for induction in patients with severe myocardial dysfunction secondary to cardiomyopathy. The use of propofol with cardiomyopathy may be a concern because cardiovascular depression has been observed possibly because of inhibition of sympathetic activity and a vasodilatory property. However, in a cardiomyopathic hamster model, there was no direct effect on myocardial contractility with propofol.100 Caution is still prudent with propofol because of the indirect inhibitory effects of sympathetic activity that many patients with cardiomyopathy and reduced left ventricular function may depend on for hemodynamic stability.
Volatile agents have long been a concern in persons with failing hearts because of their known depressant effects on myocardial contractility. The effect of currently used volatile anesthetics on intrinsic myocardial contractility is difficult to project clinically. Animal data indicate halogenated volatile agents may have more profound negative inotropic effects in cardiomyopathic muscle than healthy cardiac muscle.101 An anesthetic technique that minimizes myocardial depression is essential. There is little from a cardiovascular standpoint to support a selection of sevoflurane over isoflurane in adults. Although the failing myocardium has been thought to be more sensitive to the depressant effects of volatile agents, there is no synergistic myocardial depression in the presence of moderate left ventricular dysfunction and volatile agents.102 Desflurane, which possesses the lowest blood/gas partition coefficient of the volatile agents allowing rapid induction and emergence, would appear to have some theoretic benefit for early extubation in patients undergoing cardiac surgery. In healthy hamster papillary muscles, desflurane did not appear to have a negative inotropic effect; however, in cardiomyopathic papillary muscles, there was a profound negative inotropic effect.103 On the other hand, sevoflurane and desflurane were shown not to adversely affect the ability of the LV to respond to increased work despite their negative inotropic properties in patients undergoing CPB and cardiac surgery.104 Even with the limitations of generalizing findings of in vitro experiments to clinical situations, this difference of inotropic effect regarding healthy versus cardiomyopathic myocardium is an important distinction not present in all halogenated anesthetics and should be considered in patients with cardiomyopathy.
Invasive hemodynamic monitoring is indispensable with volatile agents and DCM. Of particular pertinence for anesthesiologists is that physical signs and symptoms of DCM may not accurately reflect physiologic parameters. Eighty percent of patients with pulmonary capillary wedge pressures greater than 25 mm Hg have no detectable rales.105 The use of more aggressive monitoring will depend partially on the operation. Patients receiving implantable defibrillators usually have severely depressed left ventricular function but are routinely managed without a PAC. Echo also is useful for patients undergoing both cardiac and noncardiac surgery. For most cardiac surgical procedures (mitral valve surgery, left ventricular assist devices), echo is considered necessary to evaluate the results of the operation. In both cardiac and noncardiac surgery, echo can provide real-time data as to how interventions such as addition of inotropes or vasodilators change ventricular function and indirectly assess CO.
Hemodynamic instability that may occur with DCM can be lessened with a low dose of inotrope and vasodilator. Acute administration of inotropes, such as dobutamine, improves hemodynamics temporarily, but tolerance begins in 3 to 4 days.77 Phosphodiesterase-3 inhibitors are useful if combined with another β-adrenergic agonist, such as milrinone and epinephrine, for short-term hemodynamic support. Afterload reduction in DCM is important because it improves regional and global indices of ventricular relaxation and ejection fraction during anesthesia when myocardial depression may be significant. It also may reduce valvular regurgitation and atrial volumes.106 The use of levosimendan, an effective calcium-sensitizing agent with vasodilatory and inotropic features, may be an excellent agent to maintain hemodynamics because it also increases myocardial performance without major changes in the oxygen consumption or effects on the diastolic function.107 Some patients with DCM may be chronically taking amiodarone, which has a negative inotropic effect that interacts with volatile agents to further reduce contractility and conduction, requiring careful titration of these anesthetic agents.108 Arrhythmogenic factors such as hypokalemia, hypomagnesemia, and sympathetic activation should be monitored and corrected. Because of the structural nature of DCM, atrial fibrillation is likely to be present in 25% of cases. Patients with atrial fibrillation and DCM show significantly diminished myocardial perfusion and perfusion reserve compared with those in normal sinus rhythm.109

Hypertrophic Cardiomyopathy
Referred to as idiopathic hypertrophic subaortic stenosis, hypertrophic obstructive cardiomyopathy, and asymmetric septal hypertrophy, among other names, the accepted name is hypertrophic cardiomyopathy (HCM). It is the most common genetic cardiac disease with marked heterogeneity in clinical expression, pathophysiology, and prognosis. The overall prevalence rate for adults in the general population is 0.2%,110 affecting men and women equally. The management continues to evolve with ever-increasing information discovered regarding HCM.
HCM is a primary myocardial abnormality with sarcomeric disarray and asymmetric left ventricular hypertrophy (Figure 22-13). The extent of sarcomeric disarray distinguishes HCM from other conditions.59 The hypertrophied muscle is composed of muscle cells with bizarre shapes and multiple intercellular connections arranged in a chaotic pattern.110 Increased connective tissue, combined with markedly disorganized and hypertrophied myocytes, contributes to the diastolic abnormalities of HCM that manifest as increased chamber stiffness, impaired and prolonged relaxation, and an unstable EP substrate that causes complex arrhythmias and sudden death. In contrast with the diastolic function, systolic function in HCM usually is normal or hyperdynamic, but eventually diminishes in the later stages of the disease.
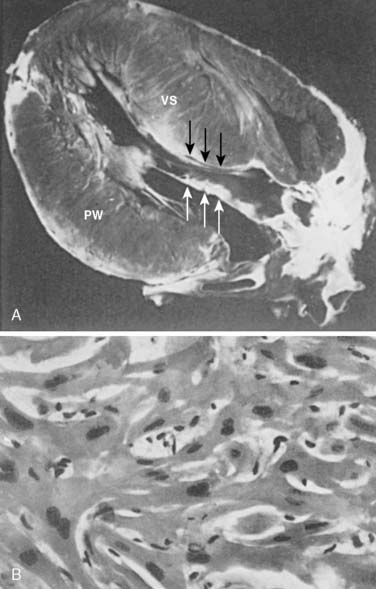
Besides diastolic dysfunction, the other major abnormality and fundamental characteristic of HCM is myocardial hypertrophy unrelated to increased systemic vascular resistance (SVR). This nonuniform, asymmetric hypertrophy typically occurs in the basal anterior ventricular septum and anterior free wall, with a disproportionate increase in the thickness of the ventricular wall relative to the posterior free wall (Figure 22-13A). Less commonly, the hypertrophy occurs at the apex, lateral wall, or concentrically. The left ventricular wall thickness is the most extensive of all cardiac conditions.110 Heart size may be deceptive because it may vary from normal to more than 100% enlarged. However, chamber enlargement is not responsible for the increase in ventricular mass but rather increases in wall thickness.
HCM is the most common genetic cardiovascular disease inherited in an autosomal dominant manner (1:500 of the general population).59 It may be caused by mutations in any of 10 genes; however, 3 genes that encode proteins of the cardiac sarcomere predominate in frequency.111 The similarity of the genes accounts for the many different expressions of HCM while resembling one disease entity. Patients without a family history of HCM may have sporadic gene mutations or simply a mild form of the disease. DNA analysis of mutant genes is the most definitive method to establish the diagnosis. The phenotype appears not only to depend on the mutation but also other modifier genes and environmental factors.111,112 Not all patients who possess a gene for HCM will manifest clinical features of the disease, reflecting incomplete genetic expression.110 A preclinical diagnosis in a patient without symptoms is possible with gene testing not routinely performed now or used to establish a treatment strategy.
HCM is unique for its range of clinical presentations from infancy to 90 years of age. Although major referral centers describe a disease with severe symptomatology, many elderly adults with mild-to-asymptomatic disease were unaccounted for, resulting in an actual annual mortality rate for HCM closer to 1% per year than previously reported to be 3% to 6% per year.110 Even left ventricular outflow tract (LVOT) obstruction may be tolerated longer than initially believed. Most patients with HCM are asymptomatic or have mild symptoms that progress slowly or not at all.63
Symptoms of HCM are nonspecific and include chest pain, palpitations, dyspnea, and syncope. Dyspnea occurs in 90% of patients secondary to diastolic abnormalities that increase filling pressures causing pulmonary congestion. Syncope occurs in only 20% of patients, but 50% may have presyncopal symptoms. The physical examination is sometimes unreliable because classic physical findings are associated with LVOT obstruction that is not present in all patients; however, a recent study observed prospectively 320 patients with HCM and their gradients.113 They found that 37% of patients had LVOT obstruction at rest and another 33% only with provocation. Symptoms usually appear in the second or third decade, but HCM is not a static disease, and left ventricular hypertrophy can occur at any age and increase or decrease dynamically throughout the person’s life.110 The ECG is abnormal in most individuals showing increased QRS voltage, ST-segment and T-wave abnormalities, QRS axis shift, and left ventricular hypertrophy with strain pattern. There is little correlation between ECG voltages and the degree of left ventricular hypertrophy.110 A chest roentgenogram may show left atrial enlargement or be normal.
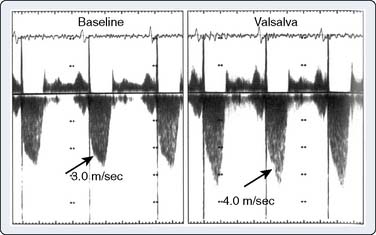
Echo is the modality of choice for the evaluation of HCM. Two-dimensional TTE usually allows the clinician to characterize the morphology of the disease and location of the hypertrophy. Doppler and color-flow imaging have typical appearances in HCM. Continuous-wave Doppler is used to quantify the gradient across the LVOT. The Doppler signal has a unique “dagger-shaped” appearance (Figure 22-14). The LVOT Doppler signal typically is obtained with an apical position of the transducer during TTE or from a deep transgastric view when TEE is utilized. If the LVOT gradient is less than expected, provocative maneuvers should be utilized in an effort to demonstrate an increased gradient. A Valsalva maneuver, by decreasing preload and ventricular filling, and inhalation of amyl nitrite are both noninvasive techniques that can be used in the conscious patient (see Figure 22-14). Mitral regurgitation usually accompanies LVOT obstruction when the obstruction is severe or symptomatic. The narrowing of the LVOT by septal hypertrophy necessitates an increase in flow velocity through the LVOT. This increase in flow velocity draws the anterior leaflet of the mitral valve into the LVOT, called the Venturi effect. The mitral leaflet and apparatus subsequently cause obstruction of flow through the LVOT, as well as mitral regurgitation. Temporally, mitral regurgitation occurs after LVOT obstruction: ejection → obstruction to left ventricular outflow → mitral regurgitation. The jet of mitral regurgitation typically is directed posterolaterally (Figure 22-15). It is important to note that if the mitral regurgitant jet is not posterolateral, there likely is another component to the mitral regurgitation such as mitral valve prolapse (MVP) or ruptured chordae. This has important ramifications in terms of planning a surgical repair. The diastolic filling pattern associated with HCM is that of severely impaired myocardial relaxation secondary to hypertrophy.110
Intraoperative TEE is essential in the care of patients undergoing septal myectomy.114 Before incision, TEE can identify the location of hypertrophy in relation to the aortic annulus, as well as the thickness of the ventricular septum, which assists the surgeon in planning the location and depth of the myectomy. The mitral valve should be closely evaluated for abnormalities that may contribute to mitral regurgitation, particularly if the regurgitant jet is directed centrally or anterior. After the myectomy, TEE is used to assess the degree of residual mitral regurgitation and evidence of continued systolic anterior motion (SAM) and LVOT obstruction. The ventricular septum also must be closely evaluated for evidence of shunting via an iatrogenic ventricular septal defect (VSD). It is common to see small shunts into the area of the LVOT from transection of coronary vessels at the site of the myectomy (Figure 22-16). It is important that these are not confused with shunting from a VSD. When an iatrogenic VSD occurs, the expected shunt would be from the LV into the RV, as opposed to the flow observed into the LV from transection of septal coronary artery branches. In addition, shunting through a VSD would be expected to occur predominantly during systole, whereas flow into the LV from septal perforators is predominantly during diastole.
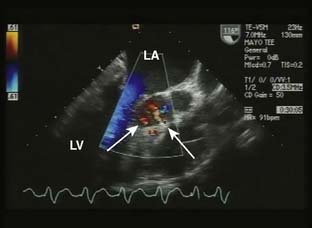
Figure 22-16 Transesophageal image from a patient after a septal myectomy for hypertrophic obstructive cardiomyopathy.
Angiography may show a left ventricular pressure gradient, decreased ventricular volume, and increased left ventricular end-diastolic pressure. Coronary artery diseases may be absent angiographically, but thallium redistribution scans demonstrate that HCM patients may experience myocardial ischemia often associated with atypical chest pain, not relieved with nitrates.115 Blunted coronary perfusion despite angiographically normal coronary arteries is characteristic of a coronary microvascular dysfunction that increases the risk for ischemia, especially in the subendocardium. More recently, the degree of LVOT obstruction and wall stress were found to exacerbate microvascular dysfunction.116 This may have implications for treatments that reduce left ventricular mass and wall stress instead of increasing diastolic filling time with medications such as β-blockers and calcium channel blockers, and partially explain the benefit of myectomy. Thallium abnormalities may represent relatively underperfused myocardium with abnormal intramyocardial coronary arteries that exist in patients with HCM. Abnormal coronary vasodilation in both nonhypertrophied and hypertrophied myocardium occur in HCM.117 This suggests that impaired coronary vasodilation has a major role in the pathogenesis of HCM and is not secondary to only myocardial hypertrophy. However, a mismatch between myocardial oxygen demand resulting from systolic and diastolic abnormalities, increased ventricular mass, and coronary circulation may exacerbate myocardial ischemia. Thus, myocardial death and scarring may then serve as a substrate for ventricular tachycardia and fibrillation.110
Two thirds of individuals with LVOT obstruction will become severely symptomatic, and there is a 10% mortality rate within 4 years of diagnosis. The LVOT is narrowed from septal hypertrophy and anterior displacement of the papillary muscles and mitral leaflets, creating a dynamic left ventricular outflow obstruction. Elongation of the mitral leaflets results in coaptation of the body of the leaflets instead of the tips. The part of the anterior leaflet distal to the coaptation is subjected to strong Venturi forces that provoke SAM, mitral septal contact, and ultimately, LVOT obstruction (Figure 22-17).111 However, studies suggest that the mechanism of SAM is more complex. SAM can occur before the opening of the aortic valve and the generation of maximum Venturi effect because of the position of the mitral leaflets on the LVOT, reflecting its importance in SAM compared with traditional explanations. Longitudinal flow in the ventricle may push the mitral valve into the LVOT.118 SAM of the anterior leaflet also may cause mitral regurgitation that is unlike that seen with intrinsic structural abnormalities of the valve. The onset and duration of mitral leaflet-septal contact determine the magnitude of the gradient and the degree of mitral regurgitation. The pressure gradient between the aorta and LV (Figure 22-18) is worsened by decreased end-diastolic volume, increased contractility, or decreased aortic outflow resistance (Figure 22-19).119 The cavity of the LV often is small in those with severe gradients (Figure 22-20).
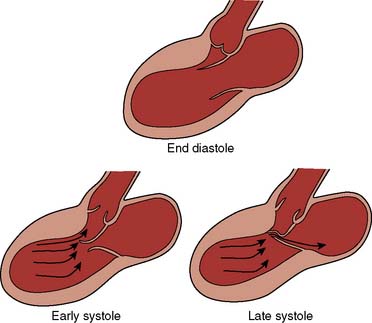
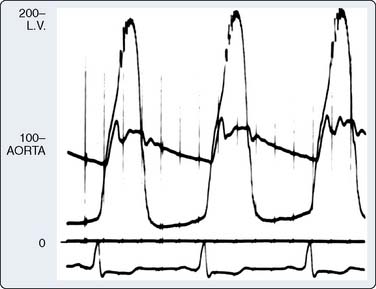
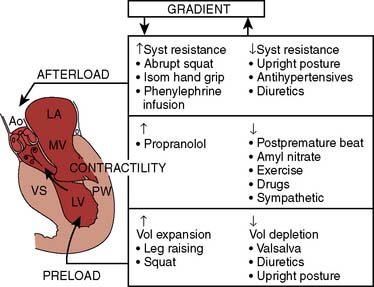
Figure 22-20 Midventricular hypertrophy.
(From Wigle ED, Rakowski H, Kimball BP, et al: Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 92:1680, 1995, by permission.)
Dynamic LVOT obstruction is not limited to HCM but may occur in cardiac tamponade or acute MI.119 It may be seen in the immediate postoperative period after aortic valve replacement for aortic stenosis or mitral valve repair. Importantly, dynamic pressure gradients do not necessarily correlate with symptoms of HCM. Significant functional limitation, disability, and sudden death may all occur even with no gradient. However, gradients exceeding 30 mm Hg are usually of physiologic and prognostic importance in HCM patients.111 The severity of symptoms is not worse once a gradient exceeds 30 mm Hg. LVOT obstruction is an independent predictor of death in HCM.120 Because LVOT obstruction is likely to be associated with increased wall stress, myocardial ischemia, cell death, and eventual fibrosis, treatment to relieve it is desirable.
The clinical course and treatment of HCM are best approached in relation to subgroups: sudden death, CHF, and atrial fibrillation with embolic stroke (Figure 22-21). Sudden death increases the annual mortality rate from 1% to 5% overall.110 Often these patients are asymptomatic or mildly symptomatic and constitute a small part of the HCM population. Sudden death is not age restricted but occurs more commonly in younger individuals, frequently in association with physical exertion. HCM is the most common cause of sudden death in otherwise healthy young individuals, reaching up to 6% per year in those 20 to 30 years of age.121 Risk stratification for sudden death is useful now that it has been found that ventricular tachycardia or ventricular fibrillation is the culprit.110,122 High-risk individuals are more likely to have these characteristics: diagnosis by 30 years of age, prior cardiac arrest, symptomatic ventricular tachycardia per Holter monitor, and family history of sudden death or syncope.121 Additional risk factors include identification of a high-risk mutant gene, unexplained syncope, abnormal blood pressure with exercise, resting LVOT obstruction of 30 mm Hg or greater, atrial fibrillation, and even coronary artery disease.111,121 Two or more risk factors are associated with a risk for sudden death of 4% to 5% per year.
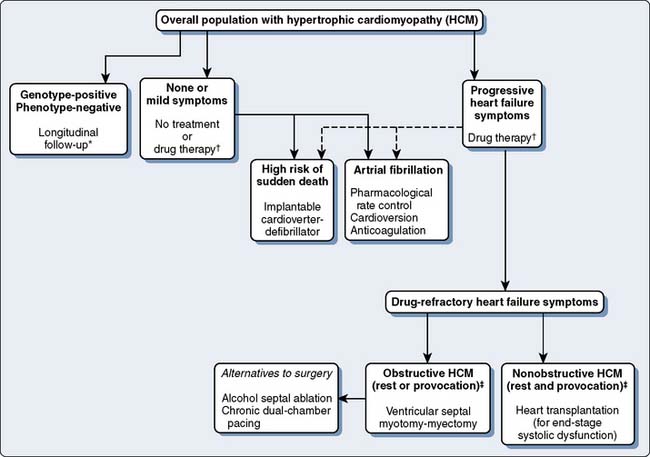
The value of EP testing as it relates to prediction of sudden death is mixed.111,123,124 Recently, routine risk stratification with EP testing was abandoned in HCM patients.111 Extreme left ventricular wall thickness has been associated with greater incidence of spontaneous ventricular arrhythmias and sudden death. Myocardial ischemia also appears to be related to sudden death in younger patients.125 In the future, genotyping may be able to reliably define the risk for sudden death, but currently there is no ability to screen patients to determine risk stratification with clinical gene tests for characteristics such as sudden death.126 Septal myectomy reduces the risk for sudden death in patients compared with those with LVOT obstruction and no surgery and those with nonobstructive HCM. The mechanism for this improved survival is indeterminate.127
The only effective modality to prevent sudden death associated with HCM currently is the implantable cardioverter-defibrillator. Pharmacologic therapy for prevention of sudden death recently was shown to reduce symptoms but not risk for sudden death.128 Symptomatic patients or those with a positive EP test appear to benefit from implantable cardioverter-defibrillator placement. A retrospective study found that 25% of high-risk cohorts required termination of lethal ventricular arrhythmias and 60% experienced some type of intervention by an implantable cardioverter-defibrillator.122 The implantable cardioverter-defibrillator has been shown in a randomized, prospective trial to be superior to drug therapy.129 However, a long-term follow-up of patients receiving implantable cardioverter-defibrillators for HCM has shown a worrisome rate of inappropriate (5.3%/year) versus appropriate (4%/year) shocks.130 The overall incidence rate of device complications was concerning at 36% during a 5-year period. Even though the implantable cardioverter-defibrillator saves lives in high-risk patients, the risk for complications should factor into the decision to implant it. Amiodarone is no longer the first line of antiarrhythmic therapy for patients at risk for sudden death as previous studies were nonrandomized case studies.63
Another subgroup is patients with CHF who develop progressive symptoms with preserved systolic function but quickly advance to end-stage CHF with impaired systolic function.111 Symptoms are primarily attributed to the diastolic abnormality; therefore, symptomatic relief is achieved by improving ventricular relaxation. Medications with negative inotropic effect generally are successful in relieving symptoms of exercise intolerance and dyspnea associated with CHF,131 but symptoms may return in up to 60% of pharmacologically treated patients.132 In general, β-blockers have been a mainstay of therapy for years in adults and are preferred over calcium channel blockers. High doses of β-blockers usually are needed for therapeutic value. Verapamil may be given if β-blockers are not tolerated or ineffective, but symptomatic improvement may be less than 50%.132 The combination of β-blockers and calcium channel blockers has not proven beneficial. β-Blockers relieve symptoms of angina and dyspnea and improve exercise tolerance by limiting the gradient associated with exercise. The gradient is not reduced at rest by these medications.111 Other beneficial effects of β-blockers include heart rate reduction, lower myocardial oxygen demand, and longer diastolic filling times, but diastolic function is neither improved nor is long-term survival prolonged. Verapamil improves diastolic function and ventricular relaxation, causing improved filling and decreased obstructive features in 50% of HCM patients. Patients with obstructive symptoms may worsen with calcium channel blockers that contain strong vasodilator properties. Disopyramide, a negative inotrope that alters calcium kinetics and produces a vasoconstrictor effect, has been recommended instead of calcium channel blockers in obstructive HCM.111 Disopyramide is the most effective agent to reduce LVOT obstruction and gradient, as well as relieve symptoms.133 Combined with β-blockers, disopyramide prolongs exercise times more successfully than verapamil.
Surgical correction of HCM is directed primarily at relieving symptoms of LVOT obstruction in the 5% of patients who are refractory to medication.111 In general, these are individuals with subaortic gradients in excess of 50 mm Hg with or without provocation and frequently associated with severe CHF.63,110 A myotomy-myectomy through a transaortic approach is the primary method used to relieve the obstruction (Figure 22-22). The muscle is excised from the proximal septum extending just beyond the mitral valve leaflets that widens the LVOT. Currently, the classic Morrow technique has been replaced by a more extensive resection that is described in more detail elsewhere.133,134 This is a technically challenging operation because of the limited exposure and precise area to excise the muscle. It usually is reserved for centers with considerable experience with myectomy. When myectomy is successful, the LVOT is widened and SAM, mitral regurgitation, and outflow gradient are all decreased. Mitral valve repair or replacement has accompanied myectomy when there is coexistent primary mitral valve structural abnormalities, but mitral valve replacement is not indicated for treating LVOT obstruction alone.118 If the valve is not repaired during myectomy, incomplete or temporary relief of obstruction may occur. Recently, Wan et al135 described the use of mitral valve repair in those with LVOT obstruction and degenerative mitral valve disease. Mitral valve repair was effective compared with replacement with low early mortality and successful treatment of both obstruction and mitral valve regurgitation. Mitral valve replacement is rare.
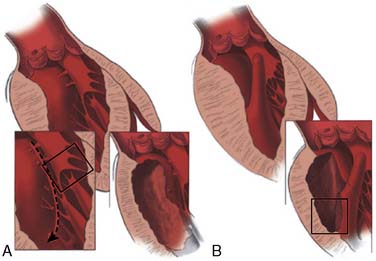
Surgery provides persistent, long-lasting relief from symptoms that exceeds any form of current medical treatment.131 Five years after myectomy, 85% of patients have improved enough to advance one NYHA functional class.136 The concomitant reduction in left atrial size may partially account for the lower incidence of atrial fibrillation after myectomy.111 Septal myectomy results in reduction in left ventricular mass that exceeds the myocardium resected over time, indicating a relief of the pressure overload.137 Myectomy also is associated with a mortality rate of less than 1% in major institutions.132 This low mortality rate is even more noteworthy because surgical patients usually have more severe disease than those treated medically. Long-term survival rates at 1, 5, and 10 years are 98%, 96%, and 83%, respectively. Surgical patients have an overall survival rate that is equivalent to the age- and sex-matched general population.127 Complications of myectomy such as complete heart block or septal perforation (0% to 2%) are rare.133 Similarly, recurrence of LVOT gradient or SAM requiring repeat myectomy is less than 2%.118
Surgery is not recommended for HCM without an obstructive component or if the individual is asymptomatic to mildly symptomatic for the following reasons: (1) Better survival with surgery versus medical therapy is not established; (2) operative mortality may exceed the risk of HCM in mildly affected patients; (3) LVOT obstruction may be compatible with longevity in selected individuals; and (4) no evidence exists that surgery halts the progression of HCM to end-stage.111 End-stage patients with nonobstructive HCM who have severely reduced systolic function are effectively treated with heart transplantation with 7-year survival rate of 90% that compares equally to those with DCM who require transplantation.138 Medical treatment for asymptomatic HCM patients is not clearly indicated.
Possible alternatives to surgery have included dual-chamber pacing and percutaneous alcohol septal ablation. Previously, atrioventricular sequential pacing had been shown to decrease subaortic gradients, possibly by causing abnormal septal motion, and thus was considered a possible treatment even though the mechanism has not been proved.131 Pacing has become the most rigorously evaluated treatment of all therapies for HCM. A series of nonrandomized and randomized studies have shown reductions in the LVOT gradient and improvement in symptoms, but others have found no differences in gradient, NYHA class, quality-of-life score, exercise duration, or maximal oxygen uptake, reflecting a significant placebo effect.139 Within groups, there appear to be responders, but no variables that identified these patients.118 Consequently, the optimal use of pacing in HCM requires continued reevaluation.131 When pacing was compared with surgical therapy, LVOT gradient was reduced to less than 20 mm Hg in only 26% of patients, compared with more than 90% of surgical patients,118 creating drastically fewer indications for pacing. Furthermore, symptoms were improved in all patients with surgery compared with only 50% with pacing. The role of pacing has been relegated to one in which there are absolute contraindications to other therapies.
Septal ablation with alcohol is a nonsurgical myocardial reduction first attempted in 1994 based on interruption of blood supply to the interventricular septum, causing infarction. This resulted in reduced septal thickness and LVOT modeling with resolution of symptoms in HCM.140 Alcohol is administered through an angioplasty balloon to the first major septal perforator of the left anterior descending coronary artery. The success rate has been good as indicated by a recent systematic review of ablation incorporating 42 studies and nearly 3000 patients.141 The study showed effective gradient reduction and improvement in NYHA class comparable with myectomy 12 months after ablation. However, 20% of patients required a second procedure, and some even required myectomy after alcohol ablation. A retrospective review of 375 patients who underwent alcohol ablation identified 20 patients who underwent subsequent surgical myectomy for recurrent dynamic LVOT obstruction.142 The surgery was successful in all of the failed ablation patients, indicating myectomy as rescue therapy for failed alcohol septal ablation. There is concern about the incidences of complications and mortality accompanying alcohol ablation. Annual mortality rate with alcohol septal ablation in the best centers in the United States is 2%, with Canada reporting 4% to 10%.118 Besides mortality, the incidence of nonfatal complications is a concern compared with surgical myectomy. Heart block necessitating pacemaker placement has been reported in between 11% and 38% of patients.143 The toxic effect of alcohol on both the coronary circulation and the myocardium has the potential to generate morbidity.131
Perhaps the greatest concern is that fewer surgeons are learning and performing myectomy because of the steep learning curve, but the relative ease of the septal ablation in comparison with myectomy has greatly increased the individuals performing ablation. Because the longest follow-up studies for alcohol ablation are 2 years, long-term benefit for this procedure is lacking. Retrospective studies do not permit any conclusion regarding the superiority of septal ablation with myectomy, so clinicians are awaiting a long-term comparative study to define its role in HCM. Because management of HCM continues to be made based on experience and consensus to a large degree, surgical myectomy remains the first option for HCM with ablation as an alternative, especially if serious comorbidity exists.111 Treatment will continue to evolve as the proper patient selection and long-term effects are evaluated.
Anesthetic Considerations
Anesthetic management of individuals with HCM is based on similar principles for those having cardiac or noncardiac surgery. The characteristic diastolic dysfunction makes the heart sensitive to changes in volume, contractility, and SVR. Because of this diastolic dysfunction, an acute increase in the pulmonary artery pressure may warn of the rapid onset of pulmonary congestion and edema. If the patient has LVOT obstruction, anesthetic management should minimize or prevent any exacerbation of obstruction and the corresponding increase in the intraventricular gradient that will affect systolic blood pressure. Induction of anesthesia is a hazardous period because the preoperative fast will reduce preload combined with a rapid decline in the venous tone, provoking an increase in LVOT obstruction. Central venous pressure monitoring is recommended to optimize and maintain preload. Hypotension may be treated temporarily with positioning (Trendelenburg), volume replacement, and/or vasoconstriction. Vasoconstrictors rather than inotropes are preferred to maintain SVR.144 Intravenously administered agents, such as narcotics, have been used successfully in HCM for induction of anesthesia. For shorter surgical procedures, propofol is popular, but its effect on hemodynamics has not been fully established. The systolic blood pressure often decreases significantly with propofol during induction of anesthesia. The mechanism of this decline in blood pressure is not completely defined but is likely an interaction of baroreflex activity, direct peripheral vasodilation, blunting of the sympathetic nervous outflow, and possibly a decrease in the myocardial contractility.145 An experimental hamster with HCM received propofol, and the myocardium was found to be unaffected compared with the healthy muscle.100 In addition, a continuous propofol infusion has been used successfully in a patient with HCM for mitral valve replacement and myectomy.144
For anesthesia maintenance, the volatile agent, halothane, is advantageous because it decreases contractility and heart rate but rarely is used today. However, halothane, in comparison with enflurane and isoflurane, has the least effect on SVR and heart rate. The potential for severe myocardial depression with volatile agents in these patients is less than once believed. There is continuing evidence in hamster models with HCM that the negative inotropic effect in the affected myocardium compared with healthy myocardium is more severe with desflurane than either isoflurane or sevoflurane.101,103 Vecuronium is the preferred muscle relaxant because it does not have histamine-releasing properties or hemodynamic effects.
Arrhythmias may complicate the anesthetic management of individuals with HCM. Ninety percent of adults with HCM have complex ventricular arrhythmias on Holter monitoring irrespective of whether HCM is obstructive or nonobstructive.111 Nonsustained ventricular tachycardia occurs in 25% to 33% of patients, whereas sustained monomorphic ventricular tachycardia is rare.146 Asymptomatic, nonsustained ventricular tachycardia tends to be benign.122 Implantable cardioverter-defibrillators need to be temporarily suspended in patients before use of cautery in the operating room (see Chapter 25).
Besides ventricular arrhythmias, there is a 30% to 50% incidence rate of paroxysmal supraventricular tachycardia in these patients.146 Atrial fibrillation is the most common supraventricular tachycardia, occurring in 25% of individuals.110 New onset of atrial fibrillation is observed in 84% of patients who report worsening symptoms.131 The combination of worsening symptoms and new onset of atrial fibrillation may rapidly deteriorate to CHF.147 Prompt cardioversion may be necessary. Junctional rhythms also are poorly tolerated in HCM because of the loss of the atrial kick. Chronic atrial fibrillation can be controlled with β-blockers and verapamil, although this combination may be deleterious to some individuals with severe LVOT obstruction and CHF.110 Amiodarone is especially effective in restoring normal sinus rhythm in HCM and reducing recurrences.

Restrictive Cardiomyopathy
RCM has included such entities as amyloidosis and eosinophilic endomyocardial disease. The 1995 WHO guidelines defined RCM as “restrictive filling and reduced diastolic volume of either or both ventricles with normal or near-normal systolic function and wall thickness.”59 Instead of being classified according to morphologic criteria, as are HCM and DCM, RCM is characterized by function. It may appear in the final stages of other cardiac conditions. Although a clinically challenging diagnosis to make, it is important to distinguish “restrictive” physiology from “constrictive” because management and treatment are decidedly different. Restrictive/constrictive physiology is not solely limited to RCM, but occurs in mitral stenosis, tricuspid stenosis, and aortic stenosis.
RCM is much less frequent than DCM or HCM.148 It is classified as myocardial (infiltrative, noninfiltrative, and storage) or endomyocardial (Box 22-1) according to cause. The compliance of the “restrictive” myocardium is reduced by one of these processes: extracellular infiltration, intracellular accumulation of abnormal substances, inflammation, or endocardial disease.148,149 Restrictive myocardial disorders are characteristically atypical in presentation and hemodynamics at times, complicating perioperative management. With idiopathic RCM, the cause is entirely unknown and lacks any identifiable histopathologic distinctiveness. Its occurrence is quite rare, with few actual series in adults.150 More extensive reviews of the many types of RCM are available.150–152
BOX 22-1 Classification of Types of Restrictive Cardiomyopathy According to Cause
From Kushwaha SS, Fallon JT, Fuster V: Medical progress: Restrictive cardiomyopathy. N Engl J Med 336:268, 1997.
Systolic function is minimally affected in RCM. Diastolic dysfunction is evident by abnormalities of ventricular relaxation and compliance. Impaired ventricular relaxation occurs before abnormal compliance. Poor compliance of the ventricle causes rapid filling in early diastole and the characteristic ventricular diastolic waveform of dip-and-plateau (square-root sign; Figure 22-23). However, the characteristic dip-and-plateau pattern may be absent in 50% of affected individuals because it depends on the degree of impaired ventricular relaxation and the level of atrial driving pressure.151 The atrial pressure waveform will have the appearance of an M or W pattern because of the rapid y descent combined with a slightly greater x descent. The rapid y descent is due to the rapid early decline of the atrial pressure corresponding to the early diastolic ventricular filling. Filling early in diastole is accentuated and impeded during the rest of diastole because of reduced ventricular distensibility. Pressure in the ventricle increases precipitously in response to small volume. Once ventricular filling is restricted, the prognosis is much worse. Both ventricles appear thick with small cavities. In contrast, the corresponding atria appear dilated despite normal left ventricular volumes. Cardiomegaly or ventricular dilation should not exist. The left-sided pulmonary venous pressures usually exceed the right-sided venous pressures by 5 mm Hg. The pulmonary artery systolic pressure also may be increased to 50 mm Hg.
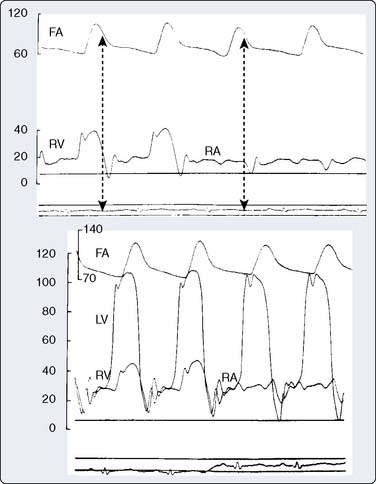
Figure 22-23 Intracardiac pressure tracings from a patient with idiopathic primary restrictive cardiomyopathy.
(From Giuliani ER, Fuster V, Gersh BJ, et al: Cardiology: Fundamentals and Practice. St. Louis, Mosby, 1991, by permission of Mayo Foundation.)
Because one or both ventricles may be involved, symptoms may predominate as right- or left-sided. The increased filling pressures that occur in early diastole of both ventricles lead to symptoms of right-sided failure manifested by increased venous pressures, peripheral edema, and ascites, or left-sided failure manifested by CHF, progressive dyspnea, and orthopnea. Both groups of symptoms may occur separately or simultaneously. Insidious onset of fatigue and exertional dyspnea are common. Symptoms and signs should be present without evidence of cardiomegaly. The jugular venous pulse fails to fall during inspiration and may even rise (Kussmaul’s sign).152 Pulsus paradoxus is infrequent and nonspecific. The liver may be enlarged and actually pulsatile. The chest roentgenogram is nondiagnostic. Pulmonary congestion and a normal or smaller than anticipated heart size suggest a diagnosis of RCM. Thromboembolic complications are common with the initial presentation of the disease. The ECG may reflect abnormalities of depolarization such as bundle branch block, low voltage, and QR or QS complexes, but these are not specific to RCM and rarely are useful to distinguish RCM from constrictive pericarditis (CP). ECG evidence of biatrial enlargement may be present.149
It is important to distinguish between RCM and CP because management of the two diseases is very different. Important distinguishing features of CP and RCM are listed in Table 22-4. Both conditions have rapid and early diastolic filling with increased filling pressures and normal filling volumes. In RCM, the impediment to filling is usually an infiltrative process involving the myocardium rather than the pericardium. Although not an absolute sign, the right ventricular and left ventricular end-diastolic pressures are increased and equal with constrictive physiology, whereas in RCM, end-diastolic pressure is usually 5 mm Hg greater on the left than on the right side because of the unequal compliance between the two ventricles.150,153 Unfortunately, symptoms and physical signs do not reliably discriminate between RCM and CP.
TABLE 22-4 Differentiation of Pericardial Constriction versus Myocardial Restriction
| Characteristic | Constrictive Pericarditis | Restrictive Cardiomyopathy |
|---|---|---|
| Jugular venous waveform | Increased with less rapid y-descent | Increased with, more rapid y-descent Large A waves |
| LAP > RAP | Absent | Almost always |
| Auscultation | Early S3 high pitched; no S4 | Late S3 low pitched; S4 in some cases |
| Mitral or tricuspid regurgitation | Frequently absent | Frequently present |
| Chest roentgenogram | Calcification of pericardium (20–30%) | Pericardial calcification rare |
| Heart size | Normal to increased | Normal to increased |
| Electrocardiogram | Conduction abnormalities rare | Conduction abnormalities common |
| Echocardiogram | Slight enlargement of atria | Major enlargement of atria |
| Right ventricular pressure waveform | Square-root pattern | Square-root pattern; dip-and-plateau often less prominent |
| Right and left heart diastolic pressures (mm Hg) | Within 5 of each other in almost all cases | Seldom within 5 of each other |
| Peak right ventricular systolic pressure (mm Hg) | Almost always < 60, sometimes < 40 | Usually > 40 sometimes > 60 |
| Discordant respiratory variation of peak ventricular systolic pressures | Right and left are out of phase with respiration | In phase |
| Paradoxical pulse | Often present | Rare |
| CT/MRI | Thickened pericardium | Rarely thickened pericardium |
| Endomyocardial biopsy | Normal or nonspecific changes | Nonspecific abnormalities |
CT, computerized axial tomography; LAP, left atrial pressure; MRI, magnetic resonance imaging; RAP, right atrial pressure.
From Hancock EW: Cardiomyopathy: Differential diagnosis of restrictive cardiomyopathy and constrictive pericarditis. Heart 86:343–349, 2001; and Chatterjee K, Alpert J: Constrictive pericarditis and restrictive cardiomyopathy: Similarities and differences. Heart Failure Monit 3:118–126, 2003.
Advances in echo and other noninvasive methods have been used to try to reliably discriminate between pericardial disease and RCM. The usual 2D echo findings are those of normal size, thickness, and preserved systolic function of the ventricles with enlargement of the atria (Figure 22-24). Classic Doppler findings are described in Table 22-5. Doppler findings may indicate the severity of RCM once the diagnosis is certain. Because the pericardium is responsible for CP and a noncompliant ventricle for RCM, some differences exist between RCM and CP regarding M-mode, 2D, and Doppler examinations.154 A pericardial thickness of 3 mm measured by echo is indicative of CP. But overall, Doppler and echo have insufficient sensitivity and specificity to provide a clinically significant differentiation between RCM and CP. More recently, tissue Doppler imaging has demonstrated some success in providing differentiation between RCM and CP by measuring E velocity to determine ventricular relaxation with 89% sensitivity and 100% specificity.155 Other noninvasive methods that may be helpful are the CT to determine the thickness of the pericardium and the MRI to diagnose infiltrative processes such as amyloidosis.149
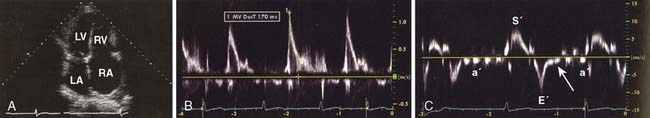
TABLE 22-5 Doppler and Echocardiographic Features Differentiating Restrictive Cardiomyopathy and Constrictive Pericarditis
| Modality | Restrictive Cardiomyopathy | Constrictive Pericarditis |
|---|---|---|
| M-mode | No septal notch | Septal notch |
| 2D | No septal shudder | Septal shudder |
| Pulsed-wave mitral and tricuspid inflow | Less variation | Prominent respiratory variation (> 10%) Significant decrease in IVRT from inspiration to expiration (usually noted in the first beat after expiration) |
| Pulsed-wave pulmonary vein | Less variation | Significant increase (> 18%) in peak diastolic velocity from inspiration to expiration |
| S/D ratio < 0.4 | S/D ratio ≥ 0.65 in inspiration | |
| Pulsed-wave hepatic veins | > 25% reduction in diastolic forward flow Prominent late diastolic flow reversal after the onset of expiration |
|
| RVSP | > 40 mm Hg | < 40 mm Hg |
| Tricuspid regurgitation | Less respiratory change | Marked inspiratory increase in peak velocity, time-velocity integral, and duration of jet |
| Tissue-Doppler mitral annulus | E′ < 8 cm/sec E/E′ > 15 |
E′ ≥ 8 cm/sec E/E′ < 15 |
| Color M-mode | Vp < 45 cm/sec | Vp ≥ 100 cm/sec |
E′, early peak velocity; IVRT, isovolumic relation time; RVSP, right ventricular systolic pressure; S/D, systolic/diastolic; Vp, propagation velocity.
From Tam JW, Shaikh N, Sutherland E: Echocardiographic assessment of patients with hypertrophic and restrictive cardiomyopathy: imaging and echocardiography. Curr Opin Cardiol 17:474, 2002.
Cardiac catheterization still is performed to differentiate between RCM and CP. During catheterization, increased left and right venous pressures with large A and V waves may be observed in RCM (see Figure 22-23). It is not uncommon to see right atrial pressures of 15 to 20 mm Hg. The ventricular pressure curve may display the diastolic dip-and-plateau pattern (square-root sign) as described previously. Recently, more advanced catheterization criteria for differentiation of RCM and CP have been devised.156 This new measurement, systolic area index, assesses the dynamic changes in ventricular pressure area during inspiration and expiration to distinguish CP from RCM. In CP, there is a dissociation between intrathoracic and intracardiac pressures that causes decreased filling in the LV during inspiration. The constricting pericardium also causes increased ventricular interaction so that the RV is better filled during inspiration. This new index has 97% sensitivity and 100% predictive accuracy in patients with documented CP compared with older catheterization indices that generated predictive accuracy for RCM and CP that was less than 75%.
Endomyocardial biopsy may differentiate RCM from CP. The biopsy in RCM rarely is normal compared with CP. Although a biopsy may identify specific causes of restrictive physiology,148 more often, only patchy endocardial and interstitial fibrosis with some myofibril hypertrophy are found.150,153 If biopsy or tests such as radionuclide angiography, Doppler, or echo do not identify either RCM or pericardial disease, exploratory surgery may be needed. Surgery is the most definitive method of differentiating RCM from CP, but the patient with RCM may not tolerate anesthesia and surgery well, so it is used sparingly.157
There is no specific treatment for RCM. Various regimens have been developed based on the specific cause of RCM158 (Table 22-6). It is important to rule out the presence of extracardiac diseases that may hamper effective treatment of RCM before it becomes intractable. Diuretics often are administered to reduce pulmonary and venous congestion, but low CO with severe hypoperfusion is a risk. With the characteristic limited stroke volume, heart rate with atrial contraction is important to support CO. Enlarging atria may increase the risk for supraventricular arrhythmias. The lower CO and enlarging atria make chronic anticoagulation important in view of the risk for thrombin formation. A surgical option for RCM is rare with the exception of endomyocardial fibrosis with eosinophilic cardiomyopathy. The fibrotic endocardium may be excised in combination with mitral or tricuspid valve replacement.
TABLE 22-6 Treatment Strategies in Restrictive Cardiomyopathy
| Etiology | Specific Symptom | Treatment |
|---|---|---|
| Idiopathic | To relieve congestion | Diuretics |
| Control of heart rate | β-Blockers, amiodarone, heart rate regulating CCBs | |
| Atrial fibrillation: to maintain sinus rhythm paroxysmal or persistent | Amiodarone, dofetilideLong-term anticoagulation | |
| To control ventricular rate | β-Blockers, heart rate–regulating calcium channel blockers, amiodarone, digoxin, atrioventricular node ablation and pacemaker | |
| Atrioventricular block | Dual-chamber pacing | |
| To enhance myocardial relaxation | Calcium channel blockers (unproven), angiotensin inhibition (unproven) | |
| Refractory heart failure | Cardiac transplantation | |
| Amyloidosis | Melphalan, prednisone, and colchicine; stem cell implant (under investigation); heart and liver transplant (in rare instances) | |
| Hemochromatosis | Phlebotomy, desferrioxamine | |
| Carcinoid heart disease | Somatostatin analogs; serotonin antagonists; valvuloplasty for severe tricuspid and pulmonary stenosis; valve replacement for severe tricuspid and pulmonary regurgitation | |
| Sarcoidosis | Corticosteroids; pacemaker for heart block; implantable cardioverter defibrillator for ventricular arrhythmia; cardiac transplant for refractory heart failure | |
| Eosinophilic endocarditis | Initial stage: corticosteroids, hydroxyurea, vincristine | |
| Endomyocardial fibrosis | Endocardiectomy with repair or replacement of tricuspid and mitral valves |
CCB, calcium channel blocker.
From Chatterjee K, Alpert J: Constrictive pericarditis and restrictive cardiomyopathy: Similarities and differences. Heart Failure Mont 3:125, 2003.
Survival with RCM depends largely on the causative factor. True idiopathic RCM has a 5-year overall survival rate of 64%, but if increasing signs of pulmonary venous congestion appear, the prognosis worsens.150 Transplantation is not always an option for adults with RCM because the processes responsible for RCM would invariably affect the newly transplanted heart.
Anesthetic Considerations
Adults with RCM rarely require cardiac surgery for reasons other than mitral or tricuspid valve replacement and transplantation.159 Occasionally, anesthesia will be administered for a scheduled pericardiectomy only to discover RCM instead of CP. Despite essentially normal ventricular systolic function, the diastolic dysfunction and filling abnormalities result in a poor CO and systemic perfusion.153 Aggressive preoperative diuretic therapy may contribute hypotension secondary to inadequate circulating blood volume. In contrast, pulmonary congestion resulting in increased airway pressures may impair oxygen delivery to the tissues.
Induction of anesthesia should avoid medications associated with decreased venous return, bradycardia, or myocardial depression. Fentanyl (30 μg/kg) or sufentanil (5 μg/kg) provides stable hemodynamics for induction and maintenance in patients with poor myocardial function. These anesthetics provide minimal hemodynamic fluctuation in patients undergoing cardiac surgery for valvular disease who have developed severe preexisting volume or pressure loads on the heart. Etomidate is an excellent alternative or adjuvant to fentanyl as an induction agent because it has minimal effect on contractility of the cardiac muscle as demonstrated in patients undergoing cardiac transplantation.96 Similarly, ketamine has been advocated for induction in patients with cardiac tamponade or CP because sympathetic activity is preserved. Ketamine is an excellent choice to use with fentanyl for induction in patients with severe myocardial dysfunction and low CO caused by cardiomyopathy. Concerns of exacerbating PAH with ketamine are unfounded if ventilation is maintained. Shorter-acting narcotics such as remifentanil may be unsuitable for patients with RCM because of a greater incidence of bradycardia and severe hypotension.94 Fentanyl-etomidate-isoflurane has been shown superior to remifentanil-sevoflurane in terms of maintaining arterial pressure and heart rate.95 Propofol may have no direct myocardial depression, but indirect inhibition of sympathetic activity and a vasodilatory property may cause hemodynamic instability in patients with RCM. Sevoflurane and desflurane are attractive agents for maintenance of anesthesia in this population because they have been shown not to adversely affect the ability of the LV to respond to increased work despite their negative inotropic properties.104
Invasive hemodynamic monitoring is important because both left and right filling pressures do not accurately reflect ventricular volumes, so small volume shifts may greatly impact CO. Inotropic support may be indicated to support hemodynamics because the risk for death from low CO is high. Despite chronic CHF, diuretics and vasodilators may be deleterious because greater filling pressures are needed to maintain CO. Some individuals may be sensitive to β-blockers and cardiac glycosides.153 Patients may be chronically taking amiodarone, which has a negative inotropic effect that interacts with volatile agents to further reduce contractility and conduction, requiring careful titration of these anesthetic agents.108

Arrhythmogenic Right Ventricular Cardiomyopathy
Formerly called arrhythmogenic right ventricular dysphasia, arrhythmogenic right ventricular cardiomyopathy (ARVC) was defined by the 1995 WHO as “progressive fibrofatty replacement of right ventricular myocardium, initially with typical regional and later global right ventricular and some left ventricular involvement, with relative sparing of the septum.”59 Evidence of a more progressive involvement of not only the RV but LV with long-term follow-up convinced the WHO to classify ARVC as a “disease of the myocardium” that incorporates the many different clinical presentations and aspects of this condition.
ARVC is familial in 30% to 50% of cases, with primarily autosomal dominant inheritance with variable expressivity and reduced penetrance.65 The incidence and prevalence are uncertain, but it usually appears during adolescence, though it may occur in younger individuals. It presents most commonly with the onset of arrhythmias ranging from premature ventricular contractions to ventricular fibrillation originating from the RV. The disease is now known to proceed through three phases: (1) concealed phase without symptoms but some EP changes that place patients at risk for sudden death; (2) overt arrhythmias; and (3) advanced stage with myocardial loss, biventricular involvement with CHF.160 Diagnosis is rare in the early stages,161 but not at autopsy.162 Distinguishing symptoms may be missing when the structural and functional changes are present or absent in the RV.163 Standard diagnostic criteria have continued to be revised.164 Postmortem examination reveals diffuse or segmental loss of myocardium, primarily in the RV, replaced with fat and fibrous tissue, and right ventricular dilation and thinning of the wall (Figure 22-25). The diagnosis also includes the functional changes associated with the RV beyond the structural alterations. These changes are believed to occur by three possible mechanisms: myoblasts differentiate to adipoblasts, apoptosis, or inflammation.161 An inflammatory process is evident within the myocardial tissue. The replacement of myocardium with fat and fibrous tissue creates an excellent environment for a fatal arrhythmia, possibly the first sign of ARVC.160 Although considered a disease of the RV, left ventricular involvement also can occur and even precede RV presentation.165 Sudden death occurs in up to 75% of patients, although it is difficult to accurately state in view of number of patients who go undiagnosed. Sudden death occurs most often during sports-related exercise, primarily from ventricular tachycardia/fibrillation.166 Although a rare disease, it accounts for 20% of sudden deaths in the young.163
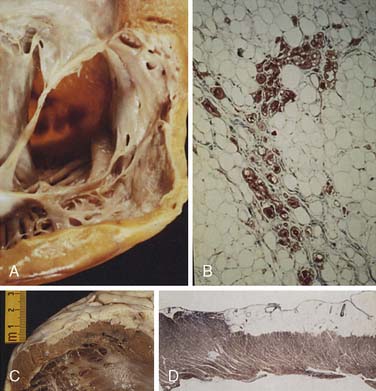
The electrical instability characteristic of ARVC is not solely responsible for the natural history of the disease. Clinical presentation may range from asymptomatic myopathic involvement to overt clinical disease with diffuse biventricular involvement.160,166 Progressive right and left ventricular dysfunction can be appreciated with serial echo examinations. Echo findings from 29 patients with ARVC have been described (Figure 22-26).167 The systolic and diastolic right ventricular outflow tract dimensions were increased on 2D echo. Regional wall motion abnormalities of the RV were common, and more than half of patients had abnormal right ventricular systolic function. Right ventricular trabeculations were also commonly found. Although there are typical echo features in ARVC, the disease may present before the appearance of these morphologic features. Recently, it has been shown that significant ventricular mechanical dyssynchrony is present in 50% of patients.168 This leads to a more dilated RV, as well as one with poorer function compared with those without it. Myocardial loss with progressive ventricular dysfunction progresses ultimately to CHF, accounting for 20% of deaths. End-stage ARVC may be difficult to distinguish from DCM because the extent of left ventricular involvement only recently has been appreciated.
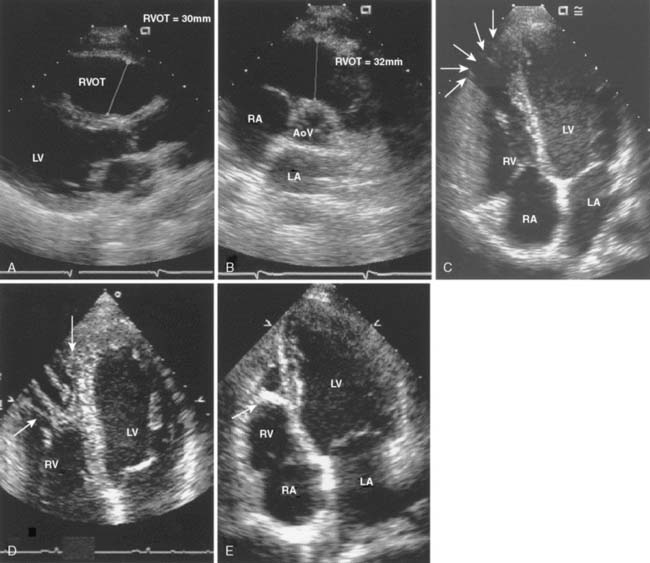
Diagnosis is based on ECG, structure, genetic studies, and arrhythmias.161 The characteristic ECG includes inverted T waves in the right precordial leads, QRS prolongation > 110 milliseconds, and extrasystoles with left bundle branch block. Diagnosis may depend on endomyocardial biopsy to reveal the distinctive changes of ARVC, yet is unrewarding if the biopsy is obtained from the septal area of the myocardium known for its lack of characteristic features.63 A new immunohistochemical analysis of a biopsy sample has proved to be highly sensitive and specific for the identification of ARVC.169 The importance of this test is that it has been used to differentiate those patients in the early stages of the disease from the more advanced state. A prospective longitudinal study examined 108 newly diagnosed patients over a 2-year period and found that their clinical profile differed from the patient with the more advanced disease. This finding may offer some options for future treatments.163
Anesthetic Considerations
Two adolescents undergoing routine operations were reported to experience development of refractory arrhythmias and sudden death after general anesthesia.162 Autopsy demonstrated typical features of ARVC. A family history of sudden death or syncope at an early age should heighten the awareness for diagnosis of ARVC and merits further evaluation.161 During the course of ARVC, arrhythmias of both a supraventricular and ventricular nature may occur at any time. Because arrhythmias are more likely in the perioperative period, noxious stimuli, hypovolemia, hypercarbia, and light anesthesia must be minimized intraoperatively and during recovery. However, general anesthesia alone does not appear to be arrhythmogenic. Houfani et al162 reported that more than 200 patients with ARVC underwent general anesthesia without a single cardiac arrest. Anesthesia has been conducted successfully with propofol, midazolam, and alfentanil.161 Acidosis may be especially detrimental because of its effect on arrhythmia generation and myocardial function.
Currently, there are no guidelines for arrhythmia prophylaxis. Amiodarone is the first line of antiarrhythmic medication during anesthesia. Empiric administration of antiarrhythmia agents is ineffective at preventing arrhythmias or sudden death in these patients.166 Sotalol appears to be the only antiarrhythmic that may reduce arrhythmias. Placement of an implantable cardioverter-defibrillator is beneficial. Link et al170 identified 12 patients with ARVC who had implantable cardioverter-defibrillators placed and determined 8 patients had appropriate discharges for ventricular tachycardia. Subsequently, it has been reported that placement of an implantable cardioverter-defibrillator is associated with an excellent prognosis in patients with ARVC.160
Mitral valve prolapse
MVP with severe mitral regurgitation is a common reason for cardiac surgery today. Knowledge about MVP has evolved greatly since the 1960s. Unrecognized as a part of the midsystolic click and systolic murmur known as Barlow disease until 1961, the finding of mitral regurgitation in 1963 by Barlow et al171 in conjunction with the ballooning of the posterior mitral leaflet on angiography resulted in the condition called Barlow syndrome or floppy-valve syndrome. It was during this time that the true cause of MVP was understood to be a degenerative condition with myxoid found on histologic examination that caused thickening, elongation, and a change in the chords. With the advent of echo in 1970, eventually a diagnosis based on 3D echo parameters of the normal mitral valve was derived in 1987 for MVP.172 In essence, only the valve that displayed prolapse in the long-axis view had “true” MVP.173 This allowed the “true” prevalence of MVP to be determined, as well as the specific process associated with the corresponding mitral regurgitation. Carpentier was further able to differentiate the degenerative changes of MVP from another causative factor that did not cause billowing of the valve or excess tissue, now referred to as “fibroelastic deficiency.”174 Currently, MVP is known to be a structural and functional disorder affecting less than 1% of the population depending on accepted criteria for diagnosis.
MVP is a multifactorial valvular abnormality that may be caused by histologic abnormalities of the valve structure and surrounding structures of the myocardium.175 It is most likely a genetically inherited disease, but the genetic defect has not been identified. Transmission appears to be in an autosomal dominant manner. As the most commonly diagnosed cardiac valve abnormality, it occurs in adults who are otherwise healthy or in association with many pathologic conditions and is equally distributed between men and women (Box 22-2).175
BOX 22-2 Conditions Associated with Mitral Valve Prolapse
From Robert A. O’Rourke (ed): Current Problems in Cardiology. 1991, p 333.
Mitral valve leaflets normally close just before ventricular systole, thus preventing regurgitation of blood into the LA. In patients with degenerative valve disease, there are two forms: Barlow disease and fibroelastic deficiency. Barlow disease is believed to result from myxomatous degeneration of the mitral valve, elongation and thinning of the chordae tendineae, and the presence of redundant and excessive valve tissue. The mechanism is unknown, but regulation of the extracellular matrix components appears to be a primary issue. Normal mitral valve leaflets may billow slightly with closure; but in MVP, redundant mitral leaflets prolapse into the LA during mid-to-late systole as the ventricle is emptied (Figure 22-27). Superior arching of the mitral leaflets above the level of the atrioventricular ring is diagnostic for MVP. Distortion or malfunction of any of the component structures of the mitral valve may cause prolapse and generate audible clicks or regurgitation associated with a murmur. If the chordae tendineae are lengthened, the valves may billow even more and progress to prolapse when valve leaflets fail to appose each other. The degree of these changes will determine the presence of mitral regurgitation. The degenerative valve changes that are responsible for progression from an asymptomatic state with murmurs and systolic clicks to dyspnea with severe mitral regurgitation occur over an average of 25 years.176
Figure 22-27 Diagrammatic representation of the functional pathology of the mitral valve mechanism.
(From Barlow JB, Pocock WA: Mitral valve prolapse enigma—two decades later. Mod Concepts Cardiovasc Dis 53:13, 1984, by permission of American Heart Association.)
Another category of MVP that was first identified by Carpentier with no billowing or excess valve tissue was fibroelastic deficiency.174 In this situation, the chords were thin and possibly ruptured, often involving a segment of the posterior leaflet. The mechanism is impaired connective tissue production caused by a deficiency of collagens, elastins, and proteoglycans, but the cause is unknown. The leaflet tissue is preserved. Unlike Barlow disease, symptoms generally arrive with chordal rupture, frequently with advancing age. This is a much less pronounced, generalized degeneration of the mitral valve than Barlow disease. Surgical repair also is less complex. Finally, even histologically normal mitral valves may prolapse.175 Normal mitral valve function depends on a number of factors including the size of the LV and mitral leaflets. Changes in these components may cause “innocent” MVP.
There are no universal criteria for diagnosis of MVP. Physical diagnosis and echo are the requirements for diagnosis of MVP. Auscultatory criteria that are strictly applied are highly sensitive for the diagnosis but lack some specificity. Typical auscultatory features are midsystolic click and late-systolic murmur. The click is related to deceleration of blood on the undersurface of the valve prolapsing into the LA. The murmur is late, with progressively increasing regurgitation that results in a crescendo systolic murmur. Certain maneuvers aid with the auscultatory diagnosis of MVP such as Valsalva, squatting, or leg raises that change the left ventricular end-diastolic volume to move the timing of the click within systole.175 Auscultatory findings may be absent despite echo determination of MVP. MVP is diagnosed more often now with 2D echo because recognition and assessment of the severity are superior. Given the saddle-shaped nature of the mitral annulus, the diagnosis of MVP is typically made from the parasternal long-axis view using TTE. MVP is defined as more than 2 -mm displacement of one or both mitral leaflets into the LA during systole. If the mitral leaflets appear thickened or myxomatous, the diagnosis is more certain. Color Doppler and continuous-wave Doppler imaging can quantify the degree of mitral regurgitation when it is present.
Most patients with MVP are asymptomatic, but symptoms may range from mild to severe. Altered autonomic function, catecholamine responsiveness, or possibly a combination of the two may account for complaints of chest pain, fatigue, palpitations, dyspnea, dizziness, syncope, and panic attacks among others. These symptoms and some clinical findings of thin body type, low blood pressure, and ECG repolarization abnormalities have been associated with MVP and termed MVP syndrome.175 However, controlled studies have identified palpitations as the only symptom linked to MVP. Nearly 50% of patients describe palpitations.177 Neuropsychiatric symptoms are no longer considered a part of MVP. The possibility of a complex of unexplained cardiovascular symptoms associated with MVP has not been completely eliminated and may exist in a few patients. The cause may be abnormal autonomic activity that leads to symptoms of vasoconstriction, enhanced β-receptor activity, and decreased plasma volume; however, a relation with MVP remains undefined.
The prognosis for most MVP patients is excellent with a normal life expectancy.175 Development of severe echo abnormalities is rare in patients with MVP. Although a benign course is typical for many patients with MVP, serious complications such as severe mitral regurgitation, infective endocarditis, sudden death, and cerebral ischemia may occur. The incidence rates of cardiovascular morbidity and mortality in MVP are just under 1% per year.178 It is a subset of patients who appear to be at increased risk for serious complications that may be defined by echo and clinical findings. These risk factors vary from reduced left ventricular systolic function to valve thickening greater than 5 mm to others that have been described.175
Mitral regurgitation is the most serious complication associated with MVP. Severe mitral regurgitation develops in approximately 2% to 4% of patients with MVP, two thirds of whom are male.177 Most patients will have mild-to-moderate mitral regurgitation that does not require surgery. MVP is the most common cause of severe mitral regurgitation, and its onset signals the need for therapeutic intervention. Irrespective of symptoms, the onset of severe mitral regurgitation can result in reduced life expectancy. Patients are usually younger than 60 years with Barlow’s form of MVP and mitral regurgitation. Mitral regurgitation in Barlow disease is not caused by the billowing of the leaflet body but from the marginal prolapse (Figure 22-28).174 The annulus is severely dilated. The posterior leaflet is affected more frequently than the anterior leaflet. Changes often are observed at the site of chordal insertion leading to rupture of the chordae and tethering of the valve leaflet. With the onset of severe mitral regurgitation, PAH, left atrial enlargement, and atrial fibrillation frequently emerge. Early repair is recommended to preserve left ventricular function and reduce the likelihood of atrial fibrillation.
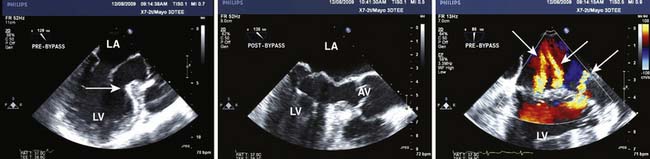
The management of MVP and mitral regurgitation without symptoms continues to be reconsidered in terms of the timing for cardiac surgery, especially in view of the risk for mitral valve repair and the improved outcomes associated with earlier surgery. Without overt symptoms, the degree of ventricular dysfunction may be worsening, unknowingly placing the patient at increased risk for permanent dysfunction and worse surgical outcome.175 Male individuals are three times more likely to require surgery than female individuals, even though there is no difference in the overall incidence of MVP between them. Noninvasive monitoring of mitral regurgitation and developing left ventricular dysfunction may help evaluate the optimal time for surgery. Flail mitral leaflet is an especially important risk for a poor surgical outcome if not addressed immediately.
Mitral valve repair is widely recommended for treatment of MVP compared with replacement. It provides long-term benefit with either causes of MVP and a low rate of recurrence.179 Mitral valve repair confers a significantly improved operative survival, as well as 5- and 10-year survival, compared with mitral valve replacement.180 The Cox maze procedure may be added to mitral valve repair safely to effectively reduce late complications of mitral valve disease.181 Advantages of mitral valve repair compared with replacement include lower risks for thromboembolism, bleeding, and infectious endocarditis, temporary anticoagulation, and better ventricular function because the valve structure is preserved.182 Most patients with degenerative mitral disease (90%)183 are able to undergo repair with low morbidity and mortality with excellent long-term outcomes according to retrospective studies.184 Recently, there has been a prospective, observational study of patients who underwent mitral valve repair or replacement for mitral regurgitation (ischemic and degenerative causes), and it found a statistically significant survival benefit in those who had repair.185 Notably, repair of the valve returns the patient to a similar life expectancy with a comparable noncardiac surgical patient.
Posterior leaflet prolapse is the most common defect in mitral regurgitation.175 The posterior leaflet repair is very low risk, but the bileaflet repair is more technically challenging, requiring more skill and advanced techniques. The anterior prolapse repair has a greater rate of reoperation and decreased survival.184 Recently, the use of robotic technology has advanced to a point that allows safe and feasible mitral repair in these circumstances, although there is no long-term follow-up.186 Surgical strategy does not involve annular plication and annuloplasty for annular dilation, but leaflet resection for excessive valve tissue to restore the normal valvular geometry (Figure 22-29). Once the repair is completed and the patient has separated from CPB, it is important to observe for SAM so it can be surgically corrected immediately. All valves cannot be successfully repaired, whereon 10% with prolapse of a degenerative mitral posterior leaflet require mitral valve replacement (see Chapter 19).
Figure 22-29 Mitral valve repair with sliding annuloplasty technique.
(From Gillinov AM, Cosgrove DM 3rd, Shiota T, et al: Cosgrove-Edwards annuloplasty system: Midterm results. Ann Thorac Surg 69:717–72, 2000, by permission.)
The association of arrhythmias and sudden death with MVP is a long-held observation. Premature atrial and ventricular beats, atrioventricular block, and supraventricular or ventricular tachyarrhythmias are common during ambulatory monitoring in adults with MVP.187 The causes of these arrhythmias are multifactorial, probably combining an anatomic substrate with some form of dysautonomia.188Mechanisms that have been proposed for arrhythmias include ventricular enlargement, hyperadrenergic states, electrolyte imbalances, and mechanical irritation of the ventricle because of traction of the chordae tendineae. Arrhythmias may be secondary to mitral regurgitation, not MVP. According to a study of individuals with nonischemic mitral regurgitation, complex arrhythmias were common and equally prevalent regardless of whether the patient had MVP.189 Ventricular tachycardia occurred in 35% of those subjects with mitral regurgitation, in contrast with only 5% of participants with MVP alone. Similarly, the risk for sudden death for patients with MVP may be related to mitral regurgitation in view of a reduction in ventricular arrhythmias that occurred with mitral valve repair or replacement.
The occurrence of sudden death with MVP in adults has been debated for years. The risk is low, with an estimated yearly rate of 40 per 10,000, but this is twice the expected rate in the population.175 Sudden death does occur within families of patients with MVP.187 The ECG is abnormal in approximately two thirds of persons with MVP, but ambulatory ECG monitoring does not show an excess of atrial or ventricular arrhythmias unless accompanied by severe mitral regurgitation.175 EP testing has been unable to identify an arrhythmogenic focus or reliably identify those at increased risk, but it is useful in the pharmacologic management of patients with ongoing complex sustainable arrhythmias. Patients with severe mitral regurgitation, flail segment, and depressed LV appear to be at greater risk for sudden death. Prophylaxis has not averted sudden death in high-risk patients with severe mitral regurgitation. An implantable cardioverter-defibrillator is a consideration in patients with inducible ventricular tachycardia or ventricular fibrillation, but these patients are rare.190 In general, most low-risk patients do not require treatment for either their symptoms or prevention of sudden death.
MVP has been reported to be associated with cerebral ischemic events, especially in the young. Mitral cusp elongation and expansion that occur with MVP may generate thrombus, vegetation, and calcification of the mitral valve. Mobile masses have been found in conjunction with cerebral events. However, a population-based cohort study involving 1079 patients found no difference in the incidence of strokes in patients younger than 45 years with MVP unless another cardiac disease process was present.191 In contrast, a more recent study of individuals in Olmsted County of Rochester, Minnesota, found the incidence rate of stroke was 0.07%/year, almost twice the normal rate.192 The strongest factors associated with increased risk in these patients with MVP included thick valves, mitral valve surgery, atrial fibrillation, and age (> 50 years). Factors that were independently associated with stroke were advanced age, coronary artery disease, CHF, and diabetes.
Bacterial endocarditis is an infrequent complication of MVP, but its incidence is three to eight times greater in these individuals than the general population.177 Male patients are three times more likely to experience development of endocarditis. Current guidelines from the American Heart Association are specific and do not recommend antibiotic prophylaxis for individuals without a prosthetic cardiac valve, complex congenital heart disease, postcardiac transplant valvular lesions, or a history of bacterial endocarditis.193 Patients with isolated clicks probably do not benefit from antibiotic prophylaxis, but cost-effectiveness with MVP has been proved nonetheless.194 Patients at greater risk for development of endocarditis (previous bacterial endocarditis, systolic murmurs, thickened leaflets, or mitral regurgitation) should receive antibiotics before undergoing procedures commonly associated with bacteremia because the risk increases to about 0.05% per year.175 The turbulent flow from the mitral regurgitation and the thickened valve tissue may cause the increased risk for infection. Patients who may require oral endotracheal intubation or fiberoptic bronchoscopy do not require antibiotic prophylaxis.176 Unfortunately, as recommendations regarding antibiotic prophylaxis have continued to be modified over the years, there is still insufficient evidence to guide the physician on the issue of prophylaxis and MVP.
Anesthetic Considerations
Anticholinergic preoperative medications are best avoided despite an increased vagal tone in MVP. A moderate anesthetic depth is desirable to minimize catecholamine levels and potential arrhythmias. Ketamine or drugs that have sympathomimetic effects must be administered with caution. Volatile anesthetics sensitize the heart to catecholamines and, thus, potentially promote arrhythmias. Isoflurane is probably less sensitizing than halothane, but autonomic imbalance may actually be more important in arrhythmogenesis than any direct effect of volatile agents in patients with MVP. These patients with MVP syndrome have been shown to possess good left ventricular function if mitral regurgitation or coronary artery disease is absent195; therefore, myocardial depression from volatile agents will be well tolerated. Narcotics such as fentanyl will block sympathetic responses and promote hemodynamic stability; however, prolonged postoperative respiratory depression is a disadvantage with higher doses. Shorter-acting narcotics such as alfentanil and remifentanil, as well as other intravenous agents such as propofol, are available to facilitate rapid extubation.196 Hypercapnia, hypoxia, and electrolyte disturbances increase ventricular excitability and should be corrected. If muscle relaxation is desired, vecuronium is an excellent choice because it does not cause tachycardia.
Patients with MVP who have mitral regurgitation or are at greater risk for complications warrant a different anesthetic approach compared with those with MVP syndrome. Patients with more severe forms of MVP may rapidly go into CHF unexpectedly and require cardiac surgery. The severity of mitral regurgitation will strongly influence anesthetic management. Routine monitoring for patients undergoing cardiac surgery should be utilized in these patients even if it is a noncardiac operation. Intraoperative TEE is placed after induction. Opioid agents provide excellent hemodynamic stability without depressing myocardial function.93 Although avoidance of ketamine has been recommended by some for patients with MVP syndrome, dosing less than 0.5 mg/kg ketamine preserves hemodynamic stability in patients who are severely ill, especially used in combination with opioids such as fentanyl. Ketamine will better preserve sympathetic activity and blood pressure in hemodynamically compromised patients. More extensive review of anesthetic management of valvular disease is provided in Chapter 19.

Mitral Valve Regurgitation and Septal Hypertrophy
The prebypass TEE confirmed the TTE findings. The basal ventricular septum measured 21 mm (see Figure 22-28). At rest, there was no evidence of SAM or LVOT obstruction. It was decided to challenge the patient with isoproterenol. With 4 mg isoproterenol the heart rate increased from 70 to 135 beats/min, and the aortic blood pressure decreased from 126/52 to 77/45. Under these conditions, the TEE showed a significant amount of SAM and turbulent flow in the LVOT with obstruction (Figure 22-30). Needles placed in the aorta and LV were transduced and revealed a gradient across the LVOT of 67 mm Hg. Based on these findings, the surgical plan was altered.
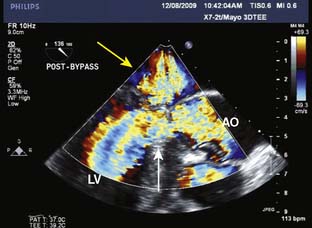
Patent foramen ovale
A patent foramen ovale (PFO) is the most common congenital anomaly. It was first recognized as a causal relation with stroke in 1877197 and has since been proved one of the mechanisms of stroke.198 Air, thrombus, or fat may travel from the RA to the LA into the systemic circulation, causing a paradoxic embolus that may affect cerebral or coronary circulations. For years, a PFO was identified in the catheterization laboratory, surgery, or autopsy. The advent of echo with its availability and diagnostic capability has increased considerably the number of PFOs that are identified inside the operating room and in the outpatient setting. The ability to so readily and safely close a PFO without surgery has created the need to develop guidelines to address the management of PFO.199
The foramen ovale is present during fetal circulation to direct better oxygenated blood from the umbilical veins through the Eustachian valve selectively into the LA. At birth, pulmonary vascular resistance declines dramatically, increasing left atrial blood return that increases the left atrial pressure more than the right atrial pressure, causing a functional closure of the foramen ovale. If the flaplike covering from the septum primum does not fuse with the septum secundum over a period of a year, there is anatomic failure of closure forming a PFO (Figure 22-31). Any condition that results in right atrial pressures exceeding left atrial pressures after birth causes a right-to-left shunt. A left-heart condition that results in left atrial pressures exceeding right atrial pressures causes a left-to-right shunt. An individual may remain asymptomatic for years with a PFO depending on the size of the shunt.
Figure 22-31 Embryologic development of the interatrial septum.
(From Hara H, Virmani R, Ladich E, et al: Patent foramen ovale: current pathology, pathophysiology, and clinical status. J Am Coll Cardiol 46:1768–1776, 2005, by permission.)
The incidence of PFO in the population has varied depending on the study and diagnostic technique. Hagen et al200 reported a 27% incidence rate of PFO in nearly 1000 autopsies. This study is considered by many to be the definitive data on incidence of PFO. Subsequent studies involving various modes of echo have reported the incidence rate of PFO in the population to be between 3% and 45%.201 A recent retrospective study reviewing more than 10 years of cardiac surgery in 13,000 patients reported an incidence rate of PFO as 17% with TEE.202 TTE is now recognized as less sensitive than TEE, possibly accounting for such variation in previous studies regarding the incidence of PFO. TEE, with its higher image resolution than other methods and 100% sensitivity and specificity with autopsy findings, has become the gold standard for diagnosis of PFO (Figure 22-32).201 Because TEE is more invasive, additional technical advances in both TEE and transcranial Doppler have improved their sensitivity, and when combined, they may be adequate for PFO screening purposes.203
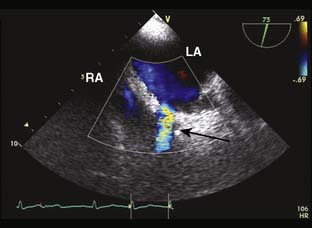
Figure 22-32 Transesophageal image from the midesophagus at 75 degrees focusing on the interatrial septum.
Although a number of conditions are associated with PFO (Box 22-3) that may merit consideration for therapy, stroke receives the most attention. Approximately 700,000 strokes occur each year in the United States, with 20% considered cryptogenic. A PFO is present in 40% to 50% of those strokes.204 In those individuals with a history of cerebral events, the prevalence rate of PFO ranges from 7% to 40% depending on the diagnostic technique.205 Several studies show a strong relation between cryptogenic stroke and PFO based on epidemiologic evidence,198 but others dispute it. Petty et al206 conducted a population-based study observing TEE in patients with cerebral ischemic events compared with control subjects who had TEE, but not for cerebral ischemic events, to eliminate bias present in other studies regarding PFO. They found that PFO was not an independent risk factor for stroke even when considering the size of the shunt. Although some recognize the PFO as an increased risk for cryptogenic stroke, it remains inconclusive.
BOX 22-3 Diseases and Risks Associated with Patent Foramen Ovale
From Meier B: Catheter-based closure of the patent foramen ovale. Circulation 120:1837–1841, 2009, by permission.
The decision to close a PFO will include individual risk factors for stroke or other complications such as hypoxia that may occur with PFO. Paradoxical embolus is more common if atrial septal aneurysm, large Eustachian valve, migraines, and older age (≥ 50 years) are present. Medium-to-large shunts in combination with any coagulation abnormalities are highly correlated with PFO and paradoxic embolus,199 and thrombus has been identified at the atrial septum in association with cryptogenic stroke.198 However, others found no relation to the size of the shunt with PFO and stroke.206
It is essential to perform a correct provocative measure to ensure right-to-left movement of air or contrast to diagnose a paradoxical embolus (Figure 22-33). Agitated saline directed through a central catheter with a Valsalva maneuver most commonly is used to confirm the diagnosis. The increased intrathoracic pressure from the Valsalva maneuver will lead to a temporary increased return of venous blood after the Valsalva has ended, so the right atrial pressure will exceed left atrial pressure briefly to allow contrast administered into the right internal jugular vein to pass to the LA.203 False-negative results may occur if the left atrial pressure is very increased so that the provocative measure does not cause right-to-left shunting.
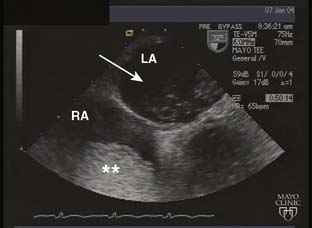
Figure 22-33 Transesophageal image from the midesophagus at 72 degrees focusing on the interatrial septum.
Management of a PFO is very dependent on the circumstances. Medical therapy basically consists of various anticoagulation regimens that may suffice for some; however, most observational and meta-analysis studies comparing medical therapy with percutaneous closure shows it to be inferior.207 Percutaneous closure of the PFO is indicated especially in those patients with recurrent paradoxic emboli. There is a reduction in the incidence of stroke.199 Another advantage over medical therapy is the lack of chronic anticoagulation.198 There are no randomized, prospective trials comparing medical therapy with percutaneous PFO closure to sufficiently answer the question of superiority between options; however, in nonrandomized trials, short-term complications appear to slightly favor medical treatment over percutaneous closure.207
Closure of PFO is performed either surgically or percutaneously. Although available in the late 1980s, percutaneous closure of PFO really became popular in 1992 after a report describing successful closure of 36 PFOs in patients with known right-to-left shunt and presumed paradoxical emboli.208 Percutaneous closure generally is performed in a catheterization laboratory with conscious sedation. Success rates (incidence of no residual shunting) have been good initially, but the methods used to evaluate success may have been flawed, accounting for the wide range of success rates between 50% and 100%.205 Recently, using transcranial Doppler to assess PFO closure, researchers found 9% of patients to have significant shunting after only 1 year after percutaneous closure.209 There are no data concerning the success rate of surgical compared with percutaneous closure. Although the percutaneous closure is being done routinely, complications occur in about 1.5% of cases. They include cardiac perforation, air embolism with placement, thrombus formation, and even death.209
Currently, it is rare to surgically close an isolated PFO, with the safety and availability of percutaneous closure. However, use of intraoperative TEE during cardiac operations has increased the number of incidentally diagnosed PFOs. A recent retrospective look at more than 10 years of cardiac operations in 13,000 patients found 17% of cases with PFO.202 Looking at the incidence of PFO year by year from 1995 to 2010, the incidence remained relatively static each year; however, the percentage of PFOs that were closed surgically increased to a maximum of 39% over that period. Not only are more PFOs closed as a percentage, but more surgeons as a percentage are choosing to close them over time.199 The causes for these trends are not certain, but a PFO diagnosed incidentally during surgery creates a dilemma for the surgeon. A majority of surgeons examine the circumstances, size of the PFO, and other criteria to decide on closure during the ongoing surgery.
The decision to close an incidental PFO during cardiac surgery is not always evident based on short- and long-term risk to the patient. Because the finding was incidental, recurrent paradoxic emboli and neurologic injury usually have not occurred. Certain conditions would almost mandate closure of the PFO, such as the insertion of a left ventricular assist device that would promote paradoxic embolus203 or the onset of severe hypoxia because of increased right-sided pressures causing a large right-to-left shunt. Surgical PFO closure does not confer additional morbidity or mortality based on retrospective review of all surgical closure of isolated PFOs from one institution that demonstrated no mortality and minimal morbidity. Recent data have confirmed that adding PFO closure during cardiac surgery did not increase mortality.202 Surgical PFO closure is done routinely through a sternotomy, but the use of new techniques of minimally invasive surgery also allow the incision to be a right thoracotomy. Closure requires CPB and cardioplegic arrest. Certain operations such as tricuspid or mitral valve repair or placement that include CPB and atriotomy require minimal deviation from the originally planned procedure to close the PFO and, thus, incur little risk. In contrast, CABG performed without CPB would entail the risk of going on CPB, aortic cross-clamping, and other complications associated with extracorporeal circulation. The decision to close the incidental PFO may depend on the long-term risk of not closing it.
Based on a survey of cardiac surgeons, 27.9% of respondents always closed PFOs if detected during CABG.203 However, there really is no evidence to indicate all PFOs found incidentally during a cardiac operation merit closure.202 A retrospective study looking at those with and without PFOs showed no differences between stroke, hospital and intensive care unit (ICU) duration, MI, and death when propensity matching was applied for the comparison.202 Long-term follow-up for 10 years also did not demonstrate any difference between those who had PFOs and those without PFOs. Outcome for those who had PFOs repaired versus not repaired found no differences in outcome except for a slight but significantly increased incidence of stroke after surgery in the repaired group that could not be explained. Other than the concern for stroke, the risk for severe desaturation after surgery if there was no PFO closure was possible, but a case series of 11 off-pump CABG with incidental PFOs that were not closed showed no adverse problems perioperatively with desaturation.210 A subset of patients may exist who do not behave well perioperatively with off-pump CABG because of a PFO.

Anesthesia
Routine care for preventing venous air should be standard for cardiac surgery. Obviously, there is a great need to be diligent with the injection of medications to remove extraneous air from entering the venous system. It is important to appreciate the potential for paradoxic embolus with any patient who requires mechanical ventilation. In situations in which the pulmonary vascular resistance may increase, such as during hypercapnia or with positive end-expiratory pressure greater than 15 mm Hg, the potential for right-to-left shunting is increased.211 Choice of anesthetic seems to have little impact on management of PFO because the patients will tolerate most regimens.

Incidental Patent Foramen Ovale Found during Coronary Artery Bypass Grafting Surgery
In the operating room, a TEE probe was placed for the purposes of monitoring ventricular function and evaluation of regional wall motion abnormalities. The prebypass examination revealed an ejection fraction of 65%, normal valves, and no regional wall motion abnormalities. The interatrial septum appeared redundant. Agitated saline was injected into the central line. After the release of sustained positive pressure ventilation, contrast was seen to enter the LA via a small PFO (see Figure 22-33). No shunt was detected by color-Doppler imaging.
Pulmonary hemorrhage
Pulmonary hemorrhage occurs in about 1.5% of patients with hemoptysis,212 but mortality may reach 85%.213 The definition of massive hemoptysis varies but commonly is characterized as more than 600 mL of expectorated blood over 24 hours or recurrent bleeding of greater than 100 mL per day for several days. Four hundred milliliters of blood in the alveolar space seriously impairs oxygenation. Pulmonary hemorrhage may stabilize only to worsen again without an obvious explanation reflecting its unpredictable nature. Notably, death is not attributable to hemodynamic instability with hemorrhage but to excessive blood in the alveoli that causes hypoventilation and refractory hypoxia. Moreover, one clot may obstruct an entire lobe of the lungs. Mortality is related to the amount of bleeding that occurs over time.213,214 If more than 600 mL blood is lost over 16 hours, the mortality rate is 45%, whereas it is only 5% if blood loss totals 600 mL over 48 hours. A delay in initiating treatment because of difficulty in isolating the location of bleeding contributes greatly to the high mortality of pulmonary hemorrhage.
Hemoptysis may occur with various diseases and circumstances (Table 22-7). In the United States, chronic inflammatory lung disease and bronchogenic carcinoma are the most common causes of hemoptysis.215 Of these causes, bronchitis (26%), lung cancer (23%), pneumonia (10%), and tuberculosis (8%) are most frequent. The inflammatory response is an important factor in the occurrence of bleeding.216 The combination of chronic infection, inflammation, and vascular growth result in hypervascularization of the bronchial circulation that ultimately erodes into the alveoli.217 Pulmonary hemorrhage also may result from vigorous suctioning of the lungs, surgery, and improper positioning of a PAC.218 Massive hemoptysis usually is an emergency because the underlying pulmonary disorder minimizes the patient’s ability to tolerate it.
| Tracheobronchial disorders |
|---|
| Amyloidosis |
| Bronchial adenoma |
| Bronchiectasis* |
| Bronchogenic carcinoma |
| Broncholithiasis |
| Bronchovascular fistula |
| Cystic fibrosis |
| Foreign body aspiration |
| Tracheobronchial trauma |
| Cardiovascular disorders |
| Congenital heart disease |
| Mitral stenosis |
| Pulmonary arteriovenous fistula |
| Septic pulmonary emboli |
| Ruptured thoracic aneurysm |
| Arteriovenous malformation |
| Localized parenchymal diseases |
| Amebiasis |
| Aspergilloma* |
| Atypical mycobacterial infection* |
| Coccidioidomycosis |
| Lung abscess |
| Mucormycosis |
| Pulmonary tuberculosis* |
| Diffuse parenchymal diseases |
| Goodpasture syndrome |
| Idiopathic pulmonary hemosiderosis |
| Polyarteritis nodosa |
| Systemic lupus erythematosus |
| Wegener’s granulomatosis |
| Other |
| Pulmonary artery rupture from a pulmonary artery catheter |
| Iatrogenic (e.g., bronchoscopy, cardiac catheterization) |
| Pulmonary hypertension |
| Pulmonary edema |
| Pulmonary infarction |
From Thompson AB, Tescheler H, Rennard SI: Pathogenesis, evaluation, and therapy for massive hemoptysis. Clin Chest Med 13:69, 1992.
It is helpful to appreciate the anatomy of the pulmonary circulation to understand the pathogenesis of pulmonary hemorrhage. The lungs have a dual blood supply. The pulmonary arterial circuit is a high-flow, low-pressure circuit. The nutritive supply of the pulmonary structures is the bronchial arteries, which originate from the aorta. Bronchial arteries extend into many areas around the lymph nodes, esophagus, and lungs, ultimately penetrating the bronchial wall to supply the bronchial mucosa. The bronchial and pulmonary circulations anastomose at several locations. Massive pulmonary hemorrhage usually involves bleeding caused by disruption of the high-pressure bronchial circulation.212 These high-pressure, tortuous bronchial arteries are found throughout the thoracic cage. They become dilated and rupture or erode or feed into the lower pressure pulmonary veins that may easily rupture. The bronchial circulation accounts for 98% of pulmonary hemorrhage, and the pulmonary circulation accounts for the remainder.214
Diagnosis of hemoptysis requires a few simple tests. Visual inspection usually can distinguish gastrointestinal bleeding from pulmonary hemorrhage. Hemoptysis often is bright red with some sputum. Hemoptysis caused by pulmonary artery rupture usually is copious (200 to 2000 mL).218 Treatment begins with conservative management and simultaneous efforts to locate the bleeding site. A chest roentgenogram may reveal an infiltrate, but neither chest roentgenogram nor physical examination has been reliable in localizing the affected lung. Recently, better noninvasive technology such as the multidetector-row CT is capable of determining rapidly and accurately the site of bleeding.215 Flexible bronchoscopy of the airways may be limited by severe bleeding that obscures the view and attempts should not persist, delaying other therapies. Rigid bronchoscopy is better suited for identification of bleeding during massive hemoptysis and the removal of any large clot that may be obstructing the airway. However, the view of the upper lobes is limited and requires general anesthesia. Instillation of epinephrine in the bronchi may facilitate better visualization by slowing the bleeding. Ultimately, angiography of the pulmonary and bronchial arteries may be necessary to localize the source of the bleeding.213 Imaging for bronchial artery bleeding begins with a thoracic aortogram to localize all the main systemic arteries to the lungs that may be bronchial or nonbronchial. Once the feeding arteries are localized, selective bronchial arteriography is then used to identify the bleeding vessels.219
Therapeutic options for bleeding depend on the extent of bleeding. Conservative treatment may suffice. Once the bleeding patient has been placed in the ICU and antibiotics administered, flexible bronchoscopy may be able to identify the source of bleeding, as well as perform techniques to minimize hemorrhage such as epinephrine flush, cold-saline lavage, and possible balloon bronchial blockers to tamponade any bleeding.220 Advancements in bronchoscopy have seen the successful use of topically applied agents consisting of oxidized regenerated cellulose mesh injected at the site of bleeding.215 In addition, the use of topically applied factor VIIa (FVIIa) has been reported in massive hemolysis secondary to a medical cause, although its use in this manner is off-label.217 FVIIa has been anecdotally reported beneficial with massive hemoptysis despite low platelet count, trauma, vasculitis, and bone marrow transplant. Other medications that have been recommended for increasing clotting to reduce bleeding are Premarin, desmopressin, vasopressin, and tranexamic acid.221
If bleeding continues to be heavy, a double-lumen endotracheal tube (ETT) is beneficial to isolate the bleeding from the unaffected lung, although placement may prove difficult in the midst of active bleeding. Misplacement of a double-lumen ETT occurs in 45% of patients after the initial attempt and 54% after patient positioning.222 If the patient was anticoagulated, airway manipulation in connection with double-lumen ETT placement may incite further mucosal damage and bleeding. A Fogarty catheter, as described by Larson and Gasior,223 may be passed within an existing ETT under bronchoscopic guidance to the affected bronchus, minimizing additional trauma to the airway caused by reintubation and protecting the uninvolved lung. The Univent ETT is an alternative to a double-lumen ETT but requires exchanging the existing ETT for the Univent tube. A single-lumen ETT should not be advanced into the right main bronchus if bleeding is believed to be in the right lung, because of the proximal location of the right upper lobe bronchus (see Chapter 24).
Continued bleeding after stabilization and conservative therapy necessitates bronchial artery embolization. First used in 1974 to stop massive hemoptysis,219 it is considered the first-line therapy for massive hemoptysis. It may stop the bleeding 75% to 98% of the time, but 16% to 20% of patients will rebleed in the following year.224–226 Bronchial artery embolization often is done simultaneously with the thoracic aortogram to identify the bleeding vessels. The offending vessels are injected with a form of polyvinyl alcohol particles, Gelfoam, and possibly dextran microspheres. A recent study found 88% of patients who underwent bronchial artery embolization had bleeding controlled immediately and another group of patients (81%) had bleeding stopped by 48 hours.227 Bronchial artery embolization has greatly advanced the success of dealing with pulmonary hemorrhage. It may require more than one procedure to be effective. Immediate recurrence of hemoptysis after embolization may be because of arteries that were not identified and not embolized. Latent hemoptysis is due to recanalization or collateralization. Although it has proved effective, it is not without complications, especially vascular complications such as coronary artery syndrome, spinal cord ischemia, and esophageal wall necrosis.219
The ability to use an array of treatment options in a multidisciplinary approach to this life-threatening condition will increase survival and reduce complications.220 Efforts to control bleeding for at least 48 hours with nonsurgical alternatives before surgery have reduced surgical mortality and postoperative complications compared with earlier implementation of surgery for massive hemoptysis in the past. If bleeding continues unabated, exsanguination may be unavoidable, so surgery is the definitive treatment. A localized bleeding site and sufficient lung function are essential to determine before surgery because resection may extend to a pneumonectomy. Postoperative mortality rate varies tremendously from 1% to 50%.212 Surgery is contraindicated in those with lung carcinoma invading the trachea, mediastinum, heart or great vessels, terminal malignancy, and progressive pulmonary fibrosis.
Introduction of the PAC enabled physicians to obtain CO, SVR, and estimates of left ventricular performance and to detect early myocardial ischemia perioperatively, greatly increasing their use. Unfortunately, with more use, a rare but often fatal cause of pulmonary hemorrhage, rupture of the pulmonary artery by the PAC in patients undergoing cardiac surgery, emerged.218 The incidence rate ranges from 0.06% to 0.2% of cases, with a corresponding mortality rate of 45% to 64% and serious morbidity.228 With respect to cardiac surgical patients, death may occur within minutes of pulmonary artery rupture or over a period of 1 to 14 days after surgery.229 The diagnosis may be initially missed because patients may be asymptomatic or only mildly symptomatic. A small amount of hemoptysis may herald the onset of severe pulmonary hemorrhage.230 If a patient is anticoagulated, bleeding may be profuse. Other signs of pulmonary artery rupture are hypotension, decreased arterial oxygenation, bronchospasm, pleural effusion, hemothorax, and pneumothorax. Pulmonary hemorrhage rarely occurs during cardiac surgery; however, when it does, it is life-threatening not only because of anticoagulation, but because of the severity of the patient’s cardiovascular disease.
Certain factors predispose patients to pulmonary artery rupture such as age older than 60, anticoagulation, and distal migration of the PAC.218 Although PAH often is present with pulmonary artery rupture, it is not a risk factor229 but simply may promote distal migration of the PAC. Chronic PAH weakens the pulmonary artery and veins; thus, a patient’s susceptibility to rupture is increased. The mechanism of pulmonary artery rupture is multifactorial, but the balloon of the catheter is instrumental.231 Maximal pressures for balloon inflation are approximately 1700 mm Hg and easily reach 1000 mm Hg with the start of inflation. PACs that reside distally will reduce the inflation pressures necessary to rupture the pulmonary artery, particularly in the elderly. Distal migration of the PAC also occurs more easily in patients undergoing hypothermic CPB because hypothermia stiffens the PAC. Eccentric inflation of the PAC balloon contributes to pulmonary artery rupture, as evidenced in both cadavers and patients.232 Inflation that distorts the balloon drives the catheter tip through the vessel wall (Figure 22-34). Manipulation of the heart also may perforate the pulmonary artery.
The extent of bleeding after pulmonary artery rupture will determine treatment. If rupture occurs during CPB, then it should be maintained to ensure adequate oxygenation and identify the site of rupture under controlled conditions. Mild bleeding may require positive end-expiratory pressure once anticoagulation is neutralized.229,233 Similarly, the balloon of a PAC may tamponade a bleeding site in the bronchus. These preceding measures are meant to be only temporary until definitive treatment is instituted. If anticoagulation has been neutralized, blood loss of 1000 mL or more over 24 hours should merit consideration for early surgical intervention if the patient has the respiratory reserve to tolerate it.213,214 If there is concern about tolerating surgery, treatment should progress to bronchial artery embolization and, ultimately, pulmonary lobectomy if bleeding is refractory.
Pericardial heart disease
The pericardium is a two-layer sac that encloses the heart and the great vessels. Its inner layer is a serous membrane (visceral pericardium) covering the surface of the heart. Its outer layer is a fibrinous sac (parietal pericardium), which is attached to the great vessels, diaphragm, and sternum. The parietal pericardium is a stiff collagenous membrane that resists acute expansion. The space between the two layers is the pericardial space. The pericardial space normally contains up to 50 mL clear fluid that is an ultrafiltrate of plasma. It gradually can dilate to accept large volumes of fluid if slowly accumulated, but if accumulated rapidly will lead to cardiac tamponade. The two layers of the pericardium are joined at the level of the great vessels and at the central tendon of the diaphragm caudally, and a serous layer extends past these junctions to line the inside of the fibrinous sac (parietal pericardium). The vagus nerve, left recurrent laryngeal nerve, and esophageal plexus innervate the pericardium, together with sympathetic contributions from the stellate ganglion, first dorsal ganglion, and other ganglia. The lateral course of the phrenic nerve on either side of the heart is an important anatomic relation because this nerve is encapsulated in the pericardium and, thus, easily can be damaged during pericardiectomy (Figure 22-35). The pericardium is not essential for life, and pericardiectomy causes no apparent disability, but it has many subtle functions that are advantageous. Foremost, it acts to minimize torsion of the heart and reduce the friction from surrounding organs. Recently, it has been shown that the pericardium also has immunologic, vasomotor, paracrine, metabolic, and fibrinolytic activities.234
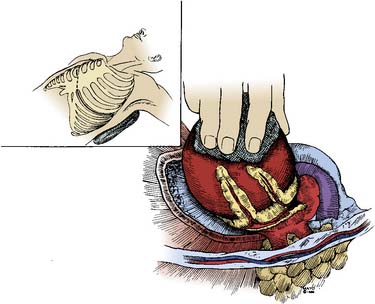
 Acute Pericarditis
Acute Pericarditis
Acute pericarditis is common, but the actual incidence is unknown because it often goes unrecognized. It generally is self-limited, lasting 6 weeks. Acute pericarditis has many causes (Box 22-4); the most common is viral (30% to 50%).235 Only 25% of cases have a defined cause, although this is improving.236 Unfortunately, acute pericarditis resembles many other conditions. Anesthesiologists encounter patients with acute pericarditis in situations of malignancy, MI, postcardiotomy syndrome, uremia, or infection when surgery is required because symptoms are incapacitating and medical therapy has failed.
BOX 22-4 Causes of Acute Pericarditis
From Oakley CM: Myocarditis, pericarditis and other pericardial diseases. Heart 84:449–454, 2000.
Acute pericarditis is characterized by fibrin deposits localized on the pericardial surface. A serous effusion may accompany the fibrinous inflammation. Consequently, the mesothelial cell layer is replaced by a fibrin membrane that has white blood cells scattered throughout it. Pericardial fluid may suggest a bacterial, neoplastic, or viral and inflammatory cause.235 Pericarditis occurs in about 20% of patients who suffer an MI, primarily within the first week.237 A distinction is made between pericarditis that occurs during the first week and Dressler syndrome, which usually appears 2 to 3 months after an MI. With acute pericarditis, pleuritic chest pain is described in the center of the chest radiating to the back and left trapezius muscle. This pain is more continuous than the intermittent pain of myocardial ischemia. Some degree of dyspnea may be present with acute pericarditis. Occasionally, pericarditis may present as right-heart failure because large and rapid accumulations of pericardial fluid will increase intrapericardial pressure sufficiently to obstruct right ventricular filling through the superior and inferior vena cava, resulting in tamponade. Atrial arrhythmias also may occur.
In general, acute pericarditis can be differentiated from MI by measurement of serum cardiac enzymes, but myocardial enzymes are sometimes found in the serum of patients with pericarditis. It also is common to see the early onset of a low-grade fever that lasts for days to weeks and the presence of a friction rub after the onset of chest pain. Pericardial friction rub is pathognomonic of pericarditis but may be heard only intermittently. Early ECG changes typical of pericarditis are ST-segment elevation in 2 or 3 standard limb leads and in most of the precordial leads. ST-T wave elevation often is present but may be confused with MI (Figure 22-36). Acute pericarditis generally results in diffuse ST-T changes, whereas MI usually is associated with more localized ST-T changes. T-wave inversions follow the acute ST-segment abnormalities as pericarditis enters the subacute phase. Q waves usually do not appear during progression of ECG changes. The echo findings in a patient with suspected acute pericarditis are variable. There may be a pericardial effusion present, the pericardium may be thickened, or it may appear completely normal. If an effusion is present, tamponade physiology must be excluded. If the pericardium appears thickened, additional evidence for constrictive physiology should be sought (see later). The chest roentgenogram may reveal a slightly enlarged cardiac silhouette with a pericardial effusion that indicates the effusion is at least 250 mL. A rapid accumulation of pericardial fluid may cause tamponade. Conversely, if it accumulates slowly, a liter or more of pericardial fluid may collect without symptoms of cardiac tamponade. Pericardial fluid also may be found with CHF, valvular disease, or endocarditis. Pericardiocentesis is performed in acute pericarditis, either to confirm the diagnosis or to relieve tamponade. It may be associated with serious complications, even cardiovascular collapse and shock. Echo should evaluate ventricular contractile function before pericardiocentesis. If repeated aspirations are required for relief of tamponade, pericardiectomy may be necessary.
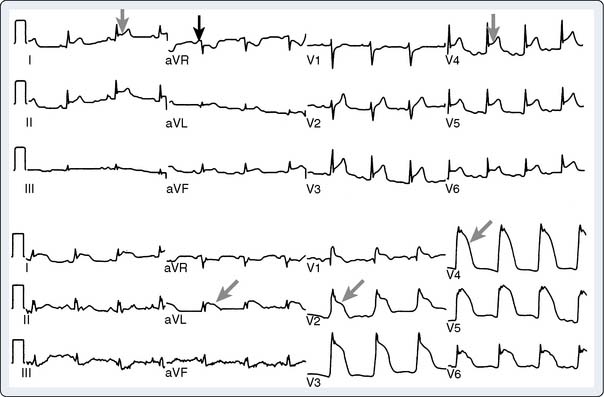
Detection of myocardially directed serum antibodies in Dressler syndrome and in postpericardiotomy syndrome suggests that these syndromes are immune-related responses. Postpericardiotomy syndrome consists of acute nonspecific pericarditis that usually begins between 10 days and 3 months after cardiac surgery or trauma. In a prospective study of 944 adult patients who underwent cardiac surgery, the incidence rate of postpericardiotomy syndrome was 17.8%, although it has been reported as high as 50% of postoperative cardiac patients.238
 Constrictive Pericarditis
Constrictive Pericarditis
CP is a dense fusion of the parietal and visceral pericardium that limits diastolic filling of the heart irrespective of cause. The changes in the pericardium can be caused by scarring, induced by a single episode of acute pericarditis or by a prolonged exposure to a recurrent or chronic inflammatory process. CP is a progressive disease. Tuberculosis was a major cause of CP, but currently most cases (33%) are idiopathic.239 The leading identifiable causes of CP are pericarditis, cardiac surgery, and mediastinal irradiation in the developed world.240Table 22-8 lists some of the causes of chronic CP. Idiopathic, neoplastic, postirradiation, or uremic pericarditis account for most cases of CP that require surgery. Eighteen percent of pericardiectomies are attributed to previous cardiac surgery,239 which may explain the increase in the number of cases of CP since the middle 1990s.241
| Cause | Percentage |
|---|---|
| Idiopathic pericarditis | 40% |
| Post-CABG | 30% |
| Tuberculosis | 10% |
| Radiation induced | 5% |
| Collagen vascular disease | 5% |
| Others (malignancy, uremia, purulent) | 5% |
CABG, coronary artery bypass grafting.
From Kabbani SS, LeWinter MM: Diastolic heart failure, constrictive, restrictive and pericardial. Cardiol Clin 18:505, 2000.
CP clinically resembles the congestive states of myocardial and chronic liver disease.234 The most frequent physical signs are jugular venous distention, hepatomegaly, and ascites suggestive of heart failure. Symptoms are nonspecific and progress over years unless the cause is radiation, cardiac surgery, or trauma that can instead develop over months. The normal filling phases of the jugular venous pulse are shown in Figure 22-37. Venous pressure waves in pericardial constriction are characterized by a prominent y descent. Kussmaul’s sign (a paradoxical increase in venous pressure with inspiration) may be present. Systemic venous congestion accounts for some of the classic symptoms of CP such as hepatic congestion and peripheral edema.151 A significant paradoxical pulse occurs in only one third of patients with chronic CP.
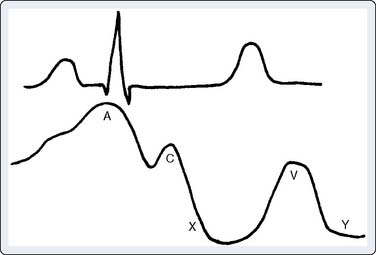
Figure 22-37 Schematic depiction of normal jugular venous pressure waves in relation to the electrocardiogram (ECG).
(From Legler D: Uncommon diseases and cardiac anesthesia. In Kaplan JA [ed]: Cardiac Anesthesia, 2nd ed. Philadelphia, WB Saunders Company, 1987, p 785, by permission.)
Characteristic ECG changes are P mitrale (broadened P wave), low QRS voltage, and T-wave inversion.241 One fourth of patients have atrial fibrillation. Cardiomegaly on chest roentgenogram is nonspecific. Pericardial calcification sometimes is apparent in lateral chest roentgenograms (< 30%),240 although less common than in the past because the incidence of tuberculosis has declined so greatly in developed nations. Although calcification is very specific for CP, it is not very sensitive. CT and MRI may demonstrate the typically thickened pericardium of CP; however, patients with surgically proven CP may have a normal-appearing pericardium with imaging tests in 28% of individuals.156
A comprehensive echo examination including 2D and Doppler imaging is an essential part of the diagnostic workup and often adequate for diagnosis. The 2D findings may include a thickened pericardium, abnormal motion of the ventricular septum, inferior vena cava dilation, and flattening of the left ventricular posterior wall during diastole.33 A key diagnostic finding in constriction is ventricular interdependence. Diastolic filling of the ventricles is reliant on each other because the overall cardiac volume is fixed by the stiffened pericardium. With inspiration, intrathoracic pressure declines, as does the pressure in the pulmonary vasculature. The thickened pericardium prevents transmission of this pressure decrease to the ventricles. Thus, filling of the LV decreases just after inspiration because of the decline in pressure within the pulmonary vasculature. Because the interpericardial space is fixed, a decrease in filling pressure to the LV allows increased filling in the RV. The result is a shift in the ventricular septum into the LV during inspiration, as well as an increase in hepatic vein diastolic flow.33 With expiration, intrathoracic pressure increases, the pressure in the pulmonary vasculature increases, and left ventricular filling is augmented with a shift of the ventricular septum into the RV during diastole. Right ventricular filling is now decreased because of the positive intrathoracic pressure, and flow reversals occur during diastole in the hepatic veins. When several cardiac cycles are viewed consecutively, the ventricular septum appears to “bounce” between the LV and RV.
Ideally, a respiratory variation of 25% or more in mitral inflow E velocity will accompany the diastolic flow reversals in the hepatic veins during expiration and support the diagnosis of pericardial constriction (Figure 22-38). However, this finding is present in only approximately 50% of patients with constriction.242,243 More recently, tissue Doppler imaging of the mitral annulus has become a valuable tool in the diagnosis of constriction. In myocardial disease, such as RCM, relaxation is impaired and the mitral annular velocity is low (< 7 cm/sec).33 However, in constriction, relaxation is normal or increased. This can be a valuable tool in differentiating between restrictive and constrictive disease, but there is no uniform acceptance of Doppler indices that differentiate CP from RCM.234 Right and left cardiac catheterization are necessary to confirm hemodynamic abnormalities in cases that are seen with Doppler imaging.151,241 Even with the Doppler echo, clinicians must be aware of the loading conditions and respiratory effort of the patient during the Doppler examination. Doppler features in CP have been more extensively reviewed.151,156,240
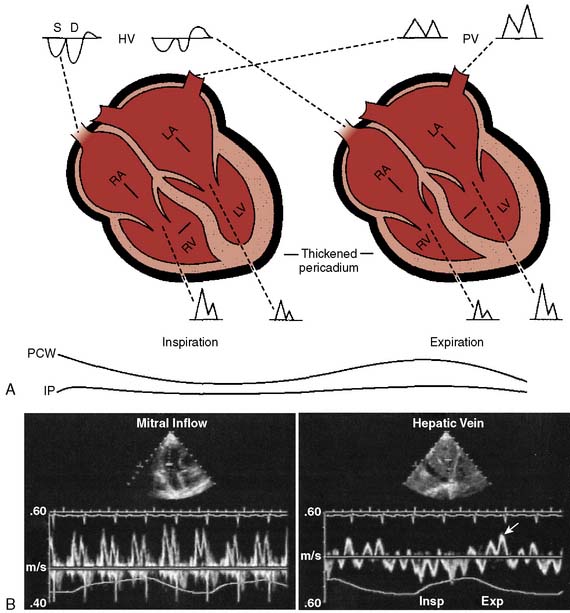
The hemodynamic changes of CP are primarily related to the isolation of the cardiac chambers from respiratory effects on thoracic pressure and a fixed end-diastolic ventricular volume.240 The pericardium limits the filling of the LV during inspiration, which leads to increased filling in the RV because the pericardium is so noncompliant. With expiration, the opposite is seen as the LV is overfilled and the RV is limited. Limitation of right ventricular diastolic filling occurs when the cardiac volume approximates the pericardial volume, usually in mid and late diastole characterized by the square-root or dip-and-plateau sign (Figure 22-39). Filling is limited by the noncompliant pericardium. The square-root sign occurs because the constricting pericardium is essentially part of the ventricular wall. When the ventricle contracts, the pericardium is deformed like a spring. As diastole begins, the spring is released and the ventricle fills rapidly, decreasing the ventricular pressure and creating the dip of the dip-and-plateau wave. As cardiac filling approaches the limit set by the fixed pericardium, the plateau of the ventricular filling curve arrives. There are marked increases of right atrial and left atrial and ventricular filling pressures. Pulmonary artery diastolic pressure, pulmonary capillary wedge pressure, and right atrial pressure are equal and elevated (“pressure plateau”) in CP because of the limitation of the pericardium. Pulmonary artery systolic and right ventricular systolic pressures range from 35 to 45 mm Hg, although PAH is rare. Most importantly, the myocardium is unaffected in CP. The systolic and early diastolic functions are normal. Differences between CP and RCM have been reviewed previously.
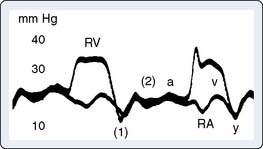
Pericardiectomy is performed for recurrent pericardial effusion and CP. Pericardial dissection for effusive pericarditis is straightforward; however, pericardiectomy for CP is a surgical challenge with an operative mortality rate of 5.9% to 11.9%.244 The 5-year survival rate is 78%.157,245 The occurrence of tricuspid regurgitation may present similar to CP with signs of right-heart failure and volume overload. In fact, a recent review found one fifth of patients presenting to surgery for CP with significant tricuspid regurgitation.244 Unfortunately, surgical treatment of tricuspid regurgitation with CP had no effect on long-term survival, but also did not increase the risk for surgery. The presence of tricuspid regurgitation may identify a subset of patients with CP who have more advanced disease and may need special attention during anesthetic management.
Persistent low CO immediately after pericardiectomy is the primary cause of morbidity and mortality, occurring in 14% to 28% of patients in the immediate postoperative period.246 The pathophysiologic mechanism of early death after pericardiectomy remains unsolved. Examination of patients who had pericardiectomy for CP with preoperative cardiac catheterization utilizing a micromanometer catheter was performed recently.246 The specialized catheter allowed nonejection indices of cardiac function to be obtained. The study compared patients with abnormalities with those with normal nonejection measurements. The most important finding was that a significant number of patients with CP had abnormal left ventricular contractility and relaxation abnormalities based on a high-fidelity manometer. More importantly, they incurred the greatest risk for operative and long-term mortality. This finding is in contrast with the accepted notion that the pericardium is the problem in these patients and the LV should be normal. Myocardial function is important as a determinant of outcome and even inotropic support. This is consistent with the clinical behavior of patients after pericardiectomy. Although patients with cardiac tamponade usually improve clinically once the pericardium is opened, improvement is not always apparent immediately after pericardiectomy. Instead, noticeable improvement in cardiac function may take weeks, but 90% of patients ultimately experience relief of symptoms with surgery.239
Median sternotomy provides excellent exposure and access for pericardiectomy, but thoracotomy in the left anterolateral position also is an option. Opinions vary regarding the extent of pericardial resection for alleviation of cardiac constriction and the need for CPB. Recently, total pericardiectomy was found to result in superior outcomes compared with partial pericardiectomy in a retrospective review.247 Removal of adherent and scarred pericardium to release both the RV and LV involves extensive manipulation of the heart and hemodynamic instability. The decision to use CPB is influenced by the surgeon’s confidence in being able to achieve complete pericardial excision with hemodynamic stability. More aggressive approaches to pericardiectomy with CPB have increased over the years as survival has improved, and the trend may continue.244 However, good results have been reported with and without CPB.247 The use of CPB entails full heparinization and may exacerbate blood loss from the many exposed cardiac surfaces. Furthermore, prolonged CPB in debilitated patients contributes to early mortality associated with pericardiectomy. An especially high-risk group of patients undergoing pericardiectomy are those with postradiation CP. They suffer not only from the effects of radiation on the myocardium that may create a more sustained restrictive effect even after pericardiectomy, but also from radiation injury to the lungs.244
Anesthetic Considerations
Anesthetic goals for managing patients with CP for pericardiectomy include minimizing bradycardia and myocardial depression, and minimizing decreases in afterload or preload. Monitoring considerations include arterial and central venous pressures. A femoral arterial catheter in patients with uremic pericarditis may preserve future potential arteriovenous fistula sites in the upper extremities. One groin site should be reserved for femoral cannulation if necessary to emergently initiate CPB. PAC monitoring is recommended because of the occurrence of low CO syndrome after surgery. Low CO syndrome develops in a subset of patients with CP irrespective of the approach or extent of pericardiectomy.247 Low CO, hypotension, and arrhythmias (atrial and ventricular) are common during chest dissection. Because of limited ventricular diastolic filling, CO is rate dependent. If myocardial function or heart rate is depressed, β-agonists or pacing will improve CO. Catastrophic hemorrhage can occur suddenly if the atrium or ventricle is perforated, so sufficient venous access is imperative. Damage to coronary arteries also may occur during dissection, so careful monitoring of the ECG for signs of ischemia is prudent. Pericardiectomy via left anterior thoracotomy requires close monitoring of oxygenation because the left lung is severely compressed during dissection. Currently, the anesthetic technique is based on achieving early extubation similar to other cardiac surgical cases; however, patients who undergo pericardiectomy for CP may benefit from remaining intubated for at least 6 to 12 hours to assess bleeding and the possibility of low CO syndrome occurring.
 Cardiac Tamponade
Cardiac Tamponade
Cardiac tamponade occurs in a variety of clinical situations, but most often in malignancies with medical patients and after pericardiocentesis.248 It is a continuum of physiologic changes that necessitate rapid diagnosis and treatment. It may be easily missed in the early stages as the signs and symptoms are subtle until it is critical. Decompensated cardiac tamponade is an emergency that requires either immediate pericardiocentesis or surgery to maintain viability.
Tamponade exists when fluid accumulation in the pericardial sac increases to an intrapericardial pressure that limits filling of the heart. The rate of fluid accumulation is the critical factor rather than the absolute volume of fluid that creates the urgency of the condition. The suspicion for cardiac tamponade must exist to increase the chance for a good outcome. Mild tamponade often is asymptomatic. One of the reasons for greater delay in the appreciation of tamponade today than 10 years ago is the tendency of clinicians to overestimate the sensitivity of clinical signs such as hypotension, pulsus paradoxus, and jugular venous distention.249 In several studies, dyspnea is the earliest and most sensitive symptom to indicate tamponade,249 and severe tamponade is accompanied by both dyspnea and chest discomfort.234 If an ECG is obtained, tamponade may show low-voltage QRS complexes, electrical alternans, and T-wave abnormalities (Figure 22-40).250 Sinus rhythm usually is present in tamponade. The chest roentogram requires at least 200 mL fluid to accumulate in the pericardium before the silhouette, known as the “water-bottle” effect, is seen.251 The classic diagnostic triad, known as Beck’s triad, of acute tamponade consists of (1) decreasing arterial pressure; (2) increasing venous pressure; and (3) a small, quiet heart. It is observed in only 10% to 40% of patients.251 Pulsus paradoxus may be present (Figure 22-41). It is a decline in systolic blood pressure of more than 12 mm Hg during inspiration caused by a reduced left ventricular stroke volume generated by increased filling of the right heart during inspiration. It is not sensitive or specific for tamponade because it may be present in those with obstructive pulmonary disease, right ventricular infarction, or CP. It may be absent if there is left ventricular dysfunction, positive-pressure breathing, atrial septal defect, or severe aortic regurgitation.
Figure 22-40 Electrical alternans in pericardial tamponade caused by the swinging of the heart within the pericardial space.
(From Aikat S, Ghaffari S: A review of pericardial diseases: Clinical, ECG and hemodynamic features and management. Cleve Clin J Med 67:909, 2000, by permission; originally modified from Longo MJ, Jaffe CC: Images in clinical medicine electrical alternans. N Engl J Med 341:2060, 1999, by permission.)
Figure 22-41 Pulsus paradoxus.
(From Reddy PS, Curtiss EI: Cardiac tamponade. Cardiol Clin 8:627–637, 1990, by permission.)
Hemodynamic monitoring may aid in the diagnosis of cardiac tamponade. As diastolic filling begins to disappear, the jugular venous pulse loses a prominent y descent. A prominent x descent remains by the decrease in intrapericardial pressure that occurs during ventricular ejection. Eventually, the pericardial pressure-volume curve becomes almost vertical, so any additional fluid greatly restricts cardiac filling and reduces diastolic compliance.252 Ultimately, the right atrial pressure, pulmonary artery diastolic pressure, and pulmonary capillary wedge pressure equilibrate (Figure 22-42). Equilibration of these pressures (within 5 mm Hg of each other) merits immediate action to rule out acute tamponade. Echo is the current method of choice and most reliable noninvasive method to detect pericardial effusion and exclude tamponade.
Figure 22-42 Hemodynamic changes in cardiac tamponade.
(From Temkin LP, Ewy GA: Cardiac catheterization. In Conahan TJ III [ed]: Cardiac Anesthesia. Menlo Park, CA, 1982, Addison-Wesley, p 1.)
The major hemodynamic changes and compensatory mechanisms of tamponade are outlined in Figure 22-43. Increased intrapericardial pressure on the heart during the cardiac cycle except for ejection is responsible for the hemodynamic changes of cardiac tamponade. Hemodynamic manifestations are due mainly to atrial rather than ventricular compression. Initially, with mild tamponade, increased atrial and pericardial pressures limit diastolic filling. Even though the intracardiac pressures are increased with tamponade, the effective preload is greatly reduced, causing lower stroke volume and CO. Sympathetic reflexes are stimulated to increase heart rate and contractility to maintain CO.241 The increasing right atrial pressure also reflexly stimulates tachycardia and peripheral vasoconstriction. Blood pressure is maintained by vasoconstriction, but CO begins to decline, as well as blood pressure, as pericardial fluid continues to increase. Once venous pressures are unable to increase to equal pericardial pressures, the decline in blood pressure is so precipitous that coronary perfusion pressure and blood flow cause ischemia, especially subendocardially.252 This resembles hypovolemic shock and will respond to fluid resuscitation, initially further confusing the diagnosis, but deterioration will soon occur if tamponade is not treated and can become fatal.234