[level-membership-for-neurology-category]
Chapter 56 Toxic and Metabolic Encephalopathies
Toxic and metabolic encephalopathies are a group of neurological disorders characterized by an altered mental status—that is, a delirium, defined as a disturbance of consciousness characterized by a reduced ability to focus, sustain, or shift attention that cannot be accounted for by preexisting or evolving dementia and that is caused by the direct physiological consequences of a general medical condition (see Chapter 4). Fluctuation of the signs and symptoms of the delirium over relatively short time periods is typical. Although the brain is isolated from the rest of the body by the blood-brain barrier, the nervous system is often affected severely by organ failure that may lead to the buildup of toxic substances normally removed from the body. This is encountered in patients with hepatic and renal failure. Damage to homeostatic mechanisms affecting the internal milieu of the brain, such as the abnormalities of electrolyte and water metabolism associated with renal failure or the syndrome of inappropriate antidiuretic hormone (SIADH) secretion, also affects brain function. In some cases, a deficiency of a critical substrate after the catastrophic failure of an organ, such as hypoglycemia caused by fulminating hepatic failure, is the precipitating factor. Frequently the history and physical examination provide information that defines the affected organ system. In other cases, the cause is evident only after laboratory data are examined.
Clinical Manifestations
Mental status abnormalities are always present and may range from subtle abnormalities, detected by neuropsychological testing, to deep coma. The level and content of consciousness reflect involvement of the reticular activating system and the cerebral cortex. Deficits in the spheres of selective attention and the ability to process information underlie many metabolic encephalopathies and affect performance on many tasks. These deficits are manifested as disorders of orientation, cognition, memory, affect, perception, judgment, and the ability to concentrate on a specific task. Evidence from studies of patients with cirrhosis suggests that metabolic encephalopathies are the result of a multifocal cortical disorder rather than uniform involvement of all brain regions. Abnormalities of psychomotor function may also be present. Among patients with coma of unknown cause, nearly two-thirds ultimately are found to have a metabolic cause. A complete discussion of coma is found in Chapter 5.
Generalized seizures occur in patients with water intoxication, hypoxia, uremia, and hypoglycemia, but only rarely as a manifestation of chronic liver failure. Seizures in patients with liver failure are generally due to alcohol or other drug withdrawal, or cerebral edema associated with acute liver failure. Focal seizures, including epilepsia partialis continua, may be seen in patients with hyperglycemia, and multifocal myoclonic seizures may occur in patients with uremia. Myoclonic status epilepticus may complicate hypoxic brain injury (see Chapter 55).
Toxic Encephalopathies
Hepatic Encephalopathy
A World Gastroenterological Association consensus statement seeks to minimize the substantial confusion in the literature and in clinical practice concerning the diagnosis of HE by using a multiaxial approach (Ferenci et al., 2002). The initial categorization addresses the presence of hepatocellular disease and portacaval shunting. Patients with acute liver disease or fulminating hepatic failure, a disorder occurring in patients with previously normal livers who exhibit neurological signs within 8 weeks of developing liver disease, form the first group. A second group consists of a small number of patients who are free of hepatocellular disease but have portacaval shunting of blood. The largest number of patients have hepatocellular disease with shunts. Further subdivisions address temporal aspects—whether HE is episodic, persistent, or minimal. Causal considerations are then applied to separate patients with precipitated HE from those with recurrent and idiopathic encephalopathy, and to identify the severity of the syndrome. The features that differentiate patients with fulminant hepatic failure from those with the much more common portal systemic encephalopathy are shown in Table 56.1.
Table 56.1 Features Distinguishing Fulminating Hepatic Failure from Chronic Hepatic Encephalopathy or Portal Systemic Encephalopathy
| Feature | Fulminating Hepatic Failure | Portal Systemic Encephalopathy |
|---|---|---|
| HISTORY | ||
| Onset | Usually acute | Varies; may be insidious or subacute |
| Mental state | Mania may evolve to deep coma | Blunted consciousness |
| Precipitating factor | Viral infection or hepatotoxin | Gastrointestinal hemorrhage, exogenous protein, drugs, uremia |
| History of liver disease | No | Usually yes |
| SYMPTOMS | ||
| Nausea, vomiting | Common | Unusual |
| Abdominal pain | Common | Unusual |
| SIGNS | ||
| Liver | Small, soft, tender | Usually large, firm, no pain |
| Nutritional state | Normal | Cachectic |
| Collateral circulation | Absent | May be present |
| Ascites | Absent | May be present |
| LABORATORY TEST | ||
| Transaminases | Very high | Normal or slightly high |
| Coagulopathy | Present | Often present |
Laboratory Evaluations
Neuropsychological tests are an underused and valuable means of diagnosing encephalopathy and monitoring the response to therapy (see Chapter 34). Sixty percent or more of all patients with cirrhosis with no overt evidence of encephalopathy exhibit significant abnormalities when given a battery of neuropsychological tests. Tests of attention, concentration, and visuospatial perception are the most likely to be abnormal. A test battery consisting of Trailmaking Tests A and B, serial dotting, line tracing, and the digit-symbol subtest of the Wechsler Adult Intelligence Scale, Revised, has been recommended for evaluating patients who may have hepatic encephalopathy. This battery is sensitive and relatively specific for the disorder, compared with other metabolic encephalopathies. Patients with alcoholic cirrhosis typically have more difficulty with memory deficits than patients with nonalcoholic cirrhosis. Even though these patients appear to be normal, the degree of impairment, particularly in the visuospatial sphere, may be severe enough to interfere with the safe operation of an automobile or other dangerous equipment. A study comparing patients with minimal encephalopathy with nonencephalopathic patients with cirrhosis and a third group with gastrointestinal disease, found that those with minimal encephalopathy performed the worst during an on-the-road driving test. Specific problems centered on handling, adaptation to road conditions, and accident avoidance. Language functions are usually normal. These data, combined with other studies showing that the quality of life is affected by these abnormalities, suggest that neuropsychological tests should be used more extensively for routine evaluation of all patients with cirrhosis, particularly those without overt evidence of HE.
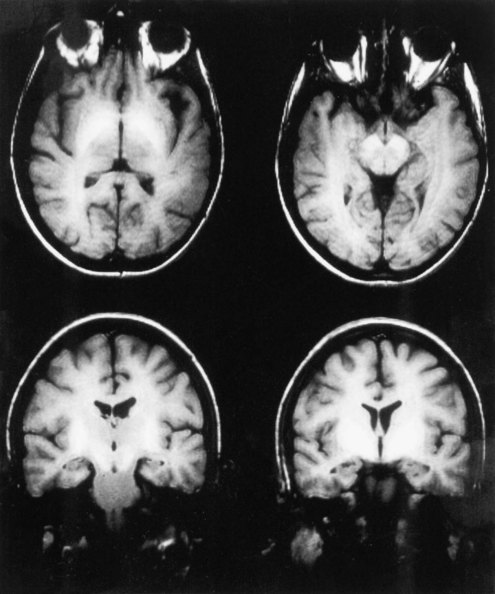
Although the diagnosis of HE is typically made on the basis of clinical criteria, neuroimaging techniques are commonly employed to exclude structural lesions. Magnetic resonance imaging (MRI) and spectroscopic studies have revealed new insights into the pathophysiology of HE (Lockwood et al., 1997). On T1-weighted images, it is common to find abnormally high signals arising in the pallidum. These are seen as whiter-than-normal areas in this portion of the brain, as shown in Fig. 56.1. In addition to these more obvious abnormalities, a systematic analysis of MR images shows that the T1 signal abnormality is widespread and found in the limbic and extrapyramidal systems, and generally throughout the white matter. A generalized shortening of the T2 signal also occurs. This abnormality is less evident on visual inspection of the images because of the generally short duration of T2 signals. These abnormalities have been linked to an increase in the cerebral manganese content. The abnormalities become more prominent with time and regress after successful liver transplantation. The unexpected finding of high T1 signals in the pallidum should suggest the possibility of liver disease.
Cerebral Blood Flow and Glucose Metabolism
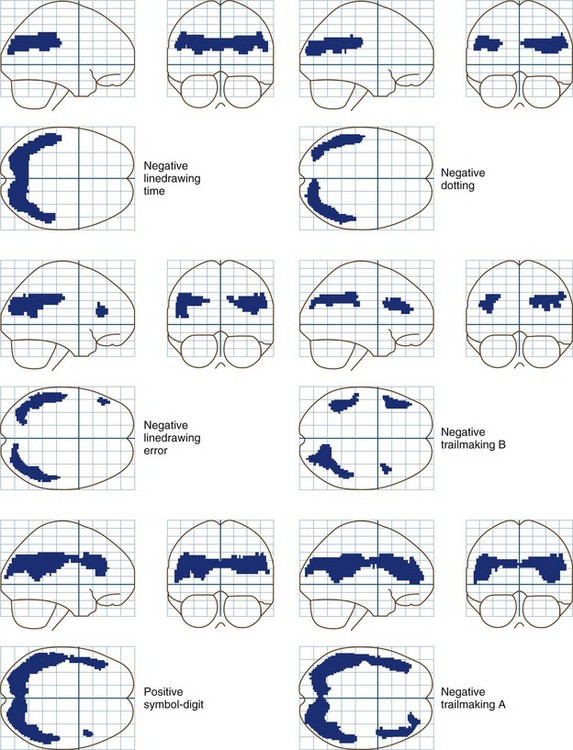
Whole-brain measurements of cerebral blood flow (CBF) and metabolism are normal in patients with grade 0 to 1 HE. Reductions occur in more severely affected patients. Sophisticated statistical techniques designed to analyze images have made it possible to identify specific brain regions in which glucose metabolism is abnormal in patients with low-grade encephalopathy and abnormal neuropsychological test scores (Lockwood et al., 2002). These positron emission tomography (PET) data show clearly that minimal forms of HE are caused by the selective impairment of specific neural systems rather than global cerebral dysfunction. Reductions occur in the cingulate gyrus, an important element in the attentional system of the brain, and in frontal and parietal association cortices. These PET data are in accord with cortical localizations based on the results of neuropsychological tests. Fig. 56.2 shows the results of correlation analyses between scores on selected neuropsychological tests and sites of reduced cerebral glucose metabolism.
Role of Ammonia
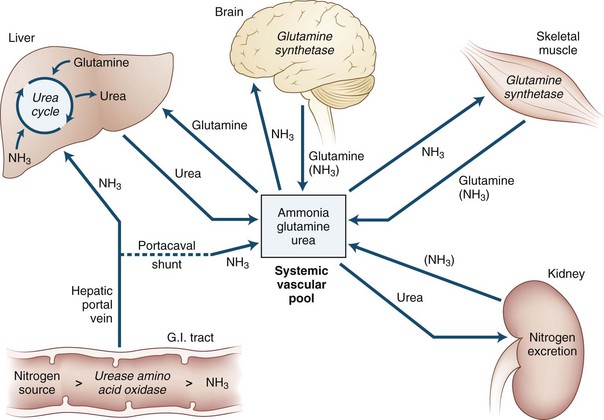
Ammonia is always extracted by the brain as arterial blood passes through the cerebral capillaries. When ammonia enters the brain, metabolic trapping reactions convert free ammonia into metabolites (Fig. 56.3). The adenosine triphosphate (ATP)-catalyzed glutamine synthetase reaction is the most important of these reactions. The blood-brain barrier is approximately 200 times more permeable to uncharged ammonia gas (NH3) than it is to the ammonium ion (NH4+); however, because the ionic form is much more abundant than the gas at physiological pH values, substantial amounts of both species appear to cross the blood-brain barrier. Because of this permeability difference and because ammonia is a weak base, relatively small changes in the pH of blood relative to the brain have a significant effect on brain ammonia extraction. As blood becomes more alkalotic, more ammonia is present as the gas and cerebral ammonia extraction increases; however, the role this has in the production of HE is not known. The permeability surface-area (PS) product of the blood-brain barrier may be affected by prolonged liver disease. However, the experimental data about this change are in conflict: one study reported an increase in the PS product, and another reported no change. An increased PS product could explain in part the toxin hypersensitivity that develops with time.
Neuropathology
The Alzheimer type II astrocyte is the neuropathological hallmark of hepatic coma. An account of the original descriptions of this change was provided in translation by Adams and Foley in 1953. In this report, they presented their own findings of this astrocyte change in the cerebral cortex and the lenticular, lateral thalamic, dentate, and red nuclei, offering the tentative proposal that the severity of these changes might be correlated with the length of coma. The cause of the astrocyte change was established by studies that reproduced the clinical and pathological characteristics of HE in primates by continuous infusions of ammonia. In studies of rats with portacaval shunts, astrocyte changes become evident after the fifth week. Before coma develops, astrocytic protoplasm increases and endoplasmic reticulum and mitochondria proliferate, suggesting that these are metabolically activated cells. After the production of coma, the more typical signs of the Alzheimer type II change became evident as mitochondrial and nuclear degeneration appeared. Norenberg (2007) suggested that HE is an astrocytic disease, although oligodendroglial cells are affected as well. More recent evidence from his laboratory has shown that ammonia affects a wide variety of astrocytic functions and aquaporin-4.
Treatment
Ideally, the management of cirrhosis should involve a cooperative effort between hepatologists, surgeons, neurologists, and psychologists, with additional input from nurses and dieticians. Practice guidelines published by the American College of Gastroenterology identify four goals: (1) provide supportive care, (2) identify and treat precipitating factors, (3) reduce the nitrogenous load from the gut, and (4) assess need for long-term therapy (Blei and Cordoba, 2001; Ferenci et al., 2002).
Lactulose
Lactulose is a mainstay for the treatment of both acute and chronic forms of HE. Its utility in the secondary prevention of HE was supported by a recent open-label placebo-controlled study of patients who had recovered from an initial episode of HE (Sharma et al., 2009). In the lactulose-treated group, 19.6% developed recurrent HE during a 1- to 14-month follow-up compared to 46.8% in the placebo group (P = 0.02). Lactulose is a synthetic disaccharide metabolized by colonic bacteria to produce acid and causes an osmotic diarrhea. A widely held but incorrect theory concerning the mechanism of action of lactulose centers on its ability to acidify the colon. Acidification presumably trapped ammonia as the charged and nonabsorbable ammonium ion, thereby preventing ammonia absorption. This theory has been questioned because lactulose treatment does not increase the fecal ammonia concentration or the total amount of ammonia excreted. The effect of lactulose is attributable to its role as a substrate in bacterial metabolism, leading to an assimilation of ammonia by bacteria or reducing deamination of nitrogenous compounds. It is probably the single most important agent in the treatment of acute and chronic encephalopathy. The usual dose of lactulose is 20 to 30 g, 3 or 4 times a day, or an amount sufficient to produce 2 or 3 stools per day. Lactulose also can be given as an enema. Lactitol, another synthetic disaccharide, is also effective. Although it is not yet available in the United States, it may have some advantages over lactulose because it can be prepared in a crystalline form that may make it more acceptable to patients who may object to the taste of lactulose preparations.
Antibiotics
Nonabsorbable antibiotics such as neomycin were among the initial treatments for HE but have been abandoned because of their renal and ototoxicity. In 2010, the U.S. Food and Drug Administration (FDA) approved oral rifaximin, 550 mg, twice daily for the treatment of HE. This nonabsorbable antibiotic had a relatively long history of use for the treatment of traveler’s diarrhea. Its efficacy was shown in a multicenter randomized, placebo-controlled, double-blind clinical trial involving 299 patients who were in remission after sustaining at least two episodes of HE (Bass et al., 2010). A breakthrough episode of HE occurred in 22.1% of the patients in the rifaximin group and in 45.9% of the patients in the placebo group, yielding a hazard ratio of 0.42 (95% confidence interval 0.28 0.64; P < 0.001). There was also a significant reduction in a secondary endpoint, the probability of rehospitalization. It is important to note that more than 90% of the patients in this trial were already receiving and continued to receive lactulose. Thus, this should be considered to be an add-on study.
Complications and Prognosis
Although studies done over 2 decades ago demonstrated that patients with hepatic coma were more likely to survive with minimal residua, this disorder still carries a substantial risk of death. Transplant-free survival at 1 year is less than 50% after an initial episode and less than 25% at 3 years. To aid in the selection of patients for transplantation, a simple rating system or MELD (Model for End-stage Liver Disease) score has been developed and validated to predict mortality. The MELD score is based on the bilirubin, serum creatinine, and the international normalized ratio (INR). The higher the MELD score, the worse the prognosis. An on-line MELD calculator and a pediatric equivalent can be found at the United Network for Organ Sharing web site (www.unos.org/resources/MeldPeldCalculator.asp?index=98).
Acute Liver Failure
Fulminant hepatic failure is usually the result of massive necrosis of hepatocytes and is defined as a syndrome in which the signs of encephalopathy develop within 8 weeks of the onset of the symptoms of liver disease in a patient with a previously normal liver. This condition has been described as “metabolic chaos” because of coexisting acid-base, renal, electrolyte, cardiac, and hematological abnormalities, usually culminating in GI bleeding, ascites, sepsis, and death frequently caused by cerebral edema. In spite of intensive treatment, patients who become comatose have an 80% to 85% mortality rate. Improvements in liver transplantation have led to better treatment and an improved likelihood of survival for these patients. Transplantation is associated with its own spectrum of neurological problems (see Chapter 49A).
ICU management focuses on the delicate balance between hypovolemia and fluid overload, treating precipitating factors, particularly if an acetaminophen overdose is present, and treating infections and coagulopathies (Trotter, 2009). The role of liver biopsy is controversial and depends on a careful consideration of risks and potential benefits. Neurological management involves the use of osmotic diuretics to reduce cerebral edema and hyperventilation to reduce cerebral blood volume. Although hyperventilation reduces cerebral blood flow, this does not appear to lead to the development of hypoxic-ischemic brain injury among survivors. Other treatments may include exchange transfusion. Substantial research efforts have been devoted to the development of artificial livers or cell-based perfusion systems designed to remove toxins from circulating blood. These treatments remain experimental.
Uremic Encephalopathy
Whereas the literature concerning encephalopathy associated with liver disease is rich and multifaceted, there are very few studies dealing with this aspect of renal failure. A recent report fills this void in part (Murray et al., 2006). Among 374 dialysis patients 55 years of age or older who were tested in the domains of memory, executive function, and language, only 12.7% were normal. Almost 14% had mild impairment, 36.1% had moderate impairment, and 37.3% had severe impairment, defined as one or more tests in two or more domains that were two or more standard deviations below age-adjusted means.
Metabolic Disturbances
Disorders of Glucose Metabolism
Clinical Aspects of Hypoglycemia
Because of the complexity of glucose homeostasis, the causes of hypoglycemia are many and varied, and a detailed discussion is beyond the scope of this chapter. In general, most authors present a physiological classification as shown in Box 56.1.
Box 56.1 Causes of Hypoglycemia
Fasting Hypoglycemia
Underproduction of Glucose
Hormone deficiencies (growth hormone, glucagon, hypoadrenalism)
Glycogen metabolism (glycogen phosphorylase, glycogen synthetase)
Hexose metabolism (glucose-6-phosphatase, fructose-1,6-biphosphatase)
Glycolysis, Krebs cycle (phosphoenolpyruvate carboxykinase, pyruvate carboxylase, malate dehydrogenase)
Alcohol and probably other drugs
Clinical Aspects of Hyperglycemia
DKA is an insulin-deficient state, and insulin is the cornerstone of therapy. In the absence of insulin, peripheral glucose uptake and glycogen formation are reduced, and glycogenolysis and lipolysis are accelerated, leading to the formation of acidic ketone bodies and hyperglycemia. When plasma glucose levels exceed the renal threshold (usually approximately 180 mg/dL), glucosuria and a forced osmotic diuresis ensue. The treatment of DKA is designed to reverse these pathophysiological abnormalities and consists of administering insulin to enhance glucose uptake, enhance glycogen formation by noncerebral tissues, and reduce the rate of ketone body formation occurring during low-insulin, high-glucagon states that promotes the entry of fatty acids into mitochondria, where they are converted to ketones. Replacing fluid and electrolytes also is required, as is treatment of precipitating factors. It is important to remember that overly vigorous treatment with rapid restoration of plasma osmolality to normal levels can lead to the development of cerebral edema (see Complications of Treatment).
Complications of Treatment
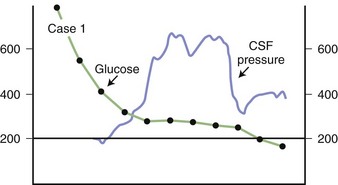
Some degree of cerebral edema attends the treatment of most patients with DKA, occasionally to the high level of 600 mm H2O CSF pressure, as shown in Fig. 56.4.
Glucose and Cardiopulmonary Resuscitation
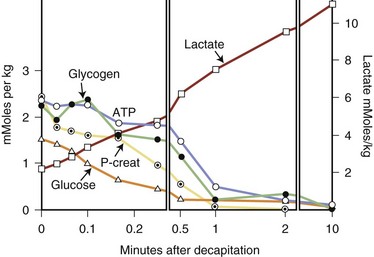
A number of studies suggest that hyperglycemia is associated with an increase in the severity of complications of cerebral ischemia and hypoxia. The presumption is that blood, and hence brain, glucose levels are higher in hyperglycemic individuals, and that this glucose produces more lactate during the hypoxic-ischemic insult. This sequence is shown in Fig. 56.5, in which the metabolic consequences of decapitation in animals are shown. Glucose is metabolized anaerobically to lactate, which with the hydrolysis of ATP causes acidosis. A large number of experimental studies suggest that cerebral acidosis is an important determinant of brain injury, including acidosis associated with lactate production during ischemia. The results of these studies have been extended to humans, in whom a less favorable outcome was suggested for stroke patients with diabetes and hyperglycemia compared with euglycemic diabetic stroke patients.
Disorders of Water and Electrolyte Metabolism
Patients with abnormalities of water and electrolyte metabolism frequently exhibit signs and symptoms of cerebral dysfunction. Typically these patients have altered states of consciousness or epileptic seizures that herald the onset of the abnormality. The vulnerability of the nervous system to abnormalities of water and electrolyte balance arises from changes in brain volume, especially the brain swelling that may be associated with water intoxication; the abnormalities are symptomatic almost immediately because the brain is enclosed by the rigid skull. The role played by electrolytes is also important in maintaining transmembrane potentials, neurotransmission, and a variety of metabolic reactions such as those involving the role of calcium and calmodulin. Although most clinicians are aware of the importance of water and electrolyte disturbances as a cause of brain dysfunction, the importance of the brain in the control of water and electrolytes is less well appreciated. Excellent reviews of these disorders have been written by Adrogué and Madias (2000).
Disordered Osmolality
Osmotic Homeostasis
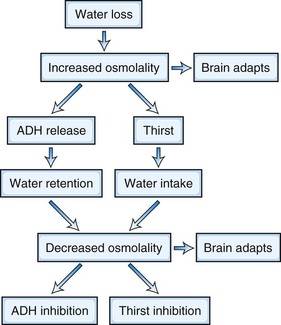
The serum, and hence whole-body osmolality, are regulated by complex neuroendocrine and renal interactions that control thirst and water and electrolyte balance. When serum osmolality increases, the brain loses volume; when osmolality falls, the brain swells. Events related to water loss are illustrated in Fig. 56.6. The brain has little protection in terms of volume changes when osmotic stress is acute. Examples of acute osmotic stress may be found in patients with heat stroke, inadvertent solute ingestion (particularly in infants), massive ingestion of water (which may be psychogenic), hemodialysis, and diabetics with nonketotic coma. Recent reports also suggest that excessive water consumption occurs in some marathon runners, leading to acute water intoxication. When osmotic stress is applied more slowly over a longer period, the predicted volume changes are smaller than would be expected. The mechanisms that underlie these protective adaptations are not known completely but involve the gain of amino acids in the case of the hyperosmolar state and the loss of potassium in the hypo-osmolar state. Experimental studies have failed to identify all of the osmotically active particles that must exist in the brain after a given osmotic stress is applied. These unidentified molecules are called idiogenic osmoles.
Hypo-Osmolality and Hyponatremia
Hypo-osmolality is almost always associated with hyponatremia. The diagnosis usually is made by laboratory testing. Conditions associated with hyponatremia are shown in Box 56.2. When hyponatremia is encountered, a measurement of serum osmolality should be performed to differentiate true from pseudo hypo-osmolality, which may be encountered in patients with lipidemic serum or in neurological patients treated with mannitol.
A large and diverse group of neurological conditions is associated with hyponatremia as a result of SIADH, as shown in Box 56.3. SIADH is characterized by hyponatremia in the face of normal or increased blood volume, normal renal function, and the absence of factors that normally operate to produce antidiuretic hormone release. The syndrome may be relatively asymptomatic, in which case water restriction is the treatment of choice. In more severe cases, hypertonic saline combined with a diuretic may be required. Overly zealous treatment may produce central pontine myelinolysis (see Therapy). Chronic syndromes have been treated successfully with a variety of drugs including the tetracycline demeclocycline, which interferes with the action of antidiuretic hormone on the renal tubules.
Box 56.3 Causes of the Syndrome of Inappropriate Antidiuretic Hormone Secretion
Hyperosmolality
Chronic hyperosmolality is associated with relative brain volume preservation as a result of the production of idiogenic osmoles, as described earlier. Administering free water at a rate that exceeds the rate at which the brain is able to rid itself of idiogenic osmoles is associated with the development of paradoxical brain edema that occurs at a time when serum glucose and electrolyte concentrations are normalized. This is illustrated by the data in Fig. 56.4, in which the CSF pressure was measured continuously as hyperglycemia due to diabetes mellitus was corrected. The increase in intracranial pressure is undoubtedly caused by adapted brain cells imbibing free water as serum osmolality decreases in response to therapy. If patients undergoing treatment for hyperosmolar states develop new neurological signs, including altered consciousness and seizures, the diagnosis of brain swelling should be considered. Mannitol treatment to restore osmolality to the prior elevated level may be required to prevent death due to brain swelling.
Drug Overdose and Toxic Exposures
The tentative diagnosis of intentional or accidental drug overdose must be considered during the course of the evaluation of almost all emergency department patients with altered behavior (see Chapters 8 and 9). Most overdoses are attributable to drugs in one of six groups that account for more than 80% of all positive laboratory results. They are, in order of decreasing frequency, ethanol, benzodiazepines, salicylates, acetaminophen, barbiturates, and tricyclic antidepressants. Box 56.4 classifies drugs into four groups based on the usefulness of toxicological information and the relationships between drug levels and symptomatology. Regional poison control centers usually are staffed by well-informed, helpful personnel and should be consulted when further information is needed or there is uncertainty about the contents of specific products. Illicit drug availability varies substantially by region and evolves constantly. So-called designer drugs are unpredictable. As benzodiazepine use has increased and replaced barbiturates used as sleeping pills, barbiturate intoxications have declined. The prevalence of overdose varies as a function of the number of prescriptions written.
Box 56.4
Characteristics of Drug Overdose
1. Toxicity predicted by drug level—specific therapy determined by toxicology:
2. Toxicity parallels drug level—supportive care required:
3. Toxicology confirms only clinical impression—clinical decisions determined by direct patient evaluation:
4. Toxicity correlates poorly with drug level—clinical decisions determined by direct patient evaluation:
Based on Mahoney, J.D., Gross, P.L., Stern, T.A., et al., 1990. Quantitative serum toxic screening in the management of suspected drug overdose. Am J Emerg Med 8, 16-22.
Miscellaneous Disorders
A similar and parallel situation has arisen among some who complain that pesticide exposure has affected their health. Because many pesticides are organophosphate cholinesterase inhibitors (OPCIs; differing from nerve agents only in potency), links between exposure and a variety of complaints have been claimed or sought. Worldwide, OPCIs are a common cause of death in agricultural workers, particularly in underdeveloped nations. In Western countries, death is less common, but accidental or intentional exposure may occur (ingestion by children, overdose among adults and agricultural workers). OPCIs may cause peripheral neuropathy, as described in Chapter 58, and a subacute condition characterized by proximal weakness and respiratory failure known as intermediate syndrome. Comparisons among control populations and OPCI-exposed subjects have shown differences in performance on certain neuropsychological tests, buttressing claims of disability and distress among exposed individuals. Epidemiological, case-control, and animal model studies all suggest that pesticide exposure may be related to the subsequent development of Parkinson disease and Alzheimer disease. The production of oxygen free radicals and oxidative stress may be the mechanism responsible for the neurodegenerative features of these disorders. The association between parkinsonism and pesticides was strengthened by an epidemiological study that included 143,000 individuals in which a 70% increase in the risk of developing Parkinson disease was found among those exposed to pesticides (Ascherio et al., 2006). Efforts to redefine pesticide tolerances, the maximal permissible concentration of pesticides in food, are complicated by predictable disagreements between pesticide manufacturers and public health groups. The pesticide industry has sponsored experiments that appear to be designed to raise tolerances. In these studies, pesticides have been administered to volunteers who are monitored for adverse effects. Ethical challenges to these studies are unresolved.
Adrogué H.J., Madias N.E. Hyponatremia. N Engl J Med. 2000;342:1493-1589.
Ascherio A., Chen H., Weisskopf M.G., et al. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197-203.
Bass N.M., Mullen K.D., Sanyal A., et al. N Engl J Med. 2010;362:1071-1081.
Blei A.T., Cordoba J. Practice guidelines: hepatic encephalopathy. Am J Gastroenterol. 2001;96:1968-1975.
Ferenci P., Lockwood A., Mullen K., et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congress of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
Lockwood A.H., Weissenborn K., Bokemeyer M., et al. Correlations between cerebral glucose metabolism and neuropsychological test performance in non-alcoholic cirrhotics. Metab Brain Dis. 2002;17:259-270.
Lockwood A.H., Weissenborn K., Butterworth R.F. An image of the brain in patients with liver disease. Curr Opin Neurol. 1997;10:525-533.
Murray A.M., Tupper D.E., Knopman D.S., et al. Cognitive impairment in hemodialysis patients is common. Neurology. 2006;67:216-223.
Norenberg M.D. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219-234.
Sharma B.C., Sharma P., Agrawal A., et al. Secondary prophylaxis of hepatic encephalopathy: an open-label randomized controlled trial of lactulose versus placebo. Gastroenterology. 2009;137:885-891.
Trotter J.F. Practical management of acute liver failure in the intensive care unit. Curr Opin Crit Care. 2009;15:163-167.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 56 Toxic and Metabolic Encephalopathies
Toxic and metabolic encephalopathies are a group of neurological disorders characterized by an altered mental status—that is, a delirium, defined as a disturbance of consciousness characterized by a reduced ability to focus, sustain, or shift attention that cannot be accounted for by preexisting or evolving dementia and that is caused by the direct physiological consequences of a general medical condition (see Chapter 4). Fluctuation of the signs and symptoms of the delirium over relatively short time periods is typical. Although the brain is isolated from the rest of the body by the blood-brain barrier, the nervous system is often affected severely by organ failure that may lead to the buildup of toxic substances normally removed from the body. This is encountered in patients with hepatic and renal failure. Damage to homeostatic mechanisms affecting the internal milieu of the brain, such as the abnormalities of electrolyte and water metabolism associated with renal failure or the syndrome of inappropriate antidiuretic hormone (SIADH) secretion, also affects brain function. In some cases, a deficiency of a critical substrate after the catastrophic failure of an organ, such as hypoglycemia caused by fulminating hepatic failure, is the precipitating factor. Frequently the history and physical examination provide information that defines the affected organ system. In other cases, the cause is evident only after laboratory data are examined.
Clinical Manifestations
Mental status abnormalities are always present and may range from subtle abnormalities, detected by neuropsychological testing, to deep coma. The level and content of consciousness reflect involvement of the reticular activating system and the cerebral cortex. Deficits in the spheres of selective attention and the ability to process information underlie many metabolic encephalopathies and affect performance on many tasks. These deficits are manifested as disorders of orientation, cognition, memory, affect, perception, judgment, and the ability to concentrate on a specific task. Evidence from studies of patients with cirrhosis suggests that metabolic encephalopathies are the result of a multifocal cortical disorder rather than uniform involvement of all brain regions. Abnormalities of psychomotor function may also be present. Among patients with coma of unknown cause, nearly two-thirds ultimately are found to have a metabolic cause. A complete discussion of coma is found in Chapter 5.
Generalized seizures occur in patients with water intoxication, hypoxia, uremia, and hypoglycemia, but only rarely as a manifestation of chronic liver failure. Seizures in patients with liver failure are generally due to alcohol or other drug withdrawal, or cerebral edema associated with acute liver failure. Focal seizures, including epilepsia partialis continua, may be seen in patients with hyperglycemia, and multifocal myoclonic seizures may occur in patients with uremia. Myoclonic status epilepticus may complicate hypoxic brain injury (see Chapter 55).
Toxic Encephalopathies
Hepatic Encephalopathy
A World Gastroenterological Association consensus statement seeks to minimize the substantial confusion in the literature and in clinical practice concerning the diagnosis of HE by using a multiaxial approach (Ferenci et al., 2002). The initial categorization addresses the presence of hepatocellular disease and portacaval shunting. Patients with acute liver disease or fulminating hepatic failure, a disorder occurring in patients with previously normal livers who exhibit neurological signs within 8 weeks of developing liver disease, form the first group. A second group consists of a small number of patients who are free of hepatocellular disease but have portacaval shunting of blood. The largest number of patients have hepatocellular disease with shunts. Further subdivisions address temporal aspects—whether HE is episodic, persistent, or minimal. Causal considerations are then applied to separate patients with precipitated HE from those with recurrent and idiopathic encephalopathy, and to identify the severity of the syndrome. The features that differentiate patients with fulminant hepatic failure from those with the much more common portal systemic encephalopathy are shown in Table 56.1.
Table 56.1 Features Distinguishing Fulminating Hepatic Failure from Chronic Hepatic Encephalopathy or Portal Systemic Encephalopathy
| Feature | Fulminating Hepatic Failure | Portal Systemic Encephalopathy |
|---|---|---|
| HISTORY | ||
| Onset | Usually acute | Varies; may be insidious or subacute |
| Mental state | Mania may evolve to deep coma | Blunted consciousness |
| Precipitating factor | Viral infection or hepatotoxin | Gastrointestinal hemorrhage, exogenous protein, drugs, uremia |
| History of liver disease | No | Usually yes |
| SYMPTOMS | ||
| Nausea, vomiting | Common | Unusual |
| Abdominal pain | Common | Unusual |
| SIGNS | ||
| Liver | Small, soft, tender | Usually large, firm, no pain |
| Nutritional state | Normal | Cachectic |
| Collateral circulation | Absent | May be present |
| Ascites | Absent | May be present |
| LABORATORY TEST | ||
| Transaminases | Very high | Normal or slightly high |
| Coagulopathy | Present | Often present |
Laboratory Evaluations
Neuropsychological tests are an underused and valuable means of diagnosing encephalopathy and monitoring the response to therapy (see Chapter 34). Sixty percent or more of all patients with cirrhosis with no overt evidence of encephalopathy exhibit significant abnormalities when given a battery of neuropsychological tests. Tests of attention, concentration, and visuospatial perception are the most likely to be abnormal. A test battery consisting of Trailmaking Tests A and B, serial dotting, line tracing, and the digit-symbol subtest of the Wechsler Adult Intelligence Scale, Revised, has been recommended for evaluating patients who may have hepatic encephalopathy. This battery is sensitive and relatively specific for the disorder, compared with other metabolic encephalopathies. Patients with alcoholic cirrhosis typically have more difficulty with memory deficits than patients with nonalcoholic cirrhosis. Even though these patients appear to be normal, the degree of impairment, particularly in the visuospatial sphere, may be severe enough to interfere with the safe operation of an automobile or other dangerous equipment. A study comparing patients with minimal encephalopathy with nonencephalopathic patients with cirrhosis and a third group with gastrointestinal disease, found that those with minimal encephalopathy performed the worst during an on-the-road driving test. Specific problems centered on handling, adaptation to road conditions, and accident avoidance. Language functions are usually normal. These data, combined with other studies showing that the quality of life is affected by these abnormalities, suggest that neuropsychological tests should be used more extensively for routine evaluation of all patients with cirrhosis, particularly those without overt evidence of HE.
Although the diagnosis of HE is typically made on the basis of clinical criteria, neuroimaging techniques are commonly employed to exclude structural lesions. Magnetic resonance imaging (MRI) and spectroscopic studies have revealed new insights into the pathophysiology of HE (Lockwood et al., 1997). On T1-weighted images, it is common to find abnormally high signals arising in the pallidum. These are seen as whiter-than-normal areas in this portion of the brain, as shown in Fig. 56.1. In addition to these more obvious abnormalities, a systematic analysis of MR images shows that the T1 signal abnormality is widespread and found in the limbic and extrapyramidal systems, and generally throughout the white matter. A generalized shortening of the T2 signal also occurs. This abnormality is less evident on visual inspection of the images because of the generally short duration of T2 signals. These abnormalities have been linked to an increase in the cerebral manganese content. The abnormalities become more prominent with time and regress after successful liver transplantation. The unexpected finding of high T1 signals in the pallidum should suggest the possibility of liver disease.
Cerebral Blood Flow and Glucose Metabolism
Whole-brain measurements of cerebral blood flow (CBF) and metabolism are normal in patients with grade 0 to 1 HE. Reductions occur in more severely affected patients. Sophisticated statistical techniques designed to analyze images have made it possible to identify specific brain regions in which glucose metabolism is abnormal in patients with low-grade encephalopathy and abnormal neuropsychological test scores (Lockwood et al., 2002). These positron emission tomography (PET) data show clearly that minimal forms of HE are caused by the selective impairment of specific neural systems rather than global cerebral dysfunction. Reductions occur in the cingulate gyrus, an important element in the attentional system of the brain, and in frontal and parietal association cortices. These PET data are in accord with cortical localizations based on the results of neuropsychological tests. Fig. 56.2 shows the results of correlation analyses between scores on selected neuropsychological tests and sites of reduced cerebral glucose metabolism.
Role of Ammonia
Ammonia is always extracted by the brain as arterial blood passes through the cerebral capillaries. When ammonia enters the brain, metabolic trapping reactions convert free ammonia into metabolites (Fig. 56.3). The adenosine triphosphate (ATP)-catalyzed glutamine synthetase reaction is the most important of these reactions. The blood-brain barrier is approximately 200 times more permeable to uncharged ammonia gas (NH3) than it is to the ammonium ion (NH4+); however, because the ionic form is much more abundant than the gas at physiological pH values, substantial amounts of both species appear to cross the blood-brain barrier. Because of this permeability difference and because ammonia is a weak base, relatively small changes in the pH of blood relative to the brain have a significant effect on brain ammonia extraction. As blood becomes more alkalotic, more ammonia is present as the gas and cerebral ammonia extraction increases; however, the role this has in the production of HE is not known. The permeability surface-area (PS) product of the blood-brain barrier may be affected by prolonged liver disease. However, the experimental data about this change are in conflict: one study reported an increase in the PS product, and another reported no change. An increased PS product could explain in part the toxin hypersensitivity that develops with time.
Neuropathology
The Alzheimer type II astrocyte is the neuropathological hallmark of hepatic coma. An account of the original descriptions of this change was provided in translation by Adams and Foley in 1953. In this report, they presented their own findings of this astrocyte change in the cerebral cortex and the lenticular, lateral thalamic, dentate, and red nuclei, offering the tentative proposal that the severity of these changes might be correlated with the length of coma. The cause of the astrocyte change was established by studies that reproduced the clinical and pathological characteristics of HE in primates by continuous infusions of ammonia. In studies of rats with portacaval shunts, astrocyte changes become evident after the fifth week. Before coma develops, astrocytic protoplasm increases and endoplasmic reticulum and mitochondria proliferate, suggesting that these are metabolically activated cells. After the production of coma, the more typical signs of the Alzheimer type II change became evident as mitochondrial and nuclear degeneration appeared. Norenberg (2007) suggested that HE is an astrocytic disease, although oligodendroglial cells are affected as well. More recent evidence from his laboratory has shown that ammonia affects a wide variety of astrocytic functions and aquaporin-4.
Treatment
Ideally, the management of cirrhosis should involve a cooperative effort between hepatologists, surgeons, neurologists, and psychologists, with additional input from nurses and dieticians. Practice guidelines published by the American College of Gastroenterology identify four goals: (1) provide supportive care, (2) identify and treat precipitating factors, (3) reduce the nitrogenous load from the gut, and (4) assess need for long-term therapy (Blei and Cordoba, 2001; Ferenci et al., 2002).
Lactulose
Lactulose is a mainstay for the treatment of both acute and chronic forms of HE. Its utility in the secondary prevention of HE was supported by a recent open-label placebo-controlled study of patients who had recovered from an initial episode of HE (Sharma et al., 2009). In the lactulose-treated group, 19.6% developed recurrent HE during a 1- to 14-month follow-up compared to 46.8% in the placebo group (P
[/not-level-membership-for-neurology-category]
