[level-membership-for-neurology-category]
Chapter 68 Sleep and Its Disorders
In the first part of this chapter, a brief overview of sleep is given, covering the definition and architecture of sleep, sleep habits and requirements, the ontogeny of sleep, dreams and sleep, the neurobiology of sleep and wakefulness, circadian rhythm and chronobiology of sleep, and functions of sleep. In the sections that follow, physiological changes in sleep are summarized, sleep deprivation, sleepiness, and causes/consequences of excessive daytime sleepiness are discussed, sleep disorders are classified, and the approach to a patient with sleep complaints is outlined. Finally, the clinical phenomenology of a variety of sleep disorders is covered, and the chapter concludes with recommendations for laboratory assessment and treatment of selected sleep disorders. For more in-depth information about the order and disorders of sleep, the reader is referred to the growing number of sleep medicine texts (Avidan and Zee, 2006; Chokroverty, 2009a; Chokroverty et al., 2003; Kryger et al., 2005; Lee-Chiong, 2006; Montagna and Chokroverty, 2011).
Definition of Sleep and Moment of Sleep Onset
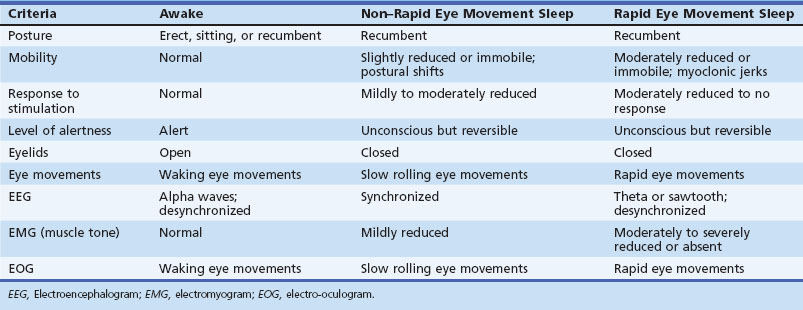
Modern researchers define sleep on the basis of both behavioral and physiological criteria (Chokroverty, 2009b) (Table 68.1). The behavioral criteria include lack of mobility or slight mobility, closed eyes, a characteristic species-specific sleeping posture, reduced response to external stimulation, quiescence, increased reaction time, elevated arousal threshold, impaired cognitive function, and a reversible unconscious state. Physiological criteria (see Sleep Architecture and Sleep Stages, later) are based on EEG, electro-oculography (EOG), and electromyography (EMG) findings as well as other physiological changes in ventilation and circulation.
Sleep Architecture and Sleep Stages
In their 1968 criteria, Rechtschaffen and Kales (RK) divided NREM sleep into stages 1, 2, 3, and 4. In 2007, this staging was modified slightly by an American Academy of Sleep Medicine (AASM) Task Force, and NREM sleep is now divided into three stages: N1, N2, and N3 (slow-wave sleep). In the modified staging criteria adopted by the AASM (Iber et al., 2007), the traditional RK stage 1 NREM sleep was labeled N1, RK stage 2 was labeled N2, and RK stages 3 and 4 were combined into one stage, N3.
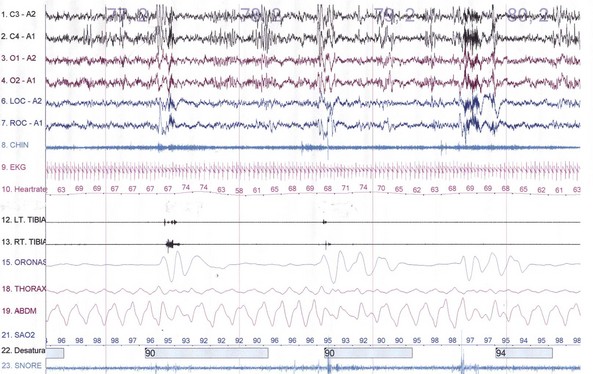
NREM sleep accounts for 75% to 80% of sleep time in adult humans. In stage N1 sleep, making up approximately 3% to 8% of sleep time, the alpha rhythms (8-13 Hz) characteristic of wakefulness (Fig. 68.1) diminish to less than 50% in an epoch (i.e., a 30-second segment of a polysomnographic (PSG) tracing with a paper or monitor speed of 10 mm/sec), and a mixture of slower theta rhythms (4-7 Hz) and beta waves (>13 Hz) appear. EMG activity decreases slightly, and slow, rolling eye movements may be recorded (Fig. 68.2). Vertex sharp waves are noted toward the end of stage N1 sleep. Stage N2 sleep begins after approximately 10 to 12 minutes of stage N1 sleep. The characteristic EEG findings of stage N2 sleep include sleep spindles (12-18 Hz, most often 14 Hz) and K complexes intermixed with vertex sharp waves (Fig. 68.3). The EEG recording contains theta activity and fewer than 20% slow waves (0.5-2 Hz). Stage N2 sleep lasts for approximately 30 to 60 minutes. During stage N3 sleep, the slow waves occupy 20% to 100% of the epoch (Fig. 68.4). Toward the end of slow-wave sleep, or N3, body movements are registered as artifacts in the PSG recordings.
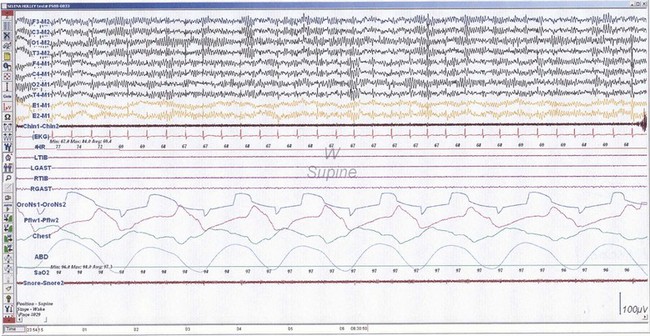
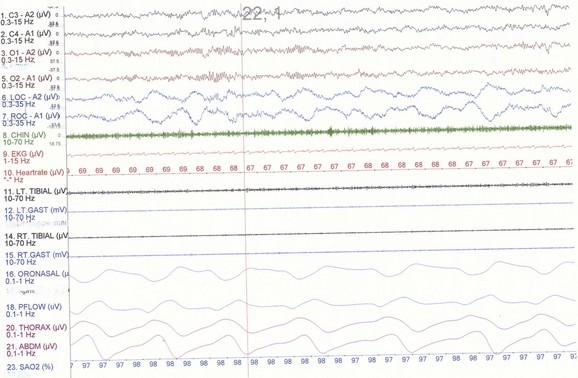
Fig. 68.2 Polysomnographic recording shows stage 1 non–rapid eye movement (NREM) sleep (N1) in an adult. Electroencephalograms (top 4 EEG channels) show a decrease of alpha activity to less than 50% and low-amplitude beta and theta activities. Left and right electro-oculograms (EOG) show slow rolling eye movements. A1, Left ear; A2, right ear; Thorax, respiratory effort (chest). Rest of montage is same as in Fig. 68.1.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.2.)
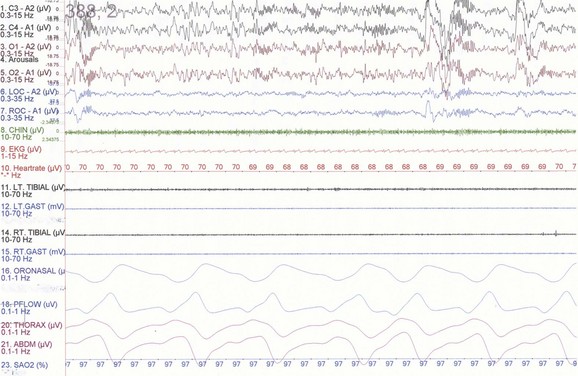
Fig. 68.3 Polysomnographic recording shows stage 2 non–rapid eye movement (NREM) sleep (N2) in an adult. Note approximately 14-Hz sleep spindles and K complexes intermixed with delta waves (0.5-2 Hz) and up to 75 µV in amplitude occupying less than 20% of the epoch. See Fig. 68.2 for description of rest of montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.3.)
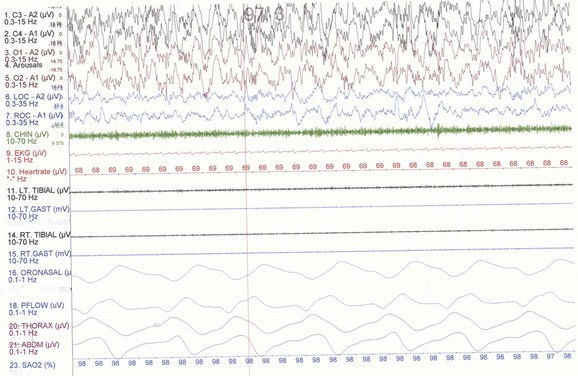
Fig. 68.4 Polysomnographic recording shows stage 4 non–rapid eye movement (NREM) sleep (N3) in an adult. Delta waves occupy more than 50% of the epoch in the traditional R&K scoring technique. See Fig. 68.2 for description of the montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.5.)
The first REM sleep (R sleep) episode is noted 60 to 90 minutes after the onset of sleep. REM sleep accounts for 20% to 25% of sleep time. Based on EEG, EMG, and EOG characteristics, REM sleep can be subdivided into two stages: tonic and phasic. However, this subdivision is not recognized in the recently modified staging. The EEG tracings during REM sleep are characterized by fast rhythms and theta activity, some of which may have a sawtooth appearance (Fig. 68.5). A desynchronized EEG, hypotonia, or atonia of the major muscle groups and depression of monosynaptic and polysynaptic reflexes are characteristics of the tonic stage. Phasic REM sleep is characterized by rapid eye movements in all directions, as well as phasic swings in blood pressure and heart rate, irregular respiration, spontaneous middle-ear muscle activity, and tongue movements. A few periods of apnea or hypopnea may arise during REM sleep. An illustration of sleep stage distribution during the night is shown in Fig. 68.6.
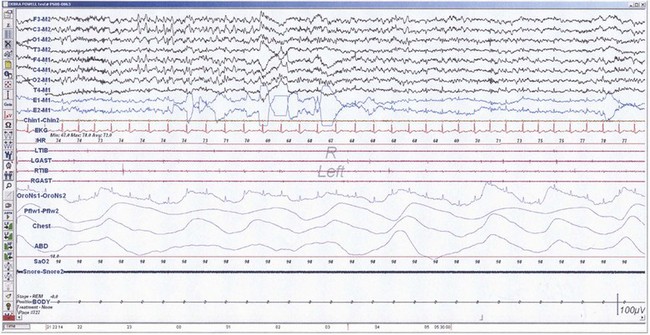
Fig. 68.5 Polysomnographic recording shows rapid eye movement (REM) sleep in an adult. Electroencephalogram (top 8 channels) shows mixed-frequency theta, low-amplitude beta, and a small amount of alpha activity. Note characteristic sawtooth waves (seen prominently in channels, 1, 2, 5, and 6 from top) of REM sleep preceding bursts of REMs in electro-oculograms (E1-M1; E2-M2). Chin electromyogram (EMG) shows marked hypotonia, whereas TIB and GAST EMG channels show very low-amplitude phasic myoclonic bursts. See Fig. 68.1 for description of the montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.6.)
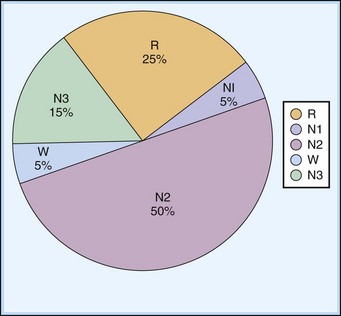
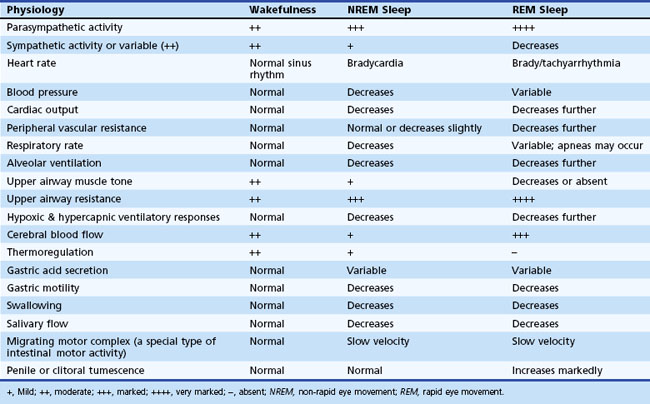
Thus, there is an orderly progression from wakefulness to sleep onset to NREM sleep and then to REM sleep. A relaxed wakefulness is characterized by a behavioral state of quiescence and a physiological state of alpha and beta frequencies on the EEG recording, waking eye movements, and increased muscle tone. Table 68.2 summarizes NREM and REM sleep states. Sleep staging and scoring address normal adult sleep and the macrostructure (Box 68.1) and microstructure (Box 68.2) of sleep. In patients with sleep disorders such as sleep apnea, parasomnias, or nocturnal seizures disrupting sleep, such assessments may be difficult.
| Sleep State | % Sleep Time |
|---|---|
| NREM sleep | 75-80 |
| N1 | 3-8 |
| N2 | 45-55 |
| N3 | 13-23 |
| REM sleep (stage R) | 20-25 |
| Tonic stage | Continuous |
| Phasic stage | Intermittent |
NREM, Non–rapid eye movement; REM, rapid eye movement.
Sleep Microstructure
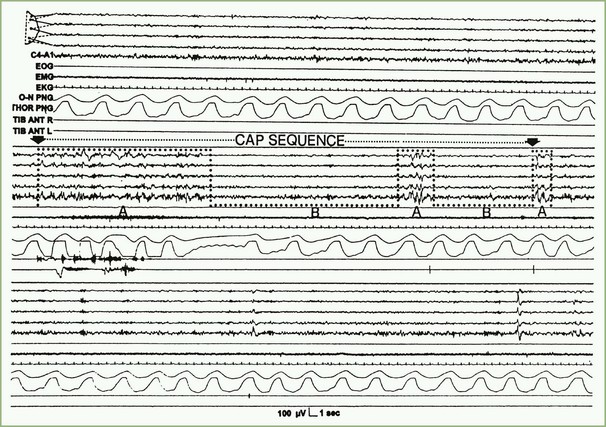
In 1992, a Task Force of the American Sleep Disorders Association (ASDA), now the AASM, offered descriptions of sleep microstructure that included momentary dynamic phenomena such as arousals and the cyclic alternating pattern (CAP), which has been described in various publications by Terzano and co-investigators (2002). Sleep microstructure also includes K complexes and sleep spindles (see Box 68.2). According to the operational definition of the ASDA Task Force, an arousal is a shift in EEG frequency lasting for 3 to 14 seconds and includes alpha, beta, or theta activities but not spindles or delta waves. The subject must be asleep for 10 consecutive seconds before an arousal can be scored. In REM sleep, arousals are scored only when accompanied by a concurrent increase in submental EMG amplitude. Unless accompanied by EEG frequency shifts, K complexes, delta waves, artifacts, and increased submental EMG activities are not counted as arousals. An arousal index is defined as the number of arousals per hour of sleep; up to 10 can be considered a normal arousal index.
In contrast to the usual pattern of arousals signifying sleep fragmentation, the CAP indicates sleep instability. The CAP is a repetitive EEG pattern that is noted mainly during NREM sleep and lasts for 2 to 60 seconds. A phase of CAP is marked by increased EEG potentials, with contributions from both synchronous high-amplitude slow and desynchronized fast rhythms in the EEG recording. A CAP cycle consists of an unstable phase (phase A) and a relatively stable phase (phase B) (Fig. 68.7). During phase A, heart rate, respiration, blood pressure, and muscle tone increase. The rate of CAP cycles and arousals increase in both older individuals and a variety of sleep disorders. A period without CAP is thought to indicate a state of sustained stability. Evaluation of periods with and without a CAP is a promising technique for understanding normal and abnormal sleep.
Ontogeny of Sleep Patterns with Age
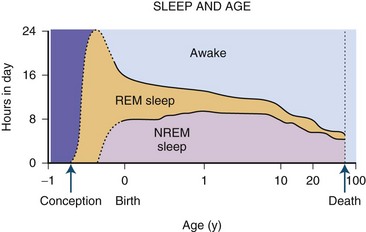
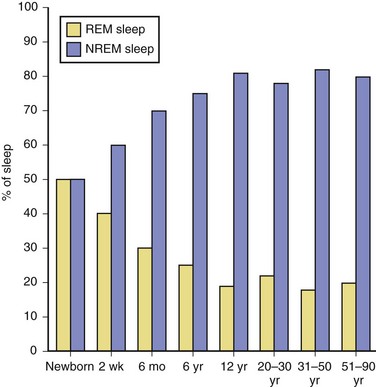
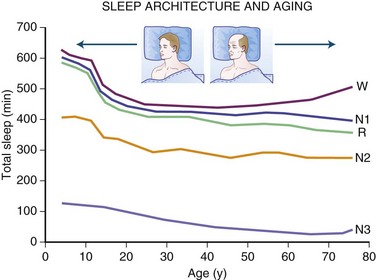
The newborn infant spends approximately 50% of the time in REM sleep, but by age 6 years this time is decreased to the normal adult pattern of 25% (Fig. 68.8). On falling asleep, a newborn baby goes immediately into REM sleep, or active sleep, which is accompanied by restless movements of the arms, legs, and facial muscles. In premature babies, it is often difficult to differentiate REM sleep from wakefulness. By age 3 months, the NREM-REM cyclical pattern of adult sleep is established. However, the duration of the NREM-REM cycle is shorter in infants, lasting for approximately 45 to 50 minutes and increasing to 60 to 70 minutes by age 5 to 10 years, and to the normal adult cyclical pattern of 90 to 100 minutes by age 10 years. Sleep spindles begin to appear at about 3 months of age; K complexes are seen by about 6 months. A characteristic feature of sleep in old age is marked attenuation of the amplitude of slow waves; therefore, during scoring of slow sleep, which depends not only on the rate but also on the amplitude of slow waves, the percentage of slow waves decreases. The other characteristic feature during old age is repeated awakenings throughout the night, including early-morning awakenings. The percentage of REM sleep in normal elderly individuals remains relatively constant, and the total duration of sleep time within 24 hours is not different from that of young adults, but elderly individuals often nap during the daytime, compensating for lost sleep during the night. Figs. 68.9 and 68.10 schematically show the evolution of sleep state distribution in newborns, infants, children, adults, and elder adults.
Sleep Habits
Depending on the sleep habit, two groups of individuals are recognized: evening types and morning types. Evening types (“owls”) have difficulty getting up early and feel tired in the morning; however, they feel fresh and energetic toward the end of the day. These people perform best in the evening; they go to sleep late and wake up late. In contrast, the morning types (“larks”) wake up early, rested and refreshed, and work efficiently in the morning. The body temperature rhythm shows two different curves in these two types of people. The body temperature reaches the evening peak an hour earlier in morning types than in evening types. Morning and evening types are most likely determined by genetic factors. Katzenberg and colleagues (1998), using the 19-item Horne-Östberg Questionnaire to determine “morningness/eveningness” in human circadian rhythms, discovered a CLOCK gene polymorphism associated with human diurnal preference. One of two H CLOCK alleles (3111C) is associated with eveningness. These findings were contradicted by a later study (Robilliard et al., 2002).
Sleep Requirements and Quantity of Sleep
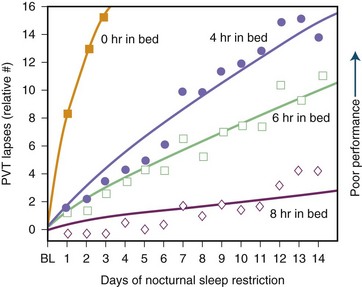
Sleep requirement is defined as the optimal amount of sleep required to remain alert and fully awake and to function adequately throughout the day. Sleep requirement for an average adult is approximately 7.5 to 8 hours regardless of environment or cultural differences. Sleep need is determined by heredity rather than by different personality traits or other psychological factors. Social or biological factors may also play a role. An important epidemiological study from 1979 (Kripke et al.) found that the chances of death from coronary artery disease, cancer, or stroke are greater for adults who sleep less than 4 hours or more than 9 hours a night when compared to those who sleep an average of 8 hours. Some more recent studies by Kripke and colleagues (2002) and others have confirmed these observations. In later studies, other confounding factors such as sleep medications may have confounded these issues. There is no clear-cut conclusion yet. There is also some controversy over whether a person can extend sleep beyond the average requirement. In 1969, Taub and Berger study showed that sleep extension beyond the average may cause exhaustion and irritability, with detriment of sleep efficiency. The overall conclusion, however, is that sleep extension has minimal effect on sleepiness and performance in absence of sleep debt, which is defined as the difference between the ideal sleep requirement and the actual duration of sleep obtained. Modern society appears to be chronically sleep deprived. Data demonstrate detrimental loss of vigilance with progressive sleep loss, confirming the possibility that sleep deprivation’s consequences are serious. Fig. 68.11 shows psychomotor vigilance task (PVT) performance lapses under varying doses of daily sleep, confirming the data of serious lapses when sleep is curtailed beyond 8 hours. A 1979 study examining 8- to 17-year-old adolescents between the years 1910 and 1963 showed a mean reduction of 1.5 hours of sleep in that time. There may, however, be a significant sampling error in this survey. A 1992 study by Bliwise and colleagues in healthy adults aged 50 to 65 showed a reduction of about 1 hour of sleep between 1959 and 1980 surveys.
Neurobiology of Sleep and Wakefulness
The neuroanatomical substrates for wakefulness, REM sleep, and NREM sleep are located in separate parts of the CNS (McCarley, 2009; Steriade and McCarley, 2005). There are no discreet sleep-promoting centers; sleep/wake states are produced by changes in interconnected systems rather than by discreet centers.
Neuroanatomical Substrates of Wakefulness
Wakefulness is controlled by the ascending reticular activating system (ARAS) containing glutamatergic, cholinergic, aminergic, and hypocretinergic neurons (Chokroverty, 2003; Steriade and McCarley, 2005). Projections from the ARAS terminating in the thalamus and thalamocortical projections to widespread areas of the cerebral cortex produce cerebral cortical activation during wakefulness. Extrathalamic projections from the brainstem reticular neurons terminate in the posterior hypothalamus and the basal forebrain regions; the latter project to the cerebral cortex (basocortical projections) to maintain wakefulness (Fig. 68.12). In the 1930s, Bremer discovered evidence of an ascending arousal system necessary for cortical arousal when he demonstrated that transection of the brainstem at the midcollicular level (i.e., cerveau isolé), but not the spinomedullary junction (i.e., encéphale isolé), produced coma in anesthetized cats. Bremer hypothesized that the resulting reduction in “cerebral tone” following the cerveau isolé was due to interruption of ascending sensory inputs—that is, a passive “deafferentation theory” of sleep (Bremer, 1937).
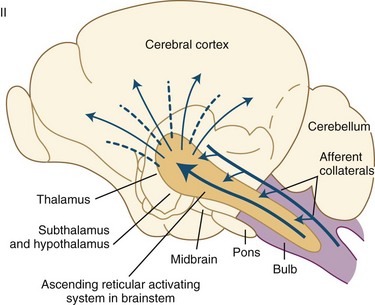
All these pathways regulating the wakefulness system use the cholinergic, noradrenergic, dopaminergic, and histaminergic neurons (Fig. 68.13). The cholinergic neurons fire at the highest rate during wakefulness and REM sleep but decrease their rates of firing at the onset of NREM sleep. Wakefulness-promoting aminergic neurons include noradrenergic neurons in the locus ceruleus, serotonergic neurons in the dorsal raphe of the brainstem, histaminergic neurons in the tuberomammillary nucleus of the hypothalamus, and possibly also dopaminergic neurons in the ventral tegmental area, substantia nigra, and ventral periaqueductal area. After a period of uncertainty regarding the role of dopamine, it has recently been confirmed that the midbrain dopaminergic system (A8-A10), particularly the dopaminergic neurons in the ventral periaqueductal gray (vPAG), play an active role in maintaining wakefulness through their widespread reciprocal connections with sleep/wake regulatory systems of neurons (Fuller and Lu, 2009). Norepinephrine-containing locus ceruleus neurons show their highest firing rates during wakefulness, their lowest during REM sleep, and intermediate rates during NREM sleep. Pharmacological studies suggest that posterior hypothalamic histaminergic neurons also help maintain wakefulness.
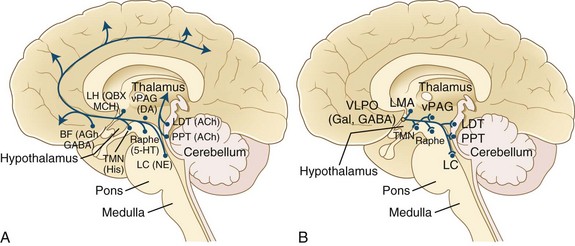
The excitatory amino acids, glutamate and aspartate, are intermingled within the ARAS and are present in many neurons projecting to the cerebral cortex, forebrain, and brainstem. These excitatory amino acids are maximally released during wakefulness. Recent discovery of hypothalamic hypocretin neurons and their widespread CNS projections have directed attention to the role of the hypocretin system in sleep/wake regulation. In 1998, deLecea and coauthors described two neuropeptides in the lateral hypothalamus and perifornical region that were termed hypocretin 1 and hypocretin 2. Independently in the same year, Sakurai and colleagues (1998) described two neuropeptides in the same region, which they named orexin A and orexin B (corresponding to hypocretin 1 and hypocretin 2, respectively). It was shown thereafter that these hypocretin systems have widespread ascending and descending projections to the locus ceruleus, dorsal raphe, ventral tegmental area, tuberomammillary nuclei of the posterior hypothalamus, laterodorsal tegmental (LDT) and pedunculopontine tegmental (PPT) nuclei, ventrolateral preoptic (VLPO) neurons in the hypothalamus, basal forebrain, limbic system (hippocampus and amygdala), cerebral cortex, thalamus (intralaminar and midline nuclei), and autonomic neurons (nucleus tractus solitarius [NTS], dorsal vagal nuclei, and intermediolateral neurons of the spinal cord). Hypocretin systems promote wakefulness mainly through excitation of tuberomammillary histaminergic, locus ceruleus noradrenergic, and midline raphe serotonergic neurons as well as dopaminergic neurons. Reduced activity of hypocretin systems may be partly responsible for inducing sleepiness. These systems also suppress REM sleep through activation of the aminergic neurons (REM-off), which in turn inhibit REM-on neurons in the LDT/PPT nuclei. As shown in Fig. 68-13, brainstem arousal centers were identified and characterized, and support was later provided for the concept of sleep-promoting circuitry in the anterior hypothalamus/preoptic area. It was later in the mid-1990s that the identity of this sleep-promoting circuitry was revealed. In these investigations (see Fig. 68.13, B), it was demonstrated that the VLPO nucleus contains sleep-active cells that contain the inhibitory neurotransmitters, γ-aminobutyric acid (GABA) and galanin (Gal).
Neuroanatomical Substrates for REM Sleep
The existence of REM sleep–generating neurons in the pontine cat has been established by transection experiments (see Fig. 68.12) through different regions of the midbrain, pons, and medulla (McCarley, 2009). A transection at the junction of the pons and midbrain produced all the physiological findings compatible with REM sleep in the section caudal to that transection, whereas in the forebrain region rostral to the section, the recording showed no signs of REM sleep. After transection between pons and medulla, structures rostral to the section showed signs of REM sleep, but structures caudal to the section had no signs of REM sleep. After transection at the junction of the spinal cord and medulla, REM sleep signs were noted in the rostral brain areas. Finally, transection at the pontomesencephalic and pontomedullary junctions produced an isolated pons that showed all the signs of REM sleep. The pons is therefore sufficient and necessary to generate all the signs of REM sleep.
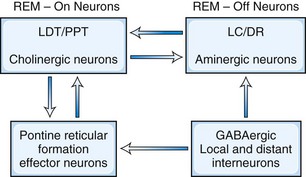
There are two main models to explain the mechanism of REM sleep. The earliest and most generally well known is the McCarley-Hobson reciprocal interaction model (Fig. 68.14) based on the reciprocal interaction of REM-on and REM-off neurons (McCarley, 2009). Neurons in the PPT and LDT nuclei in the pontomesencephalic region are cholinergic and are REM-on cells, which are responsible for REM sleep, showing highest firing rates at this stage. The REM-off cells are located in the locus ceruleus and dorsal raphe nuclei. These cells are aminergic neurons and are inactive during REM sleep. Histaminergic neurons in the tuberomammillary region of the posterior hypothalamus can also be considered REM-off cells. Thus, the cholinergic REM-on and aminergic REM-off cells are all located within the transections in the pons. LDT-PPT cholinergic neurons promote REM sleep through pontine reticular formation (PRF) effector neurons, which in turn send feedback loops to LDT-PPT neurons. Cholinergic neurons of the PPT and LDT projecting to the thalamus and basal forebrain regions, as well as to the PRF, are responsible for activation and generation of REM sleep. Aminergic cells seem to play a permissive role in the appearance of REM sleep state. In the latest modification of the reciprocal interaction model, McCarley suggested that in addition to cholinergic excitation of PRF, a reduction of GABA inhibition in the PRF also may play a role in REM sleep generation. For example, GABA levels in the PRF (as measured by microdialysis technique) are lowest during REM and intermediate between wakefulness and REM and NREM sleep. Also, injection of GABA antagonists (e.g., bicuculline) into rostral PRF produced REM sleep in cats and rats. There is evidence of GABA-ergic inhibition of locus ceruleus/dorsal raphe nuclei (REM-off neurons). The source of GABA-ergic neurons is probably both local (e.g., a subgroup of PRF GABA-ergic neurons) and distant (e.g., GABA-ergic neurons in the ventrolateral periaqueductal gray matter).
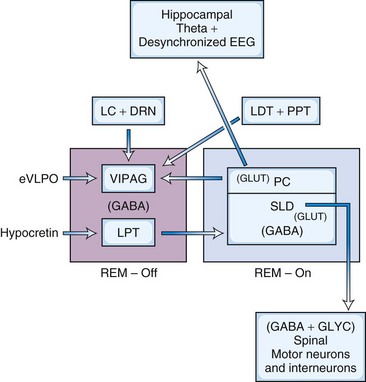
In the model (Fig. 68.15) proposed by Lu and coworkers (2006), there is reciprocal interaction between GABA-ergic REM-off neurons in ventrolateral periaqueductal gray matter (vlPAG) and lateral pontine tegmentum and GABA-ergic REM-on neurons in the sublaterodorsal nucleus (SLD; corresponding to the dorsal subceruleus or peri–locus ceruleus alpha in cats) and a dorsal extension of the SLD termed the preceruleus (PC). These mutually inhibitory neuronal populations (SLD GABA-ergic REM-on and GABA-ergic REM-off neurons in the vlPAG- lateral pontine tegmentum) serve as a flip-flop switch. Ascending glutamatergic projections from PC neurons to medial septum are responsible for EEG hippocampal theta rhythm during REM sleep. Descending glutamatergic projections from ventral SLD directly to spinal interneurons, apparently without a relay in the medial medulla, inhibit spinal ventral horns by both glycerinergic and GABA-ergic mechanisms. Cholinergic and aminergic neurons play a modulatory role and are not part of the flip-flop switch. The question arises as to how two mutually inhibitory neuronal populations (e.g., GABA-ergic REM-on and GABA-ergic REM-off neurons) stabilize the flip-flop switch permitting NREM-REM cycling. It has been suggested that inhibitory external projections from extended ventrolateral preoptic neurons (eVLPO) and excitatory projections from hypocretinergic and aminergic neurons to the REM-off neurons stabilize the switch, permitting NREM-REM cycling. McCarley (2009) suggested that this model is based on C-fos labeling, only without electrophysiological recordings. Furthermore, this model does not address how REM sleep duration progressively increases with the progression of the night. A third promising model has recently been proposed by Luppi et al. (2011).
Neuroanatomical Substrates of NREM Sleep
The contemporary theory for the mechanism of NREM sleep suggests a reciprocal interaction between two antagonistic neurons in the VLPO area of the anterior hypothalamus and wake-promoting neurons in the tuberomammillary nuclei of the posterior hypothalamus, basal forebrain, and mesopontine tegmentum (McCarley, 2009; Steriade and McCarley, 2005). Reciprocal interaction between sleep-promoting neurons in the region of the NTS and wake-promoting neurons within the ARAS of the brainstem independent of the reciprocal interaction of the neurons of the forebrain also plays a role in the generation of NREM sleep. The latter is the old reticular passive hypothesis, or the disfacilitation hypothesis, of sleep. In this model, sleep is said to result from a cascade of disfacilitation within the brainstem. The passive theory originated in the two classic preparations in cats by Bremer (1937), noted earlier in the chapter: cerveau isolé and encéphale isolé. Bremer found that in cerveau isolé (e.g., midcollicular transection), all specific sensory stimuli were withdrawn and the animals were somnolent, whereas in encéphale isolé (transection at C1 vertebral level disconnecting the entire brain from the spinal cord), these specific stimuli maintained activation of the brain and animals were awake. Moruzzi and Magoun postulated that withdrawal of generalized activation from ARAS is responsible for somnolence in cerveau isolé preparations, whereas activation of the midbrain reticular neurons causing direct excitation of the thalamocortical projections results in EEG desynchronizations and behavioral arousal. These observations support the later suggestion of Steriade and colleagues that at the onset of NREM sleep, there is a deafferentation of the brain due to blockage of afferent information, first at the thalamic level, causing the waking “open” brain to be converted to a “closed” brain owing to thalamocortical inhibition. This passive reticular theory, however, was challenged by the experiments of Batini and colleagues in 1959, producing the midpontine pretrigeminal section, which was only a few millimeters below the section that produced cerveau isolé. In this midpontine preparation, there were persistent EEG and behavioral signs of alertness, suggesting that structures located in the brainstem between cerveau isolé and midpontine pretrigeminal preparations are responsible for wakefulness. Thus an active inhibitory role of the lower brainstem hypnogenic neurons in the region of the NTS on the upper brainstem ARAS was clearly demonstrated by this preparation. Both active and passive theories partly explain the generation of NREM sleep, but the contemporary theory favors the activation of VLPO neurons and inhibition of posterior hypothalamic neurons containing histamine and hypocretin. Reduced activity of the hypocretin projections to the locus ceruleus noradrenergic, midline raphe serotonergic, mesopontine dopaminergic, and tuberomammillary hypothalamic histaminergic cells may also decrease the level of arousal, causing sleepiness.
The suprachiasmatic nuclei (SCN), the paired nuclei above the hypothalamus, function as the body clock to control circadian rhythm. The recent discovery of anatomical projections from SCN to lateral hypothalamic neurons containing hypocretin (wake-promoting) and anterior hypothalamic VLPO containing sleep-promoting neurons suggested that the SCN may also affect sleep regulation and homeostasis independent of circadian rhythm generation (Turek and Vitaterna, 2011). Finally, recent advances in neuroimaging studies, including positron emission tomography (PET) and single-photon emission computed tomography (SPECT) scans, have been able to visualize dramatic changes in function in cortical and subcortical neuronal networks in different sleep states and stages, advancing our understanding of the functional neuroanatomy of sleep/wakefulness (Dang-Vu et al., 2007). PET scans have shown marked activation of the amygdala and the anterior cingulate region (part of the limbic system) during REM sleep, which is, of course, generated by brainstem neurons. In contrast, in NREM sleep, neuroimaging techniques have shown declining function in the thalamocortical circuits, including the association cortex of the frontoparietal and temporal lobes. Thus the brainstem, hypothalamic, and forebrain sleep/wake-promoting neurons modulate functions of widespread forebrain cortical areas, keeping in balance cortical and subcortical circuits to control sleep/wake regulation.
Circadian Rhythm and Chronobiology of Sleep
The 18th-century, French astronomer de Mairan first directed our attention to the existence of circadian rhythm when he noted that in a heliotrope plant, the leaves closed at sunset and opened at sunrise even when the plant was kept in darkness (see Chokroverty, 2009a). This observation clearly pointed to a 24-hour rhythm controlled by an internal clock. The existence of such a circadian rhythm in human beings and other animals was confirmed toward the last half of the last century. The term circadian rhythm originates from the Latin circa, meaning “about,” and dies, meaning “day.” Human circadian rhythm generally has a cycle length close to 24 hours (approximately 24.2 hours) (Czeisler and Gooley, 2007). The existence of circadian rhythms independent of environmental stimuli has been clearly demonstrated by experimental isolation of humans from all environmental time cues (the German term, Zeitgeber, or “time giver”), as in a cave or underground bunker, to study free-running rhythms. The existence of environment-independent autonomous rhythms suggests that the human body also has an internal biological clock. The paired SCN of the hypothalamus above the optic chiasma are the sites of the biological clock. The master circadian clock in the SCN receives photic information from the retinohypothalamic tract, which sends signals to multiple synaptic pathways in other parts of the hypothalamus, the superior cervical ganglion, and the pineal gland where melatonin is released. The SCN contain melatonin receptors, and there is a feedback loop from the pineal gland to the SCN. Time isolation experiments have clearly shown the presence of daily rhythms in many physiological processes, such as the sleep/wake cycle, body temperature, and neuroendocrine secretion. Melatonin is secreted maximally during the night and is an important modulator of human circadian rhythm for entrainment by the light/dark cycle. The melatonin level rises fairly abruptly in the evening and then reaches its maximum level between 3:00 am and 5:00 am, after which it decreases to low levels during the daytime.
Sleep scientists have begun to identify the molecular basis of the mammalian circadian clock. A total of eight or nine genes (e.g., CLOCK, PER, Bmal) and their protein products have been identified within the circadian clock system; understanding of these is still evolving (Turek and Vitaterna, 2011). Remarkable progress has been made in the past few years in the key components of the circadian clock in both fruitflies (Drosophila) and mammals. Dysfunction of circadian rhythm results in some important human sleep disorders. The molecular mechanism of two human circadian rhythm disorders—advanced sleep phase syndrome (ASPS) and delayed sleep phase syndrome (DSPS)—has been uncovered by applying gene sequencing techniques. ASPS seems to be due to a mutation of the PER2 gene (a human homolog of the period 2 gene in Drosophila) causing advancing of the clock (i.e., alteration of the circadian timing of sleep propensity). DSPS is due to polymorphism of the PER3 gene.
Circadian, Homeostatic, and Other Sleep Factors
Sleep and wakefulness are controlled by both homeostatic and circadian factors. The duration of prior wakefulness determines the propensity to sleepiness (homeostatic factor), whereas circadian factors determine the timing, duration, and characteristics of sleep. There are two types of sleepiness: physiological and subjective (Dinges, 1995). Physiological sleepiness is the body’s propensity to sleepiness. There are two highly vulnerable periods of sleepiness: 2:00 to 6:00 am, particularly 3:00 to 5:00 am, and 2:00 to 6:00 pm, particularly 3:00 to 5:00 pm. The propensity to physiological sleepiness (e.g., midafternoon and early-morning hours) depends on circadian factors. The highest number of sleep-related accidents have been observed during these periods. Subjective sleepiness is the individual’s perception of sleepiness; it depends on several external factors such as a stimulating environment and ingestion of coffee and other caffeinated beverages.
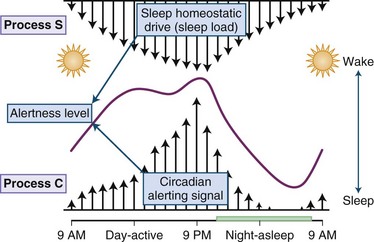
Physiological sleepiness depends on two processes: homeostatic factor and circadian phase. Homeostatic factor refers to a prior period of wakefulness and sleep debt. After a prolonged period of wakefulness, there is an increasing tendency to sleep. The recovery from sleep debt is aided by an additional amount of sleep, but this recovery is not linear. Thus, an exact number of hours of sleep are not required to repay sleep debt; rather, the body needs an adequate amount of slow-wave sleep for restoration. The interaction between the circadian and the homeostatic drive producing alertness is depicted in Fig. 68.16. The circadian factor determines the body’s propensity to maximal sleepiness between 3:00 am and 5:00 am. The second period of maximal sleepiness (3:00 to 5:00 pm) is not as strong as the first. Sleep and wakefulness and the circadian pacemaker have a reciprocal relationship: the biological clock can affect sleep and wakefulness, and sleep and wakefulness can affect the clock. There are two variables that seem to play a role in regulating the timing of sleep. First is the homeostatic sleep drive, which increases as the day progresses and the longer a person is awake. The second is timing information from the suprachiasmatic nucleus (SCN). In this two-process model, the SCN promotes wakefulness by stimulating arousal networks. The activity of the circadian system appears to oppose that of the homeostatic sleep drive, and thus the alerting mediated by the SCN increases during the day. The propensity to be awake or asleep at any time is related to the homeostatic sleep drive and the opposing SCN alerting signal. At normal bedtime, both the alerting drive and the sleep drive are at their highest level. The SCN has at least two types of melatonin receptors, MT1 and MT2, involved in the regulation of sleep. Stimulation of MT1 receptors is believed to decrease the alerting signal from the SCN, while MT2 stimulation is thought to be involved in synchronizing the circadian system.
Various sleep factors have been identified, but their role in maintaining homeostasis has not been clearly established (Krueger et al., 2011). Several cytokines, such as interleukin-1, interferon-α, and tumor necrosis factor, promote sleep. Other sleep factors increase in concentration during prolonged wakefulness and infection. It has been shown that adenosine in the basal forebrain can fulfill the major criteria for the neural sleep factor that mediates the somnogenic effect of prolonged wakefulness by acting through adenosine A1 and A2A receptors. Several other endogenous compounds may serve as sleep factors, including delta sleep-inducing peptides, muramyl peptides, cholecystokinins, arginine vasotocin, vasoactive intestinal peptides, growth hormone–releasing factors, and somatostatins.
Functions of Sleep
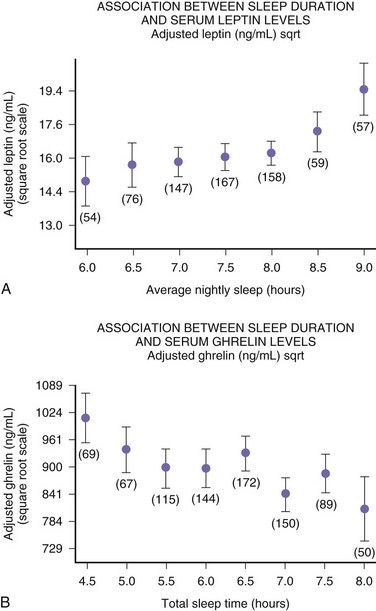
The function of sleep remains the greatest biological mystery of all time. There are several theories about the function of sleep (Box 68.3), but none are satisfactory (Crick and Mitchison, 1995; Kavanau, 1997; Mahowald et al., 1997). Sleep deprivation experiments in animals have clearly shown that sleep is necessary for survival, but from a practical point of view, complete sleep deprivation for a prolonged period cannot be conducted in humans. Sleep deprivation studies in humans have shown an impairment of performance, which demonstrates the need for sleep. The performance impairment of prolonged sleep deprivation results from a decreased motivation and frequent “microsleeps.” Overall, human sleep deprivation experiments have proven that sleep deprivation causes sleepiness and impairment of performance, vigilance, attention, concentration, and memory. Sleep deprivation may also cause some metabolic, hormonal, and immunological effects. Sleep deprivation causes immune suppression; even partial sleep deprivation reduces cellular immune responses. Studies from Van Cauter’s (Spiegel et al., 1999) group include a clearly documented elevation of cortisol level following even partial sleep loss, suggesting an alteration in hypothalamic-pituitary-adrenal axis function. This has been confirmed even in chronic sleep deprivation that causes impairment of glucose tolerance. Glucose intolerance may contribute to memory impairment as a result of decreased hippocampal function. Chronic sleep deprivation may also cause a decrement of thyrotropin concentration, increased evening cortisol level, and sympathetic hyperactivity, which may serve as risk factors for obesity, hypertension, and diabetes mellitus. Further data from the same group demonstrated that increase in ghrelin levels associated with short sleep reduced leptin. These differences in leptin and ghrelin may induce increased appetite, possibly explaining the increased BMI observed with short sleep duration in the study populations. The authors correctly indicate that in Western societies, where chronic sleep restriction is common and food is ubiquitous, alterations in appetite regulatory hormones with sleep curtailment may contribute to obesity, as shown in Fig. 68.17.
Some recent advances have been made in understanding the molecular mechanisms of memory consolidation during sleep (Stickgold and Walker, 2007). Studies have revealed that new memories can be strengthened by sleep. In other words, sleep can rescue memories that are lost during wakefulness during the daytime, and consolidated memories can be reconsolidated when they are reactivated. Memory is stored in two stages: short-term memory, which is labile, and long-term memory, which is stable and consolidated, requiring synthesis of new ribonucleic acid (RNA) and proteins by the neurons. Recent studies have clearly shown a significant contribution of sleep to memory consolidation. It has been suggested that memory consolidation occurs during both slow-wave and REM sleep. There is a further suggestion that REM-NREM sleep cycling is also important for memory consolidation.
The theory of maintenance of synaptic and neuronal network integrity is an emerging concept that is concerned with the primary function of sleep. Intermittent stimulation of neural network synapses is necessary to preserve CNS function (Kavanau, 1997).
Gene expression (Cirelli and Tononi, 2011) studied by deoxyribonucleic acid (DNA) microarray technique identified sleep- and wakefulness-related genes (brain transcripts) subserving different functions (e.g., energy metabolism, synaptic excitation, long-term potentiation, and response to cellular stress during wakefulness and protein synthesis, memory consolidation, and synaptic downscaling during sleep).
Physiological Changes in Sleep
Various physiological changes occur during NREM and REM sleep that are different from those noted during wakefulness (Chokroverty, 2009a; Chokroverty, 2002). These changes are observed in somatic and autonomic nervous systems and include changes in the respiratory, cardiovascular, and gastrointestinal systems; endocrine, renal, and sexual function; and thermoregulation (Table 68.3).
Somatic Central Nervous System
Firing rates of many neurons in the CNS decrease during NREM sleep but increase during REM sleep.
Endocrine Function Changes
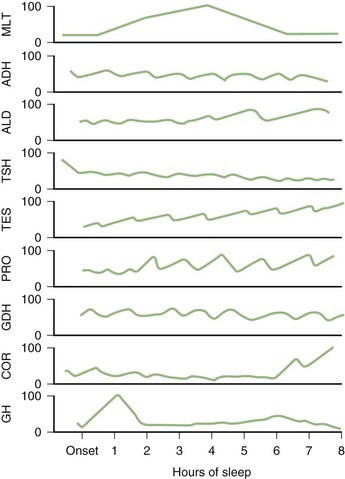
Profound changes in neuroendocrine secretions are found during sleep (Fig. 68.18). Growth hormone secretion exhibits a pulsatile increase during NREM sleep in the first one-third of the night. Prolactin secretion also rises 30 to 90 minutes after the onset of sleep. Sleep inhibits cortisol secretion. Secretion of thyroid-stimulating hormone reaches a peak in the evening and then decreases throughout the night.
Sleep Deprivation and Sleepiness
A large segment of the population working different shifts and having irregular sleep/wake schedules is chronically sleep deprived (Bonnet and Arand, 1995). The at-risk groups include doctors, nurses, firefighters, interstate truck drivers, police officers, overnight train drivers, and high school and college students. Comparing a survey conducted at the beginning of the last century and another toward the end of the century, modern America is sleep deprived by approximately 1.5 hours of sleep nightly (Harrison and Horne, 1995). This does not necessarily mean sleep requirement is smaller today, it simply means Americans are getting less sleep. However, there may have been a sampling error in these two surveys; for example, 2000 people were sampled in the earlier survey versus 311 in the later one. Increased environmental light and sound, industrialization, increasing numbers of people working in various shifts, and the advent of television and radio are cited as some of the factors responsible for this reduction of total sleep hours. Harrison and Horne (1995), however, argued that most people are not chronically sleep deprived but have the capacity to take more sleep. In one epidemiological study, sleepiness in Western society was estimated to be found in 5% to 40% of the total population (Partinen, 2011).
Total Sleep Deprivation
One of the earliest experiments in sleep deprivation in humans was in 1896 (see Chokroverty, 2009a). The effects of a 90-hour period of sleep deprivation were studied in three healthy young men. All subjects had difficulty staying awake, but they felt totally refreshed and rested after they were allowed to sleep for 10 hours. One of them, however, had sensory illusions that disappeared completely after recovery of sleep period. In 1965, a spectacular experiment was conducted when a 17-year-old California college student named Randy Gardner (see Chokroverty, 2009a) tried to set a new world record for staying awake. He remained awake for 264 hours, 12 minutes and then slept for 14 hours, 14 minutes, after which he recovered fully. Thus, the experiment concluded that it was possible to deprive people of sleep for a prolonged period without causing serious mental impairment. Other important observations include frequent occurrence of “microsleep” episodes (i.e., brief episodes of NREM sleep) and loss of performance that may have been caused by the loss of motivation. Several other later experiments conducted in humans showed no permanent adverse effects after sleep deprivation. However, sleep deprivation increases the daytime tendency to sleep, as proven by multiple sleep latency tests in such subjects. The percentage of slow-wave sleep increases considerably during the recovery sleep period after sleep deprivation. The REM sleep percentages also increase during the recovery sleep after a prolonged period of sleep deprivation, but this increment of REM sleep percentage was not shown after short periods of sleep deprivation for 4 days. These facts might suggest that different mechanisms regulate NREM and REM sleep.
Consequences of Excessive Daytime Sleepiness
The consequences of excessive daytime sleepiness (EDS) presented in Box 68.4 principally affect four key domains: (1) performance and productivity at work and school, (2) higher cerebral functions, (3) quality of life and social interactions, and (4) morbidity and mortality (Roth and Roehrs, 1996).
Higher Cerebral Functions
Sleepiness interferes with higher cerebral functions, causing impairment of short-term memory, concentration, attention, cognition, and intellectual performance. Psychometric tests document increased reaction time in patients with excessive sleepiness. These individuals make increasing numbers of errors, and they need increasing time to reach the target in reaction time tests (Dinges, 1995). Sleepiness can also impair perceptual skills and new learning. Insufficient sleep and excessive sleepiness may cause irritability, anxiety, and depression. Learning disabilities and cognitive impairment due to impaired vigilance have also been described (Roth and Roehrs, 1996).
Morbidity and Mortality
Persistent daytime sleepiness causes individuals to have an increased likelihood of accidents. Estimates by the U.S. National Highway Traffic Safety Administration showed that approximately 56,000 police-reported crashes per year resulted from drivers who were “asleep at the wheel.” New York state police estimate that 30% of all fatal crashes along the New York State Thruway occur because the driver fell asleep at the wheel. Approximately 1 million crashes annually (one-sixth of all crashes) are thought to be produced by driver inattention or lapses. Sleep deprivation and fatigue make such lapses more likely to occur. Truck drivers are especially susceptible to fatigue-related crashes (Lyznicki et al., 1998). Many truckers drive during the night while they are sleepiest. Truckers also may have a high prevalence of sleep apnea. The U.S. Department of Transportation estimates that 200,000 automobile accidents each year may be related to sleepiness. Nearly one-third of all trucking accidents that are fatal to the driver are related to sleepiness and fatigue. A general population study by Hays and colleagues (1996) involving 3962 elderly individuals reported an increased mortality risk of 1.73 in those with EDS, defined by napping most of the time.
The presence of sleep disorders (see Primary Sleep Disorders Associated with Excessive Daytime Sleepiness, later) increases the risk of crashes. Individuals with untreated insomnia, sleep apnea, and narcolepsy, as well as shift workers, have more automobile crashes than other drivers (Costa de Silva et al., 1996). A 1991 Gallup Organization national survey found that individuals with chronic insomnia reported 2.5 times as many fatigue-related automobile accidents as those without insomnia. The same 1991 Gallup survey found serious morbidity associated with untreated sleep complaints, as well as impaired ability to concentrate and accomplish daily tasks, impaired memory, and interpersonal difficulties.
A 1994 telephone survey of drivers by the New York State Task Force estimated that approximately 25% reported that they had fallen asleep at the wheel at some time. Young male drivers are especially susceptible to crashes caused by falling asleep, as documented in a study in North Carolina in 1990, 1991, and 1992 (e.g., in 55% of the 4333 crashes, the drivers were predominantly male and age 25 or younger). In the October 1995 Gallup poll, 52% of all adults surveyed said that in the past year, they had driven a car or other vehicle while feeling drowsy; 31% of adults admitted dozing off while at the wheel of a car or other vehicle, and 4% reported having had an automobile accident because of tiredness during driving. A number of national and international catastrophes involving industrial operations, nuclear power plants, and all modes of transportation have been related to sleepiness and fatigue (Dinges, 1995). Some of the more infamous examples are the Exxon Valdez oil spill in Alaska; the nuclear disaster at Chernobyl in the former Soviet Union; the near-nuclear disaster at Three Mile Island in Pennsylvania; the gas leak disaster in Bhopal, India, resulting in 25,000 deaths; and the Challenger space shuttle disaster. In postgraduate medical training, new work hours schedules have been proposed by the Institute of Medicine (IOM) and were recently accepted by the Accreditation Council for Graduate Medical Education (ACGME, 2010) and scheduled to go into affect in July 2011.
Causes of Excessive Daytime Sleepiness
Excessive sleepiness may result from both physiological and pathological causes (Box 68.5).
Box 68.5 Causes of Excessive Daytime Sleepiness
Pathological Causes of Sleepiness
Other neurological causes of EDS include bilateral paramedian thalamic and sometimes other cortical and subcortical infarcts (Bassetti and Valko, 2006), posttraumatic hypersomnolence, and multiple sclerosis (MS). Narcolepsy-cataplexy has been described in occasional patients with MS and arteriovenous malformations in the diencephalon (Clavelou et al., 1995).
Certain neurodegenerative diseases such as Alzheimer disease (AD), Parkinson disease (PD), and multisystem atrophy (MSA) also may cause EDS (Raggi and Ferri, 2010). The causes of EDS in AD include degeneration of the SCN, resulting in circadian dysrhythmia, associated sleep apnea-hypopnea, and periodic limb movements in sleep (PLMS). In PD, excessive sleepiness may be due to the associated PLMS, sleep apnea, and depression. EDS in MSA associated with cerebellar parkinsonism or parkinsonian-cerebellar syndrome and progressive autonomic deficit (Shy-Drager syndrome) may be caused by the frequent association with sleep-related respiratory dysrhythmias and possible degeneration of the reticular activating arousal systems (Chokroverty, 2009a).
Sleep disorders are being increasingly recognized as a feature of PD and other parkinsonian disorders. Although some studies have attributed the excessive daytime drowsiness and irresistible sleep episodes (sleep attacks) to antiparkinsonian medications (Ondo et al., 2001), sleep disturbances are also an integral part of PD (Arnulf et al., 2002). In one study of 303 patients with PD, 21% reported falling asleep while driving (Ondo et al., 2002). Several studies also reported a relatively high incidence (10%-20%) of symptoms of restless legs syndrome (RLS) in patients with PD (Hening et al., 2009; Krishnan et al., 2003; Ondo et al., 2002). There is also increasing awareness about the relationship between parkinsonian disorders and REM sleep behavior disorder (RBD), and RBD may be the presenting feature of PD, MSA, and other parkinsonian disorders (Boeve, 2010; Boeve et al., 2007; Postuma et al., 2010a; Raggi and Ferri, 2010).
These and other studies provide evidence supporting the notion that dopamine activity is normally influenced by circadian factors (Rye and Jankovic, 2002). For example, tyrosine hydroxylase levels fall several hours before waking, and their increase correlates with motor activity. The relationship between hypocretin and sleep disorders associated with PD is currently being explored (Baumann et al., 2005; Drouot et al., 2003; Fronczek et al., 2007; Overeem et al., 2002; Thannickal et al., 2007).
Primary Sleep Disorders Associated With Excessive Daytime Sleepiness
A number of primary sleep disorders cause excessive sleepiness (Box 68.6; see also Box 68.5). The most common cause of EDS in the general population is insufficient sleep syndrome associated with sleep deprivation. The next most common cause is obstructive sleep apnea syndrome (OSAS); narcolepsy and idiopathic hypersomnolence are other common causes of EDS. Most patients with EDS referred to the sleep laboratory have OSAS. Other causes of EDS include circadian rhythm sleep disorders, periodic limb movement disorder (PLMD), RLS, some cases of chronic insomnia, and inadequate sleep hygiene.
Box 68.6
International Classification of Sleep Disorders (ICSD-2, 2005)
II. Sleep-related breathing disorders
III. Hypersomnias of central origin not due to a circadian rhythm sleep disorder, sleep-related breathing disorder, or other cause of disturbed nocturnal sleep
IV. Circadian rhythm sleep disorders
VI. Sleep-related movement disorders
VII. Isolated symptoms, apparently normal variants, and unresolved issues
Appendix A: Sleep disorders associated with conditions classifiable elsewhere
Appendix B: Other psychiatric and behavioral disorders frequently encountered in the differential diagnosis of sleep disorders
NREM, non–rapid eye movement; REM, rapid eye movement.
Data from International Classification of Sleep Disorders (ICSD-2: Diagnostic and Coding Manual), 2005. American Sleep Disorders Association, Rochester, MN.
Classification of Sleep Disorders
International Classification of Sleep Disorders
The latest edition of the International Classification of Sleep Disorders (ICSD-2) (AASM, 2005) listed eight broad categories of disordered sleep, along with several subcategories under each category as well as appendices A and B (see Box 68.6). Appendix A and Appendix B include sleep disorders associated with other conditions, including sleep-related epilepsy and headache, fatal familial insomnia, and psychiatric and behavioral disorders that are frequently encountered in the differential diagnosis of sleep disorders. The eight broad categories consist of insomnia, sleep-related breathing disorders, hypersomnia of central origin not due to a circadian rhythm sleep disorder, sleep-related breathing disorder or other causes of disturbed nocturnal sleep, circadian rhythm sleep disorders, parasomnias, sleep-related movement disorders, isolated symptoms, apparently normal variants, and unresolved issues as well as other sleep disorders.
Approach to a Patient with Sleep Complaints
The physician must evaluate the patient first on the basis of the history and physical examination before undertaking laboratory tests, which must be determined by the clinical diagnosis (Chokroverty, 2009a). The first step in the assessment of a sleep/wakefulness disturbance is careful evaluation of the sleep complaints. The history should include information on the patient’s entire 24 hours and must include a detailed sleep history, with a sleep questionnaire as well as a sleep log or diary. It must also include psychiatric, neurological, medical, drug-alcohol, and family and past histories. The history must be followed by a physical examination to uncover medical or neurological causes of insomnia, hypersomnia, and parasomnias. Physical examination of patients with OSAS may uncover upper airway anatomical abnormalities.
It is advisable to interview the bed partner, caregiver, or parents of children to get an adequate history, particularly the history during sleep at night, which may have an effect on daytime functioning. A sleep questionnaire containing a list of pertinent questions relating to sleep complaints; sleep hygiene; sleep patterns; medical, psychiatric, and neurological disorders; and drug and alcohol use may be filled out by the patient to save time during the history taking. A sleep log kept over a 2-week period is also a valuable indicator of sleep hygiene. Such a log should include information on bedtime, arising time, daytime naps, amount of time needed to go to sleep, number of nighttime awakenings, total sleep time, and feelings on arousal (Fig. 68.19). Questions should be asked regarding the patient’s mood and naps during the daytime. In women, the relation of sleepiness to the menstrual cycle also should be ascertained.

Subjective Measures of Sleepiness
A variety of scales have been developed to assess the subjective degree of sleepiness (Chokroverty, 2009a). The Stanford Sleepiness Scale (Table 68.4) is a 7-point scale that measures subjective sleepiness but may not be reliable in patients with persistent sleepiness.
| Scale Rating | Degree of Sleepiness |
|---|---|
| 1 | Wide awake, active, and alert |
| 2 | Awake and able to concentrate but not functioning at peak |
| 3 | Relaxed, awake, and responsive but not fully alert |
| 4 | Feeling a little foggy |
| 5 | Difficulty staying awake |
| 6 | Sleepy, prefer to lie down |
| 7 | Cannot stay awake; sleep onset is imminent |
Modified with permission from Hoddes, E., Zarcone, V., Smythe, H., et al., 1973. Quantification of sleepiness: a new approach. Psychophysiology 10, 431-436.
Another scale is the visual analog scale of alertness and well-being. In this scale, subjects are asked to indicate their feelings on an arbitrary line. This scale has been used successfully in circadian rhythm disorders. The Epworth Sleepiness Scale (Table 68.5) evaluates general level of sleepiness (Johns, 1998). The patient is rated on 8 situations with a score of 0 to 3 (3 being the highest chance of dozing off). The maximum score is 24, and a score greater than 10 suggests the presence of excessive sleepiness. This scale has been weakly correlated with Multiple Sleep Latency Test (MSLT) scores, the gold-standard measurement at present for daytime sleepiness. Normal values do not necessarily imply a lack of clinically significant daytime sleepiness.
Table 68.5 Epworth Sleepiness Scale
| Eight Situations | Scores* |
|---|---|
| Sitting and reading | __ |
| Watching television | __ |
| Sitting in a public place (e.g., a theater or meeting) | __ |
| Sitting in car as a passenger for an hour without a break | __ |
| Lying down to rest in the afternoon | __ |
| Sitting and talking to someone | __ |
| Sitting quietly after a lunch without alcohol | __ |
| In a car, while stopped for a few minutes in traffic | __ |
* Scale to determine the total scores: 0, would never doze; 1, slight chance of dozing; 2, moderate chance of dozing; 3, high chance of dozing.
Modified with permission from Johns, M.W., 1991. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 14, 540-545.
Clinical Phenomenology
Insomnia
Insomnia is the most common sleep disorder affecting the population and the most common disease encountered in the practice of sleep medicine (Morin and Benca, 2009, 2011). Several population surveys of adults have determined that approximately 35% of the general public has had insomnia complaints; in 10%, this is a persistent problem (Chokroverty, 2006; Leger et al., 2000; Ohayon and Guilleminault, 2009; Partinen, 2011). The AASM (2005) defines insomnia as inability to initiate or maintain sleep, early awakening, inadequate sleep time, or poor sleep quality associated with a lack of feeling restored and refreshed in the morning, leading to poor daytime functioning. The perception of insomnia does not necessarily depend on total hours of sleep obtained, because there is considerable individual variation in sleep requirement. Nonrestorative sleep occurring three or four times a week, persisting for more than 1 month, and associated with impaired daytime functioning is the typical patient complaint. There is an increasing association of insomnia with age, female sex, low socioeconomic status, divorce, widowhood, separation, recent stress and depression, and drug or alcohol abuse (Martikainen et al., 2003).
Acute Insomnia
The category of adjustment insomnia (AASM, 2005) is the only class of acute insomnia resulting from an identifiable stressful situation. This lasts from a few days to a few weeks—3 months at most. Once the stressful event is removed and the patient adjusts to the event, the sleep disturbance resolves. Causes of acute insomnia are listed in Box 68.7.
Box 68.7 Causes of Acute Insomnia
A change of sleeping environment (most common cause of transient insomnia, the so-called first night effect)
Acute medical or surgical illnesses (including intensive care unit stays)
Stimulant medications (e.g., theophylline, beta-blockers, corticosteroids, thyroxine, bronchodilators, or withdrawal of central nervous system depressant medications)
Chronic Insomnia
Most cases of insomnia are chronic and comorbid with other conditions (Morin and Benca, 2009, 2011). Common comorbidities include psychiatric, medical, primary sleep-related (i.e., OSAS) or neurological disorders, or drug and alcohol abuse. Box 68.8 lists the causes of chronic insomnia. These comorbid insomnias were classified as secondary insomnia in the past, but this signifies causation; no definite evidence exists that these conditions are responsible for insomnia (National Institutes of Health, 2005).
Box 68.8 Causes of Chronic Insomnia
In addition to short-term effects of disturbed sleep and inadequate daytime functioning, insomniacs report impaired work performance and productivity, reduced social and physical functioning, reduced overall quality of life, and a two- to fivefold increased risk for developing subsequent depression associated with significant suicidal behavior. There are also suggestions of increased mortality and morbidity from coronary artery disease, hypertension, obesity, and diabetes mellitus (Ayas et al., 2003a, 2003b; Knutsen and Turek, 2006). A recent longitudinal sleep laboratory follow-up study for 14 years, however, found increased mortality in men with chronic insomnia with objectively measured short sleep duration that could not be explained by comorbid type 2 diabetes mellitus or hypertension (Vgontzas et al., 2010).
Inadequate Sleep Hygiene
Insomnia commonly coexists or precedes the development of a number of psychiatric illnesses. Surveys have shown that individuals with insomnia are more likely to develop new psychiatric disorders, particularly major depression, within 6 to 12 months. Anxiety disorders, depression, and schizophrenia are some of the major psychiatric disorders associated with insomnia. Medical and neurological disorders comorbid with insomnia are listed in Boxes 68.9 and 68.10. RLS, periodic limb movement disorder (PLMD), and circadian rhythm disorders are some of the other causes of chronic insomnia. In some patients, insomnia may coexist with sleep apnea.
Box 68.9 Medical Disorders Comorbid with Insomnia
Physiological Profiles of Insomnias
Chronic insomniacs may suffer from a general disorder of hyperarousal due to a functional alteration in the anatomical substrates for sleep/wake states (e.g., hyperactivity of wake-promoting neurons or hypoactivity of sleep-generating neurons that is genetically determined). Evidence of hyperarousal is provided by the following observations in these patients (Morin and Benca, 2009, 2011):



 Increased brain glucose metabolism
Increased brain glucose metabolism
 Increased EMG activity, as noted in the frontalis muscle
Increased EMG activity, as noted in the frontalis muscle
 EEG spectral analysis showing increased beta activity
EEG spectral analysis showing increased beta activity
 Decreased nocturnal melatonin and increased diurnal melatonin levels
Decreased nocturnal melatonin and increased diurnal melatonin levels
 Increased latencies on MSLT, despite disturbed nocturnal sleep
Increased latencies on MSLT, despite disturbed nocturnal sleep

Although the majority of published studies support hyperarousal in chronic insomnia, there are contradictory findings (Riemann et al., 2002; Stepanski et al., 2000; Varkevisser et al., 2005) casting doubt on the hyperarousal theory in all cases.
Mechanism of Insomnia in Neurological Diseases
Insomnia in neurological disorders may result from the following suggested mechanisms:
 Hypofunction (metabolic or structural) of the hypothalamic VLPO neurons or the lower brainstem hypnogenic neurons in the region of the NTS and dysfunction of thalamus may alter the balance between the waking and sleeping brain, causing sleeplessness.
Hypofunction (metabolic or structural) of the hypothalamic VLPO neurons or the lower brainstem hypnogenic neurons in the region of the NTS and dysfunction of thalamus may alter the balance between the waking and sleeping brain, causing sleeplessness.



 Neurological illnesses may secondarily cause depression and anxiety, which promote sleeplessness.
Neurological illnesses may secondarily cause depression and anxiety, which promote sleeplessness.

Narcolepsy and Idiopathic Hypersomnia
Since the French physician Gelineau used the term narcolepsy in 1880 to describe irresistible sleep attacks and astasia, which includes all the features of cataplexy, there have been many reports of patients with narcolepsy-cataplexy syndrome (Nishino, 2007; Scammell, 2003; Nishino and Mignot, 2011). The prevalence of narcolepsy is estimated to be approximately 1 in 2000 individuals in the United States, 1 in 600 people in Japan, and 1 in 500,000 individuals in Israel. Reports of adequate epidemiological studies in different parts of the world are, however, lacking.
Genetics of Narcolepsy
Pathogenesis Of Narcolepsy-Cataplexy Syndrome
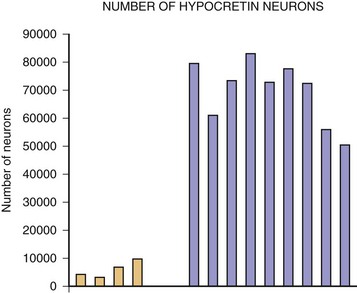
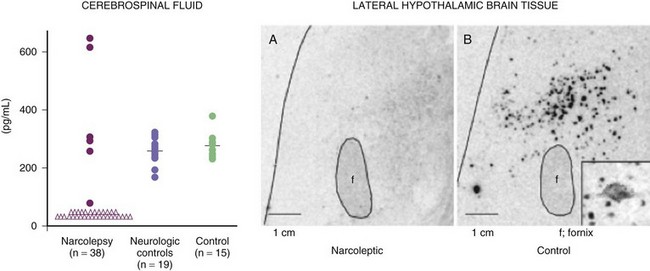
The most exciting recent development in sleep medicine is the pathogenetic role played by the recently discovered hypocretin (orexin) peptidergic systems in the lateral hypothalamus. Reports of mutation of the hypocretin receptor 2 gene (HCRTR2) in dogs (Lin et al., 1999) and pre-prohypocretin knockout mice (Chemelli et al., 1999) producing the phenotype of human narcolepsy brought narcolepsy research to the forefront of the molecular neurobiology field. Most cases of human narcolepsy and cataplexy have decreased hypocretin 1 in the cerebrospinal fluid (CSF) (Bourgin et al., 2008; Fronczek et al., 2009; Nishino, 2007). Hypocretins are noted to be depleted in narcoleptic brains at autopsy, as can be seen in Fig. 68.20 (Thannickal et al., 2000). In addition, mutation of the pre-prohypocretin gene has been identified in one child with severe narcolepsy (Peyron et al., 2000). Therefore, the contemporary theory for the pathogenesis of narcolepsy-cataplexy syndrome suggests that the condition results from a depletion (degeneration or autoimmune disorder) of the hypocretin neurons in the lateral and perifornical region of the hypothalamus, as can be appreciated in Fig. 68.21. Narcolepsy-cataplexy syndrome thus can be considered a hypocretin (orexin) deficiency syndrome. The possibility of an autoimmune disorder has been raised because of the association of narcolepsy with HLA-DQB1*0602 haplotype; however, all attempts to find evidence of autoimmune disorder in narcolepsy have so far failed (Overeem et al., 2008). There is no evidence of inflammatory processes or immune abnormalities, presence of classical autoantibodies, or an increase of oligoclonal bands in the CSF in these patients. Erythrocyte sedimentation rate, serum immunoglobulin levels, C-reactive protein, complement levels, and lymphocyte subset ratio are all found to be normal in these patients, pointing against an autoimmune disorder. On the other hand, increased levels of streptococcal antibodies in narcoleptic patients in some reports and evidence of gliosis in the lateral hypothalamus by Thannickal and colleagues (2000) indicate that further exploration for possible immune-related dysfunction in narcolepsy is needed. Finally, the presence of autoantibodies (patient serum binding to rat hypocretin neurons) in one out of nine narcolepsy-cataplexy patients keeps the autoimmune theory very much in the forefront (Knudsen et al., 2007).
Clinical Manifestations of Narcolepsy
The onset of the disorder in most cases is in adolescents and young adults, with a peak incidence between ages 15 and 30. However, rare cases have been described in children younger than age 5 and adults older than age 50. The ICSD-2 (AASM, 2005) divides narcolepsy into three types: narcolepsy with cataplexy, narcolepsy without cataplexy, and secondary narcolepsy. The major clinical manifestations of narcolepsy include narcoleptic sleep attacks (100%), cataplexy (60%-70%), sleep paralysis (25%-50%), hypnagogic hallucinations (20%-40%), disturbed night sleep (70%-80%), and automatic behavior (20%-40%) (Nishino, 2007; Nishino and Mignot, 2011; Overeem et al., 2001) (Table 68.6).
| MAJOR MANIFESTATIONS | |
| Narcoleptic sleep attacks | 100% |
| Cataplexy | 70% |
| Sleep paralysis | 25%-50% |
| Hypnagogic hallucinations | 20%-40% |
| Disturbed night sleep | 70%-80% |
| Automatic behavior | 20%-40% |
| COMORBID CONDITIONS | |
| Sleep apnea | Up to 30% |
| Periodic limb movements in sleep | 10%-60% |
| REM sleep behavior disorder | Up to 12% |
| Eating disorder (craving for food) | High prevalence, but exact percentage unknown, including night eating syndrome not related to obesity |
| Increased BMI or obesity | Exact percentage unknown |
| Increased prevalence of type 2 diabetes mellitus | Exact percentage unknown |
BMI, Body mass index; REM, Rapid eye movement.
Comorbid Conditions
In addition to the major manifestations, patients with narcolepsy may also have several important comorbid conditions (see Table 68.6): sleep apnea, PLMS, and RBD. Sleep apnea is noted in approximately 30% of narcoleptic patients; most commonly central apnea is seen, but obstructive or mixed apnea is also reported. Associated sleep apnea may aggravate sleep attacks. It is important to diagnose obstructive sleep apnea in these patients because treatment with continuous positive airway pressure (CPAP) will relieve apnea, with curtailment of EDS. RBD generally occurs in men with narcolepsy—in up to 36% of all patients with the disorder. Mostly commonly, the stimulants and tricyclic antidepressants or selective serotonin reuptake inhibitors (SSRIs) used to treat narcolepsy-cataplexy syndrome may induce or exacerbate RBD. PLMS have been noted in 10% to 60% of narcoleptic patients. There is a high prevalence (exact percentage is not known in limited studies) of both eating disorder (Droogleever-Fortuyn et al., 2008) and increased BMI (Schuld et al., 2002) not related to each other in narcoleptic patients.
Differential Diagnosis of Narcolepsy-Related Sleep Attacks
The most common conditions that should be differentiated from narcoleptic sleep attacks are illustrated in Box 68.11. Narcolepsy-related sleep attacks should be differentiated from other causes of EDS. These include sleep deprivation and insufficient sleep syndrome, OSAS, alcohol- and drug-related hypersomnolence, idiopathic hypersomnia (Aldrich, 1996), circadian rhythm sleep disorders, and other medical, neurological, and psychiatric disorders causing hypersomnolence. OSAS (see Sleep Apnea Syndrome, later) is the most common cause of EDS in patients referred to a sleep laboratory for evaluation. It is characterized by repeated episodes of obstructive and mixed apneas during NREM and REM sleep in overnight PSG recordings. These patients have prolonged daytime sleep episodes followed by fatigue and drowsiness on awakening, which contrasts with a fresh feeling in narcoleptic patients on awakening from brief sleep attacks. All patients with hypersomnolence can be excluded after careful history and physical examination and overnight PSG recording.
Differential Diagnosis of Cataplexy and Other Features of Narcolepsy
Cataplectic attacks may be mistaken for partial complex seizures, absence spells, atonic seizures, drop attacks, and syncope (Box 68.12). A partial complex seizure, however, is characterized by an altered state of consciousness, unlike cataplexy. In addition, patients with seizures may have generalized tonic-clonic movements, postictal confusion, and epileptiform discharges on EEG. Absence spells are characterized by staring with vacant expression lasting up to 30 seconds, with an altered state of alertness associated with characteristic 3-Hz spike-and-wave patterns in the EEG. Atonic seizures are accompanied by transient loss of consciousness and EEG evidence of slow spike-and-wave discharges or multiple spike-and-wave discharges.
Symptomatic or secondary narcolepsy-cataplexy may be due to diencephalic and midbrain tumors, MS, strokes, cysts, vascular malformations, encephalitis, cerebral trauma, and paraneoplastic syndrome with anti-Ma2 antibodies and may present with narcoleptic-like sleep attacks and other manifestations (Nishino, 2007; Nishino and Mignot, 2011). Symptomatic narcolepsy associated with cataplexy may develop in children affected with Niemann-Pick disease type C.
Idiopathic Hypersomnia with or without Long Sleep Time
Idiopathic hypersomnia closely resembles narcolepsy syndrome (Billiard and Dauvilliers, 2001). This disorder is characterized by EDS (presumed CNS cause but not proven) that is associated with either normal (6 to 10 hours) or prolonged (>10 hours) nocturnal sleep documented by history, actigraphy, sleep logs, or PSG (AASM, 2005). The onset of the disease is generally around the same age as narcolepsy (15 to 30 years). The sleep pattern, however, is different from that of narcolepsy. The patient generally sleeps for hours, and the sleep is not refreshing. Because of EDS, the condition may be mistaken for sleep apnea. However, the patient does not give a history of cataplexy, snoring, or repeating awakenings throughout the night. Some patients may have automatic behavior with amnesia for the events. Physical examination uncovers no abnormal neurological findings. This disabling and lifelong condition should be differentiated from other causes of EDS (see Box 68.5). There is no clear association between idiopathic hypersomnia and HLA.
Sleep Apnea Syndrome
Upper airway obstructive sleep apnea-hypopnea syndrome remains undiagnosed or underdiagnosed because of insufficient knowledge and awareness of serious consequences resulting from this disorder. OSAS causes significant morbidity and mortality and is often associated with a variety of comorbid conditions (Banno and Kryger, 2007; Guilleminault and Zupancic, 2009). Cardinal features of OSAS include habitual loud snoring, witnessed apneas during sleep, and daytime hypersomnolence as well as hyperactivity and nocturnal enuresis, particularly in children. OSAS is characterized by repetitive episodes of complete (apnea) or partial (hypopnea) upper airway obstruction during sleep, resulting in arterial oxygen desaturation and arousal from sleep.
Sleep-Disordered Breathing Terminology
The spectrum of sleep-disordered breathing (SDB) includes a variety of breathing patterns based on analysis during overnight PSG recordings. Fig. 68.22 schematically shows some patterns of SDB. Apnea, or cessation of breathing, consists of three types: obstructive, central, and mixed.
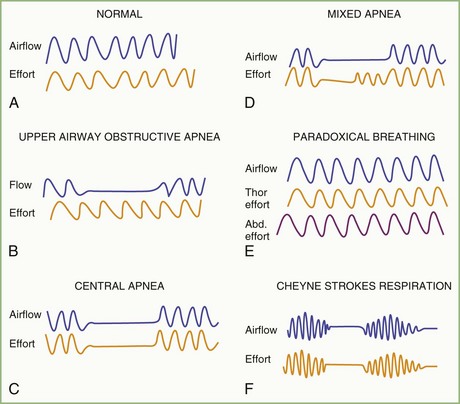
Cessation of airflow with no respiratory effort defines central apnea, during which both diaphragmatic and intercostal muscle activities as well as gas exchange through the nose or mouth are absent (see Fig. 68.22). In contrast, during obstructive apnea, airflow stops while the effort continues (Fig. 68.23; see also Fig. 68.22). In mixed apnea, there is an initial cessation of airflow with no respiratory effort (central apnea) followed by a period of upper airway obstructive sleep apnea (see Fig. 68.22).
There are two separate rules for scoring hypopnea in the AASM Scoring Manual (Iber et al., 2007). The “recommended” rule requires a reduction of nasal pressure or the alternative airflow sensor signal by 30% or more of the baseline amplitude for at least 10 seconds accompanied by oxygen desaturation of 4% or more from the pre-event baseline. The “alternative” rule requires a reduction of the signal by 50% or more of the baseline for at least 10 seconds accompanied by oxygen desaturation of 3% or more, or the event is associated with an arousal within 3 seconds of the event. In both these rules, the amplitude reduction must be present for at least 90% of the event’s duration.
Paradoxical breathing is characterized by movements of the thorax and abdomen in opposite directions, indicating increased upper airway resistance and upper airway obstruction (see Fig. 68.22). Cheyne-Stokes breathing is a type of periodic breathing characterized by cyclical changes in breathing with a crescendo-decrescendo sequence separated by central apneas or hypopneas (Fig. 68.24; see also Fig. 68.22). The Cheyne-Stokes variant pattern of breathing is distinguished by substitution of hypopnea for apnea.
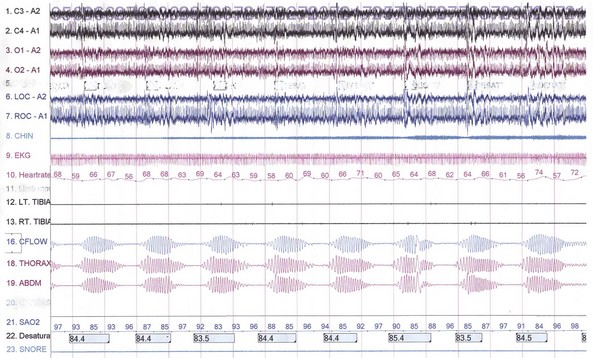
Fig. 68.24 A 500-second excerpt showing the classic Cheyne-Stokes breathing (CSB) crescendo-decrescendo pattern from an overnight polysomnographic (PSG) recording of a 69-year-old man. The presence of CSB throughout most of the non–rapid eye movement (NREM) sleep (with marked decrement or absence during REM sleep) in this patient with a history of hypertension and excessive daytime sleepiness suggests incipient left ventricular failure. The PSG montage is similar to that in Fig. 68.23.
Hypoventilation refers to a reduction of alveolar ventilation accompanied by hypoxemia and hypercapnia without any apnea or hypopnea; it may be noted in patients with neuromuscular disorders and kyphoscoliosis and those with underlying lung or chest-wall abnormalities that impair gas exchange during wakefulness. During sleep hypoventilation, the partial pressure of arterial carbon dioxide (Paco2) rises at least 10 mm above the supine awake values (Iber et al., 2007). Upper airway resistance syndrome (UARS) is characterized by repeated abnormal respiratory efforts during sleep without apnea, accompanied by recurrent arousals and EDS. The Respiratory Disturbance Index (RDI) includes all these abnormal breathing events, including respiratory effort–related arousals.
Epidemiology of Obstructive Sleep Apnea Syndrome
No study has been specifically designed to determine the incidence of OSAS in a previously healthy population. Based on a definition of at least five apneas or hypopneas per hour of sleep accompanied by EDS (Young et al., 1997), however, the prevalence of OSAS is 4% in men and 2% in women between the ages of 30 and 60. There is a strong association between OSAS and male gender, increasing age, and obesity. The condition is common in men older than 40, and among women, the incidence of OSAS is greater after menopause. Approximately 85% of patients with OSAS are men; obesity is present in about 70% of OSAS patients. There is an increased prevalence of OSAS in those with a thick neck and large abdomen. Men with a neck circumference measuring more than 17 inches and women with a neck measuring more than 16 inches are at risk for OSAS.
Race may be a factor, given that a high prevalence of SDB is noted in Pacific Islanders, Mexican Americans, and African Americans. There are also family aggregates of OSAS. Other factors with a high association are alcohol, smoking, and drug use. Other risk factors include nasal allergies or congestion, endocrine diseases (e.g., hypothyroidism, acromegaly), disorders with autonomic failure or those associated with craniofacial abnormalities (e.g., Marfan, Down, Pierre-Robin syndromes). Risk factors associated with OSAS are listed in Box 68.13.
Box 68.13 Risk Factors for Obstructive Sleep Apnea Syndrome
Clinical Manifestations
The symptoms of OSAS can be divided into two groups (Flemons, 2002; Guilleminault and Zupancic, 2009): those occurring during sleep and those occurring during waking hours (Box 68.14). Nocturnal symptoms include habitual loud snoring, choking during sleep, cessation of breathing, and abnormal motor activities during sleep (e.g., jerking and shaking movements, confusional arousals, sleep walking), severe sleep disruption, gastroesophageal reflux causing heartburn, nocturnal enuresis (noted mostly in children), and profuse sweating at night. The daytime symptoms include EDS characterized by sleep attacks lasting 0.5 to 2 hours and occurring mostly when the patient is relaxing. The prolonged duration and the nonrefreshing nature of these sleep attacks in OSAS differentiate these from narcoleptic sleep attacks. Other daytime symptoms include impairment of memory and motor skills, irritability, morning headache in some patients, automatic behavior, retrograde amnesia, and hyperactivity (in children). Impotence in men is often associated with severe and long-standing OSAS. Physical examination may reveal obesity in approximately 70% of the cases, with increased body mass index and increased neck circumference, in addition to upper airway anatomical abnormalities causing reduction of the upper airway space (e.g., low-hanging soft palate, large edematous uvula, large tonsils and adenoids—especially in children—retrognathia, micrognathia). In severe cases, physical examination may reveal evidence of congestive heart failure, cardiac arrhythmias, hypertension, and polycythemia.
Box 68.14 Symptoms and Signs in Obstructive Sleep Apnea Syndrome
Nocturnal Symptoms During Sleep
Consequences of Obstructive Sleep Apnea Syndrome
OSAS is associated with increased morbidity and mortality due to both short-term consequences (impairment of quality of life and increasing traffic- and work-related accidents), and long-term consequences from associated and comorbid conditions such as hypertension, heart failure, MI, cardiac arrhythmias, stroke due to both supratentorial and infratentorial infarctions, transient ischemic attacks, cognitive dysfunction, depression, and insomnia (Banno and Kryger, 2007; Flemons, 2002; Guilleminault and Zupancic, 2009; Marin et al., 2005; Somers et al., 2008). Several prospective longitudinal studies (Nieto et al., 2000; Peppard et al., 2000; Young et al., 1997) have shown a clear association between OSAS and systemic hypertension, which may be noted in approximately 45% of patients with OSAS. The factors that cause hypertension in OSAS include repeated hypoxemias during sleep at night, which cause increased sympathetic activity. OSAS is noted in about 30% of cases of essential hypertension. Several studies have shown improvement of hypertension or reduction of need for antihypertensive medications after effective treatment of OSAS with CPAP titration (Banno and Kryger, 2007; Faccenda et al., 2001; Hla et al., 2002; Pepperell et al., 2002). Pulmonary hypertension is also noted in approximately 15% to 20% of cases. Cardiac arrhythmias in the form of premature ventricular contractions, ventricular tachycardia, sinus pauses, and third-degree heart block as well as sudden cardiac death have been attributed to OSAS. Heart failure, mostly systolic heart failure but also diastolic heart failure (in which the studies are limited), is associated with both obstructive and central sleep apneas but more central sleep apneas, including Cheyne-Stokes breathing, than obstructive apneas (Arias et al., 2007; Javaheri, 2006; Javaheri and Somers, 2011; Somers et al., 2008). The presence of central apnea, including Cheyne-Stokes breathing, increases mortality in patients with heart failure. Cognitive dysfunction, which is noted in moderately severe to severe OSAS patients, shows improvement after satisfactory treatment with CPAP titration (Engelman et al., 2000). Recently, awareness about the presence of depression and insomnia in patients with OSAS has grown, but adequate studies have not been conducted to find the prevalence and impact of these conditions on OSAS (Aloia et al., 2005; Shepertycky et al., 2005). There has also been an increased association between OSAS and metabolic syndrome (a combination of hypertension, increased insulin resistance with type 2 diabetes, hypertriglyceridemia, and obesity) (Alam et al., 2007).
Pathogenesis of Obstructive Sleep Apnea Syndrome
Factors contributing to the pathogenesis of OSAS include local anatomical, neurological, and vascular factors, as well as familial predisposition (Banno and Kryger, 2007; Guilleminault and Zupancic, 2009). Collapse of the pharyngeal airway is the fundamental factor in OSA. During sleep, muscle tone decreases, including that of the upper airway dilator muscles which maintain upper airway patency. Decreased tone causes these muscles to relax, increasing upper airway resistance and narrowing the upper airway space. Defective upper airway reflexes may also play a role. Increasing familial occurrence of OSAS in some patients may be related to abnormal craniofacial features. In children, adenotonsilar enlargement and craniofacial dysostosis causing narrow upper airway space are important factors. Neurological factors include reduced medullary respiratory neuronal output as a result of abnormal ventilatory control. Other neurological factors include autonomic activation during sleep-related breathing events, contributing to development of hypertension and cardiac arrhythmias. Vascular factors contributing to the pathogenesis and long-term adverse consequences (Banno and Kryger, 2007; Javaheri and Somers, 2011) include increased endothelin 1 (a vasoconstrictor), reduced nitric oxide (a known vasodilator), and increased serum levels of vascular endothelial growth factor (glycoprotein responsible for vascular remodeling and atherosclerosis). It has been shown that after effective CPAP titration, these vascular abnormalities are reversed. Thus, a complex interaction of peripheral upper airway anatomical, central neural, vascular, and genetic factors contribute to the syndrome of upper airway OSAS.
Evaluation and Assessment
Evaluation and assessment are the same as for other sleep disorders. Particular attention should be paid to the detailed sleep history as well as the daytime history, and careful physical examination should be focused on specific associated and risk factors, specifically body mass index, cardiopulmonary examination, and an examination of the upper airways (neck circumference, airway size). The Mallampati classification is useful in evaluating patient airway size and gauging the possible risk for OSA (Fig. 68.25). Laboratory assessment and management are described later under Laboratory Assessment of Sleep Disorders.
Upper Airway Resistance Syndrome
Since UARS was described by Guilleminault and colleagues in 1993, there has been controversy about this entity. Some investigators have rejected it as a distinct clinical entity, whereas others have accepted it as such. One controversy concerns whether UARS can be considered as the initial stage, with progressive evolution into full-blown upper airway OSAS. In the only longitudinal study performed (in approximately 94 UARS subjects) by Guilleminault and colleagues (2006a), it was noted that about 5% of subjects had developed OSAS 4.5 years later. In the majority of patients with UARS, symptoms do not evolve into full-blown OSAS. Many patients have a multitude of symptoms of a somatic, psychiatric, or psychosomatic nature, whereas others are associated with attention-deficit hyperactivity disorder (ADHD), fibromyalgia, and chronic insomnia. UARS thus appears to be a heterogenous syndrome and could be part of a spectrum, with OSAS and obstructive hypoventilation at the far end of the spectrum and snoring at the other end. Some patients may fit with the spectrum, but the majority do not. Patients with UARS show subtle airflow limitations due to increased upper airway resistance, followed by repeated arousals during sleep at night. This subtle airflow limitation cannot be identified by the usual recording of respiration using an oronasal thermistor. Nasal pressure monitoring with a nasal cannula is more sensitive than use of a thermistor in detecting airflow limitation and increased upper airway resistance (Ayappa et al., 2000). Intraesophageal balloon recording, however, is the standard method for detecting upper airway resistance and reveals increasing efforts with increasing intraesophageal pressure leading to arousal but without any apnea or hypopnea (Fig. 68.26). These patients may or may not snore; they may have EDS and all its consequences, as seen in OSAS. Adult patients are more likely to complain of fatigue rather than sleepiness. Chronic insomnia is more commonly seen in UARS than in OSAS. UARS can sometimes be mistaken for chronic fatigue syndrome, fibromyalgia, ADHD, depression, and other psychiatric disorders. CPAP titration is widely used as the first line of therapy, but not all patients improve on CPAP. Recently it has been shown that cognitive behavioral therapy in addition to CPAP may be helpful for patients with chronic insomnia.
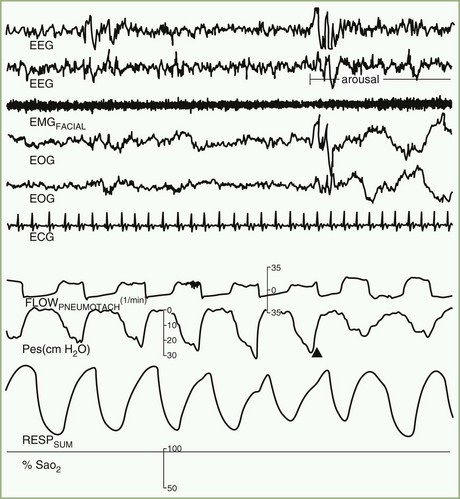
Central Sleep Apnea Syndrome
Central sleep apnea syndrome includes primary or idiopathic CSA, CSA due to Cheyne-Stokes breathing pattern, CSA due to high altitude, CSA due to central neurological disorders (particularly brainstem dysfunction), and CSA due to drug or substance abuse (e.g., use of opiates) (AASM, 2005). Primary CSAS is rare, and the patient may present with EDS and frequent awakenings due to repeated episodes of central apnea followed by arousals. The patient may also present with insomnia. Cheyne-Stokes breathing is noted in patients with congestive heart failure (Javaheri, 2006; Javaheri and Somers, 2011) and sometimes in renal failure. The presence of Cheyne-Stokes breathing in cardiac failure increases mortality. Patients with primary CSA usually are normocapnic or hypocapnic, with a Paco2 of 40 mm Hg or lower. The other important point of differentiation is that the cycle length (apnea plus ventilatory duration) is more than 45 seconds in Cheyne-Stokes breathing and less than 45 seconds in primary CSA (AASM, 2005).
Restless Legs Syndrome
Clinical Manifestations
RLS is the most common movement disorder but is uncommonly recognized and treated despite a lucid description of the entity in the middle of the last century (Hening et al., 2009). Even today, many physicians do not recognize the condition and mistake it for other superficially similar conditions. There is no single diagnostic test for RLS; therefore, the diagnosis rests entirely on clinical features and is based on the International Restless Legs Syndrome Study Group (IRLSSG) criteria first established in 1995 (Walters, 1995) and modified slightly in 2003 (Allen et al., 2003). These criteria include essential, supportive, and associated features, as listed in Box 68.15.
Box 68.15 Clinical Diagnostic Criteria for Idiopathic Restless Legs Syndrome
Essential Criteria
An urge to move the legs, usually accompanied by or caused by uncomfortable sensations in the legs
The urge to move or unpleasant sensations beginning or worsening during periods of rest or inactivity, such as lying or sitting
The urge to move or unpleasant sensations are partially or totally relieved by movement such as walking or stretching, at least as long as the activity continues.
The urge to move or unpleasant sensations are worse in the evening or night than during the day or only occur in the evening or night.
RLS is a lifelong sensory-motor neurological disorder (Earley, 2003) that often begins at a very young age but is mostly diagnosed in the middle or later years. Prevalence of RLS increases with age and plateaus for some unknown reason around age 85 to 90 (Allen et al., 2001; Earley, 2003; Earley et al., 2011). All four essential diagnostic criteria (see Box 68.15) are needed for establishing the diagnosis. The overall prevalence of RLS has been estimated at about 10% for all adult populations, particularly those in North American and European populations. Prevalence for the most severe cases is approximately 2.5% (Hening et al., 2004) but appears to be much less in some surveys from Asia (<1%-3%), suggesting possible ethnic and racial differences in RLS prevalence (Garcia-Borreguero et al., 2006). In most surveys, the prevalence is greater in women than in men, and the disease is gradually progressing. Family studies of RLS suggest an increased incidence (≈40%-50%) in first-degree relatives of idiopathic cases. A high concordance (83%) in monozygotic twins and complex segregation analysis suggest an autosomal dominant mode of inheritance (Hening et al., 2009). Linkage analysis documented significant linkage to at least six different chromosomes (12q, 14q, 9p, 2q, and 20p). Recent genome-wide association and linkage analysis have documented common variations in certain genomic regions, conferring more than 50% increase in risk to RLS-PLMS (Stefansson et al., 2007; Winkelmann et al., 2007). Four allelic variations in different genes have been identified on chromosomes 2p, 6p, 15q, and 12q.
The sensory manifestations of RLS include intense disagreeable feelings described as creeping, crawling, tingling, burning, aching, cramping, knifelike, or itching sensations. These creeping sensations occur mostly between the knees and ankles, causing an intense urge to move the limbs to relieve these feelings. Sometimes, similar symptoms occur in the arms or other parts of the body, particularly in advanced stages of the disease or when the patient develops augmentation (a hypermotor syndrome with symptoms occurring at least 2 hours earlier than the initial period, with intensification and spread to other body parts) resulting from long-standing dopaminergic medications (Allen et al., 2003; Garcia-Borreguero et al., 2007). Approximately 15% to 20% of patients complain of actual pain. Most of the movements, particularly in the early stages, are noted in the evening while the patients are resting in bed. In severe cases, however, movements may be noted in the daytime while subjects are sitting or lying down. At least 80% of RLS patients have PLMS, and many also have periodic limb movements in wakefulness. The condition may have a profound impact on sleep. Patients often seek medical attention for sleep disturbance—generally a problem of initiation, although difficulty maintaining sleep owing to associated PLMS can occur. Neurological examination is generally normal in the idiopathic form. As stated earlier, RLS often begins in childhood, but diagnosis of childhood RLS may be difficult based on the National Institutes of Health (NIH) consensus conference criteria established in 2002 (Allen et al., 2003). Some diagnostic criteria for childhood RLS have more recently been suggested. To make a correct diagnosis, RLS must be differentiated from conditions mimicking RLS (Box 68.16) or those associated (comorbid) with RLS (Chokroverty, 2009d) (Box 68.17 and 68.18). An important and often difficult condition to differentiate from RLS is akathisia (see Box 68.18).
Box 68.16
Conditions That Mimic Restless Legs Syndrome
Presenting with Excess Restlessness
Reproduced with permission from Chokroverty, S., 2009. Differential diagnosis of restless legs syndrome, in: Hening, W.A., Allen, R.P., Chokroverty, S., et al. (Eds.), Restless Legs Syndrome. Saunders, Philadelphia, p. 111.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 68 Sleep and Its Disorders
In the first part of this chapter, a brief overview of sleep is given, covering the definition and architecture of sleep, sleep habits and requirements, the ontogeny of sleep, dreams and sleep, the neurobiology of sleep and wakefulness, circadian rhythm and chronobiology of sleep, and functions of sleep. In the sections that follow, physiological changes in sleep are summarized, sleep deprivation, sleepiness, and causes/consequences of excessive daytime sleepiness are discussed, sleep disorders are classified, and the approach to a patient with sleep complaints is outlined. Finally, the clinical phenomenology of a variety of sleep disorders is covered, and the chapter concludes with recommendations for laboratory assessment and treatment of selected sleep disorders. For more in-depth information about the order and disorders of sleep, the reader is referred to the growing number of sleep medicine texts (Avidan and Zee, 2006; Chokroverty, 2009a; Chokroverty et al., 2003; Kryger et al., 2005; Lee-Chiong, 2006; Montagna and Chokroverty, 2011).
Definition of Sleep and Moment of Sleep Onset
Modern researchers define sleep on the basis of both behavioral and physiological criteria (Chokroverty, 2009b) (Table 68.1). The behavioral criteria include lack of mobility or slight mobility, closed eyes, a characteristic species-specific sleeping posture, reduced response to external stimulation, quiescence, increased reaction time, elevated arousal threshold, impaired cognitive function, and a reversible unconscious state. Physiological criteria (see Sleep Architecture and Sleep Stages, later) are based on EEG, electro-oculography (EOG), and electromyography (EMG) findings as well as other physiological changes in ventilation and circulation.
Sleep Architecture and Sleep Stages
In their 1968 criteria, Rechtschaffen and Kales (RK) divided NREM sleep into stages 1, 2, 3, and 4. In 2007, this staging was modified slightly by an American Academy of Sleep Medicine (AASM) Task Force, and NREM sleep is now divided into three stages: N1, N2, and N3 (slow-wave sleep). In the modified staging criteria adopted by the AASM (Iber et al., 2007), the traditional RK stage 1 NREM sleep was labeled N1, RK stage 2 was labeled N2, and RK stages 3 and 4 were combined into one stage, N3.
NREM sleep accounts for 75% to 80% of sleep time in adult humans. In stage N1 sleep, making up approximately 3% to 8% of sleep time, the alpha rhythms (8-13 Hz) characteristic of wakefulness (Fig. 68.1) diminish to less than 50% in an epoch (i.e., a 30-second segment of a polysomnographic (PSG) tracing with a paper or monitor speed of 10 mm/sec), and a mixture of slower theta rhythms (4-7 Hz) and beta waves (>13 Hz) appear. EMG activity decreases slightly, and slow, rolling eye movements may be recorded (Fig. 68.2). Vertex sharp waves are noted toward the end of stage N1 sleep. Stage N2 sleep begins after approximately 10 to 12 minutes of stage N1 sleep. The characteristic EEG findings of stage N2 sleep include sleep spindles (12-18 Hz, most often 14 Hz) and K complexes intermixed with vertex sharp waves (Fig. 68.3). The EEG recording contains theta activity and fewer than 20% slow waves (0.5-2 Hz). Stage N2 sleep lasts for approximately 30 to 60 minutes. During stage N3 sleep, the slow waves occupy 20% to 100% of the epoch (Fig. 68.4). Toward the end of slow-wave sleep, or N3, body movements are registered as artifacts in the PSG recordings.
Fig. 68.2 Polysomnographic recording shows stage 1 non–rapid eye movement (NREM) sleep (N1) in an adult. Electroencephalograms (top 4 EEG channels) show a decrease of alpha activity to less than 50% and low-amplitude beta and theta activities. Left and right electro-oculograms (EOG) show slow rolling eye movements. A1, Left ear; A2, right ear; Thorax, respiratory effort (chest). Rest of montage is same as in Fig. 68.1.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.2.)
Fig. 68.3 Polysomnographic recording shows stage 2 non–rapid eye movement (NREM) sleep (N2) in an adult. Note approximately 14-Hz sleep spindles and K complexes intermixed with delta waves (0.5-2 Hz) and up to 75 µV in amplitude occupying less than 20% of the epoch. See Fig. 68.2 for description of rest of montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.3.)
Fig. 68.4 Polysomnographic recording shows stage 4 non–rapid eye movement (NREM) sleep (N3) in an adult. Delta waves occupy more than 50% of the epoch in the traditional R&K scoring technique. See Fig. 68.2 for description of the montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.5.)
The first REM sleep (R sleep) episode is noted 60 to 90 minutes after the onset of sleep. REM sleep accounts for 20% to 25% of sleep time. Based on EEG, EMG, and EOG characteristics, REM sleep can be subdivided into two stages: tonic and phasic. However, this subdivision is not recognized in the recently modified staging. The EEG tracings during REM sleep are characterized by fast rhythms and theta activity, some of which may have a sawtooth appearance (Fig. 68.5). A desynchronized EEG, hypotonia, or atonia of the major muscle groups and depression of monosynaptic and polysynaptic reflexes are characteristics of the tonic stage. Phasic REM sleep is characterized by rapid eye movements in all directions, as well as phasic swings in blood pressure and heart rate, irregular respiration, spontaneous middle-ear muscle activity, and tongue movements. A few periods of apnea or hypopnea may arise during REM sleep. An illustration of sleep stage distribution during the night is shown in Fig. 68.6.
Fig. 68.5 Polysomnographic recording shows rapid eye movement (REM) sleep in an adult. Electroencephalogram (top 8 channels) shows mixed-frequency theta, low-amplitude beta, and a small amount of alpha activity. Note characteristic sawtooth waves (seen prominently in channels, 1, 2, 5, and 6 from top) of REM sleep preceding bursts of REMs in electro-oculograms (E1-M1; E2-M2). Chin electromyogram (EMG) shows marked hypotonia, whereas TIB and GAST EMG channels show very low-amplitude phasic myoclonic bursts. See Fig. 68.1 for description of the montage.
(From Chokroverty, S., 2009. Sleep Disorders Medicine: Basic Science, Technical Considerations, and Clinical Aspects, third ed. Saunders, Philadelphia, Fig. 2.6.)
Thus, there is an orderly progression from wakefulness to sleep onset to NREM sleep and then to REM sleep. A relaxed wakefulness is characterized by a behavioral state of quiescence and a physiological state of alpha and beta frequencies on the EEG recording, waking eye movements, and increased muscle tone. Table 68.2 summarizes NREM and REM sleep states. Sleep staging and scoring address normal adult sleep and the macrostructure (Box 68.1) and microstructure (Box 68.2) of sleep. In patients with sleep disorders such as sleep apnea, parasomnias, or nocturnal seizures disrupting sleep, such assessments may be difficult.
| Sleep State | % Sleep Time |
|---|---|
| NREM sleep | 75-80 |
| N1 | 3-8 |
| N2 | 45-55 |
| N3 | 13-23 |
| REM sleep (stage R) | 20-25 |
| Tonic stage | Continuous |
| Phasic stage | Intermittent |
NREM, Non–rapid eye movement; REM, rapid eye movement.
Sleep Microstructure
In 1992, a Task Force of the American Sleep Disorders Association (ASDA), now the AASM, offered descriptions of sleep microstructure that included momentary dynamic phenomena such as arousals and the cyclic alternating pattern (CAP), which has been described in various publications by Terzano and co-investigators (2002). Sleep microstructure also includes K complexes and sleep spindles (see Box 68.2). According to the operational definition of the ASDA Task Force, an arousal is a shift in EEG frequency lasting for 3 to 14 seconds and includes alpha, beta, or theta activities but not spindles or delta waves. The subject must be asleep for 10 consecutive seconds before an arousal can be scored. In REM sleep, arousals are scored only when accompanied by a concurrent increase in submental EMG amplitude. Unless accompanied by EEG frequency shifts, K complexes, delta waves, artifacts, and increased submental EMG activities are not counted as arousals. An arousal index is defined as the number of arousals per hour of sleep; up to 10 can be considered a normal arousal index.
In contrast to the usual pattern of arousals signifying sleep fragmentation, the CAP indicates sleep instability. The CAP is a repetitive EEG pattern that is noted mainly during NREM sleep and lasts for 2 to 60 seconds. A phase of CAP is marked by increased EEG potentials, with contributions from both synchronous high-amplitude slow and desynchronized fast rhythms in the EEG recording. A CAP cycle consists of an unstable phase (phase A) and a relatively stable phase (phase B) (Fig. 68.7). During phase A, heart rate, respiration, blood pressure, and muscle tone increase. The rate of CAP cycles and arousals increase in both older individuals and a variety of sleep disorders. A period without CAP is thought to indicate a state of sustained stability. Evaluation of periods with and without a CAP is a promising technique for understanding normal and abnormal sleep.
Ontogeny of Sleep Patterns with Age
The newborn infant spends approximately 50% of the time in REM sleep, but by age 6 years this time is decreased to the normal adult pattern of 25% (Fig. 68.8). On falling asleep, a newborn baby goes immediately into REM sleep, or active sleep, which is accompanied by restless movements of the arms, legs, and facial muscles. In premature babies, it is often difficult to differentiate REM sleep from wakefulness. By age 3 months, the NREM-REM cyclical pattern of adult sleep is established. However, the duration of the NREM-REM cycle is shorter in infants, lasting for approximately 45 to 50 minutes and increasing to 60 to 70 minutes by age 5 to 10 years, and to the normal adult cyclical pattern of 90 to 100 minutes by age 10 years. Sleep spindles begin to appear at about 3 months of age; K complexes are seen by about 6 months. A characteristic feature of sleep in old age is marked attenuation of the amplitude of slow waves; therefore, during scoring of slow sleep, which depends not only on the rate but also on the amplitude of slow waves, the percentage of slow waves decreases. The other characteristic feature during old age is repeated awakenings throughout the night, including early-morning awakenings. The percentage of REM sleep in normal elderly individuals remains relatively constant, and the total duration of sleep time within 24 hours is not different from that of young adults, but elderly individuals often nap during the daytime, compensating for lost sleep during the night. Figs. 68.9 and 68.10 schematically show the evolution of sleep state distribution in newborns, infants, children, adults, and elder adults.
Sleep Habits
Depending on the sleep habit, two groups of individuals are recognized: evening types and morning types. Evening types (“owls”) have difficulty getting up early and feel tired in the morning; however, they feel fresh and energetic toward the end of the day. These people perform best in the evening; they go to sleep late and wake up late. In contrast, the morning types (“larks”) wake up early, rested and refreshed, and work efficiently in the morning. The body temperature rhythm shows two different curves in these two types of people. The body temperature reaches the evening peak an hour earlier in morning types than in evening types. Morning and evening types are most likely determined by genetic factors. Katzenberg and colleagues (1998), using the 19-item Horne-Östberg Questionnaire to determine “morningness/eveningness” in human circadian rhythms, discovered a CLOCK gene polymorphism associated with human diurnal preference. One of two H CLOCK alleles (3111C) is associated with eveningness. These findings were contradicted by a later study (Robilliard et al., 2002).
Sleep Requirements and Quantity of Sleep
Sleep requirement is defined as the optimal amount of sleep required to remain alert and fully awake and to function adequately throughout the day. Sleep requirement for an average adult is approximately 7.5 to 8 hours regardless of environment or cultural differences. Sleep need is determined by heredity rather than by different personality traits or other psychological factors. Social or biological factors may also play a role. An important epidemiological study from 1979 (Kripke et al.) found that the chances of death from coronary artery disease, cancer, or stroke are greater for adults who sleep less than 4 hours or more than 9 hours a night when compared to those who sleep an average of 8 hours. Some more recent studies by Kripke and colleagues (2002) and others have confirmed these observations. In later studies, other confounding factors such as sleep medications may have confounded these issues. There is no clear-cut conclusion yet. There is also some controversy over whether a person can extend sleep beyond the average requirement. In 1969, Taub and Berger study showed that sleep extension beyond the average may cause exhaustion and irritability, with detriment of sleep efficiency. The overall conclusion, however, is that sleep extension has minimal effect on sleepiness and performance in absence of sleep debt, which is defined as the difference between the ideal sleep requirement and the actual duration of sleep obtained. Modern society appears to be chronically sleep deprived. Data demonstrate detrimental loss of vigilance with progressive sleep loss, confirming the possibility that sleep deprivation’s consequences are serious. Fig. 68.11 shows psychomotor vigilance task (PVT) performance lapses under varying doses of daily sleep, confirming the data of serious lapses when sleep is curtailed beyond 8 hours. A 1979 study examining 8- to 17-year-old adolescents between the years 1910 and 1963 showed a mean reduction of 1.5 hours of sleep in that time. There may, however, be a significant sampling error in this survey. A 1992 study by Bliwise and colleagues in healthy adults aged 50 to 65 showed a reduction of about 1 hour of sleep between 1959 and 1980 surveys.
Neurobiology of Sleep and Wakefulness
The neuroanatomical substrates for wakefulness, REM sleep, and NREM sleep are located in separate parts of the CNS (McCarley, 2009; Steriade and McCarley, 2005). There are no discreet sleep-promoting centers; sleep/wake states are produced by changes in interconnected systems rather than by discreet centers.
Neuroanatomical Substrates of Wakefulness
Wakefulness is controlled by the ascending reticular activating system (ARAS) containing glutamatergic, cholinergic, aminergic, and hypocretinergic neurons (Chokroverty, 2003; Steriade and McCarley, 2005). Projections from the ARAS terminating in the thalamus and thalamocortical projections to widespread areas of the cerebral cortex produce cerebral cortical activation during wakefulness. Extrathalamic projections from the brainstem reticular neurons terminate in the posterior hypothalamus and the basal forebrain regions; the latter project to the cerebral cortex (basocortical projections) to maintain wakefulness (Fig. 68.12). In the 1930s, Bremer discovered evidence of an ascending arousal system necessary for cortical arousal when he demonstrated that transection of the brainstem at the midcollicular level (i.e., cerveau isolé), but not the spinomedullary junction (i.e., encéphale isolé), produced coma in anesthetized cats. Bremer hypothesized that the resulting reduction in “cerebral tone” following the cerveau isolé was due to interruption of ascending sensory inputs—that is, a passive “deafferentation theory” of sleep (Bremer, 1937).
All these pathways regulating the wakefulness system use the cholinergic, noradrenergic, dopaminergic, and histaminergic neurons (Fig. 68.13). The cholinergic neurons fire at the highest rate during wakefulness and REM sleep but decrease their rates of firing at the onset of NREM sleep. Wakefulness-promoting aminergic neurons include noradrenergic neurons in the locus ceruleus, serotonergic neurons in the dorsal raphe of the brainstem, histaminergic neurons in the tuberomammillary nucleus of the hypothalamus, and possibly also dopaminergic neurons in the ventral tegmental area, substantia nigra, and ventral periaqueductal area. After a period of uncertainty regarding the role of dopamine, it has recently been confirmed that the midbrain dopaminergic system (A8-A10), particularly the dopaminergic neurons in the ventral periaqueductal gray (vPAG), play an active role in maintaining wakefulness through their widespread reciprocal connections with sleep/wake regulatory systems of neurons (Fuller and Lu, 2009). Norepinephrine-containing locus ceruleus neurons show their highest firing rates during wakefulness, their lowest during REM sleep, and intermediate rates during NREM sleep. Pharmacological studies suggest that posterior hypothalamic histaminergic neurons also help maintain wakefulness.
The excitatory amino acids, glutamate and aspartate, are intermingled within the ARAS and are present in many neurons projecting to the cerebral cortex, forebrain, and brainstem. These excitatory amino acids are maximally released during wakefulness. Recent discovery of hypothalamic hypocretin neurons and their widespread CNS projections have directed attention to the role of the hypocretin system in sleep/wake regulation. In 1998, deLecea and coauthors described two neuropeptides in the lateral hypothalamus and perifornical region that were termed hypocretin 1 and hypocretin 2. Independently in the same year, Sakurai and colleagues (1998) described two neuropeptides in the same region, which they named orexin A and orexin B (corresponding to hypocretin 1 and hypocretin 2, respectively). It was shown thereafter that these hypocretin systems have widespread ascending and descending projections to the locus ceruleus, dorsal raphe, ventral tegmental area, tuberomammillary nuclei of the posterior hypothalamus, laterodorsal tegmental (LDT) and pedunculopontine tegmental (PPT) nuclei, ventrolateral preoptic (VLPO) neurons in the hypothalamus, basal forebrain, limbic system (hippocampus and amygdala), cerebral cortex, thalamus (intralaminar and midline nuclei), and autonomic neurons (nucleus tractus solitarius [NTS], dorsal vagal nuclei, and intermediolateral neurons of the spinal cord). Hypocretin systems promote wakefulness mainly through excitation of tuberomammillary histaminergic, locus ceruleus noradrenergic, and midline raphe serotonergic neurons as well as dopaminergic neurons. Reduced activity of hypocretin systems may be partly responsible for inducing sleepiness. These systems also suppress REM sleep through activation of the aminergic neurons (REM-off), which in turn inhibit REM-on neurons in the LDT/PPT nuclei. As shown in Fig. 68-13, brainstem arousal centers were identified and characterized, and support was later provided for the concept of sleep-promoting circuitry in the anterior hypothalamus/preoptic area. It was later in the mid-1990s that the identity of this sleep-promoting circuitry was revealed. In these investigations (see Fig. 68.13, B), it was demonstrated that the VLPO nucleus contains sleep-active cells that contain the inhibitory neurotransmitters, γ-aminobutyric acid (GABA) and galanin (Gal).
Neuroanatomical Substrates for REM Sleep
The existence of REM sleep–generating neurons in the pontine cat has been established by transection experiments (see Fig. 68.12) through different regions of the midbrain, pons, and medulla (McCarley, 2009). A transection at the junction of the pons and midbrain produced all the physiological findings compatible with REM sleep in the section caudal to that transection, whereas in the forebrain region rostral to the section, the recording showed no signs of REM sleep. After transection between pons and medulla, structures rostral to the section showed signs of REM sleep, but structures caudal to the section had no signs of REM sleep. After transection at the junction of the spinal cord and medulla, REM sleep signs were noted in the rostral brain areas. Finally, transection at the pontomesencephalic and pontomedullary junctions produced an isolated pons that showed all the signs of REM sleep. The pons is therefore sufficient and necessary to generate all the signs of REM sleep.
There are two main models to explain the mechanism of REM sleep. The earliest and most generally well known is the McCarley-Hobson reciprocal interaction model (Fig. 68.14) based on the reciprocal interaction of REM-on and REM-off neurons (McCarley, 2009). Neurons in the PPT and LDT nuclei in the pontomesencephalic region are cholinergic and are REM-on cells, which are responsible for REM sleep, showing highest firing rates at this stage. The REM-off cells are located in the locus ceruleus and dorsal raphe nuclei. These cells are aminergic neurons and are inactive during REM sleep. Histaminergic neurons in the tuberomammillary region of the posterior hypothalamus can also be considered REM-off cells. Thus, the cholinergic REM-on and aminergic REM-off cells are all located within the transections in the pons. LDT-PPT cholinergic neurons promote REM sleep through pontine reticular formation (PRF) effector neurons, which in turn send feedback loops to LDT-PPT neurons. Cholinergic neurons of the PPT and LDT projecting to the thalamus and basal forebrain regions, as well as to the PRF, are responsible for activation and generation of REM sleep. Aminergic cells seem to play a permissive role in the appearance of REM sleep state. In the latest modification of the reciprocal interaction model, McCarley suggested that in addition to cholinergic excitation of PRF, a reduction of GABA inhibition in the PRF also may play a role in REM sleep generation. For example, GABA levels in the PRF (as measured by microdialysis technique) are lowest during REM and intermediate between wakefulness and REM and NREM sleep. Also, injection of GABA antagonists (e.g., bicuculline) into rostral PRF produced REM sleep in cats and rats. There is evidence of GABA-ergic inhibition of locus ceruleus/dorsal raphe nuclei (REM-off neurons). The source of GABA-ergic neurons is probably both local (e.g., a subgroup of PRF GABA-ergic neurons) and distant (e.g., GABA-ergic neurons in the ventrolateral periaqueductal gray matter).
In the model (Fig. 68.15) proposed by Lu and coworkers (2006), there is reciprocal interaction between GABA-ergic REM-off neurons in ventrolateral periaqueductal gray matter (vlPAG) and lateral pontine tegmentum and GABA-ergic REM-on neurons in the sublaterodorsal nucleus (SLD; corresponding to the dorsal subceruleus or peri–locus ceruleus alpha in cats) and a dorsal extension of the SLD termed the preceruleus (PC). These mutually inhibitory neuronal populations (SLD GABA-ergic REM-on and GABA-ergic REM-off neurons in the vlPAG- lateral pontine tegmentum) serve as a flip-flop switch. Ascending glutamatergic projections from PC neurons to medial septum are responsible for EEG hippocampal theta rhythm during REM sleep. Descending glutamatergic projections from ventral SLD directly to spinal interneurons, apparently without a relay in the medial medulla, inhibit spinal ventral horns by both glycerinergic and GABA-ergic mechanisms. Cholinergic and aminergic neurons play a modulatory role and are not part of the flip-flop switch. The question arises as to how two mutually inhibitory neuronal populations (e.g., GABA-ergic REM-on and GABA-ergic REM-off neurons) stabilize the flip-flop switch permitting NREM-REM cycling. It has been suggested that inhibitory external projections from extended ventrolateral preoptic neurons (eVLPO) and excitatory projections from hypocretinergic and aminergic neurons to the REM-off neurons stabilize the switch, permitting NREM-REM cycling. McCarley (2009) suggested that this model is based on C-fos labeling, only without electrophysiological recordings. Furthermore, this model does not address how REM sleep duration progressively increases with the progression of the night. A third promising model has recently been proposed by Luppi et al. (2011).
Neuroanatomical Substrates of NREM Sleep
The contemporary theory for the mechanism of NREM sleep suggests a reciprocal interaction between two antagonistic neurons in the VLPO area of the anterior hypothalamus and wake-promoting neurons in the tuberomammillary nuclei of the posterior hypothalamus, basal forebrain, and mesopontine tegmentum (McCarley, 2009; Steriade and McCarley, 2005). Reciprocal interaction between sleep-promoting neurons in the region of the NTS and wake-promoting neurons within the ARAS of the brainstem independent of the reciprocal interaction of the neurons of the forebrain also plays a role in the generation of NREM sleep. The latter is the old reticular passive hypothesis, or the disfacilitation hypothesis, of sleep. In this model, sleep is said to result from a cascade of disfacilitation within the brainstem. The passive theory originated in the two classic preparations in cats by Bremer (1937), noted earlier in the chapter: cerveau isolé and encéphale isolé. Bremer found that in cerveau isolé (e.g., midcollicular transection), all specific sensory stimuli were withdrawn and the animals were somnolent, whereas in encéphale isolé (transection at C1 vertebral level disconnecting the entire brain from the spinal cord), these specific stimuli maintained activation of the brain and animals were awake. Moruzzi and Magoun postulated that withdrawal of generalized activation from ARAS is responsible for somnolence in cerveau isolé preparations, whereas activation of the midbrain reticular neurons causing direct excitation of the thalamocortical projections results in EEG desynchronizations and behavioral arousal. These observations support the later suggestion of Steriade and colleagues that at the onset of NREM sleep, there is a deafferentation of the brain due to blockage of afferent information, first at the thalamic level, causing the waking “open” brain to be converted to a “closed” brain owing to thalamocortical inhibition. This passive reticular theory, however, was challenged by the experiments of Batini and colleagues in 1959, producing the midpontine pretrigeminal section, which was only a few millimeters below the section that produced cerveau isolé. In this midpontine preparation, there were persistent EEG and behavioral signs of alertness, suggesting that structures located in the brainstem between cerveau isolé and midpontine pretrigeminal preparations are responsible for wakefulness. Thus an active inhibitory role of the lower brainstem hypnogenic neurons in the region of the NTS on the upper brainstem ARAS was clearly demonstrated by this preparation. Both active and passive theories partly explain the generation of NREM sleep, but the contemporary theory favors the activation of VLPO neurons and inhibition of posterior hypothalamic neurons containing histamine and hypocretin. Reduced activity of the hypocretin projections to the locus ceruleus noradrenergic, midline raphe serotonergic, mesopontine dopaminergic, and tuberomammillary hypothalamic histaminergic cells may also decrease the level of arousal, causing sleepiness.
The suprachiasmatic nuclei (SCN), the paired nuclei above the hypothalamus, function as the body clock to control circadian rhythm. The recent discovery of anatomical projections from SCN to lateral hypothalamic neurons containing hypocretin (wake-promoting) and anterior hypothalamic VLPO containing sleep-promoting neurons suggested that the SCN may also affect sleep regulation and homeostasis independent of circadian rhythm generation (Turek and Vitaterna, 2011). Finally, recent advances in neuroimaging studies, including positron emission tomography (PET) and single-photon emission computed tomography (SPECT) scans, have been able to visualize dramatic changes in function in cortical and subcortical neuronal networks in different sleep states and stages, advancing our understanding of the functional neuroanatomy of sleep/wakefulness (Dang-Vu et al., 2007). PET scans have shown marked activation of the amygdala and the anterior cingulate region (part of the limbic system) during REM sleep, which is, of course, generated by brainstem neurons. In contrast, in NREM sleep, neuroimaging techniques have shown declining function in the thalamocortical circuits, including the association cortex of the frontoparietal and temporal lobes. Thus the brainstem, hypothalamic, and forebrain sleep/wake-promoting neurons modulate functions of widespread forebrain cortical areas, keeping in balance cortical and subcortical circuits to control sleep/wake regulation.
Circadian Rhythm and Chronobiology of Sleep
The 18th-century, French astronomer de Mairan first directed our attention to the existence of circadian rhythm when he noted that in a heliotrope plant, the leaves closed at sunset and opened at sunrise even when the plant was kept in darkness (see Chokroverty, 2009a). This observation clearly pointed to a 24-hour rhythm controlled by an internal clock. The existence of such a circadian rhythm in human beings and other animals was confirmed toward the last half of the last century. The term circadian rhythm originates from the Latin circa, meaning “about,” and dies, meaning “day.” Human circadian rhythm generally has a cycle length close to 24 hours (approximately 24.2 hours) (Czeisler and Gooley, 2007). The existence of circadian rhythms independent of environmental stimuli has been clearly demonstrated by experimental isolation of humans from all environmental time cues (the German term, Zeitgeber, or “time giver”), as in a cave or underground bunker, to study free-running rhythms. The existence of environment-independent autonomous rhythms suggests that the human body also has an internal biological clock. The paired SCN of the hypothalamus above the optic chiasma are the sites of the biological clock. The master circadian clock in the SCN receives photic information from the retinohypothalamic tract, which sends signals to multiple synaptic pathways in other parts of the hypothalamus, the superior cervical ganglion, and the pineal gland where melatonin is released. The SCN contain melatonin receptors, and there is a feedback loop from the pineal gland to the SCN. Time isolation experiments have clearly shown the presence of daily rhythms in many physiological processes, such as the sleep/wake cycle, body temperature, and neuroendocrine secretion. Melatonin is secreted maximally during the night and is an important modulator of human circadian rhythm for entrainment by the light/dark cycle. The melatonin level rises fairly abruptly in the evening and then reaches its maximum level between 3:00 am and 5:00 am, after which it decreases to low levels during the daytime.
Sleep scientists have begun to identify the molecular basis of the mammalian circadian clock. A total of eight or nine genes (e.g., CLOCK, PER, Bmal) and their protein products have been identified within the circadian clock system; understanding of these is still evolving (Turek and Vitaterna, 2011). Remarkable progress has been made in the past few years in the key components of the circadian clock in both fruitflies (Drosophila) and mammals. Dysfunction of circadian rhythm results in some important human sleep disorders. The molecular mechanism of two human circadian rhythm disorders—advanced sleep phase syndrome (ASPS) and delayed sleep phase syndrome (DSPS)—has been uncovered by applying gene sequencing techniques. ASPS seems to be due to a mutation of the PER2 gene (a human homolog of the period 2 gene in Drosophila) causing advancing of the clock (i.e., alteration of the circadian timing of sleep propensity). DSPS is due to polymorphism of the PER3 gene.
Circadian, Homeostatic, and Other Sleep Factors
Sleep and wakefulness are controlled by both homeostatic and circadian factors. The duration of prior wakefulness determines the propensity to sleepiness (homeostatic factor), whereas circadian factors determine the timing, duration, and characteristics of sleep. There are two types of sleepiness: physiological and subjective (Dinges, 1995). Physiological sleepiness is the body’s propensity to sleepiness. There are two highly vulnerable periods of sleepiness: 2:00 to 6:00 am, particularly 3:00 to 5:00 am, and 2:00 to 6:00 pm, particularly 3:00 to 5:00 pm. The propensity to physiological sleepiness (e.g., midafternoon and early-morning hours) depends on circadian factors. The highest number of sleep-related accidents have been observed during these periods. Subjective sleepiness is the individual’s perception of sleepiness; it depends on several external factors such as a stimulating environment and ingestion of coffee and other caffeinated beverages.
Physiological sleepiness depends on two processes: homeostatic factor and circadian phase. Homeostatic factor refers to a prior period of wakefulness and sleep debt. After a prolonged period of wakefulness, there is an increasing tendency to sleep. The recovery from sleep debt is aided by an additional amount of sleep, but this recovery is not linear. Thus, an exact number of hours of sleep are not required to repay sleep debt; rather, the body needs an adequate amount of slow-wave sleep for restoration. The interaction between the circadian and the homeostatic drive producing alertness is depicted in Fig. 68.16. The circadian factor determines the body’s propensity to maximal sleepiness between 3:00 am and 5:00 am. The second period of maximal sleepiness (3:00 to 5:00 pm) is not as strong as the first. Sleep and wakefulness and the circadian pacemaker have a reciprocal relationship: the biological clock can affect sleep and wakefulness, and sleep and wakefulness can affect the clock. There are two variables that seem to play a role in regulating the timing of sleep. First is the homeostatic sleep drive, which increases as the day progresses and the longer a person is awake. The second is timing information from the suprachiasmatic nucleus (SCN). In this two-process model, the SCN promotes wakefulness by stimulating arousal networks. The activity of the circadian system appears to oppose that of the homeostatic sleep drive, and thus the alerting mediated by the SCN increases during the day. The propensity to be awake or asleep at any time is related to the homeostatic sleep drive and the opposing SCN alerting signal. At normal bedtime, both the alerting drive and the sleep drive are at their highest level. The SCN has at least two types of melatonin receptors, MT1 and MT2, involved in the regulation of sleep. Stimulation of MT1 receptors is believed to decrease the alerting signal from the SCN, while MT2 stimulation is thought to be involved in synchronizing the circadian system.
Various sleep factors have been identified, but their role in maintaining homeostasis has not been clearly established (Krueger et al., 2011). Several cytokines, such as interleukin-1, interferon-α, and tumor necrosis factor, promote sleep. Other sleep factors increase in concentration during prolonged wakefulness and infection. It has been shown that adenosine in the basal forebrain can fulfill the major criteria for the neural sleep factor that mediates the somnogenic effect of prolonged wakefulness by acting through adenosine A1 and A2A receptors. Several other endogenous compounds may serve as sleep factors, including delta sleep-inducing peptides, muramyl peptides, cholecystokinins, arginine vasotocin, vasoactive intestinal peptides, growth hormone–releasing factors, and somatostatins.
Functions of Sleep
The function of sleep remains the greatest biological mystery of all time. There are several theories about the function of sleep (Box 68.3), but none are satisfactory (Crick and Mitchison, 1995; Kavanau, 1997; Mahowald et al., 1997). Sleep deprivation experiments in animals have clearly shown that sleep is necessary for survival, but from a practical point of view, complete sleep deprivation for a prolonged period cannot be conducted in humans. Sleep deprivation studies in humans have shown an impairment of performance, which demonstrates the need for sleep. The performance impairment of prolonged sleep deprivation results from a decreased motivation and frequent “microsleeps.” Overall, human sleep deprivation experiments have proven that sleep deprivation causes sleepiness and impairment of performance, vigilance, attention, concentration, and memory. Sleep deprivation may also cause some metabolic, hormonal, and immunological effects. Sleep deprivation causes immune suppression; even partial sleep deprivation reduces cellular immune responses. Studies from Van Cauter’s (Spiegel et al., 1999
[/not-level-membership-for-neurology-category]
