Chapter 54 Multiple Sclerosis and Other Inflammatory Demyelinating Diseases of the Central Nervous System
Diseases affecting central nervous system (CNS) myelin can be classified on the basis of whether a primary biochemical abnormality of myelin exists (dysmyelinating) or whether some other process damages the myelin or oligodendroglial cell (demyelinating). Demyelinating diseases in which normal myelin is disrupted include autoimmune, infectious, toxic and metabolic, and vascular processes (Box 54.1). Dysmyelinating diseases in which a primary abnormality of the formation of myelin exists include several hereditary disorders (see Box 54.1 and Chapter 62). Infectious demyelinating disease (progressive multifocal leukoencephalopathy) is discussed in Chapter 53B, toxic and metabolic demyelinating diseases in Chapter 56, and vascular demyelinating disease (Binswanger disease) in Chapter 66. The present chapter concentrates on multiple sclerosis (MS) and other inflammatory demyelinating diseases of myelin (acute disseminated encephalomyelitis [ADEM] and acute hemorrhagic leukoencephalopathy), as well as other CNS diseases that are presumably immune mediated (see Box 54.1). The paraneoplastic disorders are discussed in Chapter 52G.
MS is the most common disease caused by an inflammatory demyelinating process in the CNS. MS is a leading cause of disability in young adults (Noseworthy et al., 2000). Because MS has only a modest negative effect on longevity but potential for considerable disability over many years, the socioeconomic consequences are considerable. Pathologically, MS is characterized by multifocal areas of demyelination, axonal loss and disruption, loss of oligodendrocytes, and astroglial scarring. Certain clinical features are typical of MS, but the disease has a highly variable pace and many atypical forms. Investigative studies are often needed to confirm the diagnosis and exclude other possibilities. Understanding of the basic nature of the disease remains limited, and better control of the disease and repair of damaged CNS tissue remain goals for the future.
Pathophysiology
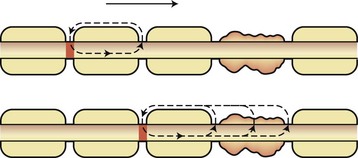
Compacted myelin is the lipid-rich plasma membrane of oligodendrocytes that provides insulation for electrical impulses traveling along axons. Myelinated axons propagate nerve impulses rapidly in a saltatory fashion, with a high safety factor for transmission (five to seven times above threshold). Current is induced by the opening of voltage-gated Na+ channels found at the nodes of Ranvier. The resultant Na+ influx creates a current that then moves toward the next node of Ranvier because current cannot flow outward in myelinated internodal segments (Fig. 54.1). K+ channel opening terminates current flow and leads to repolarization. Several types of K+ channels exist in the axon. Fast K+ channels sensitive to 4-aminopyridine are located in internodal axonal membrane and contribute to repolarization of demyelinated axons. Slow K+ channels are found at the nodes of Ranvier and have a role in modulating repetitive firing. The Na+/K+-adenosine triphosphatase (ATPase) in the axon membrane restores ionic balance after high-frequency firing. Demyelination interrupts current flow by removing the insulator of internodal axon current flow. Long segments of demyelination can result in interruption of current flow because current must flow by continuous propagation. The low density of internodal Na+ channels, at least in the early stages of demyelination, inhibits impulse propagation. If conduction does occur, it is at a much-reduced speed (5% to 10% of normal). The refractory period of demyelinated axons is prolonged, and repetitive volleys may be blocked when encountering an axon segment in a refractory period. Persistent neurological deficits or negative symptoms of MS are caused by regions in which conduction block persists, such as in regions of large plaques, whereas transient worsening of function reflects a drop below the safety threshold for conduction because of physiological changes involving the partially demyelinated axon (Uhthoff phenomenon, worsening with increased body temperature). Symptoms or signs may also arise from slowed conduction, producing temporal dispersion at time-critical synapses. Further conduction block is absolute in transected axons.
Pathology
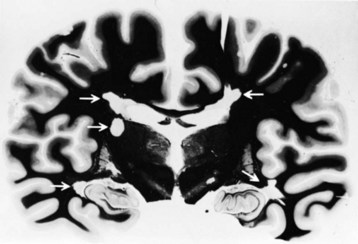
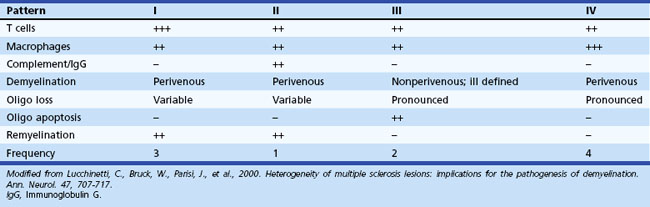
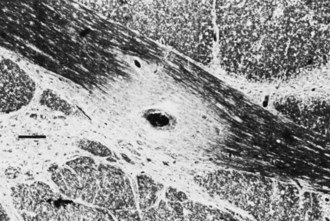
Gross examination of the brain in MS often reveals atrophy and ventricular dilatation. Plaques may be visible on the surface of the spinal cord on inspection. The cut surface of the brain reveals the plaques, which when active, appear whitish yellow or pink with somewhat indistinct borders. Older plaques appear translucent with a blue-gray discoloration and sharply demarcated margins. Individual lesions are generally small (1-2 cm) but may become confluent, generating large plaques. Plaques develop in a perivenular distribution and are seen most frequently in the periventricular white matter, brainstem, and spinal cord (Figs. 54.2 and 54.3), a finding confirmed with MRI studies. However, large numbers of small plaques, often detected only by microscopy, are found in cortical regions affecting intracortical myelinated fibers. One of the earliest features of acute MS lesions is a disruption of the blood-brain barrier (BBB) as detected by MRI studies. Disruption of the BBB appears to be a critical early step in lesion pathogenesis. The BBB depends on tight junctions between brain endothelial cells. These junctions are not disrupted; rather, a transendothelial cell vesicular transport system will become active. It can carry water, proteins, antibodies, and cytokines (and gadolinium) into the brain.
The fate of oligodendroglia in MS lesions is disputed. Consensus is that oligodendroglia numbers are reduced proportionate to myelin loss in the plaque center, whereas at the plaque edge, oligodendroglia are preserved or even increased, suggesting an attempt at remyelination. The finding of shadow plaques (areas of thinly myelinated axons) supports the concept of central remyelination (Prineas et al., 2001), and oligodendroglial precursors have been found in the adult brain.
Remyelination may involve either oligodendrocytes that previously produced myelin or maturation of progenitor cells. Such remyelination may explain the clinical finding of slow and delayed recovery from an acute attack, whereas rapid clinical recovery presumably reflects the resolution of edema, inflammation, and removal of toxic factors associated with acute plaques in which myelin destruction is minimal. Recent pathological studies demonstrate that the extent of remyelination can be quite extensive, even in patients with progressive disease (Patrikios et al., 2006).
Evidence of recurrence of activity in old plaques may be demonstrated by gadolinium-enhanced MRI. Chronic demyelinated plaques could thus result not from a single severe episode of demyelination, but rather from recurrent bouts of demyelination at the same site (Prineas et al., 2001). This could eventually exceed the ability of oligodendroglia to remyelinate.
Data derived from biopsy as well as autopsy material (Lucchinetti et al., 2000) have emphasized the heterogeneity of the MS lesion. These investigators have described four distinct pathological patterns. Some lesions appear to be chiefly inflammatory (types I and II), with retention of active oligodendrocytes derived from identifiable precursor cells and evidence of remyelination. The most common pathological pattern seen (type II) had inflammatory infiltrates and deposition of complement and immunoglobulin (Ig)G. In other patients, extensive destruction of oligodendrocytes, little replacement, and closer resemblance to a viral or toxic cell apoptosis or necrosis was found (types III and IV) (Table 54.1). All active lesions from an individual patient were of the same type. The specific target of the immune-mediated injury in MS remains undetermined. A proportionate loss of oligodendroglia and myelin would imply a primary attack against either oligodendroglia or an antigen present on oligodendroglial cell bodies and myelin. Alternatively, myelin may be the primary target of disease, and the oligodendroglia may survive demyelination, at least in the initial stages of disease. The active lesions contain T lymphocytes and macrophages in the perivascular regions and parenchyma. Most of the T cells express the α/β T-cell receptor. Both CD4+ and CD8+ T cells are present. An apparent increase in CD8+ cells occurs in the CNS when compared with the prevalence of this subset in the peripheral blood. CD4+ cells extend from the periphery of active plaques into adjacent white matter, whereas CD8+ cells predominate in the perivascular regions. Some MS lesions also have an accumulation of T cells expressing the α/β T-cell receptor, which may mediate cytolysis of CNS cells expressing heat shock proteins.
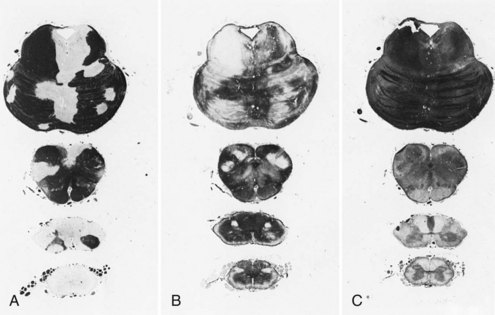
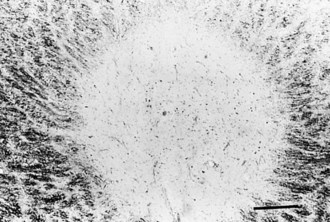
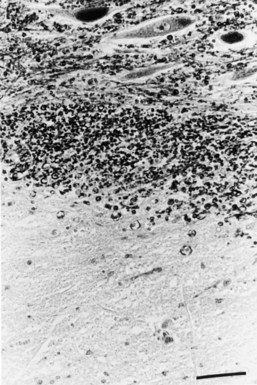
Chronic inactive plaques are hypocellular and show astrocytic proliferation with denuded axons and an absence of oligodendroglia (Figs. 54.4 and 54.5). Axonal loss also may be noted to a variable extent. Microglia and macrophages are scattered throughout the lesion. The edge of chronic plaques may still exhibit hypercellularity, suggesting continued disease activity.
Pathological differences are described between classical MS and disorders considered to be variants of the disease. Baló concentric sclerosis is characterized by alternating bands of myelinated and demyelinated fibers in white matter. Clinically, the illness is more fulminant in onset and course than typical MS and has a more inflammatory cerebrospinal fluid (CSF). Transitional forms exist with typical MS. The affected CNS structures in Devic neuromyelitis optica show more necrosis, cyst formation, and vascular proliferation than is seen in the usual MS case (Lucchinetti et al., 2002). Large tumor-like plaques found in the variant of MS known as Marburg variant may show extensive areas of inflammation and edema.
In secondary progressive MS (SPMS, defined later), ongoing low-grade demyelination was found at the borders of plaques, associated with C3d, an opsonin coupled with complement activation, and this profile may explain the slow expansion of plaques and occurrence of more diffuse inflammation leading to progressive loss of function (Kutzelnigg et al., 2005; Prineas et al., 2001).
Recent pathological studies have focused on the gray matter in MS and have found a lesion load within the cortex and deep gray structures. The nature of the intracortical plaques differs from those seen in white matter because there is less inflammation but considerable reactive microgliosis (Bo et al., 2003).
Etiology
Autoimmunity
Although the possibility of autoimmunity as the causal mechanism for MS exists, the issue is not proven. The evidence for MS being a dysimmune condition is more compelling, with alterations in immune cell repertoire and activation state both in blood and CSF of MS patients compared to others (Conlon et al., 1999; Hafler et al., 2005).
Infection
A possible role for microbial infection in the causation of MS has been a matter of ongoing debate for decades. The epidemiology of MS (see Epidemiology in this chapter and in Chapter 39) suggests an exogenous or environmental factor. Beyond epidemiology and much speculation, little direct evidence supports the concept of a role for viral infection. Innumerable efforts to culture a virus from autopsy-derived or biopsy material have yielded no consistent result. Serological data are difficult to interpret, because titers may reflect only a nonspecific tendency toward increased immune reactivity. Specific efforts to recover a known viral genome (e.g., that of human T-cell lymphotropic virus type 1 [HTLV1]) have proven negative.
Human herpesvirus 6 (HHV6), Epstein-Barr virus (EBV), and Chlamydia pneumoniae have been the focus of interest as potential triggers for MS. Several studies of serum and CSF samples have yielded varying results. For example, one early study showed 47% of MS brains were positive for HHV6, and 80% showed elevation of antibodies in the serum. A more recent study found no HHV6 DNA in any CSF sample, and the serum antibody titers were comparable to the general population. High serum levels of EBV antibodies have also been associated with an increased risk of MS. The strongest association was found for antibodies to Epstein-Barr nuclear antigen (EBNA-2); a fourfold difference in titers was associated with increased relative risk (RR) of developing MS (Ascherio et al., 2001).
Epidemiology
The epidemiology (see Chapter 39) and genetics of MS are complex topics. The interested reader is referred to the articles by Dyment et al. (2004) and Kantarci and Wingerchuk (2006).
Age of Onset
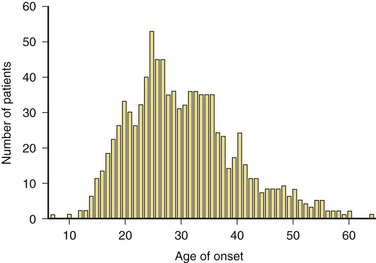
Most studies agree that the mean and median age of onset in relapsing forms of MS is age 29 to 32. The peak age of onset is approximately 5 years earlier for women than for men. Primary progressive MS (PPMS) has a mean age of onset of 35 to 39 years. The onset of MS can occur as late as the seventh decade. Perhaps as many as 5% of cases of MS have their onset before age 18. Most of these cases occur in adolescents, but a small percentage begin in the first decade of life (Fig. 54.6).
Mortality
Mortality due to MS is difficult to ascertain because of poor data collection and reporting. The U.S. Department of Health and Human Services report of deaths in the year 1992 indicates that 1900 U.S. citizens died of MS in that year, giving MS a U.S. mortality of 0.7 per 100,000. The mean age of death of all patients with MS was 58.1 years, compared with a national average of 70.5 years for all causes of death. The life expectancy of patients with MS was therefore calculated to be 82.5% of the normal lifespan. In Denmark, in an exceptionally complete survey of the country, median survival after diagnosis for men was 28 years and for women 33 years, compared with matched population death rates of 37 and 42 years, respectively. The life expectancy was 10 years shorter for MS patients than age-matched controls; this difference was becoming smaller when more recent data were studied (Bronnum-Hansen et al., 2004).
Geographical and Racial Distribution
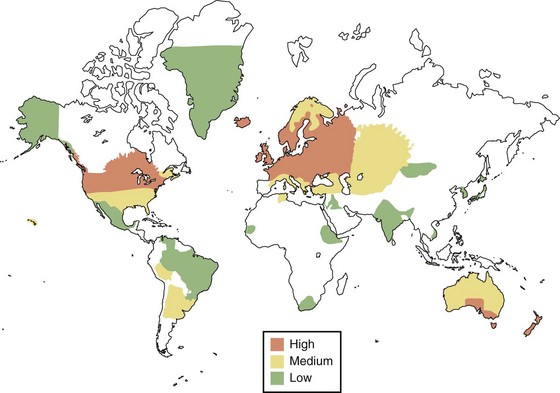
More than 250 prevalence surveys have been carried out, serving as the basis for the delineation of geographical risk for MS depicted in Fig. 54.7. High-frequency areas of the world, with current prevalence of 60 per 100,000 or more, include all of Europe (including Russia), southern Canada, the northern United States, New Zealand, and the southeastern portion of Australia. In many of these areas, the prevalence is more than 100 per 100,000, with the highest reported rate of 300 per 100,000 occurring in the Orkney Islands. In the United States, the prevalence when measured during the 1990s was approximately 350,000. Several studies suggest that the prevalence is increasing beyond what might occur because of enhanced recognition and better-appreciated diagnostic techniques.
Genetics
MS is a genetically complex disease. Many genetic and nongenetic factors of moderate effect interact to influence disease probability. Compared to the general population, a higher frequency of familial occurrence of MS suggests a strong but non-Mendelian inheritance of susceptibility. Twin studies established the importance of genetic factors: the concordance rate for a clinical diagnosis of MS in monozygotic twins is about 30%, whereas in dizygotic twins it is 2% to 5%. (Ebers et al., 1995; Willer et al., 2003). The risk is highest for siblings: 3% to 5%, or 30 to 50 times the background risk for this same population. Adoptive relatives, when raised from infancy with the patients with MS, are no more likely to develop MS than the general population. This indicates that the familial aggregation of MS is more likely to be related to shared genetic variations rather than to a shared family environment. In some studies, unaffected family members may have been found to have abnormalities on MRI, implying the risk may be even higher.
MS is associated with both MHC and human leukocyte antigen (HLA) class I A3 and B7 antigens. The class II polymorphisms, Dw2 and DR2, also show strong association, and specifically the HLA DRB1*1501 allele (Oksenberg et al., 2004). Additional HLA alleles that carry protective as well as detrimental effects with regard to MS susceptibility have been identified. HLA-A*02, for example, has a protective effect relative to MS susceptibility. In recent years, additional MS susceptibility loci outside of the MHC have been described. Specifically, the loci coding for IL-7 receptor and IL-2 receptor are strongly linked with MS susceptibility (Zuvich et al., 2010). IL-7 and IL-2 receptor signaling is critical for the differentiation of CD4− CD8− thymocytes and has a role in survival of CD4+ CD8+ cells after positive selection. This may be important not only in MS predisposition but also in disease course and outcome.
Recently an aggregate measure of MS risk that combines weighted odds ratios from 16 genetic susceptibility loci (the weighted genetic risk score [wGRS]) was created and used to predict a diagnosis of MS in three independent cohorts (De Jager et al., 2009). The results revealed that the wGRS can modestly but robustly predict MS risk. Individuals with MS have a greater “load” of susceptibility factors than healthy people, although the 16 loci assessed so far are insufficient to differentiate MS cases from healthy controls. While this tool is still in early stages of development, it is quite likely a useful application of the wGRS may be to predict individuals who are at higher risk for developing the disease, such as first-degree relatives of MS patients. Identifying individuals during the clinically silent phase of the disease could be extremely helpful in guiding the selection of those who would benefit most from early imaging screening, and eventually from early treatment.
Clinical Symptoms and Physical Findings
Cognitive Impairment
Cognitive involvement in MS was documented as early as 1877 by Charcot. He observed that patients with “multilocular sclerosis” are slow to form conceptions, have “marked enfeeblement of the memory, and blunting of intellectual and emotional faculties.” However, the subsequent era of research in MS focused primarily on physical disability. There was a paucity of rigorous studies, with a focus on cognition. In 1981, Kurtzke reported that only 5% of patients with MS suffered from cognitive impairment. The Kurtzke Expanded Disability Status Scale (EDSS) focused primarily on somatic disability measures. A decade later, data from formal neuropsychological studies indicated that cognitive involvement has been underreported in MS (Rao et al., 1991). Neuropsychological test results have shown that 34% to 65% of patients with MS have cognitive impairment (Nocentini et al., 2006).
Cognitive abnormalities affect patients across the disease spectrum and involve all MS subtypes. Some 49% to 53.7% of clinically isolated syndrome (CIS) and early relapsing-remitting multiple sclerosis (RRMS) patients show significant impairment in one or more cognitive domains that impacts their quality of life (Achiron and Barak, 2003; Glanz et al., 2007, 2010). A recent longitudinal study reported 29% of early PPMS patients (within 5 years from disease onset) as cognitively impaired (Penny et al., 2010; Ukkonen et al., 2009). The domains of verbal memory and attention/speed of information processing were the most affected, and premorbid IQ was a robust predictor of cognitive status in this cohort. A reported 19% of patients with a so-called benign MS variant have progressive cognitive disability and structural brain MRI changes similar to those with SPMS (Mesaros et al., 2009; Rovaris et al., 2009). It is increasingly recognized that low physical disability can coexist with significant cognitive disease. In a less common clinical presentation, patients may have a subacute or fulminant cognitive decline with signs of cortical dysfunction. This disability progresses rapidly and results in significant socio-occupational dysfunction, while physical disability remains minimal (Houtchens et al., [in press]).
In general, the most frequently reported abnormalities are with working memory, attention, and speed of information processing. Patients complain of memory loss, difficulties at work or with interpersonal relations, inability to multitask, and “mental fog and fatigue.” Comorbid depression, anxiety disorders, and emotional lability may further affect cognitive performance. Mild to moderate abnormalities are usually not apparent during a routine office visit, and simple screening tools for cognitive dysfunction are not yet available. A 5- to 15-minute battery of three tests assessing functions most commonly impaired in MS patients is now being validated (Portaccio et al., 2009). Longer neuropsychological testing batteries are designed to assess cognitive impairment in MS patients and are used primarily in research trials. The Brief Repeatable Battery of Neuropsychological Tests (BRB-N) (Rao et al., 1991) and Minimal Assessment of Cognitive Function in Multiple Sclerosis (MACFIMS) (Benedict et al., 2002) are the most widely used screening tools. Multiple Sclerosis Functional Composite (MSFC) also includes the Paced Auditory Serial Addition Test (PASAT) as a measure of sustained attention (Cutter et al., 1999).
Cognitive impairment in MS has been associated with a variety of MRI metrics of disease activity and progression. Early studies reported high correlations with the total T2 lesion burden and volume (Patti et al., 1998; Rao, 1989). Lesions located in frontal and parietal lobes showed stronger correlations with tasks of processing speed, attention, and verbal memory (Sperling et al., 2001). Global and regional atrophy plays a role in cognitive impairment. Thalamic atrophy appears to correlate particularly well with impairment on a variety of cognitive tests in MS patients (Houtchens et al., 2007). Contribution of cortical lesions (Bakshi et al., 2001; Zivadinov and Miagarb, 2009) and loss of cortical ribbon thickness (Calabrese et al., 2010) are important. Cognitively impaired patients have abnormalities in normal-appearing white and gray matter as measured by magnetization transfer ratio (MTR) that are not seen in cognitively intact MS patients (Amato et al., 2008).
Treatment of cognitive impairment in MS is challenging. Pharmacological therapy focuses on treatment of underlying disease as well as symptomatic manifestations of declining mental function, such as difficulties with attention, memory, and fatigue. Appropriate management of the comorbid psychiatric disease often improves cognitive performance. A recent randomized study of 469 patients showed that treatment with higher versus lower doses of subcutaneous interferon beta-1a was predictive of lower cognitive impairment at 3 years of follow-up (Patti et al., 2010). An earlier trial of intramuscular (IM) interferon beta-1a showed a 47% reduction in cognitive decline on PASAT testing over 2 years of follow-up (Fischer et al., 2000).
There is some evidence that treatment with l-amphetamine is associated with improved learning and memory in cognitively impaired MS patients (Benedict et al., 2008; Morrow et al., 2009). Modafinil may improve focused attention and dexterity and palliate fatigue. It has a favorable safety profile and can be used for symptomatic management of MS patients with severe fatigue and decreased focus. There is no convincing evidence that cholinesterase inhibitors improve memory in MS patients. A small study assessed the effects of donepezil on SPMS patients who were residents in a nursing home, documenting improvement in several cognitive domains after 8 weeks of therapy (Greene et al., 2000). There are additional anecdotal reports of efficacy in small numbers of subjects. Cognitive-behavioral therapy, family and individual counseling, strategies to improve day-to-day function, and necessary job modifications and accommodations can be of great help to MS patients suffering from cognitive decline.
Affective Disorders
Cross-sectional studies have shown some degree of affective disturbance in a significant number of patients with MS (Arnett et al., 2005). Depression is the most common manifestation and is in part secondary to the burden of having to cope with a chronic disease. However, it is more prevalent in MS than in other chronic diseases, suggesting an organic component as well. The lifetime risk of major depression in patients with MS is up to 50%, compared with 12.9% in patients with chronic medical conditions in another study. Some data indicate a comorbid association, presumably genetic, between bipolar illness and MS. Suicide rates are higher in patients with MS than in the general population or when compared to patients with other chronic illnesses (Bronnum-Hansen et al., 2005). Frontal or subcortical white-matter disease may also be a contributory causative factor. Euphoria, formerly considered to be common in MS, is actually infrequent and is usually associated with moderate or severe cognitive impairment and greater disease burden on MRI. However, emotional “dyscontrol” is quite common, and patients oscillate frequently between sad and happy states, at times without precipitant.
Cranial Nerve Dysfunction
Impairment of Visual Pathways
Optic neuritis (ON) is the most frequent type of involvement of the visual pathways, usually presenting as an acute or subacute unilateral syndrome characterized by pain in the eye accentuated by ocular movements, which is followed by a variable degree of visual loss affecting mainly central vision. Patients with ON often have a relative afferent pupillary defect (Marcus Gunn pupil; see Chapter 36). Bilateral ON may occur uncommonly. Bilateral simultaneous ON is rare in MS, and its occurrence in isolation may suggest another diagnosis such as Leber hereditary optic neuropathy or toxic optic neuropathy (see Chapter 14). In bilateral ON in MS cases, the impairment begins asymmetrically and is usually more severe in one eye. In a large ON treatment trial, 15% of placebo-treated patients developed recurrent ON within 6 to 24 months after the initial bout. Mapping of visual fields reveals a central or cecocentral scotoma (central scotoma involving the physiological blind spot). After an attack of acute ON, 90% of patients regain normal vision, typically over a period of 2 to 6 months.
Impairment of Ocular Motor Pathways
Impairment of individual ocular motor nerves is infrequent in MS. When present, the involved nerves are, in decreasing order of frequency, cranial nerves VI, III, and (rarely) IV. More frequent findings are those that reflect lesions of vestibulo-ocular connections and internuclear connections. Nystagmus is a common finding in MS (see Chapter 35). One form of nystagmus particularly characteristic of MS is acquired pendular nystagmus, in which there are rapid small-amplitude pendular oscillations of the eyes in the primary position, resembling quivering jelly. Patients frequently complain of oscillopsia (subjective oscillation of objects in the field of vision). This type of nystagmus may be seen in the presence of marked loss of visual acuity.
Impairment of Bladder, Bowel, and Sexual Functions
Clinical features uniquely characteristic of MS (Table 54.2) include bilateral INO, the Lhermitte phenomenon, and paroxysmal attacks of motor or sensory phenomena such as paroxysmal diplopia, facial paresthesia, trigeminal neuralgia, ataxia, dysarthria, and tonic spasms. These paroxysmal attacks usually respond to low doses of anticonvulsants and frequently remit after several weeks to months, usually without recurrence. Heat sensitivity (e.g., the Uhthoff phenomenon) is a well-known occurrence in MS. Fatigue is also common and is usually described as physical exhaustion unrelated to the amount of activity performed. The degree of fatigue correlates poorly with the overall severity of disease or the presence of any particular symptom or sign. In contrast to the situation with cognitive deficits, no MRI findings correlate with fatigue or depression. Fatigue is often seen in association with an acute attack, may precede the focal neurological features of the attack, and may persist long after the attack has subsided.
| Clinical Features Suggestive of Multiple Sclerosis | Clinical Features Not Suggestive of Multiple Sclerosis |
|---|---|
| Onset between ages 15 and 50 | Onset before age 10 or after age 60 |
| Involvement of multiple areas of the CNS | Involvement of the PNS |
| Optic neuritis | Hemianopsias |
| Lhermitte sign | Rigidity, sustained dystonia |
| Internuclear ophthalmoplegia | Cortical deficits such as aphasia, apraxia, alexia, neglect |
| Fatigue | Deficit developing within minutes |
| Worsening with elevated body temperature | Early dementia |
CNS, Central nervous system. PNS, peripheral nervous system.
Diagnostic Criteria
Over the years and with the advent of MRI, diagnostic criteria for MS have improved in their sensitivity and specificity. However, the cornerstone of the diagnosis of MS remains the neurological history and physical examination. In 1983, Poser and colleagues modified the diagnostic criteria previously in use. These criteria maintained the two clinical events as necessary components of the diagnosis, but they also made use of laboratory diagnostic studies including CSF analysis, evoked potentials (EP), and neuroimaging. The criteria were developed to ensure that only patients with MS were included in research studies and to improve the accuracy of MS diagnosis in clinical practice. In 2001, McDonald and colleagues proposed new criteria using MRI guidelines and timing intervals to diagnose MS. The outcome of a diagnostic evaluation was either MS, possible MS, or not MS. These latest criteria were designed for use in both practice and clinical trial settings and have most recently been expanded by a committee of the National Multiple Sclerosis Society in 2005 (Polman et al., 2005) (Table 54.3). The common thread among all MS diagnostic criteria has been the requirement for symptoms and signs that are disseminated in time and space (more than one episode involving more than one area of the CNS) (Box 54.2). In the setting of a monophasic neurological illness that is clinically consistent with MS and accompanied by typical multifocal white-matter lesions on MRI, the diagnosis of MS is almost certain. This situation is referred to as the clinically isolated syndrome. In the long-term study by Brex and colleagues (2002) that followed patients with initial demyelinating episodes for up to 14 years, in practical terms, no diagnoses were encountered other than MS or suspected MS. In addition, follow-up studies have shown that a significant percentage of patients with MRI lesions detected at onset do not progress to clinically symptomatic MS, even after many years of follow-up. The issue of the monophasic demyelinating disease is discussed later under Differential Diagnosis in this chapter. Such patients would be classified as possible MS by the latest diagnostic criteria.
Table 54.3 Revised McDonald et al. (2005) Diagnostic Criteria for Multiple Sclerosis
| Clinical (Attacks) | Objective Lesions | Additional Requirements to Make Diagnosis |
|---|---|---|
| 2 or more | 2 or more | None. Clinical evidence alone will suffice; additional evidence desirable but must be consistent with MS |
| 2 or more | 1 | Dissemination in space by MRI or 2 or more MRI lesions consistent with MS plus positive CSF or await further clinical attack implicating other site |
| 1 | 2 or more | Dissemination in time by MRI or second clinical attack |
| 1 | 1 | Dissemination in space by MRI or 2 or more MRI lesions consistent with MS plus positive CSF AND dissemination in time by MRI or second clinical attack |
| 0 (progression from onset) | 1 or more | Disease progression for 1 year (retrospective or prospective) AND 2 out of 3 of the following: |
| Positive brain MRI (9 T2 lesions or 4 or more T2 lesions with positive VEP) | ||
| Positive spinal cord MRI (2 or more focal T2 lesions) | ||
| Positive CSF |
CSF, Cerebrospinal fluid; MRI, magnetic resonance imaging; MS, multiple sclerosis; VEP, visual evoked potential.
Adapted from Polman, C.H., Reingold, S.C., Edan, G., et al., 2005. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald” criteria, Ann Neurol 58, 840-846. Courtesy of the National Multiple Sclerosis Society.
Box 54.2
Paraclinical Evidence in Multiple Sclerosis Diagnosis
What Is a Positive MRI?




These diagnostic criteria were developed through the consensus of the International Panel on the Diagnosis of MS, Courtesy of the National Multiple Society, 2006, http://www.nationalmssociety.org.
A recent new diagnostic entity of radiographically isolated syndrome (RIS) addresses the category of asymptomatic patients who have MRI-detected anomalies highly suggestive of MS (see section on MRI later in this chapter).
The European Magnetic Imaging in Multiple Sclerosis (MAGNIMS) Group proposes to simplify imaging requirements for dissemination in space and time for an MS diagnosis. To fulfill the space criteria, only one or more MRI lesions in two or more characteristic locations will be required. Lesions responsible for symptomatic presentation in spinal cord or brainstem will be excluded. For the time criteria, the requirement consists of either simultaneous presence of asymptomatic gadolinium-enhancing and nonenhancing lesions at any time, or a new T2 or T1 gadolinium-enhancing lesion on follow-up MRI scan, irrespective of the timing of the initial scan (Montalban et al., 2010). Compared to the McDonald Criteria, the MAGNIMS 2010 updated criteria have slightly lower specificity but much improved sensitivity (79.2%) in predicting conversion from CIS to clinically definite MS (CDMS).
Differential Diagnosis
The differential diagnosis of MS (Box 54.3) is quite limited in the setting of a young adult with two or more clinically distinct episodes of CNS dysfunction with at least partial resolution. Problems arise with atypical presentations, monophasic episodes, or progressive illness. The unusual nature of some sensory symptoms may result in a misdiagnosis of conversion disorder. The retrospective nature of inquiries also blurs details of prior events, making clear ascertainment of prior attacks difficult in some cases. A monophasic illness with symptoms attributable to one site of the CNS creates a large differential diagnosis that includes neoplasms, vascular events, and infections. Appropriate imaging studies may help clarify the situation, depending on the site of involvement and clinical progression. The most trouble arises with progressive CNS dysfunction, in which great care must be taken to exclude treatable etiologies (e.g., vitamin B12 deficiency, compressive spinal cord lesions, arteriovenous malformations, cavernous angiomas, Arnold-Chiari malformation), infectious causes (syphilis, HTLV-1, human immunodeficiency virus [HIV]), and hereditary disorders (adult metachromatic leukodystrophy, adrenomyeloneuropathy, spinocerebellar disorders, CADASIL).
A common error is to overinterpret multiple hyperintense lesions on MRI as equivalent to MS. Clinical symptoms must be consistent with MS. A few white-matter lesions on T2-weighted MRI scans are not infrequent, particularly in the elderly, but do not indicate a diagnosis of MS. CNS vasculitides such as systemic lupus erythematosus (SLE), Sjögren disease, polyarteritis nodosa, syphilis, retroviral diseases, and Behçet disease may all produce multifocal lesions with or without a relapsing-remitting course. SLE can present as a recurrent neurological syndrome before the systemic manifestations of this disease declare themselves. Behçet syndrome is characterized by buccogenital ulcerations in addition to the multifocal neurological findings. CNS sarcoidosis can be mistaken for MS with multifocal neurological and MRI lesions. Although uncommon, ADEM must be considered in the differential diagnosis (see Acute Disseminated Encephalomyelitis in this chapter). An MS-like phenotype associated with mitochondrial gene defects has been described. More important than features characteristic for MS are features that should prompt the clinician to reconsider the diagnosis of MS—red flags indicating that another diagnosis is more likely (Charil et al., 2006). Features that should alert the clinician to the possibility of other diseases include (1) family history of neurological disease, (2) a well-demarcated spinal level in the absence of disease above the foramen magnum, (3) prominent back pain that persists, (4) symptoms and signs that can be attributed to one anatomical site, (5) patients who are older than age 60 or younger than age 15 at onset, and (6) progressive disease (see Table 54.3).
Course
The most characteristic clinical course of MS is the occurrence of relapses (Fig. 54.8), which can be defined as the acute or subacute onset of clinical dysfunction that usually reaches its peak from days to several weeks, followed by a remission during which the symptoms and signs usually resolve partially or completely. The minimum duration for a relapse has been arbitrarily established at 24 hours. Clinical symptoms of shorter duration are less likely to represent what is considered a true relapse (i.e., new lesion formation or extension of previous lesion size). Worsening of previous clinical dysfunction can occur concurrently with fever, infection, physical activity, or metabolic upset and last for hours to a day or more and is referred to as pseudorelapse. Summaries of many studies provide an average figure of 0.4 to 0.6 relapses per year. The attack rate in the placebo group in clinical studies ranges from 0.8 to 1.2 attacks per year. In general, relapses are more frequent during the first years of the disease and tend to wane in later years.
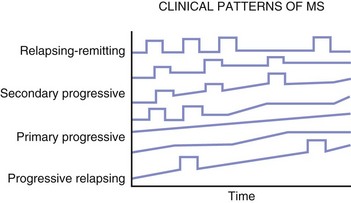
Fig. 54.8 The clinical courses of multiple sclerosis.
(Data from Lublin, F.D., Reingold, S.C., 1996. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46, 907-911.)
A course marked by relapses interspersed with periods during which the disease seems relatively dormant is termed relapsing-remitting. A small number of patients never experience a second relapse. The exact frequency of such benign disease is unknown but is thought to be about 5%. Autopsy studies found a significant number of cases with CNS pathology consistent with MS and yet no documented clinical evidence of such disease. Similarly, MRI studies have shown MS-like plaques in T2-weighted scans in patients who have never had a neurological episode. A standardization of terms has been agreed on to determine the pattern and course of the illness (Lublin and Reingold, 1996). Four categories of disease are described (see Fig. 54.8):
1. Relapsing-remitting (RR) MS: Clearly defined relapses with full recovery or with sequelae and residual deficit on recovery. The periods between disease relapses are characterized by a lack of disease progression.
2. Secondary progressive (SP) MS: Initial relapsing-remitting disease course followed by progression with or without occasional relapses, minor remissions, and plateaus.
3. Primary progressive (PP) MS: Disease progression from onset, with occasional plateaus and temporary minor improvements allowed.
4. Progressive relapsing (PR) MS: Progressive disease from onset, with clear acute relapses with or without full recovery. The periods between relapses are characterized by continuing progression.
The rate of clinical progression of MS is variable. The commonly used index of clinical disability, the Kurtzke EDSS, uses numbers ranging from 0 for normal examination and function to 10 for death caused by MS. This scale is nonlinear, with great emphasis on ambulation capabilities with scores above 4. Most MS populations have bimodal distributions of EDSS scores, with peaks at values of 1 and 6 (ambulation with unilateral assistance). In a cohort of patients followed for 25 years (in the pretreatment era), the following data emerged: 80% of the patients had reached the progressive phase by 25 years, 15% had died, 65% had reached EDSS 6 (requiring aids for walking), and 50% reached EDSS 6 within 16 years of onset. More recent studies suggest a somewhat slower course of progression (Pittock et al., 2004; Tremlett et al., 2006). The EDSS, although universally used in clinical trials, has a number of limitations. Even with special training and examiner blinding, interrater and intrarater variations in scoring are common. EDSS scores of 4 and higher depend almost entirely on the ability to walk; developing dementia, vision loss, and weakness of hands may pass undetected by the scoring once one reaches these levels. An obvious implication of these facts is that other outcome measures should be used as well, and that minor changes in EDSS score alone should not be overinterpreted. The MSFC is a clinical tool designed to avoid the problems encountered with the EDSS (Cutter et al., 1999). The MSFC consists of three parts: PASAT, nine-hole peg test (9HPT), and timed 25-foot walk (T25FW). These three measures take into account cognition, upper-extremity function, and lower-extremity function. A z-score is obtained for each measure, and a combined z-score is then derived. The MSFC has been validated in several clinical trials. The tests can be performed by a nonphysician and are highly reproducible and predictable.
Pregnancy in Multiple Sclerosis
MS preferentially affects women of childbearing age. Scant evidence and little agreement exist about the influence of pregnancy on disease activity and, conversely, the effects of MS on pregnancy outcomes. Pregnancy is recognized to induce changes in the maternal immune system, including both immunosuppression on a local level and a heightened state of immunocompetence on a global level. Several retrospective studies reported an overall increase in relapse rate during the postpartum period and a lower relapse rate during pregnancy itself (Brick et al., 1990; Weinshenker et al., 1989). Pregnancy in Multiple Sclerosis (PRIMS) was the first prospective study of 254 women (269 pregnancies). Subjects were followed for 2 years after delivery (Confavreaux et al., 1998; Vukusic et al., 2004, 2006). In the cohort, a pre-pregnancy rate of 0.7 relapses per year decreased to 0.2 per year in the third trimester. The relapse rate increased to 1.2 per year in the first 3 months postpartum. However, 72% of women did not experience any relapses during the study period. An increased relapse rate in the year before pregnancy, an increased relapse rate during pregnancy, and a higher EDSS score at the beginning of pregnancy correlated significantly with occurrence of a postpartum relapse. Epidural anesthesia and breastfeeding were not predictive of a subsequent relapse or disability progression. There are no contraindications to caesarean section or vaginal delivery in MS patients.
Prognosis




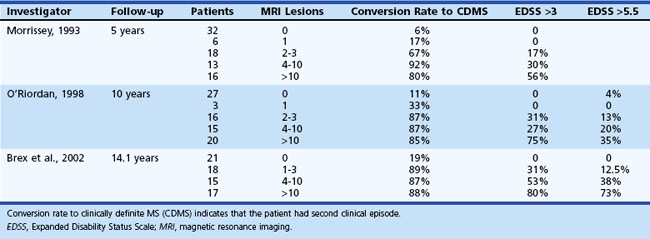
In general, patients with mild disease (EDSS score 0-3) 5 years after diagnosis only uncommonly progress to severe disease (EDSS score 6) by 10 years (7.5% of patients) and 15 years (11.5% of patients). Entities such as Devic disease, Baló concentric sclerosis, and particularly Marburg disease are more fulminant variants of MS, with early disability and even death. The risk of a second demyelinating episode (CDMS) after an initial event is detailed in Table 54.4.
Diagnostic Studies
Neuroimaging
Magnetic Resonance Imaging
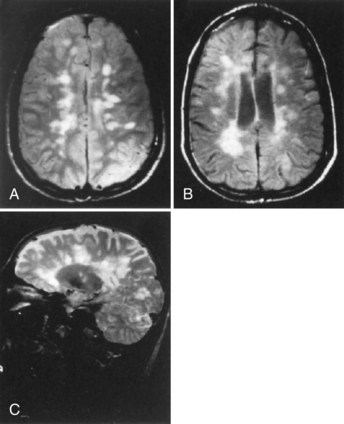
MRI is the preferred imaging modality for both making the diagnosis of MS and longitudinal follow-up of patients. MRI is based on relaxation properties of water in tissues and is sensitive to T2 (transverse) and T1 (longitudinal) relaxation rates of protons. Gadolinium diethylenetriamine pentaacetic acid (DTPA) is a paramagnetic contrast agent. It crosses the disrupted BBB, indicating increased vascular permeability in association with inflammation. Gadolinium (Gd) enhancement is best seen on spin-echo T1-weighted imaging. These fundamental properties of MR image acquisition provide a sensitive measure of pathology; areas of brain inflammation, demyelination, and loss of axons can be especially well seen with this technique. More than 95% of patients with MS have T2 and fluid-attenuated inversion recovery (FLAIR) abnormalities (Fig. 54.9). New lesions occur many times more often than new clinical attacks in RRMS. In the brain, typical lesions are located in the periventricular white matter, in and near the corpus callosum, the deep white matter, the centrum semiovale, as well as in cortical and deep gray-matter structures. Juxtacortical lesions affecting U-fibers are commonly seen in MS. Posterior optic radiation lesions are also frequently present. Posterior fossa lesions are present in the cerebellum, middle cerebral peduncles, and in areas adjacent to the fourth ventricle.
Role of Mri in Multiple Sclerosis Diagnostic Criteria
 One Gd-enhancing lesion or nine T2-hyperintense lesions.
One Gd-enhancing lesion or nine T2-hyperintense lesions.
 At least one infratentorial lesion.
At least one infratentorial lesion.


In 2005, these criteria were revised. Dissemination in space criteria were changed to allow any cord lesions to be substituted for a brain lesion. A cord lesion now carries the same weight as an infratentorial lesion. In addition, a new T2 lesion seen more than 30 days after a clinical event, and a new Gd-enhancing lesion seen more than 3 months after a clinical event could satisfy dissemination in time criteria (see Table 54.4).
Predictive Value Of Mri In Conversion To Clinically Definite Multiple Sclerosis
Studies suggest that MRI may provide some prediction of the risk of conversion to CDMS based on the presence and number of T2 lesions within 1 to 5 years following CIS. Increasingly higher numbers of T2 lesions result in greater likelihood of conversion to CDMS and higher disability scores at 5 years. Two long-term follow-up studies of 10 to 14 years show that most patients with CIS and MRI abnormalities will develop CDMS (70%-80%), while only 20% of patients with normal cerebral MRI scans will be diagnosed with MS over this time period (Brex et al., 2002; Optic Neuritis Study Group, 2003; O’Riordan et al., 1998). The concept of radiologically isolated syndrome was introduced recently (Bourdette, 2009; Lebrun, 2009; Miller, 2008) and describes a subset of individuals with no clinical symptoms or neurological events, but with imaging findings on MRI suggestive of demyelination. The reported rate of conversion to CIS in this group was 33% in 5 years. This finding may well pioneer a concept of “preclinical” MS (Okuda et al., 2009). A recent retrospective analysis of patients with RIS determined that the presence of asymptomatic spinal cord lesions carries a higher risk of conversion to CIS within 1 year (88% of patients in this category converted), and this risk is independent of the number or location of brain lesions. The utility of treating this group of patients with immunomodulating therapies—or indeed, our ability to predict and time the onset of clinical disease—may have unpredictable ethical implications. Therefore, caution should be exercised in offering brain imaging to asymptomatic individuals until further information about managing this group becomes available.
Predictive Value Of Conventional Mri Metrics In Multiple Sclerosis Progression
It is currently not possible to reliably distinguish MS subtype based on conventional MRI metrics. Few differences are appreciated in MRI T2 lesion load and volume between CIS patients and RRMS patients, although patients with benign disease have a lower rate of brain atrophy than patients with early MS (Gauthier, 2009). SPMS patients tend to have greater reduction in brain volume and diffuse changes in normal-appearing white matter (NAWM) (Bakshi, 2008). PPMS patients have fewer and smaller size lesions in the brain and greater numbers of T2 lesions in the spine, coupled with more spinal cord atrophy. Newer MRI techniques may hold promise in understanding specific imaging characteristics of MS subsets and of the clinical/MRI paradox (Fig. 54.10).
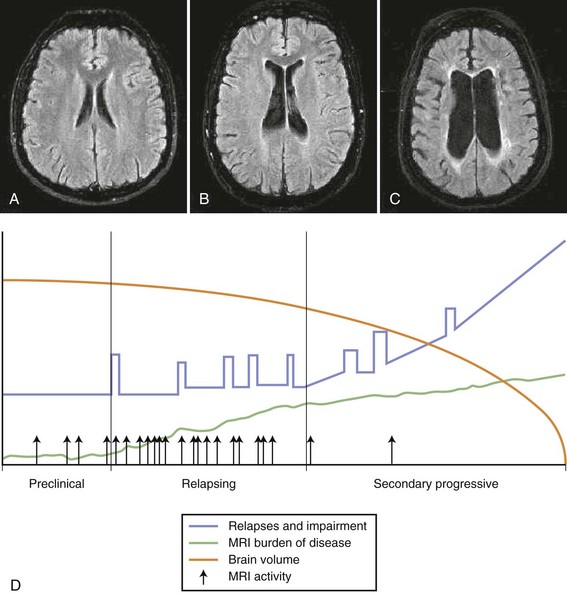
New Imaging Techniques and Approaches
Magnetic Resonance Spectroscopy
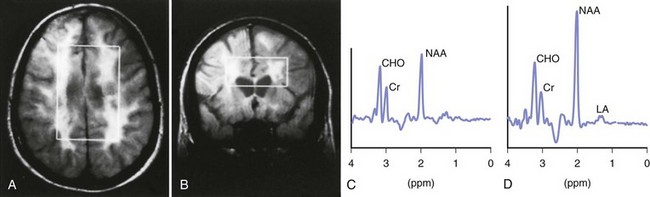
Magnetic resonance spectroscopy (MRS) is a tool that derives MRI signal from multiple metabolites. Many brain metabolite concentrations are not normal in MS patients, change over time, and can be measured with this technique. For example, N-acetylaspartate (NAA) is a marker of neuronal metabolism that is decreased in MS patients, suggesting a loss of neuronal integrity (Fig. 54.11). A high choline (Cho) peak is indicative of an increase in membrane turnover, as can be seen in demyelination and remyelination. Increased creatine (Cr) usually indicates an increase in cell density. Increased myoinositol (mI) indicates an increase in the number of glial cells. MRS gives insight into the biochemical composition of MS lesions and NAWM. In active, newly formed MS plaques, NAA is slightly decreased and Cre, Cho, and mI are increased. Lactate (Lac) peak is also increased, suggesting energy failure. Over time, the initial surge in lipids, Cho, Lac, and Cre may return to near normal. This suggests some degree of recovery. In contrast to active plaques, chronic MS lesions show considerable reduction in NAA, normal glutamate peaks, and elevation in mI. Interestingly, NAWM MRS patterns are quite similar to active acute MS plaques, and these changes can be detected some months before appearance of active and T2 bright lesions. MRS may also show changes in the earliest stages of MS. Patients with CIS have abnormalities in both NAWM and gray matter. Studies showed an early increase in mI signal even when the other metabolites remain normal. MRS measures are more robust in predicting cognitive and physical disability in MS patients in small research trials. (Bonneville, 2002; De Stefano, 1998; Mathiesen, 2006). Clinical utility of this technique, however, is limited owing to inconsistent reproducibility and lack of standardization of this tool among various platforms.
Cerebrospinal Fluid Analysis
CSF findings alone neither make nor exclude the diagnosis of MS, but they remain important in atypical clinical syndromes, atypical or nondiagnostic MRI findings, or unusual clinical manifestations such as a course of progressive neurological impairment without history of relapses. CSF does not show any gross abnormalities in MS; it is clear, colorless, and under normal pressure. Cell counts are typically normal but may be slightly elevated in 15% to 20% of patients. The predominant cells are T lymphocytes. Significant pleocytosis should raise suspicion of another etiology. Although CSF protein is also usually normal, it is common to see mild elevations of albumin in 20% to 30% of MS patients. Determining the presence of OCBs is the most important diagnostic test. Isoelectric focusing followed by immunoblot is the preferred test for OCB detection. These bands represent excess antibody produced by one or more clones of plasma cells. The pattern of banding remains relatively stable in an individual patient throughout the course of the disease. However, 20% to 30% of patients with confirmed CDMS do not have OCBs at any given point in time. Presence of OCBs in a patient with CIS predicts a higher rate of conversion to CDMS and may be used to convince a reluctant patient to start disease-modifying therapy. Myelin basic protein (MBP) in the CSF is a marker of tissue damage and has been used as a measure of CNS myelin breakdown. The levels of the protein may be quite increased in MS patients, but the specificity of these findings is not known. An abnormality in CSF IgG production (as measured by the IgG index, or as a percentage of total protein or albumin) is found in over 90% of patients with confirmed MS (Table 54.5).

Evoked Potentials
EPs are CNS electrical events generated by peripheral stimulation of a sensory organ and are useful to determine abnormal function that may be clinically unapparent. Detecting a subclinical lesion at a site remote from the region of clinical dysfunction supports the multifocal disease portion of the diagnostic criteria for MS. The three most commonly used EPs are VEPs, somatosensory evoked potentials (SSEPs), and brainstem auditory-evoked responses (BAER). The role of EPs in MS was recently reviewed, and only VEPs were thought to be useful to determine increased risk for MS (Gronseth, 2000). Using pattern shift VEPs, abnormalities (P100 wave prolongation) are detected in over 90% of patients with a history of optic neuritis, even in a setting of complete restoration of vision (Table 54.6).

Variants of Multiple Sclerosis
Devic Disease (Neuromyelitis Optica)
A combination of bilateral optic neuropathy and myelopathy characterize Devic disease, or neuromyelitis optica, which many authorities now classify as a separate entity rather than a variant of MS. The myelopathy tends to be more severe than typically occurs with MS and carries less likelihood of recovery. The neuropathological features at autopsy are those of a much more severe necrotic lesion of the cord. In some patients, the optic neuropathy and the myelopathy occur at the same time; in others one or the other component is delayed. Antibody to the aquaporin-4 water channel is present at high frequency in patients with the clinical characteristics of Devic disease (Weinshenker et al., 2006). Further recent work has expanded the clinical spectrum of neuromyelitis optica beyond the typical presentation (Wingerchuk et al., 2006).
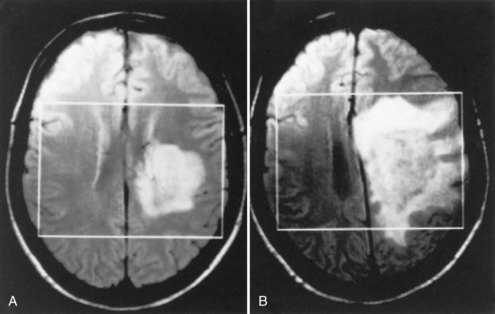
Acute Tumor-like Multiple Sclerosis (Tumefactive Multiple Sclerosis)
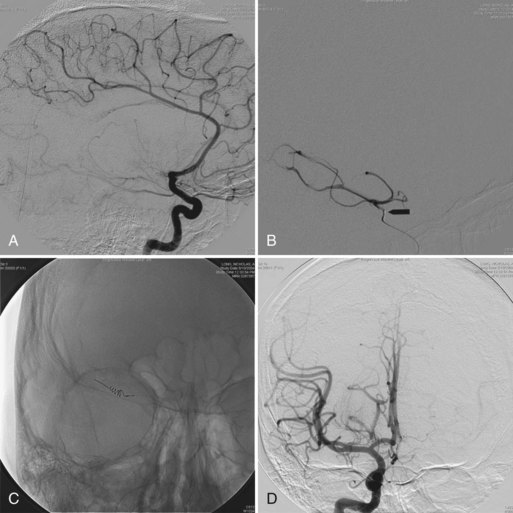
Some patients with demyelinating disease present with a large acute lesion of one hemisphere (Fig. 54.12) or rarely in other locations such as the spinal cord. Mass effect may occur, with compression of the lateral ventricle and shift across the midline. The clinical abnormalities in such patients are variable. They may be slight, even in a patient with a massive lesion, whereas confusion, hemiparesis, or neglect syndrome may be seen in another patient with a lesion that appears no different. Much of the T2 bright lesion volume is often due to edema and may be rapidly responsive to corticosteroids. (This change with corticosteroids also may occur with glioma or CNS lymphoma and is therefore not a useful diagnostic criterion.) Biopsy may be necessary to establish a correct diagnosis.
Treatment and Management
 Relief or modification of symptoms.
Relief or modification of symptoms.
 Shortening the duration or limiting the residual effects of an acute relapse.
Shortening the duration or limiting the residual effects of an acute relapse.
 Reducing the frequency of relapses.
Reducing the frequency of relapses.


Symptomatic Treatment
Cognitive Impairment
Treatment of cognitive impairment is discussed in the section on cognition earlier in this chapter.
Treatment Strategies
For some patients, MS is a disease with one or two acute neurological episodes with no further evidence of disease activity. In others, it is a chronic relapsing or progressive disease with an unpredictable clinical course that generally spans 20 to 30 years, during which time neurological disability accumulates. Treatment of MS, as with other diseases, is now based on the results of prospective well-controlled clinical trials. Most of these trials have been designed to establish efficacy but have not followed patients in controlled fashion for longer than 2 or 3 years, and have therefore only given hints about long-term results of treatment. Long-term placebo-controlled trials are not ethically possible once efficacy is established. Patients in clinical practice may differ markedly from those who have been treated in clinical trials, yet therapeutic decisions must be made, and these trials provide best evidence. Table 54.7 outlines one of the possible treatment paradigms.
| Disease Course/Stage | Treatment Options | Evidence |
|---|---|---|
| Monosymptomatic (e.g., optic neuritis)—acute attack | IV methylprednisolone, 1000 mg for 5 days, without oral taper | Class I evidence |
| Relapsing-remitting, no disease activity for several years, and/or no activity on MRI | IV corticosteroids if acute attack occurs | Class I evidence |
| Relapsing-remitting, current disease activity, and/or activity on MRI | IV corticosteroids for acute attacks, plus for prevention (1) interferon beta-1a (Avonex), 30 µg IM weekly; or (2) interferon beta-1b (Betaseron), 250 µg SQ every other day; or (3) interferonbeta-1a (Rebif), 22 or 44 µg SQ three times/week; or (4) glatiramer acetate (Copaxone), 20 mg SQ daily | Class I evidence for interferon beta-1a SQ and IM, interferon beta-1b, and glatiramer acetate. All four are FDA approved. |
| Relapsing-remitting, disease activity while on interferon or glatiramer acetate, or intolerance to either agent | Switch to natalizumab | Class I evidence |
| Relapsing-remitting, accumulating disability while on (interferon/Copaxone) | Switch to natalizumab or mitoxantrone | Class I evidence for mitoxantrone, no studies for natalizumab in this population |
| Rapidly progressing disability | Switch or add mitoxantrone | |
| Secondary progressive | Mitoxantrone | Class I and II evidence |
| Primary progressive | No established therapy |
IM, Intramuscular; IV, intravenous; MRI, magnetic resonance imaging; SQ, subcutaneous.
Disease-Modifying Treatments
Currently Available Immunomodulatory Treatments
Interferons
A second double-blind placebo-controlled study in 301 patients with relapsing-remitting disease investigated the efficacy of weekly IM injections of 6 MIU (30 µg) of interferon beta-1a (Avonex), a glycosylated recombinant interferon beta (Jacobs et al., 1996). Over 2 years, the annual exacerbation rate decreased by 29%. MRI data revealed significantly decreased T2 lesion volume and number of enhancing lesions over the 2 years of the study. There was also a 37% reduction in progression of neurological disability in the treatment group.
Another randomized double-blind placebo-controlled study of interferon beta-1a in higher doses was conducted in Europe and Canada (PRISMS Study Group, 1998). This involved 560 patients with relapsing-remitting disease given subcutaneous interferon beta-1a (Rebif). Patients were randomized to placebo, 22 µg, or 44 µg of interferon beta-1a 3 times a week for 2 years. There was a 27% reduction in the relapse rate in the group receiving 66 µg/week and a 33% reduction in the group receiving 132 µg/week. There was also a significant reduction in disability. The MRI lesion burden showed a decrease of 1.2% in the group receiving 66 µg/week, a decrease of 3.8% in the group receiving 132 µg/week, and an increase of 10.9% in the group receiving a placebo. Based on this data and a comparison trial of weekly IM interferon beta-1a versus the thrice-weekly 44 µg subcutaneous dose (Panitch et al., 2002), the latter was approved by the FDA in March 2002.
Both the EVIDENCE trial and a trial in Europe comparing high- and low-dose interferon (INCOMIN) (Durelli et al., 2002) were short-term trials and did not address the important question of which of these agents, if any, truly suppresses the development of long-term disability. It does appear, however, that there is a detectable dose-response in the use of beta interferons.
Synthetic Polymer
Glatiramer acetate (GA) (Copaxone) is a synthetic polypeptide administered by daily subcutaneous injection. In a large double-blind trial in RRMS involving 251 randomized patients (Johnson et al., 1995), the patients receiving GA had a 29% reduction in the relapse rate over 2 years. Extension data show that over 140 weeks, 41% of patients receiving placebo experienced worsening of their disability by 1.5 EDSS steps or greater, whereas only 21.6% of GA-treated patients had worsening (Johnson et al., 1998). There were also modest MRI effects, with an observed decrease in new T2 lesion load, enhancing lesions, and T1 hypointense lesions (T1 black holes.) The mechanism by which GA may work in humans is unknown but may relate to interference with antigen presentation and induction of regulatory cells (TH2) that traffic to the CNS and induce bystander suppression of immune responses.
Monoclonal Antibody
The latest agent to be approved for relapsing forms of MS is natalizumab (Tysabri), a monoclonal antibody directed against the adhesion molecule, α4-integrin. Blocking this molecule inhibits trafficking of lymphocytes from the blood into the CNS. Two well-designed trials led to FDA approval of this agent. In a placebo-controlled clinical trial (AFFIRM), natalizumab reduced relapse rate by 68% and progression of disability by 42% over 2 years (Polman et al., 2006). MRI metrics were similarly affected. In a second study (SENTINEL), significant improvements in both clinical and MRI outcomes were seen when subjects who had had an exacerbation while on interferon beta-1a once weekly were treated concomitantly with natalizumab (an add-on study), as compared to those who received a placebo add-on (Rudick et al., 2006). During the studies, the safety profile was quite good; however, during the extension phase, two cases of progressive multifocal leukoencephalopathy (PML) occurred in subjects receiving both natalizumab and interferon (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005). A third case of PML occurred in a subject who had received natalizumab in a Crohn disease study. This led to withdrawal of natalizumab from the market until July 2006, when it was reintroduced with an elaborate risk management program to try to better determine the risks of this agent and monitor its use. Enrollment in a TOUCH (Tysabri Outreach: Unified Commitment to Health) prescribing program is mandatory for those patients who are receiving natalizumab. This program is designed to monitor monthly for new neurological symptoms that may be concerning for PML. In addition, twice-yearly brain MRI screen for PML is suggested. At present, natalizumab is recommended as second-line therapy for individuals with relapsing forms of MS who have failed or not tolerated agents such as interferon or glatiramer acetate.
Treatment Considerations
The availability of disease-modifying agents makes MS one of the more unique neurological diseases. Although these therapies are only partially effective, they provide considerable benefit. As a general rule, all patients with active relapsing forms of MS should be receiving one of the immunomodulatory agents indefinitely. Substantial progress in improving on current regimens is expected in the near future. Table 54.8 outlines emerging oral and IV disease-modifying therapies.
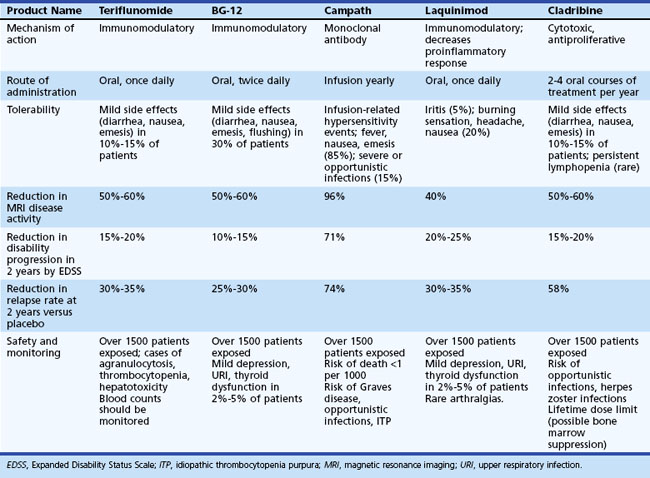
Emerging Therapies
Oral fumarate/BG-12 induces apoptosis of activated T cells. It down-regulates intracellular adhesion molecules and VCAM expression and induces antiinflammatory cytokines (Kappos et al., 2004.) Fumarate is approved for treatment of severe refractory psoriasis in several European countries. Phase II studies in RRMS have been completed and show 69% reduction in new Gd-enhancing and T2 lesions in the high-dose group. It has an acceptable safety profile. A phase III study comparing low- and high-dose BG-12 versus placebo versus Copaxone is ongoing. Over 1000 patients are enrolled, the 2-year annualized relapse rate is a primary endpoint, and results are expected in 2011.
Treatment of Progressive Disease
The most successful trial for progressing patients was performed in Europe using the chemotherapeutic agent, mitoxantrone (Novantrone), which produced a reduction in the progression of disability and a considerable decrease in relapse rate in patients with worsening RRMS, PPMS, and SPMS over a 2-year period (Hartung et al., 2002). Improvement in MRI measures of disease was also demonstrated. At 3 years, an appreciable benefit was still evident, even though the dosing had been stopped at 2 years. The toxicity profile was acceptable, but the agent has dose-related cardiac toxicity that limits lifetime dosing to 140 mg/m2. Other side effects include nausea, alopecia, and neutropenia. Secondary leukemias have been uncommonly reported.
IVIG, studied in a large European trial of SPMS, demonstrated no beneficial effect on any clinical outcome and little effect on MRI metrics (Hommes et al., 2004).
Interferon beta-1b has been used in two studies of SPMS. The first was a European multicenter controlled trial of interferon beta-1b in 718 patients with SPMS treated with either interferon beta-1b, 8 MIU subcutaneously every other day, or placebo. The study was planned for 3 years but was stopped after enrollees had completed 2 years because an interim analysis demonstrated efficacy (Kappos et al., 1998). Analysis of this study revealed that patients treated with interferon beta-1b had significant delays in progression of disability. On average, this delay amounted to 9 to 12 months. Similarly, patients receiving interferon beta-1b had a delay in time to becoming wheelchair bound of about 9 months, compared to placebo patients. Similar results were seen for relapse rate reduction, MRI analyses, hospitalizations, and steroid use. A second study of interferon beta-1b was performed in North America. In this study, SPMS patients were treated with placebo, interferon beta-1b, 8 MIU every other day, or interferon beta-1b 5 MIU/m2 every other day (this group had an average dose of 9.6 MIU). As opposed to the European study, in this study showed no effect on the primary outcome assessment of reduction in time to worsening disability was found (Kappos et al., 2004). Other outcome measures such as reduction in relapse rate, MRI activity, and lesion load favored the treated groups. Further analysis of these two studies suggested that the marked difference in their outcomes might relate in part to differences in the degree of MS disease activity between the groups. Although the entrance criteria were very similar, the actual groups differed in several ways (Kappos et al., 2004). This highlights the difficulty in trying to compare results across studies, even those that attempt to evaluate the same problem with the same type of subject.
Another European/Canadian study (King et al., 2001) of high doses of interferon beta-1a (Rebif) failed to achieve its primary outcome measure (time to confirmed worsening of EDSS). Other outcome measures such as relapse rate and MRI measures were favorable.
A study with interferon beta-1a (Avonex) dosed at 60 mg weekly (twice the standard dose of this agent in RRMS) succeeded in achieving its primary outcome measure of a significant benefit as measured by the MSFC (Cohen et al., 2002). There was also benefit on relapse rate and MRI metrics. However, no benefit was seen on time to disability as measured by EDSS; therefore, this agent did not receive regulatory approval to treat SPMS.
Acute Disseminated Encephalomyelitis
History
Acute disseminated encephalomyelitis (ADEM) is a demyelinating syndrome that often occurs in association with an immunization or vaccination (postvaccination encephalomyelitis) or systemic viral infection (parainfectious encephalomyelitis). This disorder is almost always monophasic. Recurrent cases have been described, but their nature and distinction from MS is unclear. ADEM is characterized pathologically by perivascular inflammation, edema, and demyelination within the CNS, and clinically by rapid development of focal or multifocal neurological dysfunction. The most precise clinical and pathological observations regarding ADEM are derived from case studies in which there has been a close link between the specific viral infection or vaccine and the syndrome. The syndromes arising after acute measles infection or rabies vaccine administration can be considered the prototypes of the illness (Box 54.4).
Box 54.4 Acute Disseminated Encephalomyelitis and Related Disorders
Site-Restricted Forms of Monophasic Acute Inflammatory Demyelinating Disorders That May Occur after Viral Illness or Vaccination
Laboratory Features
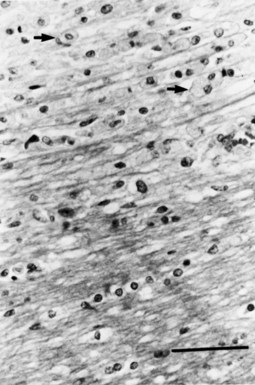
The hallmark lesions of ADEM are perivascular inflammation and surrounding demyelination within the CNS (Figs. 54.13 and 54.14). Vessel necrosis is frequently observed. The demyelinating aspect may be minimal or widespread, with coalescence of the multiple lesions. Some meningeal reaction may also be apparent. Reports of MRI studies, largely from apparent sporadic cases of ADEM, describe multifocal CNS lesions initially indistinguishable from those observed in MS. A feature of ADEM is that the majority of the T2 lesions enhance, suggesting they were of recent onset, consistent with a monophasic illness. Unlike MS, in ADEM, after several weeks, lesions show at least partial resolution without the appearance of new lesions. In some cases lesions can persist. The usual CSF formula is normal pressure, little or modest increase in cell count (<100 cells/mL), and a moderate increase in protein. Well-documented cases exist with totally normal CSF. Cases with high cell counts, including some polymorphonuclear cells and high protein values, occur and seemingly reflect a more necrotizing disease process. The high counts usually return to normal within a few days. The CSF Ig content is not usually increased, and OCB patterns occur less commonly than in MS.
1. An initial episode of what will prove to be MS (Schwarz et al., 2001). The presence of increased levels of IgG in the CSF may favor the diagnosis of MS; follow-up MRI may be needed because initial MRI scans may appear similar in the two diseases. The occurrence of a nonspecific viral illness before the onset of the clinical neurological syndrome does not discriminate between MS and ADEM because the incidence of exacerbations of MS is increased following such infections.
2. CNS vasculitis with or without systemic features (such as disseminated intravascular coagulation or serum sickness).
3. Multiple cerebral infarcts, particularly emboli from infected cardiac valves.
4. Chronic meningitis or granulomatous disease (sarcoidosis). If the main clinical feature is unifocal encephalitis, abscess or tumor has to be excluded.
Other Inflammatory Demyelinating Diseases of the Central Nervous System
Acute Hemorrhagic Leukoencephalitis
The differential diagnosis of this syndrome includes entities that present as rapidly evolving focal cerebral disorders with fever and obtundation, such as brain abscess and herpes simplex encephalitis, in addition to those syndromes considered in the section on ADEM.
Achiron A., Barak Y. Cognitive impairment in probable multiple sclerosis. Neurol Neurosurg Psychiatry. 2003;74:443-446.
Amato M.P., Portaccio M., Stromillo M.L., et al. Cognitive assessment and quantitative magnetic resonance metrics can help to identify benign multiple sclerosis. Neurology. 2008;71:632-638.
Arnett P., Ben-Zacharia A., Benedict R., et al. The Goldman Consensus statement on depression in multiple sclerosis. Mult Scler. 2005;11:328-337.
Ascherio A., Munger K.L., Lennette E.T., et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA. 2001;286(24):3083-3088.
Bakshi R., Ariyaratana S., Benedict R.H.B., et al. Fluid-attenuated inversion recovery magnetic resonance imaging detects cortical and juxtacortical multiple sclerosis lesions. Arch Neurol. 2001;58:742-748.
Benedict R.H.B., Fischer J.S., Archibald C.J, et al. Minimal neuropsychological assessment of MS patients: a consensus approach. Clin Neuropsychol. 2002;16:381-397.
Benedict R.H.B., Munschauer F., Zarevics P., et al. Effects of l-amphetamine sulfate on cognitive function in multiple sclerosis patients. J Neurol. 2008;255:848-852.
Bo L., Vedeler C.A., Nyland H., et al. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9:323-331.
Brex P.A., Ciccarelli O., O’Riordan J.I., et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346:158-164. 2002
Bronnum-Hansen H., Koch-Henriksen N., Stenager E. Trends in survival and cause of death in Danish patients with multiple sclerosis. Brain. 2004;127:844-850.
Bronnum-Hansen H., Stenager E., Nylev S.E., et al. Suicide among Danes with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2005;76:1457-1459.
Calabrese M., Rocca M.A., Atzori M., et al. A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol. 2010;67:376-383.
Charil A., Yousry T.A., Rovaris M., et al. MRI and the diagnosis of multiple sclerosis: expanding the concept of “no better explanation.”. Lancet Neurol. 2006;5:841-852.
Cohen J.A., Cutter G.R., Fischer J.S., et al. Benefit of interferon beta-1a on MSFC progression in secondary progressive MS. Neurology. 2002;59:679-687.
Conlon P., Oksenberg J.R., Zhang J., et al. The immunobiology of multiple sclerosis: an autoimmune disease of the central nervous system. Neurobiol Dis. 1999;6:149-166.
Cutter G.R., Baier M.L., Rudick R.A., et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain. 1999;122:871-882.
De Jager P.L., Chibnik L.B., Cui J., et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol. 2009;8(12):1111-1119.
Durelli L., Verdun E., Barbero P., et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet. 2002;359:1453-1460.
Dyment D.A., Ebers G.C., Sadovnick A.D. Genetics of multiple sclerosis. Lancet Neurol. 2004;3:104-110.
Ebers G.C., Sadovnick A.D., Risch N.J. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature. 1995;377:105-106.
Fischer J.S., Priore R.L., Jacobs L.D., et al. Neuropsychological effects of interferon b-1a in relapsing multiple sclerosis. Ann Neurol. 2000;48:885-892.
Glanz B.I., Healy B.C., Rintell D.J. The association between cognitive impairment and quality of life in patients with early multiple sclerosis. J Neurol Sci. 2010;290:75-79.
Glanz B.I., Holland C.M, Gautheir S.A. Cognitive dysfunction in patients with clinically isolated syndromes or newly diagnosed multiple sclerosis. Mult Scler. 2007;13(8):104-110.
Greene Y.M., Tariot P.N., Wishart H., et al. A 12-week, open trial of donepezil hydrochloride in patients with multiple sclerosis and associated cognitive impairments. J Clin Psychopharmacol. 2000;20:350-356.
Hafler D.A., Slavik J.M., Anderson D.E., et al. Multiple sclerosis. Immunol Rev. 2005;204:208-231.
Hartung H.P., Gonsette R., Konig N., et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multi-centre trial. Lancet. 2002;360:2018-2025.
Hommes O.R., Sorensen P.S., Fazekas F., et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo-controlled trial. Lancet. 2004;364:1149-1156.
Houtchens M.K., Benedict R.H.B., Killiany R., et al. Thalamic atrophy and cognition in multiple sclerosis. Neurology. 2007;69:1213-1223.
Jacobs L.D., Cookfair D.L., Rudick R.A., et alfor the Multiple Sclerosis Collaborative Research Group (MSCRG). Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285-294.
Johnson K.P., Brooks B.R., Cohen J.A., et alfor the Copolymer 1 Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology. 1995;45:1268-1276.
Johnson K.P., Brooks B.R., Cohen J.A., et alfor the Copolymer 1 Multiple Sclerosis Study Group. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Neurology. 1998;50:701-708.
Kantarci O., Wingerchuk D. Epidemiology and natural history of multiple sclerosis: new insights. Curr Opin Neurol. 2006;19:248-254.
Kappos L., Polman C., Pozzilli C., et al. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet. 1998;352:1491-1497.
Kappos L., Weinshenker B., Pozzilli C., et al. Interferon beta-1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63:1779-1787.
King J., McLeod J., Gonsette R.E., et al. Randomized controlled trial of interferon beta-1a in secondary progressive MS—clinical results. Neurology. 2001;56:1496-1504.
Kleinschmidt-DeMasters B.K., Tyler K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369-374.
Kutzelnigg A., Lucchinetti C.F., Stadelmann C., et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705-2712.
Langer-Gould A., Atlas S.W., Green A.J., et al. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375-381.
Lublin F.D., Reingold S.C. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907-911.
Lucchinetti C., Bruck W., Parisi J., et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707-717.
Lucchinetti C.F., Mandler R.N., McGavern D., et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125:1450-1461.
Mesaros S., Rocca M.A., Riccitelli G., et al. Corpus callosum damage and cognitive dysfunction in benign MS. Hum Brain Mapp. 2009;30:2656-2666.
Montalban X., Tintoré M., Swanton J., et al. MRI criteria for MS in patients with clinically isolated syndromes. Neurology. 2010;74:427-434.
Morrow S.A., Kaushik T., Zarevics P., et al. The effects of l-amphetamine sulfate on cognition in MS patients: results of a randomized controlled trial. J Neurol. 2009;256:1195-1202.
Nocentini U., Pasqualetti P., Bonavita S., et al. Cognitive dysfunction in patients with relapsing-remitting multiple sclerosis. Mult Scler. 2006;12:77-87.
Noseworthy J.H., Lucchinetti C., Rodriguez M., et al. Multiple sclerosis. N Engl J Med. 2000;343:938-952.
Oksenberg J.R., Barcellos L.F., Cree B.A. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet. 2004;74(1):160-167.
Okuda D.T., Mowry E.M., Beheshtian A., et al. Incidental MRI anomalies suggestive of multiple sclerosis. The radiologically isolated syndrome. Neurology. 2009;72:800-805.
Panitch H., Goodin D.S., Francis G., et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS—the EVIDENCE trial. Neurology. 2002;59:1496-1506.
Patrikios P., Stadelmann C., Kutzelnigg A., et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165-3172.
Patti F., Amato M.P., Bastianello S., et al. Effects of immunomodulatory treatment with subcutaneous interferon beta-1a on cognitive decline in mildly disabled patients with relapsing-remitting multiple sclerosis. Mult Scler. 2010;16:68-77.
Patti F., Failla G., Ciancio M.R., et al. Neuropsychological, neuroradiological and clinical findings in multiple sclerosis. A 3 year follow-up study. Eur J Neurol. 1995;5:283-286.
Patti F., Failla G., Ciancio M.R., et al. Neuropsychological, neuroradiological and clinical findings in multiple sclerosis. A 3 year follow-up study. Eur J Neurol. 1998;5(3):283-286.
Penny S., Khaleeli Z., Cipolotti L., et al. Early imaging predicts later cognitive impairment in primary progressive multiple sclerosis. Neurology. 2010;74:545-552.
Pittock S.J., Mayr W.T., McClelland R.L., et al. Change in MS-related disability in a population-based cohort: a 10-year follow-up study. Neurology. 2004;62:51-59.
Polman C.H., O’Connor P.W., Havrdova E., et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899-910.
Polman C.H., Reingold S.C., Edan G., et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald criteria.”. Ann Neurol. 2005;58:840-846.
Portaccio E., Stromillo M.L., Goretti B., et al. Neuropsychological and MRI measures predict short-term evolution in benign multiple sclerosis. Neurology. 2009;73:498-503.
Prineas J.W., Kwon E.E., Cho E.S., et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50:646-657.
PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498-1504.
Rao S.M., Leo G.J., Bernardin L., et al. Cognitive dysfunction in multiple sclerosis. Neurology. 1991;41:685-691.
Rao S.M., Leo G.J., Haughton V.M., et al. Correlation of magnetic resonance imaging with neuropsychological testing in multiple sclerosis. Neurology. 1989;39(2):161.
Rovaris M., Barkhof F., Calabrese M., et al. MRI features of benign MS. Neurology. 2009;72:1693-1701.
Rudick R.A., Stuart W.H., Calabresi P.A., et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911-923.
Schwarz S., Mohr A., Knauth M., et al. Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology. 2001;56:1313-1318.
Sperling R.A., Guttman C.R., Hohol M.J., et al. Regional magnetic resonance imaging lesion burden and cognitive function in multiple sclerosis: a longitudinal study. Arch Neurol. 2001;58:115-121.
Staff N.P., Lucchinetti C.F., Keegan B.M. Multiple sclerosis with predominant, severe cognitive impairment. Arch Neurol. 2009;69:1139-1143.
Tintore M., Rovira A., Rio J., et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology. 2006;67:968-972.
Tremlett H., Paty D., Devonshire V. Disability progression in multiple sclerosis is slower than previously reported. Neurology. 2006;66:172-177.
Ukkonen M., Vahvelainen T., Hamalainen P. Cognitive dysfunction in primary progressive multiple sclerosis: a neuropsychological and MRI study. Mult Scler. 2009;15:1055-1061.
Vukusic S., Confavreux C. Pregnancy and multiple sclerosis: the children of PRIMS. Clin Neurol Neurosurg. 2006;108:266-270.
Weinshenker B.G., Bass B., Rice G.P.A., et al. The natural history of multiple sclerosis: a geographically based study. Brain. 1989;112(1):133-146.
Weinshenker B.G., Wingerchuk D.M., Pittock S.J., et al. NMO-IgG: a specific biomarker for neuromyelitis optica. Dis Markers. 2006;22:197-206.
Willer C.J., Dyment D.A., Risch N.J., et al. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:12877-12882.
Wingerchuk D.M., Lennon V.A., Pittock S.J., et al. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485-1489.
Zivadinov R., Minagarb A. Evidence for gray matter pathology in multiple sclerosis: a neuroimaging approach. J Neurol Sci. 2009;282:1-4.
Zuvich R.L., McCauley J.L., Oksenberg J.R., et al. Genetic variation in the IL7RA/IL7 pathway increases multiple sclerosis susceptibility. Hum Genet. 2010;127(5):525-535.