Chapter 73 Disorders of Bones, Joints, Ligaments, and Meninges
The bones, joints, ligaments, and meninges that support and protect the tissues of the nervous system can give rise to numerous illnesses that affect the nervous system. These disorders sometimes border on other medical disciplines unfamiliar to the neurologist and hence can be enigmatic or difficult to diagnose. They may involve cognitive functions, disrupt cerebrospinal fluid (CSF) flow dynamics, or slowly compress and distort central and peripheral neural structures. They have a mixture of genetic, developmental, traumatic, degenerative, infectious, and inflammatory mechanisms. For the clinical neurologist, awareness greatly strengthens diagnostic skill, so this chapter considers many of these disorders. Chapters of overlapping interest include Chapters 24, 50C, 51E, and 60. For an expanded version of this chapter that includes figures and boxes marked “online only,” please visit www.expertconsult.com.
Heritable Disorders of Connective Tissue
Heritable disorders involving the major connective tissues of the body are among the most common human genetic diseases. We will focus on those disorders that have neurological impact (Box 73.1). Many of these are being better understood at a molecular level: osteogenesis imperfecta, Ehlers-Danlos syndrome (EDS), chondrodysplasias, Marfan syndrome, epidermolysis bullosa, and Alport syndrome to name a few.
Box 73.1 Classification of Hereditary Disorders of Connective Tissue
Skeletal, Cardiovascular, and Eye (Type I and Type III Collagen, Fibrillin, Elastin)
Classifications of heritable disorders of connective tissue can rely on pattern of inheritance, clinical features, anatomical pattern, or known molecular defects. To date, genetic or molecular classification is not always helpful clinically because there are significant genotype-to-phenotype inconsistencies, testing is not widely available, and our understanding of many disorders is incomplete. Connective tissues contain a wide range of complex macromolecules assembled in the extracellular matrix. There are at least 28 different types of collagen in bone, skin cartilage, tendons, and ligaments. Other molecules including fibrillin, elastin, proteoglycans, fibronectin, hyaluronate, osteonectin, and osteocalcin contribute to tensile strength and elasticity. Forces that control the three-dimensional organization of these components remain unknown. In growth and development, collagen fibrils in all these supporting tissues undergo repeated synthesis, degradation, and resynthesis. Nutrition, gravitational forces, trauma, other physical stresses, endocrine factors (such a glucocorticoids), and inflammation all modify these tissues (Prockop and Czarny-Ratajczak, 2008).
Osteogenesis Imperfecta
The various types of osteogenesis imperfecta (incidence approximately 1 : 30,000 births) are inherited, predominantly autosomal dominant connective tissue disorders caused by gene mutations that affect type 1 collagen. This disorder is characterized by brittle osteopenic bones and recurrent fractures (Basel and Steiner, 2009). As many as eight types are known, with wide variations in severity and associated findings such as short stature, blue sclera, progressive hearing loss, poor dentition, scoliosis, and skeletal abnormalities (osteopenia, irregular ossification, multiple fractures). Laboratory studies show a molecular defect in type I procollagen in two-thirds of patients.
Potential neurological complications of osteogenesis imperfecta include communicating hydrocephalus, basilar invagination, macrocephaly, kyphoscoliosis, skull fractures, subdural hematomas, and seizure disorder. The basilar invagination can lead to brainstem compression (Hayes et al., 1999). Spinal cord compression, syringomyelia, Chiari I malformation, Dandy-Walker cysts, leptomeningeal cysts, microcephalus, or central nervous system (CNS) tumors are rarer associations. Progressive, variably conductive and sensorineural hearing loss usually begins in the second decade and affects more than half of patients by age 30 years. Joint laxity, indistinguishable from that of EDS, can result in permanent dislocations. Rarely, osteogenesis imperfecta is complicated by cervical artery dissection resulting from fragility of large blood vessels (Grond-Ginsbach and Debette, 2009). Aortic regurgitation, floppy mitral valves, and mitral incompetence can lead to cerebrovascular complications. For unknown reasons, some patients develop a hypermetabolic state with elevated serum thyroxine levels, hyperthermia, and excessive sweating. Wide phenotypic variation is characteristic and appears to be related to the abundance of over 900 unique mutations in the type I procollagen COL1A1 and COL1A2 genes, combined with undefined stochastic or chance events during embryonic and fetal development (Prockop and Czarny-Ratajczak, 2008).
Treatment is symptomatic and tailored to the severity of symptoms, though women require special attention during pregnancy and after menopause when fractures increase. More severely affected children require more comprehensive physical therapy and orthopedic management. For severe hearing loss, stapes prosthesis placement may be successful. Neurological complications relating to potential brainstem and spinal compression require appropriate attention. Oral or intravenous bisphosphonates can increase bone mineral density but are not proven to prevent fractures in patients with osteogenesis imperfecta (Phillipi et al., 2008).
Ehlers-Danlos Syndrome
The incidence of EDS is approximately 1 : 5000. The disorder is characterized by joint hypermobility and variable skin features. At least 10 types, in addition to variants, differ in extent of skin, joint, blood vessel and other tissues involved, mode of inheritance, and molecular analysis. The vast majority of patients have joint hypermobility and minor skin changes that may be unrecognized in the context of adolescent or early adult rheumatological complaints falsely attributed to depression, neurosis, or fibromyalgia. Joint hypermobility is the major manifestation of the most common type (EDS III, or hypermobile EDS). The Beighton score, which grades joint flexibility, is an effective screening tool (Table 73.1). Skin features such as soft velvety texture, striae, and widened atrophic scarring are mild, and there is no tissue fragility.

Classic EDS is the next most prevalent form. Features in addition to the above skin changes can include easy bruising, mitral valve prolapse, hernias, flat feet, and mild to moderate scoliosis. Though laxity and repeated joint trauma can lead to degenerative arthritis, joint symptoms precede objective arthritis, so patients’ early symptoms can be erroneously discounted. Some patients have low blood pressure, pelvic and lower-extremity venous blood pooling (with dependent cyanosis) contributing to symptomatic postural orthostatic tachycardia syndrome (POTS), and irritable bowel complaints. The ligamentous laxity, with or without varying exposure to head and neck trauma, can result in a syndrome of cranial settling or craniocervical instability, mimicking the symptom complex of Chiari I malformation (Milhorat et al., 2007). Most cases of classic EDS are mild (EDS II); less than 5% are more severe (EDS I), with osteopenia, hypotonia, spontaneous aneurysms in the brain or aorta, scoliosis, dental and severe skin fragility, and irreducible large-joint dislocations.
The autosomal dominant vascular type of EDS (type IV) is rare but important to diagnose because major complications of arterial, bowel, or uterine rupture can cause premature death (Pepin et al., 2000). Patients can have skin changes, easy bruisability, and facial dysmorphism, but joint hypermobility and skin hyperextensibility are not common.
Patients with EDS often have neuromuscular symptoms that include weakness, hypotonia, myalgias, fatigability, paresthesia, and exercise intolerance (Voermans et al., 2009). They can develop axonal neuropathies, mild elevations of creatine kinase (CK), and either myopathic or neuropathic changes on electromyography (EMG). Some have mild muscle biopsy abnormalities. Compression neuropathies or brachial plexopathy are less common associations.
Chondrodysplasias
Achondroplasia
Achondroplasia is an autosomal dominant disorder of endochondral bone formation; a specific mutation of a fibroblast growth factor receptor gene (FGFR3) is present in over 90% of patients. Approximately three-fourths of cases occur because of spontaneous new mutations. The syndrome is the most common cause of short-limbed dwarfism accompanied by macrocephaly and dysplasias of the metaphyses of long bones. The mutant genotype has complete penetrance. The diagnosis can be confirmed by pathognomonic radiographic changes or by deoxyribonucleic acid (DNA) testing. Neurological complications of achondroplasia are common (Box 73.2). Young achondroplastic children should be observed for complications such as hydrocephalus, compression at the foramen magnum, thoracolumbar kyphosis, and sleep apnea. Neurological complications of spinal stenosis tend to occur later in life.
Box 73.2
Neurological Complications of Achondroplasia
Macrocrania, with or without hydrocephalus
Foramen magnum abnormalities with cervicomedullary compression
Respiratory disturbances, including sleep apnea and sudden infant death syndrome
Syringomyelia, diastematomyelia
Adapted with permission from Ruiz-Garcia, M., Tovar-Baudin, A., Del Castillo-Ruiz, V., et al., 1997. Early detection of neurological manifestations in achondroplasia. Childs Nerv Syst 13, 208-213.
Stickler Syndrome
The autosomal dominant Stickler syndrome (Rose et al., 2005) causes facial anomalies including flat cheeks, flat nasal bridge, small upper and lower jaws, pronounced upper lip groove, and palate abnormalities. The facial features can be those of the Pierre Robin syndrome, which includes a U-shaped or V-shaped cleft palate with a large tongue, predisposing these individuals to ear infections and dysphagia. Many patients have high myopia due to a small optic globe and are prone to increased ocular pressure, cataracts, and retinal detachment. Arthritis, scoliosis, and hypermobile joints are problems. Mild to severe hearing loss is common. Learning difficulties owing to hearing and sight impairments can occur if the student is not assisted within the learning environment. Essentially all patients who meet the clinical criteria have mutations in the gene for type II collagen (COL2A1), one of the components of cartilage.
Marfan Syndrome
Marfan syndrome is an autosomal dominant disorder of fibrous connective tissue with variable penetrance and variation of phenotypic expression; its most prominent features affect the skeleton, heart, great vessels, and eye. Over 90% of patients have a mutation in the fibrillin-1 gene. The classic patient is tall with inordinately long limbs and digits. Other skeletal changes are anterior chest deformity, scoliosis, thoracic lordosis, high arched palate with crowded teeth, and some ligamentous laxity. Ocular problems include myopia, flat corneas, and ectopia lentis. The major life-threatening problem is aortic aneurysm or dissection; aortic and mitral valves can also be affected. Case reports link Marfan syndrome to intracranial vascular abnormalities such as arterial dissections, giant aneurysms, or hemifacial spasm associated with vascular compression of the facial nerve. However, aneurysms or dissections are rare in patients with Marfan syndrome, and their highest risk of stroke is from cardiogenic emboli, especially if they have prosthetic heart valves or atrial fibrillation (Wityk et al., 2002). Patients with Marfan syndrome can have sleep apnea, possibly secondary to their skeletal deformities.
1. Homocystinuria, a group of autosomal recessive disorders that can cause marfanoid skeletal changes, myopia, and lens ectopia. It is important to distinguish homocystinuria from Marfan syndrome, since the former can cause mental retardation and hypercoagulability leading to strokes and because it can be treated with vitamin B6, folate and vitamin B12.
2. Loeys-Dietz aneurysm syndrome is similar to Marfan syndrome in skeletal and aortic manifestations but does not cause lens ectopia and is often associated with craniosynostosis. Neurological manifestations can include Chiari I malformation, hydrocephalus, and developmental delay. It can be due to mutations in either TGFB2 or the closely related TGFB1 genes.
3. Shprintzen-Goldberg syndrome is also characterized by marfanoid features, normal lenses, and craniosynostosis. Neurological issues can include hypotonia, Chiari I malformation, developmental delay, mental retardation, and obstructive sleep apnea.
4. Beals syndrome (congenital contractural arachnodactyly [CCA]) can cause marfanoid features, joint contracture, and some features of osteogenesis imperfecta. Like Marfan syndrome, it is autosomal dominant, but the mutated gene is for fibrillin-2 rather than fibrillin-1.
Congenital and Inherited Craniospinal Malformations and Deformities
Craniospinal malformations result from abnormalities of the bones of the skull and spinal column, connecting ligaments, or other soft tissues and may cause hydrocephalus, brain deformation, spinal cord compression or disruption, and syringobulbia or syringomyelia. (Menezes, 1997). Many of these are congenital. Magnetic resonance imaging (MRI) and computed tomographic (CT) scanning have improved the detection, understanding, and treatment of these anomalies.
Craniosynostosis
Craniosynostosis is among the most common and relatively benign abnormalities, affecting about 1 in 2500 live births. It is caused by premature closure of one or more of the sutures of the skull bones. For babies who have abnormal skull shapes, CT scans can distinguish craniosynostosis from results of fetal head position, birth trauma, or positional plagiocephaly—flattened or misshapen areas on the head that may develop due to sleeping position. Table 73.2 separates four types of craniosynostosis.
| Craniosynostosis | Suture Involved | Characteristic |
|---|---|---|
| Scaphocephaly (most common) | Sagittal | Boat shaped head; narrow side to side and elongated from front to back |
| Trigonocephaly | Metopic | Triangular forehead; eyes close set |
| Plagiocephaly | Coronal (unilateral) | Asymmetrical head and facies, right or left side, forehead and brow appear pushed back, eye appears receded |
| Brachycephaly | Coronal (bilateral) | Wide-shaped head with short skull and tall, flattened forehead |
Occipitalization of the Atlas
Occipitalization or assimilation of the atlas refers to congenital partial or complete fusion of the atlas (first cervical vertebra) to the occiput (Fig. 73.1, online only). The anterior arch of the atlas may fuse to the lower end of the clivus, or the posterior arch of the atlas may fuse to the occiput. The anomaly is often asymptomatic until early adult life but may become symptomatic after trauma. Unilateral occipitalization of the atlas is one cause of torticollis in young children. The loss of movement between the occiput and atlas increases the stresses at the atlantoaxial joint, predisposing it to gradual degeneration or traumatic dislocation. Patients with occipitalization of the atlas may have associated anomalies such as the Klippel-Feil anomaly, basilar impression, or Chiari malformation.
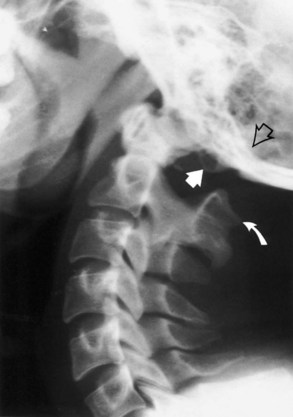
Basilar Impression
Basilar impression or invagination refers to the abnormal cephalad position of the foramen magnum (Goel et al., 1998). Several radiological lines (Chamberlain, McGregor, McRae, digastric) (Fig. 73.2, online only) and measurements can be used to make the diagnosis. Congenital basilar impression may occur in isolation or may be associated with conditions such as achondroplasia, occipital dysplasia, Down syndrome, Hurler syndrome, Klippel-Feil anomaly, and cleidocranial dysplasia. Some instances of basilar impression are familial. The skeletal anomaly is often accompanied by anomalies of the neuraxis, including Chiari I or II malformation and syringomyelia. Basilar impression can cause compression of the brainstem (Fig. 73.3, online only) or cerebellum, or (rarely) vertebral artery compression leading to vertebrobasilar ischemia. It is often asymptomatic, particularly when mild and unaccompanied by other anomalies.
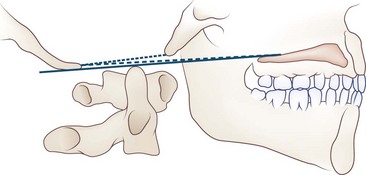
Klippel-Feil Anomaly
Patients with the Klippel-Feil anomaly (congenital synostosis of the cervical vertebrae) (Fig. 73.4, online only) have short necks, low hairlines, and limitation of cervical motion. The diagnosis is confirmed by radiographic demonstration of fused cervical vertebrae. The condition is congenital, caused by failure of normal segmentation of the cervical vertebrae between the third and eighth weeks of fetal development. Although familial instances occur, most cases are isolated and idiopathic. The anomaly can cause direct nerve root, cervical spinal cord, or vertebral or spinal artery compression. Patients may have cervical ribs, predisposing them to thoracic outlet syndrome. Neck pain is common. Hearing loss is the most common symptom of cranial neuropathy. Klippel-Feil syndrome is the anomaly most likely to cause mirror movements, particularly of the hands. Patients with mirror movements can have abnormal clefts or division of the spinal cord near the cervicomedullary junction, which can be detected by MRI or CT myelography (Royal et al., 2002). Patients with Klippel-Feil anomaly can have a wide variety of associated abnormalities of brain, spinal cord, or skeletal development, especially congenital scoliosis or Sprengel deformity with unilateral shoulder elevation. Patients may develop hydrocephalus, syringomyelia, or syringobulbia. However, many patients with Klippel-Feil anomaly have no neurological symptoms or signs.
Atlantoaxial Dislocation
Various congenital or acquired conditions can disrupt the integrity of the atlantoaxial joint, leading to its dislocation (Box 73.3). In horizontal subluxation, C1 usually moves anteriorly to C2. The movement can be assessed by measuring the separation between the dens and the anterior arch of C1 on flexion radiographs; in adults, the separation should not exceed 3.5 mm. Patients with horizontal atlantoaxial joint subluxation are likely to compress their spinal cords if the diameter of the spinal canal at the level of the dens is less than 14 mm, and they are unlikely to if the diameter is more than 17 mm. The actual relationship between the cord and the subluxing bones is best imaged with MRI or CT myelography, which should include flexion and extension views. In some patients, particularly those with acquired inflammatory disease such as rheumatoid arthritis (RA), inflamed adjacent soft tissue contributing to cord compression is best characterized by MRI.
Box 73.3 Mechanisms of Atlantoaxial Dislocation
Chiari I Malformation
In the 1890s, Chiari described four types of malformations with cerebellar tonsillar displacement. Cleland had written about them in 1883. Arnold reported a case of Chiari II malformation in 1894. In current usage, the terms Arnold-Chiari and Chiari malformation are often used interchangeably for all four types. Chiari malformations II, III, and IV are discussed later in the section on Spinal Dysraphism.
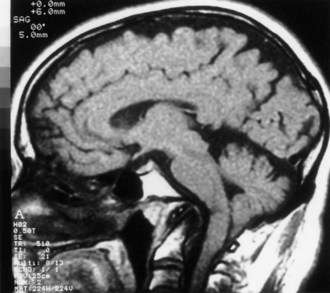
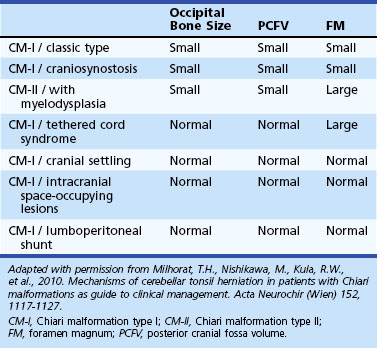
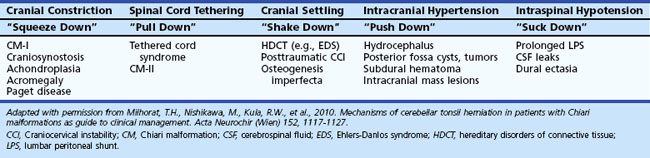
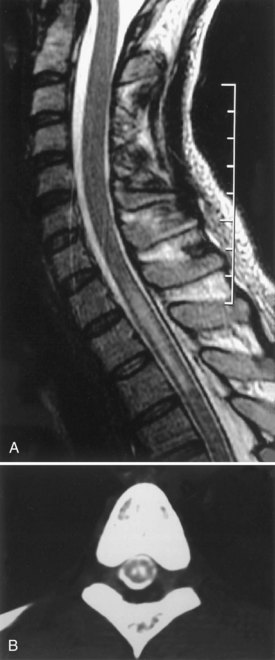
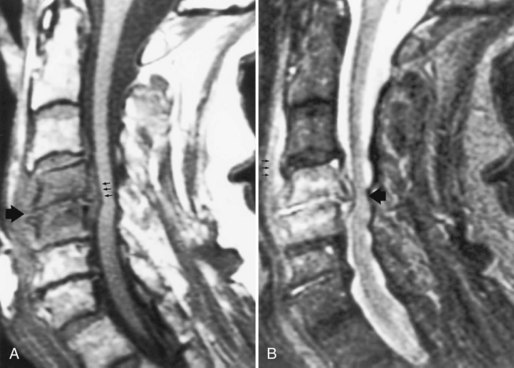
Chiari I malformation (CM-I) (Fig. 73.5), the most common type, is simply abnormal cerebellar tonsillar herniation below the foramen magnum (5 mm or more in young adults); this definition is anatomically precise but can be confusing because there are many causes of abnormal tonsillar herniation that are not malformations. Classic CM-I is a congenital mesodermal malformation resulting in a hypoplastic posterior fossa, compressing neural tissue and forcing the cerebellar tonsils down through the foramen magnum. Distinguishing classic CM-I from other mechanisms of tonsillar herniation clarifies treatment despite overlapping clinical features (Milhorat et al., 2010). Other disorders of tonsillar herniation that are unrelated to skull-base hypoplasia include hydrocephalus, intracranial mass lesions, CSF leaks, prolonged lumboperitoneal shunting, hereditary disorders of connective tissue associated with occipitoatlantoaxial joint instability and cranial settling, tethered cord syndrome, and miscellaneous conditions such as craniosynostosis, acromegaly, and Paget disease. Milhorat and colleagues have correlated measurements of the posterior cranial fossa with clinical findings in 752 patients with Chiari malformations (Milhorat et al., 2010) (Table 73.3).
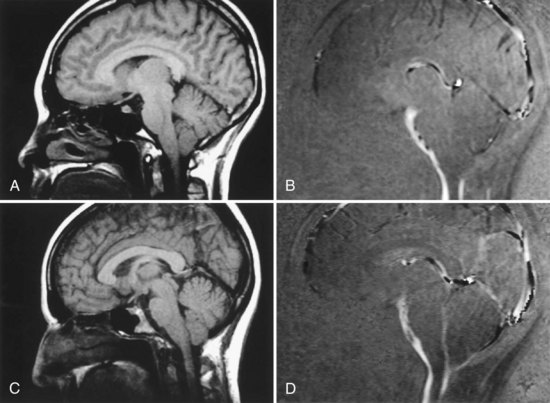
Patients with classic CM-I have a small posterior cranial fossa with constriction increasing below the Twining line, which extends from the anterior tuberculum sellae to the internal occipital protuberance. The foramen magnum is constricted transversely and has reduced outlet areas (Fig. 73.6). These findings suggest premature stenosis of the basi-exoccipital and exo-supraoccipital synchondroses (see Fig. 73.6, A) which, if normal, would permit lateral expansion of the foramen magnum during somatic growth. Failure of the foramen magnum to expand normally during development may explain the conical shape of the posterior fossa in CM-I. Patients with achondroplasia can also have a stenosed foramen magnum and small posterior fossa, but the posterior fossa constriction is more generalized than in classic CM-I, suggesting an additional pathogenic mechanism in achondroplasia. Crouzon syndrome (see Fig. 73.6, D), Apert syndrome, nonsyndromic craniosynostosis, achondroplasia, acromegaly, and Paget disease are other causes of a small posterior fossa. In each of these conditions, the constricting small posterior fossa is the apparent cause of cerebellar tonsillar herniation.
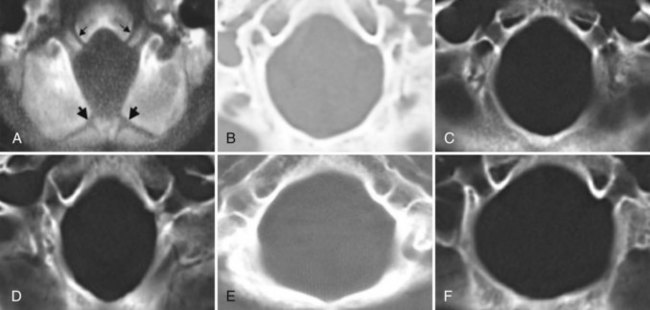
When patients with CM-I have other abnormalities like hydrocephalus, basilar impression, occipitalization of the atlas, retroflexed odontoid processes, elongated styloid processes, or C1-level spina bifida occulta, the mechanism of the tonsillar herniation varies. In contrast to CM-I, patients with tonsillar herniation due to hydrocephalus, intracranial mass lesions, occipitoatlantoaxial joint instability, or prolonged lumboperitoneal shunting have normal occipital bone size, posterior cranial fossa volume, and foramen magnum size; in these conditions, mechanisms other than a small posterior fossa cause the tonsillar herniation. In patients with hydrocephalus or intracranial mass lesions, raised intracranial pressure causes compartmental shifts that push the brain caudally. In patients with occipitoatlantoaxial joint instability, cranial settling is the main cause of the herniation. In patients undergoing prolonged lumboperitoneal shunting, overdrainage of CSF apparently creates a pressure differential between the cranial and spinal compartments, drawing cerebellar tonsils downward. When the drainage is stopped, the pressure gradient can resolve, and the herniation often reverses. Low spinal fluid pressure can also cause tonsillar herniation in patients with spinal CSF leaks, dural ectasias, and myelodysplasia. Table 73.4 outlines five distinct mechanisms of cerebellar tonsil herniation; each mechanism has its own diagnostic and therapeutic implications.
Clinical Presentation
Patients with CM-I may experience no symptoms or first have symptoms in adolescence or early adulthood (Meadows et al., 2000) (Table 73.5). At least a quarter of patients first have symptoms following relatively minor head or neck injury. Most symptomatic patients have pressing occipital headache and neck pain. Other manifestations can include visual disturbances, neuro-otological complaints, cranial nerve dysfunction, cognitive difficulties, and sleep apnea (Milhorat et al., 1999). Patients with CM-I often have associated syringomyelia (discussed later), but motor, sensory, sphincter, and reflex disturbances suggestive of myelopathy can occur whether or not a syrinx is present. Clinical severity correlates generally but not perfectly with the extent of tonsillar herniation and the degree of obstruction to CSF flow. In more severe cases, the medulla descends below the foramen magnum, in which case brainstem dysfunction is more likely to be among the clinical findings (Yamada et al., 2004).
| Symptom | % of CM-I Patients (364) |
|---|---|
| Suboccipital headache (frequent retro-orbital component); exertional and postural accentuation | 81 |
| Ocular disturbances: floaters, blurring, photophobia, diplopia | 78 |
| Acoustic and vestibular complaints: dizziness/dysequilibrium, tinnitus | 74 |
| Dysesthesias: tenderness, numbness/tingling, burning | 59 |
| Chronic fatigue | 58 |
| Bulbar and coordinative problems: | 52 |
| Dysphagia/dysarthria, sleep apnea | |
| Palpitations (23 pts with paroxysmal atrial tachycardia) | |
| Tremors, clumsiness | |
| Segmental pain | 44 |
| Impaired memory or concentration | 39 |
| Cervical pain | 34 |
| Low back pain | 24 |
| Urinary incontinence | 17 |
From Milhorat, T.M., Chou, M.W., Trinidad, E.M., et al., 1999. Chiari I malformation redefined: clinical and radiographic findings in 364 symptomatic patients. Neurosurgery 44, 1005–1017.
Deciding whether common symptoms like memory concerns, back pain, or fatigue are caused by CM-I in an individual patient is a clinical challenge. Despite speculation and public interest, there is no scientific confirmation of an association between CM-I and fibromyalgia or chronic fatigue syndrome (Garland and Robertson, 2001).
Slight extension of the tonsils below the foramen is normal in childhood, and normal values decrease with increasing age (Table 73.6). When young children have symptoms, they can have oropharyngeal dysfunction and scoliosis. Children younger than age 3 can have vomiting and gastric reflux as the sole symptoms of CM-I.
Table 73.6 Suggested Upper Limits of Normal for Position of Cerebellar Tonsils Below Foramen Magnum
| Decade of Life | Distance Below Foramen Magnum (mm) |
|---|---|
| First | 6 |
| Second or third | 5 |
| Fourth to eighth | 4 |
| Ninth | 3 |
Data used with permission from Mikulis, D.J., Diaz, O., Egglin, T.K., et al., 1992. Variance of the position of the cerebellar tonsils with age: preliminary report. Radiology 183, 725–728.
The posterior fossa or “Chiari” headache has diagnostically helpful characteristics: it is a suboccipital pressing head pain that is usually continuous, waxing and waning but not episodic, and radiating at times behind the eyes or to the vertex (Kula, 2006). The headache is almost never hemicranial and is often exaggerated more by bearing down with bowel movements, with laughter, crying, or orgasm than with cough or sneeze. Exacerbations can be explosive rather than throbbing or pounding. There is no aura, but visual sparkles and scotomas can punctuate the peaks of headache. Neck pain without radicular features is common. Patients may have evanescent hand paresthesias and other generalized musculoskeletal complaints. Many patients coincidentally have migraine headache; both types of headache can exacerbate premenstrually, but the Chiari headache rarely responds to common migraine treatments with antidepressants, beta-blockers, and triptans. An exception is topiramate, which sometimes gives relief, presumably because of carbonic anhydrase inhibitory effect lowering CSF pressure.
The malformation is best seen on T2-weighted sagittal MRI scans of the brain and cervical spine, which allow assessment of the shape of the posterior fossa, may detect accompanying syrinx, and show the extent (if any) of brainstem compression. Flow of CSF at the foramen magnum can be evaluated using phase-contrast MRI and cardiac gating to acquire images throughout the cardiac cycle (Haughton et al., 2003). Symptomatic Chiari I malformations can cause an increase in peak systolic CSF flow velocity and decreased uniformity of flow.
Management
Surgical decompression of the posterior fossa is appropriate treatment for the minority of patients who have significant functional impairment due to headache despite medical therapy or have progressive syringomyelia. It is not indicated for symptoms limited to chronic fatigue, musculoskeletal pain, or vertigo, or for prophylaxis against worsening of headache or of syringomyelia. Kula (2006) discusses treatment in detail and provides a comprehensive management algorithm.
Spinal Dysraphism
Spinal dysraphism is congenital failure of the primitive neural tube to close during fetal development and includes a number of disorders of fusion of dorsal midline structures of the spinal canal or skull (Botto et al., 1999). The neural tube normally closes during the first 3 weeks following conception. There are genetic causes, but environmental causes are important in most cases. The most extreme form is anencephaly, characterized by absence of the entire cranium at birth; the undeveloped brain lies at the base of the skull as a small vascular mass without recognizable nervous structures. Anencephaly is incompatible with life.
Spina Bifida Occulta
Spina bifida occulta is the most common and least symptomatic (usually asymptomatic) form of dysraphism. In this anomaly, the vertebral elements fail to fuse posteriorly, but the thecal and neural elements remain within the spinal canal. It is most common at posterior elements of L5-S1 and is usually noted as an incidental finding on spinal plain radiography (Fig. 73.7, online only). Cutaneous abnormalities may be associated (Box 73.4). Orthopedic foot deformities, urinary or rectal sphincter dysfunction, or focal neurological abnormalities can indicate that the spina bifida occulta is associated with compression or malformation of neural tissues or with spinal cord tethering.
Myelomeningocele and Encephalocele
In myelomeningocele and meningocele (spina bifida cystica), congenital defects of midline closure are accompanied by eventration of meninges and even of neural tissue. Spinal defects are often visible on examination of the back of the newborn (Figs. 73.8 [online only], 73.9, and 73.10). At times the skin and vertebral canal are open, and a sac of meninges is directly visible. The defect is most common in the lumbar region. If the sac contains nerve roots or spinal cord, it is a myelomeningocele; if neural elements are absent from the sac, it is a meningocele. These brain and spinal cord malformations may be associated with CSF leakage into adjacent structures, posing a risk of meningitis. Neurological deficits are directly related to the anatomical extent of the malformation and vary from insignificant to grave.
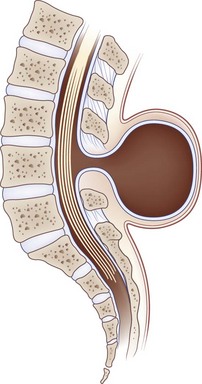
Either defect is often accompanied by hydrocephalus or by Chiari II malformation, in which the cerebellar vermis and caudal brainstem descend through an enlarged foramen magnum, (Stevenson, 2004; Tubbs and Oakes, 2004). The extent of brainstem herniation is variable, including portions of the medulla or even of the pons. Hydrocephalus and syringomyelia are common accompanying features, and patients often have various associated anomalies such as a small posterior fossa, kink in the medulla, and polymicrogyria. These infants are at risk for later development of tethered cord syndrome or spinal dermoid or epidermoid inclusion cysts. In Chiari III malformation, the displaced cerebellar and brainstem tissue extends into an infratentorial meningoencephalocele.
Tethered Cord Syndromes
Congenital abnormalities of the spinal cord or cauda equina can prevent normal cephalad movement of the conus medullaris during early life (Michelson and Ashwal, 2004) (Box 73.5). Imaging studies, such as spinal MRI showing the conus medullaris caudad to the lower endplate of L2, are evidence of tethering. A child or even an adult with these abnormalities can develop progressive neurological dysfunction due to traction on the cord or nerve roots. One presentation is lower motor neuron dysfunction in one or both legs, but patients can also have sensory loss, upper motor neuron signs, orthopedic foot deformities, or scoliosis. A tethered spinal cord can also cause isolated sphincter dysfunction as subtle as intermittent urinary incontinence.
Box 73.5
Causes of Tethered Spinal Cord
Adapted with permission from McLone, D.G., La Marca, F., 1997. The tethered spinal cord: diagnosis, significance, and management. Semin Pediatr Neurol 4, 192-208.
The so-called occult tethered cord syndrome is an area of controversy (Drake, 2006; Selden, 2006). Uncontrolled surgical series suggest that children with neurogenic voiding dysfunction and normal spinal MRI might also have cord tethering. For some children, voiding dysfunction reportedly improved after lysis of the filum terminale, which microscopically can be abnormally thickened, fatty, and fibrosis, even though the filum and conus medullaris appear normal on MRI. A few cases of cerebellar tonsillar herniation seem to be due to occult cord tethering; other features are syrinx development below the T5 level and scoliosis (Milhorat et al., 2009). In contrast to patients with classic CM-I, patients with this variation of CM-I with cord tethering have normal posterior fossa volume and an enlarged foramen magnum. Supporting the role of cord tethering as a cause of the tonsillar descent are reports of increasing herniation of the cerebellar tonsils with somatic growth, cerebellar prolapse following Chiari decompression surgery, and anatomical improvements including ascent of the conus medullaris, ascent of the cerebellar tonsils, and resolution of brainstem elongation following section of the filum terminale.
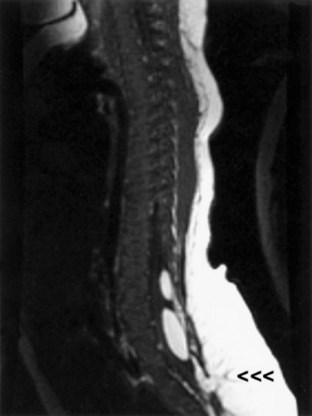
Diastematomyelia is a congenital malformation of the spinal cord characterized by sagittal division of a portion of the cord into two hemicords. In most instances, the division is located in the lower thoracic or lumbar regions. Diastematomyelia is often accompanied by skin abnormalities such as a tuft of hair at the level of the lesion. If each hemicord is enclosed in its own arachnoid sheath, the sheaths are usually separated by a bony, cartilaginous, or fibrous spur and by dura in the cleft between the two portions of the cord. The spur tethers the spinal cord, leading to progressive neurological dysfunction when the cord attempts to move rostrally during growth. The diagnosis can often be suspected on plain radiography, which shows widening of the interpeduncular distance and a posterior bony bridge at the level of the lesion. MRI scans or CT myelography can confirm the diagnosis (Fig. 73.11, online only). Surgical therapy consists of attempts to free all structures tethering the cord by removing the spurs and dura in the cleft and cutting the filum terminale if abnormally tethered.
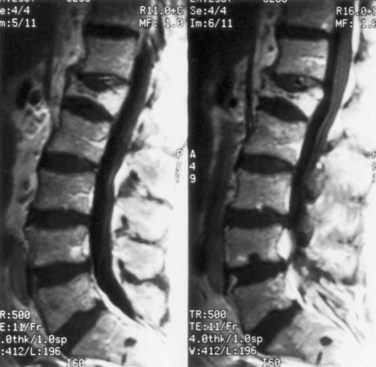
Syringomyelia and Syringobulbia
Hydromyelia is an abnormal dilation of the central spinal canal with “excess” cerebrospinal fluid contained within the ependymal lining. When fluid dissects into the surrounding white matter forming a cystic cavity or syrinx, the term syringomyelia is applied. A syrinx, then, is a cavity in the spinal cord (syringomyelia) or brainstem (syringobulbia) (Figs. 73.12, 73.13, 73.14). Hydromyelia and syringomyelia often coexist, and many physicians use the terms interchangeably.
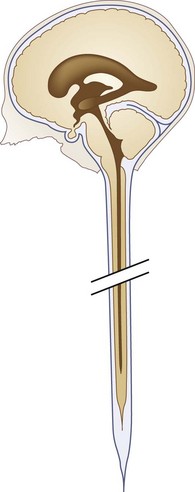
Fig. 73.12 Diagrammatic representation of persistent central canal extending throughout length of spinal cord.
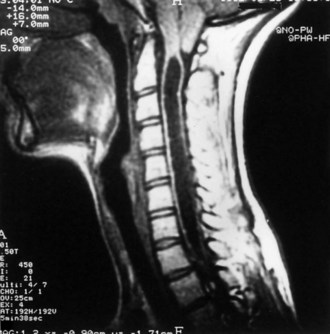
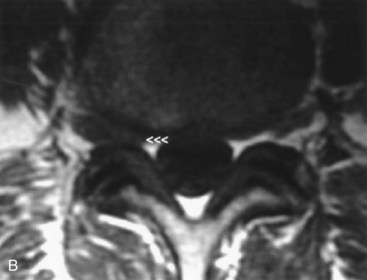
The central canal of the spinal cord is normally widely open during embryonic life and becomes atretic after birth. It is occasionally patent in the adult (see Fig. 73.12). Cervical or thoracic MRI of an adult will occasionally show an incidental asymptomatic hydromyelia, which is typically linear or fusiform on sagittal images, extends over several levels, sometimes discontinuously, and is round, central, and up to 4 mm in diameter on axial images (Batzdorf, 2005).
Clinical Presentation
Although either CT or MRI can demonstrate a syrinx, MRI is more sensitive for complete evaluation of the cord and surrounding soft tissues. CT myelography can be useful in discerning syringomyelia, which will commonly fill with contrast on delayed images because of communication with the CSF through the central canal of the spinal cord (see Fig. 73.13).
Abnormalities of the Cervicomedullary Junction
Abnormalities of the cervicomedullary junction and posterior fossa, such as Chiari anomalies types I and II and the Dandy-Walker malformation, are the most common cause of syringes; as many as 70% of all syringes are associated with CM-I. The mechanism of formation of these syringes is controversial (Di Lorenzo and Cacciola, 2005). Patients with cervical syringomyelia and no Chiari malformation often have a small posterior fossa or disturbed flow of CSF near the foramen magnum (Bogdanov et al., 2004). Syringes can extend beyond hydromyelia as an outpouching of the dilated central canal (see Figs. 73.13 and 73.14). One hypothesis is that the posterior fossa abnormalities interfere with the passage of CSF from the fourth ventricle through the foramina of Luschka and Magendie into the subarachnoid space. The consequence is transmission of bulk flow and the various pressure waves of the CSF (arterial, venous, respiratory) down the central canal of the spinal cord, leading to dissection of a syrinx into the substance of the spinal cord. Noncongenital abnormalities at the cervicomedullary junction that sometimes cause syringomyelia include arachnoiditis and meningiomas.
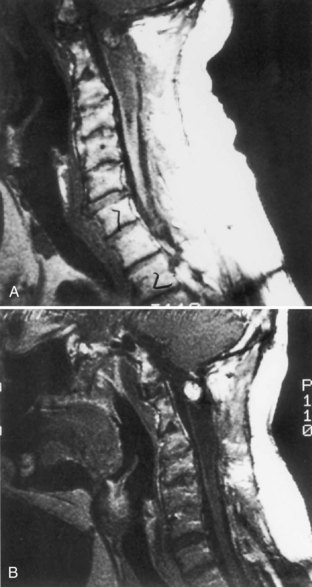
Syrinx Associated with Spinal Cord Tumors
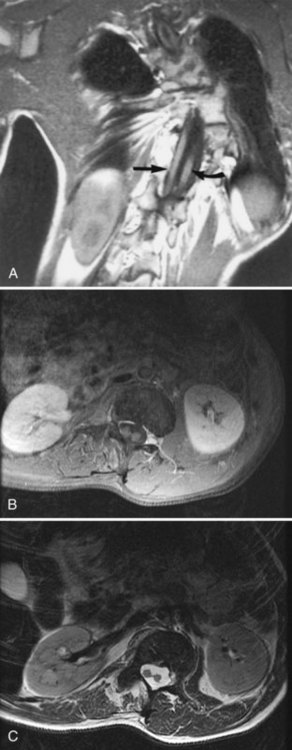
Syringes are associated with intramedullary tumors often enough that any cystic process in the spinal cord should be considered an intramedullary tumor until proven otherwise. Syringomyelia accompanies 25% to 60% of intramedullary spinal tumors; conversely, 8% to 16% of syringes are caused by tumors (Fig. 73.15, online only). Intramedullary tumors in von Hippel-Lindau syndrome and neurofibromatosis are particularly likely to be accompanied by syringes. The syrinx extends from the tumor, more often rostrally than caudally. Ependymomas, which represent up to 10% of childhood CNS tumors and affect adults as well, are particularly likely to produce syringes because of their central location. Less than 2% of extramedullary tumors in the spinal canal (e.g., meningiomas, neuromas) are associated with syringes.
Syrinx Associated with Spinal Cord Trauma
Syringes can develop as a late effect of serious spinal cord trauma (Carroll and Brackenridge, 2005). Estimates of the prevalence of syringes after trauma vary widely from 0.2% to 64%. Symptoms of ascending long-tract or segmental spinal cord dysfunction usually develop within 5 years after the acute traumatic myelopathy has stabilized, improved, or even become asymptomatic. Pain or other sensory symptoms are often prominent. Findings usually evolve gradually but occasionally worsen suddenly after events such as a cough or Valsalva maneuver. The cavities are typically eccentric and can be multiple, arising from areas of posttraumatic myelomalacia and then spreading rostrally or caudally. Severe posttraumatic spinal deformity or arachnoid scarring can also cause posttraumatic syringes.
Syrinx Associated with Other Focal Spinal Cord Pathologies
Treatment
Indications for and approaches to surgical therapy for syringes are far from standardized. In patients with Chiari I malformations, the syrinx often improves after decompression of the malformation with suboccipital craniectomy, upper cervical laminectomy, and dural grafting. When the syrinx extends from an intramedullary tumor, resection of the tumor often leads to regression of the syrinx, so no specific surgical drainage of the cavity is needed. When the syrinx extends from an area of localized arachnoiditis or other obstruction of the subarachnoid space, some patients benefit from resection of the arachnoiditis and restoration of CSF flow patterns with an expansile duraplasty; shunting or entering the cavity is a less desirable surgical approach (Batzdorf, 2005). In patients with Chiari I malformation and syrinx, pain and other sensory symptoms are more likely to improve if the foramen magnum is decompressed within 2 years of onset of the sensory symptoms (Attal et al., 2004). Among patients undergoing surgery for syrinx in the absence of Chiari malformation, slightly more than half improved or stabilized, but over a third required more than one operation (Batzdorf, 2005).
Clinical Correlations
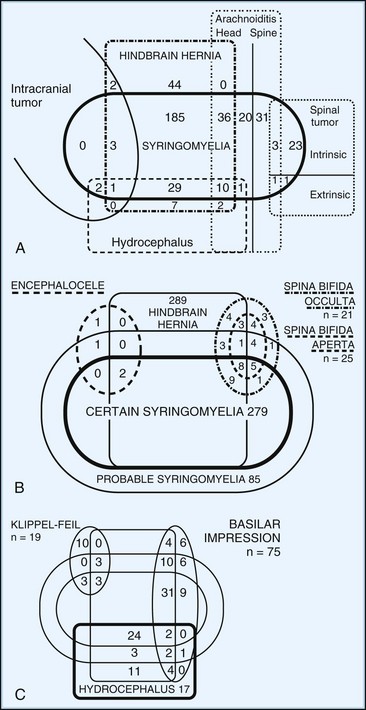
A single patient often has more than one of the conditions discussed (Fig. 73.16). Thus, a patient with one of the Chiari hindbrain malformations also may have some combination of bony abnormalities of the foramen magnum or cervical spine, syringomyelia, and meningomyelocele. The clinical manifestations of craniocervical deformities are protean depending on which neural structures and associated anomalies are involved. When a patient has these problems, diagnosis and treatment starts by analyzing each component. MRI and CT scans, especially with measurements of the foramen magnum and posterior fossa, have greatly eased the analytical process. Many patients are asymptomatic or first present with neurological complaints in adult life. Patients may have short necks or abnormal neck posture or movement, particularly if there is an element of skeletal deformity (e.g., Klippel-Feil anomaly, occipitalization of the atlas). Findings attributable to the brainstem or cerebellum may occur with Chiari malformations, compression of the brainstem (e.g., basilar impression, vertical displacement of the dens), or syringobulbia. Uncommonly, atlantoaxial disease or basilar invagination can cause compromise of vertebrobasilar circulation, causing posterior circulation strokes or transient ischemic attacks. Specific findings suggestive of disease at the foramen magnum include downbeat nystagmus or the combination of long-tract signs with lower motor neuron dysfunction in the lower cervical spinal cord; lower motor neuron dysfunction has been attributed to impaired spinal venous drainage at the foramen magnum.
Spinal Deformities and Metabolic Bone Disease
Osteoporosis
Osteoporotic vertebral compression fractures occur most commonly in the thoracic and thoracolumbar spine, especially in postmenopausal women (Fig. 73.17). By age 75 years, nearly a fourth of women have vertebral compression fractures; although these may lead to kyphosis and loss of body height, most are painless. The presence of a vertebral fracture in a postmenopausal woman or older man is a very strong predictor of subsequent fracture risk and an indication for pharmacological treatment of osteoporosis. In younger men and women, acute posttraumatic compression fractures are more likely to be painful. The pain usually is centered at the level of the compression and accompanied by loss of spinal range of motion. Pain increases with activity, decreases with bed rest, and resolves slowly, sometimes incompletely.
Many reports attest that treatment of painful compression fractures with percutaneous vertebroplasty with polymethylmethacrylate, with or without balloon kyphoplasty, can decrease the duration of pain (Chen et al., 2006; Jensen et al., 2007). In a randomized unblinded study, kyphoplasty improved symptoms and functional outcomes during the first year following treatment (Wardlaw et al., 2009). In contrast, a randomized controlled trial using blinded sham injections did not show benefit of vertebroplasty (Buchbinder et al., 2009). Furthermore, vertebroplasty has not been shown to affect the long-term outcome and may increase the incidence of compression fractures at adjacent vertebral bodies (Diamond et al., 2006). Epidural or intradural leakage of the cement causing radiculopathies or myelopathy is a rare complication of the injections.
Nonmetastatic compression fractures infrequently lead to spinal cord or nerve root compression, so if a compression fracture accompanies a focal neurological compression syndrome, a metastatic vertebral lesion should be considered. MRI features that favor a malignant cause of the compression fracture include decreased T1-weighted and increased T2-weighted signal in the vertebral body, pedicle involvement, and associated epidural or paravertebral mass (Do, 2000).
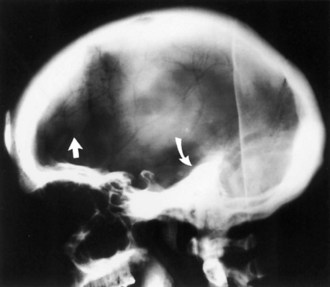
Osteopetrosis
The osteopetroses are a group of rare inherited diseases characterized by increased bone density due to impaired bone resorption (Steward, 2003) (Fig. 73.18, online only). Varied genetic defects can cause osteopetrosis, resulting in three clinical variants: infantile severe autosomal recessive, intermediate autosomal recessive, and autosomal dominant. Osteopetrosis of the skull can cause cranial neuropathies (most often optic neuropathy), basilar impression, hydrocephalus, or syringomyelia. Osteopetrosis of the spine can contribute to spinal canal stenosis with secondary compressive myelopathy. Other complications include thrombocytopenia, anemia, osteomyelitis, and fractures. Some patients can be treated by hematopoietic stem cell transplantation. Other rare sclerosing bone disorders like progressive diaphyseal dysplasia (Camurati-Engelmann disease) or endosteal hyperostosis occasionally have neurological complications (Grond-Ginsbach and Debette, 2009).
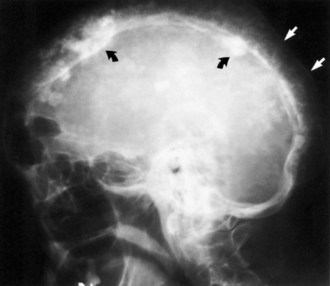
Paget Disease
Paget disease is a focal metabolic bone disease of excessive osteoclastic bony destruction coupled with reactive osteoblastic activity (Poncelet, 1999; Ralston et al., 2008) (Fig. 73.19). The incidence increases with age and varies among ethnic groups, with a high incidence (nearly 5%) in elderly whites of Northern European descent. Men are slightly more commonly affected. Paget disease appears to be caused by a combination of genetic and environmental factors, including possible roles of calcium or vitamin D deficiency, toxins, and infections, especially with paramyxovirus. Paget disease is usually asymptomatic and discovered only because of laboratory or radiographic abnormalities. However, it may cause symptoms by bone or joint distortion, fractures, compression of neurological tissue by calcification, hemorrhage, or focal ischemia due to a vascular steal by the metabolically hyperactive bony tissue. Uncommonly, neoplasms, especially osteogenic sarcoma, can develop in pagetic bone.
A few families have an autosomal dominant illness of inclusion body myositis, frontotemporal dementia, and Paget disease of bone (Kovach et al., 2001).
Diagnosis
Paget disease usually can be diagnosed by characteristic radiographic findings of mixed osteolytic and osteoblastic lesions (see Fig. 73.19). Osteolytic activity can cause well-demarcated round patches of low bone density in the skull (osteogenesis circumscripta). Osteoblastic activity leads to thickening of cortical bone and then to a general increase in bone density, often with distortion of normal organization.
Cranial Neurological Complications
Neurological complications are common in Paget disease (Rubin and Levin, 2009). Paget disease of the skull can lead to head enlargement. Patients often complain of headache. The most common focal neurological manifestation is hearing loss. Paget disease of the cribriform plate can disrupt olfaction. Other cranial mononeuropathies (e.g., optic neuropathy, trigeminal neuralgia, hemifacial spasm) are much less frequent. Perhaps a third of patients with Paget disease of the skull have some degree of basilar invagination, but symptomatic complications such as brainstem or cerebellar compression, hydrocephalus, or syringomyelia are rare (Raubenheimer et al., 2002). Patients with Paget disease of the skull occasionally develop seizures. The pagetic skull is more vulnerable to bleeding from minor trauma, which can lead to epidural hematoma.
Treatment
The potent bisphosphonates are the drugs of choice for treatment of Paget disease. Bone resorption decreases within days. Within 1 to 2 weeks of treatment, bone pain may improve. Osteoblastic bone formation and falling serum alkaline phosphatase levels occur after 1 or 2 months of therapy. Some patients experience significant neurological improvement after treatment, but improvement is often delayed 1 to 3 months. In cases with severe cord compression, surgical decompression is indicated, but drug treatment before surgery decreases the risk of operative bone hemorrhage. Patients with cranial neuropathy have less impressive responses to drug therapy. Hydrocephalus can be treated successfully with ventriculoperitoneal shunting (Roohi et al., 2005).
Scoliosis
Scoliosis can be congenital, acquired secondary to an underlying disease, or idiopathic. The most common form is idiopathic scoliosis, with or without kyphosis, that usually develops painlessly in childhood and adolescence. A few cases of acquired scoliosis are associated with tumor, spondylolisthesis, or neurological pathology such as syrinx, myelomeningocele, or Chiari I malformation. Among patients with acquired scoliosis, indications for spinal MRI include abnormal neurological examination or atypical curve features such as sudden progression, left thoracic curvature, or absent apical segment lordosis (Davids et al., 2004). Spinal cord compression is a rare complication of idiopathic scoliosis and is particularly rare if no kyphosis is present. In each patient presenting with scoliosis and myelopathy, an important consideration is whether the myelopathy caused, rather than resulted from, the scoliosis.
Scoliosis can also be caused by various neurological diseases including cerebral palsy, spinocerebellar degenerations (e.g., Friedreich ataxia), inherited neuropathies (e.g., Charcot-Marie-Tooth disease), myelopathies (e.g., syringomyelia), paralytic poliomyelitis, spinal muscular atrophy, dysautonomia (e.g., Riley-Day syndrome), and myopathies (e.g., Duchenne disease) (Berven and Bradford, 2002). Scoliosis is the most common skeletal complication of neurofibromatosis type 1. Scoliosis that develops in adulthood can often be traced to an underlying cause such as trauma, osteoporotic fracture, degenerative spondylosis, or ankylosing spondylitis; it can result in local back pain, nerve root compression, or spinal canal stenosis.
Diffuse Idiopathic Skeletal Hyperostosis
Diffuse idiopathic skeletal hyperostosis (DISH) (Forestier disease, ankylosing hyperostosis) is a syndrome of excessive calcification that develops with aging, more often in men than in women. The diagnosis is made by spinal radiographs that show “flowing” calcifications along the anterior and lateral portion of at least four contiguous vertebral bodies, without loss of disk height and without typical radiographic findings of ankylosing spondylitis (Fig. 73.20, online only). Patients are often asymptomatic but may have spinal pain or limited spinal motion. Large anterior cervical calcifications can contribute to dysphagia, hoarseness, sleep apnea, or difficulty with intubation. A rare complication is myelopathy due to spinal stenosis if the calcifications are also present within the spinal canal. Like patients with ankylosing spondylitis, patients with diffuse idiopathic skeletal hyperostosis can develop spinal fractures after relatively minor trauma.
Ossification of the Posterior Longitudinal Ligaments or Ligamentum Flavum
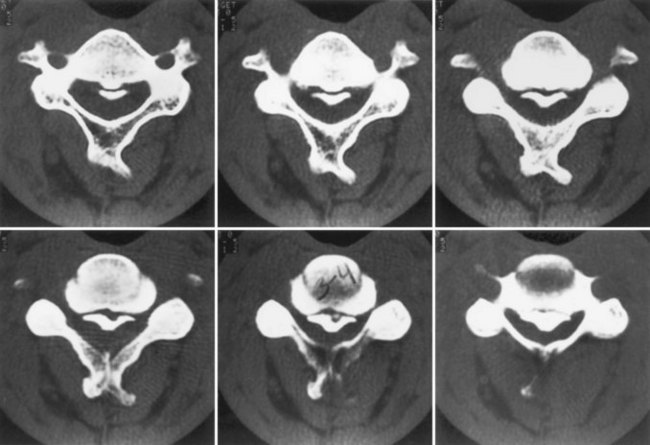
Ossification of the posterior longitudinal ligament anterior to the spinal canal (Fig. 73.21) and ossification of the ligamentum flavum posterior to the spinal canal are uncommon syndromes of acquired calcification. The posterior longitudinal ligament extends the length of the spine, separating the posterior aspects of the disks and vertebral bodies from the thecal sac. The ligamentum flavum is in the dorsal portion of the spinal canal, attaching the laminae and extending to the capsules of the facet joints and the posterior aspects of the neural foramina. Either ligament can ossify in later life, apparently independently of the usual processes of spondylosis and degenerative arthritis. Ossification of the posterior longitudinal ligament occurs more commonly in Asians than in non-Asians. It may be visible on lateral spinal radiography but is usually asymptomatic. It is better seen by CT scan, in which it is distinguished from osteophytes by favoring the middle of the vertebral bodies rather than concentrating at the endplates. Thickness of the calcification can range from 3 to 15 mm. Ossification of the posterior longitudinal ligament is most likely to be symptomatic in the cervical spine, where it can contribute to cord compression if it is thick or if the canal is further narrowed by congenital and degenerative changes.
Degenerative Disease of the Spine
Cervical Spondylosis
The cervical spinal column includes 37 joints that are continually in motion throughout life. Cervical osteoarthritis and spondylosis are ubiquitous with increasing age (Fig. 73.22, online only). These disorders can rarely be attributed to specific activities or injuries. An exception is patients with dystonia and other cervical movement disorders, who seem predisposed to premature cervical spinal degeneration. Because cervical osteoarthritis and spondylosis are so commonplace, it is usually difficult to ascertain their role in contributing to the pathogenesis of chronic neck pain or headache. Cervical spine surgery is rarely if ever indicated for treatment of headache or neck ache in the absence of cervical radiculopathy or myelopathy.
Cervical Radiculopathy
Clinical Presentation
The symptoms of cervical radiculopathy often appear suddenly (Carette and Fehlings, 2005). Although disk herniation or nerve root contusion can be caused by acute trauma, most cases become symptomatic without an identifiable preceding traumatic event. Disk herniation is more likely to be the cause in patients younger than 45 years; neural foraminal stenosis by degenerative changes is more common than disk herniation and becomes more likely with increasing age. Pain is usually in the neck, with radiation to an arm; patients may also have headache. Radiculopathic arm pain may increase with coughing or Valsalva maneuver. Arm pain may increase with neck rotation and flexion or extension to the side of the pain (Spurling sign).
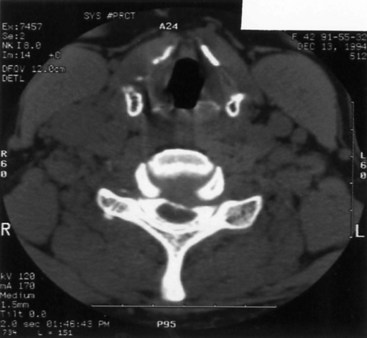
Cervical plain film radiography is of little value in diagnosing or excluding cervical radiculopathy. MRI scanning of the cervical spine is usually helpful in identifying nerve root compression in patients with cervical radiculopathy. Cervical myelography followed by CT scanning is sometimes more sensitive than MRI (Fig. 73.23). However, MRI may show nerve root compression, particularly in the neural foramina, which is invisible by CT. CT myelography is also better than MRI for distinguishing disk herniation from osteophytes. However, cervical MRI or CT myelography must be interpreted with caution because degenerative abnormalities are so commonly seen in the asymptomatic spine. EMG and nerve conduction studies can be useful in difficult diagnostic cases, both by identifying an affected motor nerve root and myotome and by helping exclude other diagnoses such as brachial plexopathy or peripheral neuropathy (Nardin et al., 1999).
Treatment
Most instances of cervical radiculopathy improve significantly over 4 to 8 weeks regardless of treatment. Various treatments such as nonsteroidal antiinflammatory drugs (NSAIDs), use of a soft cervical collar, physical therapy, or cervical traction give similar results. Patients with a typical clinical presentation and little or no neurological deficit usually can be managed with these noninvasive approaches without imaging or electrodiagnostic studies. When patients have intractable weakness or pain or have not improved with nonoperative therapy, surgical nerve root decompression is usually successful; however, there is little randomized controlled comparison of nonoperative therapy and surgery (Nikolaidis et al., 2010). Anterior cervical diskectomy, with or without fusion, or posterior cervical laminectomy are effective surgical techniques; we lack controlled trials to help choose the best surgical approach.
Cervical Spondylotic Myelopathy
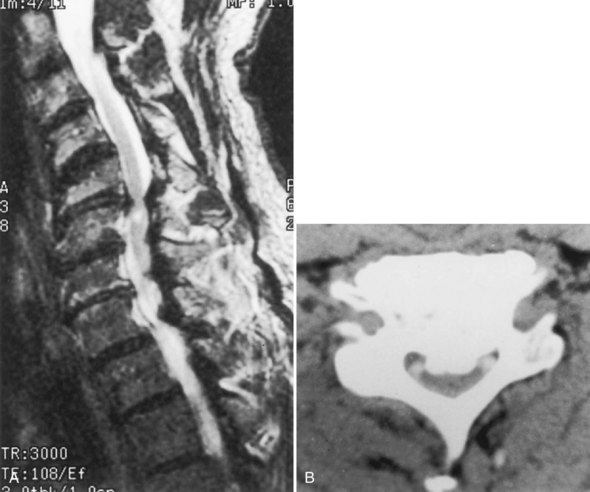
The anterior-posterior diameter of the cervical spinal cord is usually 10 mm or less. Patients rarely develop cervical spondylotic myelopathy if the congenital diameter of their spinal canal exceeds 16 mm. In congenitally narrow canals, disk protrusion, osteophytes, hypertrophy of the ligamentum flavum, ossification of the posterior longitudinal ligament, and vertebral body subluxations can combine to compress the spinal cord. MRI, CT, or myelography provide excellent images of relation between the spinal canal and the spinal cord (Fig. 73.24). MRI provides more intramedullary detail such as secondary cord edema or gliosis. CT provides better images of calcified tissues. Even with excellent cross-sectional imaging of the spinal canal, the clinical correlation between neurological deficit and cord compression is imperfect; dynamic changes in cord compression and vascular perfusion undoubtedly contribute to the pathogenesis of cervical spondylotic myelopathy.
The natural history of cervical spondylotic myelopathy is variable. Some patients have stable neurological deficits for many years without specific therapy, whereas other patients have gradual or stepwise deterioration. Some patients improve with treatments such as bed rest, soft collars, or immobilizing collars, but these treatments have not been assessed in controlled trials. Many patients with cervical spondylotic myelopathy are treated by surgical decompression, with variable surgical results (Fig. 73.25). Surgical and nonsurgical treatment results are best when the neurological deficit is mild and present less than 6 months and when the patient is younger than 70 years. Anterior cervical diskectomies are generally performed for spondylotic lesions at a limited number of levels, whereas posterior laminectomy, sometimes with an expanding laminoplasty, is generally performed for congenital spinal canal stenosis.
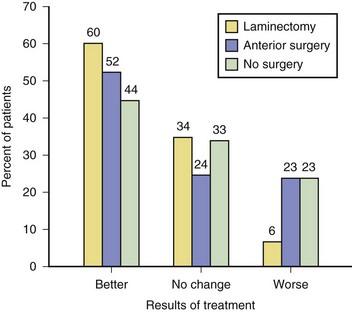
Fig. 73.25 Results of treatment of cervical spondylotic myelopathy.
(Reprinted with permission from Rosenbaum, R.B., Campbell, S.M., Rosenbaum, J.T., 1996. Clinical Neurology of Rheumatic Disease. Butterworth-Heinemann, Boston. [Data from Rowland, L.P., 1992. Surgical treatment of cervical spondylotic myelopathy: time for a controlled trial. Neurology 42, 5.])
Vertebral Artery Stroke Caused by Cervical Osteoarthritis
Compression of a vertebral artery by an osteophyte is a rare cause of stroke in the vertebrobasilar distribution (Bulsara et al., 2006). The vertebral arteries pass through foramina in the transverse processes from C6 to C2. Osteophytes from the uncinate joints can compress the arteries. The compression may occur only with head turning. However, the turning usually leaves the contralateral vertebral artery uncompressed, so ischemic symptoms are usually limited to those patients who have both osteophytic arterial compression on one side and a contralateral hypoplastic, absent, or occluded artery.
Thoracic Spondylosis
Degenerative changes are less common in the thoracic than in the lumbar or cervical spines (Vanichkachorn and Vaccaro, 2000). Thoracic osteophytes are more likely to develop on the anterior or lateral aspects of the vertebral bodies and infrequently cause clinical radiculopathy. Thoracic disk herniations are visible on MRI in many asymptomatic individuals. Thoracic disk herniations occur most often in the lower thoracic spine. These rarely cause cord or root compression and may regress spontaneously.
Thoracic myelopathy due to disk herniation probably has an annual incidence of approximately 1 case per 1 million. Most cases occur between ages 30 and 60 years. Symptoms often develop insidiously without identifiable preceding trauma. Back pain may or may not be present. Patients have some combination of motor and sensory findings of myelopathy; sphincter dysfunction is present in more severe cases. Thoracic MRI, CT, or myelography can confirm the diagnosis (Fig. 73.26). The treatment is surgical decompression.
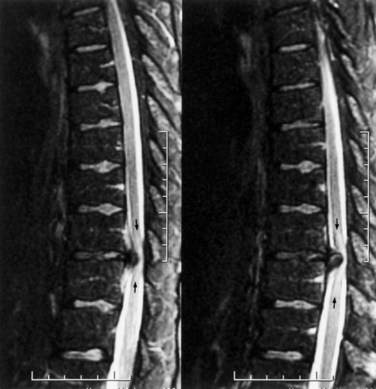
Herniation of the thoracic spinal cord through the dura is a rare cause of thoracic myelopathy (Sasani et al., 2009) (Fig. 73.27). The most common presentation is a Brown-Séquard syndrome that can slowly evolve over a few years. Thoracic spine MRI shows the spinal cord ventrally deviated, often with a kink at one level between T2 and T7, and with increased dorsal subarachnoid space. CT myelography will show similar changes and confirm that there is no subarachnoid space ventrally where the spinal cord is attached to the dura. The adjacent vertebral body may appear scalloped. Spinal cord herniation can be idiopathic, presumably due to congenital defects in the dura, or occur after trauma or thoracic spinal surgery. Rare instances are associated with thoracic disk herniation. Some patients improve after surgical reduction of the herniation.
Lumbar Spondylosis
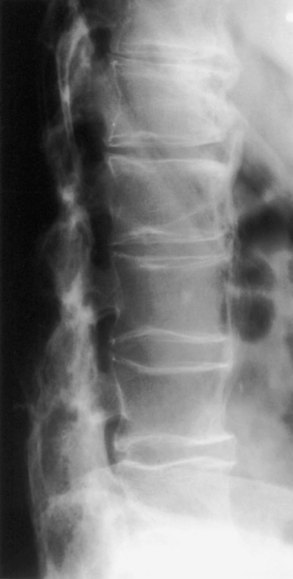
Low Back Pain
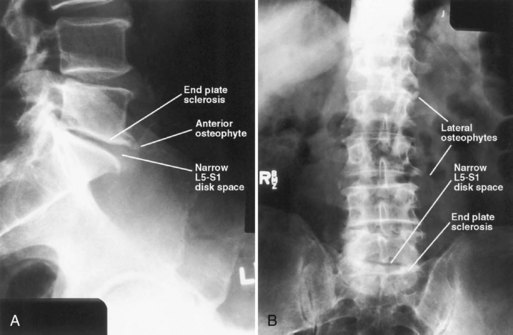
The findings of osteoarthritis and lumbar spondylosis on radiography (osteophytes, endplate sclerosis, disk-space narrowing) appear gradually with increasing age and are rarely absent by age 60 years (Fig. 73.28). The presence or absence of these findings does not correlate with symptoms and demonstrating them is of no diagnostic or therapeutic value. Therefore radiography of the lumbar spine is indicated only when alternative diagnoses such as compression fractures, neoplasia, or infections are being seriously considered. The Agency for Health Care Policy and Research has recommended that spinal radiography be reserved for patients with “red flags” for trauma, tumor, or infection (Box 73.6). Even limiting radiography to patients meeting these guidelines results in many needless radiographs. For example, back pain in a patient older than 50 years need not be an indication for imaging studies unless other findings suggest a condition more serious than nonspecific low back pain.
Box 73.6
Indications for Lumbar Spine Radiography in Patients with Acute Low Back Pain
Adapted with permission from Agency for Health Care Policy and Research, 1994. Acute Low Back Problems in Adults. Assessment and Treatment: Quick Reference Guide for Clinicians. U.S. Department of Health and Human Services, Rockville, MD.
There are many causes of lumbar disk disease, including body habitus, type and amount of physical activity, acute injury, and genetic predisposition, which is complex and polygenic with a number of known candidate genes (Kalichman and Hunter, 2008).
Lumbar Radiculopathies
Monoradiculopathy
Diagnostic Studies
Disk herniations, osteophytes, spondylolysis and spondylolisthesis, facet joint hypertrophy, and hypertrophy or calcification of intraspinal ligaments can compress nerve roots of the cauda equina within the spinal canal or in the lateral recesses and neural foramina through which the roots exit the spinal canal. The anatomical relations between the nerve roots and the surrounding tissues are well visualized by lumbar MRI or CT myelography (Fig. 73.29 (B, online only). Each technique has high sensitivity for demonstrating causes of nerve root compression. On occasion when a patient has strong clinical evidence of lumbar radiculopathy, but initial imaging studies do not show the cause of the compression, a second complementary imaging study is indicated. For example, imaging with a lumbar MRI usually is sufficient for most clinical purposes, but occasionally a patient also needs CT myelography to clarify the anatomy. Unfortunately all spinal imaging modalities frequently show anatomical abnormalities that are not the cause of symptomatic nerve root dysfunction; all imaging results must be interpreted carefully in clinical context.
Treatment
Invasive techniques such as steroids and local anesthetics injected epidurally or into facet joints are used for some patients with low back pain or radiculopathy, but we still lack consistent proof of their long-term efficacy or cost-effectiveness from randomized controlled trials (Chou et al., 2009). Lumbar epidural steroid injections may result in some improvement in radicular pain if pain is assessed 2 to 6 weeks after injections, but the injections do not have proven longer-lasting value (Armon et al., 2007).
When surgery is performed for lumbar nerve root compression, the surgical technique depends on the clinical details such as the cause of compression and the number of nerve roots compressed. In patients with lumbar radiculopathy due to disk herniation, the most common surgical approach is microsurgical diskectomy with minimal removal of the lamina. In controlled trials, this surgery provided better relief of symptoms than nonoperative therapy, based on results 2 to 3 months after treatment; however, the advantage of surgery decreased with longer follow-up (Chou et al., 2009). Perhaps 90% of patients report excellent relief of neuropathic pain after surgery. Many are able to return to physically strenuous work. However, a small proportion of patients postoperatively develop more severe chronic pain problems (failed back surgery syndrome), which particularly occurs when patients selected for surgery have neither clinical evidence of radiculopathy nor corresponding neuroimaging evidence of nerve root compression. Patients with chronic postoperative pain require careful neurological evaluation to consider such problems as surgery done at the wrong level, incomplete removal of extruded disk fragment or other matter compressing the nerve root, progression of spinal degeneration, postoperative arachnoiditis, and psychosocial issues interfering with recovery.
Lumbar Canal Stenosis
Lumbar canal stenosis results from subnormal cross-sectional area of the spinal canal due to varied anatomical changes including congenitally small canal size, degenerative osteophytes, spondylolisthesis, facet joint hypertrophy, thickening of the ligamentum flavum, disk-space narrowing, and disk herniation (Katz and Harris, 2008). It usually develops insidiously with aging and rarely becomes symptomatic before age 40 years unless due to congenital stenosis or skeletal changes like those of achondroplasia. Men are more often affected than women.
Diagnostic Studies
Spinal canal stenosis can be studied by MRI, CT, and myelography (Fig. 73.30). MRI is best at demonstrating sagittal relationships such as the role of spondylolisthesis in narrowing the canal. CT is best at studying calcified tissues and distinguishing disk from osteophyte, especially within the neural foramina. No imaging modality quantifies the extent of nerve root compression, and clinical correlations between symptoms and apparent reduction in size of the spinal canal are imperfect. In choosing which patients would benefit from decompressive surgery, one should rely more heavily on clinical findings than on the appearance of the canal in imaging studies.
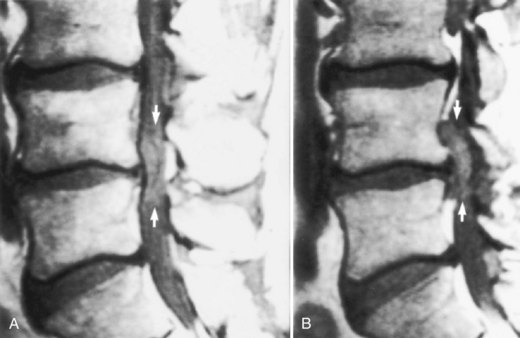
Fig. 73.30 Magnetic resonance image of patient with lumbar spinal stenosis. Midline (A) and parasagittal (B) images of lumbar spine show narrow anteroposterior dimensions of spinal canal consistent with spinal canal stenosis (see normal dimensions in Fig. 73.28). L4-L5 disk herniation (arrows) that fills entire spinal canal is actually much smaller than the herniation shown in Fig. 73.28.
(Courtesy Erik Gaensler.)
Treatment
Patients who have neurogenic intermittent claudication may have stable symptoms for many years without developing progressive neurological deficit. Some even note regression of symptoms after months of recurrent claudication. These patients may be managed with mild analgesics. Some describe decreased discomfort if they walk with a slight stoop or using a cane. Those patients with intractable leg pain or progressive neurological deficit can be treated with wide laminectomy of the stenosed spinal canal, which usually improves claudication and may help back pain (Chou et al., 2009; Weinstein et al., 2008). Those with severe or multilevel stenosis are least likely to benefit from surgery.
Infectious Diseases of the Spine
Pyogenic Vertebral Osteomyelitis and Epidural Abscess
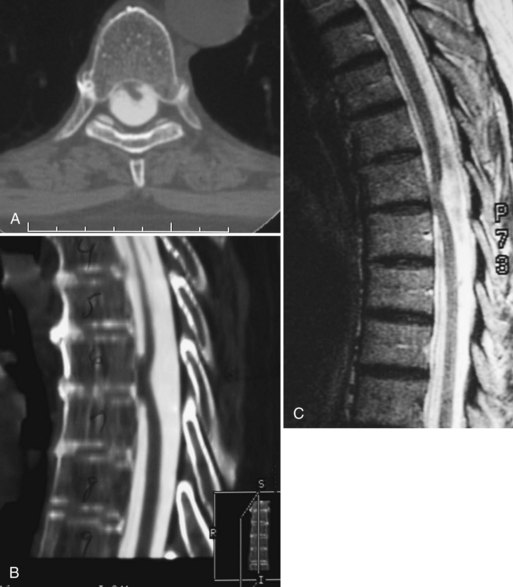
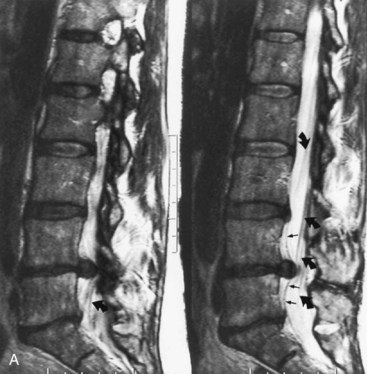
Vertebral osteomyelitis and spinal epidural abscess (see Chapter 53C) are uncommon conditions that present with focal spinal pain and tenderness (Curry et al., 2005; Zimmerli, 2010). Epidural abscesses in the anterior spinal canal are more likely than those in the posterior canal to be associated with osteomyelitis. In either location, they can cause radiculopathic pain, compromise nerve root function, or lead to spinal cord compression. Some patients with spinal epidural abscess or vertebral osteomyelitis are afebrile at presentation, but nearly all have an elevated erythrocyte sedimentation rate. Early in the infection, routine spinal radiography may be normal. If the diagnosis is being considered, an MRI scan (Fig. 73.31) of the involved area is sensitive for detecting vertebral body abnormalities and is particularly helpful to assess for epidural or paravertebral infection. Spinal CT is useful if MRI is unavailable or contraindicated.
Granulomatous Vertebral Osteomyelitis
Pott disease classically presents with destruction of vertebral bodies. Routine spine radiography results are usually abnormal by the time the diagnosis is made, and spinal deformity is a common complication. MRI or CT is needed to assess for contiguous abscess in the epidural or paraspinal spaces and to evaluate possible nerve root or spinal cord compression when the spine is deformed (Fig. 73.32). Compression of spinal cord or nerve roots can occur in vertebral TB by vertebral deformity or collapse, epidural abscess, granulation tissue, or bony sequestrum. Patients may develop delayed neurological complications after apparently successful treatment of the infection. This may be due to infarction from endarteritis obliterans, delayed degenerative bony changes, or reactivation of infection. Neurological compression is most common with thoracic vertebral disease, hence the eponym Pott paraplegia; cauda equina compression is uncommon in TB. Treatment of vertebral TB requires long-term multiple-drug antituberculous therapy. Spinal surgery may be needed, depending on the degree of spinal destruction or deformity, and is often required in cases of neurological compression.
Inflammatory Joint Disease
Rheumatoid Arthritis
Neurological Manifestations
Common neurological complications of RA are carpal tunnel syndrome and other nerve entrapments, peripheral neuropathy, and myopathy; these are discussed in Chapters 49A, 75, 76, and 79. RA can evolve to a rheumatoid vasculitis that like other medium-sized vessel vasculitides has the potential to cause ischemic mononeuritis, mononeuritis multiplex, or (rarely) stroke.
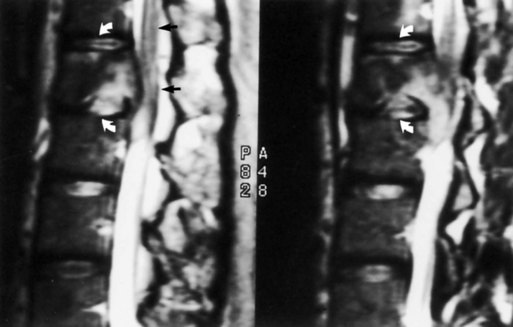
Patients with progressive RA can develop subluxation at the atlantoaxial joint. Lateral atlantoaxial joint subluxation rarely causes focal neurological dysfunction but can contribute to neck ache and headache. Horizontal atlantoaxial joint subluxation, often combined with adjoining soft-tissue pannus, can cause myelopathy, especially in patients with a smaller congenital canal diameter (Fig. 73.33, online only). The earliest neurological sign is usually hyperreflexia; assessment of gait and strength in patients with advanced RA is often difficult because of their peripheral joint pain and deformity. Vertical subluxation can lead to spinal cord or brainstem compression or rarely to vertebral artery compression or injury.
Inflammatory Spondyloarthropathies
Clinical Presentation
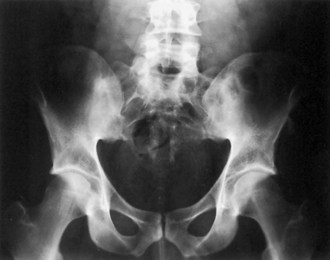
The inflammatory spondyloarthropathies include ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and the arthritis of inflammatory bowel disease. Ankylosing spondylitis is characterized by inflammatory low back pain, loss of spinal range of motion, sacroiliitis, and as it advances, radiographic evidence of sacroiliitis and spondylitis (Fig. 73.34, online only). The clinical symptoms of inflammatory spine disease are insidious onset of low back (and sometimes buttock) pain lasting more than 3 months, prominent morning stiffness, and improvement with activity. Nocturnal back pain can be present. Some patients with inflammatory back pain do not meet diagnostic criteria for an inflammatory spondyloarthropathy (Heuft-Dorenbosch et al., 2007). Most patients become symptomatic before age 40 years, and men are affected more commonly than women. Other organ systems are affected commonly in patients with inflammatory spondyloarthropathies; manifestations include uveitis, mucocutaneous lesions, peripheral arthritis, gastrointestinal disease, cardiac disease, and enthesopathy. (Entheses are sites of insertion of ligament or tendon to bone). The syndesmophytes that form where spinal ligaments join vertebral bodies are one form of enthesopathy. Examples of other sites of enthesopathy are the foot (Achilles tendonitis, plantar fasciitis, heel pain), fingers or toes (dactylitis or sausage digits), and symphysis pubis, clavicle, and ribs.
Spinal Neurological Complications
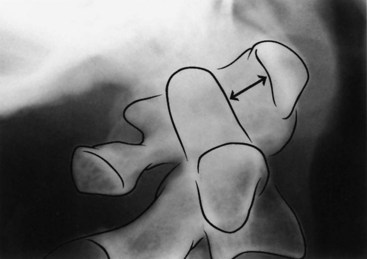
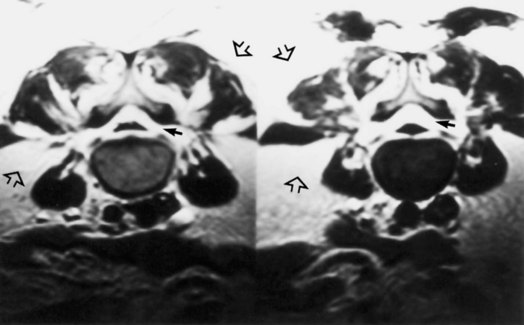
The neurological complications of the inflammatory spondyloarthropathies generally do not occur until spinal disease is clinically advanced (e.g., loss of spinal range of motion, kyphosis) and radiologically evident (e.g., vertebral body squaring, syndesmophytes). Spinal complications include atlantoaxial joint subluxation, spinal fractures and pseudoarthroses, diskovertebral destruction, spinal canal stenosis, and cauda equina syndrome due to lumbar arachnoid diverticula (Table 73.7).
Table 73.7 Spinal Complications of Ankylosing Spondylitis Based on 105 Hospitalized Patients
| Anatomically Abnormal | Neurologically Abnormal | |
|---|---|---|
| Spinal fracture | 13 | 7 |
| Diskovertebral destruction | 4 | 0 |
| Atlantoaxial subluxation | 1 | 0 |
| Spinal canal stenosis | 2 | 2 |
Data from Weinstein, P., Karpman, R.R., Gall, E.P., et al., 1982. Spinal injury, spinal fracture, and spinal stenosis in ankylosing spondylitis. J. Neurosurg. 37, 609–616. Reprinted from Rosenbaum, R.B., Campbell, S.M., Rosenbaum, J.T., 1996. Clinical Neurology of Rheumatic Disease. Butterworth-Heinemann, Boston.
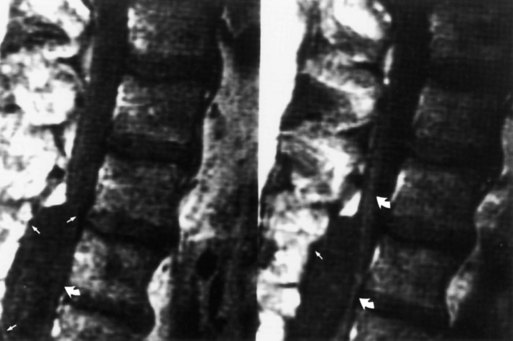
Destruction of a disk, particularly in the low lumbar or high thoracic region, is a late complication of spondylitis (Fig. 73.35, online only). The adjacent vertebral bodies also may be involved. An initiating trauma is not always identified. The destruction may be asymptomatic or painful. The pain increases with movement and decreases at rest, in contrast to typical inflammatory low back pain. An epidural inflammatory response leading to cord compression can occur.
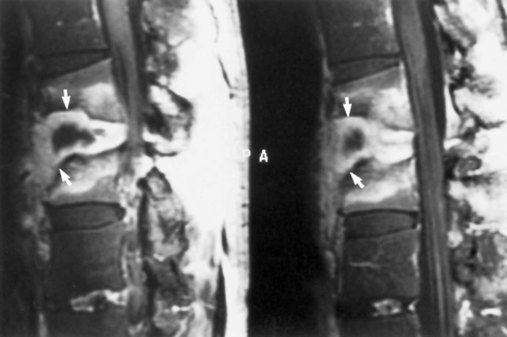
Fig. 73.35 Magnetic resonance image shows diskovertebral destruction (arrows) in a patient with ankylosing spondylitis. There is a chronic fracture in the lower thoracic spine, which is otherwise rigid because of bony fusion. Chronic hypermobility at this single nonfused segment has occurred, leading to exuberant fibrous tissue development. The fibrous tissue enhances on this post–gadolinium-enhanced T1-weighted image. This appearance can be mistaken for infectious spondylitis (see Fig. 73.31) if the presence of a bamboo spine on plain films is overlooked.
(Courtesy Erik Gaensler.)
Cauda equina syndrome with insidious evolution of leg pain, sensory loss, leg weakness, and sphincter dysfunction is a late complication of inflammatory spondyloarthropathy. Imaging studies (MRI, CT, myelography) show posterior lumbosacral arachnoid diverticula (Fig. 73.36, online only). Although arachnoiditis may play a role in development of this syndrome, the presence of the diverticula distinguishes it from most cases of chronic adhesive arachnoiditis.
Nonspinal Neurological Complications
Rare nonspinal complications of inflammatory spondyloarthropathies include brachial plexopathy or tarsal tunnel syndrome. Proximal weakness and atrophy, sometimes with mild elevations of serum CK level, often occur in advanced cases of spondylitis, suggesting an inflammatory myopathy. In patients with psoriatic arthritis, the myopathy is occasionally painful. A number of case reports detail unusual neurological illnesses in patients with reactive arthritis (Box 73.7).
Epidural Lipomatosis
Epidural lipomatosis is a non-neoplastic accumulation of fatty tissue in the thoracic or lumbar epidural space that can occur idiopathically but is more commonly a complication of chronic corticosteroid excess, obesity, or hypothyroidism (Koch et al., 2000). A typical patient has been on corticosteroids for more than 6 months and is obese and cushingoid; spinal radiography typically shows diffuse osteoporosis. Epidural lipomatosis is also a rare manifestation of the lipodystrophy that can complicate highly active antiretroviral therapy of HIV infection. Symptomatic segmental spinal cord or nerve root compression is typically associated with epidural fat thickness of more than 7 mm in the region of compression; however, the diameter of the spinal canal and other factors affect the manifestations, so the epidural fat can be thickened asymptomatically. Patients with epidural lipomatosis usually have a body mass index of more than 27.5 kg/m2 (Fig. 73.37, online only). The compressive tissue can regress when corticosteroid doses are decreased, but the compression of neurological tissue may be severe enough to require laminectomy.
Chronic Meningitis
Most cases of chronic meningitis are due to infection (see Chapter 53B), neoplasia (see Chapter 75), or sarcoidosis (see Chapter 49A). A comprehensive differential diagnosis includes Behçet syndrome, isolated CNS angiitis (see Chapter 51F), systemic lupus erythematosus, Sjögren syndrome, or Wegener granulomatosis. Some other chronic or recurring meningitic syndromes merit discussion; however, a cause is not found in many cases of chronic meningitis.
Chronic Adhesive Arachnoiditis
Adhesive arachnoiditis occasionally complicates a variety of surgical or medical violations of the thecal sac (Box 73.8, online only). Focal arachnoiditis is most common in the cauda equina following lumbar disk surgery or myelography, particularly if oil-based contrast has been used for the latter. The symptoms can include local or radicular pain, radicular paresthesia, and less commonly more severe findings of oligoradiculopathy such as motor loss or sphincter dysfunction. The diagnosis can usually be made by spinal MRI, which may show clumping of nerve roots, nodules in the subarachnoid space, loculation of spinal fluid, and local areas of enhancement. The nerve roots may clump at the periphery of the thecal sac, usually adjacent to an area of previous surgery, or in the center of the sac, usually in areas of spinal stenosis. The extent of the MRI findings correlates poorly with the severity of the clinical nerve root dysfunction. Spinal fluid may show increased CSF protein levels and mild to moderate mononuclear pleocytosis. Surgical débridement of the arachnoiditis is sometimes attempted but is usually unsuccessful and may lead to increased neurological deficit. Epidural or intrathecal corticosteroids are sometimes tried, but there is no proof of their efficacy, and there are reports of arachnoiditis caused by their use. Therefore, most treatment is aimed at symptomatic pain control, except in the unusual patient with progressive neurological deficits.
Recurrent Meningitis
Patients with recurrent attacks of acute bacterial meningitis need to be screened for dural CSF leaks or fistulas, parameningeal infections, and immunodeficiency (see Chapter 53A) (Tebruegge and Curtis, 2008). Recurrent meningitis can also be caused by chemical irritants leaked from tumors like dermoids, epidermoids, or craniopharyngiomas. Drug-induced meningitis, most common as an idiosyncratic reaction to NSAIDs, can recur with repeated drug exposures. Rarely, recurrent meningitis can complicate systemic inflammatory diseases such as systemic lupus erythematosus, Sjögren syndrome, Behçet disease, Lyme disease, familial Mediterranean fever, or sarcoidosis.
Recurrent benign lymphocytic meningitis, sometimes called Mollaret meningitis, can cause multiple self-limited attacks with symptoms like headache, fever, and meningismus; each attack lasts a few days (Shalabi and Whitley, 2006). Transient neurological features that may include seizures, hallucinations, diplopia, cranial nerve palsies, or altered consciousness can accompany the syndrome, implying that some cases are actually meningoencephalitis. The spinal fluid shows a mixed pleocytosis, sometimes including large macrophage-like cells (Mollaret cells). Most cases are due to herpes virus infections, especially HSV2. Some argue that the eponym Mollaret meningitis should be reserved for those cases without an identified causative organism.
Pachymeningitis
Pachymeningitis is a rare condition, visible on MRI scans as thickened gadolinium-enhancing dura (Kupersmith et al., 2004) (Fig. 73.38). The differential diagnosis is extensive (Box 73.9, online only). Low CSF pressure from causes such as post lumbar puncture or idiopathic intracranial hypotension can also cause dural enhancement on MRI. The enhancement when the CSF pressure is low is usually diffuse and smooth, whereas pachymeningitis can cause diffuse or localized irregular areas of enhancement. Idiopathic hypertrophic pachymeningitis refers to those cases for which cultures, serology, and dural biopsy fail to reveal a causative infection, tumor, or systemic inflammatory disease. These patients can present with headache, cranial neuropathies, ataxia, or seizures. Pachymeningitis in the spinal canal can cause radiculopathy or myelopathy. CSF may show high protein or lymphocytic pleocytosis but is sterile. Dural biopsy shows small mature lymphocytes, plasma cells, and epithelioid histiocytes. Patients can improve after treatment with corticosteroids, sometimes supplemented with drugs such as azathioprine and methotrexate.
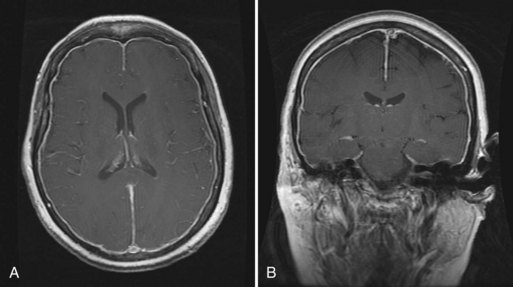
Uveomeningitis Syndromes
The combination of chronic or recurrent meningitis and uveitis has a specific differential diagnosis (Yeh et al., 2011) (Box 73.10, online only). Often, ophthalmological characterization of the uveitis can refine the differential diagnosis. For example, the uveitis of Vogt-Koyanagi-Harada syndrome is bilateral and often causes retinal elevations and retinal pigmentary changes. Vogt-Koyanagi-Harada syndrome also causes skin and hair findings such as vitiligo, poliosis, or focal alopecia.
Box 73.10
Causes of Combined Uveitis and Meningitis
Acute multifocal placoid pigmentary epitheliopathy
Human T-cell lymphotropic virus type 1 (HTLV-1) infection
Infection in immunocompromised host
Isolated central nervous system angiitis
Reprinted with permission from Rosenbaum, R.B., Campbell, S.M., Rosenbaum, J.T., 1996. Clinical Neurology of Rheumatic Disease. Butterworth-Heinemann, Boston.
Superficial Hemosiderosis
Superficial hemosiderosis is a rare disorder that causes slowly progressive cerebellar ataxia, mainly of gait, and sensorineural deafness, sometimes accompanied by manifestations of myelopathy such as spasticity, brisk reflexes, extensor plantar responses, bladder disturbance, or sensory signs (Kumar et al., 2006). Less common features include dementia, anosmia, or anisocoria, and more rarely, extraocular motor palsies, neck or backache, bilateral sciatica, or lower motor neuron signs (5%-10% each). Men are more often affected than women. The clinical diagnosis is difficult, but the neuroradiological abnormalities are striking. MRI shows a black rim around the posterior fossa structures and spinal cord and less often the cerebral hemispheres on T2-weighted images (Fig. 73.39). These paramagnetic signal changes represent encrustation of the brain surfaces with hemosiderin. The adjacent neural tissue atrophies, with accumulation of ferritin in microglia and Bergmann cells in the cerebellum. Superficial siderosis is secondary to chronic or recurrent blood leakage into the subarachnoid space. CSF analysis often shows xanthochromia, red blood cells, or elevated protein; however, not all patients have an identifiable source of bleeding. Possible sources include brain or spine trauma, neurosurgery, cerebral or spinal aneurysms or vascular malformations, cerebral amyloid angiopathy, and spinal dural defects, which are sometimes identified by fluid collections in the spine appearing as meningoceles or pseudomeningoceles or by dynamic CT myelography. Some patients with spinal dural leaks develop both superficial siderosis and intracranial hypotension (Kumar et al., 2007). Treatment relies on identifying and arresting the source of bleeding; chelation therapy does not appear to be effective.
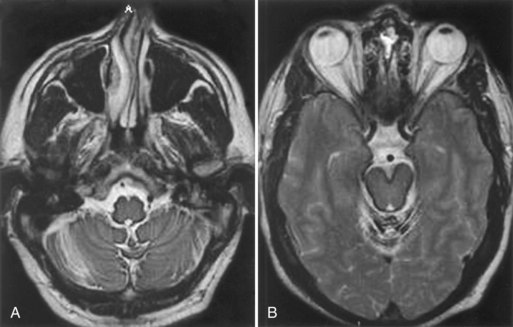
Fibromyalgia
Fibromyalgia, a syndrome defined by widespread musculoskeletal or soft-tissue pain and multiple tender points, is part of the differential diagnosis of many patients with spinal pain. The American College of Rheumatology classification criteria for the diagnosis define pain as widespread when it is bilateral, above and below the waist, and axial. To meet the classification criteria, a patient must have tenderness to palpation at 11 or more of 18 specific points (Fig. 73.40), but the validity of these tender points as diagnostic criteria has been cogently questioned. Typically, patients have multiple symptoms including fatigue, stiffness, nonrestorative sleep, headaches, and mood disorders (Bennett, 2009). Patients may have many symptoms of neurological import such as weakness, paresthesia, imbalance, and dizziness and often have cognitive complaints regarding concentration, memory, and multitasking. Nonetheless, their neurological examinations are normal unless they have a separate neurological illness. Diagnostic neurological investigations such as brain imaging, muscle biopsy, or electrodiagnostic studies are normal or show minor nonspecific abnormalities.
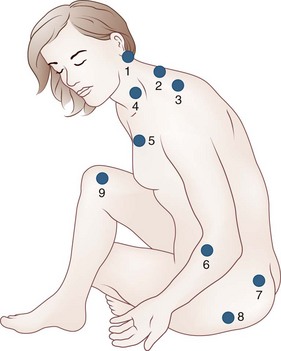
The cause of most cases of fibromyalgia is unknown. Behavioral and biological factors both contribute to the clinical presentation of the syndrome. Neuroscientific research on the pathogenesis of fibromyalgia has examined muscle, sleep, neuroendocrine function, and central pain processing, including studies using functional brain imaging (Nebel and Gracely, 2009). Symptoms and signs of fibromyalgia can occur in association with autoimmune diseases such as systemic lupus erythematosus or other systemic illness such as hypothyroidism. Focal trauma can cause localized self-limited soft-tissue myofascial pain. The pathogenic role of trauma, on-the-job injury, or workplace stress is controversial. Treatment includes a supportive doctor-patient relationship, aerobic exercise, and avoiding inactivity. Pregabalin, duloxetine, and milnacipran are now approved by the U.S. Food and Drug Administration (FDA) for treatment of fibromyalgic pain. Other drugs for chronic pain (e.g., tricyclic antidepressants) are sometimes tried off label. Small short-term controlled studies have suggested that some patients benefit from acupuncture.
Armon C., Argoff C.E., Samuels J., et al. Assessment: use of epidural steroid injections to treat radicular lumbosacral pain: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2007;68:723-729.
Attal N., Parker R., Tadi M., et al. Effects of surgery on the sensory deficits of syringomyelia and predictors of outcome: a long-term prospective study. J Neurol Neurosurg Psychiatry. 2004;75:1025-1030.
Basel D., Steiner R.D. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med. 2009;11:375-385.
Batzdorf U. Primary spinal syringomyelia. J Neurosurg Spine. 2005;3:429-435.
Bennett R.M. Clinical manifestations and diagnosis of fibromyalgia. Rheum Dis Clin North Am. 2009;35:215-232.
Berven S., Bradford D.S. Neuromuscular scoliosis: causes of deformity and principles for evaluation and management. Semin Neurol. 2002;22:167-178.
Bogdanov E.I., Heiss J.D., Mendelevich E.G., et al. Clinical and neuroimaging features of “idiopathic” syringomyelia. Neurology. 2004;62:791-794.
Botto L.D., Moore C.A., Khoury M.J., et al. Neural-tube defects. N Engl J Med. 1999;341:1509-1519.
Buchbinder R., Osborne R.H., Ebeling P.R., et al. A randomized trial of vertebroplasty for painful osteoporotic vertebral fractures. N Engl J Med. 2009;361:557-568.
Bulsara K.R., Velez D.A., Villavicencio A. Rotational vertebral artery insufficiency resulting from cervical spondylosis: case report and review of the literature. Surg Neurol. 2006;65:625-627.
Carette S., Fehlings M.G. Cervical radiculopathy. N Engl J Med. 2005;353:392-399.
Carroll A.M., Brackenridge P. Post-traumatic syringomyelia. A review of the cases presenting in a regional spine injuries unit in the north east of England over a 5-year period. Spine. 2005;30:1206-1210.
Chen Y.J., Tan T.S., Chen W.H., et al. Intradural cement leakage. A devastatingly rare complication of vertebroplasty. Spine. 2006;31:379-382.
Chou R., Baisden J., Carragee E.J., et al. Surgery for low back pain: a review of the evidence for an American Pain Society Clinical Practice Guideline. Spine (Phila Pa 1976). 2009;34:1094-1109.
Curry W.T.Jr., Hoh B.L., Amin-Hanjani S., et al. Spinal epidural abscess: clinical presentation, management, and outcome. Surg Neurol. 2005;63:364-371.
Davids J.R., Chamberlin E., Blackhurst D.W. Indications for magnetic resonance imaging in presumed adolescent idiopathic scoliosis. J Bone Joint Surg. 2004;86-A:2187-2195.
Di Lorenzo N., Cacciola F. Adult syringomyelia. J Neurosurg Sci. 2005;49:65-72.
Diamond T.H., Bryant C., Browne L., et al. Clinical outcomes after acute osteoporotic vertebral fractures: a 2-year non-randomised trial comparing percutaneous vertebroplasty with conservative therapy. Med J Aust. 2006;184:113-117.
Do H.M. Magnetic resonance imaging in the evaluation of patients for percutaneous vertebroplasty. Top Magn Reson Imaging. 2000;11:234-244.
Drake J.M. Occult tethered cord syndrome: not an indication for surgery. J Neurosurg. 2006;104:305-308.
Garland E.M., Robertson D. Chiari I malformations a cause of orthostatic intolerance symptoms: a media myth? Am J Med. 2001;111:546-552.
Goel A., Bhatjiwale M., Desai K. Basilar invagination: a study based on 190 surgically treated patients. J Neurosurg. 1998;88:962-968.
Grond-Ginsbach C., Debette S. The association of connective tissue disorders with cervical artery dissections. Curr Mol Med. 2009;9:210-214.
Haughton V.M., Korosec F.R., Medow J.E., et al. Peak systolic and diastolic CSF velocity in the foramen magnum in adult patients with Chiari I malformations and in normal control participants. Am J Neuroradiol. 2003;24:169-176.
Hayes M., Parker G., Ell J., et al. Basilar impression complicating osteogenesis imperfecta type IV: the clinical and neuroradiological findings in four cases. J Neurol Neurosurg Psychiatry. 1999;66:357-364.
Heuft-Dorenbosch L., Landewe R., Weijers R., et al. Performance of various criteria sets in patients with inflammatory back pain of short duration; the Maastricht early spondyloarthritis clinic. Ann Rheum Dis. 2007;66:92-98.
Jensen M.E., McGraw J.K., Cardella J.F., et al. Position statement on percutaneous vertebral augmentation: a consensus statement developed by the American Society of Interventional and Therapeutic Neuroradiology, Society of Interventional Radiology, American Association of Neurological Surgeons/Congress of Neurological Surgeons, and American Society of Spine Radiology. AJNR Am J Neuroradiol. 2007;28:1439-1443.
Kalichman L., Hunter D.J. The genetics of intervertebral disc degeneration. Familial predisposition and heritability estimation. Joint Bone Spine. 2008;75:383-387.
Katz J.N., Harris M.B. Clinical practice. Lumbar spinal stenosis. N Engl J Med. 2008;358:818-825.
Koch C.A., Doppman J.L., Patronas N.J., et al. Do glucocorticoids cause spinal epidural lipomatosis? When endocrinology and spinal surgery meet. Trends Endocrinol Metab. 2000;11:86-90.
Kovach M.J., Waggoner B., Leal S.M., et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. 2001;74:458-475.
Kula R.W. Chiari malformations and syringomyelia. In: Johnson R.T., Griffin J.W., McArthur J.C. Current Therapy in Neurologic Disease. Philadelphia: Mosby; 2006:92-97.
Kumar N., Cohen-Gadol A.A., Wright R.A., et al. Superficial siderosis. Neurology. 2006;66:1144-1152.
Kumar N., McKeon A., Rabinstein A.A., et al. Superficial siderosis and CSF hypovolemia: the defect (dural) in the link. Neurology. 2007;69:925-926.
Kupersmith M.J., Martin V., Heller G., et al. Idiopathic hypertrophic pachymeningitis. Neurology. 2004;62:686-694.
Meadows J., Kraut M., Guarnieri M., et al. Asymptomatic Chiari type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000;92:920-926.
Menezes A.H. Craniovertebral junction anomalies: diagnosis and management. Semin Pediatr Neurol. 1997;4:209-223.
Michelson D.J., Ashwal S. Tethered cord syndrome in childhood: diagnostic features and relationship to congenital abnormalities. Neurol Res. 2004;26:745-753.
Milhorat T.H., Bolognese P.A., Nishikawa M., et al. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and Chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7:601-609.
Milhorat T.H., Bolognese P.A., Nishikawa M., et al. Association of Chiari malformation type I and tethered cord syndrome: preliminary results of sectioning filum terminale. Surg Neurol. 2009;72:20-35.
Milhorat T.H., Chou M.W., Trinidad E.M., et al. Chiari I malformation redefined: clinical and radiologic findings for 364 symptomatic patients. Neurosurgery. 1999;44:1005-1017.
Milhorat T.H., Nishikawa M., Kula R.W., et al. Mechanisms of cerebellar tonsil herniation in patients with Chiari malformations as guide to clinical management. Acta Neurochir (Wien). 2010;152:1117-1127.
Nardin R.A., Patel M.R., Gudas T.F., et al. Electromyography and magnetic resonance imaging in the evaluation of radiculopathy. Muscle Nerve. 1999;22:151-155.
Nebel M.B., Gracely R.H. Neuroimaging of fibromyalgia. Rheum Dis Clin North Am. 2009;35:313-327.
Nikolaidis, I., Fouyas, I.P., Sandercock, P.A., et al., 2010. Surgery for cervical radiculopathy or myelopathy. Cochrane Database Syst Rev; CD001466.
Pepin M., Schwarze U., Superti-Furga A., et al. Clinical and genetic features of Ehlers-Danlos syndrome Type IV, the vascular type. N Engl J Med. 2000;342:673-680.
Phillipi, C.A., Remmington, T., Steiner, R.D., 2008. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev; CD005088.
Poncelet A. The neurologic complications of Paget’s disease. J Bone Miner Res. 1999;14(Suppl 2):88-91.
Prockop D.J., Czarny-Ratajczak M. Heritable disorders of connective tissue. In: Fauci A.S., Braunwald E., Kasper D.L., et al. Harrison’s Principles of Internal Medicine. seventeenth ed. New York: McGraw Hill Medical; 2008:2461-2469.
Ralston S.H., Langston A.L., Reid I.R. Pathogenesis and management of Paget’s disease of bone. Lancet. 2008;372:155-163.
Raubenheimer P.J., Taylor A.G., Soule S.G. Paget’s disease complicated by hydrocephalus and syringomyelia. Br J Neurosurg. 2002;16:513-516.
Roohi F., Mann D., Kula R.W. Surgical management of hydrocephalic dementia in Paget’s disease of bone: the 6-year outcome of ventriculo-peritoneal shunting. Clin Neurol Neurosurg. 2005;107:325-328.
Rose P.S., Levy H.P., Liberfarb R.M., et al. Stickler syndrome: clinical characteristics and diagnostic criteria. Am J Med Genet A. 2005;138A:199-207.
Royal S.A., Tubbs R.S., D’Antonio M.G., et al. Investigations into the association between cervicomedullary neuroschisis and mirror movements in patients with Klippel-Feil syndrome. Am J Neuroradiol. 2002;23:724-729.
Rubin D.J., Levin R.M. Neurologic complications of Paget disease of bone. Endocr Pract. 2009;15:158-166.
Sasani M., Ozer A.F., Vural M., et al. Idiopathic spinal cord herniation: case report and review of the literature. J Spinal Cord Med. 2009;32:86-94.
Selden N.R. Occult tethered cord syndrome: the case for surgery. J Neurosurg. 2006;104:302-304.
Shalabi M., Whitley R.J. Recurrent benign lymphocytic meningitis. Clin Infect Dis. 2006;43:1194-1197.
Stevenson K.L. Chiari type II malformation: past, present, and future. Neurosurg Focus. 2004;16:E5.
Steward C.G. Neurological aspects of osteopetrosis. Neuropathol Appl Neurobiol. 2003;29:87-97.
Tebruegge M., Curtis N. Epidemiology, etiology, pathogenesis, and diagnosis of recurrent bacterial meningitis. Clin Microbiol Rev. 2008;21:519-537.
Tubbs R.S., Oakes W.J. Treatment and management of the Chiari II malformation: an evidence-based review of the literature. Childs Nerv Syst. 2004;20:375-381.
Vanichkachorn J.S., Vaccaro A.R. Thoracic disk disease: diagnosis and treatment. J Am Acad Orthop Surg. 2000;8:159-169.
Voermans N.C., van Alfen N., Pillen S., et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
Wardlaw D., Cummings S.R., Van Meirhaeghe J., et al. Efficacy and safety of balloon kyphoplasty compared with non-surgical care for vertebral compression fracture (FREE): a randomised controlled trial. Lancet. 2009;373:1016-1024.
Weinstein J.N., Tosteson T.D., Lurie J.D., et al. Surgical versus nonsurgical therapy for lumbar spinal stenosis. N Engl J Med. 2008;358:794-810.
Wityk R.J., Zanferrari C., Oppenheimer S. Neurovascular complications of Marfan syndrome: a retrospective, hospital-based study. Stroke. 2002;33:680-684.
Yamada S., Won D.J., Siddiqi J., et al. Tethered cord syndrome: overview of diagnosis and treatment. Neurol Res. 2004;26:719-721.
Yeh S., Lee W.L., Rosenbaum R.B., et al. Hearing loss, uveomeningitis, and stroke in a 55-year-old man. Arthritis Care Res. 2011;63:298-306.
Zimmerli W. Clinical practice. Vertebral osteomyelitis. N Engl J Med. 2010;362:1022-1029.