[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 2
Thyroid-Stimulating Hormone
Physiology and Secretion
Ontogeny of TSH-Producing Thyrotrope Cells
Thyrotropes make up only 5% of pituitary cells in the anterior pituitary and are solely responsible for synthesizing TSH. The distinct cell types of the anterior pituitary are defined by the hormone they produce and include thyrotropes (TSH), gonadotropes (LH, FSH), corticotropes (adrenocorticotropic hormone [ACTH]), somatotropes (growth hormone [GH]), and lactotropes (prolactin [PRL]). Cell fate mapping demonstrated that the anterior pituitary develops from Rathke’s pouch, an invagination of oral ectoderm that directly contacts the hypothalamus1 at embryonic day 9.5 (e9.5). Pituitary organogenesis involves progenitor cells and their differentiation by signals that regulate proliferation, lineage commitment, and terminal differentiation.2 The key genes initiating and controlling these developmental pathways include transcription factors, secreted signaling molecules, and receptors. Distinct cell lineages emerge as a result of signaling gradients of transcription factors formed in a spatially and temporally specific manner.3
The glycoprotein hormone α–subunit (αGSU) is the first pituitary hormone gene expressed during development4 at e10.5. Wnt5a and BPM4, which are expressed in the adjacent neuroepithelium, provide the initial signals followed by expression of Hesx1, Ptx1/2, and Lhx3/4.5 TSHβ expression begins in the rostral tip of the pituitary at e12.5 and correlates with thyrotrope embryonic factor (TEF).6 By birth, TSHβ expression in the rostral tip has disappeared and another population of thyrotropes arises by e15.5 in the caudomedial region, following expression of Pit-1 (POU1F1), a pituitary-specific transcription factor.7 Both Pit-1 and TSHβ subunit expression are present in the wild-type but not in the Snell dwarf mouse, which has a mutation of the Pit-1 gene.8 These data suggest that the second population of thyrotropes, associated with Pit-1, is likely the source of mature thyrotropes. Pit-1 expression depends on the transient expression of Prophet of Pit-1 (Prop1) along with Atbf1.9
A zinc finger transcription factor, Gata2, plays a critical role in thyrotrope differentiation.3 Gata2 is transcribed in the developing anterior pituitary as early as e10.5 and persists in an expression pattern coincident with the glycoprotein hormone α-subunit. Gata2 binds and transactivates the αGSU promoter10 and acts synergistically with Pit-1 to activate the TSHβ gene.11 A ventral-dorsal gradient of Gata2 occurs early in development: the intermediate cells that express both Gata2 and Pit-1 develop into thyrotropes. The in vivo function of Gata2 in pituitary development has been examined by targeted inactivation of Gata2 in a transgenic mouse model using Cre recombinase directed by the αGSU promoter/enhancer.12 When Gata2 is ablated, mice have a decreased thyrotrope cell population at birth and lower levels of circulating TSH and FSH when adults. These studies showed that Gata2 is important for optimal thyrotrope and gonadotrope function but not for cell fate.
A recent study has uncovered the existence of an adult population of multipotent stem cells in the adult pituitary13,14 that are distinct from the embryonic precursor cells. These nestin- and Sox2-containing stem cells reside in a localized niche within the periluminal region of the gland, have the capacity to expand into all of the terminally differentiated pituitary cell types after birth, and may contribute to pituitary tumors.13
Tsh Subunit Genes
TSHβ Subunit Gene Structure
The human TSHβ subunit gene has been isolated and characterized.15 This single-copy gene is 4527 base pairs (bp) in size, and is located on the short arm of chromosome 1.16 The gene structure consists of three exons and two introns (Fig. 2-1, top panel). The first exon has 37 bp and contains the 5′-untranslated region of the gene. It is separated from the second exon by a large first intron of 3.9 kb. The coding region of the gene is contained within the second (163 bp) and third (326 bp) exons, which are separated by a 0.45 kb intron, while the 3′-untranslated region is contained within the third exon.
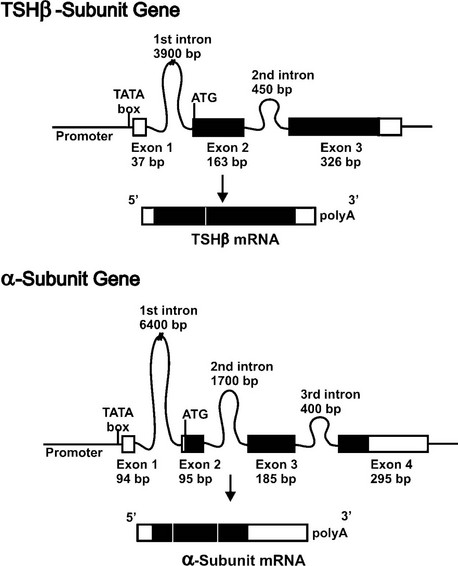
FIGURE 2-1 Structural organization of the human TSH subunit genes and mRNAs. The two panels show the TSHβ (top) and α subunit (bottom) genes. Shown are the relative locations and sizes of the exons and introns. Untranslated regions are shown as open boxes, and protein coding regions are shown in black boxes. The TATA box important for positioning the RNA transcriptional start is located in the promoter close to exon 1. Following transcription, introns are spliced out, exons precisely joined, and a polyA tail added to the 3′ end of the mature mRNA.
DNA sequences close to the transcriptional start site in the promoter of the TSHβ gene contain elements responsible for initiating transcription and regulating expression. A consensus TATA box is located 28 bp upstream of the transcriptional start site and is important for positioning RNA synthesis. Progressive 5′ deletions of the mouse TSHβ promoter linked to a luciferase reporter following expression in thyrotrope cells defined the cis-acting sequences required for expression to the first 270 bp of the promoter.17,18 Although these sequences defined the minimal promoter, other studies have shown that enhancer sequences located more than 6 kb upstream are also required for the promoter to express in pituitary thyrotropes in transgenic mice.19
Promoter deletion studies have demonstrated that the mouse TSHβ promoter from −271 to −80 is sufficient to confer thyrotrope-specific activity,17 and thyrotrope transcription factors can bind to the proximal promoter.20 Within this broad area, four regions of protein interaction have been identified using nuclear extracts from thyrotrope cells.18 Two factors, Pit-1, a homeodomain factor, and Gata2, a zinc finger transcription factor, can bind to TSHβ promoter sequences from −135 to −8821 (Fig. 2-2). Both factors can bind independently to the promoter, form a heteromeric complex with DNA, physically interact with each other, and functionally synergize to activate TSHβ promoter activity. Recently, an additional transcription factor, TRAP220 (Med1, PBP), was shown to be recruited to the TSHβ proximal promoter, where it was shown to play a role in transcriptional activation.22
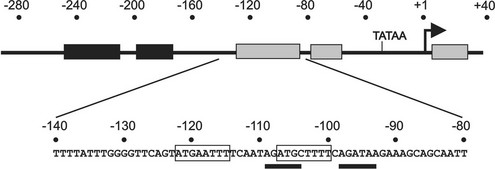
FIGURE 2-2 Functionally important regions of the TSHβ gene promoter. Top, Schematic showing regions (shaded boxes) of the TSHβ promoter that interact with transcription factors present in thyrotrope cells. Below, The DNA sequence of a key region that is critical for high level of TSHβ expression in thyrotropes. Within this region, Pit-1 sites are boxed, and sites interacting with Gata2 are underlined.
TRAP220 was originally defined as part of a transcriptional mediator complex that interacts with hormone-occupied thyroid/steroid hormone receptors.23 Mice with a single copy of this gene were hypothyroid with reduced levels of pituitary TSHβ transcripts.24 TRAP220 is recruited to the TSHβ gene by virtue of its physical interaction with both Pit-1 and Gata2, since the protein itself does not bind DNA. Co-transfections in nonpituitary cells showed that Pit-1, Gata2, or TRAP220 alone could not stimulate the TSHβ promoter. However, maximal activity resulted when all three factors were expressed. Interaction studies showed that all three factors interact with each other in vivo and in vitro. The regions of interaction were important for maximal function. Chromatin immunoprecipitation demonstrated in vivo occupancy on the proximal TSHβ promoter.22 Thus, the TSHβ gene is activated by a unique combination of transcription factors present in pituitary thyrotropes, including those that act via binding to the proximal promoter, as well as others that are recruited to the promoter via protein-protein interactions.
α Subunit Gene Structure
The human glycoprotein hormone α subunit gene is located on chromosome 6 at position 6q12-q21.25 It is present as a single copy gene that is 9635 kb in size and contains four exons and three introns and contains a consensus TATA box located 26 bp upstream of the transcriptional start site.26 The first exon (94 bp) contains 5′-untranslated sequences and is separated from the second exon by a 6.4 kilobase (kb) intron. The second exon contains 7 bp of 5′-untranslated sequence and 88 bp of the coding region. The coding sequence continues in the third (185 bp) and fourth (75 bp) exons, and the 3′-untranslated region (220 bp) is contained within the fourth exon (Fig. 2-1, bottom panel).
The α subunit gene is expressed in thyrotropes, gonadotropes, and placental cells but is differentially regulated. Cell-specific expression in each cell type is dependent on different regions of the promoter. Whereas the region downstream of −200 is sufficient for placental expression,27 gonadotropes require sequences between −225 and −200,28 and regions farther upstream appear to be critical for thyrotrope expression.29 Transgenic mouse studies have shown 480 bp of the mouse α subunit 5′-flanking DNA could target transgenic expression to both gonadotropes and thyrotropes.30 A region from −225 to −200 that binds steroidogenic factor 1 appears to be critical for gonadotrope expression,31 but not for thyrotrope expression.32 Another important sequence involves the pituitary glycoprotein hormone basal element, extending from −342 to −329, that is critical for both thyrotrope and gonadotrope expression.33 The element interacts with P-LIM, a pituitary-specific LIM-homeodomain transcription factor that is important for other pituitary cells.34 Several sequences within the region from −480 to −300 appear to be important for mouse α subunit expression in thyrotropes but not gonadotropes.29 Among these is the sequence from −434 to −421, which interacts with the developmental homeodomain transcription factor Msx1.35 Other sequences within the 480 bp promoter have been found to interact with the pituitary-specific homeodomain factor Ptx-1, and a synergism between Ptx-1 and P-LIM, mediated by the co-activator C-LIM, has been described.36 Recent studies with the mouse promoter in transgenic mice showed that an upstream DNA element located between −4.6 and −3.7 kb further enhanced expression in both thyrotropes and gonadotropes, and contained consensus binding sites for Gata, SF1, Sp1, ETS, and bHLH factors, which suggests cooperativity between factors binding both to proximal cis-acting elements and to the distal enhancer.37
Biosynthesis of TSH
Translation of TSH Subunits
The next steps in TSH biosynthesis are summarized in Fig. 2-3.38 The mRNAs for the TSHβ and α subunits are independently translated by ribosomes in the cytoplasm. The first peptide sequences consist of “signal” peptides of 20 amino acids for TSHβ and 24 amino acids for α.39 These signal peptides are hydrophobic, allowing insertion through the lipid bilayer of the membrane of the rough endoplasmic reticulum. Translation into TSHβ and α pre-subunits continues into the lumen of the rough endoplasmic reticulum, and cleavage of the signal peptide occurs before translation is completed. This results in the formation of a 118 amino acid TSHβ subunit40 and a 92 amino acid α subunit. Cleavage of TSHβ to a protein of 112 amino acids appears to be an artifact of purification. Synthesis of recombinant TSHβ subunit has resulted in two products of 112 and 118 amino acids, both of which are similarly active in vitro.41
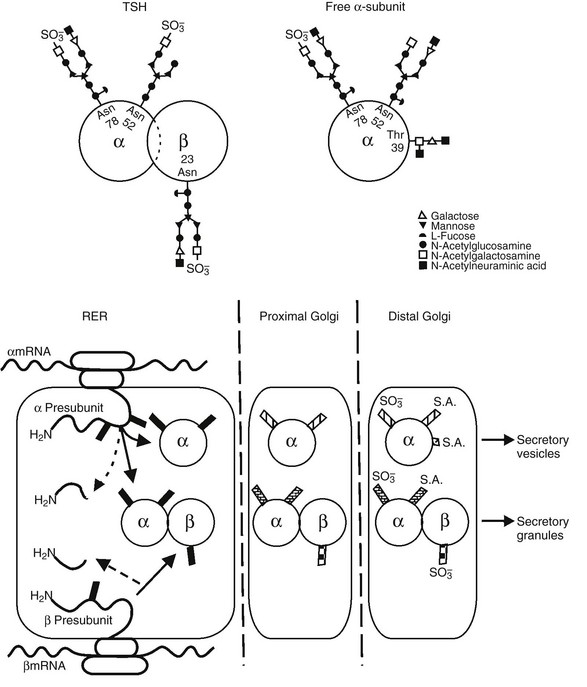
FIGURE 2-3 Top panel, Oligosaccharide chains of thyroid-stimulating hormone (TSH). Shown are typical oligosaccharide chains present on the TSH heterodimer and the free α subunit. Glycosylated asparagine (Asp) and threonine (Thr) residues are indicated. Symbols represent the oligosaccharide chain residues as indicated in the key. Bottom panel, Biosynthesis of thyroid-stimulating hormone (TSH). Schematic shown includes the processes of translation and glycosylation within the rough endoplasmic reticulum (RER) and Golgi apparatus, divided into proximal and distal. Cleavage of the aminoterminal (H2N) signal peptide and early addition of high mannose chains (black boxes), as well as the combination of α and β subunits, occur in the RER. In the proximal Golgi, oligosaccharide chains are modified (shaded boxes), and the final steps of sulfation and sialation occur in the distal Golgi apparatus. (From Weintraub BD, Gesundheit N: Thyroid-stimulating hormone synthesis and glycosylation: clinical implications. Thyroid Today 10:1–11, 1987.)
Glycosylation of TSH
Glycosylation of TSH has a significant impact on its biological activity.42 The TSHβ subunit has a single glycosylation site, the asparagine residue at position 23, whereas the α subunit is glycosylated in two sites, the asparagine residues at positions 52 and 7843 (see Fig. 2-3). Excess free α subunit is glycosylated at an additional site, the threonine residue at position 39.44 This residue is located in a region believed to be important for combination with the TSHβ subunit. It is not known whether glycosylation at this residue is a regulated step that inhibits combination with the TSHβ subunit, or whether it occurs in excess free α subunits because this site is exposed.
Extensive studies on the processes of TSH subunit glycosylation have been carried out. Glycosylation of the TSHβ and α subunits begins before translation is completed (co-translational glycosylation), and addition of the second oligosaccharide in the α subunit occurs after translation is completed (posttranslational glycosylation). The first step in this process involves the assembly of a 14-residue oligosaccharide, (glucose)3-(mannose)9-(N-acetylglucosamine)2 on a dolichol-phosphate carrier. This oligosaccharide then is transferred to asparagine residues by the enzyme olygosaccharyl transferase, which recognizes the tripeptide sequence (asparagine)-(X)-(serine or threonine).45 This mannose-rich oligosaccharide is progressively cleaved in the rough endoplasmic reticulum and Golgi apparatus. An intermediate with only six residues is produced and then other residues are added, resulting in complex oligosaccharides.46 The residues added include N-acetylglucosamine, fucose, galactose, and N-acetylgalactosamine. Oligosaccharides before the six-residue intermediate are termed high mannose and are sensitive to endoglycosidase H, which releases the oligosaccharide from the protein, whereas the intermediate and complex oligosaccharides are endoglycosidase H resistant. Complex oligosaccharides usually consist of two branches (biantennary), but sometimes three or four branches are seen, as are hybrid oligosaccharides consisting of one complex and another high-mannose branch. Sulfation and sialation occur late in the pathway, within the distal Golgi apparatus. Sulfate is bound to N-acetylgalactosamine residues, and sialic acid, or its precursor N-acetylneuraminic acid, is bound to galactoside residues.47 Thus, activation of the enzymes sulfotransferase and N-acetylgalactosamine transferase may involve important regulatory steps that affect the ratio of sulfate to sialic acid. As demonstrated with LH, it appears that sulfation increases and sialylation decreases the bioactivity of TSH,47 because the exclusively sialylated recombinant glycoprotein produced in Chinese hamster ovary cells has been found to have attenuated activity in vitro.48
Processing of complex oligosaccharides appears to occur at a slower rate for secreted glycoproteins, such as TSH, when compared with that for nonsecreted glycoproteins. For example, after an 11 minute pulse labeling with [35S]methionine and a 30 minute chase, only a few α subunits were endoglycosidase H resistant, and only 76% reached this stage after an 18 hour chase.49 Secretion was observed after a 60 minute chase, and the secreted products—TSH, free α subunit but no free β subunit—had mostly complex oligosaccharides associated with them.43 It may be important to note that many of the studies described were carried out in thyrotropic tumor tissue obtained from hypothyroid mice, and glycosylation may differ in the euthyroid as compared with the hypothyroid state. In addition, differences between species have been noted, such as the human TSH may contain more sialic acid than the bovine TSH.40
Folding, Combination, and Storage of TSH
Elucidation of the crystal structure of human CG (hCG)50 allowed the construction of a model of human TSH, as supported by other evidence.51,52 This model has greatly facilitated the interpretation of structure-function studies of the protein backbone. However, crystallization was achieved only with partly deglycosylated hCG, so it is likely that the conformation of the glycosylated protein may differ to some extent, although nuclear magnetic resonance studies suggest that the α subunit carbohydrate moieties project outward and may be freely mobile.53 Nevertheless, this model predicts that the tertiary structure of each TSH subunit consists of two hairpin loops on one side of a central knot formed by three disulfide bonds and a long loop on the opposite side. In this tertiary structure, the glycoprotein hormones share features in common with transforming growth factor β, nerve growth factor, platelet-derived growth factor, vascular endothelial growth factor, inhibin, and activin, all of which are now grouped in the family of “cystine knot” growth factors.54
Folding of nascent peptides begins before translation is completed. It has been shown that proper folding is dependent on glycosylation, because the drug tunicamycin, which prevents the initial oligosaccharide transfer to the asparagine residue, results in a peptide that does not fold properly and is degraded intracellularly.55 Site-directed mutagenesis of a single glycosylation site also disrupted processing and decreased TSH secretion in transfected Chinese hamster ovary cells.56 Folding is a critical step that allows correct internal disulfide bonding, which stabilizes the tertiary structure of the protein, allowing subunit combination.
Combination of TSH β and α subunits begins soon after translation is completed in the rough endoplasmic reticulum and continues in the Golgi apparatus.43 Subunit combination then accelerates and modifies oligosaccharide processing of the α subunit.57 In fact, studies have suggested that the conformation of the α subunit differs after combination with each type of β subunit,58 and this may affect subsequent processing. The rate of combination of TSHβ and α subunits has been examined in mouse thyrotropic tumors. After a 20 minute pulse labeling with [35S]methionine, 19% of TSHβ subunits were combined with α subunits, and this percentage increased to 61% after a additional 60 minute chase incubation.43 Recent studies have shown that the combination of the TSHβ and α subunits, as is also the case with other glycoprotein hormones, occurs after “latching” of the disulfide “seatbelt” of the β subunit, with subsequent “threading” of loop 2 and the attached oligosaccharide of the α subunit beneath that “seatbelt.”59
The sequence of the TSHβ subunit from amino acid 27 to 31 (CAGYC) is highly conserved among species and is thought to be important for combination with the α subunit. In a case of congenital hypothyroidism, a point mutation in the CAGYC region (see “Disorders of TSH Production”) results in the synthesis of altered TSHβ subunits that are unable to associate with α subunits, with consequent lack of intact TSH production.60 A lack of free circulating TSHβ subunit was also observed, suggesting that combination with α subunit is necessary for TSHβ subunit secretion. This phenomenon was also demonstrated in studies where synthesis of wild-type recombinant TSHα subunit was carried out in the presence or absence of recombinant β subunit.61 Using site-directed mutagenesis, another study showed that a mutation at residue 25 in the glycosylation recognition site, which substitutes a serine for a threonine, does not alter glycosylation but decreases TSH production by 70%, possibly because of disruption of the nearby CAGYC region.62
After TSH and free α subunit are processed in the distal Golgi apparatus, they are transported into secretory granules or vesicles.63 The secretory granules constitute a regulated secretory pathway, mainly influenced by TRH and other hypothalamic factors. These granules contain mostly TSH, whereas free α subunit is contained in the secretory vesicles that constitute a nonregulated secretory pathway.
TSH Secretion
In healthy humans, the production rate of TSH is between 100 and 400 mU/day.64,65 The distribution space of TSH is slightly greater than the plasma volume. In euthyroid subjects, the half-life of TSH in plasma is approximately 50 minutes, with a plasma clearance rate of approximately 50 mL/min. In hypothyroid subjects, TSH secretion rates increase by 10 to 15 times normal rates, and the clearance rate decreases slightly. In hyperthyroid subjects, TSH secretion is suppressed, and metabolic clearance is accelerated.
Ontogeny of TSH Levels
At 8 to 10 weeks of gestation in the human, TRH is measurable in the hypothalamus, with progressive increases in TRH levels until term. By 12 weeks of gestation, immunoreactive TSH cells are present in the human pituitary gland,66 and TSH is detectable in the pituitary and the serum.67,68 Serum and pituitary TSH levels remain low until week 18, when TSH levels increase rapidly, followed by increases in serum T4 and T3 concentrations. Fetal serum TSH and T4 concentrations continue to increase between 20 and 40 weeks of gestation. Pituitary TSH begins to respond to exogenous TRH early in the third trimester, and negative feedback control of TSH secretion develops during the last half of gestation and the first 1 to 2 months of life.69
An abrupt rise in serum TSH levels occurs within 30 minutes of birth in term infants. This is followed by an increase in serum T3 concentrations within 4 hours and a lesser increase in T4 levels within the first 24 to 36 hours. The initial increase in serum TSH levels appears to be stimulated by cooling in the extrauterine environment. Serum TSH levels fall to the adult range by 3 to 5 days after birth, and serum thyroid hormone levels stabilize by 1 to 2 months. Serum TSH levels in healthy premature infants (less than 37 weeks gestational age) are highly variable but tend to be lower at birth compared with those of term infants. TSH levels decrease slightly during the first week of life, followed by a gradual increase to normal term levels. Serum TSH levels are even lower in ill premature infants but rise toward normal levels during recovery.70,71
Patterns of TSH Secretion
TSH is secreted from the pituitary gland in a dual fashion, with secretory bursts (pulses) superimposed upon basal (apulsatile) secretion (Fig. 2-4, upper panel). Basal TSH secretion accounts for 30% to 40% of the total amount released into the circulation, and secretory bursts account for the remaining 60% to 70%. TSH pulses occur approximately every 2 to 3 hours, although considerable variability is noted among individuals.72 TSH pulses appear to directly stimulate T3 secretion from the thyroid gland, as cross-correlation analysis has shown that a free T3 peak occurs between 0.5 and 2.5 hours after a TSH peak. However, changes in free T3 levels from nadir to peak are only 11% of mean free T3 levels, probably because T3 has a long serum half-life, and most T3 does not arise from the thyroid gland.73
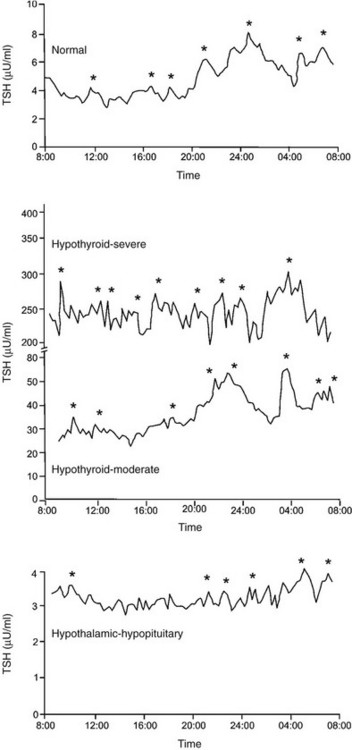
FIGURE 2-4 Serum thyroid-stimulating hormone (TSH) levels measured every 15 minutes in a healthy subject (upper panel), in two subjects with primary hypothyroidism (middle panel), and in a subject with hypothyroidism due to a craniopharyngioma (lower panel). Significant TSH pulses were located by cluster analysis, a computerized pulse detection program, and are indicated by asterisks. (Data from Sarapura VD, Samuels MH, Ridgway EC: Thyroid-stimulating hormone. In: Melmed S [ed]: The Pituitary, 2nd ed. Blackwell Science, Malden, MA, pp 187–229, 2002.)
In healthy euthyroid subjects, TSH is secreted in a circadian pattern, with nocturnal levels increasing to up to twice daytime levels72 (Fig. 2-4, upper panel). Peak TSH levels occur at between 2300 and 0500 hours in subjects with normal sleep-wake cycles, and nadir levels occur at about 1100 hours. The TSH circadian rhythm is not present in infants younger than 4 weeks old but emerges between 1 and 2 months of life and is well established in healthy children.74 The circadian variation in TSH levels is due to increased mass of TSH secreted per burst at night, as well as slightly increased frequency of bursts and more rapid increase to maximal TSH secretion within a burst.72 The nocturnal increase in TSH levels can precede the onset of sleep, and sleep deprivation enhances TSH secretion. Therefore, in contrast to other pituitary hormones with a circadian variation, the nocturnal rise in TSH levels is not sleep entrained. Instead, a sleep-related inhibition of TSH release is of insufficient magnitude to counteract the nocturnal TSH surge.
Subjects with mild primary hypothyroidism retain the nocturnal rise in TSH levels, and patients with severe primary hypothyroidism have markedly increased TSH pulse amplitude throughout the day with loss of circadian variation in TSH levels (Fig. 2-4, middle panel).72 l-Thyroxine therapy reestablishes the normal TSH circadian variation. In contrast, patients with hypothalamic-pituitary causes of hypothyroidism secrete less TSH over a 24 hour period, with loss of the nocturnal TSH surge in pulse amplitude75 (Fig. 2-4, lower panel). A similar pattern of reduced 24 hour TSH secretion occurs in critical illness.76
The origin of pulsatile and circadian TSH secretion is not known. Thyroid hormones alter TSH pulse amplitude but have little effect on pulse frequency, and therefore are unlikely to participate in TSH pulse generation. The TSH pulse generator may reside in the hypothalamus, with TRH neurons acting in concert to stimulate a burst of TSH secretion from the pituitary gland. However, constant TRH infusions do not change TSH pulse frequency in humans, which casts doubt on this theory.77 Dopamine suppresses TSH pulse amplitude but does not alter TSH pulse frequency, and therefore dopamine does not appear to control pulsatile TSH secretion. A diurnal variation in the activity of anterior pituitary 5′-monodeiodinase in the rat may control circadian TSH secretion.78 However, this has not been confirmed in the human.
Physiologic serum cortisol levels may control circadian TSH secretion, although cortisol does not appear to affect TSH pulse frequency. When subjects with adrenal insufficiency were studied under conditions of glucocorticoid withdrawal, daytime TSH levels were increased, and the usual TSH circadian rhythm was abolished. When these subjects were given physiologic doses of hydrocortisone in a pattern that mimicked normal pulsatile and circadian cortisol secretion, daytime TSH levels were decreased, and the normal TSH circadian rhythm was reestablished. Hydrocortisone infusions at the same dose given as pulses of constant amplitude throughout the 24 hour period also decreased 24 hour TSH levels, but no circadian variation was noted.79 Similarly, when healthy subjects were given metyrapone (an inhibitor of endogenous cortisol synthesis), TSH levels increased during the day, leading to abolition of the usual TSH circadian variation.80 These data suggest that the normal early morning increase in endogenous serum cortisol levels decreases serum TSH levels and leads to the observed normal circadian variation in TSH.
Regulation of TSH Biosynthesis
Hypothalamic Regulation of TSHβ Subunit Biosynthesis
Thyrotropin-releasing hormone (TRH) is a tripeptide that is secreted from the hypothalamus and transported to the pituitary via the hypothalamic-hypophyseal portal system; it is a major activator of TSH production with a significant three- to fivefold increase in the transcription of both TSHβ and α subunit mRNAs.81 TRH from maternal or fetal sources is not required for normal thyrotrope development during ontogeny, and TRH-deficient mice are not hypothyroid at birth. However, TRH is required for the postnatal maintenance of TSH activation.82
TRH binding to its receptor initiates a cascade of intracellular events. In GH3 cells, the TRH-receptor complex interacts with a guanine nucleotide binding regulatory protein (G) that then binds and activates GTP(G′). G′ binds to phospholipase C (C) and activates it (C′). C′ catalyzes the hydrolysis of phosphatidylinositol 4,5 bisphosphate, which results in the formation of two intracellular “second messengers,” inositol triphosphate (InsP3) and 1,2-diacylglycerol (1,2-DG). InsP3 diffuses from the cell-surface membrane to the endoplasmic reticulum, where it causes the release of sequestered Ca2+. This activates the movement of secretory granules to the cell surface and their exocytosis. Simultaneous with these events is a parallel activation of protein kinase C by 1,2-DG, which also leads to phosphorylation of proteins involved in exocytosis. TRH has been shown to stimulate a nuclear protein, Islet-brain-1 (IB1)/JIP-1, in the anterior pituitary gland and in cultured rat GH3 cells83 and has been implicated in the action of TRH in stimulating the TSHβ gene in thyrotropes. Studies in somatomammotrope cells, where TRH stimulates prolactin production, have suggested that phosphatidylinositol, protein kinase C, and calcium-dependent pathways may be involved,84 although TRH stimulation of the TSHβ subunit promoter may be mediated by AP1.85
Two TRH-response regions are located from −128 to −61 and from −28 to +8 of the human TSHβ promoter.86 The upstream region contains binding sites for the pituitary-specific transcription factor, Pit-1, suggesting a role for this or a similar factor in the regulation of the TSHβ subunit gene by TRH. In the rat TSHβ subunit gene, responsiveness to TRH has been localized to regions upstream of −204, where Pit-1 binding sites are also found.87 Furthermore, it has been shown that both protein kinase C and protein kinase A pathways can phosphorylate Pit-1 at two sites in response to phorbol esters and cyclic adenosine monophosphate (cAMP),88 thus altering the binding to Pit-1 transactivation elements on the human TSHβ gene.89
Dopamine acting via DA2 dopamine receptors inhibits TSHα and β subunit gene transcription by decreasing the intracellular levels of cAMP.81 Studies of the TSHβ subunit gene have localized two regions of the promoter necessary for cAMP stimulation, from −128 to −61 bp and from +3 to +8 bp. The upstream region coincides with the TRH-responsive region and contains Pit-1 binding sites. The downstream region resides within the regions responsive to T3 (+3 to +37) and TRH (−28 to +8). The downstream region also overlaps with an AP1 binding site (−1 to +6). The sequence from −1 to +6 appears to cooperate with Pit-1 in mediating responses to cAMP and TRH.85 Thus, multiple interactions between transcription factors and hormonal regulators appear to converge on sequences close to the transcriptional start site.
Peripheral Regulation of TSHβ Subunit Biosynthesis
Thyroid hormone is thought to act predominantly through a classical TR-mediated genomic model. T4 serves as a minimally active prohormone that is converted into a metabolically active T3 via a family of tissue deiodinases, termed D1, D2, and D3. These selenoprotein enzymes are membrane bound and can activate or inactivate substrate in a time- and tissue-specific manner.90 D2 is the major T4-activating deiodinase. It is present on the endoplasmic reticulum close to the nucleus and produces 3,5,3′-triiodothyronine (T3) through the removal of an iodine residue from the outer ring of thyroxine. D2 activity is rapidly lost in the presence of its substrate T4 by a ubiquitin proteasomal mechanism.91 Rat pituitary thyrotropes coexpress D2 RNA and protein, and both are increased in hypothyroidism. Murine thyrotropes in TtT-97 tumors or the TαT1 cell line have extremely high levels of D2, which accounts for the sustained production of T3 by thyrotropes even in the presence of supraphysiologic T4 levels. Serum TSH levels in normal mice are suppressed by administration of T4 or T3, although only T3 was effective in the mouse with targeted disruption of the D2 gene. The observed phenotype of pituitary resistance to T4 demonstrated the critical importance of D2 in controlling negative thyroid hormone regulation of TSH in thyrotropes.
T4 can also act, in some cases, via nongenomic mechanisms that do not involve classical nuclear TR mechanisms. T4 can bind to a cell-surface integrin αVβ3 receptor followed by activation of a mitogen-activated protein kinase cascade, which transduces the signal into a complex series of cellular and nuclear actions. These nongenomic hormone actions are likely to be contributors to basal rate setting of transcription of some genes, and they may control complex cellular events.92
TSHβ and α subunit gene transcription rates are markedly inhibited by treatment with triiodothyronine (T3). Studies using mouse TtT-97 thyrotropic tumors have demonstrated that suppression of TSHβ and α subunit mRNA transcription rates measured by nuclear run-on assays is evident by 30 minutes after treatment and is maximal by 4 hours.93 This effect was seen in the presence of the protein synthesis inhibitor cycloheximide, indicating that it did not require an intermediary protein. Other studies using mouse and rat pituitaries along with mouse thyrotropic tumors have demonstrated that steady-state mRNA levels of TSHβ and α subunits are dramatically decreased by T394 (Fig. 2-5). The mechanism of action of T3 involves interaction with nuclear receptors that act mainly at the transcriptional level. The transcriptional response to T3 is proportional to the nuclear receptor occupancy,95 and the time course of T3 nuclear binding and that of transcriptional inhibition are also in agreement96 (see Fig. 2-5).
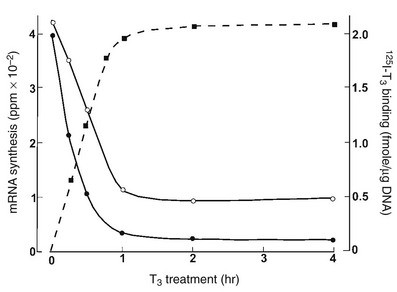
FIGURE 2-5 The effect of thyroid hormone on the transcription of the thyroid-stimulating hormone (TSH)β (black circles) and α subunit (white circles) genes. Murine thyrotropic tumor explants were incubated for up to 4 hours with 5 nmol T3 for transcription measurements or with 5 nmole 125I T3, with or without 1000-fold excess of unlabeled T3 for binding measurements. Transcription rates were measured in pools of isolated nuclei. An inverse relationship has been noted between T3 binding and TSHβ and α subunit mRNA synthesis. (Data from Shupnik MA, Ridgway EC: Triiodothyronine rapidly decreases transcription of the thyrotropin subunit genes in thyrotropic tumor explants. Endocrinology 117:1940–1946, 1985.)
The T3 inhibitory effect on the TSHβ gene requires ligand-occupied T3 receptor (TR), specifically the TRβ1 or TRβ2 isoform, because patients with thyroid hormone resistance and inappropriate secretion of TSH have abnormalities only in the TRβ, not the TRα, gene.97 TRβ interacts with specific cis-acting DNA sequences close to the transcriptional start. T3 response elements have been reported to be located between +3 and +37 of the human TSHβ gene.98 Two T3 receptor binding sites, from +3 to +13 and from +28 to +37, may mediate T3 inhibition. T3 responses can be mediated through receptor monomers, homodimers, or heterodimers involving retinoid X receptors (RXR).99 An RXR selective ligand was shown in vitro to inhibit TSHβ expression in TtT-97 thyrotropic tumor cells100 and in cultured TαT1 thyrotropes.101 This finding has been confirmed in vivo and resulted in central hypothyroidism (low T4 and low TSH) in cancer patients treated with the retinoid bexarotene.102 The RXR-selective retinoid (LG 268) decreased circulating TSH and T4 levels in mice with marked lowering of pituitary TSHβ mRNA without decreasing TRH, suggesting a direct effect on thyrotropes.101
Other, more recent studies have disputed the requirement of the negative response element located in exon 1 of the human gene, because its deletion did not eliminate T3 suppression of TSHβ promoter activity in a reconstitution system.103 These studies showed that liganded TRβ can associate with Gata2 in vitro and in vivo via direct interaction between the zinc fingers of Gata2 and the DNA binding domain of TRβ. In addition, T3-occupied TR can physically interact with TRAP220.24 Thus, it is likely that interference with the transactivation function of the Pit-1/Gata2/TRAP220 complex on the proximal TSHβ promoter plays an important role in T3 negative regulation.
Abundant information exists as to the mechanisms involved in gene stimulation by T3. Generally, TRs bind to cis-acting DNA response elements (TRE) in the absence of ligand, interact with a family of nuclear receptor corepressor molecules that recruit histone deacetylases, and locally modify chromatin structure to result in repression of the target gene.104 In the presence of T3, the corepressor complexes rapidly dissociate and are replaced by coactivator complexes that bind to TRs and increase histone methylation and acetylation locally on the chromosomal DNA, which unwinds the chromatin into an open configuration.105 Other activating transcription factors such as TRAP220 are then recruited to the TR via protein-protein interactions, which then activate RNA polymerase II–mediated transcription.
In contrast, the molecular mechanisms involved in negative T3 regulation, such as the TSH subunit genes, have not been well characterized. Liganded TRβ has been reported to recruit histone deacetylase 3 and reduce histone H4 acetylation that modifies histones, resulting in a fully repressed chromosomal state of the TSHβ gene.103 Several recent studies have demonstrated the requirement of an intact DNA binding domain of TRβ in the negative regulation of the TSHβ gene in vitro106 and in vivo.107 In one study, a combination of Pit-1 and Gata2 activated a human TSHβ (−128/+37) reporter construct along with vectors containing TRβ1 constructs in the absence or presence of T3. These investigators found that unliganded TRβ1 did not stimulate promoter activity, whereas a mutation lacking the N-terminus and DNA binding domain of TRβ1 lost the ability of T3-treated cells to negatively regulate TSHβ promoter activity. This demonstrated the importance of various modular domains that constitute the molecular structure of TRs. Moreover, using a gene targeting approach in transgenic mice and replacing the wild-type TRβ gene with a mutant that abolished DNA binding in vitro did not alter ligand and cofactor interactions.107 Homozygous mutant mice demonstrated central thyroid hormone resistance with 20-fold higher serum TSH in the face of two- to threefold higher T3 and T4 levels, which were similar to those of TRβ homozygous null mice.
Although thyrotrope cells contain all TRs—TRα1, TRβ1, and TRβ2, as well as non-T3 binding variant α2108—it is TRβ2 that is expressed predominantly in the pituitary and in T3-responsive TRH neurons109 and is most critical for the regulation of TSH.110 Moreover, TRβ2-deficient mice had a phenotype consistent with pituitary resistance to thyroid hormone, with increased TSH and thyroid hormone levels, even in the presence of TRβ1 and TRα1, showing the lack of compensation between TR isoforms.111 However, TRβ1 and TRα1 still may play a role, in that they are able to form heterodimers with TRβ2. Heterodimers of a TR and a TR accessory protein, such as RXR, may also bind to DNA,112 constituting heterodimeric complexes that may have different affinities for specific DNA sequences and different functional activities. A particular RXR isoform, RXRγ1, is uniquely expressed in thyrotropes and appears to mediate the inhibition by 9-cis-retinoic acid through a region extending from −200 to −149 of the mouse TSHβ subunit promoter, an area upstream and distinct from that mediating negative regulation by thyroid hormone.100 Other proteins that interact with TR include the coactivators, such as the glucocorticoid receptor interacting protein-1 (GRIP-1) and the steroid receptor coactivator-1 (SRC-1),113 and corepressors, such as the silencing mediator for retinoid receptors and thyroid hormone receptors (SMRT) and the nuclear receptor corepressor (NCoR).114 These coactivators and corepressors modulate the effects of many members of the steroid-thyroid hormone receptor superfamily. Their role in the regulation of TSH subunit promoters by thyroid hormone remains to be elucidated in detail.
Studies with genetic knockout mouse models in which both TRH and TRβ genes were removed have recently showed an unexpected dominant role for TRH in vivo in regulating the hypothalamic-pituitary-thyroid axis. It appears that the presence of both TRβ and TRH is necessary for a normal thyrotroph response during hypothyroidism, suggesting that unliganded TRβ stimulates TSH subunit gene expression.115
Posttranscriptional effects of T3 have also been described. T3 decreases the half-life of TSHβ subunit mRNA and decreases the size of the poly(A) tail.116 The shortening of the poly(A) tail is thought to cause mRNA instability. Leedman et al. showed that T3 increased the binding of an RNA-binding protein present in rat pituitary to the 3′-untranslated region of the rat TSHβ mRNA117 and also induced a shortening of the poly(A) tail of the mouse TSHβ mRNA from 160 to 30 nucleotides.118
Steroid hormones, specifically glucocorticoids, inhibit TSH production, but TSH subunit mRNA levels do not change significantly.119 Their major effect may be seen at the secretory level. Estrogens mildly reduce both α and TSHβ subunit mRNA in hypothyroid rats compared with euthyroid controls.120 In this study, estrogen also abolished the early rise in subunit mRNA levels seen following T3 replacement. Testosterone has been shown to increase TSHβ subunit mRNA in castrate rat pituitary and mouse thyrotropic tumor.121
Leptin and neuropeptide Y have opposite effects on TSH biosynthesis. Leptin is the product of the ob gene, which is found mainly in adipose tissue and regulates body weight and energy expenditure.122 Neuropeptide Y (NPY) is a neuropeptide that is synthesized in the arcuate nucleus of the hypothalamus and plays many roles in neuroendocrine function.123 In dispersed rat pituitary cells, leptin stimulated and NPY inhibited TSHβ mRNA levels in a dose-related manner.124 In contrast, both agents increased α subunit steady state mRNA levels.
Hypothalamic Regulation of α Subunit Biosynthesis
TRH stimulates α subunit biosynthesis through a novel mechanism. A CRE-binding protein that binds to the region from −151 to −135 of the human α subunit promoter appears to be important for TRH regulation, as well as a Pit-1–like protein that binds to a more distal region from −223 to −190.125 The CRE of the human glycoprotein hormone α subunit gene promoter consists of an 18 bp repeat and extends from −146 to −111.126 The mechanisms involved in TRH stimulation of the α subunit gene appear to involve two transcription factors, P-Lim and CREB binding protein (CBP). When stimulated with TRH, both of these factors transcriptionally cooperate to activate α subunit promoter activity caused by direct protein-protein interactions.127 Both of these factors synergistically activated the α subunit gene promoter during TRH stimulation and interact in a TRH-dependent manner. P-Lim binds to the α subunit promoter directly, but CBP does not possess a DNA binding domain, so it must be recruited to the promoter via interaction with another factor. The P-Lim/CBP binding is formed in a TRH signaling-specific manner, in contrast to forskolin, which mimics protein kinase A signaling and dissociates both the binding and the transcriptional synergy. α subunit gene expression in thyrotropes is inhibited by dopamine in coordination with expression of the TSHβ subunit gene. Its action is mediated by decreases in intracellular cAMP levels.
Peripheral Regulation of α Subunit Biosynthesis
Thyroid hormone inhibition of α subunit gene transcription is observed in thyotropes in coordination with that of the TSH β subunit. The T3 response element of the human α subunit gene promoter has been reported to be located from −22 to −7.128 Similar to the TSHβ subunit gene, the T3 response elements of human and of mouse129 and rat130 genes are located close to the transcriptional start. T3 inhibition may be mediated by different isoforms of the T3 receptor131 in combination with the corepressors SMRT and NCoR.132 Studies have suggested that mutations of the T3 response element of the human α subunit promoter that eliminate TR binding do not abrogate the inhibitory effect of T3, suggesting that protein-protein interactions may be more important than protein-DNA binding.133
Steroid hormone regulation of α subunit gene transcription is probably of limited importance. Androgen inhibition and androgen receptor (AR) binding have been localized to a region from −120 to −100. Negative regulation by estrogen was described in the gonadotropes of transgenic mice expressing a reporter gene under the control of both human and bovine promoters, but no binding of these regions to the estrogen receptor (ER) was detected, suggesting an indirect effect.134 However, other studies using rat somatomammotropes have found positive regulation by estrogen localized to the proximal 98 bp of 5′-flanking DNA of the human α subunit gene and binding of the ER to the T3 response element from −22 to −7.135 Transcriptional inhibition by glucocorticoids may be mediated by binding of the glucocorticoid receptor to sequences between −122 and −93 of the human α subunit gene.136 However, no direct binding was detected in other studies, suggesting that the GR inhibits transcription by interfering with other transactivating proteins.137
Regulation of TSH Glycosylation
Glycosylation is a regulated process that is primarily modulated by TRH and thyroid hormone.138 Primary hypothyroidism139 and TRH administration140 have been found to increase oligosaccharide addition, which results in an increased bioactivity of TSH.141 The same was noted in patients with resistance to thyroid hormone. TSH glycosylation patterns were found to differ in several pathologic states such as central hypothyroidism, TSH-producing pituitary adenomas, and euthyroid sick syndrome.142 Also observed were changes in the sulfation and sialylation of the oligosaccharide residues, which modulate bioactivity.139,143,144 Recently, thyroid hormone was shown to increase TSH bioactivity, and this was correlated with decreased sialylation.145
Regulation of TSH Secretion
As with biosynthesis, TSH secretion is a result of complex interactions between central and peripheral hormones (Fig. 2-6).
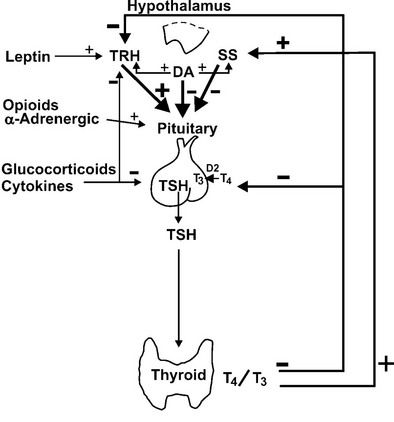
FIGURE 2-6 Hypothalamic and peripheral control of thyroid-stimulating hormone (TSH) secretion. Positive and negative stimuli are indicated at various levels within the hypothalamic-pituitary-thyroid axis. T4, thyroxine; T3, triiodothyronine; TRH, thyrotropin-releasing hormone; SS, somatostatin; DA, dopamine; D2, deiodinase type II.
Hypothalamic Regulation of TSH Secretion
TRH directly affects TSH secretion in vivo and in vitro at concentrations that exist in the pituitary portal blood.146 Immunoneutralization of TRH in animals leads to a decline in thyroid function,147 and TRH knockout mice have a reduced postnatal TSH surge, followed by impaired baseline thyroid function with a poor TSH response to hypothyroidism. Lesions of the paraventricular nucleus (PVN) decrease circulating TRH and TSH levels in normal or hypothyroid animals and cause hypothyroidism148 and electrical stimulation of this area causes TSH release. Although baseline levels of TSH are reduced in animals with lesions of the PVN, TSH levels still show appropriate responses to changes in circulating thyroid hormone levels. Thus, TRH likely determines the set point of feedback control by thyroid hormones.
Acute intravenous administration of TRH to human subjects causes a dose-related release of TSH from the pituitary. This occurs within 5 minutes and is maximal at 20 to 30 minutes. Serum TSH levels return to basal levels by 2 hours. More prolonged (2 to 4 hour) infusions of TRH lead to biphasic increases in serum TSH levels in humans and animals.149 The early phase may reflect release of stored TSH, and the later phase may reflect release of newly synthesized TSH. Interpretation of TSH responses to even more prolonged TRH infusions is complicated by the increase in serum T3 levels, with feedback to suppress further TSH release.77 Continuous TRH administration in vitro also causes desensitization of TSH responses, which may further explain decreased TSH levels with long-term TRH exposure.150
Somatostatin (SS) in humans and animals inhibits basal and TRH-stimulated TSH secretion in vivo and in vitro at concentrations that exist in the pituitary portal blood.151 In the hypothalamus, the highest concentrations of SS occur in the anterior paraventricular region.152 From this region, axonal processes of SS-containing neurons project to the median eminence. Animals that have undergone sectioning of these fibers have depletion of SS content of the median eminence and increased serum TSH levels.153 Similarly, immunoneutralization of SS in animals increases basal TSH levels and TSH responses to TRH.154 In humans, SS infusions suppress TSH pulse amplitude, slightly decrease TSH pulse frequency, and abolish the nocturnal TSH surge.155 Thus, TSH secretion probably is regulated through a simultaneous dual-control system of TRH stimulation and SS inhibition from the hypothalamus.
SS binds to specific, high-affinity receptors in the anterior pituitary gland. Five subtypes of the SS receptor (SST1 through SST5) have been identified,156 and SST1 and SST5 have been localized to thyrotropes.157 Binding of SS to its receptor inhibits adenylate cyclase via the inhibitory subunit of the guanine nucleotide regulatory protein, which lowers protein kinase A activity156 and decreases TSH secretion. SS may also exert some effects by cAMP-independent actions on intracellular calcium levels. Hypothyroidism reduces the efficacy of SS in decreasing TSH secretion in vitro, which is reversed by thyroid hormone administration.158 Additional studies in mouse thyrotropic tumors indicate that both SST1 and SST5 are markedly downregulated in hypothyroidism and are induced by thyroid hormone.157 Although short-term infusions of SS lead to pronounced suppression of TSH secretion in humans, long-term treatment with SS or its analogues does not cause hypothyroidism.159 This probably reflects compensatory mechanisms in the thyroid hormone feedback loop. GH deficiency is associated with increased TSH responses to TRH, and GH administration decreases basal and TRH-stimulated TSH secretion,160 possibly owing to GH stimulation of hypothalamic SS release.
Dopamine also inhibits basal and TRH-stimulated TSH secretion in vivo and in vitro at concentrations that exist in the pituitary portal blood.161,162 Exogenous dopamine antagonists, including those that do not penetrate the blood-brain barrier, increase TSH levels.163 In humans, dopamine infusions rapidly suppress TSH pulse amplitude, do not affect TSH pulse frequency, and abolish the nocturnal TSH surge155; administration of a dopamine antagonist has the opposite effect.164 Dopamine also has direct effects on hypothalamic hormone secretion that may indirectly affect TSH secretion. For example, dopamine and dopamine-agonist drugs stimulate both TRH and SS release from rat hypothalami.165
In the hypothalamus, dopamine is secreted by neurons in the arcuate nucleus.166 From the arcuate nucleus, neuronal processes project to the median eminence. Dopamine acts by binding to type 2 dopamine receptors (DA2) on thyrotrope cells.167 This leads to inhibition of adenylate cyclase, which decreases the synthesis and secretion of TSH. In addition, TSH may downregulate its own release through the induction of DA2 receptors on thyrotrope cells.168 The inhibitory effects of dopamine on TSH secretion vary according to sex steroids, body mass, and thyroid status. Dopamine antagonist drugs cause greater increases in serum TSH levels in women than in men. Recent studies show that obesity is associated with enhanced TSH secretion, which may be mediated via blunted central dopaminergic tone.169 Dopamine inhibition of TSH release is greater in patients with mild hypothyroidism than in normal subjects, although subjects with severe hypothyroidism may be less responsive.170 Although short-term infusions of dopamine lead to pronounced suppression of TSH secretion, long-term treatment with dopamine agonists does not cause hypothyroidism. This probably reflects compensatory mechanisms in the thyroid hormone feedback loop.
Adrenergic effects have also been reported in vivo and in vitro. α-Adrenergic activation stimulates TSH release directly from the rat pituitary gland at physiologic concentrations of catecholamines.171 α-Adrenergic agonists stimulate TSH release in rats, and blockade of norepinephrine synthesis or treatment with adrenergic receptor blockers decreases TSH levels.166,172 It is unclear whether these effects are mediated via changes in TRH and/or SS levels. In humans, data regarding adrenergic effects on TSH secretion are limited. α-Adrenergic blockade diminishes serum TSH responses to TRH.173 However, administration of epinephrine does not alter TRH-stimulated TSH secretion.174 These data suggest that endogenous adrenergic pathways do not have a major role in TSH secretion. Noradrenergic stimulation of TSH secretion is mediated by high-affinity α1-adrenoreceptors linked to adenylate cyclase.173 Therefore, dopamine and epinephrine appear to exert opposing actions on thyrotropes through opposite effects on cAMP generation.
Opioid administration to rats suppresses basal or stimulated TSH levels, and the opioid receptor antagonist naloxone reverses these effects.175 Acute opiate administration in humans may slightly stimulate TSH levels, and acute naloxone administration has little effect.176 In contrast to these acute studies, when naloxone is given over 24 hours, 24 hour TSH secretion decreases, primarily owing to a decrease in nocturnal TSH pulse amplitude.177 TSH responses to TRH are also decreased. Serum T3 levels are decreased as well, suggesting that the magnitude of TSH suppression is sufficient to affect thyroid gland function. These findings suggest that endogenous opioids may play a role in tonic stimulation of TSH secretion.
Peripheral Regulation of TSH Secretion
Thyroid hormone directly blocks pituitary secretion of TSH.178 Acute administration of T3 suppresses TSH levels within hours, and chronic administration leads to further suppression. Slight changes in serum thyroid hormone levels within the normal range alter basal and TRH-stimulated TSH levels, confirming the sensitivity of the pituitary gland to thyroid hormone feedback. Thyroid hormones alter tonic TSH secretion and TSH pulse amplitude without affecting pulse frequency, because subjects with primary hypothyroidism have a near-normal number of TSH pulses, and T4 replacement leads to a decrease in TSH pulse amplitude without much change in pulse frequency.72
In addition to direct effects on TSH secretion, thyroid hormones have other actions that affect TSH secretion. At the pituitary level, thyroid hormones decrease the number of TRH receptors179 and stimulate the activity of the pituitary TRH-degrading enzymes,180 which act in concert to decrease TRH stimulation of TSH secretion. At the hypothalamic level, TRH mRNA levels in the PVN are increased in hypothyroidism and are reduced by T3 and T4.181 Hypothalamic SS content is decreased in hypothyroid rats and is restored by T3 treatment. Finally, T3 directly stimulates SS release from hypothalamic tissue.182 These combined effects of thyroid hormones on TRH and SS decrease TRH release from the hypothalamus and indirectly decrease TSH secretion.
Glucocorticoids at pharmacologic doses or high endogenous cortisol levels (Cushing’s syndrome) suppress basal TSH levels, blunt TSH responses to TRH, and diminish the nocturnal TSH surge in humans and animals.183,184 In healthy subjects, infusions of hydrocortisone that increase serum cortisol to levels seen in mild to moderate stress suppress 24 hour TSH secretion.185,186 Glucocorticoid-induced changes in TSH levels are due to decreased TSH pulse amplitude without alteration in TSH pulse frequency, with more profound suppression of nocturnal TSH secretion and abolition of the TSH surge.
Physiologic glucocorticoid levels also affect TSH secretion.79,80 Untreated patients with adrenocortical insufficiency can have elevated serum TSH levels that resolve with steroid replacement. Complimentary studies of metyrapone (an inhibitor of cortisol synthesis) administration to healthy subjects confirm that endogenous cortisol levels suppress TSH secretion, and physiologic hydrocortisone replacement in patients with adrenal insufficiency decreases daytime TSH levels back to those seen in healthy subjects.
Glucocorticoid suppression of TSH levels may occur directly at the pituitary gland. Animal studies suggest that glucocorticoids exert direct effects on thyrotropes to impair TSH secretion, although these appear to be highly dependent on dose and time course of administration.187,188 It does not appear that glucocorticoids directly affect TSH gene transcription. In humans, TSH pulse frequency is maintained during glucorticoid administration, TSH pulse amplitude is reduced, and TSH responses to exogenous TRH are attenuated, suggesting a direct effect on TSH secretion. In addition to direct pituitary effects, it appears that glucocorticoids may have hypothalamic actions that affect TSH levels. Dexamethasone increases hypothalamic TRH levels, and circulating TRH levels are decreased.189 Reports that the proposed consensus sequence for glucocorticoid receptor binding is present in the 5′-flanking region of the TRH gene also support these data.190
Leptin is primarily a product of adipocytes, although it is also located in thyrotrophs. It regulates food intake and energy expenditure, decreasing acutely with fasting in animals and humans.191–193 Exogenous leptin administration to fed rats raises serum TSH levels, probably by increasing TRH gene expression and TRH release. Similarly, leptin administration to fasted rats or humans reverses fasting-induced decrements in TSH levels, also by increasing TRH gene expression and release. This suggests that fasting-related reductions in leptin levels play a role in suppressing TSH secretion. However, immunoneutralization of leptin increases TSH levels; therefore endogenous leptin may inhibit TSH release, at least in rats. In healthy subjects, leptin and TSH have coordinated pulsatility and circadian rhythms in plasma, with a nadir in the late morning and a peak in the early morning; however, leptin-deficient subjects have disorganized pulsatile and circadian rhythms.
Sex steroids may account for higher serum and pituitary TSH concentrations in male compared with female rats. In addition, TSH content is reduced by castration and is restored by androgen administration.121 Testosterone administration to castrated male or female rats increases basal and TRH-stimulated serum TSH levels.194 In contrast, androgen administration to intact female rats does not alter serum or pituitary levels of TSH.195 Estrogen administration to euthyroid rats does not alter serum TSH levels. In euthyroid humans, most studies suggest that changes in endogenous or exogenous sex steroid levels do not affect basal or TRH-stimulated TSH levels.196 No significant gender difference in the basal mean and pulsatile secretion of TSH or in the TSH response to TRH has been noted.197 Therefore, sex steroids do not appear to play a major regulatory role in TSH secretion in humans.
Cytokines are circulating mediators of the inflammatory response that are produced by many cells and have systemic effects on the hypothalamic-pituitary-thyroid axis. Administration of tumor necrosis factor (TNF) or interleukin-6 (IL-6) decreases serum TSH levels in healthy human subjects, and TNF and interleukin-1 (IL-1) decrease TSH levels in animals.198–200 Administration of some of these cytokines may mimic the alterations in thyroid hormone and TSH levels seen in acute nonthyroidal illness. In rats, TNF reduces hypothalamic TRH content and pituitary TSH gene transcription. IL-1 stimulates type II 5′-deiodinase activity in rat brain, which may decrease TSH secretion by increasing intrapituitary T3 levels.
Autocrine and paracrine peptides may alter regulatory pathways within the pituitary gland for TSH secretion, acting in concert with the central and peripheral factors described above.201 Peptides that have been implicated in this role include neurotensin, opioid-related peptides, galanin, substance P, epidermal growth factor (EGF), fibroblast growth factor (FGF), IL-1, and IL-6. Of particular interest is neuromedin B, a mammalian peptide that is structurally and functionally related to the amphibian peptide bombesin.202 Neuromedin B is present in high concentrations in thyrotrope cells, with levels that change according to thyroid status. Administration of neuromedin B to rodents decreases TSH levels, and intrathecal administration of neuromedin B antiserum increases TSH levels. Therefore, neuromedin B appears to act as an autocrine factor that exerts a tonic inhibitory effect on TSH secretion. Additional data suggest that neuromedin B may modulate the action of other TSH secretagogues and release inhibitors, including TRH and thyroid hormones.
Action of TSH
TSH acts on the thyroid gland by binding to the TSH receptor. An excellent review of this subject has been published recently.203 The TSH receptor is located on the plasma membranes of thyroid cells and consists of a long extracellular domain, a transmembrane domain, and a short intracellular domain. The entire extracellular domain and parts of the transmembrane domain of the TSH receptor contribute to TSH binding.
Determinants of TSH Binding
Specificity of TSH binding is conferred by the TSH β subunit. It appears that amino acid residues from 58 to 69, within the βL3 loop, and from 88 to 105, in the “seatbelt” region of the TSH β subunit,204 play an important role in binding to and activating the TSH receptor. The carboxylterminal end of the TSH β subunit contains multiple lysine residues (positions 101, 107, and 110) and a cysteine at position 105, all of which are critical for the ability to bind to the receptor.205 Congenital hypothyroidism due to biologically inactive TSH was found to result from a frameshift mutation with loss of β-cysteine105206 (see “Disorders of TSH Production”). Several regions of the α subunit are also important for TSH activity, particularly the residues α11-20 and α88-92.51,56,207 In addition, the oligosaccharide chain at position α-asparagine52 plays an important role in both binding affinity and receptor activation. A mutant TSH lacking the α-asparagine52 oligosaccharide showed increased in vitro activity, although this same mutation had the opposite effect on CG binding to its native receptor.56 However, such a mutation also increased TSH clearance and this decreased in vivo activity. In addition, the oligosaccharide chains on the TSH subunits are critically important for signal transduction.42,208 In this regard, the α subunit oligosaccharides are important for all the pathways activated by the receptor, whereas the TSHβ subunit oligosaccharide influences only the adenylate cyclase pathway.209 The mechanism by which the oligosaccharides influence signal transduction is not known. A model for the action of the glycoprotein hormones has been proposed that suggests a role for the oligosaccharides in directly modulating the influx of calcium into the target cell.210
Effects of TSH on the TSH Receptor
TSH has been shown to inhibit the expression of the rat TSH receptor by stimulating cAMP production.211 TSH receptor desensitization also has been described both in vivo and in vitro, whereby prior TSH stimulation leads to a 30% to 70% decrease in the subsequent cAMP response to TSH stimulation.212 Recent studies using recombinant TSH receptor have shown that desensitization does not occur when the receptor is expressed in nonthyroidal cells, suggesting that this phenomenon requires a cell-specific factor.213
Actions of TSH on the Thyroid Gland
The effects of TSH on the thyroid gland include changes in thyroid gland growth, cell morphology, iodine metabolism, and synthesis of thyroid hormone. The TSH receptor is coupled to the Gs protein cascade, and binding of TSH activates adenylate cyclase to produce cAMP.214,215 The Gq/phospholipase C/inositol phosphates/Ca2+ pathway is also activated216 and appears to play a role, particularly in regulating iodination.217 TSH is also able to signal through the JAK/STAT218 and mTOR/S6K1219 pathways, with important roles in thyroid cell growth. The end point of TSH action is the production of thyroid hormone by the thyroid gland. The process begins with thyroglobulin gene transcription, which in itself is able to occur independently of TSH.220 However, the transcriptional rate and possibly the mRNA stability are increased by TSH.221 TSH regulates the expression and activation of Rab5a and Rab7, which are rate-limiting catalysts of thyroglobulin internalization and transfer to lysosomes.222 TSH stimulates iodide uptake and organification, then acts on the iodinated thyroglogulin stored in the luminal colloid and stimulates its hydrolysis, resulting in release of the constituent amino acids, including the iodotyronines T3 and T4.
Extrathyroidal Actions of TSH
The ocurrence of precocious puberty in cases of severe juvenile primary hypothyroidism223 has suggested that high levels of TSH are able to cross-activate the gonadotropin receptors. This interaction has now been demonstrated using recombinant human TSH, which has been found to be capable of activating the FSH224 but not the CG/LH receptor.225
Expression of thyrotropin receptor has been reported in the brain226 and pituitary gland.227 In the brain, both astrocytes and neuronal cells were found to express TSH receptor mRNA and protein226 and to stimulate arachidonic acid release and type II 5′-iodothyronine-deiodinase activity.228 In the pituitary gland, the TSH receptor was localized to folliculo-stellate cells and may be involved in paracrine feedback inhibition of TSH secretion, which also may occur in response to TSH receptor autoantibodies.227
Expression of both TSH and its receptor has been reported in lymphocytes229 and erythrocytes,230 and this has led to speculation that TSH may have other nonclassic functions. The TSH receptor also was found to be expressed in adipocytes231 and to stimulate proliferation and inhibit differentiation of preadipocytes in vitro.232 TSH activated the nuclear factor-κB (NF-κB) pathway to induce the release of interleukin-6 (IL-6).233 Direct effects of TSH were described in bone,234 where it modulates tumor necrosis factor α (TNFα)235 and receptor activator of NF-κB ligand (RANKL)236 production to decrease osteoclast differentiation. More studies are needed to determine the physiologic significance of the extrathyroidal effects of TSH, as this may affect the safety of future treatment modalities for thyroid cancer that may attempt to target radioisotopes to the TSH receptor.237
TSH Measurements
In 1926, Uhlenhuth discovered a substance in the anterior pituitary that stimulated thyroid cells, but it was not until the 1960s that TSH was purified from pituitary glands, and in 1963 the first antibodies against TSH were developed.238,239 This provided the key reagents for measuring TSH in blood using immunologic techniques. Accurate and specific measurements of serum TSH concentrations have become the most important method for diagnosing and treating the vast majority of thyroid disorders. Initially, the radioimmunoassays were very insensitive and could detect only high levels seen in primary hypothyroidism.240 Modifications subsequently led to improved sensitivity and specificity, enabling TSH levels as low as 0.5 to 1.0 mU/L to be detected.241 These were called “first-generation assays.” One hundred percent of primary hypothyroid subjects had elevated TSH levels, but these “first-generation assays” could not accurately quantitate values within the normal range, and considerable overlap with the values found in euthyroid and hyperthyroid subjects was noted.242 Monoclonal antibody technology was applied to TSH after the first such antibody was reported in 1982.243 This new method allowed two or more antibodies with precise epitope specificity to be used in sandwich-type assays that subsequently were called immunometric assays.244,245 One or more of the monoclonal antibodies are labeled and are called the “signal antibodies.” The signal may be isotopic, chemiluminescent, or enzymatic. Another monoclonal antibody with completely different epitope specificity is attached to a solid support and is called the “capture antibody.” All antibodies are used in excess, and therefore all TSH molecules in a sample are captured and the signal generated is directly proportional to the level of TSH.
These modifications in the measurement of TSH resulted in important changes. First, the assays were highly specific with no cross-reaction to the other human glycoprotein hormones. Second, 100% of euthyroid controls have detectable and quantifiable levels of TSH residing within a normal range of approximately 0.5 to 5 mU/L. Third, little or no overlap in TSH values is seen in patients with hyperthyroidism compared with euthyroid controls. The degree to which a given assay can separate undetectable TSH levels found in hyperthyroid subjects from normal values in euthyroid controls has improved steadily.246 These improvements have resulted in progressively lower functional detection limits, defined as the lowest TSH value detected with an interassay coefficient of variation ≤20%. Thus, first-generation assays (usually radioimmunoassays) have functional detection limits of 0.5 to 1.0 mU/L, second-generation assays 0.1 to 0.2 mU/L, third-generation assays 0.01 to 0.02 mU/L, and fourth-generation assays 0.001 to 0.002 mU/L. At the present time, the most sensitive commercially available TSH assays are third-generation assays.
Although the population normal range for serum TSH levels is broad, within an individual subject TSH levels are regulated more tightly around an endogenous set point (Fig. 2-7). In a recent study of monthly sampling over a year in healthy euthyroid subjects, the significant difference in serum TSH levels on repeated testing was only 0.75 mU/L, far less than the population normal range.247 It is not clear what determines this individual set point, although studies of monozygotic and dizygotic twins suggest that it is primarily genetically determined.248 Genetic analysis has revealed a number of significant linkage peaks, but no single gene appears to have a major regulatory influence, and regulation of the TSH set point is likely polygenic.249,250 The main environmental factor that affects TSH levels in healthy euthyroid subjects appears to be iodine intake.251
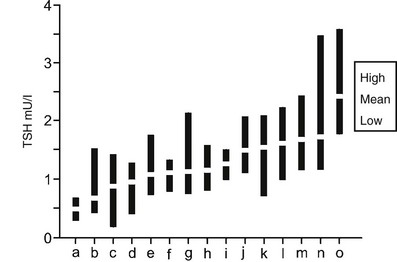
FIGURE 2-7 Serum TSH in 15 normal subjects taken monthly for 12 months. Each black bar represents the highest to lowest monthly TSH measurement, and white rectangles indicate individual mean values. Participants are sorted by increasing mean values (a through o). Laboratory reference range for TSH was 0.3 to 5.0 mU/L. Large differences were seen between individual set points, and unpredictable differences were seen in variations within individuals for the TSH assays. (Data used and figure revised from Andersen S, Pedersen KM, Bruun NH, Laurberg P: Narrow individual variations in serum T[4] and T[3] in normal subjects: a clue to the understanding of subclinical thyroid disease. J Clin Endocrinol Metab 87:1068–1072, 2002.)
Free TSH β and α Subunit Measurements
TSHβ and α subunits were purified in 1974 from human TSH, and specific antibodies to them developed.252 Radioimmunoassays were developed first, and then immunometric assays for the free α subunit. In general, free TSHβ levels are detectable only in primary hypothyroidism and therefore are of limited utility. Free α subunit levels have been useful in the evaluation of pituitary and placental disease. Free α subunit is detectable and measurable in both euthyroid and eugonadal human subjects.253 Elevated values of free α subunit are found in the sera of patients with TSH-secreting or gonadotropin-secreting pituitary tumors254–256 or choriocarcinoma,257 and with a variety of nonpituitary and nonplacental malignancies, including cancers of the lung, pancreas, stomach, prostate, and ovary.258–260
Provocative Testing of TSH
TRH directly stimulates TSH biosynthesis and secretion. Given intravenously, intramuscularly, or orally, TRH causes a reproducible rise in serum TSH levels in euthyroid subjects.261 The test is performed by giving 200 to 500 µg of TRH intravenously and measuring TSH at 0, 20 to 30, 60, 120, and 180 minutes after injection. In euthyroid subjects, an immediate release of TSH rises to peak levels approximately 20 to 30 minutes after TRH injection, usually reaching values fivefold to 10-fold higher than basal. With the use of third- and fourth-generation assays across a wide spectrum of normal and abnormal basal TSH levels, the increase in TSH after TRH stimulation has been between 2.8- and 22.9-fold246 (Fig. 2-8). In hyperthyroid subjects, the undetectable basal serum TSH levels correlate with absent TSH responses to TRH. Patients with low basal serum TSH levels secondary to pituitary insufficiency (secondary hypothyroidism) or hypothalamic disease (tertiary hypothyroidism) have absent or attenuated TSH responses to TRH.262 In contrast, patients with elevated TSH levels due to primary hypothyroidism have exuberant responses to TRH stimulation. However, elevated TSH levels in patients with pituitary TSH-secreting tumors respond less than twofold to TRH stimulation.254
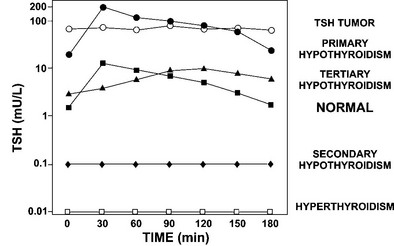
FIGURE 2-8 TSH responses to thyrotropin-releasing hormone (TRH) stimulation in patients with a variety of thyroid disorders compared with normal subjects. Serum samples of TSH were collected every 30 minutes for 3 hours. Subjects with different disorders are indicated on the right. Patients with TSH tumors, secondary hypothyroidism, and hyperthyroidism fail to respond normally to TRH stimulation. Patients with primary hypothyroidism have exuberant responses, whereas those with tertiary (hypothalamic) hypothyroidism have attenuated and delayed responses to TRH stimulation. (Data from Sarapura VD, Samuels MH, Ridgway EC: Thyroid-stimulating hormone. In: Melmed S [ed]: The Pituitary, 2nd ed. Blackwell Science, Malden, MA, pp 187–229, 2002.)
Drugs and TSH Levels
Among the most common causes of abnormal TSH levels are pharmacologic interventions that alter TSH production.263 These can be divided into those that directly affect hypothalamic-pituitary function, those that affect thyroid gland function, and those that alter the distribution of thyroid hormones between free and protein-bound thyroid hormones in plasma.
Decreased Serum TSH: Drugs That Decrease Hypothalamic-Pituitary TSH Production: Clinically, the most important drug that results in decreased serum TSH levels is exogenously administered thyroid hormone. Twenty to thirty percent of patients treated with thyroid hormone have low serum TSH levels, most fitting the diagnostic criteria for subclinical hyperthyroidism. Similarly, thyroid hormone analogues such as 3,5,3′-triiodothyroacetic acid (TRIAC) have the same potential, and the future pharmacologic development of thyroid hormone analogues with increased affinity to the TRβ receptor isoforms will have the capacity to suppress TSH secretion. These compounds decrease TSHβ gene transcription. An interesting variation has been the discovery that RXR analogues such as Bexaroten, used in the treatment of cutaneous lymphoma, can decrease TSHβ transcription, serum TSH, and T4 levels with resultant central hypothyroidism. High doses of glucocorticoids may decrease serum TSH levels through a similar mechanism. Drugs that act at a nongenomic level to decrease TSH production are somatostatin and its analogues and dopamine and its analogues, including bromocriptine, carbergoline, piribidil, levodopa, and lisuride. It is interesting to note that although these drugs acting nongenomically can acutely decrease TSH production, chronic administration usually results in compensatory mechanisms that prevent the development of clinical hypothyroidism. Growth hormone administration stimulating insulin-like growth factor (IGF)-1 production may decrease TSH levels by stimulating endogenous hypothalamic somatostatin production. Exogenous leptin administration can suppress hypothalmic TRH production. Cytokine administration (interferon and interleukins) commonly suppresses TSH levels and has been thought to mediate this action through stimulation of endogenous glucocorticoids. However, a novel alternative mechanism postulates that cytokines stimulate hypothalamic NF-κB production, and this protein directly increases deiodinase 2 gene transcription in astrocytes, leading to increased T4 to T3 conversion, TRH suppression, and central hypothyroidism.
Drugs That Increase Thyroid Hormone Production: Iodine and iodine-containing drugs such as amiodarone, many x-ray contrast agents, and antiseptics can increase thyroid gland production, particularly in susceptible individuals with nodular thyroid disease. Some of the most challenging cases of iodine-induced thyrotoxicosis involve the chronic administration of amiodarone. The resulting high levels of thyroid hormones suppress TSH production. Human chorionic gonadotropin (hCG) used in pharmacologic amounts also has the potential to increase thyroid gland hormone synthesis, because this hormone has 1% of the bioactivity of TSH against the TSH receptor. Increases in FT4 and resulting decreases in TSH levels in the first trimester of pregnancy are thought to be mediated by this mechanism; clearly, thyrotoxicosis in hydatidiform tumors is the result of very high levels of hCG.
Drugs That Increase Free Thyroid Hormones in Plasma: Salicylates and antiepileptic drugs such as Dilantin have long been known to compete with the binding of thyroid hormones to their binding proteins. They do not result in a higher free fraction of T4 and T3, nor do they suppress TSH biosynthesis. Drugs in the fenamate class of nonsteroidal antiinflammatory drugs such as fenoclofenac may also disrupt the normal partition of free T4 by interfering with T4 transport into cells that express the organic anion-transporting polypeptide 1C1, OATP1C1.264 This transporter has very high specificity for T4 and is found at the blood-brain barrier. Such a mechanism theoretically could increase free T4. Heparin has a lypolytic action, resulting in increased free fatty acids, which also can displace thyroid hormones from binding proteins; this agent can cause spuriously elevated free T4 during dialysis assays.
Increased Serum TSH: Drugs That Increase Hypothalamic-Pituitary TSH Production: Sustained increases in TSH production by direct stimulation of the hypothalamus or the pituitary are very unusual. TRH administration is the most potent but can be completely attenuated by subsequent rises in circulating thyroid hormones. Drugs from the opioid class, including morphine, apomorphine, heroin, buprenorphine, and pentazocine, all have been associated with increases in TSH levels. Theophylline and amphetamines, including ephedrine, may directly stimulate hypothalamic TRH or pituitary TSH production. Dopamine receptor antagonists domperidone, sulphuride, and metoclopramine decrease dopaminergic tone and thereby tonic suppression of TSH. Certain neuroleptics, haloperidol and chlorpromazine, have transiently increased TSH levels. Decreases in circulating glucocorticoids secondary to inhibition of adrenal gland hormone synthesis can cause high TSH levels. These drugs include aminoglutethimide, ethionamide, ketoconazole, and mitotane.
Drugs That Decrease Thyroid Hormone Production: Drugs that directly inhibit the thyroid gland, thereby increasing TSH levels, are perhaps the most important pharmacologic causes of high TSH levels. The antithyroid drugs propylthiouracil and methimazole directly inhibit thyroid hormone synthesis, thereby increasing TSH levels. All of the iodine-containing drugs and supplements can inhibit thyroid hormone production and release in susceptible individuals, particularly those with autoimmune thyroid disease. Lithium can have a similar sustained effect. Perchlorate, as a competitive inhibitor of iodine uptake by the thyroid gland, has been suspected to be a “thyroid gland disruptor” in contaminated water supplies and is certainly used in the therapy of severe hyperthyroidism.
Drugs That Decrease Free Thyroid Hormones in Plasma: Administration of estrogen has been reported to transiently decrease free thyroid hormones owing to increases in serum thyroxine binding globulin and increased TSH levels. One of the most common ways that drugs increase TSH levels is by blocking levothyroxine absorption from the intestine in patients taking this drug for hypothyroidism. Antacids and drugs that decrease gastric acid secretion inhibit thyroid hormone absortion. Iron and calcium to a lesser extent bind levothyroxine in the intestine and prevent its absorption. Drugs that block bile acid reabsorption such as cholestyramine and colestipol also inhibit levothyroxine uptake in the gut.
Disorders of TSH Production
Hypothalamic Disorders
The hypothalamus is the source of three important molecules that regulate TSH secretion: TRH, dopamine, and somatostatin. TRH is the only positive regulator of TSH production from the hypothalamus. Structural abnormalities in the hypothalamus have resulted in clinical hypothyroidism, which presumably is due to decreased or defective TRH production (Table 2-1). Exogenous administration of TRH to patients with hypothalamic hypothyroidism can restore serum thyroid hormone levels toward normal, resulting in clinical improvement.265 Hypothalamic disease has not been reported to elevate endogenous SS or dopamine, resulting in decreased TSH and hypothalamic hypothyroidism. However, administration of GH has been associated with this syndrome,160 presumably by stimulating hypothalamic SS production or enhancing the conversion of T4 to T3, both of which may result in decreased TRH production. In contrast, acute exogenous administration of both dopamine and somatostatin can decrease TSH production. It is therefore paradoxical that chronic administration of dopamine agonists or somatostatin analogues does not produce central hypothyroidism. It is likely that the exquisite sensitivity of TSH production to small changes in thyroid hormone levels may result in a correction for alterations induced by these analogues. As was outlined earlier, leptin derived from fat cells does circulate in plasma and has a variety of effects on the hypothalamus, one of which is to increase TRH production.
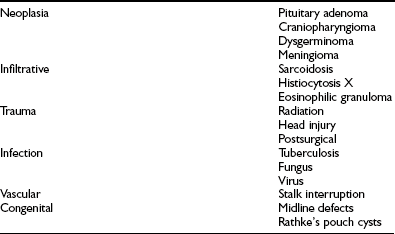
The serum levels of TSH in hypothalamic hypothyroidism may be low, normal, or even minimally elevated.262 The paradoxical finding of low serum thyroid hormone levels in association with normal or elevated basal TSH levels suggests that circulating TSH in hypothalamic hypothyroidism is biologically defective.265 In fact, TRH deficiency has been associated with differences in glycosylation patterns of the TSH molecule, which result in decreased TSH receptor binding and activation. The 24 hour secretory profile of TSH in patients with hypothalamic hypothyroidism is also abnormal.75 The frequency of TSH pulses is the same as for euthyroid controls, but the amplitude of the pulses is decreased, particularly at nighttime, resulting in loss of the normal nocturnal surge (see Fig. 2-4, bottom panel).
Pituitary Disorders: Congenital TSH Deficiency
Increases or decreases in pituitary TSH production can result directly from abnormalities in the pituitary due to congenital or acquired causes. Congenital hypothyroidism due to decreased TSH production generally is inherited as an autosomal recessive disorder, and affected individuals have severe mental and growth retardation. The molecular basis for isolated TSH deficiency has usually involved mutations in the TSHβ gene (Table 2-2). For example, a single base substitution in one family at nucleotide position 145 of the TSHβ gene altered the CAGYC region,266 a critically important contact point for the noncovalent combination of the TSHβ and α subunits. In other kindreds, a single base substitution introduced a premature stop codon, resulting in a truncated TSHβ subunit, which included only the first 11267 amino acids. Another type of mutation involves a nonsense 25 amino acid protein that results from mutation of a donor splice site and a new out-of-frame translational start point.268 In other cases, the disorder involves the production of biologically inactive TSH with loss of cysteine105 that disrupts the disulfide bridge formation important in “seatbelt” stability269,270 and is perhaps the most common of the TSHβ mutations; the less common mutation at cysteine85 disrupts the cysteine knot that is important for heterodimer formation and TSH receptor binding,268,271 resulting in a similar phenotype, except that in some cases circulating TSH was detectable.
Table 2-2
Congenital Hypothyroidism: Isolated TSHβ Defects
*Compound heterozygosity for T313del (C105Vfs114X) and C145T (Q49X) in one infant.
A more common cause of congenital TSH deficiency arises not as the result of a mutation in the TSHβ gene, but rather following defective production of a key transcription factor necessary for TSHβ gene expression. This occurs in combined pituitary hormone deficiency (CPHD), in which subjects have congenital hypothyroidism and growth retardation secondary to TSH, GH, and prolactin deficiencies.272,273 The genes for all three of these proteins are dependent on the pituitary-specific transcription factor Pit-1 for their expression. Mutations in the coding region of the Pit-1 gene alter the function of the Pit-1 protein or completely disrupt its structure. The absence of Pit-1 prevents normal pituitary development, resulting in hypoplasia of the pituitary. In heterozygotes, in which a normal allele is present, the abnormal Pit-1 protein can bind to DNA but may not be able to effect transactivation, interfering with the function of the normal Pit-1 (dominant negative mechanism). It is interesting to note that a similar combined hormone deficiency syndrome has been reported in two murine models in which the Pit-1 gene is defective: a point mutation found in the Snell dwarf (dw)8 and a major deletion in the Jackson dwarf (dwJ).274
An even more frequent cause of CPHD has been delineated by the discovery of a pituitary-specific transcription factor called “prophet of Pit-1” (PROP-1). This factor is a paired-like homeodomain protein in which a mutation in the murine species causes the Ames dwarf (df) mouse phenotype.275 Subsequently, patients with CPHD were found to have mutations in the PROP-1 gene.276,277 More than 50% of families with CPHD have been shown to contain mutations in the PROP-1 gene,278 far exceeding the prevalence of mutations in the Pit-1 gene as a cause for CPHD. The mutations all are found in the homeodomain part of the molecule. It is interesting to note that the phenotype of patients with PROP-1 mutations includes deficiencies not only of GH, prolactin, and TSH, but also of LH and FSH. Furthermore, the development of hormone deficiencies may not be neonatal but rather may occur progressively up to the age of adolescence. ACTH deficiency has been reported as a late consequence in CPHD.279
Pituitary Disorders: Acquired TSH Deficiency
The acquired causes of pituitary TSH deficiency generally relate to destructive processes in the anterior pituitary or hypothalamus. These can include infiltrative or infectious disorders, compression secondary to neoplastic processes, or active ischemic and hemorrhagic processes involving the pitutary gland. The most common cause of acquired pituitary TSH deficiency is neoplastic destruction or compression of normal anterior pituitary cells by a primary pituitary neoplasm, a craniopharyngioma, or infiltrating metastatic disease to the pituitary. Likewise, these same processes can extend into the hypothalamus, thus interrupting normal TRH production. In acquired pituitary TSH deficiency, multiple pituitary deficiencies are concomitantly associated; acquired isolated TSH deficiency is rarely, if ever, seen. When hypothyroidism is due to a pituitary disorder, the disease is called secondary hypothyroidism, and when it is due to a hypothalamic disorder (see “Hypothalamic Disorders”), it is called tertiary hypothyroidism. Most patients with acquired pituitary TSH deficiency have symptoms of hypothyroidism, as well as symptoms of LH, FSH, GH, and usually ACTH deficiency. Serum free T4 and T3 levels are low in association with a low or low/normal basal TSH level. The distinction between secondary and tertiary hypothyroidism can be challenging. A completely absent TSH and prolactin response to TRH favors secondary hypothyroidism, whereas a mildly elevated basal prolactin level (due to disrupted hypothalamic dopamine production) and normal or elevated basal TSH levels (with subnormal bioactivity) favor tertiary hypothyroidism.
Pituitary Disorders: Increased Pituitary TSH Production
TSH-Secreting Pituitary Tumors: TSHomas are rare neoplasms of the anterior pituitary. They account for less than 1% of all pituitary tumors.280 Patients have high levels of thyroid hormones in association with normal or high levels of TSH. Tumor cells are highly differentiated but synthesize the α subunit in excess of the TSHβ subunit.254 This phenomenon is useful in that a molar ratio of α subunit:TSH (ng/mL divided by µU/mL multiplied by 10) of greater than 1 supports the diagnosis of a TSH-secreting pituitary tumor when found in a hyperthyroid and eugonadal patient. For example, if such a patient had a TSH of 10 µU/mL and a free α subunit of 3 ng/mL, the calculated ratio would be 3. Because menopausal women have high gonadotropins and high free α subunit levels, use of this calculated ratio in a hyperthyroid patient would not accurately reflect free α subunit coming from thyrotrope cells. TSH-secreting tumors fail to respond to TRH stimulation and suppression by dopamine (Fig. 2-9). Another characteristic of these tumors is their failure to respond to thyroid hormone by the normal negative feedback of thyroid hormone on TSH production. In contrast, inhibition of TSH release in response to SS is preserved in these tumors (see Fig. 2-9).
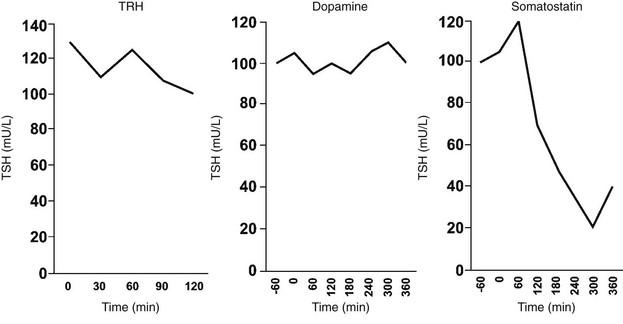
FIGURE 2-9 A patient with a TSH tumor fails to respond to TRH stimulation (500 µg intravenously) and dopamine suppression (4 µg/min for 4 hours). In contrast, TSH secretion is significantly suppressed by somatostatin (500 µg bolus followed by 250 µg/min for 4 hours). (Data from Sarapura VD, Samuels MH, Ridgway EC: Thyroid-stimulating hormone. In: Melmed S [ed]: The Pituitary, 2nd ed. Blackwell Science, Malden, MA, pp 187–229, 2002.)
Thyroid Hormone Resistance Syndromes (RTH): Another pituitary gland disorder resulting in elevated levels of serum TSH is thyroid hormone resistance (RTH).281,282 In 1967, Refetoff et al.283 were the first to describe three siblings who were clinically euthyroid or hypothyroid with goiters, stippled epiphyses, and deaf mutism. Each of the children had elevated levels of protein-bound iodide, which subsequently were shown to be associated with high serum total and free thyroid hormone levels, elevated TSH levels, and peripheral tissue responses that were refractory not only to the endogenous high levels of thyroid hormone, but also to exogenously administered supraphysiologic levels of thyroid hormone.281 RTH was found to be linked to the TRβ gene locus on chromosome 3 and then was localized to point mutations in the 9th and 10th exons of the TRβ gene, which encode for the T3 binding and adjacent hinge domains. These mutations usually disrupt normal T3 binding without altering DNA binding. RTH without TRβ mutations occurs in about 15% of cases. Because most cases of RTH are heterozygotes and are inherited as autosomal dominant traits, only half of their TRβ receptors are abnormal. TRα gene mutations have not been reported in RTH. An overwhelming majority of mutations are single nucleotide substitutions, which change a single amino acid or introduce a stop codon. Since 1967 over 1000 cases of RTH belonging to 372 families have been identified.282–294 Mutations have been found in 343 of these families, and many families have the same mutation, because only 124 different mutations have been identified.
References
1. Kouki, T, Imai, H, Aoto, K, et al. Developmental origin of the rat adenohypophysis prior to the formation of Rathke’s pouch. Development. 2001;128:959–963.
2. Zhu, X, Gleiberman, AS, Rosenfeld, MG. Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev. 2007;87:933–963.
3. Dasen, JS, O’Connell, SM, Flynn, SE, et al. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell. 1999;97:587–598.
4. Voss, JW, Rosenfeld, MG. Anterior pituitary development: short tales from dwarf mice. Cell. 1992;70:527–530.
5. Treier, M, Gleiberman, AS, O’Connell, SM, et al. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev. 1998;12:1691–1704.
6. Drolet, DW, Scully, KM, Simmons, DM, et al. TEF, a transcription factor expressed specifically in the anterior pituitary during embryogenesis, defines a new class of leucine zipper proteins. Genes and Development. 1991;5:1739–1753.
7. Simmons, DM, Voss, JW, Holloway, JM, et al. Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes and Development. 1990;4:695–711.
8. Camper, SA, Saunders, TL, Katz, RW, et al. The Pit-1 transcription factor gene is a candidate for the murine snell dwarf mutation. Genomics. 1990;8:586–590.
9. Qi, Y, Ranish, JA, Zhu, X, et al. Atbf1 is required for the Pit1 gene early activation. Proc Natl Acad Sci U S A. 2008;105:2481–2486.
10. Steger, DJ, Hecht, JH, Mellon, PL. GATA-binding proteins regulate the human gonadotropin alpha-subunit gene in the placenta and pituitary gland. Mol Cell Biol. 1994;14:5592–5602.
11. Gordon, DF, Woodmansee, WW, Black, JN, et al. Domains of Pit-1 required for transcriptional synergy with GATA-2 on the TSH beta gene. Mol Cell Endocrinol. 2002;196:53–66.
12. Charles, MA, Saunders, TL, Wood, WM, et al. Pituitary-specific gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377.
13. Gleiberman, AS, Michurina, T, Encinas, JM, et al. Genetic approaches identify adult pituitary stem cells. Proc Natl Acad Sci U S A. 2008;105:6332–6337.
14. Fauquier, T, Rizzoti, K, Dattani, M, et al. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A. 2008;105:2907–2912.
15. Wondisford, FE, Radovick, S, Moates, JM, et al. Isolation and characterization of the human thyrotropin beta-subunit gene: Differences in gene structure and promoter function from murine species. J Biol Chem. 1988;263:12538–12542.
16. Dracopoli, NC, Rettig, WJ, Whitfield, GK, et al. Assignment of the gene for the beta subunit of thyroid-stimulating hormone to the short arm of human chromosome 1. Proc Natl Acad Sci U S A. 1986;83:1822–1826.
17. Wood, WM, Kao, MY, Gordon, DF, et al. Thyroid hormone regulates the mouse thyrotropin beta subunit gene promoter in transfected primary thyrotropes. J Biol Chem. 1989;264:14840–14847.
18. Haugen, BR, McDermott, MT, Gordon, DF, et al. Determinants of thyrotrope-specific TSH-beta promoter activation: Cooperation of Pit-1 with another factor. J Biol Chem. 1996;271:385–389.
19. Camper, SA, Saunders, TL, Kendall, SK, et al. Implementing transgenic and embryonic stem cell technology to study gene expression, cell-cell interactions and gene function. Biol Reprod. 1995;52:246–257.
20. Wood, WM, Ocran, KW, Kao, MY, et al. Protein factors in thyrotropic tumor nuclear extracts bind to a region of the mouse thyrotropin beta-subunit promoter essential for expression in thyrotropes. Molecular Endocrinology. 1990;4:1897–1904.
21. Gordon, DF, Lewis, SR, Haugen, BR, et al. Pit-1 and GATA-2 interact and functionally cooperate to activate the thyrotropin beta-subunit promoter. J Biol Chem. 1997;272:24339–24347.
22. Gordon, DF, Tucker, EA, Tundwal, K, et al. 2006 MED220/thyroid receptor-associated protein 220 functions as a transcriptional coactivator with Pit-1 and GATA-2 on the thyrotropin-beta promoter in thyrotropes. Mol Endocrinol. 2006;20:1073–1089.
23. Yuan, CX, Ito, M, Fondell, JD, et al. The TRAP220 component of a thyroid hormone receptor-associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc Natl Acad Sci U S A. 1998;95:7939–7944.
24. Ito, M, Yuan, CX, Okano, HJ, et al. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol Cell. 2000;5:683–693.
25. Naylor, SL, Chin, WW, Goodman, HM, et al. Chromosomal assignment of genes encoding the alpha and beta subunits of glycoprotein hormones in man and mouse. Somat Cell Genet. 1983;9:757–770.
26. Fiddes, JC, Goodman, HM. The gene encoding the common alpha subunit of the four human glycoprotein hormones. J Mol Appl Genet. 1981;1:3–18.
27. Jameson, JL, Jaffe, RC, Deutsch, PJ, et al. The gonadotropin alpha-gene contains multiple protein binding domains that interact to modulate basal and cAMP-responsive transcription. J Biol Chem. 1988;263:9879–9886.
28. Horn, F, Windle, JJ, Barnhart, KM, et al. Tissue-specific gene expression in the pituitary: the glycoprotein hormone alpha-subunit gene is regulated by a gonadotrope-specific protein. Mol Cell Biol. 1992;12:2143–2153.
29. Sarapura, VD, Strouth, HL, Wood, WM, et al. Activation of the glycoprotein hormone alpha-subunit gene promoter in thyrotropes. Mol Cell Endocrinol. 1998;146:77–86.
30. Kendall, SK, Gordon, DF, Birkmeier, TS, et al. Enhancer-mediated high level expression of mouse pituitary glycoprotein hormone alpha-subunit transgene in thyrotropes, gonadotropes, and developing pituitary gland. Molecular Endocrinology. 1994;8:1420–1433.
31. Barnhart, KM, Mellon, PL. The orphan nuclear receptor, steroidogenic factor-1, regulates the glycoprotein hormone alpha-subunit gene in pituitary gonadotropes. Molecular Endocrinology. 1994;8:878–885.
32. Wood, WM, Dowding, JM, Sarapura, VD, et al. Functional interactions of an upstream enhancer of the mouse glycoprotein hormone alpha-subunit gene with proximal promoter sequences. Mol Cell Endocrinol. 1998;142:141–152.
33. Roberson, MS, Schoderbek, WE, Tremml, G, et al. Activation of the glycoprotein hormone alpha-subunit promoter by a LIM-homeodomain transcription factor. Mol Cell Biol. 1994;14:2985–2993.
34. Sheng, HZ, Zhadanov, AB, Mosinger, B, et al. Specification of pituitary cell lineages by the lim homeobox gene Lhx3. Science. 1996;272:1004–1007.
35. Sarapura, VD, Gordon, DF, Strouth, HL, et al. Msx1 is present in thyrotropic cells and binds to a consensus site on the glycoprotein hormone alpha-subunit promoter. Molecular Endocrinology. 1997;11:1782–1794.
36. Bach, I, Carriere, C, Ostendorff, HP, et al. A family of LIM domain-associated cofactors confer transcriptional synergism between LIM and Otx homeodomain proteins. Genes and Development. 1997;11:1370–1380.
37. Wood, WM, Dowding, JM, Gordon, DF, et al. An upstream regulator of the glycoprotein hormone alpha-subunit gene mediates pituitary cell type activation and repression by different mechanisms. J Biol Chem. 1999;274:15526–15532.
38. Weintraub, BD, Gesundheit, N. Thyroid-stimulating hormone synthesis and glycosylation: clinical implications. Thyroid Today. 1987;10:1–11.
39. Fiddes, JC, Talmadge, K. Structure, expression and evolution of the genes for the human glycoprotein hormones. Recent Prog Horm Res. 1984;40:43–78.
40. Pierce, JG, Parsons, TF. Glycoprotein hormones: Structure and function. Annu Rev Biochem. 1981;50:465–495.
41. Takata, K, Watanabe, S, Hirono, M, et al. The role of the carboxyl-terminal 6 amino acid extension of human TSH-β subunit. Biochem Biophys Res Commun. 1989;165:1035–1042.
42. Thotakura, NR, LiCalzi, L, Weintraub, BD. The role of carbohydrate in thyrotropin action assessed by a novel method of enzymatic deglycosylation. J Biol Chem. 1990;265:11527–11534.
43. Magner, JA, Weintraub, BD. Thyroid-stimulating hormone subunit processing and V combination in microsomal subfractions of mouse pituitary tumor. J Biol Chem. 1982;257:6709–6715.
44. Parsons, TF, Bloomfield, GA, Pierce, JG, Purification of an alternative form of the α-subunit of the glycoprotein hormones from bovine pituitaries and identification of its O-linked oligosaccharides. J Biol Chem. 1983;258:240–244.
45. Behrens, NH, Leloir, LF. Dolichol monophosphate glucose: an intermediate in glucose transfer in liver. Proc Natl Acad Sci USA. 1970;66:153–159.
46. Kornfeld, R, Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664.
47. Magner, JA. Thyroid Stimulating Hormone: Biosynthesis, Cell Biology, and Bioactivity. Endocrine Rev. 1990;11:354–385.
48. Thotakura, NR, Desai, RK, Bates, LG, et al. Biological activity and metabolic clearance of a recombinant human thyrotropin produced in Chinese hamster ovary cells. Endocrinology. 1991;128:341–348.
49. Weintraub, BD, Stannard, BS, Meyers, L. Glycosylation of thyroid-stimulating hormones in pituitary cells: Influence of high mannose oligosaccharide units on subunit aggregation, combination, and intracellular degradation. Endocrinology. 1983;112:1331–1345.
50. Lapthorn, AJ, Harris, DC, Littlejohn, A, et al. Crystal structure of human chorionic gonadotropin. Nature. 1994;369:455–461.
51. Szkudlinski, MW, Teh, NG, Grossmann, M, et al. Engineering human glycoprotein hormone superactive analogs. Nat Biotechnol. 1996;14:1257–1263.
52. Fairlie, WD, Stanton, PG, Hearn, MTW. The disulphide bond structure of thyroid-stimulating hormone β-subunit. Biochem J. 1996;314:449–455.
53. Weller, CT, Lustbader, J, Seshadri, K, et al. Structural and conformational analysis of glycan moieties in situ on isotopically 13C,15N-enriched human chorionic gonadotropin. Biochemistry. 1996;35:8815–8823.
54. Sun, PD, Davies, DR. The cystine-knot growth factor superfamily. Ann Rev Biophys Biomol Struct. 1995;24:269–291.
55. Weintraub, BD, Stannard, BS, Linnekin, D, et al. Relationship of glycosylation to de novo thyroid-stimulating hormone biosynthesis and secretion by mouse pituitary tumor cells. J Biol Chem. 1980;255:5715–5723.
56. Grossmann, M, Szkudlinski, MW, Tropea, JE, et al. Expression of human thyrotropin in cell lines with different glycosylation patterns combined with mutagenesis of specific glycosylation sites. Characterization of a novel role for the oligosaccharides in the in vitro and in vivo bioactivity. J Biol Chem. 1995;270:29378–29385.
57. Magner, JA, Papagiannes, E. Structures of high-mannose oligosaccharides of mouse thyrotropin: differential processing of α- versus β-subunits of the heterodimer. Endocrinology. 1987;120:10–17.
58. Weiner, RS, Dias, JA. Biochemical analyses of proteolytic nicking of the human glycoprotein hormone α-subunit and its effect on conformational epitopes. Endocrinology. 1992;131:1026–1036.
59. Xing, Y, Myers, RV, Cao, D, et al. Glycoprotein hormone assembly in the endoplasmic reticulum; I. The glycosylated end of human alpha-subunit loop 2 is threaded through a beta-subunit hole. J Biol Chem. 2004;279:35426–35436.
60. Hayashizaki, Y, Hiraoka, Y, Endo, Y, et al. Thyroid-stimulating hormone (TSH) deficiency caused by a single base substitution in the CAGYC region of the β-subunit. EMBO J. 1989;8:2291–2296.
61. Matzuk, MM, Kornmeier, CM, Whitfield, GK, et al. The glycoprotein hormone α-subunit is critical for secretion and stability of the human thyrotropin β-subunit. Mol Endocrinol. 1988;2:95–100.
62. Lash, RW, Desai, RK, Zimmerman, CA, et al. Mutations of the human thyrotropin β-subunit glycosylation site reduce thyrotropin synthesis independent of changes in glycosylation status. J Endocrinol Invest. 1992;15:255–263.
63. Kelly, RB. Pathways of protein secretion in eukaryotes. Science. 1985;230:25–32.
64. Odell, WD, Utiger, RD, Wilber, JF, et al. Estimation of the secretion rate of thyrotropin in man. J Clin Invest. 1967;46:953–959.
65. Ridgway, EC, Weintraub, BD, Maloof, F. Metabolic clearance and production rates of human thyrotropin. J Clin Invest. 1974;53:895–903.
66. Baker, BL, Jaffe, RB. The genesis of cell types in the adenohypophysis of the human fetus as observed with immunocytochemistry. Am J Anat. 1974;143:137–149.
67. Fisher, DA, Polk, DH. Development of the thyroid. Ballieres Clin Endocrinol Metab. 1989;3:67–80.
68. Fisher, DA, Klein, AH. Thyroid development and disorders of thyroid function in the newborn. N Engl J Med. 1981;304:702–711.
69. Roti, E. Regulation of thyroid stimulating hormone (TSH) secretion in the fetus and neonate. J Endocrinol Invest. 1988;11:145–155.
70. Adams, LM, Emery, JR, Clark, S, et al. Reference ranges for newer thyroid function tests in premature infants. J Pediatr. 1995;126:122–127.
71. Van Wassenaer, AG, Kolk, JH, Dekker, FW, et al. Thyroid function in very preterm infants: influences of gestational age and disease. Pediatr Res. 1997;42:604–609.
72. Samuels, MH, Veldhuis, JD, Henry, P, et al. Pathophysiology of pulsatile and copulsatile release of thyroid-stimulating hormone, luteinizing hormone, follicle-stimulating hormone, and α-subunit. J Clin Endocrinol Metab. 1990;71:425–432.
73. Russell, W, Harrison, RF, Smith, N, et al. Free triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J Clin Endocrinol Metab. 2008;93:2300–2306.
74. Mantagos, S, Koulouris, A, Makri, M, et al. Development of thyrotropin circadian rhythm in infancy. J Clin Endocrinol Metab. 1992;74:71–74.
75. Samuels, MH, Lillehei, K, Kleinschmidt-Demasters, BK, et al. Patterns of pulsatile pituitary glycoprotein secretion in central hypothyroidism and hypogonadism. J Clin Endocrinol Metab. 1990;70:391–395.
76. Van Den Barghe, G, de Zegher, F, Veldhuis, JD, et al. Thyrotropin and prolactin release in prolonged critical illness: dynamics of spontaneous secretion and effects of growth hormone-secretagogues. Clin Endocrinol (Oxf). 1997;47:599–612.
77. Samuels, MH, Henry, P, Luther, M, et al. Pulsatile TSH secretion during 48-hour continuous TRH infusions. Thyroid. 1993;3:201–206.
78. Murakimi, M, Tanaka, K, Greer, MA. There is a nyctohumeral rhythm of type II iodothyronine 5′-deiodinase activity in rat anterior pituitary. Endocrinology. 1988;123:1631–1635.
79. Samuels, MH. Effects of variations in physiological cortisol levels on thyrotropin secretion in subjects with adrenal insufficiency: a clinical research center study. J Clin Endocrinol Metab. 2000;85:1388–1393.
80. Samuels, MH. Effects of metyrapone administration on thyrotropin secretion in healthy subjects—a clinical research center study. J Clin Endocrinol Metab. 2000;85:3049–3052.
81. Shupnik, MA, Greenspan, SL, Ridgway, EC. Transcriptional regulation of thyrotropin subunit genes by thyrotropin-releasing hormone and dopamine in pituitary cell cultures. J Biol Chem. 1986;261:12675–12679.
82. Shibusawa, N, Yamada, M, Hirato, J, et al. Requirement of thyrotropin-releasing hormone for the postnatal functions of pituitary thyrotrophs: ontogeny study of congenital tertiary hypothyroidism in mice. Mol Endocrinol. 2000;14:137–146.
83. Abe, H, Murao, K, Imachi, H, et al. Thyrotropin-releasing hormone-stimulated thyrotropin expression involves islet-brain-1/c-Jun N-terminal kinase interacting protein-1. Endocrinology. 2004;145:5623–5628.
84. Shupnik, MA, Weck, J, Hinkle, PM. Thyrotropin (TSH)-releasing hormone stimulates TSH beta promoter activity by two distinct mechanisms involving calcium influx through L type Ca2+ channels and protein kinase C. Mol Endocrinol. 1996;10:90–99.
85. Kim, MK, McClaskey, JH, Bodenner, DL, et al. An AP-1-like factor and the pituitary-specific factor Pit-1 are both necessary to mediate hormonal induction of human thyrotropin beta gene expression. J Biol Chem. 1993;268:23366–23375.
86. Weintraub, BD, Wondisford, FE, Farr, EA, et al. Pre-translational and post-translational regulation of TSH synthesis in normal and neoplastic thyrotrophs. Horm Res. 1989;32:22–24.
87. Shupnik, MA, Rosenzweig, BA, Showers, MO. Interactions of thyrotropin-releasing hormone, phorbol ester, and forskolin-sensitive regions of the rat thyrotropin-beta gene. Mol Endocrinol. 1990;4:829–836.
88. Kapiloff, MS, Farkash, Y, Wegner, M, et al. Variable effects of phosphorylation of Pit-1 dictated by the DNA response elements. Science. 1991;253:786–789.
89. Steinfelder, HJ, Radovick, S, Wondisford, FE. Hormonal regulation of the thyrotropin beta-subunit gene by phosphorylation of the pituitary-specific transcription factor Pit-1. Proc Natl Acad Sci U S A. 1992;89:5942–5945.
90. St Germain, DL, Galton, VA. The deiodinase family of selenoproteins. Thyroid. 1997;7:655–668.
91. Bianco, AC, Salvatore, D, Gereben, B, et al. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89.
92. Davis, PJ, Leonard, JL, Davis, FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–218.
93. Shupnik, MA, Chin, WW, Habener, JF, et al. Transcriptional regulation of the thyrotropin subunit genes by thyroid hormone. J Biol Chem. 1985;260:2900–2903.
94. Shupnik, MA, Ridgway, EC. Thyroid hormone control of thyrotropin gene expression in rat anterior pituitary cells. Endocrinology. 1987;121:619–624.
95. Shupnik, MA, Ardisson, LJ, Meskell, MJ, et al. Triiodothyronine (T3) regulation of thyrotropin subunit gene transcription is proportional to T3 nuclear receptor occupancy. Endocrinology. 1986;118:367–371.
96. Shupnik, MA, Ridgway, EC. Triiodothyronine rapidly decreases transcription of the thyrotropin subunit genes in thyrotropic tumor explants. Endocrinology. 1985;117:1940–1946.
97. Refetoff, S. Resistance to thyroid hormone. Curr Ther Endocrinol Metab. 1997;6:132–134.
98. Bodenner, DL, Mroczynski, MA, Weintraub, BD, et al. A detailed functional and structural analysis of a major thyroid hormone inhibitory element in the human thyrotropin beta-subunit gene. J Biol Chem. 1991;266:21666–21673.
99. Glass, CK, Rosenfeld, MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141.
100. Haugen, BR, Brown, NS, Wood, WM, et al. The thyrotrope-restricted isoform of the retinoid X receptor (gamma 1) mediates 9-cis retinoic acid suppression of thyrotropin beta promoter activity. Molecular Endocrinology. 1997;11:481–489.
101. Sharma, V, Hays, WR, Wood, WM, et al. Effects of rexinoids on thyrotrope function and the hypothalamic-pituitary-thyroid axis. Endocrinology. 2006;147:1438–1451.
102. Sherman, SI, Gopal, J, Haugen, BR, et al. Central hypothyroidism associated with retinoid X receptor-selective ligands. N Engl J Med. 1999;340:1075–1079.
103. Matsushita, A, Sasaki, S, Kashiwabara, Y, et al. Essential role of GATA2 in the negative regulation of thyrotropin beta gene by thyroid hormone and its receptors. Mol Endocrinol. 2007;21:865–884.
104. Ordentlich, P, Downes, M, Evans, RM. Corepressors and nuclear hormone receptor function. Curr Top Microbiol Immunol. 2001;254:101–116.
105. Liu, Y, Xia, X, Fondell, JD, et al. Thyroid hormone-regulated target genes have distinct patterns of coactivator recruitment and histone acetylation. Mol Endocrinol. 2006;20:483–490.
106. Nakano, K, Matsushita, A, Sasaki, S, et al. Thyroid-hormone-dependent negative regulation of thyrotropin beta gene by thyroid hormone receptors: study with a new experimental system using CV1 cells. Biochem J. 2004;378:549–557.
107. Shibusawa, N, Hashimoto, K, Nikrodhanond, AA, et al. Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest. 2003;112:588–597.
108. Wood, WM, Ocran, KO, Gordon, DF, et al. Isolation and characterization of mouse complementary DNAs encoding alpha and beta thyroid hormone receptors from thyrotrope cells: the mouse pituitary specific beta-2 isoform differs at the amino terminus from the corresponding species from rat pituitary tumor cells. Molecular Endocrinology. 1991;5:1049–1061.
109. Hodin, RA, Lazar, MA, Wintman, BI, et al. Identification of a thyroid hormone receptor that is pituitary-specific. Science. 1989;244:76–79.
110. Langlois, MF, Zanger, K, Monden, T, et al. A unique role of the beta-2 thyroid hormone receptor isoform in negative regulation by thyroid hormone. Mapping of a novel amino-terminal domain important for ligand-independent activation. J Biol Chem. 1997;272:24927–24933.
111. Abel, ED, Kaulbach, HC, Campos-Barros, A, et al. Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest. 1999;103:271–279.
112. Hallenbeck, PL, Phyillaier, M, Nikodem, V. Divergent effects of 9-cis-retinoic acid receptor on positive and negative thyroid hormone receptor-dependent gene expression. J Biol Chem. 1993;268:3825–3828.
113. Weiss, RE, Xu, J, Ning, G, et al. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J. 1999;18:1900–1904.
114. Tagami, T, Gu, WX, Peairs, PT, et al. A novel natural mutation in the thyroid hormone receptor defines a dual functional domain that exchanges nuclear receptor corepressors and coactivators. Mol Endocrinol. 1998;12:1888–1902.
115. Nikrodhanond, AA, Ortiga-Carvalho, TM, Shibusawa, N, et al. Dominant role of thyrotropin-releasing hormone in the hypothalamic-pituitary-thyroid axis. J Biol Chem. 2006;281:5000–5007.
116. Krane, IM, Spindel, ER, Chin, WW. Thyroid hormone decreases the stability and the poly(A) tract length of rat thyrotropin beta-subunit messenger RNA. Molecular Endocrinology. 1991;5:469–475.
117. Leedman, PJ, Stein, AR, Chin, WW. Regulated specific protein binding to a conserved region of the 3′-untranslated region of thyrotropin beta-subunit mRNA. Mol Endocrinol. 1995;9:375–387.
118. Staton, JM, Leedman, PJ. Posttranscriptional regulation of thyrotropin beta-subunit messenger ribonucleic acid by thyroid hormone in murine thyrotrope tumor cells: a conserved mechanism across species. Endocrinology. 1998;139:1093–1100.
119. Ross, DS, Ellis, MF, Milbury, P, et al. A comparison of changes in plasma thyrotropin beta- and alpha-subunits, and mouse thyrotropic tumor thyrotropin beta- and alpha-subunit mRNA concentrations after in vivo dexamethasone or T3 administration. Metabolism. 1987;36:799–803.
120. Ahlquist, JA, Franklyn, JA, Wood, DF, et al. Hormonal regulation of thyrotrophin synthesis and secretion. Horm Metab Res Suppl. 1987;17:86–89.
121. Ross, DS. Testosterone increases TSH-beta mRNA, and modulates alpha-subunit mRNA differentially in mouse thyrotropic tumor and castrate rat pituitary. Horm Metab Res. 1990;22:163–169.
122. Friedman, JM, Halaas, JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770.
123. Fekete, C, Kelly, J, Mihaly, E, et al. Neuropeptide Y has a central inhibitory action on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2001;142:2606–2613.
124. Chowdhury, I, Chien, JT, Chatterjee, A, et al. Effects of leptin and neuropeptide-Y on transcript levels of thyrotropin beta and common alpha subunits of rat pituitary cells in vitro. Life Sci. 2004;75:2897–2909.
125. Kim, DS, Yoon, JH, Ahn, SK, et al. A 33 kDa Pit-1-like protein binds to the distal region of the human thyrotrophin alpha-subunit gene. J Mol Endocrinol. 1995;14:313–322.
126. Deutsch, PJ, Jameson, JL, Habener, JF. Cyclic AMP responsiveness of human gonadotropin-alpha gene transcription is directed by a repeated 18-base pair enhancer. J Biol Chem. 1987;262:12169–12174.
127. Hashimoto, K, Zanger, K, Hollenberg, AN, et al. cAMP response element-binding protein-binding protein mediates thyrotropin-releasing hormone signaling on thyrotropin subunit genes. J Biol Chem. 2000;275:33365–33372.
128. Chatterjee, VK, Lee, JK, Rentoumis, A, et al. Negative regulation of the thyroid-stimulating hormone alpha gene by thyroid hormone: receptor interaction adjacent to the TATA box. Proc Natl Acad Sci U S A. 1989;86:9114–9118.
129. Sarapura, VD, Wood, WM, Gordon, DF, et al. Thyrotrope expression and thyroid hormone inhibition map to different regions of the mouse glycoprotein hormone alpha-subunit promoter. Endocrinology. 1990;127:1352–1361.
130. Burnside, J, Darling, DS, Carr, FE, et al. Thyroid hormone regulation of the rat glycoprotein hormone alpha-subunit gene promoter activity. J Biol Chem. 1989;264:6886–6891.
131. Sarapura, VD, Wood, WM, Bright, TM, et al. Reconstitution of triiodothyronine inhibition in non-triiodothyronine responsive thyrotropic tumor cells using transfected thyroid hormone receptor isoforms. Thyroid. 1997;7:453–461.
132. Tagami, T, Madison, LD, Nagaya, T, et al. Nuclear receptor corepressors activate rather than suppress basal transcription of genes that are negatively regulated by thyroid hormone. Mol Cell Biol. 1997;17:2642–2648.
133. Madison, LD, Ahlquist, JA, Rogers, SD, et al. Negative regulation of the glycoprotein hormone alpha gene promoter by thyroid hormone: mutagenesis of a proximal receptor binding site preserves transcriptional repression. Mol Cell Endocrinol. 1993;94:129–136.
134. Keri, RA, Andersen, B, Kennedy, GC, et al. Estradiol inhibits transcription of the human glycoprotein hormone alpha-subunit gene despite the absence of a high affinity binding site for estrogen receptor. Mol Endocrinol. 1991;5:725–733.
135. Yarwood, NJ, Gurr, JA, Sheppard, MC, et al. Estradiol modulates thyroid hormone regulation of the human glycoprotein hormone alpha subunit gene. J Biol Chem. 1993;268:21984–21989.
136. Akerblom, IE, Slater, EP, Beato, M, et al. Negative regulation by glucocorticoids through interference with a cAMP responsive enhancer. Science. 1988;241:350–353.
137. Chatterjee, VK, Madison, LD, Mayo, S, et al. Repression of the human glycoprotein hormone alpha-subunit gene by glucocorticoids: evidence for receptor interactions with limiting transcriptional activators. Mol Endocrinol. 1991;5:100–110.
138. Persani, L. Hypothalamic thyrotropin-releasing hormone and thyrotropin biological activity. Thyroid. 1998;8:941–946.
139. Persani, L, Borgato, S, Romoli, R, et al. Changes in the degree of sialylation of carbohydrate chains modify the biological properties of circulating thyrotropin isoforms in various physiological and pathological states. J Clin Endocrinol Metab. 1998;83:2486–2492.
140. Taylor, T, Weintraub, BD. Altered thyrotropin (TSH) carbohydrate structures in hypothalamic hypothyroidism created by paraventricular nuclear lesions are corrected by in vivo TSH-releasing hormone administration. Endocrinology. 1989;125:2198–2203.
141. Menezes-Ferreira, MM, Petrick, PA, Weintraub, BD. Regulation of thyrotropin bioactivity by thyrotropin-releasing hormone and thyroid hormone. J Endocrinol. 1986;118:2125–2130.
142. Papandreou, MJ, Persani, L, Asteria Ronin, C, et al. Variable carbohydrate structures of circulating thyrotropin as studied by lectin affinity chromatography in different clinical conditions. J Clin Endocrinol Metab. 1993;77:393–398.
143. Trojan, J, Theodoropoulou, M, Usadel, KH, et al. Modulation of human thyrotropin oligosaccharide structures—enhanced proportion of sialylated and terminally galactosylated serum thyrotropin isoforms in subclinical and overt primary hypothyroidism. J Endocrinol. 1998;158:359–365.
144. Persani, L, Ferretti, E, Borgato, S, et al. Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab. 2000;85:3631–3635.
145. Oliveira, JHA, Barbosa, ER, Kasamatsu, T, et al. Evidence for thyroid hormone as a positive regulator of serum thyrotropin bioactivity. J Clin Endocrinol Metab. 2007;92:3108–3113.
146. Sheward, WJ, Harmar, AJ, Fraser, HM, et al. TRH in rat pituitary stalk blood and hypothalamus. Studies with high performance liquid chromatography. Endocrinology. 1983;113:1865–1869.
147. Fraser, HM, McNeilly, AS. Effect of chronic immunoneutralization of thyrotropin-releasing hormone on the hypothalamic-pituitary thyroid axis, prolactin and reproductive function in the ewe. Endocrinology. 1982;111:1964–1971.
148. Aizawa, T, Green, M. Delineation of the hypothalamic area controlling thyrotropin secretion in the rat. Endocrinology. 1981;109:1731–1738.
149. Chan, V, Wang, C, Yeung, TT. Thyrotropin, α and β-subunits of thyrotropin and prolactin response to four hour constant infusions of thyrotropin releasing hormone in normal subjects and patients with pituitary-thyroid disorders. J Clin Endocrinol Metab. 1979;49:127–133.
150. Sheppard, MC, Shennan, KI. Desensitization of rat anterior pituitary gland to thyrotrophin releasing hormone. Endocrinology. 1984;101:101–105.
151. Vale, W, Brazeau, P, Rivier, C, et al. Somatostatin. Recent Prog Horm Res. 1975;31:365–397.
152. Reichlin, S. Somatostatin. N Engl J Med. 1983;309:1495–1501.
153. Urman, S, Critchlow, V. Long-term elevations in plasma thyrotropin, but not growth hormone, concentrations associated with lesion-induced depletion of median eminence somatostatin. Endocrinology. 1983;112:659–664.
154. Arima, A, Schally, AV. Increase in basal and thyrotropin-releasing hormone stimulated secretion of thyrotropin by passive immunization with antiserum to somatostatin. Endocrinology. 1976;98:1069–1075.
155. Samuels, MH, Henry, P, Ridgway, EC. Effects of dopamine and somatostatin on pulsatile pituitary glycoprotein secretion. J Clin Endocrinol Metab. 1992;74:217–222.
156. Reisine, T, Bell, GI. Molecular biology of the somatostatin receptors. Endocrine Rev. 1995;16:427–442.
157. James, RA, Sarapura, VD, Bruns, C, et al. Thyroid hormone-induced expression of specific somatostatin receptor subtypes correlates with involution of the TtT-97 murine thyrotrope tumor. Endocrinology. 1997;138:719–724.
158. Ridgway, EC, Klibanski, A, Martorana, MA, et al. The effect of somatostatin on the release of thyrotropin and its subunits from bovine anterior pituitary cells in vitro. Endocrinology. 1983;112:1937–1942.
159. Page, MD, Millward, ME, Taylor, A, et al. Long-term treatment of acromegaly with a long-acting analogue of somatostatin, octreotide. Q J Med. 1990;74:189–201.
160. Lippe, BM, Van Herle, AJ, La Franchi, SH, et al. Reversible hypothyroidism in growth hormone-deficient children treated with human growth hormone. J Clin Endocrinol Metab. 1975;40:612–618.
161. Ben-Johnson, N, Oliver, C, Weiner, HJ, et al. Dopamine in hypophyseal portal plasma of the rat during the estrous cycle and throughout pregnancy. Endocrinology. 1977;100:452–458.
162. Cooper, DS, Klibanski, A, Ridgway, EC. Dopaminergic modulation of TSH and its subunits: in vivo and in vitro studies. Clin Endocrinol (Oxf). 1983;18:265–272.
163. Pourmand, M, Rodriguez-Arnao, MD, Weightman, DR, et al. Domperidone: a novel agent for the investigation of anterior pituitary function and control in man. Clin Endocrinol (Oxf). 1980;12:211–215.
164. Samuels, MH, Kramer, P. Effects of metoclopramide on fasting-induced TSH suppression. Thyroid. 1996;6:85–89.
165. Lewis, BM, Dieguez, C, Lewis, MD, et al. Dopamine stimulates release of thyrotrophin-releasing hormone from perfused intact rat hypothalamus via hypothalamic D2 receptors. J Endocrinol. 1987;115:419–424.
166. Morley, JE. Neuroendocrine control of thyrotropin secretion. Endocrine Rev. 1981;2:396–436.
167. Foord, SM, Peters, JR, Dieguez, C, et al. Dopamine receptors on intact anterior pituitary cells in culture: functional association with the inhibition of prolactin and thyrotropin. Endocrinology. 1983;112:1567–1571.
168. Foord, SM, Peters, JR, Dieguez, C, et al. TSH regulates thyrotroph responsiveness to dopamine in vitro. Endocrinology. 1985;118:1319–1324.
169. Kok, P, Roelfsema, F, Frolich, M, et al. Spontaneous diurnal thyrotropin secretion is enhanced in proportion to circulating leptin in obese premenopausal women. J Clin Endocrinol Metab. 2005;90:6185–6191.
170. Scanlon, MF, Chan, V, Heath, M, et al. Dopaminergic control of thyrotropin, α-subunit and prolactin in euthyroidism and hypothyroidism: dissociated responses to dopamine receptor blockade with metoclopramide in euthyroid and hypothyroid subjects. J Clin Endocrinol Metab. 1981;53:360–365.
171. Klibanski, A, Milbury, PE, Chin, WW, et al. Direct adrenergic stimulation of the release of thyrotropin and its subunits from the thyrotrope in vitro. Endocrinology. 1983;113:1244–1250.
172. Krulich, L, Mayfield, MA, Steele, MK, et al. Differential effects of pharmacological manipulations of central β1-and β2-adrenergic receptors on the secretion of thyrotropin and growth hormone in male rats. Endocrinology. 1982;35:139–145.
173. Zgliczynski, S, Kaniewski, M. Evidence for β-adrenergic receptor mediated TSH release in men. Acta Endocrinol (Copenh). 1980;95:172–179.
174. Rogol, AD, Reeves, GD, Varma, MM, et al. Thyroid stimulating hormone and prolactin response to thyrotropin-releasing hormone during infusion of epinephrine and propranolol in man. Neuroendocrinology. 1979;29:413–420.
175. Howlett, TA, Rees, LH. Endogenous opioid peptides and hypothalamo-pituitary function. Ann Rev Physiol. 1986;48:527–536.
176. Morley, JE, Baranetsky, NG, Wingert, TD, et al. Endocrine effects of naloxone-induced opiate receptor blockade. J Clin Endocrinol Metab. 1980;50:251–257.
177. Samuels, MH, Kramer, P, Wilson, D, et al. Effects of naloxone infusions on pulsatile thyrotropin secretion. J Clin Endocrinol Metab. 1994;78:1249–1252.
178. Larsen, PR. Thyroid-pituitary interaction. N Engl J Med. 1982;306:23–32.
179. Hinkle, PM, Goh, KBC. Regulation of thyrotropin-releasing hormone receptors and responses by L-triiodothyronine in dispersed rat pituitary cell cultures. Endocrinology. 1982;110:1725–1731.
180. Ponce, G, Charli, JL, Pasten, JA, et al. Tissue-specific regulation of pyroglutamate aminopeptidase II activity by thyroid hormones. Neuroendocrinology. 1988;48:211–214.
181. Kakucska, I, Rand, W, Lechan, RM. Thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is dependent upon feedback regulation by both triiodothyronine and thyroxine. Endocrinology. 1992;130:2845–2850.
182. Berelowitz, M, Maefa, K, Harris, S, et al. The effect of alterations in the pituitary-thyroid axis on hypothalamic content and in vitro release of somatostatin-like immunoreactivity. Endocrinology. 1980;107:24–29.
183. Brabant, G, Brabant, A, Ranft, U, et al. Circadian and pulsatile thyrotropin secretion in euthyroid man under the influence of thyroid hormone and glucocorticoid administration. J Clin Endocrinol Metab. 1987;65:83–88.
184. Bartalena, L, Martino, E, Petrini, L, et al. The nocturnal serum thyrotropin surge is abolished in patients with adrenocorticotropin (ACTH)-dependent or ACTH-independent Cushing’s syndrome. J Clin Endocrinol Metab. 1991;72:1195–1199.
185. Samuels, MH, Luther, M, Henry, P, et al. Effects of hydrocortisone on pulsatile pituitary glycoprotein secretion. J Clin Endocrinol Metab. 1994;78:211–215.
186. Samuels, MH, McDaniel, PA. Thyrotropin levels during hydrocortisone infusions that mimic fasting-induced cortisol elevations—a clinical research center study. J Clin Endocrinol Metab. 1997;82:3700–3704.
187. Pamenter, RW, Hedge, GA. Inhibition of thyrotropin secretion by physiological levels of corticosterone. Endocrinology. 1980;106:162–166.
188. Mitsuma, T, Nogimori, T. Effects of dexamethasone on the hypothalamic-pituitary-thyroid axis in rats. Acta Endocrinol (Copenh). 1982;100:51–56.
189. Mitsuma, T, Hirooka, Y, Nogimori, T. Effects of dexamethasone on TRH and TRH precursor peptide (lys-arg-gln-his-pro-gly-arg-arg) levels in various rat organs. Endocrine Regulations. 1992;26:29–34.
190. Lee, S, Sevarino, K, Roos, BA, et al. Characterization and expression of the gene encoding rat thyrotropin-releasing hormone (TRH). Ann NY Acad Sci. 1989;553:14–28.
191. Ortiga-Carvalho, TM, Oliveira, KJ, Soares, BA, et al. The role of leptin in the regulation of TSH secretion in the fed state: in vivo and in vitro studies. J Endocrinol. 2002;174:121–125.
192. Schurgin, S, Canavan, B, Koutkia, P, et al. Endocrine and metabolic effects of physiologic r-metHuLeptin administration during acute caloric deprivation in normal-weight women. J Clin Endocrinol Metab. 2004;89:5402–5409.
193. Mantzoros, CS, Ozata, M, Negrao, AB, et al. Synchronicity of frequently sampled thyrotropin (TSH) and leptin concentrations in healthy adults and leptin-deficient subjects: evidence for possible partial TSH regulation by leptin in humans. J Clin Endocrinol Metab. 2001;86:3284–3291.
194. Farbota, L, Hofman, C, Oslapas, R, et al. Sex hormone modulation of serum TSH levels. Surgery. 1987;102:1081–1087.
195. Ahlquist, JAO, Franklyn, JA, Ramsden, DB, et al. Regulation of α and thyrotropin-β subunit mRNA levels by androgens in the female rat. J Mol Endocrinol. 1990;5:1–6.
196. Erfurth, EM, Ericsson, UB. The role of estrogen in the TSH and prolactin responses to thyrotropin-releasing hormone in postmenopausal as compared to premenopausal women. Horm Metab Res. 1992;24:528–531.
197. Franklyn, JA, Ramsden, DB, Sheppard, MC. The influence of age and sex on tests of thyroid function. Ann Clin Biochem. 1985;22:502–505.
198. Hermus, RM, Sweep, CG, van der Meer, MJ, et al. Continuous infusion of interleukin-1 induces a nonthyroidal illness syndrome in the rat. Endocrinology. 1992;131:2139–2146.
199. Van der Poll, T, Romijn, JA, Wiersinga, WM, et al. Tumor necrosis factor: a putative mediator of the sick euthyroid syndrome in man. J Clin Endocrinol Metab. 1990;71:1567–1572.
200. Torpy, DJ, Tsigos, C, Lotsikas, AJ, et al. Acute and delayed effects of a single-dose injection of interleukin-6 on thyroid function in healthy humans. Metabolism. 1998;47:1289–1293.
201. Pazos-Moura, CC, Ortiga-Carvalho, TM, Gaspar de Moura, E. The autocrine/paracrine regulation of thyrotropin secretion. Thyroid. 2003;13:167–175.
202. Oliveira, KJ, Ortiga-Carvalho, TM, Cabanelas, A, et al. Disruption of neuromedin B receptor gene results in dysregulation of the pituitary-thyroid axis. J Mol Endocrinol. 2006;36:73–80.
203. Farid, NR, Szkudlinski, MW. Minireview: structural and functional evolution of the thyrotropin receptor. Endocrinology. 2004;145:4048–4057.
204. Grossmann, M, Szkudlinski, MW, Wong, R, et al. Substitution of the seat-belt region of the thyrotropin (TSH)-β subunit with the corresponding regions of the choriogonadotropin or follitropin confers luteotropic, but not follitropic, activity to chimeric TSH. J Biol Chem. 1997;272:15532–15540.
205. Leinung, MC, Bergert, ER, McCormick, DJ, et al. Synthetic analogs of the carboxyl-terminus of β-thyrotropin: the importance of basic amino acids in receptor binding activity. Biochemistry. 1992;31:10094–10098.
206. Medeiros-Neto, G, Herodotou, DT, Rajan, S, et al. A circulating biologically inactive thyrotropin caused by a mutation in the β subunit gene. J Clin Invest. 1996;97:1250–1256.
207. Grossmann, M, Szkudlinski, MW, Tropea, JE, et al. Expression of human thyrotropin in cell lines with different glycosylation patterns combined with mutagenesis of specific glycosylation sites: characterization of a novel role for the oligosaccharides in the in vitro and in vivo bioactivity. J Biol Chem. 1995;270:29378–29385.
208. Szkudlinski, MW, Thotakura, NR, Weintraub, BD. Subunit-specific functions of N-linked oligosaccharides in human thyrotropin: role of terminal residues of α- and β-subunit in metabolic clearance and bioactivity. Proc Natl Acad Sci U S A. 1995;92:9062–9066.
209. Thotakura, NR, Desai, RK, Szkudlinski, MW, et al. The role of the oligosaccharide chains of thyrotropin α- and β-subunits in hormone action. Endocrinology. 1992;131:82–88.
210. Renwick, A, Wiggin, P. An antipodean perception of the mode of action of glycoprotein hormones. FEBS Lett. 1992;297:1–3.
211. Akamizu, T, Ikuyama, S, Saji, M, et al. Cloning, chromosomal assignment, and regulation of the rat thyrotropin receptor: expression of the gene is regulated by thyrotropin agents that increase cAMP levels, and thyroid autoantibodies. Proc Natl Acad Sci U S A. 1990;87:5677–5681.
212. Field, JB, Dekker, A, Titus, G, et al. In vitro and in vivo refractoriness to thyrotropin stimulation of iodine organification and thyroid hormone secretion. J Clin Invest. 1979;64:265–271.
213. Chazenbalk, GD, Nagayama, Y, Kaufman, KD, et al. The functional expression of recombinant human thyrotropin receptors in non-thyroidal eukaryotic cells provides evidence that homologous desensitization to thyrotropin stimulation requires a cell-specific factor. Endocrinology. 1990;127:1240–1244.
214. Wolff, J, Jones, AB. The purification of bovine thyroid plasma membranes and the properties of membrane-bound adenyl cyclase. J Biol Chem. 1971;246:3939–3947.
215. Yamashita, K, Field, JB. Preparation of thyroid plasma membranes containing TSH-responsive adenyl cyclase. Biochem Biophys Res Commun. 1970;40:171–178.
216. Philip, NJ, Grollman, EF. Thyrotropin and norepinephrine stimulate the metabolism of phosphoinositides in FRTL-5 thyroid cells. FEBS Lett. 1986;202:193–196.
217. Grasberger, H, Van Sande, J, Mahameed, AH-D, et al. A familial thyrotropin (TSH) receptor mutation provides in vivo evidence that the inositol phosphates/Ca2+ cascade mediates TSH action on thyroid hormone synthesis. J Clin Endocrinol Metab. 2007;92:2816–2820.
218. Park, ES, Kim, H, Suh, JM, et al. Involvement of JAK/STAT (Janus kinase/Signal transducer and activator of transcription) in the thyrotropin signaling pathway. Mol Endocrinol. 2000;14:662–670.
219. Brewer, C, Yeager, N, Di Cristofano, A. Thyroid-stimulating hormone-initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67:8002–8006.
220. Marians, RC, Ng, L, Blair, HC, et al. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781.
221. Tosta, Z, Chabaud, O, Chebath, J. Identification of thyroglobulin mRNA sequences in the nucleus and cytoplasm of cultured thyroid cells: a fast transcriptional effect of thyrotropin. Biochem Biophys Res Commun. 1983;116:54–61.
222. van den Hove, M-F, Croizet-Berger, K, Tyteca, D, et al. Thyrotropin activates guanosine 5′-diphosphate/guanosine 5′-triphosphate exchange on the rate-limited endocytic catalyst, Rab5a, in human thyrocytes in vivo and in vitro. J Clin Endocrinol Metab. 2007;92:2803–2810.
223. Barnes, ND, Hayles, AB, Ryan, RJ. Sexual maturation in juvenile hypothyroidism. Mayo Clin Proc. 1973;48:849–856.
224. Anasti, JN, Flack, MR, Froelich, J, et al. A potential novel mechanism for precocious puberty in juvenile hypothyroidism. J Clin Endocrinol Metab. 1995;80:276–279.
225. Nagayama, Y, Yamasaki, H, Takeshita, A, et al. Thyrotropin binding specificity for the thyrotropin receptor. J Endocrinol Invest. 1995;18:283–287.
226. Crisanti, P, Omri, B, Hughes, EJ, et al. The expression of thyrotropin receptor in the brain. Endocrinology. 2001;142:812–822.
227. Prummel, MF, Brokken, LJS, Meduri, G, et al. Expression of the thyroid-stimulating hormone receptor in the folliculo-stellate cells of human anterior pituitary. J Clin Endocrinol Metab. 2000;85:4347–4353.
228. Saunier, B, Pierre, M, Jacquemin, C, et al. Evidence of cAMP-independent thyrotropin effects on astroglial cells. Eur J Biochem. 1993;218:1091–1094.
229. Peele, ME, Carr, FE, Baker, JR, et al. TSHβ subunit gene expression in human lymphocytes. Am J Med Sci. 1993;305:1–7.
230. Balzan, S, Nicolini, G, Forini, F, et al. Presence of a functional TSH receptor on human erythrocytes. Biomedicine and Pharmacotherapy. 2007;61:463–467.
231. Endo, T, Ohta, K, Haraguchi, K, et al. Cloning and functional expression of thyrotropin receptor cDNA from fat cells. J Biol Chem. 1995;270:10833–10837.
232. Haraguchi, K, Shimura, H, Kawaguchi, A, et al. Effects of thyrotropin on the proliferation and differentiation of cultured rat preadipocytes. Thyroid. 1999;9:613–619.
233. Antunes, TT, Gagnon, AM, Langille, ML, et al. Thyroid-stimulating hormone induces interleukin-6 release from human adipocytes through activation of the nuclear factor-κB pathway. Endocrinology. 2008;149:3062–3066.
234. Bassett, JHD, Williams, GR. Critical role of the hypothalamic-pituitary-thyroid axis in bone. Bone. 2008;43:418–426.
235. Hase, H, Ando, T, Eldeiry, L, et al. TNFα mediates the skeletal effects of thyroid-stiumlating hormone. Proc Natl Acad Sci U S A. 2006;103:12849–12854.
236. Martini, G, Gennari, L, De Paola, V, et al. The effects of recombinant TSH on bone turnover markers and serum osteoprotegerin and RANKL levels. Thyroid. 2007;18:455–460.
237. Morris, JC. Structure and function of the TSH receptor: its suitability as a target for radiotherapy. Thyroid. 1997;7:253–258.
238. Uhlenhuth, E, Schwartzbach, S. The anterior lobe of the hypophysis as a control mechanism of the function of the thyroid gland. Br J Exp Biol. 1927;5:1–5.
239. Bakke, JL, Lawrence, N, Arnett, F, et al. The fractionation of exogenous and endogenous thyroid-stimulating hormone from human and rat plasma and tissues. J Clin Endocrinol Metab. 1961;21:1280–1289.
240. Odell, WD, Wilber, JF, Utiger, RD. Studies of thyrotropin physiology by means of radioimmunoassay. Recent Prog Horm Res. 1967;23:47–78.
241. Ridgway, EC, Weintraub, BD, Cevallos, JL, et al. Suppression of pituitary TSH secretion in the patient with a hyperfunctioning thyroid nodule. J Clin Invest. 1973;52:2783–2792.
242. Ridgway, EC. Thyrotropin radioimmunoassays: Birth, life and demise. Mayo Clin Proc. 1988;63:1028–1034.
243. Ridgway, EC, Ardisson, LJ, Meskell, MJ, et al. Monoclonal antibody to human thyrotropin. J Clin Endocrinol Metab. 1982;55:44–48.
244. Odell, WD, Griffin, J, Zahradnik, R. Two-monoclonal-antibody sandwich-type assay for thyrotropin, with use of an avidin-biotin separation technique. Clin Chem. 1986;32:1873–1878.
245. Van Heyningen, V, Abbott, SR, Daniel, SG, et al. Development and utility of a monoclonal-antibody-based, highly sensitive immunoradiometric assay of thyrotropin. Clin Chem. 1987;33:1387–1390.
246. Spencer, CA, Schwarzbein, D, Guttler, RB, et al. Thyrotropin (TSH)-releasing hormone stimulation test responses employing third and fourth generation TSH assays. J Clin Endocrinol Metab. 1993;76:494–498.
247. Andersen, S, Pedersen, KM, Bruun, NH, et al. Narrow individual variations in serum T(4) and T(3) in normal subjects: a clue to the understanding of subclinical thyroid disease. J Clin Endocrinol Metab. 2002;87:1068–1072.
248. Hansen, PS, Brix, TH, Sørensen, TI, et al. Major genetic influence on the regulation of the pituitary-thyroid axis: a study of healthy Danish twins. J Clin Endocrinol Metab. 2004;89:1181–1187.
249. Panicker, V, Wilson, SG, Spector, TD, et al. Genetic loci linked to pituitary-thyroid axis set points: a genome-wide scan of a large twin cohort. J Clin Endocrinol Metab. 2008;93:3519–3523.
250. Arnaud-Lopez, L, Usala, G, Ceresini, G, et al. Phosphodiesterase 8B gene variants are associated with serum TSH levels and thyroid function. Am J Hum Genet. 2008;82:1270–1280.
251. Guan, H, Shan, Z, Teng, X, et al. Influence of iodine on the reference interval of TSH and the optimal interval of TSH: results of a follow-up study in areas with different iodine intakes. Clin Endocrinol (Oxf). 2008;69:136–141.
252. Kourides, IA, Weintraub, BD, Levko, MA, et al. α and β subunits of human thyrotropin: purification and development of specific radioimmunoassays. Endocrinology. 1974;94:1411–1421.
253. Kourides, IA, Weintraub, BD, Ridgway, EC, et al. Pituitary secretion of free α and β-subunit of human thyrotropin in patients with thyroid disorders. J Clin Endocrinol Metab. 1975;40:872–885.
254. Kourides, IA, Ridgway, EC, Weintraub, BD, et al. Thyrotropin-induced hyperthyroidism: use of α and β-subunit levels to identify patients with pituitary tumors. J Clin Endocrinol Metab. 1977;45:534–543.
255. Ridgway, EC, Klibanski, A, Ladenson, PW, et al. Pure α-secreting pituitary adenomas. N Engl J Med. 1981;304:1254–1259.
256. Klibanski, A, Deutsch, PJ, Jameson, JL, et al. Luteinizing hormone-secreting pituitary tumor: biosynthetic characterization and clinical studies. J Clin Endocrinol Metab. 1987;64:536–542.
257. Blackman, MR, Weintraub, BD, Rosen, SW, et al. Human placental and pituitary glycoprotein hormones and their subunits as tumor markers: a quantitative assessment. J Natl Cancer Inst. 1980;65:81–93.
258. Kahn, CR, Rosen, SW, Weintraub, BD, et al. Ectopic production of chorionic gonadotropin and its subunits by islet cell tumors: a specific marker for malignancy. N Engl J Med. 1977;197:565–569.
259. Rosen, SW, Weintraub, BD, Aaronson, SA. Nonrandom ectopic protein production by malignant cells: direct evidence in vitro. J Clin Endocrinol Metab. 1980;50:834–841.
260. Blackman, MR, Weintraub, BD, Rosen, SW, et al. Comparison of the effects of lung cancer, benign lung disease, and normal aging on pituitary-gonadal function in men. J Clin Endocrinol Metab. 1988;66:88–95.
261. Faglia, G. The clinical impact of the thyrotropin-releasing hormone test. Thyroid. 1998;8:903–908.
262. Sarapura, VD, Samuels, MH, Ridgway, EC. Thyroid- stimulating hormone. In: Melmed S, ed. The Pituitary. ed 2. Malden, MA: Blackwell Science; 2002:187–229.
263. Watanabe, S, Hayashizaki, Y, Endo, Y, et al. Production of human thyroid-stimulating hormone in Chinese hamster ovary cells. Biochem Biophys Res Commun. 1989;149:1149–1155.
264. Heuer, H, Visser, TJ. Minireview: Pathophysiological importance of thyroid hormone transporters. Endocrinology. 2009;150:1078–1083.
265. Beck-Peccoz, P, Amr, S, Menezes-Ferreira, NM, et al. Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism: effect of treatment with thyrotropin-releasing hormone. N Engl J Med. 1985;312:1085–1090.
266. Hayashizaki, Y, Hiraoka, Y, Tatsumi, K, et al. DNA analyses of five families with familial inherited thyroid stimulating hormone (TSH) deficiency. J Clin Endocrinol Metab. 1990;71:792–796.
267. Borck, G, Topaloglu, AK, Korsch, E, et al. Four new cases of congenital secondary hypothyroidism due to a splice site mutation in the thyrotropin-β gene: phenotypic variability and founder effect. J Clin Endocrinol Metab. 2004;89:4136–4141.
268. Sertedaki, A, Papadimitriou, A, Voutetakis, A, et al. Low TSH Congenital Hypothyroidism: Identification of a novel mutation of the TSH β-subunit gene in one sporadic case (C85R) and of mutation Q49stop in two siblings with congenital hypothyroidism. Pediatr Res. 2002;52:935–940.
269. Deladoey, J, Vuissoz, J-M, Domene, HM, et al. Congenital secondary hypothyroidism due to a mutation C105Vfs114X thyrotropin-β mutation: genetic study of five unrelated families from Switzerland and Argentina. Thyroid. 2003;13:553–559.
270. McDermott, MT, Haugen, BR, Black, JN, et al. Congenital isolated central hypothyroidism caused by a “hot spot” mutation in the thyrotropin-β gene. Thyroid. 2002;12:1141–1146.
271. Vuissoz, J-M, Deladoey, J, Buyukgebiz, A, et al. New autosomal recessive mutation of the TSH-β subunit gene causing central isolated hypothyroidism. J Clin Endocrinol Metab. 2001;86:4468–4471.
272. Rogol, AD, Kahn, CR. Congenital hypothyroidism in a young man with growth hormone, thyrotropin, and prolactin deficiencies. J Clin Endocrinol Metab. 1976;39:356–363.
273. Wit, JM, Drayer, NM, Jansen, M, et al. Total deficiency of GH and prolactin and partial deficiency of thyroid stimulating hormone in two Dutch families: a new variant of hereditary pituitary deficiency. Horm Res. 1989;32:170–177.
274. Behringer, RR, Mathews, LS, Palmiter, RD. Dwarf mice produced by genetic ablation of growth hormone expressing cells. Genes & Dev. 1988;2:453–461.
275. Sornson, MW, Wu, W, Dasen, JS, et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384:327–333.
276. Wu, W, Cogan, JD, Pfaffle, RW, et al. Mutations in PROP-1 cause familial combined pituitary hormone deficiency. Nature Genet. 1998;18:147–149.
277. Fluck, C, Deladoey, J, Rutishauser, K, et al. Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP-1 gene mutation resulting in the substitution of Arg→Cys at codon 120 (R120C). J Clin Endocrinol Metab. 1998;83:3727–3734.
278. Deladoey, J, Fluck, C, Buyukgebiz, A, et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1999;84:1645–1650.
279. Lamesch, C, Neumann, S, Pfäffle, R, et al. Adrenocorticotrope deficiency with clinical evidence for late onset in combined pituitary hormone deficiency caused by a homozygous 301–302delAG mutation of the PROP1 gene. Pituitary. 2002;5:163–168.
280. Beck-Peccoz, P, Persani, L. Thyrotroinomas. Endocrinol Metab Clin North Am. 2008;37:123–134.
281. Refetoff, S, Weiss, RE, Usala, SJ. The syndromes of resistance to thyroid hormone. Endocrine Rev. 1993;14:348–399.
282. Refetoff, S. Resistance to thyroid hormone: one of several defects causing reduced sensitivity to thyroid hormone. Nat Clin Pract Endocrinol Metab. 2008;4:1.
283. Refetoff, S, DeWind, LT, DeGroot, LJ. Familial syndrome combining deaf-mutism, stippled epiphyses, goiter, and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27:279–294.
284. Dacou-Voutetakis, C, Feltquate, DM, Drakopoulou, M, et al. Familial hypothyroidism caused by a nonsense mutation in the thyroid-stimulating hormone β-subunit gene. Am J Hum Genet. 1990;46:988–993.
285. Bonomi, M, Proverbio, MC, Weber, G, et al. Hyperplastic pituitary gland, high serum glycoprotein hormone α-subunit, and variable circulating thyrotropin (TSH) levels as hallmark of central hypothyroidism due to mutations of the TSHβ gene. J Clin Endocrinol Metab. 2001;86:1600–1604.
286. Sertedaki, A, Papadimitriou, A, Voutetakis, A, et al. Low TSH congenital hypothyroidism: Identification of a novel mutation of the TSH β-subunit gene in one sporadic case (C85R) and of mutation Q49stop in two siblings with congenital hypothyroidism. Pediatr Res. 2002;52:935–940.
287. Pohlenz, J, Dumitrescu, A, Aumann, U, et al. Congenital secondary hypothyroidism caused by exon skipping due to a homozygous donor splice site mutation in the TSHβ-subunit gene. J Clin Endocrinol Metab. 2002;87:336–339.
288. Karges, B, LeHeup, B, Schoenle, E, et al. Compound heterozygous and homozygous mutations of the TSHβ gene as a cause of congenital central hypothyroidism in Europe. Horm Res. 2004;62:149–155.
289. Doeker, BM, Pfaffle, RW, Pohlenz, J, et al. Congenital central hypothyroidism due to a homozygous mutation in the thyrotropin β-subunit gene follows an autosomal recessive inheritance. J Clin Endocrinol Metab. 1998;83:1762–1765.
290. Brumm, H, Pfeufer, A, Biebermann, H, et al. Congenital central hypothyroidism due to homozygous thyrotropin beta 313deltaT mutation is caused by a founder effect. J Clin Endocrinol Metab. 2002;87:4811–4816.
291. Partsch, CJ, Riepe, FG, Krone, N, et al. Initially elevated TSH and congenital central hypothyroidism due to a homozygous mutation of the TSH beta subunit gene: case report and review of the literature. Exp Clin Endocrinol Diabetes. 2006;114:227–234.
292. Heinrichs, C, Parma, J, Scherberg, NH, et al. Congenital central hypothyroidism caused by a homozygous mutation in the TSH-beta subunit gene. Thyroid. 2000;10:387–391.
293. Domene, HM, Gruneiro-Papendieck, L, Chiesa, A, et al. The C105fs114X is the prevalent thyrotropin beta-subunit gene mutation in Argentinean patients with congenital central hypothyroidism. Horm Res. 2004;61:41–46.
294. Felner, EI, Dickson, BA, White, PC. Hypothyroidism in siblings due to a homozygous mutation of the TSH-beta subunit gene. J Pediatr Endocrinol Metab. 2004;17:669–672.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 2
Thyroid-Stimulating Hormone
Physiology and Secretion
Ontogeny of TSH-Producing Thyrotrope Cells
Thyrotropes make up only 5% of pituitary cells in the anterior pituitary and are solely responsible for synthesizing TSH. The distinct cell types of the anterior pituitary are defined by the hormone they produce and include thyrotropes (TSH), gonadotropes (LH, FSH), corticotropes (adrenocorticotropic hormone [ACTH]), somatotropes (growth hormone [GH]), and lactotropes (prolactin [PRL]). Cell fate mapping demonstrated that the anterior pituitary develops from Rathke’s pouch, an invagination of oral ectoderm that directly contacts the hypothalamus1 at embryonic day 9.5 (e9.5). Pituitary organogenesis involves progenitor cells and their differentiation by signals that regulate proliferation, lineage commitment, and terminal differentiation.2 The key genes initiating and controlling these developmental pathways include transcription factors, secreted signaling molecules, and receptors. Distinct cell lineages emerge as a result of signaling gradients of transcription factors formed in a spatially and temporally specific manner.3
The glycoprotein hormone α–subunit (αGSU) is the first pituitary hormone gene expressed during development4 at e10.5. Wnt5a and BPM4, which are expressed in the adjacent neuroepithelium, provide the initial signals followed by expression of Hesx1, Ptx1/2, and Lhx3/4.5 TSHβ expression begins in the rostral tip of the pituitary at e12.5 and correlates with thyrotrope embryonic factor (TEF).6 By birth, TSHβ expression in the rostral tip has disappeared and another population of thyrotropes arises by e15.5 in the caudomedial region, following expression of Pit-1 (POU1F1), a pituitary-specific transcription factor.7 Both Pit-1 and TSHβ subunit expression are present in the wild-type but not in the Snell dwarf mouse, which has a mutation of the Pit-1 gene.8 These data suggest that the second population of thyrotropes, associated with Pit-1, is likely the source of mature thyrotropes. Pit-1 expression depends on the transient expression of Prophet of Pit-1 (Prop1) along with Atbf1.9
A zinc finger transcription factor, Gata2, plays a critical role in thyrotrope differentiation.3 Gata2 is transcribed in the developing anterior pituitary as early as e10.5 and persists in an expression pattern coincident with the glycoprotein hormone α-subunit. Gata2 binds and transactivates the αGSU promoter10 and acts synergistically with Pit-1 to activate the TSHβ gene.11 A ventral-dorsal gradient of Gata2 occurs early in development: the intermediate cells that express both Gata2 and Pit-1 develop into thyrotropes. The in vivo function of Gata2 in pituitary development has been examined by targeted inactivation of Gata2 in a transgenic mouse model using Cre recombinase directed by the αGSU promoter/enhancer.12 When Gata2 is ablated, mice have a decreased thyrotrope cell population at birth and lower levels of circulating TSH and FSH when adults. These studies showed that Gata2 is important for optimal thyrotrope and gonadotrope function but not for cell fate.
A recent study has uncovered the existence of an adult population of multipotent stem cells in the adult pituitary13,14 that are distinct from the embryonic precursor cells. These nestin- and Sox2-containing stem cells reside in a localized niche within the periluminal region of the gland, have the capacity to expand into all of the terminally differentiated pituitary cell types after birth, and may contribute to pituitary tumors.13
Tsh Subunit Genes
TSHβ Subunit Gene Structure
The human TSHβ subunit gene has been isolated and characterized.15 This single-copy gene is 4527 base pairs (bp) in size, and is located on the short arm of chromosome 1.16 The gene structure consists of three exons and two introns (Fig. 2-1, top panel). The first exon has 37 bp and contains the 5′-untranslated region of the gene. It is separated from the second exon by a large first intron of 3.9 kb. The coding region of the gene is contained within the second (163 bp) and third (326 bp) exons, which are separated by a 0.45 kb intron, while the 3′-untranslated region is contained within the third exon.
FIGURE 2-1 Structural organization of the human TSH subunit genes and mRNAs. The two panels show the TSHβ (top) and α subunit (bottom) genes. Shown are the relative locations and sizes of the exons and introns. Untranslated regions are shown as open boxes, and protein coding regions are shown in black boxes. The TATA box important for positioning the RNA transcriptional start is located in the promoter close to exon 1. Following transcription, introns are spliced out, exons precisely joined, and a polyA tail added to the 3′ end of the mature mRNA.
DNA sequences close to the transcriptional start site in the promoter of the TSHβ gene contain elements responsible for initiating transcription and regulating expression. A consensus TATA box is located 28 bp upstream of the transcriptional start site and is important for positioning RNA synthesis. Progressive 5′ deletions of the mouse TSHβ promoter linked to a luciferase reporter following expression in thyrotrope cells defined the cis-acting sequences required for expression to the first 270 bp of the promoter.17,18 Although these sequences defined the minimal promoter, other studies have shown that enhancer sequences located more than 6 kb upstream are also required for the promoter to express in pituitary thyrotropes in transgenic mice.19
Promoter deletion studies have demonstrated that the mouse TSHβ promoter from −271 to −80 is sufficient to confer thyrotrope-specific activity,17 and thyrotrope transcription factors can bind to the proximal promoter.20 Within this broad area, four regions of protein interaction have been identified using nuclear extracts from thyrotrope cells.18 Two factors, Pit-1, a homeodomain factor, and Gata2, a zinc finger transcription factor, can bind to TSHβ promoter sequences from −135 to −8821 (Fig. 2-2). Both factors can bind independently to the promoter, form a heteromeric complex with DNA, physically interact with each other, and functionally synergize to activate TSHβ promoter activity. Recently, an additional transcription factor, TRAP220 (Med1, PBP), was shown to be recruited to the TSHβ proximal promoter, where it was shown to play a role in transcriptional activation.22
FIGURE 2-2 Functionally important regions of the TSHβ gene promoter. Top, Schematic showing regions (shaded boxes) of the TSHβ promoter that interact with transcription factors present in thyrotrope cells. Below, The DNA sequence of a key region that is critical for high level of TSHβ expression in thyrotropes. Within this region, Pit-1 sites are boxed, and sites interacting with Gata2 are underlined.
TRAP220 was originally defined as part of a transcriptional mediator complex that interacts with hormone-occupied thyroid/steroid hormone receptors.23 Mice with a single copy of this gene were hypothyroid with reduced levels of pituitary TSHβ transcripts.24 TRAP220 is recruited to the TSHβ gene by virtue of its physical interaction with both Pit-1 and Gata2, since the protein itself does not bind DNA. Co-transfections in nonpituitary cells showed that Pit-1, Gata2, or TRAP220 alone could not stimulate the TSHβ promoter. However, maximal activity resulted when all three factors were expressed. Interaction studies showed that all three factors interact with each other in vivo and in vitro. The regions of interaction were important for maximal function. Chromatin immunoprecipitation demonstrated in vivo occupancy on the proximal TSHβ promoter.22 Thus, the TSHβ gene is activated by a unique combination of transcription factors present in pituitary thyrotropes, including those that act via binding to the proximal promoter, as well as others that are recruited to the promoter via protein-protein interactions.
α Subunit Gene Structure
The human glycoprotein hormone α subunit gene is located on chromosome 6 at position 6q12-q21.25 It is present as a single copy gene that is 9635 kb in size and contains four exons and three introns and contains a consensus TATA box located 26 bp upstream of the transcriptional start site.26 The first exon (94 bp) contains 5′-untranslated sequences and is separated from the second exon by a 6.4 kilobase (kb) intron. The second exon contains 7 bp of 5′-untranslated sequence and 88 bp of the coding region. The coding sequence continues in the third (185 bp) and fourth (75 bp) exons, and the 3′-untranslated region (220 bp) is contained within the fourth exon (Fig. 2-1, bottom panel).
The α subunit gene is expressed in thyrotropes, gonadotropes, and placental cells but is differentially regulated. Cell-specific expression in each cell type is dependent on different regions of the promoter. Whereas the region downstream of −200 is sufficient for placental expression,27 gonadotropes require sequences between −225 and −200,28 and regions farther upstream appear to be critical for thyrotrope expression.29 Transgenic mouse studies have shown 480 bp of the mouse α subunit 5′-flanking DNA could target transgenic expression to both gonadotropes and thyrotropes.30 A region from −225 to −200 that binds steroidogenic factor 1 appears to be critical for gonadotrope expression,31 but not for thyrotrope expression.32 Another important sequence involves the pituitary glycoprotein hormone basal element, extending from −342 to −329, that is critical for both thyrotrope and gonadotrope expression.33 The element interacts with P-LIM, a pituitary-specific LIM-homeodomain transcription factor that is important for other pituitary cells.34 Several sequences within the region from −480 to −300 appear to be important for mouse α subunit expression in thyrotropes but not gonadotropes.29 Among these is the sequence from −434 to −421, which interacts with the developmental homeodomain transcription factor Msx1.35 Other sequences within the 480 bp promoter have been found to interact with the pituitary-specific homeodomain factor Ptx-1, and a synergism between Ptx-1 and P-LIM, mediated by the co-activator C-LIM, has been described.36 Recent studies with the mouse promoter in transgenic mice showed that an upstream DNA element located between −4.6 and −3.7 kb further enhanced expression in both thyrotropes and gonadotropes, and contained consensus binding sites for Gata, SF1, Sp1, ETS, and bHLH factors, which suggests cooperativity between factors binding both to proximal cis-acting elements and to the distal enhancer.37
Biosynthesis of TSH
Translation of TSH Subunits
The next steps in TSH biosynthesis are summarized in Fig. 2-3.38 The mRNAs for the TSHβ and α subunits are independently translated by ribosomes in the cytoplasm. The first peptide sequences consist of “signal” peptides of 20 amino acids for TSHβ and 24 amino acids for α.39 These signal peptides are hydrophobic, allowing insertion through the lipid bilayer of the membrane of the rough endoplasmic reticulum. Translation into TSHβ and α pre-subunits continues into the lumen of the rough endoplasmic reticulum, and cleavage of the signal peptide occurs before translation is completed. This results in the formation of a 118 amino acid TSHβ subunit40 and a 92 amino acid α subunit. Cleavage of TSHβ to a protein of 112 amino acids appears to be an artifact of purification. Synthesis of recombinant TSHβ subunit has resulted in two products of 112 and 118 amino acids, both of which are similarly active in vitro.41
FIGURE 2-3 Top panel, Oligosaccharide chains of thyroid-stimulating hormone (TSH). Shown are typical oligosaccharide chains present on the TSH heterodimer and the free α subunit. Glycosylated asparagine (Asp) and threonine (Thr) residues are indicated. Symbols represent the oligosaccharide chain residues as indicated in the key. Bottom panel, Biosynthesis of thyroid-stimulating hormone (TSH). Schematic shown includes the processes of translation and glycosylation within the rough endoplasmic reticulum (RER) and Golgi apparatus, divided into proximal and distal. Cleavage of the aminoterminal (H2N) signal peptide and early addition of high mannose chains (black boxes), as well as the combination of α and β subunits, occur in the RER. In the proximal Golgi, oligosaccharide chains are modified (shaded boxes), and the final steps of sulfation and sialation occur in the distal Golgi apparatus. (From Weintraub BD, Gesundheit N: Thyroid-stimulating hormone synthesis and glycosylation: clinical implications. Thyroid Today 10:1–11, 1987.)
Glycosylation of TSH
Glycosylation of TSH has a significant impact on its biological activity.42 The TSHβ subunit has a single glycosylation site, the asparagine residue at position 23, whereas the α subunit is glycosylated in two sites, the asparagine residues at positions 52 and 7843 (see Fig. 2-3). Excess free α subunit is glycosylated at an additional site, the threonine residue at position 39.44 This residue is located in a region believed to be important for combination with the TSHβ subunit. It is not known whether glycosylation at this residue is a regulated step that inhibits combination with the TSHβ subunit, or whether it occurs in excess free α subunits because this site is exposed.
Extensive studies on the processes of TSH subunit glycosylation have been carried out. Glycosylation of the TSHβ and α subunits begins before translation is completed (co-translational glycosylation), and addition of the second oligosaccharide in the α subunit occurs after translation is completed (posttranslational glycosylation). The first step in this process involves the assembly of a 14-residue oligosaccharide, (glucose)3-(mannose)9-(N-acetylglucosamine)2 on a dolichol-phosphate carrier. This oligosaccharide then is transferred to asparagine residues by the enzyme olygosaccharyl transferase, which recognizes the tripeptide sequence (asparagine)-(X)-(serine or threonine).45 This mannose-rich oligosaccharide is progressively cleaved in the rough endoplasmic reticulum and Golgi apparatus. An intermediate with only six residues is produced and then other residues are added, resulting in complex oligosaccharides.46 The residues added include N-acetylglucosamine, fucose, galactose, and N-acetylgalactosamine. Oligosaccharides before the six-residue intermediate are termed high mannose and are sensitive to endoglycosidase H, which releases the oligosaccharide from the protein, whereas the intermediate and complex oligosaccharides are endoglycosidase H resistant. Complex oligosaccharides usually consist of two branches (biantennary), but sometimes three or four branches are seen, as are hybrid oligosaccharides consisting of one complex and another high-mannose branch. Sulfation and sialation occur late in the pathway, within the distal Golgi apparatus. Sulfate is bound to N-acetylgalactosamine residues, and sialic acid, or its precursor N-acetylneuraminic acid, is bound to galactoside residues.47 Thus, activation of the enzymes sulfotransferase and N-acetylgalactosamine transferase may involve important regulatory steps that affect the ratio of sulfate to sialic acid. As demonstrated with LH, it appears that sulfation increases and sialylation decreases the bioactivity of TSH,47 because the exclusively sialylated recombinant glycoprotein produced in Chinese hamster ovary cells has been found to have attenuated activity in vitro.48
Processing of complex oligosaccharides appears to occur at a slower rate for secreted glycoproteins, such as TSH, when compared with that for nonsecreted glycoproteins. For example, after an 11 minute pulse labeling with [35S]methionine and a 30 minute chase, only a few α subunits were endoglycosidase H resistant, and only 76% reached this stage after an 18 hour chase.49 Secretion was observed after a 60 minute chase, and the secreted products—TSH, free α subunit but no free β subunit—had mostly complex oligosaccharides associated with them.43 It may be important to note that many of the studies described were carried out in thyrotropic tumor tissue obtained from hypothyroid mice, and glycosylation may differ in the euthyroid as compared with the hypothyroid state. In addition, differences between species have been noted, such as the human TSH may contain more sialic acid than the bovine TSH.40
Folding, Combination, and Storage of TSH
Elucidation of the crystal structure of human CG (hCG)50 allowed the construction of a model of human TSH, as supported by other evidence.51,52 This model has greatly facilitated the interpretation of structure-function studies of the protein backbone. However, crystallization was achieved only with partly deglycosylated hCG, so it is likely that the conformation of the glycosylated protein may differ to some extent, although nuclear magnetic resonance studies suggest that the α subunit carbohydrate moieties project outward and may be freely mobile.53 Nevertheless, this model predicts that the tertiary structure of each TSH subunit consists of two hairpin loops on one side of a central knot formed by three disulfide bonds and a long loop on the opposite side. In this tertiary structure, the glycoprotein hormones share features in common with transforming growth factor β, nerve growth factor, platelet-derived growth factor, vascular endothelial growth factor, inhibin, and activin, all of which are now grouped in the family of “cystine knot” growth factors.54
Folding of nascent peptides begins before translation is completed. It has been shown that proper folding is dependent on glycosylation, because the drug tunicamycin, which prevents the initial oligosaccharide transfer to the asparagine residue, results in a peptide that does not fold properly and is degraded intracellularly.55 Site-directed mutagenesis of a single glycosylation site also disrupted processing and decreased TSH secretion in transfected Chinese hamster ovary cells.56 Folding is a critical step that allows correct internal disulfide bonding, which stabilizes the tertiary structure of the protein, allowing subunit combination.
Combination of TSH β and α subunits begins soon after translation is completed in the rough endoplasmic reticulum and continues in the Golgi apparatus.43 Subunit combination then accelerates and modifies oligosaccharide processing of the α subunit.57 In fact, studies have suggested that the conformation of the α subunit differs after combination with each type of β subunit,58 and this may affect subsequent processing. The rate of combination of TSHβ and α subunits has been examined in mouse thyrotropic tumors. After a 20 minute pulse labeling with [35S]methionine, 19% of TSHβ subunits were combined with α subunits, and this percentage increased to 61% after a additional 60 minute chase incubation.43 Recent studies have shown that the combination of the TSHβ and α subunits, as is also the case with other glycoprotein hormones, occurs after “latching” of the disulfide “seatbelt” of the β subunit, with subsequent “threading” of loop 2 and the attached oligosaccharide of the α subunit beneath that “seatbelt.”59
The sequence of the TSHβ subunit from amino acid 27 to 31 (CAGYC) is highly conserved among species and is thought to be important for combination with the α subunit. In a case of congenital hypothyroidism, a point mutation in the CAGYC region (see “Disorders of TSH Production”) results in the synthesis of altered TSHβ subunits that are unable to associate with α subunits, with consequent lack of intact TSH production.60 A lack of free circulating TSHβ subunit was also observed, suggesting that combination with α subunit is necessary for TSHβ subunit secretion. This phenomenon was also demonstrated in studies where synthesis of wild-type recombinant TSHα subunit was carried out in the presence or absence of recombinant β subunit.61 Using site-directed mutagenesis, another study showed that a mutation at residue 25 in the glycosylation recognition site, which substitutes a serine for a threonine, does not alter glycosylation but decreases TSH production by 70%, possibly because of disruption of the nearby CAGYC region.62
After TSH and free α subunit are processed in the distal Golgi apparatus, they are transported into secretory granules or vesicles.63 The secretory granules constitute a regulated secretory pathway, mainly influenced by TRH and other hypothalamic factors. These granules contain mostly TSH, whereas free α subunit is contained in the secretory vesicles that constitute a nonregulated secretory pathway.
TSH Secretion
In healthy humans, the production rate of TSH is between 100 and 400 mU/day.64,65 The distribution space of TSH is slightly greater than the plasma volume. In euthyroid subjects, the half-life of TSH in plasma is approximately 50 minutes, with a plasma clearance rate of approximately 50 mL/min. In hypothyroid subjects, TSH secretion rates increase by 10 to 15 times normal rates, and the clearance rate decreases slightly. In hyperthyroid subjects, TSH secretion is suppressed, and metabolic clearance is accelerated.
Ontogeny of TSH Levels
At 8 to 10 weeks of gestation in the human, TRH is measurable in the hypothalamus, with progressive increases in TRH levels until term. By 12 weeks of gestation, immunoreactive TSH cells are present in the human pituitary gland,66 and TSH is detectable in the pituitary and the serum.67,68 Serum and pituitary TSH levels remain low until week 18, when TSH levels increase rapidly, followed by increases in serum T4 and T3 concentrations. Fetal serum TSH and T4 concentrations continue to increase between 20 and 40 weeks of gestation. Pituitary TSH begins to respond to exogenous TRH early in the third trimester, and negative feedback control of TSH secretion develops during the last half of gestation and the first 1 to 2 months of life.69
An abrupt rise in serum TSH levels occurs within 30 minutes of birth in term infants. This is followed by an increase in serum T3 concentrations within 4 hours and a lesser increase in T4 levels within the first 24 to 36 hours. The initial increase in serum TSH levels appears to be stimulated by cooling in the extrauterine environment. Serum TSH levels fall to the adult range by 3 to 5 days after birth, and serum thyroid hormone levels stabilize by 1 to 2 months. Serum TSH levels in healthy premature infants (less than 37 weeks gestational age) are highly variable but tend to be lower at birth compared with those of term infants. TSH levels decrease slightly during the first week of life, followed by a gradual increase to normal term levels. Serum TSH levels are even lower in ill premature infants but rise toward normal levels during recovery.70,71
Patterns of TSH Secretion
TSH is secreted from the pituitary gland in a dual fashion, with secretory bursts (pulses) superimposed upon basal (apulsatile) secretion (Fig. 2-4, upper panel). Basal TSH secretion accounts for 30% to 40% of the total amount released into the circulation, and secretory bursts account for the remaining 60% to 70%. TSH pulses occur approximately every 2 to 3 hours, although considerable variability is noted among individuals.72 TSH pulses appear to directly stimulate T3 secretion from the thyroid gland, as cross-correlation analysis has shown that a free T3 peak occurs between 0.5 and 2.5 hours after a TSH peak. However, changes in free T3 levels from nadir to peak are only 11% of mean free T3 levels, probably because T3 has a long serum half-life, and most T3 does not arise from the thyroid gland.73
FIGURE 2-4 Serum thyroid-stimulating hormone (TSH) levels measured every 15 minutes in a healthy subject (upper panel), in two subjects with primary hypothyroidism (middle panel), and in a subject with hypothyroidism due to a craniopharyngioma (lower panel). Significant TSH pulses were located by cluster analysis, a computerized pulse detection program, and are indicated by asterisks. (Data from Sarapura VD, Samuels MH, Ridgway EC: Thyroid-stimulating hormone. In: Melmed S [ed]: The Pituitary, 2nd ed. Blackwell Science, Malden, MA, pp 187–229, 2002.)
In healthy euthyroid subjects, TSH is secreted in a circadian pattern, with nocturnal levels increasing to up to twice daytime levels72 (Fig. 2-4, upper panel). Peak TSH levels occur at between 2300 and 0500 hours in subjects with normal sleep-wake cycles, and nadir levels occur at about 1100 hours. The TSH circadian rhythm is not present in infants younger than 4 weeks old but emerges between 1 and 2 months of life and is well established in healthy children.74 The circadian variation in TSH levels is due to increased mass of TSH secreted per burst at night, as well as slightly increased frequency of bursts and more rapid increase to maximal TSH secretion within a burst.72 The nocturnal increase in TSH levels can precede the onset of sleep, and sleep deprivation enhances TSH secretion. Therefore, in contrast to other pituitary hormones with a circadian variation, the nocturnal rise in TSH levels is not sleep entrained. Instead, a sleep-related inhibition of TSH release is of insufficient magnitude to counteract the nocturnal TSH surge.
Subjects with mild primary hypothyroidism retain the nocturnal rise in TSH levels, and patients with severe primary hypothyroidism have markedly increased TSH pulse amplitude throughout the day with loss of circadian variation in TSH levels (Fig. 2-4, middle panel).72 l-Thyroxine therapy reestablishes the normal TSH circadian variation. In contrast, patients with hypothalamic-pituitary causes of hypothyroidism secrete less TSH over a 24 hour period, with loss of the nocturnal TSH surge in pulse amplitude75 (Fig. 2-4, lower panel). A similar pattern of reduced 24 hour TSH secretion occurs in critical illness.76
The origin of pulsatile and circadian TSH secretion is not known. Thyroid hormones alter TSH pulse amplitude but have little effect on pulse frequency, and therefore are unlikely to participate in TSH pulse generation. The TSH pulse generator may reside in the hypothalamus, with TRH neurons acting in concert to stimulate a burst of TSH secretion from the pituitary gland. However, constant TRH infusions do not change TSH pulse frequency in humans, which casts doubt on this theory.77 Dopamine suppresses TSH pulse amplitude but does not alter TSH pulse frequency, and therefore dopamine does not appear to control pulsatile TSH secretion. A diurnal variation in the activity of anterior pituitary 5′-monodeiodinase in the rat may control circadian TSH secretion.78 However, this has not been confirmed in the human.
Physiologic serum cortisol levels may control circadian TSH secretion, although cortisol does not appear to affect TSH pulse frequency. When subjects with adrenal insufficiency were studied under conditions of glucocorticoid withdrawal, daytime TSH levels were increased, and the usual TSH circadian rhythm was abolished. When these subjects were given physiologic doses of hydrocortisone in a pattern that mimicked normal pulsatile and circadian cortisol secretion, daytime TSH levels were decreased, and the normal TSH circadian rhythm was reestablished. Hydrocortisone infusions at the same dose given as pulses of constant amplitude throughout the 24 hour period also decreased 24 hour TSH levels, but no circadian variation was noted.79 Similarly, when healthy subjects were given metyrapone (an inhibitor of endogenous cortisol synthesis), TSH levels increased during the day, leading to abolition of the usual TSH circadian variation.80 These data suggest that the normal early morning increase in endogenous serum cortisol levels decreases serum TSH levels and leads to the observed normal circadian variation in TSH.
Regulation of TSH Biosynthesis
Hypothalamic Regulation of TSHβ Subunit Biosynthesis
Thyrotropin-releasing hormone (TRH) is a tripeptide that is secreted from the hypothalamus and transported to the pituitary via the hypothalamic-hypophyseal portal system; it is a major activator of TSH production with a significant three- to fivefold increase in the transcription of both TSHβ and α subunit mRNAs.81 TRH from maternal or fetal sources is not required for normal thyrotrope development during ontogeny, and TRH-deficient mice are not hypothyroid at birth. However, TRH is required for the postnatal maintenance of TSH activation.82
TRH binding to its receptor initiates a cascade of intracellular events. In GH3 cells, the TRH-receptor complex interacts with a guanine nucleotide binding regulatory protein (G) that then binds and activates GTP(G′). G′ binds to phospholipase C (C) and activates it (C′). C′ catalyzes the hydrolysis of phosphatidylinositol 4,5 bisphosphate, which results in the formation of two intracellular “second messengers,” inositol triphosphate (InsP3) and 1,2-diacylglycerol (1,2-DG). InsP3 diffuses from the cell-surface membrane to the endoplasmic reticulum, where it causes the release of sequestered Ca2+. This activates the movement of secretory granules to the cell surface and their exocytosis. Simultaneous with these events is a parallel activation of protein kinase C by 1,2-DG, which also leads to phosphorylation of proteins involved in exocytosis. TRH has been shown to stimulate a nuclear protein, Islet-brain-1 (IB1)/JIP-1, in the anterior pituitary gland and in cultured rat GH3 cells83 and has been implicated in the action of TRH in stimulating the TSHβ gene in thyrotropes. Studies in somatomammotrope cells, where TRH stimulates prolactin production, have suggested that phosphatidylinositol, protein kinase C, and calcium-dependent pathways may be involved,84 although TRH stimulation of the TSHβ subunit promoter may be mediated by AP1.85
Two TRH-response regions are located from −128 to −61 and from −28 to +8 of the human TSHβ promoter.86 The upstream region contains binding sites for the pituitary-specific transcription factor, Pit-1, suggesting a role for this or a similar factor in the regulation of the TSHβ subunit gene by TRH. In the rat TSHβ subunit gene, responsiveness to TRH has been localized to regions upstream of −204, where Pit-1 binding sites are also found.87 Furthermore, it has been shown that both protein kinase C and protein kinase A pathways can phosphorylate Pit-1 at two sites in response to phorbol esters and cyclic adenosine monophosphate (cAMP),88 thus altering the binding to Pit-1 transactivation elements on the human TSHβ gene.89
Dopamine acting via DA2 dopamine receptors inhibits TSHα and β subunit gene transcription by decreasing the intracellular levels of cAMP.81 Studies of the TSHβ subunit gene have localized two regions of the promoter necessary for cAMP stimulation, from −128 to −61 bp and from +3 to +8 bp. The upstream region coincides with the TRH-responsive region and contains Pit-1 binding sites. The downstream region resides within the regions responsive to T3 (+3 to +37) and TRH (−28 to +8). The downstream region also overlaps with an AP1 binding site (−1 to +6). The sequence from −1 to +6 appears to cooperate with Pit-1 in mediating responses to cAMP and TRH.85 Thus, multiple interactions between transcription factors and hormonal regulators appear to converge on sequences close to the transcriptional start site.
Peripheral Regulation of TSHβ Subunit Biosynthesis
Thyroid hormone is thought to act predominantly through a classical TR-mediated genomic model. T4 serves as a minimally active prohormone that is converted into a metabolically active T3 via a family of tissue deiodinases, termed D1, D2, and D3. These selenoprotein enzymes are membrane bound and can activate or inactivate substrate in a time- and tissue-specific manner.90 D2 is the major T4-activating deiodinase. It is present on the endoplasmic reticulum close to the nucleus and produces 3,5,3′-triiodothyronine (T3) through the removal of an iodine residue from the outer ring of thyroxine. D2 activity is rapidly lost in the presence of its substrate T4 by a ubiquitin proteasomal mechanism.91 Rat pituitary thyrotropes coexpress D2 RNA and protein, and both are increased in hypothyroidism. Murine thyrotropes in TtT-97 tumors or the TαT1 cell line have extremely high levels of D2, which accounts for the sustained production of T3 by thyrotropes even in the presence of supraphysiologic T4 levels. Serum TSH levels in normal mice are suppressed by administration of T4 or T3, although only T3 was effective in the mouse with targeted disruption of the D2 gene. The observed phenotype of pituitary resistance to T4 demonstrated the critical importance of D2 in controlling negative thyroid hormone regulation of TSH in thyrotropes.
T4 can also act, in some cases, via nongenomic mechanisms that do not involve classical nuclear TR mechanisms. T4 can bind to a cell-surface integrin αVβ3 receptor followed by activation of a mitogen-activated protein kinase cascade, which transduces the signal into a complex series of cellular and nuclear actions. These nongenomic hormone actions are likely to be contributors to basal rate setting of transcription of some genes, and they may control complex cellular events.92
TSHβ and α subunit gene transcription rates are markedly inhibited by treatment with triiodothyronine (T3). Studies using mouse TtT-97 thyrotropic tumors have demonstrated that suppression of TSHβ and α subunit mRNA transcription rates measured by nuclear run-on assays is evident by 30 minutes after treatment and is maximal by 4 hours.93 This effect was seen in the presence of the protein synthesis inhibitor cycloheximide, indicating that it did not require an intermediary protein. Other studies using mouse and rat pituitaries along with mouse thyrotropic tumors have demonstrated that steady-state mRNA levels of TSHβ and α subunits are dramatically decreased by T394 (Fig. 2-5). The mechanism of action of T3 involves interaction with nuclear receptors that act mainly at the transcriptional level. The transcriptional response to T3 is proportional to the nuclear receptor occupancy,95 and the time course of T3 nuclear binding and that of transcriptional inhibition are also in agreement96 (see Fig. 2-5).
FIGURE 2-5 The effect of thyroid hormone on the transcription of the thyroid-stimulating hormone (TSH)β (black circles) and α subunit (white circles) genes. Murine thyrotropic tumor explants were incubated for up to 4 hours with 5 nmol T3 for transcription measurements or with 5 nmole 125I T3, with or without 1000-fold excess of unlabeled T3 for binding measurements. Transcription rates were measured in pools of isolated nuclei. An inverse relationship has been noted between T3 binding and TSHβ and α subunit mRNA synthesis. (Data from Shupnik MA, Ridgway EC: Triiodothyronine rapidly decreases transcription of the thyrotropin subunit genes in thyrotropic tumor explants. Endocrinology 117:1940–1946, 1985.)
The T3 inhibitory effect on the TSHβ gene requires ligand-occupied T3 receptor (TR), specifically the TRβ1 or TRβ2 isoform, because patients with thyroid hormone resistance and inappropriate secretion of TSH have abnormalities only in the TRβ, not the TRα, gene.97 TRβ interacts with specific cis-acting DNA sequences close to the transcriptional start. T3 response elements have been reported to be located between +3 and +37 of the human TSHβ gene.98 Two T3 receptor binding sites, from +3 to +13 and from +28 to +37, may mediate T3 inhibition. T3 responses can be mediated through receptor monomers, homodimers, or heterodimers involving retinoid X receptors (RXR).99 An RXR selective ligand was shown in vitro to inhibit TSHβ expression in TtT-97 thyrotropic tumor cells100 and in cultured TαT1 thyrotropes.101 This finding has been confirmed in vivo and resulted in central hypothyroidism (low T4 and low TSH) in cancer patients treated with the retinoid bexarotene.102 The RXR-selective retinoid (LG 268) decreased circulating TSH and T4 levels in mice with marked lowering of pituitary TSHβ mRNA without decreasing TRH, suggesting a direct effect on thyrotropes.101
Other, more recent studies have disputed the requirement of the negative response element located in exon 1 of the human gene, because its deletion did not eliminate T3 suppression of TSHβ promoter activity in a reconstitution system.103 These studies showed that liganded TRβ can associate with Gata2 in vitro and in vivo via direct interaction between the zinc fingers of Gata2 and the DNA binding domain of TRβ. In addition, T3-occupied TR can physically interact with TRAP220.24 Thus, it is likely that interference with the transactivation function of the Pit-1/Gata2/TRAP220 complex on the proximal TSHβ promoter plays an important role in T3 negative regulation.
Abundant information exists as to the mechanisms involved in gene stimulation by T3. Generally, TRs bind to cis-acting DNA response elements (TRE) in the absence of ligand, interact with a family of nuclear receptor corepressor molecules that recruit histone deacetylases, and locally modify chromatin structure to result in repression of the target gene.104 In the presence of T3, the corepressor complexes rapidly dissociate and are replaced by coactivator complexes that bind to TRs and increase histone methylation and acetylation locally on the chromosomal DNA, which unwinds the chromatin into an open configuration.105 Other activating transcription factors such as TRAP220 are then recruited to the TR via protein-protein interactions, which then activate RNA polymerase II–mediated transcription.
In contrast, the molecular mechanisms involved in negative T3 regulation, such as the TSH subunit genes, have not been well characterized. Liganded TRβ has been reported to recruit histone deacetylase 3 and reduce histone H4 acetylation that modifies histones, resulting in a fully repressed chromosomal state of the TSHβ gene.103 Several recent studies have demonstrated the requirement of an intact DNA binding domain of TRβ in the negative regulation of the TSHβ gene in vitro106 and in vivo.107 In one study, a combination of Pit-1 and Gata2 activated a human TSHβ (−128/+37) reporter construct along with vectors containing TRβ1 constructs in the absence or presence of T3. These investigators found that unliganded TRβ1 did not stimulate promoter activity, whereas a mutation lacking the N-terminus and DNA binding domain of TRβ1 lost the ability of T3-treated cells to negatively regulate TSHβ promoter activity. This demonstrated the importance of various modular domains that constitute the molecular structure of TRs. Moreover, using a gene targeting approach in transgenic mice and replacing the wild-type TRβ gene with a mutant that abolished DNA binding in vitro did not alter ligand and cofactor interactions.107 Homozygous mutant mice demonstrated central thyroid hormone resistance with 20-fold higher serum TSH in the face of two- to threefold higher T3 and T4 levels, which were similar to those of TRβ homozygous null mice.
Although thyrotrope cells contain all TRs—TRα1, TRβ1, and TRβ2, as well as non-T3 binding variant α2108—it is TRβ2 that is expressed predominantly in the pituitary and in T3-responsive TRH neurons109 and is most critical for the regulation of TSH.110 Moreover, TRβ2-deficient mice had a phenotype consistent with pituitary resistance to thyroid hormone, with increased TSH and thyroid hormone levels, even in the presence of TRβ1 and TRα1, showing the lack of compensation between TR isoforms.111 However, TRβ1 and TRα1 still may play a role, in that they are able to form heterodimers with TRβ2. Heterodimers of a TR and a TR accessory protein, such as RXR, may also bind to DNA,112 constituting heterodimeric complexes that may have different affinities for specific DNA sequences and different functional activities. A particular RXR isoform, RXRγ1, is uniquely expressed in thyrotropes and appears to mediate the inhibition by 9-cis-retinoic acid through a region extending from −200 to −149 of the mouse TSHβ subunit promoter, an area upstream and distinct from that mediating negative regulation by thyroid hormone.100 Other proteins that interact with TR include the coactivators, such as the glucocorticoid receptor interacting protein-1 (GRIP-1) and the steroid receptor coactivator-1 (SRC-1),113 and corepressors, such as the silencing mediator for retinoid receptors and thyroid hormone receptors (SMRT) and the nuclear receptor corepressor (NCoR).114 These coactivators and corepressors modulate the effects of many members of the steroid-thyroid hormone receptor superfamily. Their role in the regulation of TSH subunit promoters by thyroid hormone remains to be elucidated in detail.
Studies with genetic knockout mouse models in which both TRH and TRβ genes were removed have recently showed an unexpected dominant role for TRH in vivo in regulating the hypothalamic-pituitary-thyroid axis. It appears that the presence of both TRβ and TRH is necessary for a normal thyrotroph response during hypothyroidism, suggesting that unliganded TRβ stimulates TSH subunit gene expression.115
Posttranscriptional effects of T3 have also been described. T3 decreases the half-life of TSHβ subunit mRNA and decreases the size of the poly(A) tail.116 The shortening of the poly(A) tail is thought to cause mRNA instability. Leedman et al. showed that T3 increased the binding of an RNA-binding protein present in rat pituitary to the 3′-untranslated region of the rat TSHβ mRNA117 and also induced a shortening of the poly(A) tail of the mouse TSHβ mRNA from 160 to 30 nucleotides.118
Steroid hormones, specifically glucocorticoids, inhibit TSH production, but TSH subunit mRNA levels do not change significantly.119 Their major effect may be seen at the secretory level. Estrogens mildly reduce both α and TSHβ subunit mRNA in hypothyroid rats compared with euthyroid controls.120 In this study, estrogen also abolished the early rise in subunit mRNA levels seen following T3 replacement. Testosterone has been shown to increase TSHβ subunit mRNA in castrate rat pituitary and mouse thyrotropic tumor.121
Leptin and neuropeptide Y have opposite effects on TSH biosynthesis. Leptin is the product of the ob gene, which is found mainly in adipose tissue and regulates body weight and energy expenditure.122 Neuropeptide Y (NPY) is a neuropeptide that is synthesized in the arcuate nucleus of the hypothalamus and plays many roles in neuroendocrine function.123 In dispersed rat pituitary cells, leptin stimulated and NPY inhibited TSHβ mRNA levels in a dose-related manner.124 In contrast, both agents increased α subunit steady state mRNA levels.
Hypothalamic Regulation of α Subunit Biosynthesis
TRH stimulates α subunit biosynthesis through a novel mechanism. A CRE-binding protein that binds to the region from −151 to −135 of the human α subunit promoter appears to be important for TRH regulation, as well as a Pit-1–like protein that binds to a more distal region from −223 to −190.125 The CRE of the human glycoprotein hormone α subunit gene promoter consists of an 18 bp repeat and extends from −146 to −111.126 The mechanisms involved in TRH stimulation of the α subunit gene appear to involve two transcription factors, P-Lim and CREB binding protein (CBP). When stimulated with TRH, both of these factors transcriptionally cooperate to activate α subunit promoter activity caused by direct protein-protein interactions.127 Both of these factors synergistically activated the α subunit gene promoter during TRH stimulation and interact in a TRH-dependent manner. P-Lim binds to the α subunit promoter directly, but CBP does not possess a DNA binding domain, so it must be recruited to the promoter via interaction with another factor. The P-Lim/CBP binding is formed in a TRH signaling-specific manner, in contrast to forskolin, which mimics protein kinase A signaling and dissociates both the binding and the transcriptional synergy. α subunit gene expression in thyrotropes is inhibited by dopamine in coordination with expression of the TSHβ subunit gene. Its action is mediated by decreases in intracellular cAMP levels.
Peripheral Regulation of α Subunit Biosynthesis
Thyroid hormone inhibition of α subunit gene transcription is observed in thyotropes in coordination with that of the TSH β subunit. The T3 response element of the human α subunit gene promoter has been reported to be located from −22 to −7.128 Similar to the TSHβ subunit gene, the T3 response elements of human and of mouse129 and rat130 genes are located close to the transcriptional start. T3 inhibition may be mediated by different isoforms of the T3 receptor131 in combination with the corepressors SMRT and NCoR.132 Studies have suggested that mutations of the T3 response element of the human α subunit promoter that eliminate TR binding do not abrogate the inhibitory effect of T3, suggesting that protein-protein interactions may be more important than protein-DNA binding.133
Steroid hormone regulation of α subunit gene transcription is probably of limited importance. Androgen inhibition and androgen receptor (AR) binding have been localized to a region from −120 to −100. Negative regulation by estrogen was described in the gonadotropes of transgenic mice expressing a reporter gene under the control of both human and bovine promoters, but no binding of these regions to the estrogen receptor (ER) was detected, suggesting an indirect effect.134 However, other studies using rat somatomammotropes have found positive regulation by estrogen localized to the proximal 98 bp of 5′-flanking DNA of the human α subunit gene and binding of the ER to the T3 response element from −22 to −7.135 Transcriptional inhibition by glucocorticoids may be mediated by binding of the glucocorticoid receptor to sequences between −122 and −93 of the human α subunit gene.136 However, no direct binding was detected in other studies, suggesting that the GR inhibits transcription by interfering with other transactivating proteins.137
Regulation of TSH Glycosylation
Glycosylation is a regulated process that is primarily modulated by TRH and thyroid hormone.138 Primary hypothyroidism139 and TRH administration140 have been found to increase oligosaccharide addition, which results in an increased bioactivity of TSH.141 The same was noted in patients with resistance to thyroid hormone. TSH glycosylation patterns were found to differ in several pathologic states such as central hypothyroidism, TSH-producing pituitary adenomas, and euthyroid sick syndrome.142 Also observed were changes in the sulfation and sialylation of the oligosaccharide residues, which modulate bioactivity.139,143,144 Recently, thyroid hormone was shown to increase TSH bioactivity, and this was correlated with decreased sialylation.145
Regulation of TSH Secretion
As with biosynthesis, TSH secretion is a result of complex interactions between central and peripheral hormones (Fig. 2-6).
FIGURE 2-6 Hypothalamic and peripheral control of thyroid-stimulating hormone (TSH) secretion. Positive and negative stimuli are indicated at various levels within the hypothalamic-pituitary-thyroid axis. T4, thyroxine; T3, triiodothyronine; TRH, thyrotropin-releasing hormone; SS, somatostatin; DA, dopamine; D2, deiodinase type II.
Hypothalamic Regulation of TSH Secretion
TRH directly affects TSH secretion in vivo and in vitro at concentrations that exist in the pituitary portal blood.146 Immunoneutralization of TRH in animals leads to a decline in thyroid function,147 and TRH knockout mice have a reduced postnatal TSH surge, followed by impaired baseline thyroid function with a poor TSH response to hypothyroidism. Lesions of the paraventricular nucleus (PVN) decrease circulating TRH and TSH levels in normal or hypothyroid animals and cause hypothyroidism148
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
