[level-membership-for-anesthesiology-category]
Chapter 52 Thyroid emergencies
Thyroid emergencies are a rare cause for admission to critical care. However, mortality is high unless specific treatment is provided in an expeditious manner. Abnormal thyroid function tests are commonly encountered during critical illness; numerous factors must be considered before interpreting these findings as indicating thyroid disease.
BASIC PHYSIOLOGY
Thyroid hormones affect the function of virtually every organ system and must be constantly available for these functions to continue. The two biologically active hormones are tetraiodothyronine (thyroxine or T4) and triiodothyronine (T3). These are synthesised by incorporating iodine into tyrosine residues, a process which occurs in thyroglobulin contained within the lumena of the thyroid gland. Stimulation of hormone release by thyroid-stimulating hormone (TSH) results in endocytosis of thyroglobulin from the lumen into the follicular cells, followed by hydrolysis to form T4 and T3, which are released into the circulation.1
Both T4 and T3 contain two iodine atoms on their inner (tyrosine) ring. They differ in that T4 contains two further iodine atoms on its outer (phenol) ring, whereas T3 contains only one. T4 is produced solely by the thyroid gland whereas the majority of T3 is synthesised peripherally by the removal of one iodine atom (deiodination) from the outer ring of T4. If deiodination of an inner-ring iodine atoms occurs, the metabolically inert reverse-T3 (rT3) is formed. This is produced in preference to T3 during starvation and many non-thyroidal illnesses, and the ratio of inactive (rT3) to active T3 synthesis appears to play an important role in the control of metabolism.2 Numerous factors can affect the peripheral deiodination process (Table 52.1). Both T4 and T3 are highly protein-bound in the serum, predominantly to thyroid-binding globulin (TBG), but to a lesser extent to albumin and prealbumin. Changes in concentration of these serum-binding proteins have a large effect on total T4 and T3 serum concentrations. Such protein changes do not, however, affect the concentration of free hormone or their rates of metabolism. The serum-binding proteins act as both a store and a buffer to allow an immediate supply of the metabolically active free T4 (fT4) and free T3 (fT3). In addition, protein binding reduces the glomerular filtration and renal excretion of the hormones.
Table 52.1 States associated with decreased deiodination of thyroxine to triiodothyronine
The regulation of thyroid function is predominantly determined by three main mechanisms, the latter two providing physiological control. Firstly, availability of iodine is crucial for the synthesis of the thyroid hormones. Dietary iodide is absorbed and rapidly distributed in the extracellular fluid, which also contains iodide released from the thyroid gland and from peripheral deiodination processes. This becomes trapped within thyroid follicular cells, from which it is actively transported into the lumen to be oxidised into iodine and subsequently combined with tyrosine.3 Other ions, such as perchlorate and pertechnetate, share this follicular cell active transport mechanism, acting as competitive inhibitors for the process.
Secondly, thyroid hormone release is controlled by a closed feedback loop with the anterior pituitary. Diminished levels of circulating hormones trigger secretion of TSH which acts on the follicular cells of the thyroid gland, causing them to release thyroglobulin-rich colloid from the lumen, which is hydrolysed to form T4 and T3 for systemic release. Increased levels of T4 and T3 cause diminished TSH secretion, resulting in the follicular cells becoming flat and allowing increased capacity for colloid storage. As a result, less thyroglobulin is mobilised and hydrolysed with less T4 and T3 release. The degree to which TSH is secreted in response to changes in circulating thyroid hormones is dependent on the hypothalamic hormone thyrotrophin-releasing hormone (TRH), which is itself modulated by feedback from the thyroid hormones. TRH secretion is inhibited by dopamine, glucocorticoids and somatostatin.
THYROID CRISIS (THYROID STORM)
Thyrotoxic storm is arguably the most serious complication of hyperthyroidism, with reported mortality ranging from 10 to 75% in hospitalised patients.4,5 Crisis most commonly occurs as a result of unrecognised or poorly controlled Graves disease; however, other underlying diseases may be the cause.6,7 Females outnumber males. Laboratory findings are inconsistent due to acute disruption of the normal steady state of the circulating hormones, and there is no definitive value that separates thyrotoxicosis from thyroid storm. The latter is a clinical diagnosis and a scoring system has been proposed to guide the likelihood of the diagnosis (Figure 52.1).8 Precipitating factors are not always present, though many have been identified (Table 52.2).
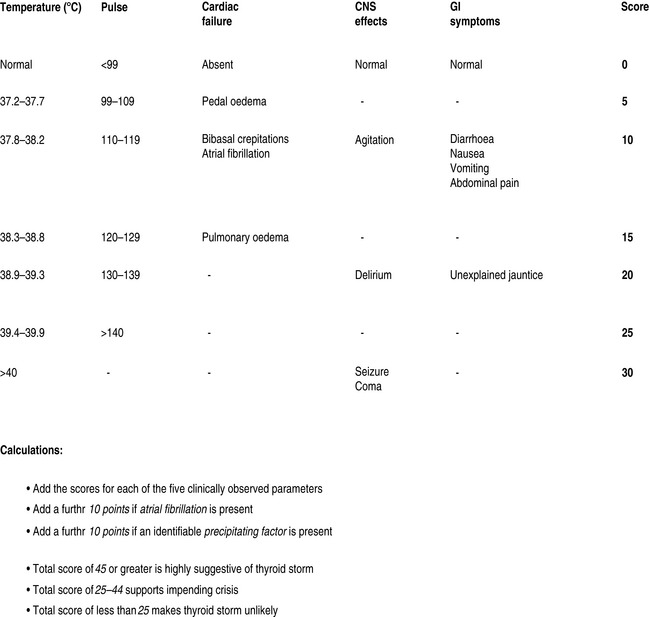
Figure 52.1 Severity assessment in thyroid crisis. CNS, central nervous system; GI, gastrointestinal.
(Adapted from Tewari K, Balderston KD, Carpenter SE et al. Papillary thyroid carcinoma manifesting as thyroid storm of pregnancy: case report. Am J Obstet Gynecol 1998; 179: 818–19.)
Table 52.2 Factors associated with precipitating thyroid storm
CLINICAL PRESENTATION (Table 52.3)
The classic signs of thyroid crisis include fever, tachycardia, tremor, diarrhoea, nausea and vomiting.9 However presentation is extremely variable and may range from apathetic hyperthyroidism (apathy, depression, hyporeflexia and myopathy)10 to multiple-organ dysfunction.11,12 Differential diagnosis includes sepsis and other causes of hyperpyrexia such as adrenergic and anticholinergic syndromes.
Table 52.3 Clinical features of hyperthyroidism/thyroid crisis









FEVER
This is the most characteristic feature. Temperature may rise above 41°C. There have been suggestions that pyrexia is present in all cases of thyroid storm,13 though normothermia has been reported.11 Pyrexia is rare in uncomplicated thyrotoxicosis and should always raise suspicion of thyroid storm. It is not clear whether this febrile response is due to alteration of central thermoregulation or elevation of basal metabolic thermogenesis beyond the body’s ability to lose heat.
CARDIOVASCULAR FEATURES
Fluid requirements may be substantial in some patients, whereas diuresis may be required in those with severe cardiac congestion. Cardiac decompensation can occur in young patients with no known antecedent cardiac disease. Systolic hypertension with widened pulse pressure is common initially; however hypotension supervenes later. Cardiogenic shock with vascular collapse is a preterminal sign.14 Arrhythmias are common and include atrial and polymorphic ventricular tachycardias. The latter may result from prolonged QTc interval, the presence of which may complicate and contraindicate the use of amiodarone therapy.
NEUROMUSCULAR FEATURES
Tremor is a common early sign, but as ‘storm’ progresses, central nervous dysfunction evolves with progression to encephalopathy or even coma.15 Thyroid storm has been reported in association with status epilepticus and cerebrovascular accident.16 Weakness may be a feature, particularly with apathetic thyrotoxicosis.10 Thyrotoxic myopathy and rhabdomyolysis may be present,11,17 the latter being differentiated from the former by its association with markedly elevated creatine phosphokinase levels. A number of other syndromes of neuromuscular weakness have been described, including hypokalaemic periodic paralysis18 and myasthenia gravis.19
GASTROINTESTINAL FEATURES
Diarrhoea, nausea and vomiting are common, though the patient may present with symptoms of an acute abdomen.20 Severe abdominal tenderness should raise the possibility of an abdominal emergency. Liver function tests may be abnormal due to congestion or necrosis and tenderness over the hepatic area may be present. Hepatosplenomegaly may be present. The presence of jaundice is a poor prognostic sign.14
LABORATORY FINDINGS
Numerous abnormalities may be found:
MANAGEMENT
β-ADRENERGIC BLOCKADE
This is the mainstay of controlling adrenergic symptoms.21 Intravenous propranolol titrated in 0.5–1-mg increments, while monitoring cardiovascular response, diminishes the systemic hypersensitivity to catecholamines. In addition it inhibits peripheral conversion of T4 to T3.22 Concurrent administration of enteral propranolol is the norm, with doses as high as 60–120 mg 4–6-hourly often being necessary due to enhanced elimination during thyroid crisis.23 An alternative regimen uses intravenous esmolol with a loading dose of 250–500 μg/kg followed by infusion at 50–100 μg/kg per min. This allows rapid titration of β-blockade while minimising adverse reactions.24 β1-selective antagonists such as metoprolol may be of use, particularly in the presence of reactive airways and heart failure. These agents do not inhibit conversion of T4 to T3 to the extent of non-selective β-antagonists and should be combined with other therapy. Patients who exhibit resistance to β-adrenergic blockade may be successfully treated with reserpine or guanethidine, though their onset of action is slow and side-effects may be significant.
DIGOXIN
Control of heart rate and rhythm may result in significant improvement in cardiac performance. Electrolyte imbalance, particularly hypokalaemia and hyomagnesaemia, should be corrected prior to drug therapy. Relative resistance to digoxin may occur due to increased renal clearance25 and increased Na/K-ATPase units in cardiac muscle.26 Arterial thromboembolic phenomena are common (10–40%) in thyrotoxicosis-related atrial fibrillation. This may be due to a procoagulant state or an increased incidence of mitral valve prolapse. Anticoagulation is controversial given the increased sensitivity to warfarin and potential for bleeding; however it should be considered in the management of these patients.
AMIODARONE
Amiodarone has theoretical benefits in thyrotoxicosis as it inhibits peripheral conversion of T4 to T3 and reduces the concentration of T3-induced adrenoceptors.27 It does however cause profound (and sometimes physiologically irrelevant) changes to thyroid function tests and should not therefore be used as the first-line agent. The presence of prolonged QTc interval should be excluded before commencing therapy.
IODINE
The release of preformed glandular thyroid hormones is inhibited by administering either inorganic iodine or lithium. Enterally administered iodides include Lugol’s solution and sodium or potassium iodide. Intravenous infusion of sterile sodium iodide may be used at a dose of 1 g 12-hourly; however, this is not always available commercially. If not, it may be prepared by the hospital pharmacy. Iodine therapy should not commence without prior thionamide administration. Used alone, it will enrich hormone stores within the thyroid gland and exacerbate thyrotoxicosis.
LITHIUM
Lithium carbonate has a similar, though weaker, action to iodine and can be used in patients with iodine allergy. An initial dose regime of 300 mg 6-hourly has been used with subsequent dosage adjusted to maintain serum drug levels at about 1 mmol/l.28 Renal and neurological toxicity tends to limit its use.
STEROIDS
Glucocorticoids reduce T4-to-T3 conversion and may modulate any autoimmune process underlying the thyroid crisis (e.g. Graves disease). In addition, relative glucocorticoid deficiency may be a feature of the crisis. An adrenocorticotrophic hormone (ACTH) stimulation test is desirable prior to administration of hydrocortisone (100 mg 6–8-hourly); alternatively dexamethasone (4 mg 6-hourly) can be administered until the test has been performed. Combination therapy with iodides can produce rapid results. Glucocorticoids are the most effective treatment for type 2 amiodarone-induced thyrotoxicosis.29
MYXOEDEMA COMA
Myxoedema coma is the extreme manifestation of hypothyroidism which, although rare, carries a mortality ranging from 30% to 60%. The term is a misnomer in that the majority of patients present with neither the non-pitting oedema, known as myxeodema, nor coma.30 The condition should be considered in any patient presenting with reduced level of consciousness and hypothermia. The crisis occurs most commonly in elderly women with long-standing undiagnosed or undertreated hypothyroidism in whom an additional significant stress is experienced. Numerous precipitating factors have been identified (Table 52.4).









CLINICAL PRESENTATION (TABLE 52.5)
Myxoedema coma can be defined when decreased mental status, hypothermia and clinical features of hypothyroidism are present (see Table 52.5). When these features are present, diagnosis is straightforward; however, asymptomatic or atypical presentation (e.g. decreased mobility) may occur.31





















CARDIOVASCULAR FEATURES
Diastolic hypertension is due to increased systemic vascular resistance and blood volume reduction;32 however myxoedema is associated with bradycardia and impaired myocardial contractility, with reduced cardiac output and hypotension a common feature. While pericardial effusions may occur, tamponade is uncommon. Creatine phosphokinase may be elevated, though this more commonly originates from skeletal rather than cardiac muscle. Acute coronary syndrome must nevertheless be excluded as a precipitant of the crisis. Electrocardiographic changes include bradycardia, decreased voltage, non-specific ST and T changes, varying types of block and prolonged QT interval. All of the cardiovascular abnormalities are reversible with thyroid hormone treatment.33
RESPIRATORY FEATURES
Hypothyroidism causes numerous respiratory alterations (see Table 52.5). There is a propensity to respiratory alkalosis, particularly during artificial ventilation. This is due to low metabolic rate, which may be compounded by iatrogenic hyperventilation.34 Diaphragmatic weakness may occur due to abnormalities of the phrenic nerve; as a result, exercise tolerance may be significantly reduced. These abnormalities improve with thyroid hormone replacement, though full recovery can take several months.
LABORATORY FINDINGS
Thyroid function tests will reveal low T4 and T3; TSH is raised in primary and low in secondary and tertiary hypothyroidism. Hyponatraemia is common and usually develops due to free water retention resulting from excess vasopressin secretion or impaired renal function.35 It may be severe and can contribute to diminished mental function. Although total body water is increased, intravascular volume is usually decreased. Hypoglycaemia may result from hypothyroidism alone, or as a result of concurrent adrenal insufficiency (Schmidt’s syndrome). The mechanism is probably reduced gluconeogenesis, but infection and starvation may contribute. Azotaemia and hypophosphataemia are common; renal function may be severely abnormal due to low cardiac output and vasoconstriction. Mild leukopenia and normocytic anaemia are frequently present, though macrocytic and pernicious anaemia due to autoimmune dysfunction may occur. Arterial blood gases often reveal respiratory acidosis, hypoxia and hypercapnia.
MANAGEMENT
The mainstay of therapy consists of thyroid hormone replacement, steroid replacement and supportive measures. Once clinical diagnosis has been made or is suspected, blood should be collected for thyroid function and plasma cortisol tests. This should be followed by thyroid hormone treatment, which should not be delayed to await laboratory results. Consideration should be given to identifying and treating precipitating factors and complications of the crisis.
THYROID HORMONE THERAPY
Some experts favour administration of T3 as it is biologically more active; has more rapid onset of action; and bypasses the impaired deiodination of T4 to T3 which occurs in hypothyroidism and non-thyroidal illness. High serum T3 concentration has, however, been associated with increased mortality36 and T3 is expensive and may be difficult to obtain. T3 may be administered orally or intravenously and has been combined with T4 therapy.37
Most authorities recommend use of T4 alone38,39 as the delayed conversion to T3 allows more gradual replacement of the deficient hormone. Bioavailability of orally administered T4 is unpredictable given the high incidence of gastrointestinal dysfunction; therefore intravenous administration is more frequently used. A loading dose of intravenous levothyroxine 100–500 mcµg is recommended38 as this saturates the binding proteins. This should be followed by 50–100 mcµg daily until conversion to the bioequivalent oral formulation is possible. The doses must be adjusted to allow for patient age, weight and cardiovascular risk factors; the lower doses should be administered to patients who are elderly, frail or have comorbidities (particularly cardiovascular disease).
SUPPORTIVE THERAPY
Numerous alterations in respiratory physiology occur, including hypoventilation and altered response to arterial oxygen and carbon dioxide tensions. In addition, anatomical changes to the airway, delayed gastric empyting and increased sensitivity to sedative drugs may be present. These changes should be considered when mechanical ventilation is required, particularly during intubation and the weaning process.34
NON-THYROIDAL ILLNESS
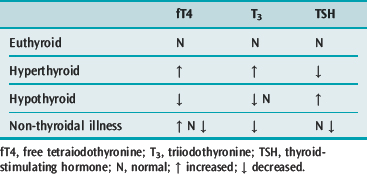
Non-thyroidal illness describes the phenomenon of starved or systemically unwell patients with abnormal serum thyroid function tests but with no apparent thyroidal illness. Low T3, T4 and TSH are commonly found; the degree of abnormality correlates with the severity of illness. Variants of these hormone levels are well described. Low serum T3 is a frequent finding and occurs due to downregulation of the monodeiodinase enzyme that converts T4 to T3, while rT3 may increase due to increased activity of the T4-to-rT3 monodeiodinase.40
Serum T4 is also commonly low in the critically ill. This is due to a decrease in the concentration of thyroid hormone-binding proteins and the presence of inhibitors that reduce T4 binding to these proteins. There is also the suggestion that T4 entry into cells may be impaired.41 Free T4 levels should be normal in less severe illness; however, in severe illness, the level may be low due to inadequate correction during the fT4 assay.42
Low serum TSH levels were previously thought to be associated with the euthyroid state; however more recent work suggests that acquired transient central hypothyroidism is present in these patients.43 Cytokines such as tumour necrosis factor-α are known to inhibit TSH secretion. Such phenomena are probably evolutionary adaptations to conserve protein and energy during severe illness. Elevated TSH may also occur in non-thyroidal illness, but few of these patients prove to have hypothyroidism following recovery from their acute illness.
Changes in thyroid function test are well described during starvation, sepsis, bone marrow transplantation, surgery, myocardial infarction, coronary artery bypass surgery and probably any critical illness. However, replacement of thyroid hormone in these patients is of no benefit and may be harmful;44 hence thyroid function should not be assessed in critically ill patients unless there is a strong clinical indication to do so. A number of specific non-thyroidal illnesses are associated with abnormal thyroid function tests. These include: some psychiatric illnesses; hepatic disease; nephritic syndrome; acromegaly; acute intermittent porphyria; and Cushing’s syndrome.45 Where assays are performed, TSH should not be interpreted in isolation as low values will not discriminate between true thyroid and non-thyroidal disease. If low T4 is also present, non-thyroidal illness is likely. If T4 is elevated then hyperthyroidism is the likely diagnosis, though elevated T4 has been documented in non-thyroidal illness.
Given the alterations in these hormones and difficulties with their assay during critical illness, thyroid hormone replacement should not be undertaken on the strength of thyroid function test results alone: additional laboratory and clinical indications must also be present (Table 52.6).
1 Kopp P. Thyroid hormone synthesis. In: Braverman LE, Utiger RD, editors. The Thyroid: Fundamental and Clinical Text. 9th edn. Philadelphia: Lippincott Williams and Wilkins; 2005:52.
2 Marshall W, Bangert S. The thyroid gland. In: Marshall W, Bangert S, editors. Clinical Chemistry. 5th edn. Mosby: Elsevier; 2004:161-175.
3 Spitzweg C, Heufelder AE, Morris JC. Thyroid iodine transport. Thyroid. 2000;10:321.
4 Dillman WH. Thyroid storm. Curr Ther Endocrinol Metab. 1997;6:81-85.
5 Tietgens ST, Leinung MC. Thyroid storm. Med Clin North Am. 1995;79:169-184.
6 Tewari K, Balderston KD, Carpenter SE, et al. Papillary thyroid carcinoma manifesting as thyroid storm of pregnancy: case report. Am J Obstet Gynecol. 1998;179:818-819.
7 Naito Y, Sone T, Kataoka K, et al. Thyroid storm due to functioning metastatic thyroid carcinoma in a burn patient. Anaesthesiology. 1997;87:433-435.
8 Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am. 1993;22:263.
9 Waldstein SS, Slodki SJ, Kaganiec GI. A clinical study of thyroid storm. Ann Intern Med. 1960;52:626-642.
10 Yi-Sun Y, Chong-Jen Y, Fen-Yu T. Apathetic hyperthyroidism associated with thyroid storm. Geriatr Gerontol Int. 2004;4:255-258.
11 Jiang Y-Z, Hutchinson A, Bartelloni P, et al. Thyroid storm presenting as multiple organ dysfunction syndrome. Chest. 2000;118:877-879.
12 Rufener S, Arunachalam V, Ajluni R, et al. Thyroid storm precipitated by infection. Endocrinologist. 2005;15:111-114.
13 Mazzaferri MEL, Skillman TG. Thyroid storm: a review of 22 episodes with special emphasis on the use of guanethadine. Arch Intern Med. 1969;124:684-690.
14 Wartofsky L. Thyroid storm. In: Wass JAH, Shalet SM, Gale E, editors. Oxford Textbook of Endocrinology and Diabetes. Oxford: Oxford University Press; 2002:481-485.
15 Aiello DP, DuPlessis AJ, Pattishall EGIII, et al. Thyroid storm presenting with coma and seizures. Clin Pediatr. 1989;28:571-574.
16 Lee TG, Ha CK, Lim BH. Thyroid storm presenting as status epilepticus and stroke. Postgrad Med J. 1997;73:61.
17 Lichstein DM, Arteaga RB. Rhabdomyolysis associated with hyperthyroidism. Am J Med Science. 2006;332:103-105.
18 Dias Da Silva MR, Cerutti JM, et al. A mutation in the KCNE3 potassium channel gene is associated with susceptibility to thyrotoxic hypokalaemic periodic paralysis. J Clin Endocrinol Metab. 2002;87:4881-4884.
19 Marino M, Ricciardi R, Pinchera A, et al. Mild clinical expression of myasthenia gravis associated with autoimmune thyroid diseases. J Clin Endocrinol Metab. 1997;82:438-443.
20 Bhattacharyya A, Wiles PG. Thyrotoxic crisis presenting as acute abdomen. J R Soc Med. 1997;90:681-682.
21 Das G, Kreiger M. Treatment of thyrotoxic storm with intravenous administration of propranolol. Ann Intern Med. 1969;70:985.
22 Perrild H, Hansen JM, Skovsted L, et al. Different effects of propranolol, alprenolol, sotalol, atenolol, and metoprolol on serum T3 and rT3 in hyperthyroidism. Clin Endocrinol. 1983;18:139.
23 Feely J, Forrest A, Gunn A, et al. Propranolol dosage in thyrotoxicosis. J Clin Endocrinol Metab. 1980;51:658-661.
24 Brunette DD, Rothong C. Emergency department management of thyrotoxic crisis with esmolol. Am J Emerg Med. 1991;9:232.
25 Shenfield GM, Thompson J, Horn DB. Plasma and urinary digoxin in thyroid dysfunction. Eur J Clin Pharmacol. 1977;12:437-443.
26 Chaudhury S, Ismail-Beigi F, Gick GG, et al. Effect of thyroid hormone on the abundance of Na,K adenosine triphosphate-subunit messenger ribonucleic acid. Mol Endocrinol. 1987;1:83-89.
27 Perret G, Yin YL, Nicolas P, et al. Amiodarone decreases cardiac beta-adrenoceptors through an antagonistic effect on 3,5,3′triodothyronine. J Cardiovasc Pharmacol. 1992;19:541-545.
28 Boehm TM, Burman KD, Barnes S, et al. Lithium and iodine combination therapy for thyrotoxicosis. Acta Endocrinol (Copenh). 1980;94:174-183.
29 Bartalena L, Brogioni S, Grasso L, et al. Treatment of amiodarone-induced thyrotoxicosis, a difficult challenge: results of a prospective study. J Clin Endocrinol Metab. 1996;81:2930-2933.
30 Nicoloff JT, LoPresti JS. Myxedema coma. A form of decompensated hypothyroidism. Endocrinol Metab Clin North Am. 1993;22:279-290.
31 Mintzer MJ. Hypothyroidism and hyperthyroidism in the elderly. J Fla Med Assoc. 1992;79:231-235.
32 Streeten DHP, Anderson GHJr, Howard T, et al. Effects of thyroid function on blood pressure. Hypertension. 1988;11:78.
33 Shenoy MM, Goldman JM. Hypothyroid cardiomyopathy. Echocardiographic documentation of reversibility. Am J Med Sci. 1987;294:1.
34 Behnia M, Clay A, Farber M. Management of myedematous respiratory failure: review of ventilation and weaning principles. Am J Med Sci. 2000;320:368-373.
35 Iwasaki Y, Oisa Y, Yamauchi K, et al. Osmoregulation of plasma vasopressin in myxedema. J Clin Endocrinol Metab. 1990;70:751.
36 Hylander B, Rosenqvist U. Treatment of myxoedema coma – factors associated with fatal outcome. Acta Endocrinol (Copenh). 1985;108:65.
37 Wartofsky L. Myxedema coma. In: Braverman LE, Utiger RD, editors. The Thyroid. Philadelphia: Lippincott-Raven; 1996:871.
38 Rodriguez I, Fluiters E, Perez-Mendez LF, et al. Factors associated with mortality of patients with myxoedema coma: prospective study of 11 cases treated in a single institution. J Endocrinol. 2004;180:347.
39 Smallridge RC. Metabolic and anatomic thyroid emergencies: a review. Crit Care Med. 1992;20:276-291.
40 Peeters RP, Wouters PJ, Kaptein E, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003;88:3202.
41 De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrin Metab. 1999;84:151-164.
42 Chopra IJ, Solomon DH, Hepner GW, et al. Misleading low free T4 index (FT4I) and usefulness of reverse T3 (rT3) measurement in nonthyroidal illness. Ann Intern Med. 1979;90:905.
43 Chopra IJ. Euthyroid sick syndrome: Is it a misnomer? J Clin Endocrinol Metab. 1997;82:329.
44 Utiger RD. Altered thyroid function in nonthyroidal illness and surgery. To treat or not to treat? N Engl J Med. 1995;333:1562.
45 Ross DS. Thyroid function in nonthyroidal illness. Up To Date. 2006. version 14.2
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 52 Thyroid emergencies
Thyroid emergencies are a rare cause for admission to critical care. However, mortality is high unless specific treatment is provided in an expeditious manner. Abnormal thyroid function tests are commonly encountered during critical illness; numerous factors must be considered before interpreting these findings as indicating thyroid disease.
BASIC PHYSIOLOGY
Thyroid hormones affect the function of virtually every organ system and must be constantly available for these functions to continue. The two biologically active hormones are tetraiodothyronine (thyroxine or T4) and triiodothyronine (T3). These are synthesised by incorporating iodine into tyrosine residues, a process which occurs in thyroglobulin contained within the lumena of the thyroid gland. Stimulation of hormone release by thyroid-stimulating hormone (TSH) results in endocytosis of thyroglobulin from the lumen into the follicular cells, followed by hydrolysis to form T4 and T3, which are released into the circulation.1
Both T4 and T3 contain two iodine atoms on their inner (tyrosine) ring. They differ in that T4 contains two further iodine atoms on its outer (phenol) ring, whereas T3 contains only one. T4 is produced solely by the thyroid gland whereas the majority of T3 is synthesised peripherally by the removal of one iodine atom (deiodination) from the outer ring of T4. If deiodination of an inner-ring iodine atoms occurs, the metabolically inert reverse-T3 (rT3) is formed. This is produced in preference to T3 during starvation and many non-thyroidal illnesses, and the ratio of inactive (rT3) to active T3 synthesis appears to play an important role in the control of metabolism.2 Numerous factors can affect the peripheral deiodination process (Table 52.1). Both T4 and T3 are highly protein-bound in the serum, predominantly to thyroid-binding globulin (TBG), but to a lesser extent to albumin and prealbumin. Changes in concentration of these serum-binding proteins have a large effect on total T4 and T3 serum concentrations. Such protein changes do not, however, affect the concentration of free hormone or their rates of metabolism. The serum-binding proteins act as both a store and a buffer to allow an immediate supply of the metabolically active free T4 (fT4) and free T3 (fT3). In addition, protein binding reduces the glomerular filtration and renal excretion of the hormones.
Table 52.1 States associated with decreased deiodination of thyroxine to triiodothyronine
The regulation of thyroid function is predominantly determined by three main mechanisms, the latter two providing physiological control. Firstly, availability of iodine is crucial for the synthesis of the thyroid hormones. Dietary iodide is absorbed and rapidly distributed in the extracellular fluid, which also contains iodide released from the thyroid gland and from peripheral deiodination processes. This becomes trapped within thyroid follicular cells, from which it is actively transported into the lumen to be oxidised into iodine and subsequently combined with tyrosine.3 Other ions, such as perchlorate and pertechnetate, share this follicular cell active transport mechanism, acting as competitive inhibitors for the process.
Secondly, thyroid hormone release is controlled by a closed feedback loop with the anterior pituitary. Diminished levels of circulating hormones trigger secretion of TSH which acts on the follicular cells of the thyroid gland, causing them to release thyroglobulin-rich colloid from the lumen, which is hydrolysed to form T4 and T3 for systemic release. Increased levels of T4 and T3 cause diminished TSH secretion, resulting in the follicular cells becoming flat and allowing increased capacity for colloid storage. As a result, less thyroglobulin is mobilised and hydrolysed with less T4 and T3 release. The degree to which TSH is secreted in response to changes in circulating thyroid hormones is dependent on the hypothalamic hormone thyrotrophin-releasing hormone (TRH), which is itself modulated by feedback from the thyroid hormones. TRH secretion is inhibited by dopamine, glucocorticoids and somatostatin.
THYROID CRISIS (THYROID STORM)
Thyrotoxic storm is arguably the most serious complication of hyperthyroidism, with reported mortality ranging from 10 to 75% in hospitalised patients.4,5 Crisis most commonly occurs as a result of unrecognised or poorly controlled Graves disease; however, other underlying diseases may be the cause.6,7 Females outnumber males. Laboratory findings are inconsistent due to acute disruption of the normal steady state of the circulating hormones, and there is no definitive value that separates thyrotoxicosis from thyroid storm. The latter is a clinical diagnosis and a scoring system has been proposed to guide the likelihood of the diagnosis (Figure 52.1).8 Precipitating factors are not always present, though many have been identified (Table 52.2).
Figure 52.1 Severity assessment in thyroid crisis. CNS, central nervous system; GI, gastrointestinal.
(Adapted from Tewari K, Balderston KD, Carpenter SE et al. Papillary thyroid carcinoma manifesting as thyroid storm of pregnancy: case report. Am J Obstet Gynecol 1998; 179: 818–19.)
Table 52.2 Factors associated with precipitating thyroid storm
CLINICAL PRESENTATION (Table 52.3)
The classic signs of thyroid crisis include fever, tachycardia, tremor, diarrhoea, nausea and vomiting.9 However presentation is extremely variable and may range from apathetic hyperthyroidism (apathy, depression, hyporeflexia and myopathy)10 to multiple-organ dysfunction.11,12 Differential diagnosis includes sepsis and other causes of hyperpyrexia such as adrenergic and anticholinergic syndromes.
Table 52.3 Clinical features of hyperthyroidism/thyroid crisis
FEVER
This is the most characteristic feature. Temperature may rise above 41°C. There have been suggestions that pyrexia is present in all cases of thyroid storm,13 though normothermia has been reported.11 Pyrexia is rare in uncomplicated thyrotoxicosis and should always raise suspicion of thyroid storm. It is not clear whether this febrile response is due to alteration of central thermoregulation or elevation of basal metabolic thermogenesis beyond the body’s ability to lose heat.
CARDIOVASCULAR FEATURES
Fluid requirements may be substantial in some patients, whereas diuresis may be required in those with severe cardiac congestion. Cardiac decompensation can occur in young patients with no known antecedent cardiac disease. Systolic hypertension with widened pulse pressure is common initially; however hypotension supervenes later. Cardiogenic shock with vascular collapse is a preterminal sign.14 Arrhythmias are common and include atrial and polymorphic ventricular tachycardias. The latter may result from prolonged QTc interval, the presence of which may complicate and contraindicate the use of amiodarone therapy.
NEUROMUSCULAR FEATURES
Tremor is a common early sign, but as ‘storm’ progresses, central nervous dysfunction evolves with progression to encephalopathy or even coma.15 Thyroid storm has been reported in association with status epilepticus and cerebrovascular accident.16 Weakness may be a feature, particularly with apathetic thyrotoxicosis.10 Thyrotoxic myopathy and rhabdomyolysis may be present,11,17 the latter being differentiated from the former by its association with markedly elevated creatine phosphokinase levels. A number of other syndromes of neuromuscular weakness have been described, including hypokalaemic periodic paralysis18 and myasthenia gravis.19
GASTROINTESTINAL FEATURES
Diarrhoea, nausea and vomiting are common, though the patient may present with symptoms of an acute abdomen.20 Severe abdominal tenderness should raise the possibility of an abdominal emergency. Liver function tests may be abnormal due to congestion or necrosis and tenderness over the hepatic area may be present. Hepatosplenomegaly may be present. The presence of jaundice is a poor prognostic sign.14
LABORATORY FINDINGS
Numerous abnormalities may be found:
MANAGEMENT
β-ADRENERGIC BLOCKADE
This is the mainstay of controlling adrenergic symptoms.21 Intravenous propranolol titrated in 0.5–1-mg increments, while monitoring cardiovascular response, diminishes the systemic hypersensitivity to catecholamines. In addition it inhibits peripheral conversion of T4 to T3.22 Concurrent administration of enteral propranolol is the norm, with doses as high as 60–120 mg 4–6-hourly often being necessary due to enhanced elimination during thyroid crisis.23 An alternative regimen uses intravenous esmolol with a loading dose of 250–500 μg/kg followed by infusion at 50–100 μg/kg per min. This allows rapid titration of β-blockade while minimising adverse reactions.24 β1
[/not-level-membership-for-anesthesiology-category]
