Thyroid Disorders
DIAGNOSTIC APPROACH TO THYROID DISEASE
Pathogenesis of the Laboratory Findings
Nonthyroidal Illness Syndrome Among Cardiac Patients
HYPOTHYROIDISM AND MYXEDEMA COMA
HYPERTHYROIDISM, THYROID STORM, THYROCARDIAC CRISIS, AND CORONARY ARTERY SPASM
Thyroid Physiology
Cellular Effects of Thyroid Hormone
Thyroid hormone action is initiated by the binding of nuclear hormone receptors activated by 3,5,3′-triiodothyronine (T3) to nuclear thyroid hormone response elements, in which the hormone-receptor complex acts as a transcription factor.1 In addition, T3 exerts rapid nontranscriptional extranuclear effects that are especially important in cardiovascular physiology.2 Transporters necessary for cellular uptake of thyroid hormone recently have been recognized.3 Thyroxine (T4) acts as a prohormone for T3 and itself is only weakly interactive with thyroid hormone receptors. The compound 3,3′,5′-triiodothyronine(reverse T3) is almost devoid of metabolic activity (Fig. 60.1).
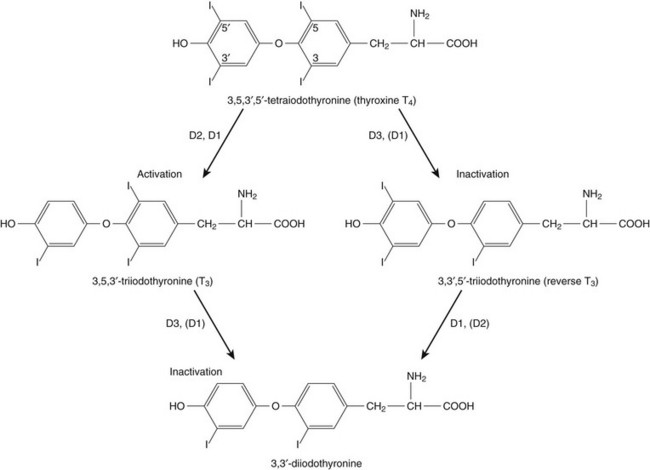
Peripheral and Intrathyroidal Conversions of T4 and T3
Normally about 80% of circulating T3 and probably greater than 90% of circulating reverse T3 are derived from circulating T4. Peripheral production of T3 is modulated by the availability of T4, by peripheral cellular uptake of T4, and by the activities of the enzymes identified as iodothyronine selenodeiodinase 1 (D1), selenodeiodinase 2 (D2), and selenodeiodinase 3 (D3). The enzymes affecting peripheral levels of circulating thyroid hormone and tissue levels of T3 either convert T4 to T3, also removing reverse T3 by deiodination (the D1 and D2 enzymes), or deiodinate and thus inactivate T3 (the D3 enzyme) and convert T4 to reverse T3 (the D3 enzyme)4,5 (see Fig. 60.1).
Hormone Transport
Circulating T4 is carried about 49% to 64% by thyroxine-binding globulin (TBG), 12% to 13% by transthyretin, and 7% to 9% by albumin. T3 is carried 80% by TBG, 9% by transthyretin, and 11% by albumin.6 A small fraction is transported by lipoproteins. About 0.03% of circulating T4 and 0.3% of T3 is free or unbound. TBG is produced by the liver, and transthyretin by the liver and choroid plexus. The approximate half-lives of circulating transport proteins are transthyretin, 2 days; TBG, 5 days; and albumin, 15 days.6 Recent research suggests that TBG allows targeted delivery of T4 to tissues, for example, at sites of inflammation.7 The unbound fraction of circulating hormone gains access to peripheral tissues and pituitary, determines metabolic status, and participates in feedback inhibition of the pituitary.
Diagnostic Approach to Thyroid Disease
History, Physical Examination, and Record Review
In critical illness, ambiguity of thyroid function tests is commonplace. The obstacles to laboratory assessment augment the importance of record review, history, and physical examination. When overt thyroid dysfunction is present, the history and physical examination usually yield multisystemic positive findings.8
Case Finding by Screening
In a meta-analysis of earlier studies, the frequency of thyroid disease ascertainable by screening hospitalized patients was similar to that among outpatients, about 1% to 2%.9 In the hospital the relatively low case-finding rate and the confounding effect of nonthyroidal illness have been viewed as impediments to general screening of unselected patients, except possibly among elderly women.10,11 However, one study in which TSH and free T4 index were performed on sera drawn at the time of admission from 364 consecutive patients suggested that the rate of nonthyroidal illness syndrome (7.4%) was exceeded by the combined rates of unsuspected thyroidal failure (5.8%), subclinical hypothyroidism (6%), and hyperthyroidism (2%).12 Among critical care patients, the case-finding rate for thyroid disease during screening is not known with confidence. Clinical suspicion of thyroid disease based on patient symptoms or findings should, of course, result in testing.
Assays and Imaging
Among critically ill patients, the TSH assay taken alone may yield misleading results, and the utility of most commercial methods for determining free thyroid hormone levels is limited.13–17 Thyroid function test results may change on a daily basis. The best course of action is not to screen with a single test but to order a potentially useful battery of tests from the outset.
Thyroid-Stimulating Hormone
Reasons for misleading TSH results, some of which apply to critically ill patients, include nonequilibrium conditions in which thyroid status has recently fluctuated, acute psychiatric illness, nonthyroidal illness syndrome, central causes of hyperthyroidism or hypothyroidism, and the effects of medication.18–20 The distribution of TSH results in nonthyroidal illness syndrome may overlap with the range seen in thyrotoxicosis. Conversely, in mild cases of subclinical thyrotoxicosis, for example, in nodular thyroid disease at the earliest stages of autonomy, TSH suppression may be minimal. The indication for measuring TSH is to support a diagnosis of primary hypothyroidism or hyperthyroidism.
Estimates of Free Thyroxine
Although with some exceptions most free T4 assays perform well in the ambulatory setting, in critically ill patients some commercial assays for free T4 yield low results that cannot be verified by an equilibrium dialysis method.14,17 The indication for ordering a free T4 rather than total T4 assay is to assess thyroid function in the presence of suspected abnormalities of thyroid hormone transport or to clarify the significance of an abnormal total T4 result in a patient whose clinical condition appears euthyroid. A normal result of a free T4 determination together with a normal TSH is reassuring.
Total Thyroxine
The indication for ordering a total T4 assay is to discount hyperthyroidism in the presence of misleading free T4 elevations in euthyroid patients, to provide reassurance when considered together with an estimate of thyroid hormone binding that central hypothyroidism may be absent, or to demonstrate the presence and quantitate the severity of hypothyroidism or hyperthyroidism.13
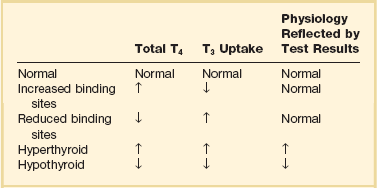
T3 Uptake or Thyroxine-Binding Globulin and Calculated Free T4 Index
The T3 uptake, together with a determination of total T4, is used in index methods to provide information about hormone transport and to help estimate free T4. In critically ill patients with hypothyroxinemia, the free T4 index frequently is misleading. Nevertheless, when hypothyroxinemia by a free hormone estimate is present but its interpretation is uncertain, for example, when the TSH is normal or low, it is advantageous to review a second independent assay such as the T3 resin uptake or a direct measurement of TBG to see whether qualitatively the result is consistent with reduced T4 binding sites on circulating transport proteins. Elevation of T3 uptake accompanied by low total T4 suggests reduced hormone binding to transport proteins, consistent with nonthyroidal illness syndrome. Nonelevated T3 uptake or low T3 resin uptake accompanied by hypothyroxinemia in the face of nonthyroidal illness suggests the possibility of hypothyroidism (Table 60.1).
Free T3
Free T3 sometimes is found to be normal among patients with nonthyroidal illness syndrome having low total T3, helping offset suspicion of secondary hypothyroidism due to organic pituitary or hypothalamic disease. Methodologic limitations in critically ill patients may result in variability of findings between assay systems. Chopra and colleagues reported that by direct equilibrium dialysis radioimmunoassay in nonthyroidal illness syndrome serum free T3 concentration was normal in approximately 83% of patients with low total T3.21–23 The indication for ordering the free T3 test is to confirm or exclude T3 toxicosis in patients suspected of having thyroid transport abnormalities, such that reliance upon total T3 might be inappropriate. Free T3 also may help identify amiodarone-induced thyrotoxicosis.
“Best Panel” for Critically Ill Patients
For confirmation of a suspected diagnosis of thyroid dysfunction, a panel rather than a single test is recommended at the outset. The TSH should not be used as monoscreening in the hospital (Fig. 60.2). The initial “best panel” is whichever has the faster turnaround time or weekend availability, or both, at the laboratory used by the institution. The initial panel should include either a calculated free T4 index (utilizing T3 uptake or TBG together with total T4) or a free T4 estimate by any other method having rapid turnaround time, together with total T4 and TSH. These initial panels will yield unambiguous results in most cases of critical illness that are caused or complicated by preexisting clinically significant primary hypothyroidism or hyperthyroidism. Furthermore, the findings of normal TSH and normal or elevated free T4 with normal total T4 suggest euthyroidism. The isolated finding of free T4 elevation may result from sepsis or the effects of furosemide, heparin or enoxaparin.
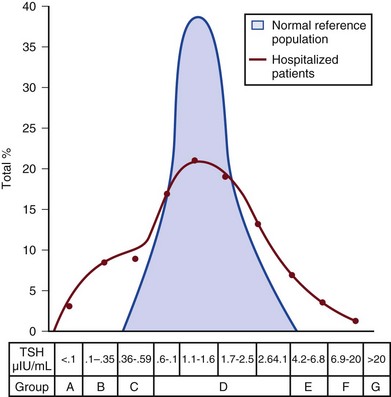
If the free T4 and TSH considered as a hormone pair appear discordant (both values high or both values low), and if the explanation or management is not straightforward, a consultation should be obtained. However, in the setting of nonthyroidal illness, if low T4, low free T4 estimate, and normal or low TSH are demonstrated during screening, to discount suspicion of secondary hypothyroidism it is sometimes helpful to order TSH, free T4 by equilibrium dialysis, and morning cortisol (Box 60.1).
Medication Effects
When unexpected thyroid function test results are reported, a medication review should be conducted. Drugs may alter the results of thyroid function tests either in vivo or in vitro without affecting thyroid function. Additionally, drugs not designed to treat hypothyroidism or hyperthyroidism may alter thyroid function.15–17 Periodic monitoring before and during long-term use should occur during use of a drug that is recognized to be a potential cause of thyroid dysfunction. Medication effects are summarized in Table 60.2.
Table 60.2
Thyroid Function Test Abnormalities Induced by Medication in Euthyroid Patients
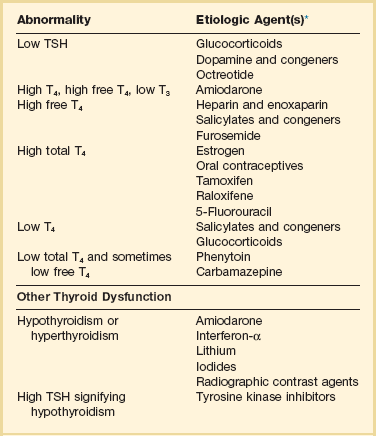
T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
TSH elevation signifying hypothyroidism or TSH suppression signifying hyperthyroidism may result, or patients may remain euthyroid during treatment with any of the following: interferon-α,24,25 lithium,26,27 iodinated contrast agents,28 iodine,29 and amiodarone.30–35 During use of iodine-containing medications, hyperthyroidism of the Jod-Basedow type due to the iodine content of the drug may fail to self-resolve. However, when the mechanism of hyperthyroidism is destructive thyroiditis, as may be seen during some cases of interferon-α-induced hyperthyroidism and type 2 amiodarone-induced thyrotoxicosis, a possible sequence is hyperthyroidism followed by hypothyroidism.31,32 The prevalence of amiodarone-induced hypothyroidism has been variably reported, but it may be seen in up to 22% of treated patients from iodine-sufficient regions,35 and its occurrence may be increased in the presence of positive antithyroid antibodies.30,33 The combined findings of elevation of total and free T4 together with reduction of T3 occur in euthyroid persons receiving amiodarone; these findings taken alone, when seen together with a normal TSH, do not indicate thyroid dysfunction (Box 60.2). High TSH signifying hypothyroidism may be caused by tyrosine kinase inhibitors including sunitinib, imatinib, motesanib, and sorafenib.36–41
Nonthyroidal Illness Syndrome
Nonthyroidal illness syndrome is usually recognized as a constellation of laboratory findings of uncertain clinical significance, discovered on thyroid function testing among patients having acute medical or surgical illness, the more pronounced abnormalities being associated with worse prognosis, and characterized by resolution after recovery from illness (Fig. 60.3). Although the severity of illness often makes clinical assessment difficult, patients usually appear clinically euthyroid. Laboratory findings of nonthyroidal illness syndrome may occur with psychiatric illness, starvation, congestive heart failure, acute and chronic renal failure, acquired immunodeficiency syndrome (AIDS), postoperative status, trauma, and in general with critical illness.19,42–62
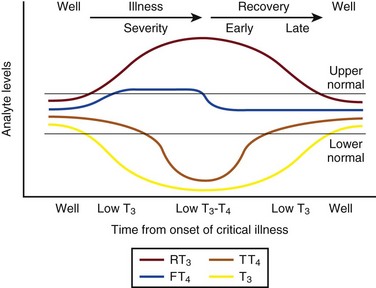
Figure 60.3 Nonthyroidal illness syndrome. The time course of concentration of circulating iodothyronine levels are shown qualitatively during evolution of nonthyroidal illness syndrome. A characteristic sequence involves first the development of a low total T3, initially with normal T4 or sometimes with high free T4. Characteristically, as T3 falls, reverse T3 (RT3) rises. With duration of illness, as thyroid hormone transport protein concentrations decline, concentrations of total T3 and total T4 (TT4) both are affected, and total T4 levels fall. Later, during the greatest severity of the illness, there may be low thyroid-stimulating hormone (TSH) and low free T4, a predictor of higher mortality risk. During recovery, TSH overshoots the normal range (not shown) as T4 rises. Finally, after recovery, tests become normal. (Adapted with permission from Figure 156: In turn, Chopra had reproduced this from Nicoloff et al.)
High free T4 without elevation of total T4, seen in the early stages of nonthyroidal illness, may be associated with use of certain drugs or with sepsis. Otherwise, during development of nonthyroidal illness syndrome, reduction of circulating T3 is one of the earliest and most consistently observed findings, possibly seen in over 70% of patients, depending upon the duration and severity of the illness.42,43 The decline of T3 often is accompanied by a rise of reverse T3. Patients with chronic renal failure who have low T3 do not invariably have high reverse T3 levels,44,45 and patients with HIV may have low reverse T3.52
Incidence
Among hospitalized patients, intrinsic thyroid disease is less common than nonthyroidal illness syndrome.9,11,47 The prevalence of nonthyroidal illness syndrome among patients with psychiatric disease has been reported to be about 10%, often manifesting as elevated TSH or elevated T4.19,55 Abnormalities of thyroid function tests interpreted as euthyroid sick syndrome were found in 51.5% of elderly patients undergoing emergency surgery.57 Hypothyroxinemia without TSH elevation was reported in 22% of critically ill patients in one study.46 In another study of intensive care unit patients, depending upon the duration of illness and patient outcome, the findings of low total T3 approached 70% to 80%, and about half of patients had low total T4.59
Pathogenesis of the Laboratory Findings
In order to understand the mechanisms for observed alterations of iodothyronine molecules (T4, T3, and reverse T3) during nonthyroidal illness, under conditions of health and illness with and without specific interventions, there would need to be comparative human data on each of the following: tissue-specific functions of thyroid hormone; actual tissue levels of T4 and T3; cellular uptake of thyroid hormones; activity of each of the deiodinase enzymes in each tissue of interest, including hypothalamus and pituitary, thyroid, liver, muscle, and other tissues; tissue-specific direct and indirect mechanisms of regulation of each deiodinase enzyme, including enzyme-regulating effects of iodothyronines, cytokines, and drugs; contribution of each tissue and each of the three deiodinase enzymes to circulating levels of T3; the role of transport proteins in targeted delivery of thyroid hormone in health if any and in illness; and any adaptative or maladaptative effects of the alterations of iodothyronine availability seen in illness. Much of our present knowledge derives from experimental animal models and is complicated by possible ambiguity of experimental assay results for deiodinase enzyme activity and by the necessarily inferential nature of human data.5,7,63–96
Alterations of T3 and reverse T3 usually precede reductions of T4. Peripherally, there is reduced production of T3 by 5′-deiodination of T4, reduced removal of reverse T3 by 5′-deiodination, and increased removal of T3 by deiodination (see Fig. 60.1). The reduction in circulating T3 is believed to result from a combination of mechanisms, including changes of cellular uptake of T4, reduced activity of deiodinase enzymes that normally catalyze peripheral conversion of T4 to T3, inactivation of T3 by deiodination resulting from activity of the D3 enzyme, and possibly decline of hypothalamic TRH resulting from an enzymatically mediated increase of T3 production in the tanycytes.5 The role of cytokines in mediating some of these changes has been explored.71,76–79,81,84 Because T3 may amplify its own production at the tissue level by activating the D2 enzyme, a decline of central TSH drive to the thyroid indirectly could reduce peripheral production of T3. With acknowledgment that the finding of free T3 abnormalities may be method-dependent, studies suggest that in some but not all patients having low total T3, the results of free T3 assays may be normal.21–23
Elevations of free T4 sometimes occur in nonthyroidal illness, often accompanied by low or low normal total T4, and without TSH suppression. The isolated finding of free T4 elevation in vivo at least in some instances may represent artifact, such as could be introduced by nonselective beta blockers, furosemide, or free fatty acids (see previously, under “Assays and Imaging”). Mechanisms leading to high free T4 in the early stages of sepsis have been controversial. Circulating inhibitors of binding of T4 to transport proteins may be present such as nonesterified fatty acids.65,67–69,72 Additionally, cleavage by elastases or consumption of the TBG molecule by serine proteases may occur at sites of inflammation, possibly leading to release of free T4 to the circulation and permitting targeted delivery of T4 at sites of inflammation.82,83,85
Patients with both low T4 and low T3 generally have disease of greater severity and longer duration. Early studies of hormone kinetics in the setting of low T4 were consistent with reduced binding to vascular sites.64,66 A low serum albumin level often, but not always, permits the caregiver to predict that TBG will be low. Hypoalbuminemia is highly associated with nonthyroidal illness syndrome.57,60 Reduced concentration of transport proteins is a prevalent finding among cases of nonthyroidal illness presenting with low T4 and low T3.49 Inhibitors of hormone binding to transport proteins also may result in low total concentrations of circulating thyroid hormone.
The more severely ill patients with nonthyroidal illness syndrome, usually those having low T4 levels, as well as low T3, may have low circulating levels of TSH.51,80,81,87,88 Central production of TRH and TSH may be reduced by inflammatory mediators. In a human autopsy study, reduced TRH gene expression was demonstrated among patients with an antemortem finding of low T3.80 In animal studies, it has been suggested that central conversion of T4 to the active hormone T3 in hypothalamic tanycytes under the action of the D2 enzyme, by producing local hyperthyroidism under conditions of illness, may act as a negative feedback signal that reduces hypothalamic release of TRH and pituitary release of TSH.5,88,92,94,96 Therefore, in prolonged or severe nonthyroidal illness syndrome, a central mechanism may contribute to the findings of low T4 and low T3.
Investigationally, after TNF-α administration, as during recovery following nonthyroidal illness, there may be temporary overshoot of TSH into the mildly elevated range, accompanied by rising T4.51,81
Presently it is unknown whether nonthyroidal illness syndrome is adaptive or maladaptive. In starvation, low T3 syndrome may promote protein sparing.97 Human trials have not been conducted with a sufficient number of randomized, critically ill human subjects to resolve the question of whether thyroid hormone therapy is beneficial. In a nonrandomized study of patients with sepsis, T3 was used to reduce dopamine dependence.98 Treatment with T3 for human burn injury showed no benefit.99 In a small study of critically ill patients with low T3 and low T4, therapy with T4 did not correct the low T3 or improve the prognosis,100 and T4 for nonthyroidal illness may increase mortality rate among acute renal failure patients.101
Nonthyroidal Illness Syndrome Among Cardiac Patients
Low T3 or low T3 and T4 levels are observed in the setting of advanced heart failure, after revascularization, and after myocardial infarction, providing a rationale for therapeutic trials of treatment with thyroid hormone among cardiac patients.102–121 Among patients with severely impaired left ventricular performance, the use of intravenous T3 as an alternative to standard therapy (such as dopamine) and the compatibility or usefulness of T3 in combination with other inotropic and vasodilating regimens requires further research. Correction of any reversible ischemia should first be achieved. Use of intravenous T3 shows promise as an inotrope and vasodilator.104,111 In the setting of advanced heart failure, coronary bypass or valve surgery, correction of congenital heart lesions, or in the treatment of transplantation donors and recipients, the benefits that have been attributed to intravenous T3 therapy include improvement of cardiac index with reduction of systemic vascular resistance,104,111 a reduction in postoperative episodes of atrial fibrillation,107 reduction in estimated mortality rate among high-risk patients,109 a reduced requirement for inotropic support and mechanical devices,112,113 a lower incidence of postoperative myocardial ischemia,113 improved cardiac allograft function,102 and improved neuroendocrine profile with improved ventricular performance.121 Whether the apparently beneficial cardiac effects of T3 administration are pharmacologic effects or at least partially the effects of physiologic replacement of a true hormone deficiency are unclear. Overall, the use of T3 for cardiac indications has not gained widespread acceptance.
Approach to Management and Therapeutic Alternatives
It is unknown whether the spontaneously developing alterations of tissue exposure to thyroid hormone during nonthyroidal illness are advantageous or disadvantageous to the patient. Although there is interest in evaluating T3 therapy for nonthyroidal illness syndrome, it is expected that differences in outcome resulting from such treatment will be small and difficult to demonstrate.62,122–125 Proposed approaches have not been adequately studied for safety or efficacy in critically ill patients having nonthyroidal illness syndrome. A special niche may exist for the use of T3 in the treatment of cardiac patients who require hemodynamic support. As a precaution, it is noted that high levels of T3 have been identified as a risk factor for coronary events.126 Although future research may bring about changes in the standard of care, at the present time for nonthyroidal illness syndrome most experts recommend observation without thyroid hormone treatment, with reevaluation of thyroid function tests after recovery. The philosophy of nontreatment would imply that there is no obligation to order thyroid tests unless thyroid disease is suspected.
Hypothyroidism and Myxedema Coma
Hypothyroidism
Incidence
In contrast to the high frequency of finding the laboratory manifestations of nonthyroidal illness syndrome in the intensive care unit, among outpatients the prevalence of spontaneous hypothyroidism is relatively low, found in 1% to 2% in iodine-replete communities, and more commonly found in women than in men.141–143 An age-related increase of incidence of hypothyroidism exists, and the prevalence is higher in women. The appearance of overt hypothyroidism is predicted by prior isolated TSH elevation and positive antithyroid antibodies. Hypothyroidism can be induced by iodine, amiodarone, lithium, or tyrosine kinase inhibitors.
Pathophysiology
Patients with severe hypothyroidism have reduced calorigenesis and oxygen consumption. Many metabolic processes proceed at a markedly reduced rate. Glycosaminoglycan metabolism is impeded, resulting in widespread tissue deposition of hyaluronan (hyaluronic acid). A contributory factor in the production of generalized edema is transcapillary albumin escape.144 Slowing of the metabolism of lipoproteins results in secondary hyperlipidemia. Hypercholesterolemia is common. Hypometabolism affects conversion of carotene to vitamin A and the rate of removal of vitamin K.
Reduced ventilatory responses to hypoxia and hypercapnia appear to have a dominantly central mechanism, and there may be upper airway obstruction.145–150 The effusions of myxedema contain high concentrations of protein and cholesterol. Pericardial effusion is more characteristic than pericardial tamponade.151–154 Vasoconstriction with or without hypertension exists. The mechanisms of resistance to catecholamine effects are complex and controversial. There is reduced responsiveness to adrenergic stimuli but actual elevation of circulating norepinephrine concentration.155–157 Myocardial contractility, oxygen consumption, ejection time, diastolic ventricular compliance, stroke volume, heart rate, and cardiac index are reduced and systemic vascular resistance is increased.115,119,120 The left ventricular end-diastolic pressure is not necessarily elevated in myxedema and the cardiac index may increase in response to exercise.158–161 Nevetheless, when TSH is higher than 10 µIU/mL, there is an increased risk for congestive heart failure even at the stage of subclinical hypothyroidism.162–164 An increased occurrence of coronary artery disease also may exist.165–170 Hypomotility of the bowels is common. There may be coexistent iron losses as a result of menorrhagia or gastrointestinal bleeding. Malabsorption of vitamin B12 and folic acid may occur. A reduction in atrial natriuretic factor production may occur,171 as well as a reduction of the glomerular filtration rate. The kidney cannot excrete a water load effectively, but antidiuretic hormone deficiency cannot be consistently implicated when hyponatremia and defective intrarenal mechanisms are suspected.172–174 Calcium loading can result in hypercalcemia. Hyperuricemia commonly results from underexcretion of uric acid. Pituitary overproduction of prolactin but retarded responsiveness of the pituitary-adrenal axis to appropriate challenges may occur in myxedema.
Clinical Manifestations
Hypertension may be attributable to myxedema. The thyroid is often atrophic but may be goitrous. The following characteristics often permit clinical diagnosis: a deep, husky quality of the voice; slow mode of speech; involuntary blepharoptosis; torpid expression; facial bloating; eyelid and infraorbital edema; sallowness, facial pallor; hearing loss; bradycardia, distant muffled heart tones; cool, dry, coarse skin; nonpitting edema of the supraclavicular fossae, hands, legs, and feet144; bruising; and the delayed relaxation of deep tendon reflexes. The patient may present with adynamic ileus.
Diagnostic Approach
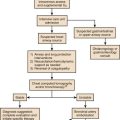
The TSH level of patients with untreated primary hypothyroidism can be lowered into the normal range by critical illness, but only rarely.175 Sometimes the severity of TSH elevation is blunted by the myxedema itself, with extreme hypothyroxinemia accompanied by TSH levels that are elevated but may be less than 20 µIU/mL. In most cases of advanced hypothyroidism the TSH will be above the normal reference range and the T4 and free T4 will be low. Therefore, in general, during nonthyroidal illness the laboratory diagnosis of coexistent primary hypothyroidism is straightforward (see Fig. 60.2).
Milder cases of hypothyroidism potentially can be confused with the recovery phase of nonthyroidal illness syndrome, when transitory TSH elevation commonly occurs. The patient should be examined for goiter. The finding of subnormal free T4, positive antithyroid peroxidase antibodies, or TSH above 20 µIU/mL sometimes signifies intrinsic thyroid disease.18,51 Outpatient reassessment should occur.
In critically ill patients the diagnosis of secondary hypothyroidism is not straightforward. When a low free T4 level by equilibrium dialysis and low TSH level are present, the question arises of whether the findings signify central hypothyroidism for any reason other than nonthyroidal illness syndrome. The most pressing immediate need would be to recognize and treat cortisol deficiency or pituitary mass effect. History of prior reproductive dysfunction, examination of cranial nerve function and mental status, and measurements of cortisol and other pituitary and target gland hormones, including follicle-stimulating hormone (FSH) and luteinizing hormone (LH) among postmenopausal women, may suggest preexisting pituitary dysfunction with or without tumor or the new occurrence of pituitary apoplexy or may help provide evidence of intactness of pituitary function. However, some critically ill patients lacking intrinsic hypothalamic or pituitary disease may have features of “eugonadal sick” syndrome as well as nonthyroidal illness syndrome. Measurement of cortisol or free cortisol may be of special value.54 The diagnosis of pituitary tumor or apoplexy is confirmed by pituitary magnetic resonance imaging (MRI) or CT scanning.
Approach to Management
Replacement therapy for hypothyroidism can be provided as levothyroxine (thyroxine, T4) or liothyronine (triiodothyronine, T3), each of which is available for oral or intravenous administration, but T4 is the preferred hormone for the treatment of ambulatory patients and most hospitalized patients.176–191 Normally, production of T3 from T4 occurs rapidly. Because of the prolonged half-life of T4, after each daily dose or after short-term interruption of chronic T4 therapy there are stable blood levels of T4 and T3.178,179 An ambulatory hypothyroid patient, when treated with T4 in dosage sufficient to maintain euthyroidism (normal TSH), often has normal T3 but blood levels of T4 slightly above the mean for a euthyroid patient.183 A patient whose T4 dose requirement was established before hospitalization generally should be maintained on the same dose, if it can be given orally. The oral absorption of T4 is impeded by intestinal disease or concomitant administration of iron, sucralfate, cholestyramine, colestipol, calcium, and other drugs.185 Enteric administration of T4 should be separated from these drugs by at least 2 to 4 hours.
When T4 therapy is given for overt hypothyroidism, whole body oxygen consumption and myocardial work load increase. If coronary artery disease is present, the myocardial demand for increased oxygen consumption may not be met. The risks of angina, arrhythmia, or myocardial infarction indicate the need that introduction of thyroid hormone treatment of older patients should be cautious and gradual, using starting doses less than full replacement, making incremental small doses until full replacement is achieved by biochemical parameters, with assessment of tolerance before each adjustment, and with willingness to aim for less than full replacement in case of poor tolerance. Despite the need for a cautious approach in patients who may have heart disease, the long-range goal is to reduce risk for dyslipidemia, heart failure, and coronary atherosclerosis.192–198 Even younger patients sometimes may experience discomfort if replacement is introduced too quickly, experiencing myopathic symptoms that may worsen at first but eventually will resolve if treatment continues, or, rarely, cardiac symptoms.198 In case of intolerance during initiation, the risk for abandonment of treatment may be reduced if a temporary T4 dose reduction is made, with intent to work back more gradually toward full replacement. An uncommon outcome during initiation of treatment in children is increased pseudotumor cerebri.
To initiate therapy for overt hypothyroidism, patients with abrupt development of hypothyroidism or young patients may be started on full replacement doses of T4. To reduce the risk of discomfort during initiation, young patients with longstanding severe untreated hypothyroidism may be started at a dose of levothyroxine 0.05 mg/day. Older patients or those with coronary artery disease should start with levothyroxine 0.025 mg/day. Increments of levothyroxine 0.0125 to 0.025 mg for older patients are made at about 3-week intervals until it is estimated that the patient is close to full replacement, and then the free T4 and TSH levels are rechecked. After a dosage adjustment of T4 therapy, 6 weeks is necessary before biochemical reevaluation will reflect a steady-state condition. After upward titration of the dose, the average adult requirement for hypothyroidism is about 0.112 mg/day levothyroxine orally. For elderly patients the dose is lower than for younger patients,180,183 and for subclinical or early hypothyroidism, the dose of levothyroxine necessary to normalize the TSH may be as low as 0.05 to 0.075 mg/day.
It may be stated anecdotally that atrial arrhythmias are no contraindication to providing replacement therapy for hypothyroidism.195 Development of hypothyroidism during lithium or amiodarone therapy does not require drug discontinuation but may require thyroid hormone replacement.
Subclinical hypothyroidism refers to persistent TSH elevation with normal free T4 and absence of characteristic symptoms of hypothyroidism. Mild or subclinical hypothyroidism is not likely to present short-term risks to a critically ill patient. However, evidence suggests increased long-term morbidity, including heart failure, from untreated subclinical hypothyroidism.162,164 Therefore, follow-up in the ambulatory setting is appropriate to determine whether TSH elevation is persistent.
The hypothyroidism associated with medication use may remit if the initiating agent is later withdrawn, so that determination of a treatment plan for hypothyroidism in part depends upon the drug and in part depends upon the severity of symptoms and duration of intended treatment with the drug held responsible for the development of hypothyroidism (Fig. 60.4).
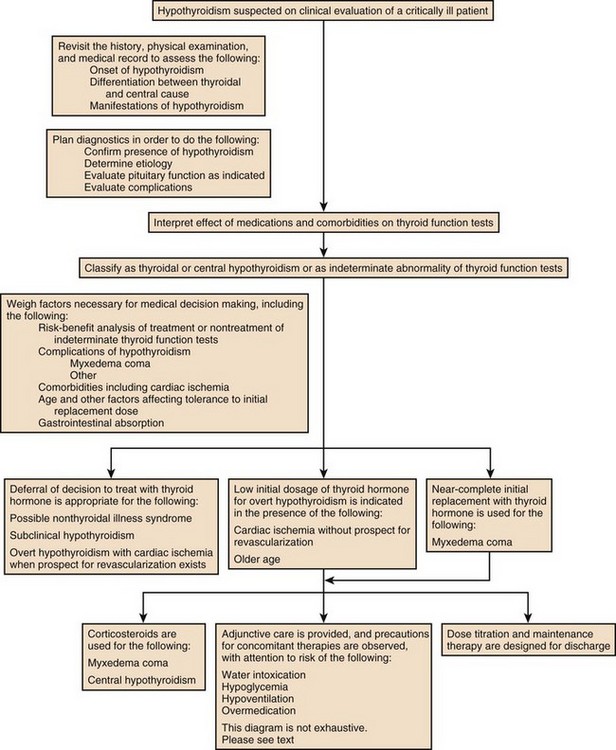
Figure 60.4 Approach to management for hypothyroidism.
Continuation of Established Thyroid Hormone Therapy During NPO Status
For prolonged NPO (nil per os, nothing by mouth) status or during continuous administration of substances that may impair absorption of levothyroxine or for other indications, intravenous levothyroxine may be provided daily to substitute for enteral administration, in reduced amount compared to the ambulatory daily dose, with subsequent monitoring.* The fractional gastrointestinal absorption of a tablet of levothyroxine has been reported to be about 81%, considerably higher than the earlier estimate of 48%.183 However, concomitant administration of other substances, including calcium carbonate, may diminish absorption.186 Intestinal malabsorption and, anecdotally, development of severe right-sided heart failure may result in unusually high dose requirements for oral levothyroxine therapy. More than half of patients receiving enteral levothyroxine may develop subclinical or overt hypothyroidism after 2 to 3 weeks if a previously established levothyroxine dose is maintained concomitantly with continuous enteral feedings.189 Owing to the long half-life of levothyroxine and delayed tissue response to dosage adjustments, manifestations of myxedema evolve only slowly after interruption of therapy, so that interruption of levothyroxine during short-term NPO status generally is inconsequential. The American Association of Clinical Endocrinologists and the American Thyroid Association (AACE-ATA) guideline recommends that “Patients resuming L-thyroxine therapy after interruption (less than 6 weeks) and without an intercurrent cardiac event or marked weight loss may resume their previously employed full replacement doses.”191 For patients whose oral intake will be curtailed for a prolonged interval, the usual enteral dose of levothyroxine should be reduced by 20% to 40% to arrive at a dose for intravenous therapy.176,183,188
Preparation of the Patient with Untreated Hypothyroidism for Emergency Surgery or Coronary Revascularization
Subclinical hypothyroidism has not been shown to increase operative risk.200 For patients with overt hypothyroidism, there is increased risk of perioperative complications such as sensitivity to analgesics and anesthesia, prolonged ventilator dependence, hypotension, water intoxication, and iatrogenic myxedema coma. Elective surgery should be deferred until euthyroidism is attained. For emergency surgery, younger patients without coronary disease should be prepared as if they already had myxedema coma, using a preoperative intravenous bolus of levothyroxine, depending on age and transport protein status, as described later (see “Myxedema Coma”), and providing hydrocortisone coverage, with other precautions as described earlier (see “Precautions in the Care of the Hypothyroid Patient”). The risk of undiagnosed coronary insufficiency has to be considered in determining the preoperative levothyroxine replacement regimen of older patients. Emergency surgery should be deferred for 24 to 48 hours after initiation of thyroid hormone treatment if possible.188,201,202
Patients who are candidates for correction of reversible myocardial ischemia generally should undergo revascularization before one attempts to treat their hypothyroidism. The risks of immediate thyroid hormone replacement before noncardiac surgery should be weighed against the probably acceptable risks of successful operation without levothyroxine pretreatment.188,194,203–212
Prognosis
New onset of angina, myocardial infarction, or sudden death may occur within days or weeks after initiation of treatment for hypothyroidism.192,193 Secondary hyperlipidemia and most clinical manifestations of juvenile hypothyroidism and adult myxedema are reversible after therapy, although some features, such as anemia, may require months for correction.
Myxedema Coma
History and Incidence
Our present-day knowledge of myxedema coma derives from isolated case reports and small retrospective series.213–234 Historically, the low doses of thyroid hormone normally used to initiate treatment of uncomplicated hypothyroidism, when administered enterally for myxedema coma, failed to prevent fatalities. The mortality rate was probably higher than 80%. In 1964 it was demonstrated that intravenous replacement with 500 µg levothyroxine, a dose calculated to nearly replete body stores of T4, improved the rate of survival.218 Liothyronine (triiodothyronine, T3) for intravenous injection later became commercially available.
Pathogenesis
Myxedema coma arising in the community can generally be divided into episodes that arise spontaneously and those that arise in connection with a precipitating illness or event. Those arising spontaneously tend to occur during the colder months of the year. Precipitating factors may include congestive heart failure, pneumonia or other infection, bleeding, administration of hypotonic fluids, sedative and analgesic drugs, or anesthesia and surgery. The particular risk of hypoventilation probably is increased by the presence of heart failure, obesity, pleural or other restrictive disease, chronic obstructive lung disease, neuromuscular disease, or exposure to drugs that reduce respiratory drive.146,147
Clinical Manifestations and Diagnosis
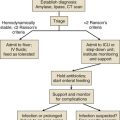
Myxedema coma presents with a constellation of findings including physical evidence of advanced hypothyroidism, stupor, bradycardia, hypotension, hypothermia, alveolar hypoventilation, obstipation, or ileus, and sometimes water intoxication or hypoglycemia.224 Patients are often elderly. The condition if untreated progresses to fatal hypotension.
In the cases of myxedema coma arising spontaneously in the community, stupor progresses over several days, and families report that the number of hours spent sleeping has gradually increased to include most of a 24-hour period. Seizures have been reported.221 In history taking it is important to ask whether the patient with suspected myxedema coma formerly was diagnosed with hypothyroidism or formerly was treated with radioactive iodine or surgery for overactive thyroid. On physical examination overt manifestations of myxedema are apparent. The patient often can be aroused and will make monosyllabic responses to questioning before lapsing back into stupor. Breathing is stertorous. Some patients with an infectious process may not have a fever. The most ominous sign of impending myxedema coma for a hypothyroid patient under inpatient observation is progressive hypothermia.
In contrast to patients with nonthyroidal illness syndrome, the laboratory evaluation will demonstrate low free T4 and high TSH in the majority of true cases of myxedema coma. Myxedema coma resulting from pituitary failure is uncommon.227
Therapeutic Alternatives
Theoretical controversy continues to exist on whether to include intravenous liothyronine in the initial treatment plan. Some authorities recommend using both hormones at the outset, especially if coexistent illnesses are present that might impede conversion of T4 to T3.227,234 Additional arguments in favor of including liothyronine relate to the delayed conversion of T4 that is seen in hypometabolic patients and the more rapid effect on tissues when T3 therapy is used.181 In combination therapy, the recommended intravenous doses are approximately 10 µg of liothyronine initially and 10 µg of liothyronine every 8 to 12 hours on the first day, combined with an initial loading dose of about 200 to 250 µg levothyroxine. This treatment is followed by approximately 100 µg of levothyroxine daily intravenously on the second day and 50 µcg levothyroxine daily thereafter. On the other hand, it has been speculated that some cases of fatality were caused by relatively high T3 levels attained early in therapy.225 Conventional treatment with levothyroxine alone has not been shown to be inferior and is generally effective.
Prognosis
Patient findings associated with fatality have included old age, cerebrovascular bleeding, or myocardial infarction during treatment213,225,226,229 or suspected coronary events after recovery.218 In a series of 11 cases the level of consciousness, Glasgow score, and APACHE II score were predictive of death.232 Treatment factors associated with fatality may include overly gradual oral regimen of replacement of thyroid hormone, high replacement doses of thyroid hormone [liothyronine (triiodothyronine, T3) doses = 75 µg/day, levothyroxine doses = 500 µg/day], or high measured levels of T3 during treatment.213,225,229 Within 6 to 36 hours most patients treated with T4 in sufficient dosage experience a rise of temperature and blood pressure; improvement of mentation; and through peripheral conversion of T4, correction of low T3 levels.
Hyperthyroidism, Thyroid Storm, Thyrocardiac Crisis, and Coronary Artery Spasm
Hyperthyroidism
Prevalence and Incidence
The age-related incidence of hyperthyroidism depends on the cause of hyperthyroidism.141–143,235,236 The prevalence of overt hyperthyroidism is between 0.5% and 2% in women, and is 10 times more common in women than in men in iodine replete areas, with an annual incidence rate of 0.4 per 1000 women and 0.1 per 1000 men.141,143 In Sweden the overall incidences of Graves’ disease, toxic multinodular goiter, and toxic adenoma were 17.7, 5.4, and 2.7 per 100,000 persons per year, respectively, but the peak age-specific incidence of toxic multinodular goiter and toxic adenoma occurred in the 80-plus age group: 31.5 per 100,000 persons per year.235 In the ambulatory setting among adult patients, the overall prevalence of subclinical hyperthyroidism (isolated TSH suppression) is 0.5% to 6.3%, with variability dependent on population under study, inclusion or exclusion of patients with thyroid disease, and definition of threshold TSH for inclusion.141–143
Pathogenesis
In susceptible individuals, especially those from iodine-deficient regions of the world or with preexisting nodular thyroid disease, hyperthyroidism can be produced by iodine (Jod-Basedow phenomenon). Hyperthyroidism can be produced by glandular destruction, as in thyroiditis, characterized by having low radioactive iodine uptake. Common causes of low-uptake hyperthyroidism include silent thyroiditis, postpartum thyroiditis, subacute thyroiditis, and exogenous thyroid hormone. Substances implicated in drug-induced forms of destructive thyroiditis causing hyperthyroidism include amiodarone, lithium, interferon-α, interleukin 2, iodine, and iodinated contrast agents.236 Two different mechanisms of hyperthyroidism are possible during amiodarone therapy: iodine-induced thyrotoxicosis and drug-induced thyroiditis.
In general, intense activation of the TSH receptor complex results in an increased ratio of T3 to T4 resulting from direct thyroidal T3 secretion. The augmentation of thyroidal T3 release of hyperthyroid patients may result in part from enhancement of the thyroidal type 2 deiodinase activity.237 The altered ratio of T3 to T4 is not observed in destructive hyperthyroidism.238
Clinical Manifestations
Intubation and central lines may prevent adequate palpation of the thyroid. If thyroidal bruit is detected on auscultation of the upper poles of the lateral lobes of the thyroid in a hyperthyroid patient, the classification of the cause of hyperthyroidism as Graves’ disease essentially is assured. The eyes, nails, and skin should be examined for evidence of orbitopathy, onycholysis, fine skin quality (smooth elbows), or dermopathy (“pretibial myxedema”). The remainder of the physical examination is likely to yield findings that are suggestive of hyperthyroidism but nonspecific, such as hyperhidrosis, atrial arrhythmia, hyperdynamic heart, precordial lift, systolic ejection murmur, S3 gallop, restlessness, hyperkinesia, agitation, or briskness of Achilles reflexes. In severe cases, adrenal reserve may be insufficient.239 A different spectrum of symptoms and signs has been reported in the elderly, who may appear apathetic or depressed, and among whom the average number of thyrotoxic symptoms may be as low as two, with dominance of weight loss, proximal myopathy, tremor, and cardiac manifestations such as heart failure, sinus tachycardia, or atrial fibrillation.240–243
Diagnostic Approach
T4 Hyperthyroidism
The finding of high T4 without high T3, although atypical of hyperthyroidism, sometimes occurs after surgery, during exposure to substances that block conversion of T4 to T3, among the elderly or the critically ill, or sporadically. During the acute illness, differentiation between euthyroid hyperthyroxinemia and hyperthyroidism may be difficult.244–248
Approach to Management
For those having subclinical hyperthyroidism, the 2011 Hyperthyroidism Management Guidelines of the American Thyroid Association and the American Association of Clinical Endocrinologists suggest that persistent findings of TSH at 0.1 µIU/mL or below over 3 to 6 months should lead to determination of cause, with a decision for treatment or observation determined by patient characteristics.162,164,236
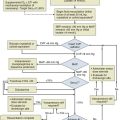
For those critically ill patients having overt hyperthyroidism classifiable as a probable high-uptake form, generally treatment should consist of antithyroid medication (Fig. 60.5). Management of most aspects of overt hyperthyroidism are addressed in the guideline, including many of the issues important to critical care medicine.236 The drugs used in treatment of hyperthyroidism include the thionamides propylthiouracil and the longer-acting drug methimazole.249–259 Thionamides inhibit the coupling of moieties on TG and organification of iodide, thus preventing storage and synthesis of thyroid hormone. By inhibiting 5′-monodeiodination of T4 by the D1 enzyme, propylthiouracil reduces peripheral conversion of T4 to T3.249,260 To initiate therapy it is common practice to start with a relatively higher dose than will be required for maintenance and to choose a dosage on the basis of the apparent clinical severity of hyperthyroidism and size of the thyroid gland. Methimazole can cause a reversible cholestatic picture of hepatic injury. Propylthiouracil, however, can cause irreversible hepatic necrosis resulting in fatality.256 Both drugs can cause neutropenia or agranulocytosis.261 A rare adverse effect of propylthiouracil is antineutrophil cytoplasmic antibody–positive vasculitis. Methimazole taken in the first trimester of pregnancy has been associated with aplasia cutis and choanal atresia of the neonate. When thionamide drug therapy is chosen, except for hyperthyroidism during the first trimester of pregnancy, for thyroid storm, or for patients having a history of a minor reaction to methimazole, because of the difference in severity of potential hepatotoxicity, the preferred drug for virtually every patient is methimazole rather than propylthiouracil.236 For uncomplicated hyperthyroidism the initial daily dosage might be propylthiouracil 50 to 150 mg every 8 hours or methimazole 10 mg twice daily. After control is achieved, methimazole usually is converted to once-daily therapy and, after gradual dose reduction, sometimes is given in doses as little as 5 mg daily. Once control is achieved, methimazole may be given as a single daily dose. Propylthiouracil may be reduced to as little as 50 mg two or three times daily but must be administered in a divided dosage to prevent escape.
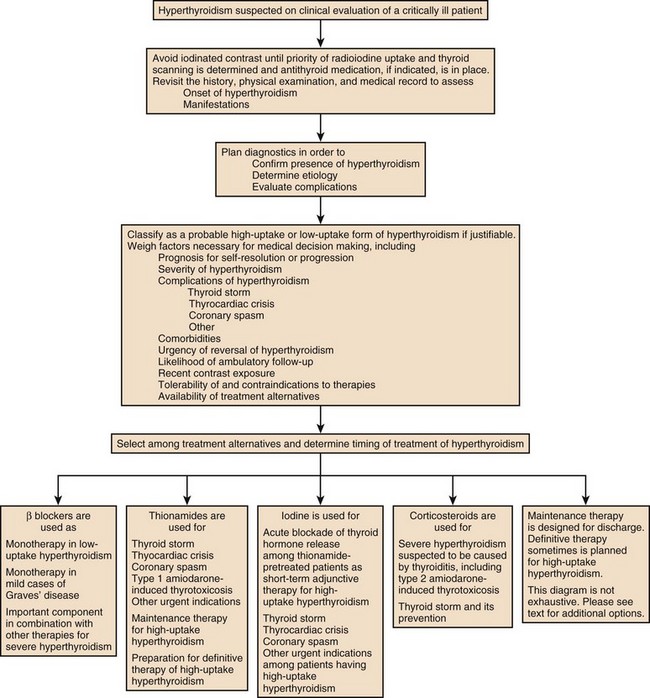
Figure 60.5 Approach to management for hyperthyroidism.
During hyperthyroidism beta-blocker therapy is the preferred method of controlling tachycardia and peripheral manifestations of sensitivity to catecholamines. Among the beta blockers, there is the greatest experience with propranolol. The metabolism of propranolol is enhanced, and blood levels are highly variable in hyperthyroidism. Most patients requiring enteral propranolol for hyperthyroidism should begin with 10 to 40 mg every 6 hours. Other beta blockers have been used, and some advocate cardioselective agents.262–278 Definitive treatment of hyperthyroidism consists of radioactive iodine or surgery. Use of radioiodine therapy is inappropriate for low-uptake hyperthyroidism.
Classification and Management of Amiodarone-Induced Thyrotoxicosis
Hyperthyroidism may occur in up to 6% of amiodarone-treated patients in iodine-sufficient regions, and in up to 9.6% or more of treated patients in iodine-deficient regions.30,236 A regimen recommended for detection of thyroid dysfunction during amiodarone therapy is to check before and at 1 and 3 months following initiation, and then at 3- to 6-month intervals thereafter.236 Amiodarone-induced thyrotoxicosis may be recognized by characteristic clinical and laboratory findings, including tachycardia, weight loss, proximal muscle weakness, low TSH, and high or high normal T3 and free T3.279–292 Classification as type 1 or type 2 amiodarone-induced thyrotoxicosis is based on the mechanism of the hyperthyroidism. Type 1 amiodarone-induced thyrotoxicosis is a Jod-Basedow type of hyperthyroidism consequent to the high iodine content of amiodarone, likely to be associated with preexisting nodular goiter or, less commonly, Graves’ disease. On color flow Doppler examination, the gland may appear hypervascular. Despite the iodine exposure from amiodarone, the radioiodine uptake, though relatively low, may be measurable, and technetium MIBI (methoxyisobutylisonitrile) scanning may show areas of uptake. Type 2 amiodarone-induced thyrotoxicosis, now by far the more common type, results from a drug-related destructive thyroiditis, often occurring in a patient having no prior history of known thyroid disease and not having thyroidal enlargement, sometimes recognized by markedly diminished vascularity on color flow Doppler examination.284,289 Radioiodine uptake is negligible or absent. When the differentiation can be made, type 1 and type 2 amiodarone-induced thyrotoxicosis are addressed by different management strategies.291 In type 1 amiodarone-induced thyrotoxicosis, thionamide therapy is used. Because there is unlikely to be a full response during continuation of amiodarone, the usual recommendation is to interrupt amiodarone for type 1 amiodarone-induced thyrotoxicosis. Once the iodine stores from the previous treatment have dissipated, radioactive iodine may be administered to ablate the thyroid in preparation for reinstitution of amiodarone, especially if hyperthyroidism still is demonstrable. Sometimes surgery is the best option available for the treatment of resistant cases of type 1 amiodarone-induced thyrotoxicosis.279 Postoperatively amiodarone may be reinstituted.
For type 2 amiodarone-induced hyperthyroidism, the course of the hyperthyroidism may be transitory, with subsequent development of hypothyroidism among some patients.31,32 Glucocorticoids promote resolution of type 2 amiodarone-induced thyrotoxicosis.280,290 During glucocorticoid therapy it is not necessary to interrupt amiodarone.288,292 A typical starting dose is 30 mg prednisone daily.292 Because there may be mixed types or indeterminate presentations in which the classification of the amiodarone-induced thyrotoxicosis may be uncertain, in actual practice thionamides and glucocorticoids both may be used during initiation of treatment. In this country, perchlorate, another option, is not available.
Preparation of the Thyrotoxic Patient for Emergency Nonthyroidal Surgery
Elective surgery should be deferred until euthyroidism is attained. In deciding whether to operate, the risk of perioperative fatality due to hyperthyroidism has to be weighed against the strength of the medical indications for immediate surgery. Before emergency surgery, the risk of thyroid storm obligates the caregiver to prepare the patient with antithyroid treatment.293–295 For newly diagnosed patients with endogenous hyperthyroidism requiring rapid preparation for emergency surgery, preoperatively propylthiouracil 200 mg every 4 hours or methimazole 20 mg every 4 hours has been recommended.294 At least 2 hours after the first dose of propylthiouracil, the patient should receive iodide. Ipodate or iopanoic acid (as described later in the section on treatment of thyroid storm) are potentially useful but unavailable in many countries. In the absence of contraindications, propranolol orally in a dosage up to 20 to 40 mg four times per day or more should be started. The regimen should be continued in the immediate postoperative period by use of a nasogastric tube if necessary. By the time of discharge methimazole would be the preferred thionamide, and the dose should be tapered. Glucocorticoids also may have a role in preoperative preparation, initially 100 mg hydrocortisone every 8 hours.
Alternatives for Treatment
For patients who cannot take oral medications, antithyroid drugs can be prepared for rectal administration.251,252 Patients with a history of minor skin reactions to one thionamide may be treated with the other. Patients with a history of hepatocellular jaundice during propylthiouracil therapy may be treated with methimazole.254,257 Lithium has been used for the control of hyperthyroidism in patients unable to use propylthiouracil or methimazole, starting at a dose of 300 mg twice per day.254,296–299 The ability of the radiographic contrast agents iopanoic acid and ipodate to inhibit thyroid hormone release, peripheral conversion of T4, and possibly nuclear binding of T3 has been exploited in certain hyperthyroid states including cases of destructive hyperthyroidism and in thionamide intolerance.300–305 The use of radiographic contrast agents usually should be adjunctive to thionamides and beta-blocker therapy. Instances of escape after prolonged use of iodinated substances have been reported.302 Therefore, if a radiographic contrast agent is used as an antithyroid medication without antecedent and concomitant thionamide medication, some patients will require thyroidectomy as soon as reasonable control has been obtained.295 Recommended daily doses are ipodate 500 to 1000 mg orally or iopanoic acid 500 mg twice daily. These iodinated oral cholecystographic agents have not been clinically available in the United States recently.
For patients with contraindications to the use of beta blockers, older regimens included guanethidine306 or reserpine,307 which are seldom used at present. For control of heart rate in patients with reversible airway disease, diltiazem is an alternative to beta blockers.308,309 Iodine has been used to reduce vascularity of the thyroid and inhibit thyroid function. Brief treatment with iodine may hasten clinical recovery by acutely blocking release of thyroid hormone (see section “Thyroid Storm and Thyrocardiac Crisis”). Thionamide administration must precede use of iodine, and must be continued during treatment. Accidental gastrointestinal injuries have been reported.310–312
Prognosis
Socioeconomic factors coupled with a lapse in care may account for some of the long-term morbidity and mortality risks associated with hyperthyroidism.313 Some cardiovascular abnormalities may persist after treatment of hyperthyroidism. Some but not all epidemiologic studies suggest that an increased overall mortality risk is associated with a history of hyperthyroidism, even after radioiodine treatment.314–322
Thyroid Storm
Thyroid storm is a potentially lethal complication of hyperthyroidism, managed in the critical care setting.231,236,323–327 In a hyperthyroid patient the diagnosis of this emergency is based on the clinical presentation.*
History and Incidence
In the present day, thyroid storm is seen after the development of intercurrent illness or infection, operative intervention for nonthyroidal illness, or infrequently after exposure to medications that exacerbate either the severity of hyperthyroidism or patient sensitivity to thyroid hormone excess. Initially this entity was recognized as a complication of thyroidectomy for hyperthyroidism that had been performed in unprepared patients.328 In an early series, two thirds of the patients died329; postmortem examinations were not illuminating. After it was recognized that patients should be rendered euthyroid before thyroidectomy, the incidence of storm after thyroidectomy declined, and the literature began to focus on nonsurgical precipitating factors.331,332 As early as 1960, after the use of reserpine and glucocorticoids was added to therapy with antithyroid medication and iodide, it was reported that the mortality rate from thyroid storm had been reduced from 60% or 70% nearly to 25%.332 In 1966 the importance of initiating thionamide therapy before iodide administration was emphasized.334 By employing guanethidine as reported in 1969306 or reserpine therapy reported in 1970,307 the mortality rate of thyroid storm in small series was as low as 7% and 0%, respectively. By the mid-1970s the use of propranolol had largely replaced earlier methods of attaining sympathetic blockade.262,264,266,337
Thyroid storm is a relatively infrequent reason for admission to critical care units. Even before the existence of modern antithyroid therapy, the estimated incidence of thyroid storm among patients hospitalized for hyperthyroidism was only 7%.332
Pathogenesis
Graves’ hyperthyroidism is the most common thyroid disorder identified with thyroid storm, but storm has also been associated with toxic multinodular goiter, amiodarone-induced hyperthyroidism,282 and rarely exogenous thyroid hormone overdosage.330,345,347,363 Precipitating factors precede the development of thyroid storm in the majority of cases. Identifiable events accompanying thyroid storm may include infection,335 surgery,328,329,335,372 administration of radioactive iodine,331,341,357 recent exposure to iodine or iodinated contrast dye, or withdrawal of inorganic iodide,329,354 trauma,48,361,368 parturition,333 diabetic ketoacidosis,358,366 status epilepticus and stroke,351 pulmonary embolism,332 pseudoephedrine administration,346 other precipitating events, discontinuation of antithyroid medication,331 or a lapse in care.340 In the absence of thionamide therapy, administration of iodine-containing compounds floods the gland with iodine, but inhibition of hormone release by iodine may be incomplete or temporary, and thyroid storm may occur during continued administration of iodine.329 Recently instituted or incompletely effective thionamide therapy does not necessarily protect against storm induced by radiation thyroiditis.344 Reliance on lithium297 or propranolol265 as monotherapy for preoperative preparation may be responsible for thyroid storm.
In thyroid storm the T3 and total T4 levels are not different from levels in uncomplicated cases of hyperthyroidism, but the dialyzable fraction of T4 (the percentage that is free) is higher.338,339 This finding has been taken to support the idea that the medical illness may cause acute unbinding of thyroid hormone from transport proteins. Relative adrenal insufficiency may be present.
Clinical Manifestations and Diagnosis
The definition of thyroid storm has come to include a constellation of fever greater than 100° F, tachycardia out of proportion to fever, and exaggerated manifestations of thyrotoxicosis affecting at least two other of the following systems: cardiac, gastrointestinal, or neurologic.332 A clinical scoring system for identifying thyroid storm has been endorsed in which identification of a precipitating factor is one diagnostic criterion.236,324 The system also identifies a range of scores that is used to classify a patient’s presentation as impending thyroid storm. Cardiac findings may include marked tachycardia out of proportion to fever, atrial arrhythmia, heart failure, shock, ventricular tachycardia, and ventricular fibrillation.352,364,369,371 Gastrointestinal symptoms include hyperdefecation, diarrhea, vomiting, abdominal pain, acute abdomen,349,350,370 and jaundice or liver failure.353,355,356,375 Hepatic failure usually is a result of right-sided heart failure. Rhabdomyolysis may occur.342 Neurologic features include tremulousness, agitation, hyperkinesia, muscle weakness, delirium or psychosis, apathy, prostration, seizures, or obtundation. Brisk contraction and relaxation time often are observed when deep tendon reflexes are elicited. Fever is the hallmark of the condition, sometimes as high as 106° F. As a practical matter, even if another underlying cause of fever is identified, any febrile patient (temperature >100° F) who has known hyperthyroidism with marked tachycardia or exaggerated manifestations of hyperthyroidism is best treated as having impending thyroid storm.
No specific laboratory finding defines the presence of thyroid storm, but rather, thyroid storm is defined by the characteristic clinical picture, which will be accompanied by laboratory findings of TSH suppression and generally free T4 elevation. Measurement of T3 is not necessary unless free T4 levels are normal.336 Rarely, in the presence of coexisting illness, the T4 may be elevated but total T3 may be normal.247
Approach to Management
Although thionamides block the synthesis of thyroid hormone, this effect will not benefit the patient until several weeks later, when thyroid glandular stores of preformed hormone have been discharged. To block hormone release acutely, iodide is administered. Thionamide pretreatment with a loading dose is essential to induce a biosynthetic blockade that will prevent flooding of the gland with iodide, with the attendant future risk of exacerbation of hyperthyroidism. Glucocorticoid therapy is employed to correct potential relative adrenal insufficiency.239 Beta blockers combat cardiac sensitivity to catecholamines and reduce central and peripheral neurologic manifestations of hyperthyroidism. Propylthiouracil, glucocorticoids, and propranolol block conversion of T4 to T3.
Treatment of thyroid storm requires thionamide, beta blocker, and glucocorticoid doses higher than usually required for ordinary care. There is no comparative evidence that would support specific titration rules or a preference among differing dose ranges that have been recommended for thionamides and beta blockers.231,236,323–326,337 Nevertheless, although incomplete response to beta blockers could require upward dose titration, caution should be exercised in the initial dosing, and tolerance to the initial dose should be demonstrated.
A reasonable starting regimen is an initial propylthiouracil loading dose of 600 mg, and thereafter a continued dose of 200 mg every 4 hours, or 1200 mg daily in divided dosage, given orally or by nasogastric tube. The literature suggests that the initial propylthiouracil loading dose may be as high as 1000 mg, with a continued dose of 250 mg every 4 hours, or 1500 mg daily. Propylthiouracil inhibits peripheral conversion of T4 to T3 and therefore is favored over methimazole in thyroid storm. Alternatively, methimazole 60 to 80 mg per day in divided dosage may be used.324,325,337 These doses are reduced as the patient begins to respond. Beginning 1 to 2 hours after the loading dose of propylthiouracil or methimazole, saturated solution of potassium iodide, 5 drops every 6 hours, is given, or Lugol’s solution, 30 drops daily in divided dosage three to four times a day. Hydrocortisone is administered in a dose of 100 mg every 8 hours. Beta-blocker therapy is considered to be one of the mainstays of therapy, useful for control of tachyarrhythmias and other manifestations of the characteristic increased sensitivity to catecholamines. Although the literature on beta-blocker therapy during thyroid storm acknowledges use of or requirement for extremely high doses in some cases, nevertheless adverse effects including hypotension or cardiorespiratory arrest also may be seen in conjunction with beta-blocker therapy.277,278 For patients having evidence of heart failure or hypotension or developing these findings during treatment, beta blockers must be given during hemodynamic monitoring with great caution, and alternative methods of controlling heart rate and rhythm should be considered. Despite evidence that extremely high doses may be required by some patients, a starting oral dose of propranolol as low as 20 to 40 mg every 6 hours has been recommended.325 The propranolol dose requirement may be as high 480 mg daily, given as 60 to 80 mg propranolol every 4 hours, or 80 to 120 mg every 6 hours.231,323,326 With monitoring of blood pressure and rhythm, while awaiting the effects of orally administered propranolol, 0.5 to 1 mg propranolol intravenously may be cautiously provided; the dose may be increased to 2 to 3 mg over 15 minutes if tolerated.324 Digoxin is used for atrial fibrillation. The underlying cause of thyroid storm must be identified and treated, cultures made, and in general empiric antibiotics may be considered until infection is excluded. Because salicylates may promote unbinding of thyroid hormone from transport proteins, for control of fever, acetaminophen is preferred, and cooling blankets may be required. Hemodynamic monitoring should be provided as appropriate. Fluid and electrolyte replacement are necessary, and administration of vitamins is prudent.
Therapeutic Alternatives
For thyroid storm propylthiouracil normally is administered enterally, but the rectal route for administration has been described.253,259 Lithium is not a substitute for thionamide therapy unless unacceptable intolerance to thionamide therapy has been demonstrated. The initial dose of lithium in this setting is 300 mg every 6 hours.325 The route of administration of iodide may be sublingual or rectal by retention enemas.253,350 Ipodate therapy 500 mg once or twice daily and iopanoic acid are effective, but these agents presently have become unavailable in many countries.305 For patients at risk of cardiac decompensation, the rapid-acting drug esmolol may replace propranolol as initial intravenous therapy, used while awaiting the effects of orally administered cautious doses of propranolol.275 Adjuncts to therapy, reserved for resistant cases, include peritoneal dialysis, plasma exchange, or plasmapheresis.* Thyroidectomy also has been used successfully for iodine-induced storm and failure of medical management.354,359,362
Prognosis
Clinical markers suggesting a poor prognosis include delay in therapy, coma, jaundice, and shock. Normalization of temperature, drop of heart rate, and improved mental status are favorable signs. With treatment as described earlier, a sharp reduction of circulating thyroid hormone concentration is observable by the second hospital day, but it may be several days before adequate stabilization of clinical manifestations is seen.325
Thyrocardiac Crisis and Coronary Artery Spasm
In most cases of overt hyperthyroidism and in thyroid storm, cardiac manifestations are one component of a multisystem presentation. Chest discomfort is not an uncommon complaint among patients with hyperthyroidism, even among the young, and usually has benign significance. Characteristic anginal symptoms are uncommon. Sometimes, however, cardiac manifestations dominate the clinical presentation, especially in the elderly or in patients whose cardiac events emerge suddenly.* Whereas the full evolution of most manifestations of hyperthyroidism emerge over an extended period, the sudden onset of thyrocardiac effects may complicate any form of thyrotoxicosis, even fleeting or recent-onset thyrotoxicosis, such as that resulting from subacute thyroiditis or overdosage of thyroid hormone, and may occur among young patients. In the absence of other criteria permitting a diagnosis of thyroid storm, thyrocardiac symptoms may present with a medical crisis.
Prevalence and Incidence of Thyrocardiac Disease
Thyrocardiac crises without storm are relatively more common than thyroid storm.376–379 Preexisting heart disease and age older than 60 are risk factors for manifestations of thyrocardiac disease.377,379,380 Among 462 patients with hyperthyroidism who had no associated organic heart disease and who were referred for radioactive iodine, a 1958 study reported the prevalence to be atrial fibrillation 10%, congestive heart failure with atrial fibrillation 5%, and congestive heart failure with sinus rhythm 0.6%.377 In a study of unselected hyperthyroid patients, the prevalence was atrial fibrillation 9% and congestive heart failure 6%.379 In the Framingham Heart Study, for persons with TSH levels 0.1 µIU/mL or less compared with persons with normal TSH, the relative risk of atrial fibrillation was 3.1 (95% confidence interval, 1.7 to 5.5). Among patients with TSH suppression of less severity (i.e., >0.1 µIU/mL), the risk for atrial fibrillation was comparable with the risk among people with normal TSH.381 After emergency presentation with atrial fibrillation, screening for hyperthyroidism may be justified by the possibility of detecting unrecognized cases of hyperthyroidism.382,385 No body of literature exists on predictors of myocardial infarction, ventricular arrhythmia, or sudden death. The occurrence of coronary ischemic events during hyperthyroidism had been considered to be surprisingly low. In an early series of unselected hyperthyroid patients, 2 of 200 died as a result of myocardial infarction.379 In one study, high levels of T3 on admission were associated with the presence of a coronary event and also predicted subsequent coronary events over a 3-year follow-up period.126 Isolated case reports document the occurrence of sudden death or coronary spasm even among young patients.
Pathophysiology
Structural and regulatory proteins in the heart are encoded by genes regulated by T3.115 Despite the low levels of circulating catecholamines, the physiology of a hyperthyroid patient resembles a hyperadrenergic state.386 Sinus tachycardia is the most common arrhythmia in uncomplicated hyperthyroidism. On examination there are characteristically tachycardia at rest, widened pulse pressure, and systolic hypertension. The patient with hyperthyroid heart disease has a high left ventricular ejection fraction at rest, high cardiac output, subnormal response to exercise, and sometimes rate-related heart failure.376 Physiologic abnormalities include decreased systemic vascular resistance; increased blood volume; increased work load; increased contractility; shortening of systolic contraction and diastolic relaxation times; and reduction of contractile reserve. Hyperthyroidism reduces serum cholesterol.
Clinical Manifestations
Thyrocardiac crises are potentially seen without the complete picture of thyroid storm—in particular, without fever. Thyrocardiac crises otherwise can be divided into several major problems, including atrial arrhythmias and thromboembolism*; heart failure and manifestations of cardiomyopathy376,380,399–410; the rare complications of conduction disturbance,411 ventricular arrhythmia,412 and sudden death413–415; and coronary artery spasm. Coronary artery spasm is increasingly recognized as a potentially reversible complication of uncontrolled hyperthyroidism, sometimes seen in young women, infrequently complicated by myocardial infarction.416–428
Approach to Management
If a thyrocardiac crisis is present, β-blockade is part of the therapy unless contraindications are present. In treating atrial fibrillation digoxin is used, and for congestive heart failure digoxin and furosemide are used. If some degree of systolic cardiac dysfunction may be present, β-blockade may result in hypotension and should be instituted with careful monitoring. For patients with tachycardia-related congestive heart failure, the use of beta blockers in doses equivalent to 20 mg propranolol every 6 hours may be well tolerated, and the dose may be titrated up to 40 or 80 mg every 6 hours. However, in low-output heart failure or heart disease of other causes complicated by hyperthyroidism, administration of beta blockers must be approached with caution if these agents are used at all.380 Diltiazem also must be used with caution because it may have negative inotropic effects. Thionamides should be given in relatively high dosage, such as such as 20 mg methimazole daily every 8 hours. Two hours after the first dose of methimazole, SSKI (potassium iodide oral solution) 5 drops every 6 hours may be started. Iodide is tapered to 2 drops three times per day before discharge and is discontinued after 7 to 14 days. Patients in atrial fibrillation resulting from hyperthyroidism generally should receive anticoagulation therapy according to the same criteria as other patients. For thyrocardiac crises, treatment with glucocorticoids is seldom indicated. A reduction of methimazole dose usually is indicated once the initial response is assured.
After medical stabilization, definitive therapy with radioactive iodine usually is offered to patients who have experienced thyrocardiac symptoms.378 Radiation thyroiditis or interruption of antithyroid medication to permit radioiodine administration can cause patient destabilization. Pretreatment with thionamide medication usually should be offered for about 2 months in the absence of contraindications. For patients in atrial fibrillation, the frequency of spontaneous conversion to sinus rhythm is greater if relatively large ablative doses of radioactive iodine are used, sufficient to bring about prompt development of hypothyroidism.391 Thionamide therapy must be withheld temporarily to permit radioiodine treatment, but if antithyroid medication is reinstituted after radioiodine treatment, any recurrence of hyperthyroidism is usually corrected within 4 to 6 weeks.
Patients who do not spontaneously convert to sinus rhythm generally should be rendered euthyroid before elective cardioversion is attempted; otherwise, there is a greater risk of relapse of atrial fibrillation after cardioversion. It has been suggested that because no spontaneous conversions occurred more than 16 weeks after attainment of euthyroidism, the ideal timing for elective cardioversion is at that time.390,396 Hyperthyroid patients are more sensitive to warfarin than others. The immediate risks of anticoagulation sometimes must be weighed against the desirability of postponing elective cardioversion. The guidelines of the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy recommended that because the incidence of thromboembolic events in patients with thyrotoxic atrial fibrillation appeared similar to other causes of atrial fibrillation, antithrombotic therapies should be chosen based on the presence of validated stroke risk factors.398
Prognosis
Even in young adult patients, atrial fibrillation may be complicated by thromboembolism resulting in fatal or disabling cerebrovascular accidents.387,388,395
About 37% to 61% of patients may experience spontaneous conversion of atrial fibrillation after attainment of euthyroidism, mostly within 6 weeks.377,390 Absence of congestive heart failure is a favorable prognostic predictor of spontaneous conversion. Other patients may be successfully cardioverted. The absence of other heart disease, short duration of atrial fibrillation, and younger age predict successful conversion and maintenance of sinus rhythm.
As a result of treatment of hyperthyroidism, a patient with heart failure caused by thyrotoxic heart disease experiences improved ability to augment cardiac output during exercise.399 With treatment of thyrotoxicosis alone, about 41% of patients with congestive heart failure and thyrotoxicosis who receive radioactive iodine experience relief of heart failure.377 For the rare patient having hyperthyroidism and anginal chest pain caused by coronary artery spasm, isolated published case reports emphasize reversibility of anginal symptoms after successful treatment of hyperthyroidism.
Lipid status should be reevaluated after correction of hyperthyroidism.
References
1. Cheng, SY, Leonard, JL, Davis, PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010; 31(2):139–170.
2. Hiroi, Y, Kim, HH, Ying, H, et al. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006; 103(38):14104–14109.
3. Jansen, J, Friesema, EC, Milici, C, Visser, TJ. Thyroid hormone transporters in health and disease. Thyroid. 2005; 15(8):757–768.
4. Bianco, AC, Salvatore, D, Gereben, B, et al. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002; 23(1):38–89.
5. Gereben, B, Zavacki, AM, Ribich, S, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008; 29(7):898–938.
6. Benvenga, S. Thyroid hormone transport proteins and the physiology of hormone binding. In: Braverman LE, Cooper DS, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2012:93–103.
7. Zhou, A, Wei, Z, Read, RJ, Carrell, RW. Structural mechanism for the carriage and release of thyroxine in the blood. Proc Natl Acad Sci U S A. 2006; 103(36):13321–13326.
8. White, GH, Walmsley, RN. Can the initial clinical assessment of thyroid function be improved? Lancet. 1978; 2(8096):933–935.
9. Attia, J, Margetts, P, Guyatt, G. Diagnosis of thyroid disease in hospitalized patients: A systematic review. Arch Intern Med. 1999; 159(7):658–665.
10. Simons, RJ, Simon, JM, Demers, LM, Santen, RJ. Thyroid dysfunction in elderly hospitalized patients. Effect of age and severity of illness. Arch Intern Med. 1990; 150(6):1249–1253.
11. Small, M, Buchanan, L, Evans, R. Value of screening thyroid function in acute medical admissions to hospital. Clin Endocrinol. 1990; 32(2):185–191.
12. DeGroot, LJ, Mayor, G. Admission screening by thyroid function tests in an acute general care teaching hospital. Am J Med. 1992; 93(5):558–564.
13. Stockigt, JR. Free thyroid hormone measurement. A critical appraisal. Endocrinol Metab Clin North Am. 2001; 30(2):265–289.
14. Sapin, R, d’Herbomez, M. Free thyroxine measured by equilibrium dialysis and nine immunoassays in sera with various serum thyroxine-binding capacities. Clin Chem. 2003; 49(9):1531–1535.
15. UK Guidelines for the Use of Thyroid Function Tests. www.acb.org.uk, 2006. [Accessed on 03/24/12 available at].
16. Dufour, DR. Laboratory tests of thyroid function: Uses and limitations. Endocrinol Metab Clin North Am. 2007; 36(3):579–594.
17. Soldin, OP. Measuring serum thyroid-stimulating hormone, thyroid hormones, thyroid-directed antibodies, and transport proteins. In: Braverman LE, Cooper DS, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2012:279–297.
18. Spencer, C, Eigen, A, Shen, D, et al. Specificity of sensitive assays of thyrotropin (TSH) used to screen for thyroid disease in hospitalized patients. Clin Chem. 1987; 33(8):1391–1396.
19. Chopra, IJ, Solomon, DH, Huang, TS. Serum thyrotropin in hospitalized psychiatric patients: Evidence for hyperthyrotropinemia as measured by an ultrasensitive thyrotropin assay. Metabolism. 1990; 39(5):538–543.
20. Nicoloff, JT, Spencer, CA. Clinical review 12: The use and misuse of the sensitive thyrotropin assays. J Clin Endocrinol Metab. 1990; 71(3):553–558.
21. Sapin, R, Schlienger, JL, Kaltenbach, G, et al. Determination of free triiodothyronine by six different methods in patients with non-thyroidal illness and in patients treated with amiodarone. Ann Clin Biochem. 1995; 32(Pt 3):314–324.
22. Faber, J, Siersbaek-Nielsen, K. Serum free 3,5,3′-triiodothyronine (T3) in non-thyroidal somatic illness, as measured by ultrafiltration and immunoextraction. Int J Clin Chem. 1996; 256(2):115–123.
23. Chopra, IJ. Simultaneous measurement of free thyroxine and free 3,5,3′-triiodothyronine in undiluted serum by direct equilibrium dialysis/radioimmunoassay: Evidence that free triiodothyronine and free thyroxine are normal in many patients with the low triiodothyronine syndrome. Thyroid. 1998; 8(3):249–257.
24. Carella, C, Mazziotti, G, Amato, G, et al. Clinical review 169: Interferon-alpha-related thyroid disease: Pathophysiological, epidemiological, and clinical aspects. J Clin Endocrinol Metab. 2004; 89(8):3656–3661.
25. Tomer, Y, Blackard, JT, Akeno, N. Interferon alpha treatment and thyroid dysfunction. Endocrinol Metab Clin North Am. 2007; 36(4):1051–1066.
26. Kleiner, J, Altshuler, L, Hendrick, V, Hershman, JM. Lithium-induced subclinical hypothyroidism: Review of the literature and guidelines for treatment. J Clin Psychiatry. 1999; 60(4):249–255.
27. Miller, KK, Daniels, GH. Association between lithium use and thyrotoxicosis caused by silent thyroiditis. Clin Endocrinol. 2001; 55(4):501–508.
28. Rhee, CM, Bhan, I, Alexander, EK, Brunelli, SM. Association between iodinated contrast media exposure and incident hyperthyroidism and hypothyroidism. Arch Intern Med. 2012; 172(2):153–159.
29. Braverman, LE. Effects of iodine on thyroid function in man. Trans Am Clin Climatol Assoc. 1991; 102:143–151.
30. Amico, JA, Richardson, V, Alpert, B, Klein, I. Clinical and chemical assessment of thyroid function during therapy with amiodarone. Arch Intern Med. 1984; 144(3):487–490.
31. Roti, E, Minelli, R, Gardini, E, et al. Thyrotoxicosis followed by hypothyroidism in patients treated with amiodarone. A possible consequence of a destructive process in the thyroid. Arch Intern Med. 1993; 153(7):886–892.
32. Alyan, O, Arda, K, Ozdemir, O, et al. Differential diagnosis and clinical course of amiodarone-induced thyroid dysfunction. Med Sci Monit. 2003; 9(9):PI117–122.
33. Trifanescu, R, Fica, S, Barbu, C, et al. Amiodarone-induced thyroid dysfunction in cardiac patients from areas with iodine deficiency. Rom J Intern Med. 2004; 42(3):595–605.
34. Basaria, S, Cooper, DS. Amiodarone and the thyroid. Am J Med. 2005; 118(7):706–714.
35. Martino, E, Safran, M, Aghini-Lombardi, F, et al. Environmental iodine intake and thyroid dysfunction during chronic amiodarone therapy. Ann Intern Med. 1984; 101(1):28–34.
36. Desai, J, Yassa, L, Marqusee, E, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med. 2006; 145(9):660–664.
37. Mannavola, D, Coco, P, Vannucchi, G, et al. A novel tyrosine-kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Metab. 2007; 92(9):3531–3534.
38. Sherman, SI, Wirth, LJ, Droz, JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med. 2008; 359(1):31–42.
39. Vetter, ML, Kaul, S, Iqbal, N. Tyrosine kinase inhibitors and the thyroid as both an unintended and an intended target. Endocr Pract. 2008; 14(5):618–624.
40. Wolter, P, Stefan, C, Decallonne, B, et al. The clinical implications of sunitinib-induced hypothyroidism: A prospective evaluation. Br J Cancer. 2008; 99(3):448–454.
41. Illouz, F, Laboureau-Soares, S, Dubois, S, et al. Tyrosine kinase inhibitors and modifications of thyroid function tests: A review. Eur J Endocrinol. 2009; 160(3):331–336.
42. Carter, JN, Eastman, CJ, Corcoran, JM, Lazarus, L. Effect of severe, chronic illness on thyroid function. Lancet. 1974; 2(7887):971–974.
43. Bermudez, F, Surks, MI, Oppenheimer, JH. High incidence of decreased serum triiodothyronine concentration in patients with nonthyroidal disease. J Clin Endocrinol Metab. 1975; 41(1):27–40.
44. Kosowicz, J, Malczewska, B, Czekalski, S. Serum reverse triiodothyronine (3,3′,5′-l-triiodothyronine) in chronic renal failure. Nephron. 1980; 26(2):85–89.
45. Kaptein, EM, Levitan, D, Feinstein, EI, et al. Alterations of thyroid hormone indices in acute renal failure and in acute critical illness with and without acute renal failure. Am J Nephrol. 1981; 1(3-4):138–143.
46. Slag, MF, Morley, JE, Elson, MK, et al. Hypothyroxinemia in critically ill patients as a predictor of high mortality. JAMA. 1981; 245(1):43–45.
47. Kaplan, MM, Larsen, PR, Crantz, FR, et al. Prevalence of abnormal thyroid function test results in patients with acute medical illnesses. Am J Med. 1982; 72(1):9–16.
48. Melmed, S, Geola, FL, Reed, AW, et al. A comparison of methods for assessing thyroid function in nonthyroidal illness. J Clin Endocrinol Metab. 1982; 54(2):300–306.
49. Chopra, IJ, Hershman, JM, Pardridge, WM, Nicoloff, JT. Thyroid function in nonthyroidal illnesses. Ann Intern Med. 1983; 98(6):946–957.
50. Zaloga, GP, Chernow, B, Smallridge, RC, et al. A longitudinal evaluation of thyroid function in critically ill surgical patients. Ann Surg. 1985; 201(4):456–464.
51. Hamblin, PS, Dyer, SA, Mohr, VS, et al. Relationship between thyrotropin and thyroxine changes during recovery from severe hypothyroxinemia of critical illness. J Clin Endocrinol Metab. 1986; 62(4):717–722.
52. LoPresti, JS, Fried, JC, Spencer, CA, Nicoloff, JT. Unique alterations of thyroid hormone indices in the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989; 110(12):970–975.
53. Chu, SH, Huang, TS, Hsu, RB, et al. Thyroid hormone changes after cardiovascular surgery and clinical implications. Ann Thorac Surg. 1991; 52(4):791–796.
54. Rosen, HN, Greenspan, SL, Landsberg, L, Faix, JD. Distinguishing hypothyroxinemia due to euthyroid sick syndrome from pituitary insufficiency. Isr J Med Sci. 1994; 10:746–750.
55. Nader, S, Warner, MD, Doyle, S, Peabody, CA. Euthyroid sick syndrome in psychiatric inpatients. Biol Psychiatry. 1996; 40(12):1288–1293.
56. Chopra, IJ. Nonthyroidal illness syndrome or euthyroid sick syndrome? Endocr Pract. 1996; 2(1):45–52.
57. Girvent, M, Maestro, S, Hernandez, R, et al. Euthyroid sick syndrome, associated endocrine abnormalities, and outcome in elderly patients undergoing emergency operation. Surgery. 1998; 123(5):560–567.
58. Langton, JE, Brent, GA. Nonthyroidal illness syndrome: Evaluation of thyroid function in sick patients. Endocrinol Metab Clin North Am. 2002; 31(1):159–172.
59. Ray, DC, Macduff, A, Drummond, GB, et al. Endocrine measurements in survivors and non-survivors from critical illness. Intensive Care Med. 2002; 28(9):1301–1308.
60. Hama, S, Kitaoka, T, Shigenobu, M, et al. Malnutrition and nonthyroidal illness syndrome after stroke. Metabolism. 2005; 54(6):699–704.
61. Cerillo, AG, Storti, S, Mariani, M, et al. The non-thyroidal illness syndrome after coronary artery bypass grafting: A 6-month follow-up study. Clin Chem Lab Med. 2005; 43(3):289–293.
62. Adler, SM, Wartofsky, L. The nonthyroidal illness syndrome. Endocrinol Metab Clin North Am. 2007; 36(3):657–672.
63. Carter, JN, Eastmen, CJ, Corcoran, JM, Lazarus, L. Inhibition of conversion of thyroxine to triiodothyronine in patients with severe chronic illness. Clin Endocrinol. 1976; 5(6):587–594.
64. Kaptein, EM, Grieb, DA, Spencer, CA, et al. Thyroxine metabolism in the low thyroxine state of critical nonthyroidal illnesses. J Clin Endocrinol Metab. 1981; 53(4):764–771.
65. Chopra, IJ, Solomon, DH, Teco, GN, Eisenberg, JB. An inhibitor of the binding of thyroid hormones to serum proteins is present in extrathyroidal tissues. Science. 1982; 215(4531):407–409.
66. Kaptein, EM, Robinson, WJ, Grieb, DA, Nicoloff, JT. Peripheral serum thyroxine, triiodothyronine and reverse triiodothyronine kinetics in the low thyroxine state of acute nonthyroidal illnesses. A noncompartmental analysis. J Clin Invest. 1982; 69(3):526–535.
67. Oppenheimer, JH, Schwartz, HL, Mariash, CN, Kaiser, FE. Evidence for a factor in the sera of patients with nonthyroidal disease which inhibits iodothyronine binding by solid matrices, serum proteins, and rat hepatocytes. J Clin Endocrinol Metab. 1982; 54(4):757–766.
68. Chopra, IJ, Teco, GN, Mead, JF, et al. Relationship between serum free fatty acids and thyroid hormone binding inhibitor in nonthyroid illnesses. J Clin Endocrinol Metab. 1985; 60(5):980–984.
69. Chopra, IJ, Huang, TS, Beredo, A, et al. Evidence for an inhibitor of extrathyroidal conversion of thyroxine to 3,5,3′-triiodothyronine in sera of patients with nonthyroidal illnesses. J Clin Endocrinol Metab. 1985; 60(4):666–672.
70. Sarne, DH, Refetoff, S. Measurement of thyroxine uptake from serum by cultured human hepatocytes as an index of thyroid status: Reduced thyroxine uptake from serum of patients with nonthyroidal illness. J Clin Endocrinol Metab. 1985; 61(6):1046–1052.
71. van der Poll, T, Romijn, JA, Wiersinga, WM, Sauerwein, HP. Tumor necrosis factor: A putative mediator of the sick euthyroid syndrome in man. J Clin Endocrinol Metab. 1990; 71(6):1567–1572.
72. Mendel, CM, Laughton, CW, McMahon, FA, Cavalieri, RR. Inability to detect an inhibitor of thyroxine-serum protein binding in sera from patients with nonthyroid illness. Metabolism. 1991; 40(5):491–502.
73. Arem, R, Wiener, GJ, Kaplan, SG, et al. Reduced tissue thyroid hormone levels in fatal illness. Metabolism. 1993; 42(9):1102–1108.
74. Lim, CF, Bernard, BF, de Jong, M, et al. A furan fatty acid and indoxyl sulfate are the putative inhibitors of thyroxine hepatocyte transport in uremia. J Clin Endocrinol Metab. 1993; 76(2):318–324.
75. Lim, CF, Docter, R, Visser, TJ, et al. Inhibition of thyroxine transport into cultured rat hepatocytes by serum of nonuremic critically ill patients: Effects of bilirubin and nonesterified fatty acids. J Clin Endocrinol Metab. 1993; 76(5):1165–1172.
76. Stouthard, JM, van der Poll, T, Endert, E, et al. Effects of acute and chronic interleukin-6 administration on thyroid hormone metabolism in humans. J Clin Endocrinol Metab. 1994; 79(5):1342–1346.
77. van der Poll, T, Van Zee, KJ, Endert, E, et al. Interleukin-1 receptor blockade does not affect endotoxin-induced changes in plasma thyroid hormone and thyrotropin concentrations in man. J Clin Endocrinol Metab. 1995; 80(4):1341–1346.
78. Boelen, A, Schiphorst, MC, Wiersinga, WM. Relationship between serum 3,5,3′-triiodothyronine and serum interleukin-8, interleukin-10 or interferon gamma in patients with nonthyroidal illness. J Endocrinol Invest. 1996; 19(7):480–483.
79. Davies, PH, Black, EG, Sheppard, MC, Franklyn, JA. Relation between serum interleukin-6 and thyroid hormone concentrations in 270 hospital in-patients with non-thyroidal illness. Clin Endocrinol. 1996; 44(2):199–205.
80. Fliers, E, Guldenaar, SE, Wiersinga, WM, Swaab, DF. Decreased hypothalamic thyrotropin-releasing hormone gene expression in patients with nonthyroidal illness. J Clin Endocrinol Metab. 1997; 82(12):4032–4036.
81. Feelders, RA, Swaak, AJ, Romijn, JA, et al. Characteristics of recovery from the euthyroid sick syndrome induced by tumor necrosis factor alpha in cancer patients. Metabolism. 1999; 48(3):324–329.
82. Afandi, B, Schussler, GC, Arafeh, AH, et al. Selective consumption of thyroxine-binding globulin during cardiac bypass surgery. Metabolism. 2000; 49(2):270–274.
83. Jirasakuldech, B, Schussler, GC, Yap, MG, et al. A characteristic serpin cleavage product of thyroxine-binding globulin appears in sepsis sera. J Clin Endocrinol Metab. 2000; 85(11):3996–3999.
84. Michalaki, M, Vagenakis, AG, Makri, M, et al. Dissociation of the early decline in serum T(3) concentration and serum IL-6 rise and TNF-alpha in nonthyroidal illness syndrome induced by abdominal surgery. J Clin Endocrinol Metab. 2001; 86(9):4198–4205.
85. Janssen, OE, Golcher, HM, Grasberger, H, et al. Characterization of T(4)-binding globulin cleaved by human leukocyte elastase. J Clin Endocrinol Metab. 2002; 87(3):1217–1222.
86. Peeters, RP, Wouters, PJ, Kaptein, E, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003; 88(7):3202–3211.
87. Fekete, C, Gereben, B, Doleschall, M, et al. Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: Implications for the nonthyroidal illness syndrome. Endocrinology. 2004; 145(4):1649–1655.
88. Lechan, RM, Fekete, C. Feedback regulation of thyrotropin-releasing hormone (TRH): Mechanisms for the non-thyroidal illness syndrome. J Endocrinol Invest. 2004; 27(6 Suppl):105–119.
89. den Brinker, M, Joosten, KF, Visser, TJ, et al. Euthyroid sick syndrome in meningococcal sepsis: The impact of peripheral thyroid hormone metabolism and binding proteins. J Clin Endocrinol Metab. 2005; 90(10):5613–5620.
90. Peeters, RP, Kester, MHA, Wouters, PJ, et al. Increased thyroxine sulfate levels in critically ill patients as a result of a decreased hepatic type I deiodinase activity. J Clin Endocrinol Metab. 2005; 90(12):6460–6465.
91. Peeters, RP, van der Geyten, S, Wouters, PJ, et al. Tissue thyroid hormone levels in critical illness. J Clin Endocrinol Metab. 2005; 90(12):6498–6507.
92. Bianco, AC, Kim, BW. Deiodinases: Implications of the local control of thyroid hormone action. J Clin Invest. 2006; 116(10):2571–2579.
93. Debaveye, Y, Ellger, B, Mebis, L, et al. Regulation of tissue iodothyronine deiodinase activity in a model of prolonged critical illness. Thyroid. 2008; 18(5):551–560.
94. Huang, SA, Bianco, AC. Reawakened interest in type III iodothyronine deiodinase in critical illness and injury. Endocrinol Metab. 2008; 4(3):148–155.
95. Heemstra, KA, Soeters, MR, Fliers, E, et al. Type 2 iodothyronine deiodinase in skeletal muscle: Effects of hypothyroidism and fasting. J Clin Endocrinol Metab. 2009; 94(6):2144–2150.
96. Warner, MH, Beckett, GJ. Mechanisms behind the non-thyroidal illness syndrome: An update. J Endocrinol. 2010; 205(1):1–13.
97. Gardner, DF, Kaplan, MM, Stanley, CA, Utiger, RD. Effect of triiodothyronine replacement on the metabolic and pituitary responses to starvation. N Engl J Med. 1979; 300:579–584.
98. Hesch, RD, Husch, M, Kodding, R, et al. Treatment of dopamine-dependent shock with triiodothyronine. Endocr Res Commun. 1981; 8(4):229–237.
99. Becker, RA, Vaughan, GM, Ziegler, MG, et al. Hypermetabolic low triiodothyronine syndrome of burn injury. Crit Care Med. 1982; 10(12):870–875.
100. Brent, GA, Hershman, JM. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J Clin Endocrinol Metab. 1986; 63(1):1–8.
101. Acker, CG, Singh, AR, Flick, RP, et al. A trial of thyroxine in acute renal failure. Kidney Int. 2000; 57(1):293–298.
102. Novitzky, D, Cooper, DK, Chaffin, JS, et al. Improved cardiac allograft function following triiodothyronine therapy to both donor and recipient. Transplantation. 1990; 49(2):311–316.
103. Moruzzi, P, Doria, E, Agostoni, PG, et al. Usefulness of L-thyroxine to improve cardiac and exercise performance in idiopathic dilated cardiomyopathy. Am J Cardiol. 1994; 73(5):374–378.
104. Klemperer, JD, Klein, I, Gomez, M, et al. Thyroid hormone treatment after coronary-artery bypass surgery. N Engl J Med. 1995; 333(23):1522–1527.
105. Klemperer, JD, Zelano, J, Helm, RE, et al. Triiodothyronine improves left ventricular function without oxygen wasting effects after global hypothermic ischemia. J Thorac Cardiovasc Surgery. 1995; 109(3):457–465.
106. Bennett-Guerrero, E, Jimenez, JL, White, WD, et al. Cardiovascular effects of intravenous triiodothyronine in patients undergoing coronary artery bypass graft surgery. A randomized, double-blind, placebo-controlled trial. Duke T3 study group. JAMA. 1996; 275(9):687–692.
107. Klemperer, JD, Klein, IL, Ojamaa, K, et al. Triiodothyronine therapy lowers the incidence of atrial fibrillation after cardiac operations. Ann Thorac Surgery. 1996; 61(5):1323–1327.
108. Moruzzi, P, Doria, E, Agostoni, PG. Medium-term effectiveness of L-thyroxine treatment in idiopathic dilated cardiomyopathy. Am J Med. 1996; 101(5):461–467.
109. Novitzky, D, Fontanet, H, Snyder, M, et al. Impact of triiodothyronine on the survival of high-risk patients undergoing open heart surgery. Cardiology. 1996; 87(6):509–515.
110. Opasich, C, Pacini, F, Ambrosino, N, et al. Sick euthyroid syndrome in patients with moderate-to-severe chronic heart failure. Eur Heart J. 1996; 17(12):1860–1866.
111. Hamilton, MA, Stevenson, LW, Fonarow, GC, et al. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am J Cardiol. 1998; 81(4):443–447.
112. Chowdhury, D, Parnell, VA, Ojamaa, K, et al. Usefulness of triiodothyronine (T3) treatment after surgery for complex congenital heart disease in infants and children. Am J Cardiol. 1999; 84(9):1107–1110.
113. Mullis-Jansson, SL, Argenziano, M, Corwin, S, et al. A randomized double-blind study of the effect of triiodothyronine on cardiac function and morbidity after coronary bypass surgery. J Thorac Cardiovasc Surgery. 1999; 117(6):1128–1134.
114. Chowdhury, D, Ojamaa, K, Parnell, VA, et al. A prospective randomized clinical study of thyroid hormone treatment after operations for complex congenital heart disease. J Thorac Cardiovasc Surgery. 2001; 122(5):1023–1025.
115. Klein, I, Ojamaa, K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001; 344(7):501–509.
116. Ascheim, DD, Hryniewicz, K. Thyroid hormone metabolism in patients with congestive heart failure: The low triiodothyronine state. Thyroid. 2002; 12(6):511–515.
117. Iervasi, G, Pingitore, A, Landi, P, et al. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation. 2003; 107(5):708–713.
118. Rays, J, Wajngarten, M, Gebara, OC, et al. Long-term prognostic value of triiodothyronine concentration in elderly patients with heart failure. Am J Geriatr Cardiol. 2003; 12(5):293–297.
119. Fazio, S, Palmieri, EA, Lombardi, G, Biondi, B. Effects of thyroid hormone on the cardiovascular system. Recent Prog Horm Res. 2004; 59:31–50.
120. Klein, I, Danzi, S. Thyroid disease and the heart. Circulation. 2007; 116(15):1725–1735.
121. Pingitore, A, Galli, E, Barison, A, et al. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J Clin Endocrinol Metab. 2008; 93(4):1351–1358.
122. De Groot, LJ. Dangerous dogmas in medicine: The nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999; 84(1):151–164.
123. Stathatos, N, Wartofsky, L. The euthyroid sick syndrome: Is there a physiologic rationale for thyroid hormone treatment? J Endocrinol Invest. 2003; 26(12):1174–1179.
124. De Groot, LJ. Non-thyroidal illness syndrome is a manifestation of hypothalamic-pituitary dysfunction, and in view of current evidence, should be treated with appropriate replacement therapies. Criti Care Clin. 2006; 22(1):57–86.
125. Farwell, AP. Thyroid hormone therapy is not indicated in the majority of patients with the sick euthyroid syndrome. Endocr Pract. 2008; 14(9):1180–1187.
126. Peters, A, Ehlers, M, Blank, B, et al. Excess triiodothyronine as a risk factor of coronary events. Arch Intern Med. 2000; 160(13):1993–1999.
127. McLarty, DG, Ratcliffe, WA, McColl, K, et al. Letter: Thyroid-hormone levels and prognosis in patients with serious non-thyroidal illness. Lancet. 1975; 2(7928):275–276.
128. Kaptein, EM, Weiner, JM, Robinson, WJ, et al. Relationship of altered thyroid hormone indices to survival in nonthyroidal illnesses. Clin Endocrinol. 1982; 16(6):565–574.
129. Phillips, RH, Valente, WA, Caplan, ES, et al. Circulating thyroid hormone changes in acute trauma: Prognostic implications for clinical outcome. J Trauma. 1984; 24(2):116–119.
130. Silberman, H, Eisenberg, D, Ryan, J, et al. The relation of thyroid indices in the critically ill patient to prognosis and nutritional factors. Surgery Gynecol Obstet. 1988; 166(3):223–228.
131. Custro, N, Scafidi, V, Costanzo, G, Notarbartolo, A. Prospective study on thyroid function anomalies in severely ill patients. Ann Ital Med Int. 1992; 7(1):13–18.
132. Maldonado, LS, Murata, GH, Hershman, JM, Braunstein, GD. Do thyroid function tests independently predict survival in the critically ill? Thyroid. 1992; 2(2):119–123.
133. Hamilton, MA. Prevalence and clinical implications of abnormal thyroid hormone metabolism in advanced heart failure. Ann Thorac Surgery. 1993; 56(1 Suppl):S48–S52.
134. Jarek, MJ, Legare, EJ, McDermott, MT, et al. Endocrine profiles for outcome prediction from the intensive care unit. Crit Care Med. 1993; 21(4):543–550.
135. Loh, KC, Eng, PC. Prevalence and prognostic relevance of sick euthyroid syndrome in a medical intensive care unit. Ann Acad Med Singapore. 1995; 24(6):802–806.
136. Rothwell, PM, Lawler, PG. Prediction of outcome in intensive care patients using endocrine parameters. Crit Care Med. 1995; 23(1):78–83.
137. Leon-Sanz, M, Lorente, JA, Larrodera, L, et al. Pituitary-thyroid function in patients with septic shock and its relation with outcome. Eur J Med Res. 1997; 2(11):477–482.
138. Scoscia, E, Baglioni, S, Eslami, A, et al. Low triiodothyronine (T3) state: A predictor of outcome in respiratory failure? Results of a clinical pilot study. Eur J Endocrinol. 2004; 151(5):557–560.
139. Pingitore, A, Landi, P, Taddei, MC, et al. Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am J Med. 2005; 118(2):132–136.
140. Bello, G, Pennisi, MA, Montini, L, et al. Nonthyroidal illness syndrome and prolonged mechanical ventilation in patients admitted to the ICU. Chest. 2009; 135(6):1448–1454.
141. Canaris, GJ, Manowitz, NR, Mayor, G, Ridgway, EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000; 160(4):526–534.
142. Hollowell, JG, Staehling, NW, Flanders, WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002; 87(2):489–499.
143. Vanderpump, MP. The epidemiology of thyroid disease. Br Med Bull. 2011; 99:39–51.
144. Parving, HH, Hansen, JM, Nielsen, SL, et al. Mechanisms of edema formation in myxedema—Increased protein extravasation and relatively slow lymphatic drainage. N Engl J Med. 1979; 301(9):460–465.
145. Wilson, WR, Bedell, GN. The pulmonary abnormalities in myxedema. J Clin Invest. 1960; 39:42–55.
146. Weg, JG, Calverly, JR, Johnson, C. Hypothyroidism and alveolar hypoventilation. Arch Intern Med. 1965; 115:302–306.
147. Zwillich, CW, Pierson, DJ, Hofeldt, FD, et al. Ventilatory control in myxedema and hypothyroidism. N Engl J Med. 1975; 292(13):662–665.
148. Rajagopal, KR, Abbrecht, PH, Derderian, SS, et al. Obstructive sleep apnea in hypothyroidism. Ann Intern Med. 1984; 101(4):491–494.
149. Ladenson, PW, Goldenheim, PD, Ridgway, EC. Prediction and reversal of blunted ventilatory responsiveness in patients with hypothyroidism. Am J Med. 1988; 84(5):877–883.
150. Duranti, R, Gheri, RG, Gorini, M, et al. Control of breathing in patients with severe hypothyroidism. Am J Med. 1993; 95(1):29–37.
151. Gordon, AH. Pericardial effusion in myxedema. Trans Assoc Am Physicians. 1935; 50:272–276.
152. Boivin, J-M. Histoire d’un épanchement péricardique récidivant asséché par l’extrait thyroïdien. Arch Mal Coeur. 1945; 38:289–292.
153. Davis, PJ, Jacobson, S. Myxedema with cardiac tamponade and pericardial effusion of “gold paint” appearance. Arch Intern Med. 1967; 120(5):615–619.
154. Karu, AK, Khalife, WI, Houser, R, VanderWoude, J. Impending cardiac tamponade as a primary presentation of hypothyroidism: Case report and review of literature. Endocr Pract. 2005; 11(4):265–271.
155. Polikar, R, Kennedy, B, Ziegler, M, et al. Plasma norepinephrine kinetics, dopamine-beta-hydroxylase, and chromogranin-A, in hypothyroid patients before and following replacement therapy. J Clin Endocrinol Metab. 70(1), 1990.
156. Manhem, P, Bramnert, M, Hallengren, B, et al. Increased arterial and venous plasma noradrenaline levels in patients with primary hypothyroidism during hypothyroid as compared to euthyroid state. J Endocrinol Invest. 1992; 15(10):763–765.
157. Bramnert, M, Hallengren, B, Lecerof, H, et al. Decreased blood pressure response to infused noradrenaline in normotensive as compared to hypertensive patients with primary hypothyroidism. Clin Endocrinol. 1994; 40(3):317–321.
158. Amidi, M, Leon, DF, DeGroot, WJ, et al. Effect of the thyroid state on myocardial contractility and ventricular ejection rate in man. Circulation. 1968; 38(2):229–239.
159. Crowley, WF, Ridgway, EC, Bough, EW, et al. Noninvasive evaluation of cardiac function in hypothyroidism. N Engl J Med. 1977; 1977:1–6.
160. Wieshammer, S, Keck, FS, Waitzinger, J, et al. Acute hypothyroidism slows the rate of left ventricular diastolic relaxation. Can J Physiol Pharmacol. 1989; 67(9):1007–1010.
161. Bengel, FM, Nekolla, SG, Ibrahim, T, et al. Effect of thyroid hormones on cardiac function, geometry, and oxidative metabolism assessed noninvasively by positron emission tomography and magnetic resonance imaging. J Clin Endocrinol Metab. 2000; 85(5):1822–1827.
162. Biondi, B, Palmieri, EA, Lombardi, G, Fazio, S. Effects of subclinical thyroid dysfunction on the heart. Ann Intern Med. 2002; 137(11):904–914.
163. Rodondi, N, Bauer, DC, Cappola, AR, et al. Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health Study. J Am Coll Cardiol. 2008; 52(14):1152–1159.
164. Nanchen, D, Gussekloo, J, Westendorp, RG, et al. Subclinical thyroid dysfunction and the risk of heart failure in older persons at high cardiovascular risk. J Clin Endocrinol Metab. 2012; 97(3):852–861.
165. Vanhaelst, L, Neve, P, Chailly, P, Bastenie, PA. Coronary-artery disease in hypothyroidism. Observations in clinical myxoedema. Lancet. 1967; 2(7520):800–802.
166. Mya, MM, Aronow, WS. Subclinical hypothyroidism is associated with coronary artery disease in older persons. J Gerontol A Biol Sci Med Sci. 2002; 57(10):M658–M659.
167. Auer, J, Berent, R, Weber, T, et al. Thyroid function is associated with presence and severity of coronary atherosclerosis. Clin Cardiol. 2003; 26(12):569–573.
168. Roos, A, Zoet-Nugteren, SK, Berghout, A. Evaluation of cardiac ischaemia in cardiac asymptomatic newly diagnosed untreated patients with primary hypothyroidism. Neth J Med. 2005; 63(3):97–102.
169. Asvold, BO, Bjoro, T, Nilsen, TI, et al. Thyrotropin levels and risk of fatal coronary heart disease: The HUNT study. Arch Intern Med. 2008; 168(8):855–860.
170. Rodondi, N, den Elzen, WP, Bauer, DC, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010; 304(12):1365–1374.
171. Zimmerman, RS, Gharib, H, Zimmerman, D, et al. Atrial natriuretic peptide in hypothyroidism. J Clin Endocrinol Metab. 1987; 64(2):353–355.
172. Iwasaki, Y, Oiso, Y, Yamauchi, K, et al. Osmoregulation of plasma vasopressin in myxedema. J Clin Endocrinol Metab. 1990; 70(2):534–539.
173. Hierholzer, K, Finke, R. Myxedema. Kidney Int Suppl. 1997; 59:S82–S89.
174. Baajafer, FS, Hammami, MM, Mohamed, GE. Prevalence and severity of hyponatremia and hypercreatininemia in short-term uncomplicated hypothyroidism. J Endocrinol Invest. 1999; 22(1):35–39.
175. Hooper, MJ. Diminished TSH secretion during acute non-thyroidal illness in untreated primary hypothyroidism. Lancet. 1976; i:48–49.
176. Hays, MT. Absorption of oral thyroxine in man. J Clin Endocrinol Metab. 1968; 28(6):749–756.
177. Ridgway, EC, McCammon, JA, Benotti, J, Maloof, F. Acute metabolic responses in myxedema to large doses of intravenous L-thyroxine. Ann Intern Med. 1972; 77(4):549–555.
178. Saberi, M, Utiger, RD. Serum thyroid hormone and thyrotropin concentrations during thyroxine and triiodothyronine therapy. J Clin Endocrinol Metab. 1974; 39(5):923–927.
179. Stock, JM, Surks, MI, Oppenheimer, JH. Replacement dosage of l-thyroxine in hypothyroidism. A re-evaluation. N Engl J Med. 1974; 290(10):529–533.
180. Rosenbaum, RL, Barzel, US. Levothyroxine replacement dose for primary hypothyroidism decreases with age. Ann Intern Med. 1982; 96(1):53–55.
181. Ladenson, PW, Goldenheim, PD, Ridgway, EC. Rapid pituitary and peripheral tissue responses to intravenous L-triiodothyronine in hypothyroidism. J Clin Endocrinol Metab. 1983; 56(6):1252–1259.
182. Kaptein, EM, Quion-Verde, H, Swinney, RS, et al. Acute hemodynamic effects of levothyroxine loading in critically ill hypothyroid patients. Arch Intern Med. 1986; 146(4):662–666.
183. Fish, LH, Schwartz, HL, Cavanaugh, J, et al. Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism. Role of triiodothyronine in pituitary feedback in humans. N Engl J Med. 1987; 316(13):764–770.
184. Hays, MT, Nielsen, KR. Human thyroxine absorption: Age effects and methodological analyses. Thyroid. 1994; 4(1):55–64.
185. Choe, W, Hays, MT. Absorption of oral thyroxine. Endocrinologist. 1995; 5:222–228.
186. Singh, N, Singh, PN, Hershman, JM. Effect of calcium carbonate on the absorption of levothyroxine. JAMA. 2000; 283(21):2822–2825.
187. Singh, N, Weisler, SL, Hershman, JM. The acute effect of calcium carbonate on the intestinal absorption of levothyroxine. Thyroid. 2001; 11(10):967–971.
188. Stathatos, N, Wartofsky, L. Perioperative management of patients with hypothyroidism. Endocrinol Metab Clin North Am. 2003; 32(2):503–518.
189. Dickerson, RN, Maish, GO, 3rd., Minard, G, Brown, RO. Clinical relevancy of the levothyroxine-continuous enteral nutrition interaction. Nutr Clin Pract. 2010; 25(6):646–652.
190. Vinagre, ALM, de Souza, MVL. Levothyroxine absorption and difficult management of hypothyroid patients in the intensive care unit: Two case reports and a literature review. Rev Bras Ter Intensiva. 2010; 23(2):242–248.
191. Garber, JR, Cobin, RH, Gharib, H, et al. Clinical Practice Guidelines for Hypothyroidism in Adults: Co-sponsored by American Association of Clinical Endocrinologists and the American Thyroid Association. Endocr Pract. 2012; 1–207.
192. Smyth, CJ. Angina pectoris and myocardial infarction as complications of myxedema: With especial reference to the danger of treatment with thyroid preparations. Am Heart J. 1938; 15:652–660.
193. Keating, FR, Jr., Parkin, TW, Selby, JB, Dickinson, LS. Treatment of heart disease associated with myxedema. Prog Cardiovasc Dis. 1961; 3:364–381.
194. Levine, HD. Compromise therapy in the patient with angina pectoris and hypothyroidism. A clinical assessment. Am J Med. 1980; 69(3):411–418.
195. Polikar, R, Feld, GK, Dittrich, HC, et al. Effect of thyroid replacement therapy on the frequency of benign atrial and ventricular arrhythmias. J Am Coll Cardiol. 1989; 14(4):999–1002.
196. Perk, M, O’Neill, BJ. The effect of thyroid hormone therapy on angiographic coronary artery disease progression. Can J Cardiol. 1997; 13(3):273–276.
197. Fadeyev, VV, Sytch, J, Kalashnikov, V, et al. Levothyroxine replacement therapy in patients with subclinical hypothyroidism and coronary artery disease. Endocr Pract. 2006; 12(1):5–17.
198. Taguchi, T, Iwasaki, Y, Asaba, K, et al. Myxedema coma and cardiac ischemia in relation to thyroid hormone replacement therapy in a 38-year-old Japanese woman. Clin Ther. 2007; 29(12):2710–2714.
199. Hays, MT, Nielsen, KR. Human thyroxine absorption: Age effects and methodological analyses. Thyroid. 1994; 4(1):55–64.
200. Murkin, JM. Anesthesia and hypothyroidism: A review of thyroxine physiology, pharmacology, and anesthetic implications. Anesth Analg. 1982; 61(4):371–383.
201. Weinberg, AD, Brennan, MD, Gorman, CA, et al. Outcome of anesthesia and surgery in hypothyroid patients. Arch Intern Med. 1983; 143(5):893–897.
202. Ladenson, PW, Levin, AA, Ridgway, EC, Daniels, GH. Complications of surgery in hypothyroid patients. Am J Med. 1984; 77(2):261–266.
203. Nelson, JC, Palmer, FJ, Bowyer, AF. The successful treatment of myxedema and coronary artery disease in patients intolerant of thyroid hormone. Med Arts Sci. 1974; 28(3):15–22.
204. Paine, TD, Rogers, WJ, Baxley, WA, Russell, RO, Jr. Coronary arterial surgery in patients with incapacitating angina pectoris and myxedema. Am J Cardiol. 1977; 40(2):226–231.
205. Hay, I, Duick, DS, Vlietstra, RE, et al. Thyroxine therapy in hypothyroid patients undergoing coronary revascularization: A retrospective analysis. Ann Intern Med. 1981; 95(4):456–457.
206. Finlayson, DC, Kaplan, JA. Myxoedema and open heart surgery: Anaesthesia and intensive care unit experience. Can Anaesth Soc J. 1982; 29(6):543–549.
207. Myerowitz, PD, Kamienski, RW, Swanson, DK, et al. Diagnosis and management of the hypothyroid patient with chest pain. J Thorac Cardiovasc Surgery. 1983; 86(1):57–60.
208. Holvoet, G, Gillebert, TC, Piessens, J, De Geest, H. Coronary artery surgery in patients with myxoedema. Acta Cardiol. 1984; 39(2):139–145.
209. Drucker, DJ, Burrow, GN. Cardiovascular surgery in the hypothyroid patient. Arch Intern Med. 1985; 145(9):1585–1587.
210. Yamamoto, K, Sugimoto, Y, Taki, Y, et al. [Anesthetic management of hypothyroid patients for coronary artery bypass grafting]. Masui. Jpn J Anesthesiol. 1990; 39(12):1683–1689.
211. Kawasuji, M, Sawa, S, Tsujiguchi, H, Iwa, T. Coronary artery bypass surgery in patients with angina pectoris and hypothyroidism. Eur J Cardiothorac Surgery. 1991; 5(5):230–234.
212. Sherman, SI, Ladenson, PW. Percutaneous transluminal coronary angioplasty in hypothyroidism. Am J Med. 1991; 90(3):367–370.
213. Anderson, A, Hausmann, W. Triiodothyronine in myxedema coma. Lancet. 1956; 2:999.
214. Nickerson, JF, Hill, SR, McNeil, JH, Barker, SB. Fatal myxedema with and without coma. Ann Intern Med. 1960; 53:475.
215. Nordqvist, P, Dhuner, KG, Stenberg, K, Orndahl, G. Myxoedema coma and carbon dioxide-retention. Acta Med Scand. 1960; 166:189–194.
216. Catz, B, Russell, S. Myxedema, shock and coma. Seven survival cases. Arch Intern Med. 1961; 108:407–417.
217. Winawer, SJ, Rosen, SM, Cohn, H. Myxedema coma with ventricular tachycardia. Arch Intern Med. 1963; 111:647–650.
218. Holvey, DN, Goodner, CJ, Nicoloff, JT, Dowling, JT. Treatment of myxedema coma with intravenous thyroxine. Arch Intern Med. 1964; 113:89–96.
219. Hohl, RD, Nixon, RK. Myxedema ileus. Arch Intern Med. 1965; 115:145–150.
220. Domm, BM, Vassallo, CL. Myxedema coma with respiratory failure. Am Rev Respir Dis. 1973; 107(5):842–845.
221. Woods, KL, Holmes, GK. Myxoedema coma presenting in status epilepticus. Postgrad Med J. 1977; 53(615):46–48.
222. Pereira, VG, Haron, ES, Lima-Neto, N, Medeiros-Neto, GA. Management of myxedema coma: Report on three successfully treated cases with nasogastric or intravenous administration of triiodothyronine. J Endocrinol Invest. 1982; 5(5):331–334.
223. McCulloch, W, Price, P, Hinds, CJ, Wass, JA. Effects of low dose oral triiodothyronine in myxoedema coma. Intensive Care Med. 1985; 11(5):259–262.
224. Hermansen, K, Johannsen, LG, Rasmussen, OB. Hypoglycaemic coma in severe primary hypothyroidism. Acta Med Scand. 1985; 218(3):345–346.
225. Hylander, B, Rosenqvist, U. Treatment of myxoedema coma—Factors associated with fatal outcome. Acta Endocrinol. 1985; 108(1):65–71.
226. Arlot, S, Debussche, X, Lalau, JD, et al. Myxoedema coma: Response of thyroid hormones with oral and intravenous high-dose L-thyroxine treatment. Intensive Care Med. 1991; 17(1):16–18.
227. MacKerrow, SD, Osborn, LA, Levy, H, et al. Myxedema-associated cardiogenic shock treated with intravenous triiodothyronine. Ann Intern Med. 1992; 117(12):1014–1015.
228. Appoo, JJ, Morin, JF. Severe cerebral and cardiac dysfunction associated with thyroid decompensation after cardiac operations. J Thorac Cardiovasc Surgery. 1997; 114(3):496.
229. Yamamoto, T, Fukuyama, J, Fujiyoshi, A. Factors associated with mortality of myxedema coma: Report of eight cases and literature survey. Thyroid. 1999; 9(12):1167–1174.
230. Benvenga, S, Squadrito, S, Saporito, F, et al. Myxedema coma of both primary and secondary origin, with non-classic presentation and extremely elevated creatine kinase. Horm Metab Res. 2000; 32(9):364–366.
231. Sarlis, NJ, Gourgiotis, L. Thyroid emergencies. Rev Endocr Metab Disord. 2003; 4(2):129–136.
232. Rodriguez, I, Fluiters, E, Perez-Mendez, LF, et al. Factors associated with mortality of patients with myxoedema coma: Prospective study in 11 cases treated in a single institution. J Endocrinol. 2004; 180(2):347–350.
233. Jansen, HJ, Doebe, SR, Louwerse, ES, et al. Status epilepticus caused by a myxoedema coma. Neth J Med. 2006; 64(6):202–205.
234. Wartofsky, L. Myxedema coma. In: Braverman LE, Cooper DS, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2012:600–605.
235. Berglund, J, Christensen, SB, Hallengren, B. Total and age-specific incidence of Graves’ thyrotoxicosis, toxic nodular goitre and solitary toxic adenoma in Malmo 1970-74. J Intern Med. 1990; 227(2):137–141.
236. Bahn, RS, Burch, HB, Cooper, DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: Management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011; 17(3):456–520.
237. Salvatore, D, Tu, H, Harney, JW, Larsen, PR. Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest. 1996; 98(4):962–968.
238. Amino, N, Yabu, Y, Kuro, R, et al. T3/T4 ratio in thyroid disease. Lancet. 1979; 1(8107):107.
239. Tsatsoulis, A, Johnson, EO, Kalogera, CH, et al. The effect of thyrotoxicosis on adrenocortical reserve. Eur J Endocrinol. 2000; 142(3):231–235.
240. Thomas, FB, Mazzaferri, EL, Skillman, TG. Apathetic thyrotoxicosis: A distinctive clinical and laboratory entity. Ann Intern Med. 1970; 72(5):679–685.
241. Tibaldi, JM, Barzel, US, Albin, J, Surks, M. Thyrotoxicosis in the very old. Am J Med. 1986; 81(4):619–622.
242. Nordyke, RA, Gilbert, FI, Jr., Harada, AS. Graves’ disease. Influence of age on clinical findings. Arch Intern Med. 1988; 148(3):626–631.
243. Boelaert, K, Torlinska, B, Holder, RL, Franklyn, JA. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: A large cross-sectional study. J Clin Endocrinol Metab. 2010; 95(6):2715–2726.
244. Birkhauser, M, Burer, T, Busset, R, Burger, A. Diagnosis of hyperthyroidism when serum-thyroxine alone is raised. Lancet. 1977; 2(8028):53–56.
245. Engler, D, Donaldson, EB, Stockigt, JR, Taft, P. Hyperthyroidism without triiodothyronine excess: An effect of severe non-thyroidal illness. J Clin Endocrinol Metab. 1978; 46(1):77–82.
246. Gavin, LA, Rosenthal, M, Cavalieri, RR. The diagnostic dilemma of isolated hyperthyroxinemia in acute illness. JAMA. 1979; 242(3):251–253.
247. Topliss, DJ, Stockigt, JR. Normal serum T3 after abdominal operation in severe hyperthyroidism. N Engl J Med. 1979; 300(2):92–93.
248. Caplan, RH, Pagliara, AS, Wickus, G. Thyroxine toxicosis. A common variant of hyperthyroidism. JAMA. 1980; 244(17):1934–1938.
249. Oppenheimer, JH, Schwartz, HL, Surks, MI. Propylthiouracil inhibits the conversion of L-thyroxine to L-triiodothyronine. An explanation of the antithyroxine effect of propylthiouracil and evidence supporting the concept that triiodothyronine is the active thyroid hormone. J Clin Invest. 1972; 51(9):2493–2497.
250. Cooper, DS, Saxe, VC, Meskell, M, et al. Acute effects of propylthiouracil (PTU) on thyroidal iodide organification and peripheral iodothyronine deiodination: Correlation with serum PTU levels measured by radioimmunoassay. J Clin Endocrinol Metab. 1982; 54(1):101–107.
251. Nabil, N, Miner, DJ, Amatruda, JM. Methimazole: An alternative route of administration. J Clin Endocrinol Metab. 1982; 54(1):180–181.
252. Walter, RM, Jr., Bartle, WR. Rectal administration of propylthiouracil in the treatment of Graves’ disease. Am J Med. 1990; 88(1):69–70.
253. Yeung, SC, Go, R, Balasubramanyam, A. Rectal administration of iodide and propylthiouracil in the treatment of thyroid storm. Thyroid. 1995; 5(5):403–405.
254. Waseem, M, Seshadri, KG, Kabadi, UM. Successful outcome with methimazole and lithium combination therapy for propylthiouracil-induced hepatotoxicity. Endocr Pract. 1998; 4(4):197–200.
255. Jongjaroenprasert, W, Akarawut, W, Chantasart, D, et al. Rectal administration of propylthiouracil in hyperthyroid patients: Comparison of suspension enema and suppository form. Thyroid. 2002; 12(7):627–631.
256. Ruiz, JK, Rossi, GV, Vallejos, HA, et al. Fulminant hepatic failure associated with propylthiouracil. Ann Pharmacother. 2003; 37(2):224–228.
257. Cooper, DS. Antithyroid drugs. N Engl J Med. 2005; 352(9):905–917.
258. Hodak, SP, Huang, C, Clarke, D, et al. Intravenous methimazole in the treatment of refractory hyperthyroidism. Thyroid. 2006; 16(7):691–695.
259. Zweig, SB, Schlosser, JR, Thomas, SA, et al. Rectal administration of propylthiouracil in suppository form in patients with thyrotoxicosis and critical illness: Case report and review of literature. Endocr Pract. 2006; 12(1):43–47.
260. Mandel, SJ, Berry, MJ, Kieffer, JD, et al. Cloning and in vitro expression of the human selenoprotein, type I iodothyronine deiodinase. J Clin Endocrinol Metab. 1992; 75(4):1133–1139.
261. Watanabe, N, Narimatsu, H, Noh, JY, et al. Antithyroid drug-induced hematopoietic damage: A retrospective cohort study of agranulocytosis and pancytopenia involving 50,385 patients with Graves’ disease. J Clin Endocrinol Metab. 2012; 97(1):E49–53.
262. Das, G, Krieger, M. Treatment of thyrotoxic storm with intravenous administration of propranolol. Ann Intern Med. 1969; 70(5):985–988.
263. Grossman, W, Robin, NI, Johnson, LW, et al. Effects of beta blockade on the peripheral manifestations of thyrotoxicosis. Ann Intern Med. 1971; 74(6):875–879.
264. Abrams, JJ, Sandler, J. Propranolol for thyroid storm. N Engl J Med. 1977; 297:671.
265. Eriksson, M, Rubenfeld, S, Garber, AJ, Kohler, PO. Propranolol does not prevent thyroid storm. N Engl J Med. 1977; 296(5):263–264.
266. Hellman, R, Kelly, KL, Mason, WD. Propranolol for thyroid storm. N Engl J Med. 1977; 297(12):671–672.
267. Verhoeven, RP, Visser, TJ, Doctor, R, et al. Plasma thyroxine, 3,3′,5-triiodothyronine and 3,3′,5′-triiodothyronine during beta-adrenergic blockade in hyperthyroidism. J Clin Endocrinol Metab. 1977; 44(5):1002–1005.
268. McDevitt, DG, Nelson, JK. Comparative trial of atenolol and propranolol in hyperthyroidism. Br J Clin Pharmacol. 1978; 6(3):233–237.
269. Murchison, LE, How, J, Bewsher, PD. Comparison of propranolol and metoprolol in the management of hyperthyroidism. Br J Clin Pharmacol. 1979; 8(6):581–587.
270. Rubenfeld, S, Silverman, VE, Welch, KM, et al. Variable plasma propranolol levels in thyrotoxicosis. N Engl J Med. 1979; 300(7):353–354.
271. Feely, J, Forrest, A, Gunn, A, et al. Propranolol dosage in thyrotoxicosis. J Clin Endocrinol Metab. 1980; 51(3):658–661.
272. Feely, J, Stevenson, IH, Crooks, J. Increased clearance of propranolol in thyrotoxicosis. Ann Intern Med. 1981; 94(4 pt 1):472–474.
273. Peden, NR, Isles, TE, Stevenson, IH, Crooks, J. Nadolol in thyrotoxicosis. Br J Clin Pharmacol. 1982; 13(6):835–840.
274. Mooradian, A, Morley, JE, Simon, G, Shafer, RB. Propranolol-induced hyperthyroxinemia. Arch Intern Med. 1983; 143:2193–2195.
275. Brunette, DD, Rothong, C. Emergency department management of thyrotoxic crisis with esmolol. Am J Emerg Med. 1991; 9(3):232–234.
276. Nieswandt, J, Wagner, S, Schlegel, J, et al. [Cardiopulmonary parameters in hyperthyroidism]. Med Klin (Munich). 1999; 94(1):9–14.
277. Ashikaga, H, Abreu, R, Schneider, RF. Propanolol administration in a patient with thyroid storm. Ann Intern Med. 2000; 132(8):681–682.
278. Dalan, R, Leow, MK. Cardiovascular collapse associated with beta blockade in thyroid storm. Exp Clin Endocrinol Diabetes. 2007; 115(6):392–396.
279. Brennan, MD, van Heerden, JA, Carney, JA. Amiodarone-associated thyrotoxicosis (AAT): Experience with surgical management. Surgery. 1987; 102(6):1062–1067.
280. Bonnyns, M, Sterling, I, Renard, M, et al. Dexamethasone treatment of amiodarone-induced thyrotoxicosis (AIT) with or without persistent administration of the drug. Acta Cardiol. 1989; 44(3):235–243.
281. Bartalena, L, Brogioni, S, Grasso, L, et al. Treatment of amiodarone-induced thyrotoxicosis, a difficult challenge: Results of a prospective study. J Clin Endocrinol Metab. 1996; 81(8):2930–2933.
282. Samaras, K, Marel, GM. Failure of plasmapheresis, corticosteroids and thionamides to ameliorate a case of protracted amiodarone-induced thyroiditis. Clin Endocrinol. 1996; 45(3):365–368.
283. Daniels, GH. Amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2001; 86(1):3–8.
284. Eaton, SE, Euinton, HA, Newman, CM, et al. Clinical experience of amiodarone-induced thyrotoxicosis over a 3-year period: Role of colour-flow Doppler sonography. Clin Endocrinol. 2002; 56(1):33–38.
285. Bogazzi, F, Martino, E, Dell’Unto, E, et al. Thyroid color flow doppler sonography and radioiodine uptake in 55 consecutive patients with amiodarone-induced thyrotoxicosis. J Endocrinol Invest. 2003; 26(7):635–640.
286. Bartalena, L, Wiersinga, WM, Tanda, ML, et al. Diagnosis and management of amiodarone-induced thyrotoxicosis in Europe: Results of an international survey among members of the European Thyroid Association. Clin Endocrinol. 2004; 61(4):494–502.
287. Hermida, JS, Tcheng, E, Jarry, G, et al. Radioiodine ablation of the thyroid to prevent recurrence of amiodarone-induced thyrotoxicosis in patients with resistant tachyarrhythmias. Europace. 2004; 6(2):169–174.
288. Uzan, L, Guignat, L, Meune, C, et al. Continuation of amiodarone therapy despite type II amiodarone-induced thyrotoxicosis. Drug Safety. 2006; 29(3):231–236.
289. Bogazzi, F, Bartalena, L, Dell’Unto, E, et al. Proportion of type 1 and type 2 amiodarone-induced thyrotoxicosis has changed over a 27-year period in Italy. Clin Endocrinol. 2007; 67(4):533–537.
290. Bogazzi, F, Tomisti, L, Rossi, G, et al. Glucocorticoids are preferable to thionamides as first-line treatment for amiodarone-induced thyrotoxicosis due to destructive thyroiditis: A matched retrospective cohort study. J Clin Endocrinol Metab. 2009; 94(10):3757–3762.
291. Bogazzi, F, Bartalena, L, Martino, E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010; 95(6):2529–2535.
292. Eskes, SA, Endert, E, Fliers, E, et al. Treatment of amiodarone-induced thyrotoxicosis type 2: A randomized clinical trial. J Clin Endocrinol Metab. 2012; 97(2):499–506.
293. Baeza, A, Aguayo, J, Barria, M, Pineda, G. Rapid preoperative preparation in hyperthyroidism. Clin Endocrinol. 1991; 35(5):439–442.
294. Langley, RW, Burch, HB. Perioperative management of the thyrotoxic patient. Endocrinol Metab Clin North Am. 2003; 32(2):519–534.
295. Panzer, C, Beazley, R, Braverman, L. Rapid preoperative preparation for severe hyperthyroid Graves’ disease. J Clin Endocrinol Metab. 2004; 89(5):2142–2144.
296. Lazarus, JH, Richards, AR, Addison, GM, Owen, GM. Treatment of thyrotoxicosis with lithium carbonate. Lancet. 1974; 2(7890):1160–1163.
297. Reed, J, Bradley, EL, 3rd. Postoperative thyroid storm after lithium preparation. Surgery. 1985; 98(5):983–986.
298. Dickstein, G, Shechner, C, Adawi, F, et al. Lithium treatment in amiodarone-induced thyrotoxicosis. Am J Med. 1997; 102(5):454–458.
299. Benbassat, CA, Molitch, ME. The use of lithium in the treatment of hyperthyroidism. Endocrinologist. 1998; 8:383–387.
300. Wu, SY, Chopra, IJ, Solomon, DH, Johnson, DE. The effect of repeated administration of ipodate (Oragrafin) in hyperthyroidism. J Clin Endocrinol Metab. 1978; 47(6):1358–1362.
301. Roti, E, Robuschi, G, Manfredi, A, et al. Comparative effects of sodium ipodate and iodide on serum thyroid hormone concentrations in patients with Graves’ disease. Clin Endocrinol. 1985; 22(4):489–496.
302. Wang, YS, Tsou, CT, Lin, WH, Hershman, JM. Long term treatment of Graves’ disease with iopanoic acid (Telepaque). J Clin Endocrinol Metab. 1987; 65(4):679–682.
303. Arem, R, Munipalli, B. Ipodate therapy in patients with severe destruction-induced thyrotoxicosis. Arch Intern Med. 1996; 156(15):1752–1757.
304. Tomaski, SM, Mahoney, EM, Burgess, LP, et al. Sodium ipodate (Oragrafin) in the preoperative preparation of Graves’ hyperthyroidism. Laryngoscope. 1997; 107(8):1066–1070.
305. Braga, M, Cooper, DS. Clinical review 129: Oral cholecystographic agents and the thyroid. J Clin Endocrinol Metab. 2001; 86(5):1853–1860.
306. Mazzaferri, EL, Skillman, TG. Thyroid storm. A review of 22 episodes with special emphasis on the use of guanethidine. Arch Intern Med. 1969; 124(6):684–690.
307. Dillon, PT, Babe, J, Meloni, CR, Canary, JJ. Reserpine in thyrotoxic crisis. N Engl J Med. 1970; 283(19):1020–1023.
308. Roti, E, Montermini, M, Roti, S, et al. The effect of diltiazem, a calcium channel-blocking drug, on cardiac rate and rhythm in hyperthyroid patients. Arch Intern Med. 1988; 148(9):1919–1921.
309. Milner, MR, Gelman, KM, Phillips, RA, et al. Double-blind crossover trial of diltiazem versus propranolol in the management of thyrotoxic symptoms. Pharmacotherapy. 1990; 10(2):100–106.
310. Wartofsky, L, Ransil, BJ, Ingbar, SH. Inhibition by iodine of the release of thyroxine from the thyroid glands of patients with thyrotoxicosis. J Clin Invest. 1970; 49(1):78–86.
311. Myung Park, J, Seok Lee, I, Young Kang, J, et al. Acute esophageal and gastric injury: Complication of Lugol’s solution. Scand J Gastroenterol. 2007; 42(1):135–137.
312. Kinoshita, H, Yasuda, M, Furumoto, Y, et al. Severe duodenal hemorrhage induced by Lugol’s solution administered for thyroid crisis treatment. Intern Med. 2010; 49(8):759–761.
313. Sherman, SI, Simonson, L, Ladenson, PW. Clinical and socioeconomic predispositions to complicated thyrotoxicosis: A predictable and preventable syndrome? Am J Med. 1996; 101(2):192–198.
314. Franklyn, JA, Maisonneuve, P, Sheppard, MC, et al. Mortality after the treatment of hyperthyroidism with radioactive iodine. N Engl J Med. 1998; 338(11):712–718.
315. Parle, JV, Maisonneuve, P, Sheppard, MC, et al. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: A 10-year cohort study. Lancet. 2001; 358(9285):861–865.
316. Osman, F, Gammage, MD, Franklyn, JA. Hyperthyroidism and cardiovascular morbidity and mortality. Thyroid. 2002; 12(6):483–487.
317. Franklyn, JA, Sheppard, MC, Maisonneuve, P. Thyroid function and mortality in patients treated for hyperthyroidism. JAMA. 2005; 294(1):71–80.
318. Nyirenda, MJ, Clark, DN, Finlayson, AR, et al. Thyroid disease and increased cardiovascular risk. Thyroid. 2005; 15(7):718–724.
319. Osman, F, Franklyn, JA, Holder, RL, et al. Cardiovascular manifestations of hyperthyroidism before and after antithyroid therapy: A matched case-control study. J Am Coll Cardiol. 2007; 49(1):71–81.
320. Brandt, F, Green, A, Hegedus, L, Brix, TH. A critical review and meta-analysis of the association between overt hyperthyroidism and mortality. Eur J Endocrinol. 2011; 165(4):491–497.
321. Ross, DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011; 364(6):542–550.
322. Waring, AC, Harrison, S, Samuels, MH, et al. Thyroid function and mortality in older men: A prospective study. J Clin Endocrinol Metab. 2012; 97(3):862–870.
323. Smallridge, RC. Metabolic and anatomic thyroid emergencies: A review. Crit Care Med. 1992; 20(2):276–291.
324. Burch, HB, Wartofsky, L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clinics North Am. 1993; 22(2):263–277.
325. Tietgens, ST, Leinung, MC. Thyroid storm. Med Clin North Am. 1995; 79(1):169–184.
326. Nayak, B, Burman, K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am. 2006; 35(4):663–686.
327. Wartofsky, L. Thyrotoxic storm. In: Braverman LE, Cooper DS, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. 10th ed. Philadelphia: Lippincott Williams & Wilkins; 2012:481–486.
328. Lahey, FH. The crisis of exophthalmic goiter. N Engl J Med. 1928; 199:255–257.
329. McArthur, JW, Rawson, RW, Means, JH, Cope, O. Thyrotoxic crisis; an analysis of the thirty-six cases at the Massachusetts General Hospital during the past twenty-five years. JAMA. 1947; 134(10):868–874.
330. Levy, RP, Gilger, WG. Acute thyroid poisoning; report of a case. N Engl J Med. 1957; 256(10):459–460.
331. Lamberg, BA. The medical thyroid crisis. Acta Med Scand. 1959; 164:479–496.
332. Waldstein, SS, Slodki, SJ, Kaganiec, GI, Bronsky, D. A clinical study of thyroid storm. Ann Intern Med. 1960; 52:626–642.
333. Kamm, ML, Weaver, JC, Page, EP, Chappell, CC. Acute thyroid storm precipitated by labor. Report of a case. Obstet Gynecol. 1963; 21:460–463.
334. Ingbar, SH. Management of emergencies. IX. Thyrotoxic storm. N Engl J Med. 1966; 274(22):1252–1254.
335. Nelson, NC, Becker, WF. Thyroid crisis: Diagnosis and treatment. Ann Surg. 1969; 170(2):263–273.
336. Jacobs, HS, Mackie, DB, Eastman, CJ, et al. Total and free triiodothyronine and thyroxine levels in thyroid storm and recurrent hyperthydroidism. Lancet. 1973; 2(7823):236–238.
337. Mackin, JF, Canary, JJ, Pittman, CS. Thyroid storm and its management. N Engl J Med. 1974; 291(26):1396–1398.
338. Brooks, MH, Waldstein, SS, Bronsky, D, Sterling, K. Serum triiodothyronine concentration in thyroid storm. J Clin Endocrinol Metab. 1975; 40(2):339–341.
339. Brooks, MH, Waldstein, SS. Free thyroxine concentrations in thyroid storm. Ann Intern Med. 1980; 93(5):694–697.
340. Menon, V, McDougall, WW, Leatherdale, BA. Thyrotoxic crisis following eclampsia and induction of labour. Postgrad Med J. 1982; 58(679):286–287.
341. McDermott, MT, Kidd, GS, Dodson, LE, Jr., Hofeldt, FD. Radioiodine-induced thyroid storm. Case report and literature review. Am J Med. 1983; 75(2):353–359.
342. Bennett, WR, Huston, DP. Rhabdomyolysis in thyroid storm. Am J Med. 1984; 77(4):733–735.
343. Tajiri, J, Katsuya, H, Kiyokawa, T, et al. Successful treatment of thyrotoxic crisis with plasma exchange. Crit Care Med. 1984; 12(6):536–537.
344. Braithwaite, SS, Brooks, MH, Collins, S, Bermes, EW. Plasmapheresis: An adjunct to medical management of severe hyperthyroidism. J Clin Apheresis. 1986; 3(2):119–123.
345. Binimelis, J, Bassas, L, Marruecos, L, et al. Massive thyroxine intoxication: Evaluation of plasma extraction. Intensive Care Med. 1987; 13(1):33–38.
346. Wilson, BE, Hobbs, WN. Case report: Pseudoephedrine-associated thyroid storm: Thyroid hormone-catecholamine interactions. Am J Med Sci. 1993; 306(5):317–319.
347. Redahan, C, Karski, JM. Thyrotoxicosis factitia in a post-aortocoronary bypass patient. Can J Anaesth. 1994; 41(10):969–972.
348. Yoshida, D. Thyroid storm precipitated by trauma. J Emerg Med. 1996; 14(6):697–701.
349. Bhattacharyya, A, Wiles, PG. Thyrotoxic crisis presenting as acute abdomen. J R Soc Med. 1997; 90(12):681–682.
350. Cansler, CL, Latham, JA, Brown, PM, Jr., et al. Duodenal obstruction in thyroid storm. South Med J. 1997; 90(11):1143–1146.
351. Lee, TG, Ha, CK, Lim, BH. Thyroid storm presenting as status epilepticus and stroke. Postgrad Med J. 1997; 73(855):61.
352. Ureta-Raroque, SS, Abramo, TJ. Adolescent female patient with shock unresponsive to usual resuscitative therapy. Pediatr Emerg Care. 1997; 13(4):274–276.
353. Ho, SC, Eng, PH, Ding, ZP, et al. Thyroid storm presenting as jaundice and complete heart block. Ann Acad Med Singapore. 1998; 27(5):748–751.
354. Weber, C, Scholz, GH, Lamesch, P, Paschke, R. Thyroidectomy in iodine induced thyrotoxic storm. Exp Clin Endocrinol Diabetes. 1999; 107(7):468–472.
355. Choudhary, AM, Roberts, I. Thyroid storm presenting with liver failure. J Clin Gastroenterol. 1999; 29(4):318–321.
356. Jiang, YZ, Hutchinson, KA, Bartelloni, P, Manthous, CA. Thyroid storm presenting as multiple organ dysfunction syndrome. Chest. 2000; 118(3):877–879.
357. Kadmon, PM, Noto, RB, Boney, CM, et al. Thyroid storm in a child following radioactive iodine (RAI) therapy: A consequence of RAI versus withdrawal of antithyroid medication. J Clin Endocrinol Metab. 2001; 86(5):1865–1867.
358. Lee, HL, Yu, E, Guo, HR. Simultaneous presentation of thyroid storm and diabetic ketoacidosis. Am J Emerg Med. 2001; 19(7):603–604.
359. Reichmann, I, Frilling, A, Hormann, R, et al. [Early operation as a treatment measure in thyrotoxic crisis]. Chirurgia. 2001; 72(4):402–407.
360. De Keulenaer, BL, Lahaye, FJ, Schepens, DR, et al. Thyroid storm presenting with no fever and an absolute adrenal insufficiency. Intensive Care Med. 2002; 28(8):1192.
361. Vora, NM, Fedok, F, Stack, BC, Jr. Report of a rare case of trauma-induced thyroid storm. Ear Nose Throat J. 2002; 81(8):570–572.
362. Scholz, GH, Hagemann, E, Arkenau, C, et al. Is there a place for thyroidectomy in older patients with thyrotoxic storm and cardiorespiratory failure? Thyroid. 2003; 13(10):933–940.
363. Yoon, SJ, Kim, DM, Kim, JU, et al. A case of thyroid storm due to thyrotoxicosis factitia. Yonsei Med J. 2003; 44(2):351–354.
364. Jao, YT, Chen, Y, Lee, WH, Tai, FT. Thyroid storm and ventricular tachycardia. South Med J. 2004; 97(6):604–607.
365. Kokuho, T, Kuji, T, Yasuda, G, Umemura, S. Thyroid storm-induced multiple organ failure relieved quickly by plasma exchange therapy. Ther Apheresis Dial. 2004; 8(4):347–349.
366. Lin, CH, Chen, SC, Lee, CC, et al. Thyroid storm concealing diabetic ketoacidosis leading to cardiac arrest. Resuscitation. 2004; 63(3):345–347.
367. Petry, J, Van Schil, PE, Abrams, P, Jorens, PG. Plasmapheresis as effective treatment for thyrotoxic storm after sleeve pneumonectomy. Ann Thorac Surg. 2004; 77(5):1839–1841.
368. Kanbay, M, Sengul, A, Guvener, N. Trauma induced thyroid storm complicated by multiple organ failure. Chin Med J (Engl). 2005; 118(11):963–965.
369. Ngo, SY, Chew, HC. When the storm passes unnoticed—A case series of thyroid storm. Resuscitation. 2007; 73(3):485–490.
370. Leow, MK, Chew, DE, Zhu, M, Soon, PC. Thyrotoxicosis and acute abdomen—Still as defying and misunderstood today? Brief observations over the recent decade. Q J Med. 2008; 101(12):943–947.
371. Daly, MJ, Wilson, CM, Dolan, SJ, et al. Reversible dilated cardiomyopathy associated with post-partum thyrotoxic storm. Q J Med. 2009; 102(3):217–219.
372. Bish, LT, Bavaria, JE, Augoustides, J. Thyroid storm after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2010; 140(5):e67–69.
373. Koball, S, Hickstein, H, Gloger, M, et al. Treatment of thyrotoxic crisis with plasmapheresis and single pass albumin dialysis: A case report. Artif Organs. 2010; 34(2):E55–58.
374. Vyas, AA, Vyas, P, Fillipon, NL, et al. Successful treatment of thyroid storm with plasmapheresis in a patient with methimazole-induced agranulocytosis. Endocr Pract. 2010; 16(4):673–676.
375. Kandil, E, Khalek, MA, Thethi, T, et al. Thyroid storm in a patient with fulminant hepatic failure. Laryngoscope. 2011; 121(1):164–166.
376. Graettinger, JS, Muenster, JJ, Selverstone, LA, Campbell, JA. A correlation of clinical and hemodynamic studies in patients with hyperthyroidism with and without congestive heart failure. J Clin Invest. 1959; 38(8):1316–1327.
377. Sandler, G, Wilson, GM. The nature and prognosis of heart disease in thyrotoxicosis. A review of 150 patients treated with 131 I. Q J Med. 1959; 28:347–369.
378. Delit, C, Silver, S, Yohalem, SB, Segal, RL. Thyrocardiac disease and its management with radioactive iodine I-131. JAMA. 1961; 176:262–267.
379. Summers, VK, Surtees, SJ. Thyrotoxicosis and heart disease. Acta Med Scand. 1961; 169:661–671.
380. Ikram, H. The nature and prognosis of thyrotoxic heart disease. Q J Med. 1985; 54(213):19–28.
381. Sawin, CT, Geller, A, Kaplan, MM, et al. Low serum thyrotropin (thyroid-stimulating hormone) in older persons without hyperthyroidism. Arch Intern Med. 1991; 151(1):165–168.
382. Krahn, AD, Klein, GJ, Kerr, CR, et al. How useful is thyroid function testing in patients with recent-onset atrial fibrillation? The Canadian Registry of Atrial Fibrillation Investigators. Arch Intern Med. 1996; 156(19):2221–2224.
383. Fadel, BM, Ellahham, S, Ringel, MD, et al. Hyperthyroid heart disease. Clin Cardiol. 2000; 23(6):402–408.
384. Toft, AD, Boon, NA. Thyroid disease and the heart. Heart. 2000; 84(4):455–460.
385. Barbisan, JN, Fuchs, FD, D’Agord Schaan, B. Prevalence of thyroid dysfunction in patients with acute atrial fibrillation attended at a cardiology emergency room. Sao Paulo Med J. 2003; 121(4):159–162.
386. Levey, GS, Klein, I. Catecholamine-thyroid hormone interactions and the cardiovascular manifestations of hyperthyroidism. Am J Med. 1990; 88(6):642–646.
387. Staffurth, JS, Gibberd, MC, Fui, SN. Arterial embolism in thyrotoxicosis with atrial fibrillation. BMJ. 1977; 2(6088):688–690.
388. Bar-Sela, S, Ehrenfeld, M, Eliakim, M. Arterial embolism in thyrotoxicosis with atrial fibrillation. Arch Intern Med. 1981; 141(9):1191–1192.
389. Forfar, JC, Toft, AD. Thyrotoxic atrial fibrillation: An underdiagnosed condition? BMJ (Clin Res Ed). 1982; 285(6346):909–910.
390. Nakazawa, HK, Sakurai, K, Hamada, N, et al. Management of atrial fibrillation in the post-thyrotoxic state. Am J Med. 1982; 72(6):903–906.
391. Scott, GR, Forfar, JC, Toft, AD. Graves’ disease and atrial fibrillation: The case for even higher doses of therapeutic iodine-131. BMJ (Clin Res Ed). 1984; 289(6442):399–400.
392. Petersen, P, Hansen, JM. Stroke in thyrotoxicosis with atrial fibrillation. Stroke. 1988; 19(1):15–18.
393. Sawin, CT, Geller, A, Wolf, PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994; 331(19):1249–1252.
394. Montereggi, A, Marconi, P, Olivotto, I, et al. Signal-averaged P-wave duration and risk of paroxysmal atrial fibrillation in hyperthyroidism. Am J Cardiol. 1996; 77(4):266–269.
395. Feroze, M, May, H. Apathetic thyrotoxicosis. Int J Clin Pract. 1997; 51(5):332–333.
396. Nakazawa, H, Lythall, DA, Noh, J, et al. Is there a place for the late cardioversion of atrial fibrillation? A long-term follow-up study of patients with post-thyrotoxic atrial fibrillation. Eur Heart J. 2000; 21(4):327–333.
397. Frost, L, Vestergaard, P, Mosekilde, L. Hyperthyroidism and risk of atrial fibrillation or flutter: A population-based study. Arch Intern Med. 2004; 164(15):1675–1678.
398. Singer, DE, Albers, GW, Dalen, JE, et al. Antithrombotic therapy in atrial fibrillation: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004; 126(3 Suppl):429S–456S.
399. Forfar, JC, Muir, AL, Sawers, SA, Toft, AD. Abnormal left ventricular function in hyperthyroidism: Evidence for a possible reversible cardiomyopathy. N Engl J Med. 1982; 307(19):1165–1170.
400. Magner, JA, Clark, W, Allenby, P. Congestive heart failure and sudden death in a young woman with thyrotoxicosis. West J Med. 1988; 149(1):86–91.
401. Riaz, K, Forker, AD, Isley, WL, et al. Hyperthyroidism: A “curable” cause of congestive heart failure—Three case reports and a review of the literature. Congest Heart Fail. 2003; 9(1):40–46.
402. Ma, RC, Cheng, AY, So, WY, et al. Thyrotoxicosis and pulmonary hypertension. Am J Med. 2005; 118(8):927–928.
403. Merce, J, Ferras, S, Oltra, C, et al. Cardiovascular abnormalities in hyperthyroidism: A prospective Doppler echocardiographic study. Am J Med. 2005; 118(2):126–131.
404. Soroush-Yari, A, Burstein, S, Hoo, GW, Santiago, SM. Pulmonary hypertension in men with thyrotoxicosis. Respiration. 2005; 72(1):90–94.
405. Marvisi, M, Zambrelli, P, Brianti, M, et al. Pulmonary hypertension is frequent in hyperthyroidism and normalizes after therapy. Eur J Intern Med. 2006; 17(4):267–271.
406. Paran, Y, Nimrod, A, Goldin, Y, Justo, D. Pulmonary hypertension and predominant right heart failure in thyrotoxicosis. Resuscitation. 2006; 69(2):339–341.
407. Siu, CW, Yeung, CY, Lau, CP, et al. Incidence, clinical characteristics and outcome of congestive heart failure as the initial presentation in patients with primary hyperthyroidism. Heart. 2007; 93(4):483–487.
408. Hwang, SH, Hong, KH, Noh, HM, et al. Improvement of severe tricuspid regurgitation with right heart failure associated with thyrotoxicosis due to Graves’ disease. J Cardiovasc Ultrasound. 2009; 17(1):22–24.
409. Hsieh, MH, Chen, CC, Wang, TY, Chang, CT. Chylous ascites as a manifestation of thyrotoxic cardiomyopathy in a patient with untreated Graves’ disease. Thyroid. 2010; 20(6):653–655.
410. Yue, WS, Chong, BH, Zhang, XH, et al. Hyperthyroidism-induced left ventricular diastolic dysfunction: Implication in hyperthyroidism-related heart failure. Clin Endocrinol. 2011; 74(5):636–643.
411. Muggia, AL, Stjernholm, M, Houle, T. Complete heart block with thyrotoxic myocarditis. Report of a case. N Engl J Med. 1970; 283(20):1099–1100.
412. Nadkarni, PJ, Sharma, M, Zinsmeister, B, et al. Thyrotoxicosis-induced ventricular arrhythmias. Thyroid. 2008; 18(10):1111–1114.
413. Bhasin, S, Wallace, W, Lawrence, JB, Lesch, M. Sudden death associated with thyroid hormone abuse. Am J Med. 1981; 71(5):887–890.
414. Herman, GE, Kanluen, S, Monforte, J, et al. Fatal thyrotoxic crisis. Am J Forensic Med Pathol. 1986; 7(2):174–176.
415. Hanterdsith, B, Mahanupab, P. Sudden and unexpected death in a young Thai female due to poorly controlled Graves’ disease: A case report. Am J Forensic Med Pathol. 2010; 31(3):253–254.
416. Resnekov, L, Falicov, RE. Thyrotoxicosis and lactate-producing angina pectoris with normal coronary arteries. Br Heart J. 1977; 39(10):1051–1057.
417. Symmes, JC, Lenkei, SC, Berman, ND. Myocardial infarction, hyperthyroidism and normal coronary arteries: Report of two cases. Can Med Assoc J. 1977; 117(5):489–491.
418. Wei, JY, Genecin, A, Greene, HL, Achuff, SC. Coronary spasm with ventricular fibrillation during thyrotoxicosis: Response to attaining euthyroid state. Am J Cardiol. 1979; 43(2):335–339.
419. Featherstone, HJ, Stewart, DK. Angina in thyrotoxicosis. Thyroid-related coronary artery spasm. Arch Intern Med. 1983; 143(3):554–555.
420. Nakano, T, Konishi, T, Futagami, Y, Takezawa, H. Myocardial infarction in Graves’ disease without coronary artery disease. Jpn Heart J. 1987; 28(3):451–456.
421. Nakano, T, Konishi, T, Takezawa, H. Vasospastic angina in thyrotoxicosis—Case reports. Angiology. 1987; 38(9):717–722.
422. Bergeron, GA, Goldsmith, R, Schiller, NB. Myocardial infarction, severe reversible ischemia, and shock following excess thyroid administration in a woman with normal coronary arteries. Arch Intern Med. 1988; 148(6):1450–1453.
423. Carey, D, Hurst, JW, Jr., Silverman, ME. Coronary spasm and cardiac arrest after coronary arteriography in unsuspected thyrotoxicosis. Am J Cardiol. 1992; 70(7):833–834.
424. Moliterno, D, DeBold, CR, Robertson, RM. Case report: Coronary vasospasm—Relation to the hyperthyroid state. Am J Med Sci. 1992; 304(1):38–42.
425. Phull, PS, Collins, CE, Norell, MS, Thomas, DJ. Variant angina in thyrotoxicosis. Br J Clin Pract. 1993; 47(1):17–18.
426. Masani, ND, Northridge, DB, Hall, RJ. Severe coronary vasospasm associated with hyperthyroidism causing myocardial infarction. Br Heart J. 1995; 74(6):700–701.
427. Choi, YH, Chung, JH, Bae, SW, et al. Severe coronary artery spasm can be associated with hyperthyroidism. Coron Artery Dis. 2005; 16(3):135–139.
428. Kuang, X-H, Zhang, S-Y. Hyperthyroidism-associated coronary spasm: A case of non-ST segment elevation myocardial infarction with thyrotoxicosis. J Geriatr Cardiol. 2011; 8:258–259.