[level-membership-for-anesthesiology-category]
Chapter 74 Thermal disorders
Body temperature is normally very tightly controlled by a balance between heat production and heat loss, through a complex feedback mechanism involving the thermoregulatory centre in the hypothalamus. In the intensive care unit (ICU), fever (pyrexia) is usually due to resetting of the thermoregulatory set-point at a higher level by activation of heat-conserving mechanisms, whereas hyperthermia is due to failure of effector mechanisms to maintain body temperature at the normal set-point.
THERMOREGULATION
The three major components of the thermoregulatory system are:
Humoral mediators from the circulation act to alter temperature primarily via the organum vasculosum of the lamina terminalis (OVLT), an area of fenestrated capillaries in the hypothalamus that permits cytokine access to neuronal receptors. Cytokines appear to be the endogenous pyrogens, with interleukin-6 (IL-6) and prostaglandin-E2 (PGE2) being a final common pathway. In addition to elevating body temperature, several cytokines also reduce the thermoregulatory set-point, and are known as endogenous cryogens.1
FEVER IN THE ICU
Fever is defined by a regulated hyperthermia, that is, it is a regulated elevation in the preoptic set-point temperature. Endogenous pyrogens as well as other mediators inhibit warm-sensitive neurones that normally facilitate heat loss and suppress heat production. This elevates the set-point temperature for all thermoregulatory responses and activates cold defences such as vasoconstriction and shivering, which decrease heat loss and increase metabolic heat production respectively. The set-point temperature returns to normal when pyrogen concentrations decrease, triggering heat loss by vasodilatation and sweating.2
Fever may reflect a wide variety of pathological processes including infection, inflammation, trauma, malignancy and connective tissue diseases (Table 74.1), necessitating a systematic and comprehensive diagnostic approach.3 It is often assumed that a patient presenting with a fever should be treated, regardless of the presence or absence of other symptoms. However, the evidence that anti-fever treatments lead to an improvement in morbidity or mortality, or even patient comfort, is lacking.4
| System | Infectious aetiology | Non-infectious aetiology |
|---|---|---|
| Cardiovascular | Endocarditis | Myocardial infarction |
| Catheter-related infection | Deep-vein thrombosis | |
| Pacemaker infection | Pericarditis | |
| Respiratory | Pneumonia | Atelectasis |
| Empyema | Chemical pneumonitis | |
| Sinusitis | Pulmonary emboli | |
| Alimentary | Abdominal abscess | Inflammatory bowel disease |
| Biliary infection | Acalculous cholecystitis | |
| Peritonitis | Pancreatitis | |
| Diverticulitis | Ischaemic colitis | |
| Viral hepatitis | Non-viral hepatitis | |
| Antibiotic-related colitis | Gastrointestinal haemorrhage | |
| Renal | Pyelonephritis | |
| Urinary tract infection | ||
| Central nervous | Meningitis | Cerebral haemorrhage/infarct |
| Encephalitis | Seizures | |
| Rheumatological | Septic arthritis | Connective tissue disease |
| Osteomyelitis | ||
| Gout | Vasculitis | |
| Endocrine | Adrenocortical insufficiency | |
| Alcohol and drug withdrawal | ||
| Hyperthyroidism | ||
| Skin/soft tissue | Cellulitis | Burns |
| Decubitus ulcer | Intramuscular injections | |
| Wound infections | ||
| Haematoma | ||
| Other | Parotitis | Drug fever |
| Pharyngitis | Transfusion reaction | |
| Otitis media | Neoplasms |
The development of fever in response to infection may be a protective adaptive response, and appears to be a phylogenetically preserved evolutionary response because of its survival value.5 In mammalian models, increasing body temperature results in enhanced resistance to infection. In humans, retrospective clinical trials have shown a positive correlation between maximum temperature on the day of bacteraemia and increased survival in patients with Gram-negative bacteraemia and spontaneous bacterial peritonitis.6 Also, septic patients with hypothermia have a poorer outcome than those who develop fever, although this causality is less clear. Both local and systemic hyperthermia has been used to facilitate cancer treatment. The protective effects of fever result from increased immune and cytokine functions.7–10
Temperature elevation has been shown to enhance:
In addition, elevated temperatures inhibit some pathogens, such as Streptococcus pneumoniae.11
Moderate fever is a common occurrence in ICU patients, but approximately half of these are non-infectious in origin.12–14 The presence of fever frequently results in the performance of diagnostic tests and exposes the patient to unnecessary invasive procedures and inappropriate use of antibiotics.15
Whilst very high fevers (> 40°C) are dangerous, it is less clear whether moderate elevation of body temperature is detrimental and, indeed, may be protective.4 Moreover, artificially lowering the temperature of a febrile patient may mask the signs of infection and make diagnosis and monitoring more difficult. Any decision to adopt anti-fever measures, physical or pharmacological, must take into consideration the variable response by this patient population. Antipyretics may be ineffective.The usual concern about external cooling measures inducing peripheral vasoconstriction, reducing heat loss and making the pyrexia worse by shivering and hypermetabolism, may not be observed in sedated ICU patients.16 The most likely cause for this response is the drugs used to maintain sedation.17,18
Pyrexia is associated with a number of deleterious physiological effects. Cardiac output, oxygen consumption, carbon dioxide production and energy expenditure are all increased, particularly in the presence of shivering. Oxygen consumption is increased on average by 10%/°C.2 These changes are poorly tolerated by patients with limited cardiorespiratory reserve, and this group of patients would probably derive benefit from cooling measures. Other patient groups that require special consideration include those with immunosuppression, prosthetic implants and acute brain injury. Recent trials of therapeutic moderate hypothermia and traumatic brain injury indicate that hypothermia is a complicated treatment that is likely to benefit only a subgroup of patients with traumatic brain injury.19–21
HEAT STROKE
Exertional heat stroke is a consequence of prolonged, intense exercise in warm humid environments, often seen in athletes and military recruits. Classic heat stroke is commonly seen in sedentary, elderly patients with underlying illnesses during heat waves. Factors predisposing to heat stroke are listed in Table 74.2. About 80% of heat stroke deaths occur in people aged 50 years and older, because of the diminished ability of the older body to compensate for increased core temperatures. Heat stroke is estimated to be the cause of approximately 1700 deaths each year in the USA.22 The European heat wave of 2003 was responsible for > 14 000 excess deaths within 2 weeks in France alone, of which a third were attributed to heat stroke, hyperthermia or dehydration.23,24 A high mortality rate of > 62% was reported for this cohort, which is higher than that for leading killers in ICUs such as acute respiratory distress syndrome (ARDS) and septic shock.25 Furthermore, there is a late mortality contributed by survivors who have sustained neurological injury. A study of former heat stroke patients suggests that susceptible individuals have a poorer physiological response to heat stress in terms of core temperature, heart rate and sweat response.
| Age | Elderly |
| Environmental | High ambient temperature and humidity |
| Heat waves | |
| Poor ventilation | |
| Behavioural | Lack of acclimatisation |
| Salt and water deprivation | |
| Obesity | |
| Underlying conditions | Infection/fever |
| Diabetes | |
| Malnutrition | |
| Alcoholism | |
| Hyperthyroidism | |
| Impaired sweat production | |
| Healed burns | |
| Ectodermal dysplasia | |
| Impaired sweating | |
| Cardiovascular disease | |
| Fatigue | |
| Potassium deficiency | |
| Drugs | Anticholinergics |
| Antiparkinsonians | |
| Antihistamines | |
| Butyrophenones | |
| Phenothiazines | |
| Tricyclics | |
| Diuretics | |
| Sympathomimetics |
PATHOGENESIS
The pathogenesis of multiple organ failure in heat stroke is complex. Although direct cellular damage from increased temperature constitutes the initiating insult,26 the precise sequence of injury and responsible mediators are poorly understood. At the cellular level, thermal injury results in increased membrane permeability, which in turn stimulates membrane enzymes such as Na+K+-ATPase to maintain membrane integrity. This ATP-consuming enzyme activity is also responsible for nerve impulse conduction, which ismarkedly curtailed when ATP is depleted. This results in tissue oedema, reduced oxygen extraction and neuronal injury. High temperatures ameliorate ATP synthesis leading to fatigue.
Recent evidence suggests that the pathways for tissue injury in heat stroke share many features with that of sepsis, endotoxaemia and systemic inflammation. Increased levels of circulating endotoxin and cytokines have been identified in patients with heat stroke.27,28 The use of anti-endotoxin antibodies in primate models of heat stroke suggests that endotoxin at least in part mediates the tissue injury associated with hyperthermia. There was also a significant correlation between plasma IL-6 concentration and the severity of heat stroke. Since this cytokine is known to modulate the hypothalamic set-point, the ramifications of such a response in an already hyperthermic patient are obvious.
Activation of coagulation factors29 and release of endothelin and adhesion molecules30,31 from activated or injured endothelium have also been demonstrated in heat stroke. These recent observations lead to the speculation that certain mediators that are implicated in the pathogenesis of acute organ injury are also elevated in heat stress, but become intense when heat stroke develops and are not normalised upon cooling.
CLINICAL PRESENTATION
Dehydration follows excessive insensible losses although sweating is generally absent in the terminal stages of classic heat stroke, leaving a hot, dry skin. Hypovolaemia is a consequence of dehydration and fluid redistribution, and results in reduced organ perfusion. A severe metabolic (lactic) acidosis is present. The major biochemical abnormalities include hyperglycaemia, hypophosphataemia, and raised serum enzymes and acute phase proteins (Table 74.3). Haematological findings include leukocytosis, thrombocytopenia, and activation of coagulation and fibrinolysis.
Table 74.3 Biochemical differences between classic and exertional heat stroke
| Classic heat stroke | Exertional heat stoke | |
|---|---|---|
| Arterial gases | Mixed respiratory alkalosis | Severe metabolic acidosis |
| Serum electrolytes | Na+, Mg2+, Ca2+ are usually normal | HyperkalaemiaHypocalcaemia |
| Hypophosphataemia | Hyperphosphataemia | |
| Blood glucose | Hyperglycaemia | Hypoglycaemia |
| Creatinine kinase | Moderately increased | Markedly increased |
| Hepatic enzymes | Markedly increased | Moderately increased |
| Acute phase proteins | Markedly increased | Moderately increased |
Exertional heat stroke differs slightly in that additional findings include rhabdomyolysis and acute renal failure that is associated with hyperkalaemia, hyperphosphataemia and hypocalcaemia (Table 74.3).
MANAGEMENT
Evaporation is considerably more effective. Pharmacological treatment with antipyretic agents, or dantrolene,32 is ineffective. Prevention of vasoconstriction and shivering by overcooling is important because of the danger of subsequent rebound hyperthermia. Core and skin temperature monitoring is useful, but measurement of rectal temperature should be avoided because it lags considerably during cooling. Cooling can be stopped when core temperature reaches below 39°C. However, despite cooling, about 25% of patients experience failure of one or more organ systems.
OUTCOME
The largest study reported of heat stroke patients in intensive care suggests an alarmingly high mortality of > 60%,23 although this diminishes substantially with early recognition and aggressive treatment. The incidence of permanent neurological deficit remains at 7–15%. Variables associated independently with reduced hospital survival include:
DRUG-INDUCED HYPERTHERMIAS
The numerous causes of hyperthermia are listed in Table 74.4. This section will review the relatively common causes of drug-induced hyperthermias, including malignant hyperthermia, neuroleptic malignant syndrome, and the sympathomimetic and anticholinergic syndromes.
| Disorders of excessive heat production | Exertional hyperthermia |
| Heat stroke (exertional) | |
| Malignant hyperthermia | |
| Neuroleptic malignant syndrome | |
| Lethal catatonia | |
| Thyrotoxicosis | |
| Phaeochromocytoma | |
| Salicylate intoxication | |
| Sympathomimetic drug abuse | |
| Delirium tremens | |
| Seizures | |
| Tetanus | |
| Disorders of diminished heat dissipation | Heat stroke (classic) |
| Dehydration | |
| Autonomic dysfunction | |
| Anticholinergic poisoning | |
| Neuroleptic malignant syndrome | |
| Disorders of hypothalamic function | Cerebrovascular accidents |
| Encephalitis | |
| Trauma | |
| Granulomatous diseases | |
| Neuroleptic malignant syndrome |
MALIGNANT HYPERTHERMIA
CLINICAL PRESENTATION
The earliest sign of an impending MH crisis is an unexplained rise in end-tidal CO2 and heart rate, and not necessarily an increase in body temperature. This classic crisis of acute fulminant MH with its multiplicity of marked metabolic and muscle anomalies and sympathetic stimulation is unmistakable, but now rare. The pattern of presentation has altered since its first description in 1960, as anaesthetic techniques and succinylcholine use have changed.
The more gradual appearance of signs following exposure to triggering agents may be more difficult to diagnose as the signs may be subtle, non-specific and have variable intensity, incidence and temporal association (Table 74.5).33 Apart from the classic crisis, other forms of MH – including smouldering, recurring, delayed and abortive – may also occur. Other conditions that may mimic MH include inadequate levels of anaesthesia or analgesia, sepsis, ischaemia or anaphylaxis. Early diagnosis is important as immediate treatment is associated with improved outcome. Masseter muscle spasm (MMS) has been associated with MH.34 When MMS is the only presenting sign, the incidence of MH susceptibility is likely to be low. However, this incidence is increased if MMS is associated with other muscle or metabolic signs.
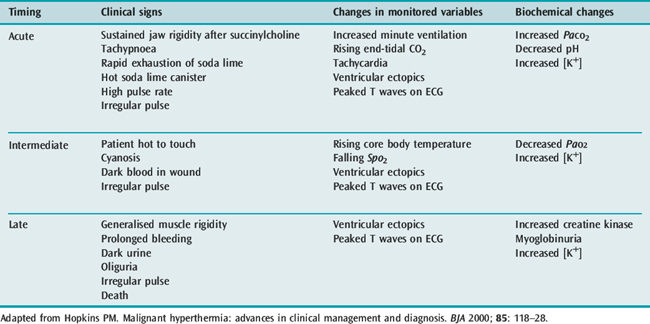
Halothane is the most potent of the contemporary volatile anaesthetic agents at inducing sustained contractures in isolated muscle strips from MH patients, and has formed the basis of diagnostic testing for MH for 30 years. There may be a difference amongst the volatile anaesthetics in their relative potency to trigger an MH reaction. Succinylcholine will cause an increase in the calcium concentration in the cytosol of normal muscle, and it appears that this release of calcium is exaggerated in MH muscle. Non-depolarising muscle relaxants are generally accepted to be safe in MH. A list of safe and implicated drugs is shown in Table 74.6.
| Contraindicated drugs | Safe drugs |
|---|---|
| Halothane | Nitrous oxide |
| Enflurane | Barbiturates |
| Isoflurane | Propofol |
| Desflurane | Etomidate |
| Sevoflurane | Ketamine |
| Succinylcholine | Opiates |
| Verapamil | Amide/ester local anaesthetics |
| Nifedipine | Noradrenaline (norepinephrine) |
| Diltiazem | Adrenaline (epinephrine) |
| Dopamine | |
| Dobutamine |
MOLECULAR GENETICS OF MH
MH is a heterogenetic disorder that is autosomal dominant with variable penetration and expression. Recent advances in molecular genetics have supported the concept for a role of the RYR1 gene in the pathogenesis of MH. The RYR1 gene is found at position 13.1 on chromosome 19 q and codes for the ryanodine receptor that regulates calcium transport across the calcium release channel. However, only 50% of MH families show genetic linkage to the RYR1 gene, and more than 17 mutations at this locus have been described so far. The mutation does not always segregate with in vitro contracture testing within a family. Furthermore, RYR1 linkage is not found in some MH families. Although all known mutations share the final common pathway of excessive calcium release from the SR, the genetic diversity of this disease precludes DNA-based diagnosis at present.
MANAGEMENT
NEUROLEPTIC MALIGNANT SYNDROME
PATHOGENESIS
A common pathogenesis of NMS and MH has been suggested by virtue of similar clinical features (hyperthermia, rigidity, raised serum muscle enzymes), abnormal in vitro contracture tests and, interestingly, successful treatmentwith dantrolene in both. However, conflicting results have been reported regarding the prevalence of MH susceptibility among NMS patients. Neuroleptic agents induce muscle contracture in vitro; however, no difference was found in response to four neuroleptic drugs between muscle from NMS patients and normal individuals.35
CLINICAL PRESENTATION
A predictable progression of symptoms may be identified in many patients with NMS where mental status changes and rigidity precede hyperthermia and autonomic dysfunction:36
SEROTONERGIC SYNDROME
Clinical features of the serotonergic syndrome include encephalopathy, hyperreflexia, nausea and vomiting, and autonomic instability. Sweating and mild increases in temperature are evident in 50% of cases. Rhabdomyolysis, hyperkalaemia, renal failure, DIC and seizures are all rare but reported findings. The serotonergic syndrome is usually self-limited, and resolves uneventfully once the offending drug has been withdrawn. However, rarely severe forms require more aggressive supportive therapy analogous to the treatment of NMS. Specific treatments including the use of serotonin antagonists have beenproposed; however, these cannot be recommended at present due to lack of well-designed studies.
MANAGEMENT
The benefit of adding specific pharmacotherapies in addition to supportive measures is unclear, but potential use cannot be excluded. Treatments with bromocriptine (a dopamine agonist) and dantrolene have led to a faster resolution of symptoms than with supportive therapy alone.37 However, the place of dantrolene in the treatment of NMS is less well defined. Bromocriptine seems to be well tolerated by psychotic patients despite being a strong central dopamine agonist. The drug is effective within 24 h, with a reduction in rigidity followed by resolution of temperature and normalisation of blood pressure. Similarly, amantadine, as well as a combination of levodopa-carbidopa, has also been reported to be effective. Anticholinergic drugs have little effect on muscle rigidity or hyperthermia. Non-specific adjuncts, such as the use of benzodiazepines, have been reported to be useful in agitated patients. Electroconvulsive therapy (ECT) may be of value in selected patients, e.g. those with refractory NMS, those who remain catatonic or those with ECT-responsive psychotic symptoms.
SYMPATHOMIMETIC AND ANTICHOLINERGIC SYNDROMES
SYMPATHOMIMETIC POISONING
Central thermoregulatory disturbances from sympathomimetics may arise from complex interactions of these neurotransmitters in the brainstem and hypothalamus. The syndrome of hyperthermia associated with sympathomimetic poisoning bears similarity to that of heat stroke, MH, NMS and serotonin syndrome. This may reflect the final common pathway associated with the consequences of severe hyperthermia. Sympathomimetics such as MDMA (‘ecstasy’) have been shown to induce marked hyperthermia through central mechanisms involving 5-HT2 receptors, with subsequent rhabdomyolysis, DIC and multiorgan failure.38 Hyperkinetic muscle action, motor excitability and seizures may contribute to the rise in core temperature. Furthermore, elevated levels of both cocaine and amphetamine result in peripheral vasoconstriction, thus impairing heat dissipation. Mortality appears to be related to the extent and duration of hyperthermia.
ANTICHOLINERGIC POISONING
whereas peripheral signs of toxicity include:
Muscular hyperactivity from agitation, restlessness and convulsions in combination with impaired sweating mayresult in hyperthermia and rhabdomyolysis. Excessive temperatures can lead to multiorgan failure as described for other types of heat illness.
HYPOTHERMIA
Hypothermia is defined as a core body temperature below 35 °C. In the UK, hypothermia accounts for 1% of winter admissions, particularly amongst the elderly population. The leading causes of hypothermia in the USA are exposure due to alcoholism, drug addiction, mental illness, or accidents involving immersion in cold water. The mortality rate from accidental hypothermia varies according to its severity, but averages 21% when core temperature is decreased to 28–32°C.39 However, a core temperature of 32 °C or less in trauma victims is associated with a mortality rate near 100%, and any hypothermia is considered a poor prognostic sign.40,41
Hypothermia is traditionally classified as:
The causes and predisposing conditions of hypothermia are listed in Table 74.7; however, the most frequent causes appear to be exposure, hypoglycaemia and the use of depressant drugs including alcohol. An impaired thermoregulatory system together with a reduced functional reserve makes the elderly more susceptible.
Table 74.7 Causes and predisposing conditions of hypothermia
| Age | Extremes of age |
| Environmental | Exposure to cold |
| Immersion | |
| Poor living conditions | |
| Drugs | Anaesthetic agents |
| Phenothiazines | |
| Barbiturates | |
| Alcohol | |
| Central nervous system disorders | Cerebrovascular accidents |
| Trauma | |
| Spinal cord transections | |
| Brain tumours | |
| Wernicke’s encephalopathy | |
| Alzheimer’s and Parkinson’s disease | |
| Mental illness | |
| Endocrine dysfunction | Hypoglycaemia |
| Diabetic ketoacidosis | |
| Hyperosmolar coma | |
| Panhypopituitarism | |
| Hypoadrenalism | |
| Hypothyroidism | |
| Trauma | Major trauma |
| Debility | Severe cardiac, renal, hepatic impairment |
| Malnutrition, sepsis | |
| Skin disorders | Burns |
| Exfoliative dermatitis |
Administration of anaesthesia impairs the ability to maintain thermal homeostasis, decreases heat production and causes heat loss due to vasodilatation and exposure. General anaesthesia also alters the threshold for thermoregulatory vasoconstriction and shivering in the non-paralysed patient.42
PATHOGENESIS AND CLINICAL PRESENTATION
Shivering occurs in the early phase of hypothermia and is characterised by intense heat and energy production from the metabolism of stored fuels. Non-shivering thermogenesis is probably only of importance in children. Metabolic processes slow by approximately 6%/°C, and the metabolic rate is reduced by half at 28°C.43 A mixed respiratory and metabolic acidosis results from hypoventilation and reduced tissue perfusion, leading to lactate accumulation from anaerobic metabolism. Hepatic function is depressed, affecting most enzymatic and detoxifying processes. There is a high risk of developing pancreatitis.
There is generalised cerebral depression as the metabolism of the brain declines with a fall in core temperature. This adaptation is neuroprotective and may improve the chances of survival even after prolonged hypothermic arrest. Cerebral blood flow falls as a consequence of reduced cardiac output and increased blood viscosity at a rate of 7%/°C drop in temperature.44 Confusion can cause illogical behaviour (e.g. aggression) and paradoxical undressing. Coma, pupillary dilatation, absence of tendon reflexes and rigidity are present below 28°C. Cerebral electrical activity ceases below 20°C.
MANAGEMENT
Once the diagnosis of hypothermia has been made, further heat loss must be prevented and rewarming started with close monitoring to avoid complications. Individual management should be modified according to aetiology and severity of hypothermia, as well as the functional reserve of the patient. Severe hypothermia, especially in the immersion victim, can mimic death, with apnoea, cardiac standstill, coma, unreactive pupils, and a silent electrocardiogram and electroencephalogram. Successful resuscitation of such patients has been reported and death should not be assumed until resuscitation has failed in an adequately warmed patient (at least 35°C).45
Recommendation for basic and advanced life support for hypothermic patients is according to the principles of Advanced Cardiorespiratory Life Support (ACLS). Aggressive rewarming should be continued during resuscitation until the core temperature is at least 35°C. A resuscitation protocol based on core temperature and cardiac monitoring may be used.46 If core temperature is unknown or known to be above 28°C, cardiopulmonary resuscitation (CPR) should be instituted for apparent cardiac arrest. If the patient is known to be severely hypothermic (< 28°C) but maintains sinus rhythm, chest compression may precipitate ventricular fibrillation (VF) and may be best withheld.
Sodium bicarbonate should be avoided because of paradoxical intracellular acidosis, and severe alkalosis on rewarming may induce refractory VF and left shift of the oxyhaemoglobin dissociation curve, thereby reducing tissue oxygen uptake. Progressive cardiac depression during rewarming (‘recovery shock’, ‘afterdrop’) may be due to further cooling of blood as it redistributes from the core to the relatively colder periphery.47
REWARMING
Various methods of rewarming have been employed depending upon the severity of the hypothermia (Table 74.8). Careful monitoring and supportive therapy are mandatory during rewarming.
| Passive | Warm environment > 30°C (rate 0.5–1.0 °C/hour) |
| Insulating cover (warm blanket) | |
| Active, external | Conduction methods |
| Warmed pads, blanket | |
| Convective methods (rate at 2–3°C/hour) | |
| Hot air blower (e.g. Bair Hugger) | |
| Radiant methods | |
| Active, core | Humidified warm inspired gases (rate 0.5–1.5°C/hour) |
| Warmed intravenous fluids | |
| Body cavity lavage (rate 2–3°C/hour) | |
| Gastric irrigation | |
| Pleural irrigation | |
| Peritoneal dialysis | |
| Extracorporeal methods | |
| Haemodialysis, continuous arteriovenous or venovenous rewarming (rate 5°C/hour) | |
| Cardiopulmonary bypass (rate up to 10°C/ hour) |
Patients with moderate or severe hypothermia should be treated with active rewarming, which consists of active external rewarming or active core rewarming. Patients with moderate hypothermia and no evidence of circulatory collapse can initially be treated with active external rewarming techniques. These include use of immersion, radiant heat, forced air and electric blankets. Convective (forced air) warming at 43°C has been shown to increase body temperature by 2–3°C/hour, and is extremely effective in both preventing and treating hypothermia, as well as preventing shivering in the postoperative period.48,49
Peritoneal lavage with heated dialysate, and closed pleural irrigation using warm sterile saline (temperature 40–42°C) through large bore tubes may also be attempted.50,51 These methods are equally effective in raising temperature (2–3°C/h), but impossible in patients with thoracoabdominal injuries.
Haemodialysis and continuous arteriovenous or venovenous rewarming techniques are extremely effective in raising body temperature (5 °C/h). The advantages of this technique include non-requirement of heparinisation if heparin-bonded tubing is used, rapid reversal of hypothermia, decreased total fluid requirements, decreased organ failure, decreased length of ICU stay and decreased early mortality.52 Patients with severe cardiovascular dysfunction may not tolerate high arteriovenous fistula flows.
The best method for rewarming patients with severe hypothermia who have haemodynamic instability involves the use of cardiopulmonary bypass. Its advantages include the highest rewarming rate (up to 10°C/h), control of rewarming rate, oxygenation, fluid composition and haemodynamic support.53 However, associated risks include heparinisation, haemolysis and air embolism.
1 Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1996;71:93-127.
2 Lenhardt R, Kurz A, Sessler DI. Thermoregulation and hyperthermia. Acta Anaesthesiol Scand Suppl. 1996;40:34-38.
3 O’Grady WP, Barie PS, Bartlett JG, et al. Practice guidelines for evaluating new fever in critically ill adult patients? Clin Infect Dis. 1998;26:1042-1059.
4 Gozolli V, Schottker P, Suter PM, et al. Is it worth treating fever in intensive care unit patients. Arch Int Med. 2001;161:121-123.
5 Kluger MJ, Kozak W, Conn CA, et al. The adaptive value of fever. Infect Dis Clin North Am. 1996;10:1-20.
6 Weinstein MR, Iannini PB, Stratton CW, et al. Spontaneous bacterial peritonitis: a review of 28 cases with emphasis on improved survival and factors influencing prognosis. Am J Med. 1978;64:592-598.
7 Azocar J, Yunis EJ, Essex M. Sensitivity of human natural killer cells to hyperthermia. Lancet. 1982;1:16-17.
8 Biggar W, Bohn DJ, Kent G, et al. Neutrophil migration in vitro and in vivo during hyperthermia. Infect Immunol. 1984;46:857-859.
9 Jampel HD, Duff GW, Gershon RK, et al. Fever and immunoregulation: III. Hyperthermia augments the primary in vitro humoral immune response. J Exp Med. 1983;157:1229-1238.
10 van Oss CJ, Absolom DR, Moore LL, et al. Effect of temperature on chemotaxis, phagocyte engulfment, digestion and oxygen consumption of human polymorphonuclear leucocytes. J Reticuloendothel Soc. 1980;27:561-565.
11 Styrt B, Sugarman B. Antipyresis and fever. Arch Intern Med. 1990;150:1589-1597.
12 Cunha BA, Shea KW. Fever in the intensive care unit. Infect Dis Clin North Am. 1996;10:185-209.
13 Circiumaru B, Baldock G, Cohen J. A prospective of fever in the intensive care unit. Intensive Care Med. 1999;25:668-673.
14 Cunha BA. Intensive care, not intensive antibiotics. Heart Lung. 1994;23:361-362.
15 Marik PE. Fever in the ICU. Chest. 2000;117:855-869.
16 Poblette B, Romand JA, Pilchard C, et al. Metabolic effects of intravenous propacetamol, metamizol or external cooling in critically ill febrile sedated patients. BJA. 1997;78:123-127.
17 Kurz A, Go JC, Sessler DI. Alfentanil slightly increases the sweating threshold and markedly reduces the vasoconstrictor and shivering thresholds. Anesthesiology. 1995;83:293-299.
18 Kurz A, Sessler DI, Annadata R, et al. Midazolam minimally impairs thermoregulatory control. Anesth Analg. 1995;81:393-398.
19 Hindman BJ, Todd MM, Gelb AW, et al. Mild hypothermia as a protective therapy during intracranial aneurysm surgery: a randomized prospective pilot trial. Neurosurgery. 1999;44:23-33.
20 Marion DW, Penrod LE, Kelsey SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997;336:540-546.
21 Bernard SA, Jones BM, Buist M. Experience with prolonged induced hypothermia in severe head injury. Crit Care. 1999;3:167-172.
22 Centers for Disease Control. Heat related illness and deaths: United States, 1994–95. MMWR. 1995;44:465-468.
23 Hemon D, Jorgla E. The heat wave in France in August 2003. Rev Epidemiol Sante Publique. 2004;52:3-5.
24 Kosatsky T. The 2003 European heat waves. Eur Surveill. 2005;10:1-5.
25 Missett B, De Jonghe B, Bastuji-Garin S, et al. Mortality of patients with heatstroke admitted to intensive care units during the 2003 heat wave in France: a national multiple-center risk factor study. Crit Care Med. 2006;34:1087-1092.
26 Sminia P, van der Zee J, Wondergem J, et al. Effect of hyperthermia on the central nervous system: a review. Int J Hyperthermia. 1994;10:1-30.
27 Bouchama A, Parhar RS, El-Yazigi A, et al. Endotoxaemia and release of TNF and IL-1α in acute heat stroke. J Appl Physiol. 1991;70:2640-2644.
28 Bouchama A, Al-Sedairy S, Siddiqui S, et al. Elevated pyrogenic cytokines in heat stroke. Chest. 1993;104:1498-1502.
29 Bouchama A, Bridley F, Hammami MM, et al. Activation of coagulation and fibrinolysis in heat stroke. Thrombosis Haemostasis. 1996;76:909-915.
30 Bouchama A, Hammami MM, Hay A, et al. Evidence for endothelial cell activation/injury in heatstroke. Crit Care Med. 1996;24:1173-1178.
31 Hammami MM, Bouchama A. Levels of soluble L-selectin and E-selectin in heatstroke and heatstress. Chest. 1998;114:949-950.
32 Bouchama A, Cafege A, deVol EB, et al. Ineffectiveness of dantrolene sodium in the treatment of heatstroke. Crit Care Med. 1991;19:176-180.
33 Hopkins PM. Malignant hyperthermia: advances in clinical management and diagnosis. BJA. 2000;85:118-128.
34 Ramirez JA, Cheetham ED, Laurence AS. Succinylcholine, masseter spasm and later malignant hyperthermia. Anaesthesia. 1998;53:1111-1116.
35 Reyford HG, Cordonnier C, Adnet P, et al. The in vitro exposure of muscle strips from patients with neuroleptic malignant syndrome cannot be correlated with the clinical features. J Neurol Sci. 1990;98:527.
36 Velamoor VR, Norman R, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182:168-213.
37 Rosenberg MR, Green M. Neuroleptic malignant syndrome: a review of response to therapy. Arch Intern Med. 1989;149:1927-1931.
38 McKenna DJ, Peroutka SJ. Neurochemistry and neurotoxicity of 3,4 methylenediaoxymetamphetamine (MDMA, ‘ecstasy’). J Neurochem. 1990;54:14-22.
39 Danzl D, Pozos RS, Auerbach PS, et al. Multicentre hypothermia survey. Ann Emerg Med. 1987;16:1042-1055.
40 Jurkovic GJ, Greiser WB, Luterman A, et al. Hypothermia in trauma victims: an ominous predictor of survival. J Trauma. 1987;27:1019-1024.
41 Smith CE. Focus on: Perioperative hypothermia. Trauma and hypothermia. Curr Anaesth Crit Care. 2001;12:87-95.
42 Sessler DI. Consequences and treatment of perioperative hypothermia. Anesthesiol Clin North Am. 1994;12:425-456.
43 Corneli HM. Accidental hypothermia. J Pediatr. 1992;120:671-679.
44 Morley-Forster PK. Unintentional hypothermia in the operating room. Can Anaesth Soc J. 1986;33:516-527.
45 Larach MG. Accidental hypothermia. Lancet. 1995;345:493-498.
46 Zell SC, Kurtz KJ. Severe experimental hypothermia: a resuscitation protocol. Ann Emerg Med. 1985;4:339-345.
47 Giesbrecht GG, Bristow GK. A second post-cooling afterdrop: more evidence for a convective mechanism. J Appl Physiol. 1992;73:1253-1258.
48 Smith CE, Yamat RA. Avoiding hypothermia in the trauma patient. Curr Opin Anaesthesiol. 2000;13:167-174.
49 Smith CE, Patel N. Hypothermia in adult trauma patients: anaesthetic considerations. Part II. Prevention and treatment. Am J Anesthesiol. 1997;24:29-36.
50 Otto KJ, Metzler MH. Rewarming from experimental hypothermia: comparison of heated aerosol inhalation, peritoneal lavage and pleural lavage. Crit Care Med. 1988;16:869-875.
51 Brunette DD, Biros M, Mlinek EJ, et al. Internal cardiac massage and mediastinal irrigation in hypothermic cardiac arrest. Am J Emerg Med. 1992;10:32-34.
52 Gentilello LM, Jurkovic GJ, Stark MS, et al. Is hypothermia in the victim of major trauma protective or harmful? Ann Surg. 1997;226:439-449.
53 Walpoth BH, Walpoth-Aslan BN, Mattle HP, et al. Outcome of survivors of accidental deep hypothermia and circulatory arrest treated with extracorporeal blood warming. N Engl J Med. 1997;337:1500-1505.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 74 Thermal disorders
Body temperature is normally very tightly controlled by a balance between heat production and heat loss, through a complex feedback mechanism involving the thermoregulatory centre in the hypothalamus. In the intensive care unit (ICU), fever (pyrexia) is usually due to resetting of the thermoregulatory set-point at a higher level by activation of heat-conserving mechanisms, whereas hyperthermia is due to failure of effector mechanisms to maintain body temperature at the normal set-point.
THERMOREGULATION
The three major components of the thermoregulatory system are:
Humoral mediators from the circulation act to alter temperature primarily via the organum vasculosum of the lamina terminalis (OVLT), an area of fenestrated capillaries in the hypothalamus that permits cytokine access to neuronal receptors. Cytokines appear to be the endogenous pyrogens, with interleukin-6 (IL-6) and prostaglandin-E2 (PGE2) being a final common pathway. In addition to elevating body temperature, several cytokines also reduce the thermoregulatory set-point, and are known as endogenous cryogens.1
FEVER IN THE ICU
Fever is defined by a regulated hyperthermia, that is, it is a regulated elevation in the preoptic set-point temperature. Endogenous pyrogens as well as other mediators inhibit warm-sensitive neurones that normally facilitate heat loss and suppress heat production. This elevates the set-point temperature for all thermoregulatory responses and activates cold defences such as vasoconstriction and shivering, which decrease heat loss and increase metabolic heat production respectively. The set-point temperature returns to normal when pyrogen concentrations decrease, triggering heat loss by vasodilatation and sweating.2
Fever may reflect a wide variety of pathological processes including infection, inflammation, trauma, malignancy and connective tissue diseases (Table 74.1), necessitating a systematic and comprehensive diagnostic approach.3 It is often assumed that a patient presenting with a fever should be treated, regardless of the presence or absence of other symptoms. However, the evidence that anti-fever treatments lead to an improvement in morbidity or mortality, or even patient comfort, is lacking.4
| System | Infectious aetiology | Non-infectious aetiology |
|---|---|---|
| Cardiovascular | Endocarditis | Myocardial infarction |
| Catheter-related infection | Deep-vein thrombosis | |
| Pacemaker infection | Pericarditis | |
| Respiratory | Pneumonia | Atelectasis |
| Empyema | Chemical pneumonitis | |
| Sinusitis | Pulmonary emboli | |
| Alimentary | Abdominal abscess | Inflammatory bowel disease |
| Biliary infection | Acalculous cholecystitis | |
| Peritonitis | Pancreatitis | |
| Diverticulitis | Ischaemic colitis | |
| Viral hepatitis | Non-viral hepatitis | |
| Antibiotic-related colitis | Gastrointestinal haemorrhage | |
| Renal | Pyelonephritis | |
| Urinary tract infection | ||
| Central nervous | Meningitis | Cerebral haemorrhage/infarct |
| Encephalitis | Seizures | |
| Rheumatological | Septic arthritis | Connective tissue disease |
| Osteomyelitis | ||
| Gout | Vasculitis | |
| Endocrine | Adrenocortical insufficiency | |
| Alcohol and drug withdrawal | ||
| Hyperthyroidism | ||
| Skin/soft tissue | Cellulitis | Burns |
| Decubitus ulcer | Intramuscular injections | |
| Wound infections | ||
| Haematoma | ||
| Other | Parotitis | Drug fever |
| Pharyngitis | Transfusion reaction | |
| Otitis media | Neoplasms |
The development of fever in response to infection may be a protective adaptive response, and appears to be a phylogenetically preserved evolutionary response because of its survival value.5 In mammalian models, increasing body temperature results in enhanced resistance to infection. In humans, retrospective clinical trials have shown a positive correlation between maximum temperature on the day of bacteraemia and increased survival in patients with Gram-negative bacteraemia and spontaneous bacterial peritonitis.6 Also, septic patients with hypothermia have a poorer outcome than those who develop fever, although this causality is less clear. Both local and systemic hyperthermia has been used to facilitate cancer treatment. The protective effects of fever result from increased immune and cytokine functions.7–10
Temperature elevation has been shown to enhance:
In addition, elevated temperatures inhibit some pathogens, such as Streptococcus pneumoniae.11
Moderate fever is a common occurrence in ICU patients, but approximately half of these are non-infectious in origin.12–14 The presence of fever frequently results in the performance of diagnostic tests and exposes the patient to unnecessary invasive procedures and inappropriate use of antibiotics.15
Whilst very high fevers (> 40°C) are dangerous, it is less clear whether moderate elevation of body temperature is detrimental and, indeed, may be protective.4 Moreover, artificially lowering the temperature of a febrile patient may mask the signs of infection and make diagnosis and monitoring more difficult. Any decision to adopt anti-fever measures, physical or pharmacological, must take into consideration the variable response by this patient population. Antipyretics may be ineffective.The usual concern about external cooling measures inducing peripheral vasoconstriction, reducing heat loss and making the pyrexia worse by shivering and hypermetabolism, may not be observed in sedated ICU patients.16 The most likely cause for this response is the drugs used to maintain sedation.17,18
Pyrexia is associated with a number of deleterious physiological effects. Cardiac output, oxygen consumption, carbon dioxide production and energy expenditure are all increased, particularly in the presence of shivering. Oxygen consumption is increased on average by 10%/°C.2 These changes are poorly tolerated by patients with limited cardiorespiratory reserve, and this group of patients would probably derive benefit from cooling measures. Other patient groups that require special consideration include those with immunosuppression, prosthetic implants and acute brain injury. Recent trials of therapeutic moderate hypothermia and traumatic brain injury indicate that hypothermia is a complicated treatment that is likely to benefit only a subgroup of patients with traumatic brain injury.19–21
HEAT STROKE
Exertional heat stroke is a consequence of prolonged, intense exercise in warm humid environments, often seen in athletes and military recruits. Classic heat stroke is commonly seen in sedentary, elderly patients with underlying illnesses during heat waves. Factors predisposing to heat stroke are listed in Table 74.2. About 80% of heat stroke deaths occur in people aged 50 years and older, because of the diminished ability of the older body to compensate for increased core temperatures. Heat stroke is estimated to be the cause of approximately 1700 deaths each year in the USA.22 The European heat wave of 2003 was responsible for > 14 000 excess deaths within 2 weeks in France alone, of which a third were attributed to heat stroke, hyperthermia or dehydration.23,24 A high mortality rate of > 62% was reported for this cohort, which is higher than that for leading killers in ICUs such as acute respiratory distress syndrome (ARDS) and septic shock.25 Furthermore, there is a late mortality contributed by survivors who have sustained neurological injury. A study of former heat stroke patients suggests that susceptible individuals have a poorer physiological response to heat stress in terms of core temperature, heart rate and sweat response.
| Age | Elderly |
| Environmental | High ambient temperature and humidity |
| Heat waves | |
| Poor ventilation | |
| Behavioural | Lack of acclimatisation |
| Salt and water deprivation | |
| Obesity | |
| Underlying conditions | Infection/fever |
| Diabetes | |
| Malnutrition | |
| Alcoholism | |
| Hyperthyroidism | |
| Impaired sweat production | |
| Healed burns | |
| Ectodermal dysplasia | |
| Impaired sweating | |
| Cardiovascular disease | |
| Fatigue | |
| Potassium deficiency | |
| Drugs | Anticholinergics |
| Antiparkinsonians | |
| Antihistamines | |
| Butyrophenones | |
| Phenothiazines | |
| Tricyclics | |
| Diuretics | |
| Sympathomimetics |
PATHOGENESIS
The pathogenesis of multiple organ failure in heat stroke is complex. Although direct cellular damage from increased temperature constitutes the initiating insult,26 the precise sequence of injury and responsible mediators are poorly understood. At the cellular level, thermal injury results in increased membrane permeability, which in turn stimulates membrane enzymes such as Na+K+-ATPase to maintain membrane integrity. This ATP-consuming enzyme activity is also responsible for nerve impulse conduction, which ismarkedly curtailed when ATP is depleted. This results in tissue oedema, reduced oxygen extraction and neuronal injury. High temperatures ameliorate ATP synthesis leading to fatigue.
Recent evidence suggests that the pathways for tissue injury in heat stroke share many features with that of sepsis, endotoxaemia and systemic inflammation. Increased levels of circulating endotoxin and cytokines have been identified in patients with heat stroke.27,28 The use of anti-endotoxin antibodies in primate models of heat stroke suggests that endotoxin at least in part mediates the tissue injury associated with hyperthermia. There was also a significant correlation between plasma IL-6 concentration and the severity of heat stroke. Since this cytokine is known to modulate the hypothalamic set-point, the ramifications of such a response in an already hyperthermic patient are obvious.
Activation of coagulation factors29 and release of endothelin and adhesion molecules30,31 from activated or injured endothelium have also been demonstrated in heat stroke. These recent observations lead to the speculation that certain mediators that are implicated in the pathogenesis of acute organ injury are also elevated in heat stress, but become intense when heat stroke develops and are not normalised upon cooling.
CLINICAL PRESENTATION
Dehydration follows excessive insensible losses although sweating is generally absent in the terminal stages of classic heat stroke, leaving a hot, dry skin. Hypovolaemia is a consequence of dehydration and fluid redistribution, and results in reduced organ perfusion. A severe metabolic (lactic) acidosis is present. The major biochemical abnormalities include hyperglycaemia, hypophosphataemia, and raised serum enzymes and acute phase proteins (Table 74.3). Haematological findings include leukocytosis, thrombocytopenia, and activation of coagulation and fibrinolysis.
Table 74.3 Biochemical differences between classic and exertional heat stroke
| Classic heat stroke | Exertional heat stoke | |
|---|---|---|
| Arterial gases | Mixed respiratory alkalosis | Severe metabolic acidosis |
| Serum electrolytes | Na+, Mg2+, Ca2+ are usually normal | HyperkalaemiaHypocalcaemia |
| Hypophosphataemia | Hyperphosphataemia | |
| Blood glucose | Hyperglycaemia | Hypoglycaemia |
| Creatinine kinase | Moderately increased | Markedly increased |
| Hepatic enzymes | Markedly increased | Moderately increased |
| Acute phase proteins | Markedly increased | Moderately increased |
Exertional heat stroke differs slightly in that additional findings include rhabdomyolysis and acute renal failure that is associated with hyperkalaemia, hyperphosphataemia and hypocalcaemia (Table 74.3).
MANAGEMENT
Evaporation is considerably more effective. Pharmacological treatment with antipyretic agents, or dantrolene,32 is ineffective. Prevention of vasoconstriction and shivering by overcooling is important because of the danger of subsequent rebound hyperthermia. Core and skin temperature monitoring is useful, but measurement of rectal temperature should be avoided because it lags considerably during cooling. Cooling can be stopped when core temperature reaches below 39°C. However, despite cooling, about 25% of patients experience failure of one or more organ systems.
OUTCOME
The largest study reported of heat stroke patients in intensive care suggests an alarmingly high mortality of > 60%,23 although this diminishes substantially with early recognition and aggressive treatment. The incidence of permanent neurological deficit remains at 7–15%. Variables associated independently with reduced hospital survival include:
DRUG-INDUCED HYPERTHERMIAS
The numerous causes of hyperthermia are listed in Table 74.4. This section will review the relatively common causes of drug-induced hyperthermias, including malignant hyperthermia, neuroleptic malignant syndrome, and the sympathomimetic and anticholinergic syndromes.
| Disorders of excessive heat production | Exertional hyperthermia |
| Heat stroke (exertional) | |
| Malignant hyperthermia | |
| Neuroleptic malignant syndrome | |
| Lethal catatonia | |
| Thyrotoxicosis | |
