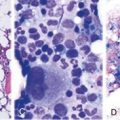
Chapter 28 The Polycythemias
Table 28-1 Differential Diagnosis of the Polycythemias
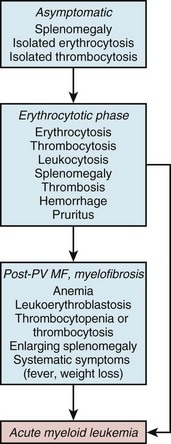
Table 28-2 International Working Group for Myelofibrosis Research and Recommended Treatment Criteria for Post-PV MF
WHO, World Health Organization.
Adapted from Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A consensus statement from the international working group for myelofibrosis research and treatment. Leukemia 22:437, 2008.
Table 28-3 Risk Stratification in Polycythemia Vera Based on Thrombotic Risk
| Risk Category | Age >60 Years or History of Thrombosis | Cardiovascular Risk Factors* |
|---|---|---|
| Low | No | No |
| Intermediate | No | Yes |
| High | Yes |
*Hypertension, hypercholesterolemia, diabetes, and smoking. Extreme thrombocytosis (platelet count >1500 × 109 L−1) is a risk factor for bleeding. Its role as a risk factor for thrombosis is uncertain. An increasing leukocyte count has been identified as a novel risk factor for thrombosis, but confirmation is required.
Data from Finazzi G, Barbui T: How I treat patients with polycythemia vera. Blood 109:5104, 2007.
General Principles of Therapy
1. The etiology of erythrocytosis must be correctly categorized to be certain the patient has PV. This will avoid inappropriate exposure of patients with nonmalignant disorders to potential leukemogenic agents. To this end, a major contribution is played by incorporating a JAK2V617F and exon 12 JAK2 mutational determinations in the diagnostic workup, and in this way, early phases of PV can be recognized as well. In individuals with erythrocytosis without a JAK2 mutation, search for mutations in VHL, PHD2, HIF-1, and HIF-2 should be considered.
2. Therapy should be individualized.
3. Initially, the hematocrit should be reduced to 45% as soon as possible. The speed of phlebotomy will depend on patients’ general medical conditions (250-500 mL every other day). Elderly patients with compromised cardiovascular or pulmonary systems should be more carefully phlebotomized (twice a week), or smaller volumes of blood should be removed.
4. Hematocrit levels should be maintained at 45%.
5. The use of chemotherapeutic agents should be avoided in low-risk PV patients, and treatment with hydroxyurea or INF should be reserved for high-risk patients. Be on the look-out for toxicities from each of these agents.
6. Hyperuricemia is treated with allopurinol (100-300 mg/day).
7. Pruritus is treated empirically with cyproheptadine 4 to 16 mg/day or, if unsuccessful, INF-α therapy 3.0 × 106 units subcutaneously three times a week. A role of selective serotonin uptake inhibitors (paroxetine 20 mg/day or fluoxetine 10 mg/day) or with phototherapy has been proposed. Dramatic relief occurs with the institution of JAK2 inhibitors, and these agents should be used as a component of an experimental trial or if the patients have features that justify the diagnosis of post-PV MF syndrome.
8. Elective surgery or dental procedures should be delayed until hematocrit and platelet counts have been normalized for more than 2 months. Aspirin should be withdrawn at least 1 week before surgery. If emergency surgery is contemplated, phlebotomy and cytapheresis should be pursued.
9. Women and men who are contemplating having children should be treated by phlebotomy plus low-dose aspirin therapy (81 mg/day or with INF-α) to avoid teratogenic effects of chemotherapy. Such avoidance will also prevent deleterious effects on fertility. During pregnancy, therapy is frequently not necessary; if it is, phlebotomy plus low-dose aspirin should be exclusively used. If phlebotomy control is inadequate, treatment with INF-α should be pursued.
Algorithm for Management of Patients With Polycythemia Vera
Low-Risk Young Patients (Age <60 years) and No History of Thrombosis, Platelet Count <1.5 × 106 mm−3
Severe pruritus refractory to histamine antagonists
High-Risk Patients (Age >60 Years), Previous Thrombosis, Platelet Count >1.5 × 106 mm−3
Phlebotomy to hematocrit of 42% in females and 45% in males
Aspirin (81 mg/day) to be given only in patients with platelet counts <1.5 × 106 mm−2
Myelosuppressive therapy with hydroxyurea 30 mg/kg orally for 1 week
Consider pegylated INF 45 to 180 µg/wk or add busulphan 4-6 mg/d orally for 4 to 8 weeks.
If on bisulphan, stop when blood counts are normalized or platelet count is lower than 300,000 mm−3.