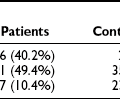The Metabolic Syndrome
Linkage of Metabolic Syndrome to Coronary Heart Disease and Type 2 Diabetes
Linkage of the Metabolic Syndrome to Other Disorders
Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis
Treatment of the Metabolic Syndrome
The Thrifty Genotype and the Increasing Incidence of the Metabolic Syndrome
For the purposes of this review, we will define the metabolic syndrome as a state of metabolic dysregulation characterized by insulin resistance, hyperinsulinemia and a predisposition to type 2 diabetes, atherosclerotic vascular disease, hypertension, and many other disorders, including Alzheimer’s disease and certain cancers1–3 (Fig. 18-1). Affected individuals are typically obese or overweight or show more subtle manifestations of increased adiposity such as an increase in abdominal fat4 or fat cell size.5,6 In addition, they may have a decreased capacity for exercise7,8 and show evidence of low-grade inflammation9–11 and a procoagulant state.12,13 Type 2 diabetes develops when they are no longer able to sustain the high insulin levels required for near normal glucose homeostasis.14 As will be discussed later, the metabolic syndrome is diagnosed clinically on the basis of abnormalities in plasma glucose and lipids and increases in blood pressure and waist circumference, which independently might not be sufficient to lead to therapeutic intervention.
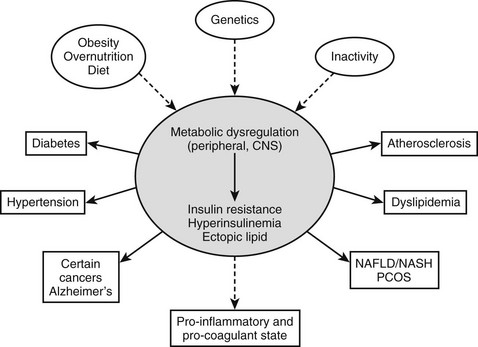
FIGURE 18-1 The metabolic syndrome, current view. A combination of overnutrition, inactivity, and genetic factors interact to produce a state of metabolic dysregulation that leads to insulin resistance, hyperinsulinemia, and a proinflammatory state. In individuals who are genetically predisposed, this in turn leads to one or more of the indicated disorders and often others. Nearly all of these disorders are associated with an increased risk of coronary heart disease. A considerable body of evidence suggests that the metabolic dysregulation involves cellular lipid metabolism and may be mediated by abnormalities in the regulation of the fuel-sensing enzyme AMPK. NAFLD/NASH, Non-alcohol fatty liver disease/non-alcoholic steatohepatitis; PCOS, polycystic ovary syndrome.
From a clinical perspective, the importance of the metabolic syndrome is attributable principally to three factors: (1) It antedates the multiple disorders with which it is associated, (2) it may be a target for the prevention and therapy of these disorders, and (3) it is extremely common. Based on recent adult treatment panel (ATP III) diagnostic guidelines15 (see following discussion), it has been estimated in the population evaluated in the National Health and Nutrition Survey (NHANES) study that upwards of 50 million people in the United States above the age of 20 have the metabolic syndrome, including 40% of all individuals above the age of 60 (Fig. 18-2).16 Likewise, a high prevalence of the metabolic syndrome has been found in many other countries,1,17 and its prevalence is increasing in adolescents and children in parallel with the pandemic of obesity in this population.18–20 This chapter will update the reader on the present status of the metabolic syndrome (formally referred to as syndrome X or the insulin resistance syndrome).14,21 Emphasis will be placed on describing novel mechanisms and factors that are transforming our previous conceptions about its pathogenesis and treatment. In addition, proposed guidelines for the diagnosis of the metabolic syndrome will be discussed, as will the special problems that need to be considered when attempting to treat and/or prevent it. For other perspectives, the reader is referred to several excellent recent reviews.15,22–24
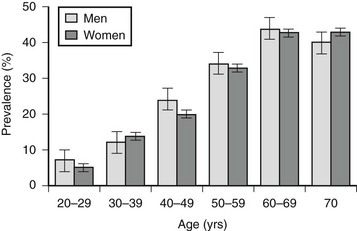
FIGURE 18-2 Prevalence of the metabolic syndrome in the United States according to age. Based on National Health and Nutrition Examination Survey (NHANES) data and ATP III criteria (see Table 18-3). (Data from Ford ES, Giles WH, Dietz WH: Prevalence of the metabolic syndrome among U.S. adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287:356–359, 2002.)
History
The clustering of the major components of the metabolic syndrome, such as obesity, type 2 diabetes, hypertension, and dyslipidemia, has long been recognized25; however; its delineation as a distinct entity took place only after its linkage to insulin resistance, hyperinsulinemia, and cardiovascular disease became more apparent.21,26,27 Insulin resistance has been defined as a state (of a cell, tissue, system, or body) in which greater than normal amounts of insulin are required to elicit a normal biological response.28 In humans, it is currently diagnosed on the basis of high levels of plasma insulin, either fasting or during a glucose tolerance test, or by a decreased rate of glucose infusion during a euglycemic-hyperinsulinemic clamp.8 The much greater prevalence of insulin resistance in patients with type 2 rather than type 1 diabetes was appreciated over 60 years ago, based on their substantially higher insulin requirement29,30 and diminished response to exogenous insulin.31 Shortly after the development of the insulin immunoassay by Yalow and Berson,32 this suspicion was confirmed,33,34 and other disorders associated with insulin resistance and hyperinsulinemia were identified, including coronary heart disease and several of its risk factors,21,35–40 as well as obesity itself (see Fig. 18-1). In general, most adults with insulin resistance and hyperinsulinemia are obese (BMI > 29) or overweight (BMI 25 to 29). However, a significant percentage are normal weight by BMI but show increases in abdominal fat (central obesity)4,8 and in intrahepatic and intramyocellular triglyceride41 and/or enlarged fat cells.5,42 The presence of central obesity has been shown to correlate strongly with an individual’s predisposition to most of the diseases indicated in Fig. 18-1, including coronary heart disease,43,44 and this is the reason waist circumference is one of the diagnostic criteria for the metabolic syndrome, as will be discussed later.
Hyperinsulinemia and insulin resistance are also present in normal-weight offspring of people with type 2 diabetes,7,45–47 hypertension,48 and hypertriglyceridemia8,49,50 and in individuals at increased risk for coronary heart disease,51–54 suggesting that they are early markers or pathogenetic factors for these disorders. Studies such as these, plus the presence of a high rate of ischemic heart disease in patients with type 2 diabetes at the time of diagnosis (20% to 50%)55–57 and to a somewhat lesser extent individuals with impaired glucose tolerance,58 have led to the suggestion that treatment of the metabolic syndrome at an early stage may be needed for preventing coronary heart disease.8,22,39,59,60
Pathophysiology
Insulin Resistance and Compensatory Hyperinsulinemia
The presence of insulin resistance in otherwise normal offspring of people with type 2 diabetes, hypertriglyceridemia, and hypertension has led to the notion that it is a causal factor for the metabolic syndrome or an early pathogenetic event.21 According to this widely held view, insulin resistance affects a number of organs (e.g., muscle, liver, adipose tissue), and hyperinsulinemia due to increased insulin secretion by the pancreatic β cell and decreased insulin degradation by the liver is a compensatory phenomenon.14,21,35 The observation that therapies that increase insulin sensitivity and lower plasma insulin levels, such as lifestyle modification (diet and exercise)61–63 and treatment with metformin61 or thiazolidinediones,64–65 prevent or delay the onset of diabetes in individuals with glucose intolerance is compatible with this notion, as is the efficacy of these therapies in some people with other disorders associated with the metabolic syndrome, such as nonalcoholic fatty liver disease66,67 and the polycystic ovary syndrome.69,70 Left unexplained by this hypothesis, however, is the molecular mechanism by which insulin resistance develops initially and how it leads to hyperinsulinemia. Also, the possibility that hyperinsulinemia is the more primary of the two events or occurs simultaneously with the insulin resistance has not been ruled out.70
The Insulin Signaling Cascade
Hypothetically, the metabolic syndrome could be related to genetic abnormalities in the insulin signaling cascade. In keeping with this possibility, mutations of IRS1 and IRS2, the initial targets of the insulin receptor tyrosine kinase, have been shown to lead to insulin resistance and diabetes in transgenic mice.71,72 Evidence that these or other genetic defects in the insulin signaling cascade are common in humans with the metabolic syndrome or type 2 diabetes and account for observed signaling defects73 is still lacking, however.
The Lipid Theory
Insulin resistance and hyperinsulinemia in humans and experimental animals have been linked to obesity and dysregulation of cellular lipid metabolism in a wide variety of circumstances.73–76 Early studies focused on free fatty acids (FFA) released from adipose tissue and assumed that insulin resistance in skeletal muscle was in some way the result of elevated plasma FFA levels. More recently, it has become apparent that insulin resistance is associated with alterations in lipid metabolism in tissues other than skeletal muscle, and that its appearance is affected by a number of newly discovered hormones and intracellular regulatory mechanisms. In addition, it has been demonstrated that the metabolic syndrome occurs in people who lack adipose tissue as well as in those with excess adiposity, and that in both groups, it is associated with triglyceride deposition in ectopic sites such as muscle, liver, and visceral fat. In this section, we will attempt to review the current status of this increasingly complex but intriguing area. Three distinct but often interrelated mechanisms that have been put forth to explain the link between altered lipid metabolism and components of the metabolic syndrome will be discussed.
Excess Free Fatty Acids
Over 40 years ago, Randle and his co-workers77 first demonstrated that elevated circulating free fatty acid levels diminish insulin-stimulated glucose utilization by a perfused rat heart preparation. They presented evidence that this effect occurred within minutes, and that it was associated with enhanced mitochondrial fat oxidation that led to both inhibition of glucose oxidation at the pyruvate dehydrogenase step and an increase in the cytosolic concentration of citrate. They also demonstrated that the increase in citrate inhibited glycolysis at phosphofructokinase, and that the secondary increase in glucose 6-phosphate inhibited hexokinase and secondarily diminished insulin-stimulated glucose uptake78 (Fig. 18-3). It was suggested that a similar mechanism might account for the insulin resistance observed in humans with obesity or type 2 diabetes, in both of whom plasma FFA levels were known to be elevated.79 Over the next 25 years, most investigators were unable to reproduce these findings in skeletal muscle, however,80,81 and for this reason the contribution of elevated plasma FFA levels to the insulin resistance observed in most humans with obesity, type 2 diabetes, and other metabolic syndrome–associated disorders remained unclear.
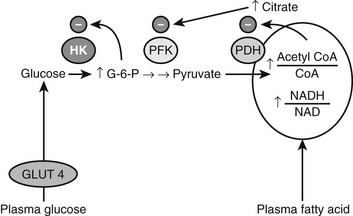
FIGURE 18-3 Inhibition of glucose uptake and oxidation by fatty acids as described in heart muscle by Randle, Garland, Hales, and Newsholme (1964, 1965). See text for details. (Adapted from Shulman GI: Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176, 2000.)
In 1991, Boden et al.82 demonstrated that raising plasma FFA (by infusing a lipid emulsion with heparin to activate lipoprotein lipase activity) in humans during a euglycemic-hyperinsulinemic clamp inhibits insulin-stimulated glucose uptake and its incorporation into glycogen by leg muscle. Importantly, they found that this effect required 4 to 6 hours and not minutes to become evident, and that it was not accompanied by an increase in citrate. Subsequent investigations by Shulman’s laboratory in which 31P magnetic resonance spectroscopy (MRS) was used to measure intracellular glucose-6-phosphate noninvasively found that its concentration was reduced, suggesting that free fatty acids principally inhibit insulin-stimulated glucose transport or phosphorylation activity83 and not the phosphofructokinase reaction, as suggested by Randle. More recently, studies by the same group, in which 13C MRS was used to assess intracellular free glucose concentrations, revealed that insulin-stimulated glucose transport and not phosphorylation was the step inhibited by high fatty acids.84
Other studies have demonstrated that insulin resistance in human muscle, caused by infusing lipids to increase plasma FA, is associated with impaired insulin signaling,73 increases in the concentrations of muscle triglyceride, long-chain fatty acyl CoA85 and diacylglycerol, and increases in protein kinase C (PKC) activity and the translocation of various PKCs from the cytosol to a membrane fraction.76,86–88 Another finding was a decrease in IKBα abundance, suggesting activation of NF-κB and proinflammatory events.88 As will be discussed later, similar abnormalities have been found in rodent liver following a sustained exposure to fatty acids,89–92 in rodent muscle in a wide variety of states associated with insulin resistance,76,86,87 and in liver and muscle of massively obese, insulin-resistant humans with type 2 diabetes.93,94 Thus the intracellular changes produced by an excess of free fatty acids are associated with insulin resistance in many tissues.
A still unanswered question is whether an increase in plasma FFA is an early pathogenetic event in the metabolic syndrome. Elevated concentrations of plasma FFA, attributable to increased adipose tissue mass, and the relative insensitivity of large fat cells and visceral fat to insulin35,85 are present in people with obesity and type 2 diabetes, and they appear to contribute to insulin resistance when these disorders are established.74,85,95,96 On the other hand, even some massively obese individuals remain insulin sensitive, suggesting that other factors are involved97 (see discussion in Adipose Tissue). Also unclear is whether plasma FFA are elevated in individuals with the metabolic syndrome in its very early stages. Only modest increases in plasma FFA, if any, have been observed in normal-weight, insulin-resistant individuals who are at risk for developing diabetes because of family history.45,98
Altered Fatty Acid Metabolism: Malonyl Coenzyme A, Mitochondria, and Adenosine Monophosphate–Activated Protein Kinase
A second abnormality in lipid metabolism that could lead to insulin resistance is a disturbance in fatty acid metabolism in which the oxidation of cytosolic long-chain fatty acyl CoA (FACoA) by mitochondria is impaired and its esterification and metabolism by other non-mitochondrial processes is enhanced.76,99 This could occur if either the intrinsic ability of mitochondria to oxidize fatty acid is decreased or the activity of carnitine palmitoyl transferase 1, the enzyme that regulates the transfer of cytosolic long-chain fatty acyl CoA into mitochondria, is diminished.
Malonyl CoA: Altered fatty acid partitioning between the mitochondria and cytoplasm as a cause of insulin resistance was first suggested by studies in denervated rat muscle, in which enhanced diacylglycerol (DAG) synthesis and PKC activation were observed,100 and later by similar findings in muscle of obese, insulin-resistant KKAy mice.101 In these and other instances, insulin resistance in rodent muscle correlated with an increase in the concentration of malonyl CoA,76 an allosteric inhibitor of carnitine palmitoyltransferase. As shown in Figure 18-4, an increase in malonyl CoA by decreasing the oxidation of cytosolic FACoA would increase its availability for the formation of DAG, triglycerides, ceramide, and possibly other factors linked to insulin resistance. In keeping with such a mechanism, it has been demonstrated by McGarry99,102 that treatment with etomoxir, a pharmacologic CPT1 inhibitor, concurrently increases triglyceride accumulation and causes insulin resistance in rat skeletal muscle. Also supporting this notion are a number of other findings, including the following: mice lacking functional acetyl CoA carboxylase 2 (ACC2, the isoform that generates the malonyl CoA that regulates CPT1) are more insulin sensitive than control rats103,104; the administration of an ACC inhibitor diminishes obesity and insulin resistance in fat-fed rats105; and antisense oligonucleotides directed at hepatic ACC1 and ACC2 diminish both hepatic steatosis and insulin resistance in mice fed a high-fat diet.106 Finally, low rates of fatty acid oxidation have been reported in pre-obese humans,107,108 Zucker diabetic rats,109 and interleukin-6 (IL-6) knockout mice110 prior to the onset of diabetes and obesity. Malonyl CoA was not assayed in any of these studies; however, in the two rodent models, a decrease in the activity of AMP-activated protein kinase (AMPK) was found, suggesting that its concentration was elevated111,112 (see section on AMPK).
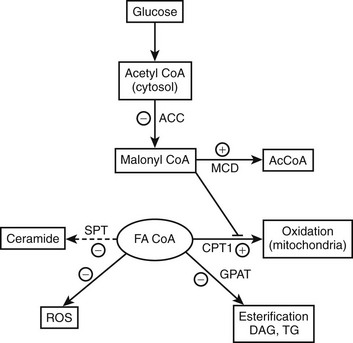
FIGURE 18-4 AMPK concurrently regulates multiple aspects of fatty acid metabolism. By phosphorylating and inhibiting ACC and activating MCD, AMPK diminishes the concentration of malonyl CoA. This relieves the inhibition of CPT1 by malonyl CoA and results in both increased fatty acid oxidation and a decrease in the availability of cytosolic FACoA for TG, DAG, and ceramide synthesis, and possibly lipid peroxidation and protein acylation. Conversely, AMPK activation independently decreases the expression of GPAT, fatty acid synthase (not shown), and SPT, decreasing respectively the synthesis of glycerolipids, fatty acids (de novo), and ceramide (de novo). A decrease in AMPK produces the opposite effects. The basis for the ability of AMPK to inhibit oxidant stress (ROS generation) in some settings is not known. Whether AMPK activation enhances or inhibits a process or an enzyme in this scheme is denoted by plus and minus signs, respectively. Not shown in the diagram is that AMPK achieves these changes in some instances by phosphorylating the indicated enzyme and/or in other instances by regulating its expression. In addition, AMPK can enhance mitochondrial biogenesis and function by enhancing the expression of the transcriptional coactivator PGC1α. ACC, Acetyl CoA carboxylase; CPT1, carnitine palmitoyltransferase 1; DAG, diacylglycerol; FA CoA, cytosolic long chain fatty acyl CoA; GPAT, glycerophosphate acyltransferase; MCD, malonyl CoA decarboxylase; ROS, reactive O2 species. (Adapted from Ruderman N, Prentki M: AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 3:340–351, 2004.)
Mitochondrial Dysfunction: Altered fatty acid partitioning in muscle and other tissues could also occur if fatty acid oxidation is depressed as a consequence of mitochondrial dysfunction. Decreases in mitochondrial function, and in some instances number and size, have been found in muscle of individuals with type 2 diabetes associated with obesity,113 in lean insulin-resistant elderly individuals,114 and in lean insulin-resistant offspring of diabetic parents.7 In the latter case, the reduction in mitochondrial function could be attributed to a similar reduction in mitochondrial content.115 Likewise, decreases in the mRNA for PGC1α, a transcriptional coactivator that enhances genes for mitochondrial biogenesis and function, have been observed in some116 but not all115 studies of nondiabetic first-degree relatives of patients with type 2 diabetes, as well as in individuals with type 2 diabetes and impaired glucose tolerance.117 Whether these mitochondrial changes are hereditary or secondary to metabolic events (e.g., lipotoxic changes due to abnormalities in intracellular lipid metabolism) or abnormalities in AMPK regulation (see next section) remains to be determined. However, given the key role for increased intracellular fatty acid metabolites in mediating insulin resistance, decreased mitochondrial function leading to decreased fatty acid oxidation will certainly exacerbate the problem. Also to be determined is whether the changes observed in the offspring of diabetic parents reflect a difference in muscle fiber type, since the ratio of mitochondrial-rich type 1 fibers to glycolytic type 2 fibers may be decreased in these individuals.7,118
AMP-Activated Protein Kinase: AMPK (Box 18-1) is a fuel-sensing enzyme that appears to play a key role in regulating both cellular metabolism and mitochondrial function. In addition, an increasing body of evidence has suggested that its dysregulation could be a cause of the metabolic syndrome (animal studies) as well as a target for its prevention and therapy (human and animals studies).3,24,75
As shown in Fig. 18-4, when AMPK is activated (e.g., during exercise or in some tissues by caloric deprivation), it phosphorylates and inhibits acetyl CoA carboxylase (ACC), the enzyme that catalyzes the synthesis of malonyl CoA. By a still undetermined mechanism, it activates malonyl CoA decarboxylase, an enzyme that catalyzes malonyl CoA degradation.119 In addition, AMPK concurrently inhibits the use of cytosolic FACoA for diacylglycerol, triglyceride, and ceramide synthesis, and at least in endothelial cells120 and adipocytes (see Adipose Tissue), it diminishes the generation of lipid peroxides and the activation of NF-κB caused by elevated concentrations of fatty acids.75,120,121(and unpublished data) Thus AMPK appears to protect cells against lipotoxicity and to do so by multiple actions. In addition to modulating these events acutely, AMPK chronically regulates the effects of transcriptional regulators (e.g., SREBP1C) that govern the synthesis of acetyl CoA carboxylase and other key enzymes. Of specific relevance to this chapter, decreases in AMPK activity and increases in malonyl CoA are both associated with insulin resistance in muscle (Table 18-1) and liver in many situations, and failure to activate AMPK in the fat cell during lipolysis is associated with increases in oxidative stress and inflammation.121(and unpublished observations) In addition, hormones and other factors that decrease AMPK in peripheral tissues, such as glucocorticoids,122 TNF-α,123 and resistin,124 cause insulin resistance. Conversely, numerous endogenous hormones (e.g., adiponectin and leptin) and pharmacologic agents (e.g., metformin, thiazolidinediones, α-lipoic acid) that diminish insulin resistance have been demonstrated to activate AMPK and diminish the concentration of malonyl CoA in experimental animals, as do exercise and treatment with the AMPK activator AICAR (5-aminoimidazole-4-carboxamide riboside)75 (Table 18-2).
Table 18-1
AMPK, Malonyl CoA, and Other Abnormalities Associated With Insulin Resistance in Muscle

Data from Ruderman N, Prentki M: AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 3:340–351, 2004 and from the laboratories of the authors and those of Turinsky, Kraegen, Caro, Boden, and Shoelson. Many of these changes have also been demonstrated in liver in insulin-resistant obese humans, fat-fed and glucose-infused rats, and fa/fa rats. Whether AMPK activity is depressed and the concentration of malonyl CoA elevated in skeletal muscle of obese insulin-resistant humans is controversial (see Bandyopadhy & Olefsky: Diabetes 2006; DeFillipis & Mandarino, Am J Physiol (Endo) 2008). Studies, primarily in vitro, suggest that similar events occur in the liver, pancreatic β cell, and cultured vascular endothelium.
Table 18-2
Effect of Therapies That Activate AMPK in Humans and/or Rodents on Various Manifestations of the Metabolic Syndrome
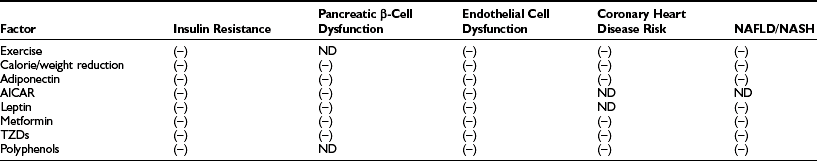
Data from Ruderman N, Prentki M: AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov 3:340–351, 2004.
Closely linked to mitochondrial theories of insulin resistance is PGC1α (PPARγ-coactivator 1α), a transcriptional coactivator whose expression is increased when AMPK is activated (e.g., by exercise or AICAR).125–128 In skeletal muscle, and likely in other tissues, PGC1α regulates mitochondrial biogenesis and the expression of multiple genes governing oxidative phosphorylation, including those for citric acid cycle enzymes and electron transport proteins.116,117,129 PGC1α polymorphisms have been reported in populations with type 2 diabetes in Denmark130 and Japan.131 Also, modest but coordinated decreases in the expression of many PGC1α-mediated genes have been reported in muscle of overweight/obese middle-aged individuals with type 2 diabetes or impaired glucose tolerance.116,117 A decreased capacity for exercise (VO2 max) was also observed in these subjects, in keeping with the findings of earlier studies in similar patient groups.118,132–134
In keeping with its effect on PGC1α, it has been shown that AMPK is necessary for mitochondrial biogenesis.128 Whether the various hormones (e.g., adiponectin, leptin, catecholamines) and pharmacologic agents (thiazolidinediones, metformin) reported to activate AMPK in peripheral tissues also increase PGC1α expression and stimulate mitochondrial biogenesis and enzyme activity in skeletal muscle and other tissues, or whether agents that decrease AMPK (e.g., endocannabinoids and glucocorticoids) have the opposite effect, are active areas for study. Also requiring study is the relative role of Ca2+ calmodulin–dependent protein kinase, which like AMPK is activated during exercise and increases PGC1α expression.135
Adipose Tissue
Overview: A number of lines of investigation have linked abnormalities in adipose tissue to the pathogenesis of the metabolic syndrome. First, as already noted, elevated plasma FFA levels, attributable to an increase in their release from adipocytes in obese or centrally obese individuals, correlate with the presence of insulin resistance in most patients.85,136 Second, when the function of the adipocyte as a store for lipid is impaired,137,138 as it is in many insulin-resistant obese individuals and people with various lipodystrophies (see later discussion), fatty acids are deposited as triglyceride in ectopic sites such as muscle, liver, and visceral fat, and this is associated with insulin resistance, inflammation, and other manifestations of cellular dysfunction (lipotoxicity).139 Recent studies suggest that markedly obese individuals with diminished levels of the lipid droplet proteins CIDE A and perilipin in adipose tissue have an impaired ability to deposit lipid in their adipocytes, a higher rate of lipolysis, and they are insulin resistant.97 In contrast, in comparably obese individuals with normal levels of these lipid droplet proteins, increased lipolysis and insulin resistance were not observed. Thus differences in the capacity of the adipocyte to store lipid may be a key determinant of whether obesity leads to the metabolic syndrome. Finally, the adipocyte can release hormones such as leptin and adiponectin that in multiple tissues activate AMPK, diminish ectopic lipid, and enhance insulin action. In addition, it can release proinflammatory cytokines such as TNF-α,140 resistin,124 and plasminogen activator inhibitor (PAI)12 that could cause insulin resistance.141 As already noted, diminished AMPK activity, which results in large increases in oxidative stress and possibly inflammatory changes in the adipocyte when lipolysis is stimulated, might contribute to the latter.121(and unpublished data)
Adiponectin: Of the various adipokines, adiponectin, also referred to as ACRP30, has been most closely linked to insulin resistance in humans. It is produced exclusively or at least predominantly in adipose tissue and is present in the circulation in trimeric, hexameric, and high-molecular-weight (HMW) forms. The biological relevance of the three oligomers142,143 and still other forms of adiponectin is incompletely understood. These considerations aside, there is abundant evidence that low immunoassayable adiponectin levels in plasma (accounted for mainly by the HMW form) are present in obese individuals and in individuals at risk for type 2 diabetes144,145 and coronary heart disease,146 even in the absence of overt obesity. In addition, polymorphisms of the adiponectin gene have been associated with the metabolic syndrome in some populations and a predisposition to type 2 diabetes in others.144
In keeping with these findings in humans, in mice fed a high-fat diet, genetically knocking out adiponectin causes glucose intolerance and insulin resistance147; and conversely, overexpression of full-length148 or globular149 adiponectin attenuates the severity of atherosclerosis in apoE knockout mice. In addition, the administration of full-length (HMW) adiponectin and the globular subunit have been shown respectively to diminish hepatic lipid accumulation in ob/ob mice with non-alcoholic fatty liver disease (NAFLD)150 and insulin resistance in fat-fed rats.151
Adiponectin, like exercise, activates AMPK and stimulates AMPK-mediated events such as glucose transport and fatty acid oxidation in muscle152,153 and inhibition of glucose production by liver.154 In addition, adiponectin has antiinflammatory actions.155 Whether the insulin-sensitizing effect of adiponectin is AMPK-mediated has not been proven definitively; however, treatment with thiazolidinediones increases plasma adiponectin, and the insulin-sensitizing effect of these agents and their ability to activate AMPK are both markedly attenuated in adiponectin knockout mice.156,157
Leptin: Since its discovery by Friedman and his co-workers,158 interest in leptin has for the most part focused on its role as an appetite suppressant. However, it has also been recognized for some time that leptin increases oxidative metabolism and fatty acid oxidation in peripheral tissues, owing both to a direct action159 and to an effect on the hypothalamus that appears to be mediated by the sympathetic nervous system.160 As first reported by Minokoshi and Kahn and their co-workers, both the direct and centrally-mediated effects of leptin on peripheral tissues are associated with AMPK activation,161 whereas its action on hypothalamic nuclei is associated with a decrease in AMPK activity.162
When leptin is lacking or its receptor is not functioning, lipid accumulates in many tissues, and cellular damage may result. Unger et al.163,164 have reported that in the Zucker diabetic fatty rat (ZDF), such ectopic lipid accumulation occurs in liver, muscle, and the pancreatic β cell, and that it antedates the presence of diabetes and pancreatic β-cell apoptosis. They coined the term lipotoxicity to describe this phenomenon and have proposed that a major action of leptin is to prevent the accumulation of ectopic lipid and related events (e.g., increased ceramide and oxidant stress) that cause lipotoxicity (apoptosis, mitochondrial dysfunction, inflammation, insulin resistance). Of particular note for this review, decreases in AMPK activity and an increased concentration of malonyl CoA have been found in tissues of the fa/fa and the ZDF rat, rodents with a functionally deficient leptin receptor, and the ob/ob mouse, which does not synthesize leptin.112 Furthermore, treatment with the AMPK activator, AICAR,112 as well as the TZD troglitazone165 and other means of AMPK activation including exercise,166 prevent the ectopic lipid accumulation, pancreatic β-cell damage, and the development of diabetes in the ZDF rat.
Vascular Endothelial Cells
An impressive case has been made that atherogenesis is essentially an inflammatory response to a variety of risk factors, and the consequences of this response include acute coronary and cerebrovascular syndromes.167 An early site at which this inflammatory response appears to occur is the endothelial cell168; indeed, increases in NF-κB expression have been observed in endothelium at sites predisposed to atherosclerotic plaque formation169 and in endothelial cells exposed to elevated concentrations of glucose170 and free fatty acids.171 Likewise, impaired endothelium-dependent relaxation and increases in circulating adhesion molecules (VCAM1, ICAM, selectins), markers of cellular dysfunction and incipient atherosclerotic vascular disease, have been observed in humans with type 2 diabetes and the metabolic syndrome172,173 and in normal individuals in whom plasma FFA levels are increased by a lipid infusion.174 Conversely, endothelial cell dysfunction is diminished in humans by factors that diminish the proinflammatory state, including exercise and caloric restriction175,176 and by treatment with thiazolidinediones.172,177 As already noted, all of these interventions have been reported to activate AMPK in rodents. Studies with endothelial cells in culture also support such a protective role for AMPK. Thus increases in oxidative stress and NF-κB-mediated gene expression observed in cultured endothelium (HUVEC) incubated with palmitate are inhibited by AICAR and other AMPK activators.120,178,179 Also, AICAR and (where studied) expression of a constitutively active AMPK have been shown to inhibit apoptosis, mitochondrial dysfunction, DAG synthesis, and the development of insulin resistance (diminished Akt activation) in HUVEC incubated in a high-glucose medium.180 Finally, it has recently been demonstrated that the administration of atorvastatin by gavage activates AMPK and eNOS in the rat aorta,181 and it has a similar effect when incubated with cultured endothelial cells (HUVEC). Whether this accounts for the anti-atherogenic effect of statins attributed to its antiinflammatory action is an intriguing possibility.182
Liver
Changes similar to those in muscle and the endothelial cell occur in the liver in insulin-resistant states. Thus, as in muscle, an association between hepatic lipid deposition and insulin resistance has been clearly demonstrated in humans.183,184 Also, in rats infused with a lipid emulsion to increase plasma FFA levels during a euglycemic-hyperinsulinemic clamp89,185 or with short-term fat feeding,91 the development of hepatic insulin resistance is associated with increases in DAG content, PKC activation, and a decrease in IKBα abundance—changes almost identical to those observed in human muscle.88 Similar alterations in PKC have been noted in the livers of massively obese, insulin-resistant humans93 and fat-fed rats with hepatic steatosis.91 Also, knockdown of PKCe expression by antisense oligonucleotides protects rats from fat-induced hepatic insulin resistance.186 Finally, as will be discussed later, the metabolic and inflammatory changes observed in the liver of patients with NAFLD/NASH closely resemble those attributable to lipotoxicity in other organs.75
Pancreatic β Cell
Insulin resistance in muscle and liver does not initially result in hyperglycemia because it is accompanied by hyperinsulinemia. As noted previously, it is unclear whether the hyperinsulinemia is compensatory or results from the same factors that cause insulin resistance, in which event it might occur at the same time or even precede it.70,187 In this context, it is noteworthy that increases in plasma FFA have been shown acutely to increase insulin secretion in certain settings, whereas chronic increases in the concentration of saturated fatty acids and glucose cause dysfunction and damage to the β cell and ultimately result in apoptosis.75,99,180,188 Work from a number of laboratories163,164,189,190 has both delineated the events that lead to these phenomena in the β cell and revealed their similarity to the events observed in endothelium and other cells when exposed to high concentrations of FFA or glucose. As already noted, in the ZDF rat, the leptin-receptor deficient rodent characterized by Unger,163 the activity of AMPK in multiple tissues is depressed, and treatment with troglitazone, AICAR, or caloric deprivation prevent or at least markedly attenuate the development of β-cell damage and dysfunction and hyperglycemia.112,165 Likewise, it has been demonstrated that AMPK activation prevents the apoptosis and mitochondrial dysfunction observed in pancreatic β cells when incubated with saturated fatty acids at a high glucose concentration.188 Finally, although theories linking triglyceride accumulation to β-cell dysfunction are attractive and have led to interesting hypotheses,22,177,191,192 to our knowledge there have been no definitive studies showing that triglyceride accumulates in human islets in patients with type 2 diabetes.
Molecular Mechanisms of Insulin Resistance and Cellular Dysfunction According to the Lipid Theory
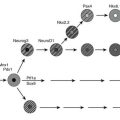
Based on studies reviewed in the preceding sections, a model can be proposed in which insulin resistance and cellular dysfunction are due to an increase in intracellular fatty acid metabolites such as diacylglycerol87,88,193 and cytosolic LCCoA, possibly secondary to dysregulation of AMPK (Figs. 18-5 and 18-6). According to this scheme, such changes activate a serine–threonine kinase cascade that includes conventional and/or novel protein kinase C isoforms,87,88,194,195 IKKB,196,197 and Jun-activated kinase (JNK-1), one or more of which phosphorylate serine residues on IRS-1 (in muscle).85 Similar changes in IRS1 in response to hyperglycemia may also result from activation of the mTOR/p70s6k signaling mechanism.198 Serine phosphorylation of IRS-1 in turn impairs its ability to associate with P13-kinase, leading to a diminished activation by insulin of Akt and PKC zeta, glucose transport, glycogen synthesis, and other insulin-stimulated downstream events. Recent studies demonstrating that transgenic mice with a muscle-specific alteration in IRS-1 Ser→Ala are protected from fat-induced insulin resistance in skeletal muscle strongly support this hypothesis.199 Similar changes appear to occur in liver, except that the inhibitory actions of insulin on gluconeogenesis and glycogenolysis are impaired,85,91,186 and IRS1 and IRS2 may be differentially affected.200 Also possibly involved in this chain of events are increases in oxidative and ER stress, ceramide synthesis, NF-κB activation, and NF-κB-mediated gene expression that could explain, at least in part, the proinflammatory state associated with the metabolic syndrome.73,75,201 Interestingly, the hallmark of this insulin-resistant state is an increase in intracellular triglyceride in liver and muscle that can be quantified noninvasively with magnetic resonance imaging.202,203 Triglyceride accumulation in muscle and liver is generally regarded as a marker of lipid-induced insulin resistance and cellular dysfunction rather than a cause.139 On the other hand, by providing an additional source of intracellular free fatty acids, it could play a more pathogenetic role.
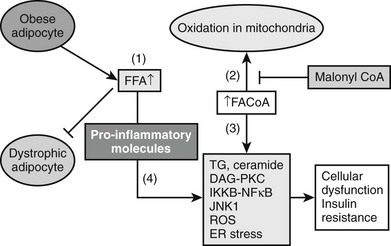
FIGURE 18-5 The pathogenesis of the metabolic syndrome—the lipid hypothesis. According to the proposed scheme, cellular dysfunction and insulin resistance in the setting of the metabolic syndrome can result from multiple events that alter lipid metabolism, including (1) increased plasma FFA and inflammatory cytokines (4) secondary to impaired triglyceride storage and enhanced lipolysis in the fat cell; (2) increased FACoA and inappropriately normal or decreased mitochondrial fatty acid oxidation in liver, muscle, and possibly other tissues due to increases in the concentration of malonyl CoA and other factors (see Fig. 18-4); and (3) increased esterification of FACoA to form triglyceride (TG), diacylglycerol (DAG), and in some tissues ceramide, in increased amounts. By mechanisms only partially understood, these abnormalities can in turn lead to activation of various protein kinase C isoforms, oxidant stress (ROS) and endoplasmic reticulum (ER) stress, and activation of the IKKB-NF-κB system. As described in the text, a decrease in AMPK activity could predispose to all of these abnormalities and AMPK activation could prevent them (see also Figs. 18-4 and 18-6).
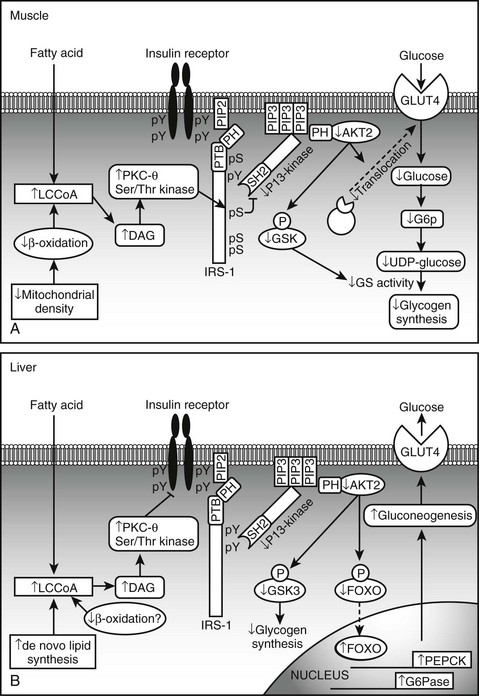
FIGURE 18-6 Proposed mechanism of fatty acid induced insulin resistance in skeletal muscle (A) and liver (B). A, Muscle. Increases is intramyocellular LCCoA and DAG, due to increased fatty acid delivery and/or decreased mitochondrial fatty acid oxidation, trigger a serine/threonine kinase (Ser/Thr) cascade initiated by nPKCs and possibly involving IKKB and/or JNK-1. This ultimately induces serine/threonine phosphorylation of critical IRS-1 sites in muscle, that inhibit IRS-1 tyrosine phosphorylation and activation of PI 3-kinase. This in turn results in reduced insulin-stimulated glucose transport and glycogen synthesis. B, Liver. Increases in intracellular DAG, due to enhanced lipogenesis and/or decreased mitochondrial fatty acid oxidation, activate PKCs (ε and/or possibly δ), which bind to and inactivate the insulin receptor kinase. This results in reduced insulin-stimulated IRS-1 and IRS-2 tyrosine phosphorylation, and decreased insulin activation of PI 3-kinase and AKT2. This in turn leads to diminished GSK3 and FOXO phosphorylation, which respectively decrease insulin-stimulated liver glycogen synthesis and hepatic gluconeogenesis. DAG, Diacylglycerol; FOXO, forkhead box protein O; GLUT, glucose transporter; G6P, glucose-6-phosphate; GSK3, glycogen synthase kinase 3; IRS, insulin receptor substrate; IKKB, IκB kinase β; JNK-1, Jun-activated kinase 1; LCCoA, long-chain acylcoenzyme A; PEPCK, phosphoenolpyruvate carboxykinase; PI 3-kinase, phosphatidylinositol-3-kinase; PTB, phosphotyrosine-binding domain; PH, pleckstrin homology domain; SH2, src homology domain. (Adapted from Savage DB, Petersen KF, Shulman GI: Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev 87:507–520, 2007.)
The Hypothalamus, Food Intake, and Insulin Resistance
Leptin, which activates AMPK in peripheral tissues by a direct action, diminishes AMPK activity in the hypothalamus. This in turn leads to decreased food intake and activation of the sympathetic nervous system and secondary to this, further activation of AMPK in peripheral tissues.198 Conversely, glucocorticoids increase AMPK in the hypothalamus, leading to an increase in food intake, and they decrease AMPK and cause insulin resistance in peripheral tissues [see discussion of Cushing’s Syndrome in reference 122, Christ-Crain et al., 2008]. Various antipsychotic drugs have also been found to activate AMPK in the hypothalamus, and like glucocorticoids, they increase food intake and cause insulin resistance.204 Why some agents that decrease or increase AMPK activity in the hypothalamus have opposite effects on peripheral tissues is not known. On the other hand, the possibility that a drug that activates or inhibits AMPK activity in peripheral tissues has the opposite effect in the hypothalamus or elsewhere in the central nervous system needs to be considered in evaluating its clinical efficacy and side effects.
Other Theories of Insulin Resistance
Alternative theories to explain the origin of insulin resistance and cellular dysfunction in the metabolic syndrome have focused on glucocorticoid excess and the cellular conversion of cortisone to cortisol by 11-β-dehydrogenase,205 mutations of PPARγ,206 alterations in muscle capillarity,35 and increased flux through the hexosamine pathway.207,208 Perhaps the greatest attention, however, has been given to the theories in which the combination of oxidative and ER stress, IKKB and NF-κB activation, and proinflammatory changes play a central role. As noted by numerous investigators, all three have been linked to insulin resistance in humans and experimental animals in a wide variety of disorders, which they often antedate.9,10,173,201,209 Because of this, it has been suggested that an abnormality of the innate immune system could be a proximal event in the pathogenesis of the metabolic syndrome.10 Although this possibility cannot be disproved, and it is unquestioned that proinflammatory events are an integral component of the metabolic syndrome, a number of observations including the following suggest that dysregulation of lipid metabolism that can lead to oxidative stress and inflammation is likely to be a more primary factor: (1) the close correlation of the metabolic syndrome with obesity, central obesity, ectopic lipid deposition and elevated plasma FFA levels; (2) the presence of the metabolic syndrome and ectopic lipid deposition in humans and experimental animals deficient in peripheral adipose tissue (lipodystrophy) and the reversal of these abnormalities in rodents by the implantation of fat137,138,210 and in humans and/or rodents by the administration of leptin,211,212 adiponectin,144 and in some instances thiazolidinediones, all of which activate AMPK75; (3) the observation that elevating plasma FFA levels in humans and rodents by itself acutely (hours) leads to insulin resistance and proinflammatory changes85,88,185; and (4) the very early occurrence of alterations in cellular lipid metabolism and mitochondrial function in normal-weight, normoglycemic, young offspring of diabetic parents.7 To our knowledge, evidence of a proinflammatory state has not been reported in the latter group. These considerations aside, the possibility that proinflammatory changes leading to alterations in lipid metabolism are the cause of the metabolic syndrome has not been ruled out; indeed, studies in rodents suggest that TNF-α-induced inflammation can lead to a decrease in AMPK activity in skeletal muscle.123 In addition, it has recently been demonstrated that treatment with the antiinflammatory drug salsalate improves glycemic status and reduces systemic inflammation in obese adults at risk for developing type 2 diabetes,213 and a multicenter trial to test the efficacy of this drug is currently in progress. Since therapies targeting lipid metabolism, oxidative stress, and in some instances inflammatory events have all been shown to diminish insulin resistance in specific situations,92,185,201,209 what is clear is that these events are almost certainly interrelated.
Diagnosis
No single definitive diagnostic test for the metabolic syndrome is yet available. Historically, it has been diagnosed based on the presence of general or abdominal obesity, dyslipidemia, hypertension, and impaired fasting glucose or glucose intolerance in various combinations. In addition, the presence of premature coronary heart disease, type 2 diabetes in its early stages, and other disorders associated with insulin resistance have sometimes been considered diagnostic criteria. Several organizations have published standards for diagnosis. The criteria adopted by the National Cholesterol Education Program, Adult Treatment Panel (ATP III) and by the World Health Organization (WHO) (Tables 18-3 and 18-4) have recently been reviewed.214 They differ principally in that the WHO criteria place more emphasis on measures of insulin resistance, the presence of microalbuminuria, and the use of the glucose tolerance test, whereas the ATP III emphasizes abdominal obesity and risk factors for cardiovascular disease such as dyslipidemia and hypertension. A more recent set of criteria proposed by the International Diabetes Federation is similar to that of ATP III but with somewhat lower cutoff points for blood pressure and fasting glucose.17 In part because of its relative simplicity for clinical purposes, the ATP guidelines appear to be in widest use. On the other hand, they probably underestimate the prevalence of insulin resistance in the general population.215 Also of note, modified criteria have been developed for different ethnic groups, since in some of them (e.g., South Asians) the metabolic syndrome is not as closely associated with obesity (based on BMI) as it is in Caucasians17,216 (see Table 18-3).
Table 18-3
ATP III Diagnostic Criteria for the Metabolic Syndrome

*American Diabetes Association has recently lowered fasting glucose level to 100 mg/dL.
Data from Grundy SM, Brewer HB, Cleeman JI et al: Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on scientific issues related to definition. Circulation 109:433–438, 2004.
Table 18-4
Insulin resistance, identified by one of the following:
Or for those with normal fasting glucose levels (<110 mg/dL), glucose uptake below the lowest quartile for background population under investigation under hyperinsulinemic, euglycemic, conditions
Plus any two of the following:
Antihypertensive medication and/or high blood pressure (≥140 mm Hg systolic or ≥90 mm Hg diastolic)
Plasma triglycerides ≥ 150 mg/dL (≥1.7 mmol/L)
HDL cholesterol < 35 mg/dL (<0.9 mmol/L) in men or <39 mg/dL (1.0 mmol/L) in women
BMI > 30 kg/m2 and/or waist : hip ratio > 0.9 in men, > 0.85 in women
Urinary albumin excretion rate ≥ 20 µg/min or albumin : creatinine ratio ≥ 30 mg/g
Adapted from Grundy SM, Brewer HB, Cleeman JI et al: Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on scientific issues related to definition. Circulation 109:433–438, 2004.
Linkage of Metabolic Syndrome to Coronary Heart Disease and Type 2 Diabetes
The notion that the metabolic syndrome, or its surrogate markers hyperinsulinemia and insulin resistance, antedate and contribute to the pathogenesis of coronary heart disease, diabetes, and at least some cases of hypertension was proposed many years ago.21,35 Coronary heart disease in the setting of the metabolic syndrome can to a great extent be attributed to the dyslipidemia present in people with the metabolic syndrome (increased dense LDL, diminished HDL cholesterol, and hypertriglyceridemia)217 as well as to elevations in blood pressure and blood glucose and the presence of a procoagulant, proinflammatory state22,214 In addition, some studies suggest that hyperinsulinemia and insulin resistance, as well as hyperglycemia, may be independent risk factors.51 Whether elevated FFA levels or a dysregulation of intracellular fatty acid metabolism contribute to atherosclerosis by directly altering the function of endothelium (see Vascular Endothelial Cells) or other cells in the vascular wall remains to be determined. Relevant to this discussion, low levels of adiponectin are associated with an increased risk of coronary heart disease in humans,146 whereas as noted earlier, overexpression of adiponectin or its globular subunit diminishes the severity of atherosclerosis in ApoE -/- mice.148,149
More definitive evidence that the metabolic syndrome per se predisposes to coronary heart disease and cerebrovascular disease has recently been reported. Thus a two- to fourfold increase in subsequent cardiovascular events has been described in men and women with the metabolic syndrome (modified WHO criteria) even in the absence of type 2 diabetes or impaired glucose tolerance.218–220 Qualitatively, similar results have been obtained when the metabolic syndrome was defined by ATP III criteria221,222 (Fig. 18-7). In a compilation of multiple studies, the presence of the metabolic syndrome had a greater impact on the risk for developing diabetes (fivefold) than of ASCVD (twofold).22,177,192 In addition, where studied, the rate of cardiovascular events was higher in patients who had diabetes and the metabolic syndrome than in individuals with only the metabolic syndrome.22,223
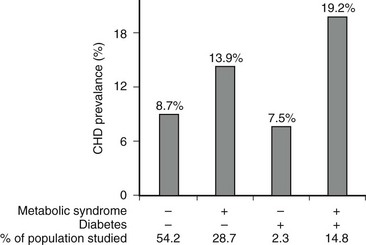
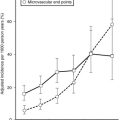
FIGURE 18-7 Age-adjusted prevalence of coronary heart disease (CHD) in the U.S. population over 50 years of age categorized by the presence of the metabolic syndrome (ATP III criteria) and diabetes. Data are from the NHANES study. The complete absence of an increase in CHD incidence in the diabetic patients without the metabolic syndrome should be viewed with caution because of the small size of this group. (Data from Alexander CM, Landsman PB, Teutsch SM, Haffner SM: NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 52(5):1210–1214, 2003.)
Linkage of the Metabolic Syndrome to Other Disorders
Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis
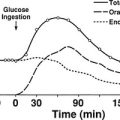
It is estimated that nearly 20,000,000 people in the United States have the diagnosis of nonalcoholic fatty liver disease (NAFLD), and approximately 10% of these individuals develop nonalcoholic steatohepatitis (NASH), a disorder characterized by mitochondrial dysfunction, increases in oxidative stress and cell cytokines and a predisposition to cirrhosis (approximately 20% of patients with NASH), and less commonly hepatocellular carcinoma.67,150,224–226 NAFLD is also seen with increasing frequency in children and adolescents in parallel with the increasing prevalence of obesity in these populations.227 Presumably, mildly abnormal liver function tests in the presence of obesity or type 2 diabetes or other manifestations of insulin resistance would help to identify individuals with NAFLD at an early point in time. Recent studies in young, lean, insulin-resistant individuals have demonstrated that insulin resistance in skeletal muscle, as reflected by decreased glycogen synthesis, can promote atherogenic dyslipidemia by changing the pattern of ingested carbohydrate away from skeletal muscle glycogen synthesis into hepatic de novo lipogenesis, resulting in an increase in plasma triglyceride concentrations and a reduction in plasma high-density lipoprotein concentrations (Fig. 18-8). Furthermore, insulin resistance in these subjects was independent of changes in the plasma concentrations of TNF-α, IL-6, high-molecular-weight adiponectin, resistin, retinol-binding protein 4, or intraabdominal obesity, suggesting that these factors do not play a primary role in causing insulin resistance in the early stages of the metabolic syndrome.228
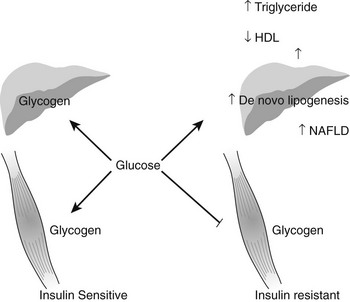
FIGURE 18-8 Schematic of whole-body energy distribution after high-carbohydrate mixed meals in young insulin-sensitive and insulin-resistant individuals. (Adapted from Petersen KF, Dufour S, Savage DB et al: The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 104:12587–12594, 2007.)
Polycystic Ovary Syndrome
Another common disorder that becomes more prevalent in the presence of the metabolic syndrome is polycystic ovary syndrome (PCOS).68,69 PCOS is characterized by genetically determined increases in ovarian androgen production and disordered gonadotropin secretion that may be exaggerated by hyperinsulinemia or insulin resistance. Perhaps because of its association with the metabolic syndrome and often although not always with obesity, PCOS is a leading risk factor for glucose intolerance and type 2 diabetes in adolescent and premenopausal women.229 It may also increase their risk for premature cardiovascular disease.230 PCOS is associated with increases in such inflammatory factors as plasminogen activator inhibitor (PAI), c-reactive protein (CRP), and TNF-α.68 Like NAFLD/NASH, type 2 diabetes, and other disorders associated with the metabolic syndrome, PCOS often responds to treatments that activate AMPK and/or lower the concentration of malonyl CoA, such as diet and exercise, metformin, and thiazolidinediones68 (see Table 18-2 and section on Treatment).
Certain Cancers
The reason for the link between the metabolic syndrome; obesity; type 2 diabetes; and cancers of the colon, breast, liver, and other sites is less clear. It has generally been held that insulin-like growth factor 1 (IGF-1) could be a factor, since insulin both stimulates the synthesis of IGF-1 by liver and inhibits the synthesis of its binding protein IGFBP-1.231 Another possibility relates to the fact that an upstream kinase that activates AMPK is LKB1,232,233 a tumor suppressor that is deficient in people with Peutz-Jeghers syndrome and places them at increased risk for developing carcinomas of the colon, stomach, and pancreas and adenocarcinomas of the lung. The possibility that LKB1 and AMPK link the metabolic syndrome to these cancers and are targets for their prevention or therapy warrants consideration.234–236
Alzheimer’s Disease
Alzheimer’s disease and other disorders associated with dementia are more common in individuals with the metabolic syndrome and type 2 diabetes. As reviewed elsewhere,237 Alzheimer’s disease appears to be associated with insulin resistance as well as inflammation and mitochondrial dysfunction in the brain. Also of note, physical activity, which has been shown to activate AMPK in many tissues in rodents,119 has demonstrated efficacy for preventing and treating Alzheimer’s disease in both humans and experimental animals.3,237 Likewise, benefits from thiazolidinedione treatment have been reported in some studies.238
Cushing’s Syndrome and Related Disorders
Patients with primary Cushing’s syndrome or Cushing’s syndrome due to therapy with glucocorticoids typically demonstrate central obesity, insulin resistance, hyperinsulinemia, and a predisposition to diabetes and hypertension.239 In addition, like other patients with this clustering of events, they are at increased risk of coronary heart disease.240 The value of treatments aimed at AMPK and malonyl CoA in people with Cushing’s syndrome, when its primary cause cannot be corrected, has to our knowledge not been evaluated. It is noteworthy, however, that glucocorticoids diminish AMPK in peripheral tissues in rodents and that AMPK activity is decreased in visceral fat of patients with Cushing’s syndrome.122 The possibility that an increase in cellular 11-β-dehydrogenase activity resulting in local increases in cortisol could be a cause of the metabolic syndrome205 has already been discussed.
Lipodystrophy
Patients with primary lipodystrophy and lipodystrophy secondary to drug therapy (protease inhibitors in HIV patients) appear to be subject to the same lipotoxicity observed in people with the metabolic syndrome for other reasons. Two factors appear to contribute to an insulin-resistant state in these patients: (1) a decrease in peripheral fat cells that causes more plasma FFA and lipoprotein triglyceride to be shunted to ectopic sites, and (2) plasma leptin and adiponectin levels are very low. The importance of the latter is suggested by successes in treating some patients with leptin211,212 or with thiazolidinediones.241 Interestingly, in the latter study the beneficial effect of rosiglitazone was accompanied by an increase in adiponectin but not of subcutaneous fat mass.
Hyperalimentation
As recently suggested by Unger,242 hyperalimentation can cause lipotoxic damage and by inference, the metabolic syndrome in some individuals. He hypothesized that at one extreme are normal individuals who eat enormous amounts of food for an extended period and become hyperinsulinemic and insulin resistant when the ability of their adipose tissue to deposit the excess lipid in their diet is exceeded.243 At the other extreme are hyperalimented patients with an extensive loss of subcutaneous adipose tissue due to third-degree burns. Such burn patients are historically very insulin resistant; however, it remains to be determined if their burns are associated with the predicted increase in ectopic lipid deposition and, if so, whether their insulin resistance responds to pharmacologic agents that enhance insulin sensitivity.
Additional Disorders
The metabolic syndrome has also been associated with an increased prevalence of such disorders as gout, sleep apnea, gallstones, and chronic kidney disease.192 Like other metabolic syndrome–related disorders, they are associated with obesity, type 2 diabetes, and a predisposition to atherosclerotic cardiovascular disease.
Treatment of the Metabolic Syndrome
The demonstration that the metabolic syndrome increases the risk for developing type 2 diabetes, coronary heart disease, and other disorders in both otherwise normal individuals and patients with type 2 diabetes strongly suggests that it warrants treatment. Less clear is what therapies should be used in a given circumstance and when they should be started (see Table 18-3).
Lifestyle Modification (Weight Loss and Physical Activity)
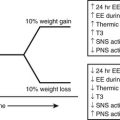
The treatment of the metabolic syndrome with the aim of preventing cardiovascular disease has recently been reviewed by a clinical conference jointly sponsored by the American Heart Association; National Heart, Lung and Blood Institute; and the American Diabetes Association (AHA/NHLBI/ADA). The consensus was that lifestyle modifications consisting of diet for the treatment of obesity and overweight and physical activity were the first line of therapy.214,244,245 The efficacy of this approach for preventing disease has not been assessed specifically in patients with the metabolic syndrome diagnosed by ATP III or WHO criteria; however, exercise has been shown to reverse the defects in insulin-stimulated glucose transport and muscle glycogen synthesis in young, lean, insulin-resistant offspring of parents with type 2 diabetes,46 and in several prospective studies,61–63 the combination of diet and exercise has proven quite effective in delaying or preventing the onset of diabetes in patients with impaired glucose tolerance (most of whom probably had the metabolic syndrome). Also, numerous epidemiologic studies have shown a 30% to 50% decrease in the risk of developing coronary heart disease, as well as type 2 diabetes, with the maintenance of a physically active compared to a sedentary lifestyle.245 It must be emphasized, however, that the incidence of diabetes in the treated patients with impaired glucose tolerance in these studies was still higher than in the general population,61,63 suggesting that to be maximally effective, lifestyle changes may have to be introduced even earlier. Whether lifestyle changes have a similar effect on coronary heart disease is less certain. However, in follow-up studies of patients in the U.S. and Finnish diabetes prevention program who were insulin resistant, diet and exercise diminished nontraditional risk factors for coronary heart disease, such as c-reactive protein and fibrinogen, as well as some of the more classic risk factors associated with the metabolic syndrome.246,247
Drug Therapy
When recommended therapeutic goals are not achieved with diet and exercise, pharmacologic therapy is necessary. The recommendations of the AHA/NHLBI/ADA at different stages of the metabolic syndrome, based on Framingham risk scores for developing ASCVD, have been presented elsewhere.22,223 Suffice it to say, they vary with an individual’s risk for developing coronary heart disease and cerebrovascular disease over given time intervals. Thus in an individual already at risk for these disorders because of diabetes, the use of statins and other agents to diminish plasma cholesterol has a lower goal (LDL-C < 100 mg/dL) than in someone with isolated hypercholesterolemia. Likewise, certain antihypertensive agents such as angiotension-converting enzyme (ACE) inhibitors248 and fibric-acid derivatives249 may be of special benefit in patients with type 2 diabetes or at high risk for developing it.
In patients with diabetes, metformin and thiazolidinediones (TZDs) are routinely used because of their blood glucose–lowering actions and in the case of the thiazolidinediones, their insulin-sensitizing effect (Table 18-5). To what extent they and other oral agents should be used as second-line therapy for the metabolic syndrome in the absence of diabetes or impaired glucose tolerance is still a matter for debate. In 2004, an AHA/NHLBI/ADA conference on the metabolic syndrome244 concluded that “there is insufficient evidence to recommend these drugs for anything other than their glucose-lowering action at this time”. On the other hand, as already noted, both metformin and TZDs have shown some efficacy in treating NAFLD and PCOS, and in the United Kingdom prospective diabetes study,250 chronic metformin therapy was associated with a decrease in new cardiovascular events in obese individuals with type 2 diabetes that was still evident 10 years after this initial finding.251
Table 18-5
Recommended Treatments for the Metabolic Syndrome in Adults, Children, and Adolescents
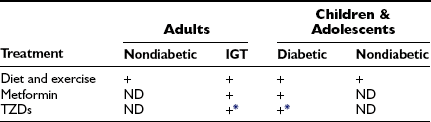
+, Currently recommended treatment; ND, as yet no definitive evidence for or against use.
*See text for discussion of controversy concerning TZD use.
Data from Alberti G, Zimmet P, Shaw J, Bloomgarden Z, Kaufman F, Silink M: Type 2 diabetes in the young: the evolving epidemic: the International Diabetes Federation Consensus Workshop. Diabetes Care 27:1798–1811, 2004.
The picture with respect to the TZDs is less clear. TZDs have shown efficacy equal to that of lifestyle change and better than that of metformin in diminishing progression from impaired glucose tolerance to overt diabetes,61,65 and they have shown utility in treating NAFLD.226 However, a controversial meta-analysis252 concluded that the TZD rosiglitazone increases the incidence of myocardial infarction in patients with established type 2 diabetes. In contrast, in a second meta-analysis by the same group253 and in a large prospective study,254 a different TZD, pioglitazone, was found to cause a modest decrease in cardiovascular events and/or death due to myocardial infarction. Pioglitazone has also been demonstrated to decrease the progression of atherosclerosis and diminish plaque size in patients with type 2 diabetes.255 In part based on these findings, a special panel of the Joint American and European Diabetes Associations recently recommended that rosiglitazone not be used for the therapy of type 2 diabetes.256 Whether pioglitazone should be used in patients with impaired glucose tolerance and the metabolic syndrome to prevent the development of type 2 diabetes when lifestyle change is unsuccessful remains to be determined.
The role of statins in the treatment of the metabolic syndrome is somewhat clearer. A recent subgroup analysis of statin trials revealed that this class of agents reduces the risk for cardiovascular events in people with the metabolic syndrome.257 To what extent this is because of their ability to diminish LDL cholesterol and the levels of all apolipoprotein B–containing lipoproteins and to what extent their antiinflammatory action182,258 is uncertain. Recently it has been demonstrated that treatment with rosuvastatin (for 1.9 years) reduces the incidence of major cardiovascular events by 50% in apparently healthy 60- to 70-year-old people, and that it does so independently of hyperlipidemia and other indices of metabolic syndrome and of plasma cholesterol.182 Presumably, statins would have the same or an even greater effect in people with the metabolic syndrome and evidence of inflammation; however, this remains to be proven. The NHLBI/AHA/ADA panel244 recommended that cigarette smoking be discouraged in all individuals with the metabolic syndrome. It also suggested that low-dose aspirin for primary prevention of coronary heart disease in patients with the metabolic syndrome is promising.257 The subject of treatment of the metabolic syndrome, based on the risk for coronary heart disease by Framingham criteria, has recently been reviewed.22,259
Special Considerations
The magnitude of the problem is such that the prevention of diseases associated with the metabolic syndrome will almost certainly require governmental and societal, as well as individual, change. In such an effort, a primary target will be the prevention of obesity and central adiposity through increasing physical activity and promoting healthy eating.18 It has recently been suggested that for children at risk for developing type 2 diabetes, school-based programs and governmental actions that focus on lifestyle will be required.18 It is highly likely that similar programs will be required for adults.
Children and Adolescents
Recent studies indicate that the prevalence of the metabolic syndrome is increasing dramatically in children and adolescents in parallel with the increase in obesity and type 2 diabetes. In adolescents aged 12 to 19 years, in the NHANES study (1988-1994), the metabolic syndrome, diagnosed by ATPIII criteria, was present in 4% of the total population, 6.8% of those classified as overweight (BMI = 85th to 95th percentile), and 28% of those categorized as obese (BMI > 95th percentile).260 Furthermore, since the incidence of obesity has been increasing in the 10 years since these data where obtained, these are likely to be underestimates of its prevalence at the present time.
Where studied, the metabolic syndrome in children and adolescents has been associated with insulin resistance, central adiposity, dyslipidemia, elevations in blood pressure, and increases in intramyocellular and intrahepatic lipid, much as in adults, although with different absolute measurements.19,20 In addition, as in adults, the prevalence of NAFLD227 and PCOS68 are increased, as are plasma levels of the proinflammatory markers, c-reactive protein and IL-6, and immunoassayable adiponectin in plasma is diminished.20 Also, the prevalence of cardiovascular diseases in these individuals later in life may be increased.261–263 Current treatment recommendations for children with type 2 diabetes, most of whom have the metabolic syndrome, have recently been discussed critically by an International Diabetes Federation Consensus Workshop.18
Low-Birth-Weight Infants
There is abundant evidence that a low birth weight, independent of gestational age, predicts the development of type 2 diabetes, coronary heart disease, central obesity, and other aspects of the metabolic syndrome in middle age in some individuals.264,265 Recent reports suggest that the greatest risk is in infants with low birth weight who gain weight rapidly in childhood.266 It has been suggested that an altered intrauterine environment (e.g., poor nutrition) could be responsible for the low birth weight in these children. The nature of alteration is not known, nor is it clear how it makes these infants more likely to develop the metabolic syndrome in later life.
The Thrifty Genotype and the Increasing Incidence of the Metabolic Syndrome
The recent increase in the prevalence of the metabolic syndrome and type 2 diabetes has been attributed to the epidemic of obesity that began in the second half of the 20th century and continues unabated. As reviewed elsewhere,267 this is almost certainly a reflection of environmental factors, most notably the increased availability of food and the decrease in physical activity that occurred in many industrialized societies during this time period. In 1962, Neel268 raised the question of why diabetes (type 2), which has adverse effects on health, persisted in humans during the course of evolution. He suggested that it could be related to the existence of a “thrifty gene” that predisposes individuals to obesity, and secondarily diabetes, but had survival value for our hunter-gatherer ancestors. More specifically, he proposed that in the feast-famine environment of early humans “individuals exceptionally efficient in the uptake and utilization of food and its storage as fat” would have had a selective advantage. Neel further suggested that in our modern environment of calorie surplus, such a gene (or genes) which protected us from death when food sources were scarce might make us prone to obesity and diabetes (and the metabolic syndrome). Over the years, there have been many candidate thrifty genes suggested, including UCP2, UCP3, and B3-adrenergic receptors. Genes governing the AMPK/malonyl CoA fuel sensing network, which has been linked to the regulation of both food intake103,162,269 and energy expenditure,103,108 have also been suggested. This is an attractive notion, since a decreased ability to oxidize fatty acids appears to be characteristic of pre-obese humans.76,107 For the same reason, genes that regulate mitochondrial biogenesis also need to be considered.7 In addition, decreased mitochondrial content/activity, be it acquired or inherited, might promote obesity by two mechanisms. First, by decreasing daily energy expenditure, even by a small amount, it could lead to progressive weight gain. For instance, a 50-calorie-per-day reduction in energy expenditure in a typical adult, if not accompanied by a comparable decrease in food intake, would lead to a weight gain of approximately 5 lb/year. Second, a decrease in mitochondrial activity in skeletal muscle, by predisposing an individual to altered cellular lipid metabolism (as reflected by an increase in intramyocellular triglyceride), like alterations in the AMPK/malonyl CoA network,75 can lead to insulin resistance and hyperinsulinemia.114 The latter in turn could promote obesity by increasing lipogenesis in liver and adipose tissue and by inhibiting lipolysis until a new, more obese, steady state is achieved.
Concluding Remarks
1. The metabolic syndrome is becoming increasingly common. Upwards of 50,000,000 people above the age of 20 are affected in the United States, and the diagnosis is becoming increasingly common in children and adolescents.
2. In both adolescents and adults, the metabolic syndrome substantially increases the risk for type 2 diabetes, coronary heart disease, and other associated disorders.
3. The prevalence of the metabolic syndrome is markedly increased in people who lack adipose tissue, as well as in people who are obese. In both populations, a common occurrence is ectopic lipid deposition in muscle, liver, and often visceral fat.
4. An increasing body of evidence suggests that a likely pathogenetic mechanism in most patients is an abnormality of cellular lipid metabolism that causes lipotoxic changes including: insulin resistance, oxidant and ER stress, inflammation, and mitochondrial dysfunction in one or more tissues. Proposed causes of these abnormalities in cell lipid metabolism are elevated plasma free fatty acid levels, dysregulation of the AMPK, and/or malonyl CoA and primary mitochondrial defects.
5. Genetic, metabolic, and functional changes attributable to the metabolic syndrome are present in lean offspring of diabetic parents. Because of this and the fact that it antedates most of the diseases with which it is associated, the metabolic syndrome appears to be an excellent target for disease prevention.
6. Lifestyle modifications, consisting of diet to induce weight loss and exercise, have been shown to diminish many of the abnormalities of the metabolic syndrome and to prevent or delay progression from impaired glucose tolerance to type 2 diabetes in prospective studies. Because of this and their safety and low cost, diet and exercise are the first line of therapy for children and adolescents with the metabolic syndrome and for adults without associated diseases. The role of “insulin-sensitizing” agents such as thiazolidinediones and metformin in individuals who are not hyperglycemic remains to be determined.
7. By virtue of the number of people affected and its many potential consequences, the metabolic syndrome is a major public health problem. As such, it will require governmental and societal as well as individual change in children and adolescents and possibly for adults.
References
1. Eckel, RH, Grundy, SM, Zimmet, PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428.
2. Reaven, G. Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation. 2002;106:286–288.
3. Richter, E, Ruderman, N. AMPK and the biochemistry of exercise: implications for human health and disease. The Biochemical Journal. 2009. [in press].
4. Kissebah, AH, Vydelingum, N, Murray, R, et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab. 1982;54:254–260.
5. Bernstein, RS, Grant, N, Kipnis, DM. Hyperinsulinemia and enlarged adipocytes in patients with endogenous hyperlipoproteinemia without obesity or diabetes mellitus. Diabetes. 1975;24:207–213.
6. Ruderman, NB, Schneider, SH, Berchtold, P. The “metabolically-obese,” normal-weight individual. Am J Clin Nutr. 1981;34:1617–1621.
7. Petersen, KF, Dufour, S, Befroy, D, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671.
8. Ruderman, N, Chisholm, D, Pi-Sunyer, X, et al. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47:699–713.
9. Festa, A, D’Agostino, R, Jr., Howard, G, et al. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation. 2000;102:42–47.
10. Pickup, JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823.
11. Schmidt, MI, Duncan, BB, Sharrett, AR, et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652.
12. Juhan-Vague, I, Alessi, MC, Vague, P. Increased plasma plasminogen activator inhibitor 1 levels. A possible link between insulin resistance and atherothrombosis. Diabetologia. 1991;34:457–462.
13. Sakkinen, P, Geffken, D, Cushman, M, et al. Association between physical activity and markers of inflammation in a healthy elderly population. Am J Epidemiol. 2001;153:242–250.
14. Reaven, GM. Insulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular disease. J Clin Endocrinol Metab. 2003;88:2399–2403.
15. Grundy, SM, Cleeman, JI, Daniels, SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752.
16. Ford, ES, Giles, WH, Dietz, WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359.
17. Grundy, SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28:629–636.
18. Alberti, G, Zimmet, P, Shaw, J, et al. Type 2 diabetes in the young: the evolving epidemic: the international diabetes federation consensus workshop. Diabetes Care. 2004;27:1798–1811.
19. Weiss, R, Dufour, S, Taksali, SE, et al. Prediabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet. 2003;362:951–957.
20. Weiss, R, Dziura, J, Burgert, TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–2374.
21. Reaven, GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607.
22. Grundy, SM. Metabolic syndrome: a multiplex cardiovascular risk factor. J Clin Endocrinol Metab. 2007;92:399–404.
23. Kahn, R, Buse, J, Ferrannini, E, et al. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28:2289–2304.
24. Viollet, B, Mounier, R, Leclerc, J, et al. Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes Metab. 2007;33:395–402.
25. Dieterle, P, Fehm, H, Stroder, W, et al. Asymptomatic diabetes mellitus in hypertensive patients of normal weight. Glucose tolerance and serum levels of insulin and nonesterified fatty acids in essential hypertension. Ger Med Mon. 1968;13:478–483.
26. Modan, M, Halkin, H, Almog, S, et al. Hyperinsulinemia. A link between hypertension obesity and glucose intolerance. J Clin Invest. 1985;75:809–817.
27. Stern, MP. The insulin resistance syndrome. In: Alberti KGMM, ed. International Textbook of Diabetes Mellitus. New York: John Wiley & Sons Ltd.; 1997:255–283.
28. Flier, JS. An overview of insulin resistance. In: Moller DE, ed. Insulin Resistance. Chichester, U.K.: Wiley; 1993:1–8.
29. Himsworth, H. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Lancet. 1936;1:127.
30. Himsworth, HP, Kerr, RB. Insulin sensitive and insulin-insensitive types of diabetes mellitus. Clin Sci. 1939;4:119–152.
31. Himsworth, HP. The syndrome of diabetes mellitus and its causes. Lancet. 1949;1:465–473.
32. Yalow, RS, Berson, SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39:1157–1175.
33. Berson, SA, Yalow, RS. Plasma insulin in health and disease. Am J Med. 1961;31:874–881.
34. Karam, JH, Grodsky, GM, Forsham, PH. Excessive insulin response to glucose in obese subjects as measured by immunochemical assay. Diabetes. 1963;12:197–204.
35. Bjorntorp, P. The relationship between obesity and diabetes. In: Alberti KGMM, ed. International Textbook of Diabetes Mellitus. New York: John Wiley & Sons Ltd.; 1997:612–627.
36. Davidson, PC, Albrink, MJ. Insulin resistance in hyperglyceridemia. Metabolism. 1965;14:1059–1070.
37. DeFronzo, RA, Ferrannini, E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194.
38. Malherbe, C, Heller, F, de Gasparo, M, et al. Insulin response during prolonged glucose infusion. J Clin Endocrinol Metab. 1970;30:535–538.
39. Ruderman, NB, Berchtold, P, Schneider, S. Obesity-associated disorders in normal-weight individuals: some speculations. Int J Obes. 1982;6(Suppl 1):151–157.
40. Ruderman, NB, Haudenschild, C. Diabetes as an atherogenic factor. Prog Cardiovasc Dis. 1984;26:373–412.
41. Petersen, KF, Dufour, S, Feng, J, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A. 2006;103:18273–18277.
42. Bays, H, Mandarino, L, DeFronzo, RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab. 2004;89:463–478.
43. Lapidus, L, Bengtsson, C, Larsson, B, et al. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow up of participants in the population study of women in Gothenburg, Sweden. Br Med J (Clin Res Ed). 1984;289:1257–1261.
44. Larsson, B, Svardsudd, K, Welin, L, et al. Abdominal adipose tissue distribution, obesity, and risk of cardiovascular disease and death:13 year follow up of participants in the study of men born in 1913. Br Med J (Clin Res Ed). 1984;288:1401–1404.
45. Beck-Nielsen, H, Groop, LC. Metabolic and genetic characterization of prediabetic states. Sequence of events leading to non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:1714–1721.
46. Perseghin, G, Price, TB, Petersen, KF, et al. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med. 1996;335:1357–1362.
47. Rothman, DL, Magnusson, I, Cline, G, et al. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A. 1995;92:983–987.
48. Ferrari, P, Weidmann, P, Shaw, S, et al. Altered insulin sensitivity, hyperinsulinemia, and dyslipidemia in individuals with a hypertensive parent. Am J Med. 1991;91:589–596.
49. Ruderman, NB, Schneider, SH, Berchtold, P. The “metabolically-obese,” normal-weight individual. Am J Clin Nutr. 1981;34:1617–1621.
50. Werbin, B, Tamir, I, Heldenberg, D, et al. Immunoreactive insulin and glucose response to oral glucose in offspring of patients with endogenous hypertriglyceridaemia. Clin Chim Acta. 1977;76:35–40.
51. Despres, JP, Lamarche, B, Mauriege, P, et al. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med. 1996;334:952–957.
52. Ducimetiere, P, Eschwege, E, Papoz, L, et al. Relationship of plasma insulin levels to the incidence of myocardial infarction and coronary heart disease mortality in a middle-aged population. Diabetologia. 1980;19:205–210.
53. Pyorala, K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: results from two population studies in Finland. Diabetes Care. 1979;2:131–141.
54. Welborn, TA, Wearne, K. Coronary heart disease incidence and cardiovascular mortality in Busselton with reference to glucose and insulin concentrations. Diabetes Care. 1979;2:154–160.
55. Nesto, RW, Phillips, RT, Kett, KG, et al. Angina and exertional myocardial ischemia in diabetic and nondiabetic patients: assessment by exercise thallium scintigraphy. Ann Intern Med. 1988;108:170–175.
56. UKPDS. Complications of newly diagnosed type 2 diabetes patients and their association with different clinical and biochemical risk factors. Diab Res. 1990;13:1–11.
57. Uusitupa, MI, Niskanen, LK, Siitonen, O, et al. Ten-year cardiovascular mortality in relation to risk factors and abnormalities in lipoprotein composition in type 2 (non-insulin-dependent) diabetic and non-diabetic subjects. Diabetologia. 1993;36:1175–1184.
58. Jarrett, RJ, McCartney, P, Keen, H. The Bedford survey: ten year mortality rates in newly diagnosed diabetics, borderline diabetics and normoglycaemic controls and risk indices for coronary heart disease in borderline diabetics. Diabetologia. 1982;22:79–84.
59. Haffner, SM, Stern, MP, Hazuda, HP, et al. Cardiovascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA. 1990;263:2893–2898.
60. Panzer, C, Brieke, A, Ruderman, NB. Prevention of type 2 diabetes and its macrovascular complications: whom, when and how should we treat? Current Opinions in Endocrinology and Diabetes. 2003;10:219–236.
61. Knowler, WC, Barrett-Connor, E, Fowler, SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
62. Pan, XR, Li, GW, Hu, YH, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance, The Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544.
63. Tuomilehto, J, Lindstrom, J, Eriksson, JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350.
64. Buchanan, TA, Xiang, AH, Peters, RK, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes. 2002;51:2796–2803.
65. Gerstein, HC, Yusuf, S, Bosch, J, et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105.
66. Neuschwander-Tetri, BA, Brunt, EM, Wehmeier, KR, et al. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38:1008–1017.
67. Neuschwander-Tetri, BA, Caldwell, SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219.
68. Dhindsa, G, Bhatia, R, Dhindsa, M, et al. Insulin resistance, insulin sensitization and inflammation in polycystic ovarian syndrome. J Postgrad Med. 2004;50:140–144.
69. Sam, S, Dunaif, A. Polycystic ovary syndrome: syndrome XX? Trends Endocrinol Metab. 2003;14:365–370.
70. McGarry, JD. What if Minkowski had been ageusic? An alternative angle on diabetes. Science. 1992;258:766–770.
71. Almind, K, Accili, D, Kahn, CR. Knockout mice as a tool to the understanding of diabetes. In: LeRoith D, Taylor SI, Olefsky JM, eds. Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia: Lippincott; 2004:245–254.
72. White, MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422.
73. Shulman, GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176.
74. Reaven, GM. The fourth musketeer–from Alexandre Dumas to Claude Bernard. Diabetologia. 1995;38:3–13.
75. Ruderman, N, Prentki, M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3:340–351.
76. Ruderman, NB, Saha, AK, Vavvas, D, et al. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol. 1999;276:E1–E18.
77. Randle, PJ, Garland, PB, Hales, CN, et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789.
78. Randle, PJ, Newsholme, EA, Garland, PB. Regulation of glucose uptake by muscle. 8. Effects of fatty acids, ketone bodies and pyruvate, and of alloxan-diabetes and starvation, on the uptake and metabolic fate of glucose in rat heart and diaphragm muscles. Biochem J. 1964;93:652–665.
79. Randle, PJ, Garland, PB, Newsholme, EA, et al. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann N Y Acad Sci. 1965;131:324–333.
80. Randle, PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14:263–283.
81. Ruderman, NB, Toews, CJ, Shafrir, E. Role of free fatty acids in glucose homeostasis. Arch Intern Med. 1969;123:299–313.
82. Boden, G, Jadali, F, White, J, et al. Effects of fat on insulin-stimulated carbohydrate metabolism in normal men. J Clin Invest. 1991;88:960–966.
83. Roden, M, Price, TB, Perseghin, G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865.
84. Dresner, A, Laurent, D, Marcucci, M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259.
85. Boden, G, Shulman, GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest. 2002;32(Suppl 3):14–23.
86. Ellis, BA, Poynten, A, Lowy, AJ, et al. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol Endocrinol Metab. 2000;279:E554–E560.
87. Griffin, ME, Marcucci, MJ, Cline, GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274.
88. Itani, SI, Ruderman, NB, Schmieder, F, et al. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011.
89. Boden, G, She, P, Mozzoli, M, et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes. 2005;54:3458–3465.
90. Lam, TK, van de Werve, G, Giacca, A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol. 2003;284:E281–E290.
91. Samuel, VT, Liu, ZX, Qu, X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353.
92. Shoelson, SE, Lee, J, Goldfine, AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801.
93. Considine, R, Nyce, MR, Allen, LE, et al. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: an alteration not due to hyperglycemia. J Clin Invest. 1995;95:2938–2944.
94. Cortright, RN, Azevedo, JL, Jr., Zhou, Q, et al. Protein kinase C modulates insulin action in human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;278:E553–E562.
95. Boden, G. Free fatty acids as a target for therapy. Current Opinions in Endocrinology and Diabetes. 2004.
96. Reaven, G. Insulin resistance and its consequences type 2 diabetes mellitus and the insulin resistance syndrome. In: Leroith D, Taylor SI, Olefsky JM, eds. Diabetes melliltus: a fundamental and clinical text. Lippincott Williams & Wilkins; 2004:899–915.
97. Puri, V, Ranjit, S, Konda, S, et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci U S A. 2008;105:7833–7838.
98. Perseghin, G, Ghosh, S, Gerow, K, et al. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1001–1009.
99. McGarry, JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18.
100. Heydrick, SJ, Ruderman, NB, Kurowski, TG, et al. Enhanced stimulation of diacylglycerol and lipid synthesis by insulin in denervated muscle. Altered protein kinase C activity and possible link to insulin resistance. Diabetes. 1991;40:1707–1711.
101. Saha, AK, Kurowski, TG, Colca, JR, et al. Lipid abnormalities in tissues of the KKAy mouse: effects of pioglitazone on malonyl-CoA and diacylglycerol. Am J Physiol. 1994;267:E95–E101.
102. Dobbins, RL, Szczepaniak, LS, Bentley, B, et al. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130.
103. Abu-Elheiga, L, Matzuk, MM, Abo-Hashema, KAH, et al. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616.
104. Choi, CS, Savage, DB, Abu-Elheiga, L, et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A. 2007;104:16480–16485.
105. Harwood, HJ, Jr., Petras, SF, Shelly, LD, et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem. 2003;278:37099–37111.
106. Savage, DB, Choi, CS, Samuel, VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824.
107. Ravussin, E, Swinburn, BA. Energy expenditure and obesity. Diab Rev. 1996;4:403–422.
108. Ruderman, NB, Saha, AK, Kraegen, EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171.
109. Etgen, GJ, Oldham, BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism. 2000;49:684–688.
110. Faldt, J, Wernstedt, I, Fitzgerald, SM, et al. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology. 2004;145:2680–2686.
111. Kelly, M, Keller, C, Avilucea, PR, et al. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004;320:449–454.
112. Yu, X, McCorkle, S, Wang, M, et al. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: prevention of diabetes and ectopic lipid deposition. Diabetologia. 2004;47:2012–2021.
113. Kelley, DE, He, J, Menshikova, EV, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950.
114. Petersen, KF. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142.
115. Morino, K, Petersen, KF, Dufour, S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593.
116. Patti, ME, Butte, AJ, Crunkhorn, S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471.
117. Mootha, VK, Lindgren, CM, Eriksson, KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately down-regulated in human diabetes. Nat Genet. 2003;34:267–273.
118. Nyholm, B, Mengel, A, Nielsen, S, et al. Insulin resistance in relatives of NIDDM patients: the role of physical fitness and muscle metabolism. Diabetologia. 1996;39:813–822.
119. Park, H, Kaushik, VK, Constant, S, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–32577.
120. Ruderman, NB, Cacicedo, JM, Itani, S, et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans. 2003;31:202–206.
121. Gauthier, MS, Miyoshi, H, Souza, SC, et al. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J Biol Chem. 2008;283:16514–16524.
122. Christ-Crain, M, Kola, B, Lolli, F, et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. FASEB J. 2008;22:1672–1683.
123. Steinberg, GR, Michell, BJ, van Denderen, BJ, et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4:465–474.
124. Banerjee, RR, Rangwala, SM, Shapiro, JS, et al. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–1198.
125. Bergeron, R, Ren, JM, Cadman, KS, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346.
126. Jager, S, Handschin, C, St-Pierre, J, et al. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022.
127. Suwa, M, Nakano, H, Kumagai, S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–968.
128. Zong, H, Ren, JM, Young, LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987.
129. Wu, Z, Puigserver, P, Andersson, U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124.
130. Ek, J, Andersen, G, Urhammer, SA, et al. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to type II diabetes mellitus. Diabetologia. 2001;44:2220–2226.
131. Hara, K, Tobe, K, Okada, T, et al. A genetic variation in the PGC-1 gene could confer insulin resistance and susceptibility to type II diabetes. Diabetologia. 2002;45:740–743.
132. Eriksson, KF, Lindgarde, F. Impaired glucose tolerance in a middle-aged male urban population: a new approach for identifying high-risk cases. Diabetologia. 1990;33:526–531.
133. Schneider, SH, Amorosa, LF, Khachadurian, AK, et al. Studies on the mechanism of improved glucose control during regular exercise in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1984;26:355–360.
134. Schneider, SH, Khachadurian, AK, Amorosa, LF, et al. Ten-year experience with an exercise-based outpatient life-style modification program in the treatment of diabetes mellitus. Diabetes Care. 1992;15:1800–1810.
135. Wu, H, Kanatous, SB, Thurmond, FA, et al. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352.
136. Saloranta, C, Groop, L. Interactions between glucose and FFA metabolism in man. Diab Metab Rev. 1996;12:15–36.
137. Gavrilova, O, Marcus-Samuels, B, Graham, D, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–278.
138. Kim, JK, Gavrilova, O, Chen, Y, et al. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460.
139. Unger, RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165.
140. Hotamisligil, GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867.
141. Gustafson, B, Hammarstedt, A, Andersson, CX, et al. Inflamed adipose tissue: a culprit underlying the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2276–2283.
142. Rajala, MW, Scherer, PE. Minireview: The adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773.
143. Tsao, TS, Tomas, E, Murrey, HE, et al. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J Biol Chem. 2003;278:50810–50817.
144. Scherer, PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55:1537–1545.
145. Lindsay, RS, Funahashi, T, Hanson, RL, et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet. 2002;360:57–58.
146. Pischon, T, Girman, CJ, Hotamisligil, GS, et al. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737.
147. Maeda, N, Shimomura, I, Kishida, K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737.
148. Okamoto, Y, Kihara, S, Ouchi, N, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–2770.
149. Yamauchi, T, Kamon, J, Waki, H, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278:2461–2468.
150. Yu, AS, Keeffe, EB. Nonalcoholic fatty liver disease. Rev Gastroenterol Disord. 2002;2:11–19.
151. Fruebis, J, Tsao, TS, Javorschi, S, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98:2005–2010.
152. Tomas, E, Tsao, TS, Saha, AK, et al. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A. 2002;99:16309–16313.
153. Yamauchi, T, Kamon, J, Minokoshi, Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295.
154. Combs, TP, Berg, AH, Obici, S, et al. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest. 2001;108:1875–1881.
155. Kadowaki, T, Yamauchi, T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451.
156. Kubota, N, Terauchi, Y, Kubota, T, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–8755.
157. Nawrocki, AR, Rajala, MW, Tomas, E, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660.
158. Zhang, Y, Proenca, R, Maffei, M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432.
159. Muoio, DM, Seefeld, K, Witters, LA, et al. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J. 1999;338(Pt 3):783–791.
160. Friedman, JM, Halaas, JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770.
161. Minokoshi, Y, Kim, YB, Peroni, OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343.
162. Minokoshi, Y, Alquier, T, Furukawa, N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574.
163. Unger, RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–336.
164. Unger, RH, Orci, L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15:312–321.
165. Higa, M, Zhou, YT, Ravazzola, M, et al. Troglitazone prevents mitochondrial alterations, beta cell destruction, and diabetes in obese prediabetic rats. Proc Natl Acad Sci U S A. 1999;96:11513–11518.
166. Pold, R, Jensen, LS, Jessen, N, et al. Long-term AICAR administration and exercise prevents diabetes in ZDF rats. Diabetes. 2005;54:928–934.
167. Pearson, TA, Mensah, GA, Alexander, RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511.
168. Libby, P. Inflammation in atherosclerosis. Nature. 2002;420:868–874.
169. Collins, T, Cybulsky, MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264.
170. Nishikawa, T, Edelstein, D, Du, XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790.
171. Hennig, B, Meerarani, P, Ramadass, P, et al. Fatty acid-mediated activation of vascular endothelial cells. Metabolism. 2000;49:1006–1013.
172. Haffner, SM, Greenberg, AS, Weston, WM, et al. Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation. 2002;106:679–684.
173. Meigs, JB, Hu, FB, Rifai, N, et al. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA. 2004;291:1978–1986.
174. Steinberg, HO, Baron, AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634.
175. Esposito, K, Pontillo, A, Di Palo, C, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA. 2003;289:1799–1804.
176. Hamdy, O, Ledbury, S, Mullooly, C, et al. Lifestyle modification improves endothelial function in obese subjects with the insulin resistance syndrome. Diabetes Care. 2003;26:2119–2125.
177. Hu, FB. Obesity and Cardiovascular Disease Obesity, Epidemiology. New York: Oxford University Press; 2008.
178. Cacicedo, JM, Yagihashi, N, Keaney, JF, Jr., et al. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–1209.
179. Kukidome, D, Nishikawa, T, Sonoda, K, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127.
180. Ido, Y, Carling, D, Ruderman, N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–167.
181. Sun, W, Lee, TS, Zhu, M, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–2662.
182. Ridker, PM, Danielson, E, Fonseca, FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207.
183. Petersen, KF, Dufour, S, Befroy, D, et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608.
184. Seppala-Lindroos, A, Vehkavaara, S, Hakkinen, AM, et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023–3028.
185. Lam, TK, Yoshii, H, Haber, CA, et al. Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab. 2002;283:E682–E691.
186. Samuel, VT, Liu, ZX, Wang, A, et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745.
187. Poitout, V, Robertson, RP. Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342.
188. El-Assaad, W, Buteau, J, Peyot, ML, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–4163.
189. Prentki, M, Joly, E, El-Assaad, W, et al. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51:S405–413.
190. Robertson, RP, Harmon, J, Tran, PO, et al. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587.
191. Bakker, S, Jzerman, R, Teerlink, T, et al. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis. 2000;148:17–21.
192. Hu, FB. Metabolic consequence of obesity, Obesity, Epidemiology. New York: Oxford University Press; 2008.
193. Yu, C, Chen, Y, Cline, GW, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236.
194. Laybutt, DR, Schmitz-Peiffer, C, Saha, AK, et al. Muscle lipid accumulation and protein kinase C activation in the insulin-resistant chronically glucose-infused rat. Am J Physiol. 1999;277:E1070–E1076.
195. Schmitz-Peiffer, C, Browne, CL, Oakes, ND, et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes. 1997;46:169–178.
196. Hundal, RS, Petersen, KF, Mayerson, AB, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–1326.
197. Yuan, M, Konstantopoulos, N, Lee, J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IKKbeta. Science. 2001;293:1673–1677.
198. Kahn, BB, Alquier, T, Carling, D, et al. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25.
199. Morino, K, Neschen, S, Bilz, S, et al. Muscle-specific IRS-1 Ser→Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes. 2008;57:2644–2651.
200. Brown, MS, Goldstein, JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96.
201. Evans, JL, Goldfine, ID, Maddux, BA, et al. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622.
202. Jacob, S, Machann, J, Rett, K, et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119.
203. Krssak, M, Falk Petersen, K, Dresner, A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116.
204. Kim, SF, Huang, AS, Snowman, AM, et al. From the cover: antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci U S A. 2007;104:3456–3459.
205. Walker, BR. Is “Cushing’s disease of the omentum” an affliction of mouse and men? Diabetologia. 2004;47:767–769.
206. Savage, DB, Tan, GD, Acerini, CL, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes. 2003;52:910–917.
207. Hawkins, M, Hu, M, Yu, J, et al. Discordant effects of glucosamine on insulin-stimulated glucose metabolism and phosphatidylinositol 3-kinase activity. J Biol Chem. 1999;274:31312–31319.
208. McClain, DA, Crook, ED. Hexosamines and insulin resistance. Diabetes. 1996;45:1003–1009.
209. Gregor, ME, Yang, L, Fabbrini, E, et al. Endoplasmic reticulum stress is reduced in tissue of obese subjects after weight loss. Diabetes. 2009;58:693–700.
210. Garg, A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234.
211. Oral, EA, Simha, V, Ruiz, E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578.
212. Petersen, KF, Oral, EA, Dufour, S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350.
213. Fleischman, A, Shoelson, SE, Bernier, R, et al. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. 2008;31:289–294.
214. Grundy, SM, Brewer, HB, Jr., Cleeman, JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438.
215. Cheal, KL, Abbasi, F, Lamendola, C, et al. Relationship to insulin resistance of the adult treatment panel III diagnostic criteria for identification of the metabolic syndrome. Diabetes. 2004;53:1195–1200.
216. McKeigue, PM, Pierpoint, T, Ferrie, JE, et al. Relationship of glucose intolerance and hyperinsulinaemia to body fat pattern in south Asians and Europeans. Diabetologia. 1992;35:785–791.
217. Brunzell, JD, Ayyobi, AF. Dyslipidemia in the metabolic syndrome and type 2 diabetes mellitus. Am J Med. 2003;115(Suppl 8A):24S–28S.
218. Isomaa, B, Almgren, P, Tuomi, T, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689.
219. Isomaa, B, Henricsson, M, Almgren, P, et al. The metabolic syndrome influences the risk of chronic complications in patients with type II diabetes. Diabetologia. 2001;44:1148–1154.
220. Lakka, HM, Laaksonen, DE, Lakka, TA, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–2716.
221. Alexander, CM, Landsman, PB, Teutsch, SM, et al. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes. 2003;52(5):1210–1214.
222. Sattar, N, Gaw, A, Scherbakova, O, et al. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Circulation. 2003;108:414–419.
223. Grundy, SM. Cardiovascular and metabolic risk factors: how can we improve outcomes in the high-risk patient? Am J Med. 2007;120:S3–S8.
224. Green, RM. NASH–hepatic metabolism and not simply the metabolic syndrome. Hepatology. 2003;38:14–17.
225. Marchesini, G, Brizi, M, Bianchi, G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850.
226. Stefan, N, Kantartzis, K, Haring, HU. Causes and metabolic consequences of fatty liver. Endocr Rev. 2008;29:939–960.
227. Roberts, EA. Nonalcoholic steatohepatitis in children. Curr Gastroenterol Rep. 2003;5:253–259.
228. Petersen, KF, Dufour, S, Savage, DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104:12587–12594.
229. Legro, RS, Kunselman, AR, Dodson, WC, et al. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165–169.
230. Solomon, CG, Hu, FB, Dunaif, A, et al. Menstrual cycle irregularity and risk for future cardiovascular disease. J Clin Endocrinol Metab. 2002;87:2013–2017.
231. Calle, EE, Thun, MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378.
232. Hawley, SA, Boudeau, J, Reid, JL, et al. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28.
233. Woods, A, Johnstone, SR, Dickerson, K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008.
234. Luo, Z, Saha, AK, Xiang, X, et al. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci. 2005;26:69–76.
235. Motoshima, H, Goldstein, BJ, Igata, M, et al. AMPK and cell proliferation–AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71.
236. Xiang, X, Saha, AK, Wen, R, et al. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004;321:161–167.
237. Kramer, AF, Erickson, KI. Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci. 2007;11:342–348.
238. Watson, GS, Craft, S. Insulin resistance, inflammation, and cognition in Alzheimer’s disease: lessons for multiple sclerosis. J Neurol Sci. 2006;245:21–33.
239. Morris, D. Cushing’s syndrome. In: DeGroot LJ, ed. Endocrinology. ed 5. Philadelphia: Elsevier; 2006:429–464.
240. Seely, WW, Williams, GH. The cardiovascular system and endocrine disease. In: Becker KL, ed. Principles and Practice of Endocrinology and Metabolism. Philadelphia: Lippincott Williams & Wilkins; 2001:1857–1864.
241. Sutinen, J, Hakkinen, AM, Westerbacka, J, et al. Rosiglitazone in the treatment of HAART-associated lipodystrophy—a randomized double-blind placebo-controlled study. Antivir Ther. 2003;8:199–207.
242. Unger, R. How adipocytes integrate surplus caloric intake with caloric storage: lessons from Morgan Spurlock and some French geese, Current Opinion in Endocrinology. Diabetes and Obesity. 2004;11:251–257.
243. Sims, EA, Danforth, E, Jr., Horton, ES, et al. Endocrine and metabolic effects of experimental obesity in man. Recent Prog Horm Res. 1973;29:457–496.
244. Grundy, SM, Hansen, B, Smith, SC, Jr., et al. Clinical management of metabolic syndrome: report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation. 2004;109:551–556.
245. Skerrett, PJ, Manson, JE. Exercise and diabetes prevention: reduction in risk of coronary heart disease in diabetes. In: Ruderman N, Devlin JT, Schneider SH, Kriska A, eds. Handbook of diabetes in exercise. Alexandria VA: American Diabetes Association; 2002:155–182.
246. Orchard, TJ, Temprosa, M, Goldberg, R, et al. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: the Diabetes Prevention Program randomized trial. Ann Intern Med. 2005;142:611–619.
247. Lindstrom, J, Ilanne-Parikka, P, Peltonen, M, et al. Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: follow-up of the Finnish Diabetes Prevention Study. Lancet. 2006;368:1673–1679.
248. Yusuf, S, Gerstein, H, Hoogwerf, B, et al. Ramipril and the development of diabetes. JAMA. 2001;286:1882–1885.
249. Robins, SJ, Rubins, HB, Faas, FH, et al. Insulin resistance and cardiovascular events with low HDL cholesterol: the Veterans Affairs HDL Intervention Trial (VA-HIT). Diabetes Care. 2003;26:1513–1517.
250. UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352:854–865.
251. Holman, RR, Paul, SK, Bethel, MA, et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589.
252. Nissen, SE, Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471.
253. Lincoff, AM, Wolski, K, Nicholls, SJ, et al. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188.
254. Dormandy, JA, Charbonnel, B, Eckland, DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROACTIVE Study (Prospective Pioglitazone Clinical Trial in Macrovascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289.
255. Nissen, SE, Nicholls, SJ, Wolski, K, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA. 2008;299:1561–1573.
256. Nathan, DM, Buse, JB, Davidson, MB, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care. 2008;29:1963–1972.
257. Grundy, SM, Cleeman, JI, Merz, CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239.
258. Ridker, PM, Rifai, N, Clearfield, M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–1965.
259. Rosenzweig, JL, Ferrannini, E, Grundy, SM, et al. Primary prevention of cardiovascular disease and type 2 diabetes in patients at metabolic risk: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3671–3689.
260. Cook, S, Weitzman, M, Auinger, P, et al. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med. 2003;157:821–827.
261. Berenson, GS, Srinivasan, SR, Bao, W, et al. Associations between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338:1650–1656.
262. Must, A, Jacques, PF, Dallal, GE, et al. Long-term morbidity and mortality of overweight adolescents, A follow-up of the Harvard Growth Study of 1922 to 1935. N Engl J Med. 1992;327:1350–1355.
263. Steinberger, J, Daniels, SR. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation. 2003;107:1448–1453.
264. Barker, DJ, Hales, CN, Fall, CH, et al. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67.
265. Phillips, DI. Birth weight and the future development of diabetes. A review of the evidence. Diabetes Care. 1998;21(Suppl 2):B150–B155.
266. Bhargava, SK, Sachdev, HS, Fall, CH, et al. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N Engl J Med. 2004;350:865–875.
267. Zimmet, P, Alberti, KG, Shaw, J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787.
268. Neel, JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–362.
269. Hu, Z, Cha, SH, Chohnan, S, et al. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc Natl Acad Sci U S A. 2003;100:12624–12629.
270. Kemp, BE, Stapleton, D, Campbell, DJ, et al. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003;31:162–168.
271. Hardie, DG, Scott, JW, Pan, DA, et al. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120.
272. Sanders, MJ, Grondin, PO, Hegarty, BD, et al. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148.
273. Towler, MC, Hardie, DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341.