[level-membership-for-surgery-category]
Chapter 1 The Hypothalamic-Pituitary-Ovarian Axis and Control of the Menstrual Cycle
INTRODUCTION
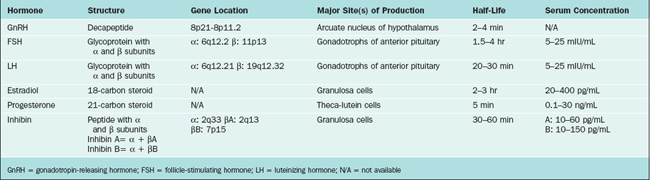
The hypothalamus and pituitary gland form a unit that exerts control over a wide range of endocrine organs, including the gonads. This chapter describes the hypothalamic-pituitary-ovarian axis and control of the menstrual cycle, which is modulated by the central nervous system, other endocrine systems, and the environment. Key hormones in the hypothalamic-pituitary-ovarian axis include gonadotropin-releasing hormone (GnRH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), estradiol, and progesterone (Table 1-1). Supporting roles are also played by inhibin, activin, follistatin, and endorphins.
THE HYPOTHALAMUS
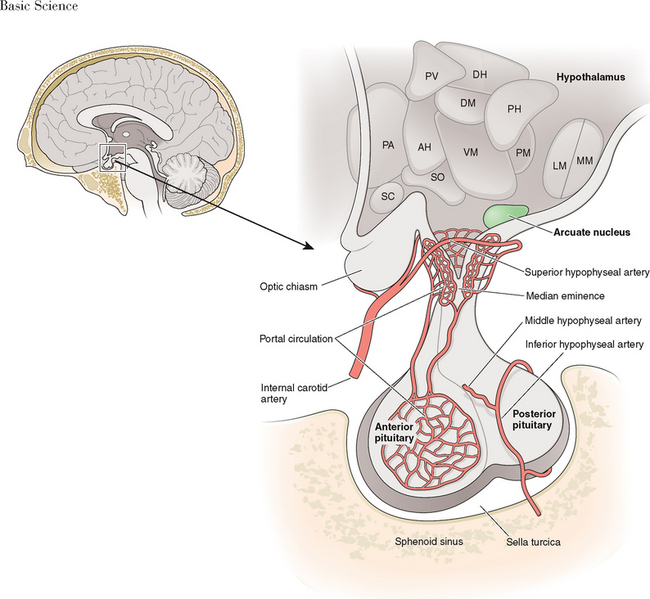
The hypothalamus forms the lower part of the lateral wall and the floor of the third ventricle, and weighs approximately 10 grams. The hypothalamus is typically divided into eight specific nuclei (consistently clustered groups of neurons) and three areas (less clustered, less distinctly demarcated neurons), as illustrated in Figure 1-1. From a reproductive standpoint, the most important of these are the arcuate nucleus and the preoptic area, the principal sites of GnRH-producing neurons.1 The arcuate nucleus is located in the medial basal hypothalamus and is the most proximal of all the hypothalamic nuclei to the optic chiasm and the pituitary stalk. The arcuate nucleus is also the site of dopamine-secreting neurons that function to inhibit pituitary prolactin secretion and neurons that secrete growth hormone-releasing hormone.
GnRH
GnRH is the primary hypothalamic regulator of pituitary reproductive function. Two human forms of GnRH (GnRH-I and GnRH-II) have been identified.2,3 Both are decapeptides and are the products of different genes. At least 20 other types of GnRH have been identified in fish, amphibians and protochordates, but none of these are believed to be present in humans.4,5
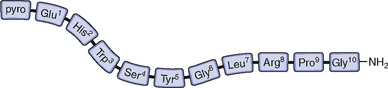
GnRH-I was first characterized and synthesized in 1971 by Andrew Schally and Roger Guillemin, an accomplishment for which both men ultimately received the Nobel prize.6–9 The structure of GnRH-I is common to all mammals, and its action is similar in males and females (Fig. 1-2). GnRH-I is synthesized from a much larger, 92-amino acid precursor peptide that contains GnRH-associated peptide.10 GnRH-I then travels along an axonal pathway called the tuberoinfundibular tract to the median eminence of the hypothalamus, where it is released into the portal circulation in a pulsatile fashion. The half-life of GnRH-I is very short (2 to 4 minutes) because it is rapidly cleaved between amino acids 5 and 6, 6 and 7, and 9 and 10. Because of its short half-life and rapid dilution in the peripheral circulation, serum GnRH-I levels are difficult to measure and do not correlate with pituitary action.
GnRH-I has three principal actions on anterior pituitary gonadotrophs: (1) synthesis and storage of gonadotropins, (2) movement of gonadotropins from the reserve pool to a point where they can be readily released, and (3) direct secretion of gonadotropins. GnRH-I pulses occur in response to intrinsic rhythmic activity within GnRH neurons in the arcuate nucleus. Pulsatile release of GnRH-I from the median eminence within a critical frequency and amplitude results in normal gonadotropin secretion.11,12 Continuous, rather than pulsatile, exposure to GnRH-I results in suppression of FSH and LH secretion and suppression of gonadotropin gene transcription.13,14
In the absence of gonadal feedback, the GnRH pulse frequency is approximately once per hour.15 During the menstrual cycle the frequency and amplitude of GnRH pulses vary in response to hypothalamic feedback (Table 1-2).16 In general, the follicular phase is characterized by high-frequency, low-amplitude pulses, and the luteal phase is characterized by lower-frequency, higher-amplitude pulses.17,18 However, considerable variability exists both between and within individuals.19 In humans, GnRH-I pulse frequency and amplitude are best approximated by measuring LH pulse frequency and amplitude.
Table 1-2 Menstrual Cycle Variation in LH Pulse Frequency and Amplitude
| Cycle Phase | Mean Frequency (minutes) | Mean Amplitude (mIU/mL) |
|---|---|---|
| Early follicular | 90 | 6.5 |
| Mid-follicular | 50 | 5 |
| Late follicular | 60–70 | 7 |
| Early luteal | 100 | 15 |
| Mid-luteal | 150 | 12 |
| Late luteal | 200 | 8 |
GnRH-II is most highly expressed outside of the brain, in tissues that include kidneys, bone marrow, and prostate. This is in contrast to GnRH-I, which is not expressed in high levels outside the brain. Although GnRH-II can induce release of both FSH and LH, it appears to have a wide array of physiologic functions outside the brain including regulation of cellular proliferation and mediation of ovarian and placental hormone secretion.20
Initial attempts in the mid-1990s to identify estrogen receptors in GnRH neurons were unsuccessful.21,22 However, subsequent use of more sophisticated techniques identified estrogen receptors α and β in the arcuate nucleus.23–26 Both receptors mediate estrogen action on GnRH neurons in vivo.27,28 The GnRH gene contains a hormone response element for the estrogen–estrogen receptor complex.29 Transcription of GnRH-I and GnRH-II is differentially regulated by estrogen.30 The regulatory role of estradiol on GnRH is complex. Estrogen inhibits GnRH gene expression/biosynthesis, but secretion of GnRH may be increased, decreased, or unaffected.31,32
The activity of the hypothalamus is further modulated by nervous stimuli from higher brain centers. GnRH neurons exhibit many connections to each other and to other neurons. Some of the neurotransmitters that modulate GnRH secretion are outlined in Table 1-3. The effects of these neurotransmitters help explain the mechanism by which certain physical or clinical conditions may affect the menstrual cycle (Table 1-4).
Table 1-3 Neurotransmitter Effects on GnRH Release
| Neurotransmitter | Effect |
|---|---|
| Dopamine | Inhibits GnRH release |
| Endorphin | Inhibits GnRH release |
| Serotonin | Inhibits GnRH release |
| Norepinephrine, epinephrine | Stimulates GnRH release |
Table 1-4 Mechanisms for Oligo/amenorrhea in Various Clinical Conditions
| Hyperprolactinemia | Elevated dopamine suppresses GnRH |
| Hypothyroidism | Elevated TRH increases prolactin, which in turn increases dopamine which, then suppresses GnRH |
| Stress | Increased corticotropin (ACTH) results in increased endorphins (both are derived from the same peptide precursor); endorphins suppress GnRH |
| Exercise | Increased endorphins suppress GnRH |
TRH = thyrotropin-releasing hormone; GnRH = gonadotropin-releasing hormone.
Cells that produce GnRH originate embryologically from the olfactory area.33 GnRH neurons, like olfactory epithelial cells of the nasal cavity, have cilia.34 During embryogenesis GnRH neurons migrate from the medial olfactory placode to the arcuate nucleus of the hypothalamus.35 The common origin of GnRH and olfactory neurons is demonstrated by Kallmann’s syndrome, where GnRH deficiency is associated with anosmia. Kallmann’s syndrome is believed to be caused by a variety of gene defects that affect neuronal cell migration.36
The common origin of GnRH and olfactory neurons is also suggestive of the relationship between pheromones and menstrual cyclicity. Pheromones are small airborne chemicals that when secreted externally by one individual may be perceived by other individuals of the same species, producing a change in sexual or social behavior. It is well recognized that women who work or live together often develop synchrony of their menstrual cycles.37 Moreover, it has been shown that odorless compounds from the axillae of cycling women can alter the cycle characteristics of recipient women.38 Presumably, these alterations occur through olfactory GnRH-mediated mechanisms.
The GnRH Receptor
The GnRH-I receptor is a G-protein receptor that utilizes inositol triphosphate and diacylglycerol as second messengers to stimulate protein kinase, release of calcium ions, and cyclic adenosine monophosphate (cAMP) activity. The GnRH-I receptor is encoded by a gene on chromosome 14q21.1 and is expressed in many parts of the body outside of the brain, including ovarian follicles and the placenta. In humans it appears that GnRH-II signaling occurs through the GnRH-I receptor.5 Although a GnRH-II receptor is present in many mammalian species, its functional capacity is limited due to a frame shift and a stop codon. GnRH receptors are regulated by many substances, including GnRH itself, inhibin, activin, estrogen, and progesterone.
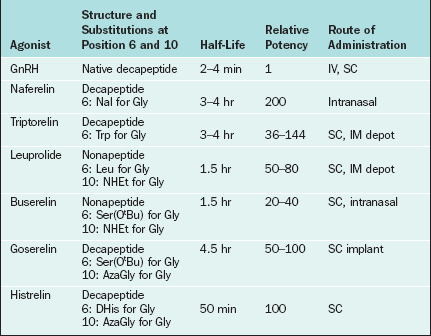
Changes to the amino acid sequence of GnRH can extend its half-life to hours or days and can change its biologic activity from an agonist to an antagonist. All of the GnRH agonists currently available extend their half-life by substitutions at amino acid 6 and sometimes amino acid 10 of native GnRH (Table 1-5). Continuous activation of the GnRH receptor results in desensitization due to phosphorylation and conformational change of the receptor, uncoupling from G proteins, internalization of the receptor via endocytosis, and decreased receptor synthesis.39,40 On administration all GnRH agonists increase gonadotropin secretion (the flare effect). However, after 7 to 14 days GnRH receptor desensitization occurs and pituitary suppression is achieved.
In contrast, GnRH antagonists directly inhibit gonadotropin secretion. Structurally, GnRH antagonists are characterized by multiple amino acid substitutions to the natural GnRH decapeptide. The commercially available GnRH antagonists cetrorelix and ganirelix have large amino acid additions to position 1 of native GnRH. GnRH antagonists compete for and occupy pituitary GnRH receptors, thus competitively blocking endogenous GnRH–GnRH receptor binding. In contrast to GnRH agonists, there is no flare effect with GnRH antagonists. Because receptor loss does not occur, a constant supply of antagonist is necessary to ensure that all GnRH receptors are continuously occupied. Thus, the therapeutic dosage range for antagonists is typically higher than that for agonists (mg versus μg).
THE PITUITARY
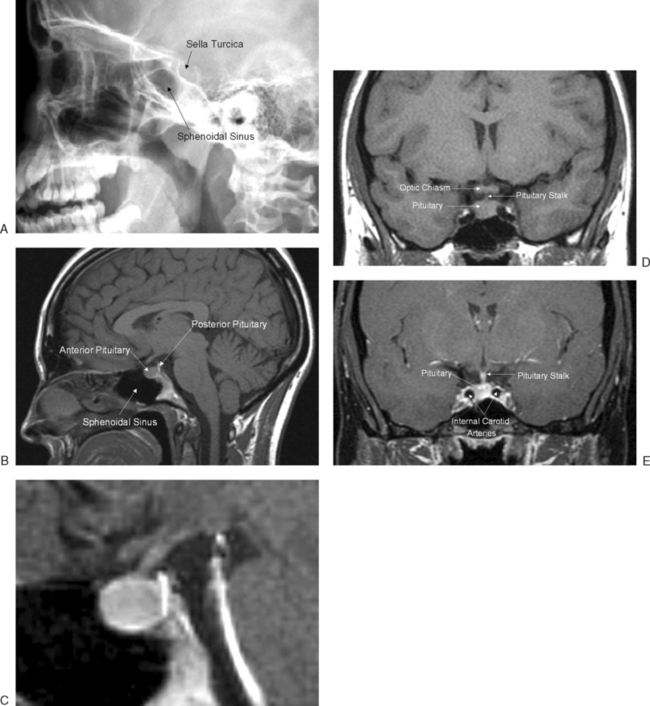
The pituitary gland measures approximately 15 × 10 × 6 mm and weighs approximately 500 to 900 mg. The pituitary gland lies immediately beneath the third ventricle and just above the sphenoidal sinus in a bony cavity called the sella turcica (Turkish saddle). It consists of anterior and posterior lobes, each having different embryologic origins, functions, and control mechanisms (Fig. 1-3).
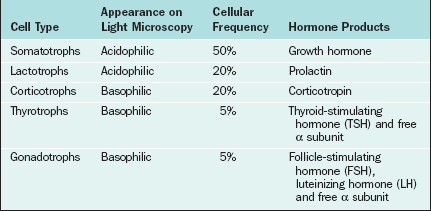
The Anterior Pituitary
The major cell types of the anterior pituitary are outlined in Table 1-6. The anterior pituitary is a classic endocrine gland in that it is composed of secretory cells of epithelial origin supported by connective tissue rich in blood and lymphatic capillaries. In accordance with their active synthetic function the endocrine cells are characterized by prominent nuclei and prolific mitochondria, endoplasmic reticulum, Golgi bodies, and secretory vesicles. Synthesis of gonadotropins takes place in the rough endoplasmic reticulum, after which the hormones are packaged within the Golgi apparatus and stored as secretory granules. In response to GnRH the secretory granules are extruded from the cell membrane. The endothelial lining of capillary sinusoids is fenestrated, facilitating the passage of pituitary hormones into the sinusoids.
Gonadotropins
FSH
The sialic acid content of FSH, LH, TSH, and hCG varies, and these differences are largely responsible for variations in half-life of the glycoprotein hormones. The liver is the major site of clearance for gonadotropins. Sialic acid prevents hepatic clearance; thus, the greater the sialic acid content, the longer the half-life.41 hCG, with 20 sialic acid residues, has the longest half-life (about 24 hours), whereas LH (1 to 2 sialic acid residues) has the shortest half-life (20 to 30 minutes). Addition of sialic acid residues in urinary-derived commercially available gonadotropins (e.g., hMG) is responsible for their longer half-life (30 hours).
In gonadotrophs of the anterior pituitary, GnRH signaling leads to transcription of the α and β subunits for both FSH and LH. The GnRH-dependent availability of the β subunits is the rate-limiting step in gonadotropin synthesis. Although both FSH and LH require GnRH stimulation, synthesis of the FSH β subunit also requires the presence of activin.42,43
FSH and LH Receptors
In contrast, FSH receptors are found exclusively on granulosa cells in the ovary. FSH binds to receptors on the cell surface of granulosa cells in antral follicles. Like LH, FSH acts via the cAMP-dependent protein kinase pathway.44 In response to FSH the androgens produced as a result of LH stimulation are then aromatized to estrogens in granulosa cells.
Opioid Modulation of Pituitary Hormones
Opioids (i.e., endogenous opiates) are natural occurring sedative narcotics produced in the brain whose structure and function are similar to opium. Opioids include enkephalins, endorphins, and dynorphins; they modulate every pituitary hormone by acting on the hypothalamus. An important action of opioids is to inhibit gonadotropin secretion by suppressing GnRH release.45
Opioid tone is an important regulator of menstrual cyclicity.46–49 Endorphins are at a nadir in the early follicular phase (menstruation) and gradually rise to peak levels in the luteal phase in response to the rise in estrogen and progesterone. It is believed that opioids mediate the negative feedback of ovarian steroids on gonadotropin release, particularly in the luteal phase.50
Endogenous opioids appear to play a central role in hypothalamic amenorrhea. Treatment of women suffering from this condition with an opioid receptor antagonist (e.g., naltrexone) results in the return to ovulatory menstrual patterns and even conception in some cases.51,52 Women with stress-related amenorrhea demonstrate increased hypothalamic corticotropin-releasing hormone and pituitary corticotropin, which manifests as hypercortisolism.53 The corticotropin precursor peptide, pro-opiomelanocortin, is also the precursor for endorphin synthesis. It is hypothesized that stress-related amenorrhea is the result of GnRH inhibition secondary to increased production of endogenous opioids. Opioids also rise during exercise (“runners’ high”), and this may contribute to hypothalamic amenorrhea in athletes.54,55
Ovarian Peptide Hormone Feedback on Gonadotropin Secretion
The ovary secrets two polypeptide hormones that inhibit or stimulate FSH secretion by the anterior pituitary. Inhibin and activin act as opposing nonsteroidal gonadal hormones that regulate FSH synthesis and secretion by the pituitary. They also have paracrine effects within the ovary, where they modulate follicle growth and steroidogenesis. Follistatin is a binding protein that modulates the effects of activin but not inhibin.
Inhibin and Activin
Inhibin and activin are members of the transforming growth factor-β (TGF-β) superfamily of ligands, which includes müllerian inhibiting substance (MIS; see Chapter 2). Like the gonadotropins, inhibin and activin are comprised of two subunits. Inhibin is comprised of an α and β subunit and has been isolated in two forms containing different β subunits, Inhibin-A and Inhibin-B. Activin is comprised of two beta subunits identical to those found in inhibin.
Inhibin is secreted by granulosa cells in response to FSH.56 However, mRNA for inhibin has also been found in pituitary gonadotrophs. Inhibin selectively inhibits FSH but not LH secretion.57 Thus, a negative feedback loop is created where FSH stimulates inhibin and in turn inhibin suppresses FSH.
Inhibin B is predominantly secreted in the follicular phase of the menstrual cycle, whereas inhibin A is predominantly secreted in the luteal phase.58 Peak levels of inhibin B in the follicular phase are in the range of 50 to 100 pg/mL. Peak levels of inhibin A in the luteal phase are between 40 and 60 pg/mL.
OVARIAN STEROIDOGENESIS DURING THE MENSTRUAL CYCLE
Ovarian steroidogenesis during the menstrual cycle occurs in granulosa and theca cells (Table 1-7 and Fig. 1-4). Before ovulation, theca cells are separated from granulosa cells in the same follicle by a basal membrane. Thus, granulosa cells of preovulatory follicles do not have a blood supply. However, at the time of the LH surge the preovulatory follicle undergoes luteinization with disappearance of the basal membrane and capillary invasion of the granulosa cells. Theca cells become theca-lutein cells, and granulosa cells become granulosa-lutein cells.
Table 1-7 Site of Synthesis of Major Steroidogenic Products of the Ovary
| Cell Type | Major Steroid Hormone Products |
|---|---|
| Theca cells | Androgens (androstenedione, DHEA, testosterone)* |
| Granulosa cells | Estrogens (estradiol, estrone) |
| Theca-lutein cells | Progestogens (progesterone, 17-hydroxyprogesterone)** |
| Granulosa-lutein cells | Estrogens (estradiol, estrone) |
Two-Cell Theory
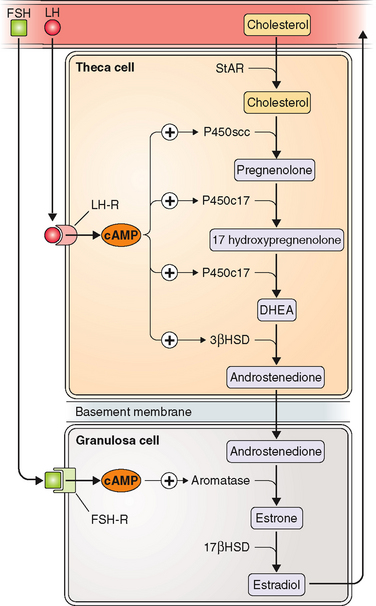
The two-cell, two-gonadotropin theory of ovarian steroidogenesis holds that follicular estrogen/androgen production is compartmentalized.59 Ovarian theca cells produce androgens in response to LH. These androgens may then be aromatized to estrogens in granulosa cells appropriately stimulated by FSH. FSH receptors are present only on granulosa cells, and early in the follicular phase LH receptors are present only on theca cells.60 The enzyme P450c17 (17-hydroxylase and 17,20-lyase) is only present in theca cells. Thus, only theca cells have the ability to convert 21-carbon steroids to 19-carbon steroids. In contrast, aromatase is only present in granulosa cells. Thus, in the ovary only granulosa cells have the ability to aromatize androgens to estrogens (Table 1-8 and Fig. 1-5). Supporting evidence for the two-cell theory includes the fact that women with hypogonadotropic hypogonadism may develop follicles in response to treatment with recombinant FSH, but do not significantly elevate androgen or estrogen levels unless LH is added to the stimulation regimen.61
| Enzyme | Location | Function |
|---|---|---|
| P450c17 | Theca cells only | Converts 21-carbon steroids (progesterone/pregnenolone) to 19-carbon steroids (androstenedione, DHEA) |
| Aromatase | Granulosa cells only | Converts 19-carbon steroids (androstenedione/testosterone) to 18-carbon steroids (estrone, estradiol) |
Estrogens
Estrogen is largely bound to carrier proteins in serum. Approximately 60% of estradiol is bound to albumin, 38% is bound to sex hormone-binding globulin (SHBG), and 2% to 3% is free. It had previously been thought that only the free hormone was active and could enter cells, but recent evidence suggests that hormone transport and hormone availability may be more complex.62
Estrogen Receptors
There are two known estrogen receptors, ERα and ERβ. Both contain a steroid-binding domain, a DNA-binding domain, a hinge region, and a transcription-activation functional domain. The negative feedback of estradiol on FSH secretion is a direct effect of estradiol coupled to its receptor repressing transcription of the FSH β subunit.63 Negative feedback of estradiol on FSH may also be modulated by the estrogen-associated decline in pituitary expression of activin.64
Estrogen Metabolism
The liver conjugates estrogens to form glucuronides and sulfates, about 80% of which are then excreted into the urine and 20% of which are excreted in the bile. In the liver, circulating estradiol is rapidly converted to estrone by 17β-hydroxysteroid dehydrogenase. Estrone may then be further metabolized in the liver to 16α-hydroxyestrone and then estriol. Estriol is then converted to estriol 3-sulfate-16-glucuronide before excretion by the kidney.
Progesterone
At high concentrations progesterone inhibits both FSH and LH secretion by negative feedback on both the hypothalamus and pituitary.65 Progesterone also slows the GnRH pulse generator; hence the decline in GnRH pulse frequency in the luteal phase. However, at low concentrations, and only after previous exposure to estrogen, progesterone stimulates LH release.66
Androgens
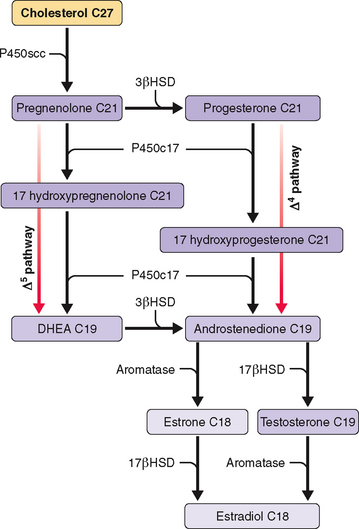
Side chain cleavage of cholesterol to pregnenolone is the starting point and rate-limiting step in steroidogenesis. In the ovary cholesterol side chain cleavage is regulated by LH. Low-density lipoprotein (LDL) cholesterol is the principal source of cholesterol for steroidogenesis in the human ovary.67 Increased cAMP production due to LH stimulation of adenylate cyclase increases transcription of LDL receptor mRNA and LDL uptake. cAMP-activated steroidogenic acute regulatory protein then increases the intracellular transport of cholesterol to the inner mitochondrial membrane, where side chain cleavage occurs.68
In the preovulatory follicle the preferred pathway for androgen/estrogen synthesis involves conversion of pregnenolone to 17-hydroxypregnenolone, the so-called Δ5 pathway (see Fig. 1-4). Ovarian theca cells have the enzymatic capability to convert pregnenolone to androgens, but lack the ability to aromatize androstenedione or testosterone into estrogens. Only granulosa cells, under the influence of FSH, can aromatize androgens to estrogens. In contrast to the preovulatory follicle, the corpus luteum prefers the Δ4 pathway, the initial conversion of pregnenolone to progesterone.
Androgen Receptors
The androgen receptor is similar to the estrogen receptor. It activates target gene expression via a similar sequence of ligand binding, nuclear translocation, DNA binding, and complex formation with coregulators and general transcription factors. Although the androgen receptor is known to play a central role in the development of male sex organs and secondary sexual characteristics, its physiologic roles in female reproduction remain unclear. Recent studies in androgen-deficient animals suggest that androgen receptors play an important role in granulosa cell development.69 These animals also exhibit reduced fertility with defective folliculogenesis, reduced corpus luteum formation, and reduced uterine response to gonadotropins.
Androgen Metabolism
Androstenedione is produced in equal amounts by the ovaries and adrenal glands. The serum concentrations mirror estradiol levels and range from 0.5 to 3 ng/mL. Androstenedione levels increase in the mid- to late follicular phase and are maximal just before the LH surge. They decline at the time of ovulation, reach a second peak in the midluteal phase, and are lowest around the time of menstruation. Serum testosterone concentrations, in contrast, vary to a much lesser extent during the menstrual cycle and exhibit only a transient periovulatory increase. Testosterone concentrations range from 1.5 to 60 ng/dL.
CONTROL OF THE MENSTRUAL CYCLE
Normal menstrual cycles, termed eumenorrhea, normally range in length between 24 to 35 days, with menstrual bleeding lasting 3 to 7 days. The average amount of blood loss is approximately 30 mL.70 Heavy, prolonged, or irregular menses are referred to as abnormal uterine bleeding and are considered in length in Chapter 21.
Normal menstrual cycles result from a relatively precise interaction of the hypothalamus, pituitary, and ovaries. Under the influence of pituitary gonadotropins, the ovary undergoes cyclic changes providing for the development and release of a mature oocyte and production of ovarian hormones that prepare the endometrial lining for implantation. LH stimulates androgen production in theca cells; FSH promotes follicle development and aromatization of androgens to estrogens in granulosa cells (see Fig. 1-5). In turn, estrogens lead to proliferation of the endometrial lining and the induction of endometrial receptors for both estrogen and progesterone.71
Follicular Phase
The purpose of the follicular phase is to develop a single mature follicle to release a mature oocyte at ovulation. The presence of sufficient FSH also leads to expression of LH receptors on mature granulosa cells of preovulatory follicles. Thus, in the late follicular phase LH can sustain follicular endocrine activity, even in the absence of FSH.72 The follicular phase is variable in duration, but the other three phases are relatively constant, averaging 14 ± 2 days.
Gonadotropins
FSH levels rise in the early follicular phase due to the lack of negative inhibition from estradiol and inhibin (Fig. 1-6). FSH stimulates follicle growth and estrogen production.73 Through binding of FSH to its receptor, granulosa cells in developing follicles attain the ability to aromatize androstenedione to estrone and testosterone to estradiol. Importantly, receptors for FSH are not detected on granulosa cells until the preantral stage.74 Moreover, both in vitro and in vivo administration of FSH to granulosa cells can cause upregulation or downregulation of granulosa cell FSH receptors.75 Without functional FSH follicle growth and ovarian estrogen production cannot occur.76
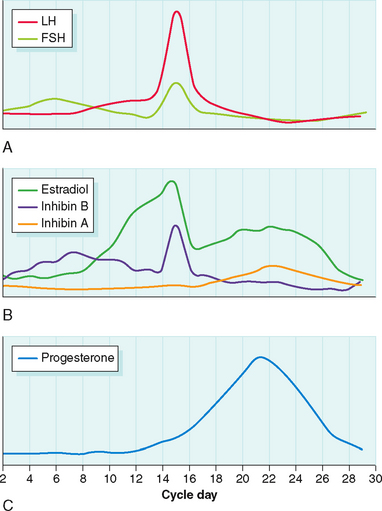
Ovarian Hormones
Estradiol levels rise as the dominant follicle emerges. FSH and estrogen synergistically exert a mitogenic effect on granulosa cells, stimulating their proliferation. This in turn increases the FSH receptor content of the follicle, enhancing the ability of the follicle to respond to FSH and produce estrogen. Curiously, not every granulosa cell must express FSH receptors to respond to the gonadotropin signal. Gap junctions between cells allow cells with receptors to transmit protein kinase activation to their neighbors.77
Within the follicular fluid of follicles greater than 8 mm in diameter, the concentration of FSH, estradiol, and progesterone are all extremely high. Within smaller antral follicles, androgens predominate in the follicular fluid. The role of androgens in the follicle is dose-dependent. At low levels androgens provide a substrate for aromatization. However, at higher levels androgens are converted in granulosa cells by 5α-reductase to more active forms such as dihydrotestosterone (DHT) that cannot be aromatized to estrogens.78,79 Granulosa cells have androgen receptors.80 Activation of granulosa cell androgen receptors inhibits aromatase activity and also inhibits FSH induction of granulosa LH receptors.81 Follicles exposed to excessive androgens eventually become atretic.82,83 In contrast, follicles with the highest estrogen-to-androgen ratios and the highest estrogen concentrations are most likely to contain a competent oocyte.84
Inhibin B levels begin to rise almost immediately after the rise in FSH levels. By the midfollicular phase the rise in estradiol and inhibin B levels causes FSH levels to decline. Inhibin B levels peak approximately 4 days after the FSH peak.47 In the late follicular phase inhibin B levels fall, mirroring the decline in FSH levels.
Ovulatory Phase
GnRH
GnRH plays a supporting role for the midcycle gonadotropin surge, but it does not trigger the surge.85 There is no change in GnRH pulse frequency during the midcycle gonadotropin surge.86 Rather, ovarian steroid feedback to the primed anterior pituitary triggers the LH surge.87 Whereas estrogen inhibits the secretion of pituitary gonadotropins, it facilitates their synthesis and storage. Estrogen also increases the expression of GnRH receptors.88,89 Thus, in the mid- to late follicular phase each pulse of GnRH is met with a greater gonadotropin response.90,91 When the estradiol level in the circulation meets a critical level for a sufficiently long period of time, the inhibitory action of estradiol on LH secretion changes to a stimulatory one. The LH surge is accompanied by a surge of GnRH in both portal and peripheral blood.92 However, as demonstrated by women with hypogonadotropic hypogonadism treated with a pulsatile GnRH pump, ovulation and pregnancy may occur in the absence of any change in GnRH pulse frequency or amplitude.93 Moreover, the LH surge ends before there is a decline in the GnRH signal.94
Gonadotropins
Both FSH and LH levels peak just before ovulation. The initiation of the gonadotropin surge is dependent on attaining serum estradiol levels of at least 200 pg/mL for at least 2 days.95 In natural cycles this level of estradiol is typically not attained until the dominant follicle reaches a mean diameter of 15 mm.96
Ovulation typically occurs from mature follicles 34 to 36 hours after the onset of the LH surge.97 The peak of LH and FSH occurs 10 to 12 hours before ovulation.98 The LH surge usually lasts 48 to 50 hours and must be maintained for at least 14 hours for full maturation of the oocyte to occur.99 The mechanism that turns off the LH surge is unknown. It may simply reflect the depletion in pituitary LH content.
Ovarian Hormones
Just prior to ovulation the follicle becomes vascularized.100 Angiogenesis is mediated by LH and a variety of other factors, including vascular endothelial growth factor.101–103 Prostaglandins reach peak levels in the follicular fluid.104 Proteolytic enzymes digest collagen in the follicular wall, resulting in distensibility and thinning just before ovulation.105 Progesterone rises in the follicular fluid. FSH, LH, and progesterone all serve to increase the activity of proteolytic enzymes. There is a rapid increase in follicular fluid volume, but due to increased elasticity there is little to no change in intrafollicular pressure. Finally, a protrusion (stigma) appears on the follicular wall, and it is at this site that ovulation ultimately occurs.
Interestingly, spontaneous lueteinization occurs in the absence of LH when granulosa cells are removed from follicles and cultured in vitro. Similarly, cumulus-enclosed oocytes removed from developing follicles before the LH surge will spontaneously resume meiosis.106,107 These findings have led to speculation that substances functioning as oocyte maturation inhibitors or luteinization inhibitors must exist within each follicle. Further support for this hypothesis lies in the fact that cumulus cells lack LH receptors.
Luteal Phase
GnRH and Gonadotropins
GnRH pulse frequency declines but pulse amplitude increases in the luteal phase. Changes in GnRH pulse frequency correlate with the duration of exposure to progesterone; changes in pulse amplitude correlate with progesterone levels.17
Luteinizing hormone and FSH levels reach a nadir during the luteal phase in response to the elevation in estrogen, progesterone, and inhibin A. Nevertheless, function of the corpus luteum is dependent on continued low-level pituitary gonadotropin secretion throughout the luteal phase.108 LH pulses stimulate pulses of progesterone secretion from the corpus luteum.17,109 Moreover, reducing LH pulse frequency and amplitude with a GnRH agonist in the luteal phase shortens the luteal phase itself.110
Ovarian Hormones
Granulosa-lutein cells can now make progesterone directly from LDL via side chain cleavage and 3β-hydroxysteroid dehydrogenase. Levels of mRNA for side chain cleavage and 3β-hydroxysteroid dehydrogenase are maximal at the time of ovulation and in the early luteal phase.111 The induction of LDL receptor expression in granulosa cells occurs in response to the LH surge and is an early feature of luteinization.112 Progesterone secretion correlates with the number of LH receptors and adenylate cyclase activity.113 Progesterone levels peak during the midluteal phase.
In granulosa-lutein cells inhibin production switches from inhibin B to inhibin A. Thus inhibin B levels decline to their nadir during the luteal phase, whereas inhibin A levels reach their peak. Secretion of inhibin A by granulosa-lutein cells is controlled by LH.114 Inhibin A, like inhibin B, suppresses FSH levels.115
Luteal–Follicular Transition
GnRH
A progressive and rapid increase in GnRH pulse frequency occurs during the luteal–follicular transition. GnRH pulse frequency, as estimated by LH pulse frequency, increases from 3 pulses per 24 hours (midluteal phase) to 14 pulses per 24 hours.18
Gonadotropins
FSH and LH levels rise from their nadir due to the decline in negative feedback from estradiol and inhibin and the rise in activin.116 The rise in FSH initiates recruitment of gonadotropin-responsive follicles for the next menstrual cycle. This recruitment of antral follicles actually begins at least 2 days before the onset of menstrual bleeding. In fact, an increase in FSH bioactivity can be measured back to the midluteal phase.117 During the luteal–follicular transition both inhibin A and B levels are at a nadir.118 In contrast, activin levels begin to increase in the late luteal phase and peak at the time of menstruation.119 Activin plays an important role as gonadotropin responses to GnRH require the presence of activin.43
Ovarian Hormones
In the absence of pregnancy corpus luteum function declines approximately 10 days after ovulation. The exact mechanisms for luteolysis are unclear. Luteolysis involves apoptosis and expression of matrix metalloproteinases.120,121 Luteolysis may also be mediated by nitric oxide.122 Nitric oxide induces apoptosis in the human corpus luteum.123 One of the final signs of luteolysis is ovarian production of prostaglandin F2α, which inhibits luteal steroidogenesis. Thus, unless the corpus luteum is rescued by the hCG of pregnancy, estrogen, progesterone, and inhibin levels fall as the luteal–follicular transition occurs.
1 Rance NE, Young WS3rd, McMullen NT. Topography of neurons expressing luteinizing hormone-releasing hormone gene transcripts in the human hypothalamus and basal forebrain. J Comp Neurol. 1994;339:573-586.
2 White RB, Eisen JA, Kasten TL, Fernald RD. Second gene for gonadotropin-releasing hormone in humans. Proc Natl Acad Sci USA. 1998;95:305-309.
3 Cheng CK, Leung PCK. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II and their receptors in humans. Endocr Rev. 2005;26:283-306.
4 Morgan K, Millar RP. Evolution of GnRH ligand precursors and GnRH receptors in protochordate and vertebrate species. Gen Comp Endocrinol. 2004;139:191-197.
5 Millar RP, Lu ZL, Pawson AJ, et al. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235-275.
6 Schally AV, Arimura A, Baba Y, et al. Isolation and properties of the FSH and LH-releasing hormone. Biochem Biophys Res Commun. 1971;43:393-399.
7 Schally AV, Arimura A, Kastin AJ, et al. Gonadotropin-releasing hormone: One polypeptide regulates secretion of luteinizing and follicle-stimulating hormones. Science. 1971;173:1036-1038.
8 Arimura A, Matsuo H, Baba Y, Schally AV. Ovulation induced by synthetic luteinizing hormone-releasing hormone in the hamster. Science. 1971;174:511-512.
9 Amoss M, Burgus R, Blackwell R, et al. Purification, amino acid composition and N-terminus of the hypothalamic luteinizing hormone releasing factor (LRF) of ovine origin. Biochem Biophys Res Commun. 1971;44:205-210.
10 Nikolics K, Mason AJ, Szonyi E, et al. A prolactin-inhibiting factor within the precursor for human gonadotropin-releasing hormone. Nature. 1985;316:511-517.
11 Knobil E. The neuroendocrine control of the menstrual cycle. Recent Prog Horm Res. 1980;36:53-88.
12 Mais V, Kazer RR, Cetel NS, et al. The dependency of folliculogenesis and corpus luteum function on pulsatile gonadotropin secretion in cycling women using a gonadotropin-releasing hormone antagonist as a probe. J Clin Endocrinol Metab. 1986;62:1250-1255.
13 Belchetz PE, Plant TM, Nakai Y, et al. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science. 1978;202:631-633.
14 Haisenleder DJ, Dalkin AC, Ortolano GA, et al. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: Evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128:509-517.
15 Marshall JC, Dalkin AC, Haisenleder DJ, et al. Gonadotropin-releasing hormone pulses: regulators of gonadotropin synthesis and ovulatory cycles. Recent Prog Horm Res. 1991;47:155-189.
16 Reame N, Sauder SE, Kelch RP, Marshall JC. Pulsatile gonadotropin secretion during the human menstrual cycle: Evidence for altered frequency of gonadotropin-releasing hormone secretion. J Clin Endocrinol Metab. 1984;59:328-337.
17 Filicori M, Santoro N, Merriam GR, Crowley WFJr. Characterization of the physiological pattern of episodic gonadotropin secretion throughout the human menstrual cycle. J Clin Endocrinol Metab. 1986;62:1136-1144.
18 Hall JE, Schoenfeld DA, Martin KA, Crowley WFJr. Hypothalamic gonadotropin-releasing hormone secretion and follicle-stimulating hormone dynamics during the luteal-follicular transition. J Clin Endocrinol Metab. 1992;74:600-607.
19 Veldhuis JD, Evans WS, Johnson ML, et al. Physiological properties of the luteinizing hormone pulse signal: Impact of intensive and extended venous sampling paradigms on its characterization in healthy men and women. J Clin Endocrinol Metab. 1986;62:881-891.
20 Kauffman AS. Emerging functions of gonadotropin-releasing hormone II in mammalian physiology and behaviour. J Neuroendocrinol. 2004;16:794-806.
21 Sullivan KA, Witkin JW, Ferin M, Silverman AJ. Gonadotropin-releasing hormone neurons in the rhesus macaque are not immunoreactive for the estrogen receptor. Brain Res. 1995;685:198-200.
22 Herbison AE, Horvath TL, Naftolin F, Leranth C. Distribution of estrogen receptor-immunoreactive cells in monkey hypothalamus: Relationship to neurones containing luteinizing hormone-releasing hormone and tyrosine hydroxylase. Neuroendocrinology. 1995;61:1-10.
23 Kallo I, Butler JA, Barkovics-Kallo M, et al. Oestrogen receptor beta-immunoreactivity in gonadotropin releasing hormone-expressing neurones: Regulation by oestrogen. J Neuroendocrinol. 2001;13:741-748.
24 Hrabovszky E, Steinhauser A, Barabas K, et al. Estrogen receptor-beta immunoreactivity in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2001;142:3261-3264.
25 Hrabovszky E, Shughrue PJ, Merchenthaler I, et al. Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2000;141:3506-3509.
26 Skynner MJ, Sim JA, Herbison AE. Detection of estrogen receptor alpha and beta messenger ribonucleic acids in adult gonadotropin-releasing hormone neurons. Endocrinology. 1999;140:5195-5201.
27 Abraham IM, Han SK, Todman MG, et al. Estrogen receptor beta mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci. 2003;23:5771-5777.
28 Dorling AA, Todman MG, Korach KS, Herbison AE. Critical role for estrogen receptor alpha in negative feedback regulation of gonadotropin-releasing hormone mRNA expression in the female mouse. Neuroendocrinology. 2003;78:204-209.
29 Radovick S, Ticknor CM, Nakayama Y, et al. Evidence for direct estrogen regulation of the human gonadotropin-releasing hormone gene. J Clin Invest. 1991;88:1649-1655.
30 Chen A, Zi K, Laskar-Levy O, Koch Y. The transcription of the hGnRH-I and hGnRH-II genes in human neuronal cells is differentially regulated by estrogen. J Mol Neurosci. 2002;18:67-76.
31 Matagne V, Lebrethon MC, Gerard A, Bourguignon JP. In vitro paradigms for the study of GnRH neuron function and estrogen effects. Ann NY Acad Sci. 2003;1007:129-142.
32 Krajewski SJ, Abel TW, Voytko ML, Rance NE. Ovarian steroids differentially modulate the gene expression of gonadotropin-releasing hormone neuronal subtypes in the ovariectomized cynomolgus monkey. J Clin Endocrinol Metab. 2003;88:655-662.
33 Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature. 1989;338:161-164.
34 Jennes L, Stumpf WE, Sheedy ME. Ultrastructural characterization of gonadotropin-releasing hormone (GnRH)-producing neurons. J Comp Neurol. 1985;232:534-547.
35 Silverman AJ, Jhamandas J, Renaud LP. Localization of luteinizing hormone-releasing hormone (LHRH) neurons that project to the median eminence. J Neurosci. 1987;7:2312-2319.
36 Waldstreicher J, Seminara SB, Jameson JL, et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J Clin Endocrinol Metab. 1996;81:4388-4395.
37 McClintock MK. Menstrual synchorony and suppression. Nature. 1971;229:244-245.
38 Stern K, McClintock MK. Regulation of ovulation by human pheromones. Nature. 1998;392:177-179.
39 Suarez-Quian CA, Wynn PC, Catt KJ. Receptor-mediated endocytosis of GnRH analogs: Differential processing of gold-labeled agonist and antagonist derivatives. J Steroid Biochem. 1986;24:183-192.
40 Schvartz I, Hazum E. Internalization and recycling of receptor-bound gonadotropin-releasing hormone agonist in pituitary gonadotropes. J Biol Chem. 1987;262:17046-17050.
41 Morell AG, Gregoriadis G, Scheinberg IH, et al. The role of sialic acid in determining the survival of glycoproteins in the circulation. J Biol Chem. 1971;246:1461-1467.
42 Weiss J, Guendner MJ, Halvorson LM, Jameson JL. Transcriptional activation of the follicle-stimulating hormone beta-subunit gene by activin. Endocrinology. 1995;136:1885-1891.
43 Besecke LM, Guendner MJ, Schneyer AL, et al. Gonadotropin-releasing hormone regulates follicle-stimulating hormone-beta gene expression through an activin/follistatin autocrine or paracrine loop. Endocrinology. 1996;137:3667-3673.
44 Richards JS, Hedin L. Molecular aspects of hormone action in ovarian follicular development, ovulation, and luteinization. Annu Rev Physiol. 1988;50:441-463.
45 Goodman RL, Parfitt DB, Evans NP, et al. Endogenous opioid peptides control the amplitude and shape of gonadotropin-releasing hormone pulses in the ewe. Endocrinology. 1995;136:2412-2420.
46 Genazzani AR, Genazzani AD, Volpogni C, et al. Opioid control of gonadotrophin secretion in humans. Hum Reprod. 1993;8(Suppl 2):151-153.
47 Whisnant SC, Havern RL, Goodman RL. Endogenous opioid suppression of luteinizing hormone pulse frequency and amplitude in the ewe: Hypothalamic sites of action. Neuroendocrinology. 1991;54:587-593.
48 Genazzani AR, Petraglia F. Opioid control of luteinizing hormone secretion in humans. J Steroid Biochem. 1989;33:751-755.
49 Ferin M. The role of endogenous opioid peptides in the regulation of the menstrual cycle. J Steroid Biochem. 1989;33(4B):683-685.
50 Maruncic M, Casper RF. The effect of luteal phase estrogen antagonism on luteinizing hormone pulsatility and luteal function in women. J Clin Endocrinol Metab. 1987;64:148-152.
51 Wildt L, Sir-Petermann T, Leyendecker G, et al. Opiate antagonist treatment of ovarian failure. Hum Reprod. 1993;8(Suppl 2):168-174.
52 Wildt L, Leyendecker G, Sir-Petermann T, Waibel-Treber S. Treatment with naltrexone in hypothalamic ovarian failure: Induction of ovulation and pregnancy. Hum Reprod. 1993;8:350-358.
53 Suh BY, Liu JH, Berga SL, et al. Hypercortisolism in patients with functional hypothalamic-amenorrhea. J Clin Endocrinol Metab. 1988;66:733-739.
54 De Cree C. Endogenous opioid peptides in the control of the normal menstrual cycle and their possible role in athletic menstrual irregularities. Obstet Gynecol Surv. 1989;44:720-732.
55 Harber VJ, Sutton JR. Endorphins and exercise. Sports Med. 1984;1:154-171.
56 Bicsak TA, Tucker EM, Cappel S, et al. Hormonal regulation of granulosa cell inhibin biosynthesis. Endocrinology. 1986;119:2711-2719.
57 Rivier C, Rivier J, Vale W. Inhibin-mediated feedback control of follicle-stimulating hormone secretion in the female rat. Science. 1986;234:205-208.
58 Groome NP, Illingworth PJ, O’Brien M, et al. Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996;81:1401-1405.
59 McNatty KP, Smith DM, Makris A, et al. The intraovarian sites of androgen and estrogen formation in women with normal and hyperandrogenic ovaries as judged by in vitro experiments. J Clin Endocrinol Metab. 1980;50:755-763.
60 Kobayashi M, Nakano R, Ooshima A. Immunohistochemical localization of pituitary gonadotrophins and gonadal steroids confirms the “two-cell, two-gonadotrophin” hypothesis of steroidogenesis in the human ovary. J Endocrinol. 1990;126:483-488.
61 Schoot DC, Coelingh Bennink HJ, Mannaerts BM, et al. Human recombinant follicle-stimulating hormone induces growth of preovulatory follicles without concomitant increase in androgen and estrogen biosynthesis in a woman with isolated gonadotropin deficiency. J Clin Endocrinol Metab. 1992;74:1471-1473.
62 Mendel CM. The free hormone hypothesis. Distinction from the free hormone transport hypothesis. J Androl. 1992;13:107-116.
63 Miller CD, Miller WL. Transcriptional repression of the ovine follicle-stimulating hormone-beta gene by 17 beta-estradiol. Endocrinology. 1996;137:3437-3446.
64 Baratta M, West LA, Turzillo AM, Nett TM. Activin modulates differential effects of estradiol on synthesis and secretion of follicle-stimulating hormone in ovine pituitary cells. Biol Reprod. 2001;64:714-719.
65 Wildt L, Hutchison JS, Marshall G, et al. On the site of action of progesterone in the blockade of the estradiol-induced gonadotropin discharge in the rhesus monkey. Endocrinology. 1981;109:1293-1294.
66 Liu JH, Yen SS. Induction of midcycle gonadotropin surge by ovarian steroids in women: A critical evaluation. J Clin Endocrinol Metab. 1983;57:797-802.
67 Carr BR, MacDonald PC, Simpson ER. The role of lipoproteins in the regulation of progesterone secretion by the human corpus luteum. Fertil Steril. 1982;38:303-311.
68 Clark BJ, Soo SC, Caron KM, et al. Hormonal and developmental regulation of the steroidogenic acute regulatory protein. Mol Endocrinol. 1995;9:1346-1355.
69 Hu YC, Wang PH, Yeh S, et al. Subfertility and defective folliculogenesis in female mice lacking androgen receptor. Proc Natl Acad Sci USA. 2004;101:11209-11214.
70 Hallberg L, Hogdahl AM, Nilsson L, Rybo G. Menstrual blood loss—a population study. Variation at different ages and attempts to define normality. Acta Obstet Gynecol Scand. 1966;45:320-351.
71 Lessey BA, Killam AP, Metzger DA, et al. Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab. 1988;67:334-340.
72 Sullivan MW, Stewart-Akers A, Krasnow JS, et al. Ovarian responses in women to recombinant follicle-stimulating hormone and luteinizing hormone (LH): A role for LH in the final stages of follicular maturation. J Clin Endocrinol Metab. 1999;84:228-232.
73 Yong EL, Baird DT, Hillier SG. Mediation of gonadotrophin-stimulated growth and differentiation of human granulosa cells by adenosine-3′,5′-monophosphate: One molecule, two messages. Clin Endocrinol (Oxf). 1992;37:51-58.
74 Oktay K, Briggs D, Gosden RG. Ontogeny of follicle-stimulating hormone receptor gene expression in isolated human ovarian follicles. J Clin Endocrinol Metab. 1997;82:3748-3751.
75 LaPolt PS, Tilly JL, Aihara T, et al. Gonadotropin-induced up- and down-regulation of ovarian follicle-stimulating hormone (FSH) receptor gene expression in immature rats: Effects of pregnant mare’s serum gonadotropin, human chorionic gonadotropin, and recombinant FSH. Endocrinology. 1992;130:1289-1295.
76 Matthews CH, Borgato S, Beck-Peccoz P, et al. Primary amenorrhoea and infertility due to a mutation in the beta-subunit of follicle-stimulating hormone. Nat Genet. 1993;5:83-86.
77 Fletcher WH, Greenan JR. Receptor mediated action without receptor occupancy. Endocrinology. 1985;116:1660-1662.
78 McNatty KP, Makris A, Reinhold VN, et al. Metabolism of androstenedione by human ovarian tissues in vitro with particular reference to reductase and aromatase activity. Steroids. 1979;34:429-443.
79 Hillier SG, van den Boogaard AM, Reichert LEJr, van Hall EV. Intraovarian sex steroid hormone interactions and the regulation of follicular maturation: Aromatization of androgens by human granulosa cells in vitro. J Clin Endocrinol Metab. 1980;50:640-647.
80 Hild-Petito S, West NB, Brenner RM, Stouffer RL. Localization of androgen receptor in the follicle and corpus luteum of the primate ovary during the menstrual cycle. Biol Reprod. 1991;44:561-568.
81 Jia XC, Kessel B, Welsh THJr, Hsueh AJ. Androgen inhibition of follicle-stimulating hormone-stimulated luteinizing hormone receptor formation in cultured rat granulosa cells. Endocrinology. 1985;117:13-22.
82 Erickson GF, Magoffin DA, Dyer CA, Hofeditz C. The ovarian androgen producing cells: A review of structure/function relationships. Endocr Rev. 1985;6:371-399.
83 Chabab A, Hedon B, Arnal F, et al. Follicular steroids in relation to oocyte development and human ovarian stimulation protocols. Hum Reprod. 1986;1:449-454.
84 Andersen CY. Characteristics of human follicular fluid associated with successful conception after in vitro fertilization. J Clin Endocrinol Metab. 1993;77:1227-1234.
85 Knobil E, Plant TM, Wildt L, et al. Control of the rhesus monkey menstrual cycle: Permissive role of hypothalamic gonadotropin-releasing hormone. Science. 1980;207:1371-1373.
86 Adams JM, Taylor AE, Schoenfeld DA, et al. The midcycle gonadotropin surge in normal women occurs in the face of an unchanging gonadotropin-releasing hormone pulse frequency. J Clin Endocrinol Metab. 1994;79:858-864.
87 Nakai Y, Plant TM, Hess DL, et al. On the sites of the negative and positive feedback actions of estradiol in the control of gonadotropin secretion in the rhesus monkey. Endocrinology. 1978;102:1008-1014.
88 Gregg DW, Nett TM. Direct effects of estradiol-17 β on the number of gonadotropin-releasing hormone receptors in the ovine pituitary. Biol Reprod. 1989;40:288-293.
89 Bauer-Dantoin AC, Weiss J, Jameson JL. Roles of estrogen, progesterone, and gonadotropin-releasing hormone (GnRH) in the control of pituitary GnRH receptor gene expression at the time of the preovulatory gonadotropin surges. Endocrinology. 1995;136:1014-1019.
90 Adams TE, Norman RL, Spies HG. Gonadotropin-releasing hormone receptor binding and pituitary responsiveness in estradiol-primed monkeys. Science. 1981;213:1388-1390.
91 Menon M, Peegel H, Katta V. Estradiol potentiation of gonadotropin-releasing hormone responsiveness in the anterior pituitary is mediated by an increase in gonadotropin-releasing hormone receptors. Am J Obstet Gynecol. 1985;151:534-540.
92 Xia L, Van Vugt D, Alston EJ, et al. A surge of gonadotropin-releasing hormone accompanies the estradiol-induced gonadotropin surge in the rhesus monkey. Endocrinology. 1992;131:2812-2820.
93 Leyendecker G, Wildt L, Hansmann M. Pregnancies following chronic intermittent (pulsatile) administration of Gn-RH by means of a portable pump (“Zyklomat”)—a new approach to the treatment of infertility in hypothalamic amenorrhea. J Clin Endocrinol Metab. 1980;51:1214-1216.
94 Caraty A, Antoine C, Delaleu B, et al. Nature and bioactivity of gonadotropin-releasing hormone (GnRH) secreted during the GnRH surge. Endocrinology. 1995;136:3452-3460.
95 Young JR, Jaffe RB. Strength–duration characteristics of estrogen effects on gonadotropin response to gonadotropin-releasing hormone in women. II. Effects of varying concentrations of estradiol. J Clin Endocrinol Metab. 1976;42:432-442.
96 Cahill DJ, Wardle PG, Harlow CR, Hull MG. Onset of the preovulatory luteinizing hormone surge: Diurnal timing and critical follicular prerequisites. Fertil Steril. 1998;70:56-59.
97 Hoff JD, Quigley ME, Yen SS. Hormonal dynamics at midcycle: A reevaluation. J Clin Endocrinol Metab. 1983;57:792-796.
98 Pauerstein CJ, Eddy CA, Croxatto HD, et al. Temporal relationships of estrogen, progesterone, and luteinizing hormone levels to ovulation in women and infrahuman primates. Am J Obstet Gynecol. 1978;130:876-886.
99 Zelinski-Wooten MB, Hutchison JS, Chandrasekher YA, et al. Administration of human luteinizing hormone (hLH) to macaques after follicular development: Further titration of LH surge requirements for ovulatory changes in primate follicles. J Clin Endocrinol Metab. 1992;75:502-507.
100 McClure N, Macpherson AM, Healy DL, et al. An immunohistochemical study of the vascularization of the human graafian follicle. Hum Reprod. 1994;9:1401-1405.
101 Wulff C, Wilson H, Wiegand SJ, et al. Prevention of thecal angiogenesis, antral follicular growth, and ovulation in the primate by treatment with vascular endothelial growth factor Trap R1R2. Endocrinology. 2002;143:2797-2807.
102 Wulff C, Wilson H, Largue P, et al. Angiogenesis in the human corpus luteum: Localization and changes in angiopoietins, tie-2, and vascular endothelial growth factor messenger ribonucleic acid. J Clin Endocrinol Metab. 2000;85:4302-4309.
103 Dickson SE, Fraser HM. Inhibition of early luteal angiogenesis by gonadotropin-releasing hormone antagonist treatment in the primate. J Clin Endocrinol Metab. 2000;85:2339-2344.
104 Lumsden MA, Kelly RW, Templeton AA, et al. Changes in the concentration of prostaglandins in preovulatory human follicles after administration of hCG. J Reprod Fertil. 1986;77:119-124.
105 Yoshimura Y, Santulli R, Atlas SJ, et al. The effects of proteolytic enzymes on in vitro ovulation in the rabbit. Am J Obstet Gynecol. 1987;157:468-475.
106 Edwards RG. Maturation in vitro of human ovarian oocytes. Lancet. 1965;2(7419):926-929.
107 Edwards RG. Maturation in vitro of mouse, sheep, cow, pig, rhesus monkey and human ovarian oocytes. Nature. 1965;208:349-351.
108 Hutchison JS, Zeleznik AJ. The rhesus monkey corpus luteum is dependent on pituitary gonadotropin secretion throughout the luteal phase of the menstrual cycle. Endocrinology. 1984;115:1780-1786.
109 Filicori M, Butler JP, Crowley WFJr. Neuroendocrine regulation of the corpus luteum in the human. Evidence for pulsatile progesterone secretion. J Clin Invest. 1984;73:1638-1647.
110 Sheehan KL, Casper RF, Yen SS. Luteal phase defects induced by an agonist of luteinizing hormone-releasing factor: A model for fertility control. Science. 1982;215:170-172.
111 Bassett SG, Little-Ihrig LL, Mason JI, Zeleznik AJ. Expression of messenger ribonucleic acids that encode for 3 β-hydroxysteroid dehydrogenase and cholesterol side-chain cleavage enzyme throughout the luteal phase of the macaque menstrual cycle. J Clin Endocrinol Metab. 1991;72:326-362.
112 Brannian JD, Shiigi SM, Stouffer RL. Gonadotropin surge increases fluorescent-tagged low-density lipoprotein uptake by macaque granulosa cells from preovulatory follicles. Biol Reprod. 1992;47:355-360.
113 Rojas FJ, Moretti-Rojas I, Balmaceda JP, Asch RH. Regulation of gonadotropin-stimulable adenylyl cyclase of the primate corpus luteum. J Steroid Biochem. 1989;32:175-182.
114 McLachlan RI, Cohen NL, Vale WW, et al. The importance of luteinizing hormone in the control of inhibin and progesterone secretion by the human corpus luteum. J Clin Endocrinol Metab. 1989;68:1078-1085.
115 Molskness TA, Woodruff TK, Hess DL, et al. Recombinant human inhibin-A administered early in the menstrual cycle alters concurrent pituitary and follicular, plus subsequent luteal, function in rhesus monkeys. J Clin Endocrinol Metab. 1996;81:4002-4006.
116 le Nestour E, Marraoui J, Lahlou N, et al. Role of estradiol in the rise in follicle-stimulating hormone levels during the luteal-follicular transition. J Clin Endocrinol Metab. 1993;77:439-442.
117 Christin-Maitre S, Taylor AE, Khoury RH, et al. Homologous in vitro bioassay for follicle-stimulating hormone (FSH) reveals increased FSH biological signal during the mid- to late luteal phase of the human menstrual cycle. J Clin Endocrinol Metab. 1996;81:2080-2088.
118 Roseff SJ, Bangah ML, Kettel LM, et al. Dynamic changes in circulating inhibin levels during the luteal-follicular transition of the human menstrual cycle. J Clin Endocrinol Metab. 1989;69:1033-1039.
119 Muttukrishna S, Fowler PA, George L, et al. Changes in peripheral serum levels of total activin A during the human menstrual cycle and pregnancy. J Clin Endocrinol Metab. 1996;81:3328-3334.
120 Shikone T, Yamoto M, Kokawa K, et al. Apoptosis of human corpora lutea during cyclic luteal regression and early pregnancy. J Clin Endocrinol Metab. 1996;81:2376-2380.
121 Young KA, Hennebold JD, Stouffer RL. Dynamic expression of mRNAs and proteins for matrix metalloproteinases and their tissue inhibitors in the primate corpus luteum during the menstrual cycle. Mol Hum Reprod. 2002;8:833-840.
122 Friden BE, Runesson E, Hahlin M, Brannstrom M. Evidence for nitric oxide acting as a luteolytic factor in the human corpus luteum. Mol Hum Reprod. 2000;6:397-403.
123 Vega M, Urrutia L, Iniguez G, et al. Nitric oxide induces apoptosis in the human corpus luteum in vitro. Mol Hum Reprod. 2000;6:681-687.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 1 The Hypothalamic-Pituitary-Ovarian Axis and Control of the Menstrual Cycle
INTRODUCTION
The hypothalamus and pituitary gland form a unit that exerts control over a wide range of endocrine organs, including the gonads. This chapter describes the hypothalamic-pituitary-ovarian axis and control of the menstrual cycle, which is modulated by the central nervous system, other endocrine systems, and the environment. Key hormones in the hypothalamic-pituitary-ovarian axis include gonadotropin-releasing hormone (GnRH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), estradiol, and progesterone (Table 1-1). Supporting roles are also played by inhibin, activin, follistatin, and endorphins.
THE HYPOTHALAMUS
The hypothalamus forms the lower part of the lateral wall and the floor of the third ventricle, and weighs approximately 10 grams. The hypothalamus is typically divided into eight specific nuclei (consistently clustered groups of neurons) and three areas (less clustered, less distinctly demarcated neurons), as illustrated in Figure 1-1. From a reproductive standpoint, the most important of these are the arcuate nucleus and the preoptic area, the principal sites of GnRH-producing neurons.1 The arcuate nucleus is located in the medial basal hypothalamus and is the most proximal of all the hypothalamic nuclei to the optic chiasm and the pituitary stalk. The arcuate nucleus is also the site of dopamine-secreting neurons that function to inhibit pituitary prolactin secretion and neurons that secrete growth hormone-releasing hormone.
GnRH
GnRH is the primary hypothalamic regulator of pituitary reproductive function. Two human forms of GnRH (GnRH-I and GnRH-II) have been identified.2,3 Both are decapeptides and are the products of different genes. At least 20 other types of GnRH have been identified in fish, amphibians and protochordates, but none of these are believed to be present in humans.4,5
GnRH-I was first characterized and synthesized in 1971 by Andrew Schally and Roger Guillemin, an accomplishment for which both men ultimately received the Nobel prize.6–9 The structure of GnRH-I is common to all mammals, and its action is similar in males and females (Fig. 1-2). GnRH-I is synthesized from a much larger, 92-amino acid precursor peptide that contains GnRH-associated peptide.10 GnRH-I then travels along an axonal pathway called the tuberoinfundibular tract to the median eminence of the hypothalamus, where it is released into the portal circulation in a pulsatile fashion. The half-life of GnRH-I is very short (2 to 4 minutes) because it is rapidly cleaved between amino acids 5 and 6, 6 and 7, and 9 and 10. Because of its short half-life and rapid dilution in the peripheral circulation, serum GnRH-I levels are difficult to measure and do not correlate with pituitary action.
GnRH-I has three principal actions on anterior pituitary gonadotrophs: (1) synthesis and storage of gonadotropins, (2) movement of gonadotropins from the reserve pool to a point where they can be readily released, and (3) direct secretion of gonadotropins. GnRH-I pulses occur in response to intrinsic rhythmic activity within GnRH neurons in the arcuate nucleus. Pulsatile release of GnRH-I from the median eminence within a critical frequency and amplitude results in normal gonadotropin secretion.11,12 Continuous, rather than pulsatile, exposure to GnRH-I results in suppression of FSH and LH secretion and suppression of gonadotropin gene transcription.13,14
In the absence of gonadal feedback, the GnRH pulse frequency is approximately once per hour.15 During the menstrual cycle the frequency and amplitude of GnRH pulses vary in response to hypothalamic feedback (Table 1-2).16 In general, the follicular phase is characterized by high-frequency, low-amplitude pulses, and the luteal phase is characterized by lower-frequency, higher-amplitude pulses.17,18 However, considerable variability exists both between and within individuals.19 In humans, GnRH-I pulse frequency and amplitude are best approximated by measuring LH pulse frequency and amplitude.
Table 1-2 Menstrual Cycle Variation in LH Pulse Frequency and Amplitude
| Cycle Phase | Mean Frequency (minutes) | Mean Amplitude (mIU/mL) |
|---|---|---|
| Early follicular | 90 | 6.5 |
| Mid-follicular | 50 | 5 |
| Late follicular | 60–70 | 7 |
| Early luteal | 100 | 15 |
| Mid-luteal | 150 | 12 |
| Late luteal | 200 | 8 |
GnRH-II is most highly expressed outside of the brain, in tissues that include kidneys, bone marrow, and prostate. This is in contrast to GnRH-I, which is not expressed in high levels outside the brain. Although GnRH-II can induce release of both FSH and LH, it appears to have a wide array of physiologic functions outside the brain including regulation of cellular proliferation and mediation of ovarian and placental hormone secretion.20
Initial attempts in the mid-1990s to identify estrogen receptors in GnRH neurons were unsuccessful.21,22 However, subsequent use of more sophisticated techniques identified estrogen receptors α and β in the arcuate nucleus.23–26 Both receptors mediate estrogen action on GnRH neurons in vivo.27,28 The GnRH gene contains a hormone response element for the estrogen–estrogen receptor complex.29 Transcription of GnRH-I and GnRH-II is differentially regulated by estrogen.30 The regulatory role of estradiol on GnRH is complex. Estrogen inhibits GnRH gene expression/biosynthesis, but secretion of GnRH may be increased, decreased, or unaffected.31,32
The activity of the hypothalamus is further modulated by nervous stimuli from higher brain centers. GnRH neurons exhibit many connections to each other and to other neurons. Some of the neurotransmitters that modulate GnRH secretion are outlined in Table 1-3. The effects of these neurotransmitters help explain the mechanism by which certain physical or clinical conditions may affect the menstrual cycle (Table 1-4).
Table 1-3 Neurotransmitter Effects on GnRH Release
| Neurotransmitter | Effect |
|---|---|
| Dopamine | Inhibits GnRH release |
| Endorphin | Inhibits GnRH release |
| Serotonin | Inhibits GnRH release |
| Norepinephrine, epinephrine | Stimulates GnRH release |
Table 1-4 Mechanisms for Oligo/amenorrhea in Various Clinical Conditions
| Hyperprolactinemia | Elevated dopamine suppresses GnRH |
| Hypothyroidism | Elevated TRH increases prolactin, which in turn increases dopamine which, then suppresses GnRH |
| Stress | Increased corticotropin (ACTH) results in increased endorphins (both are derived from the same peptide precursor); endorphins suppress GnRH |
| Exercise | Increased endorphins suppress GnRH |
TRH = thyrotropin-releasing hormone; GnRH = gonadotropin-releasing hormone.
Cells that produce GnRH originate embryologically from the olfactory area.33 GnRH neurons, like olfactory epithelial cells of the nasal cavity, have cilia.34 During embryogenesis GnRH neurons migrate from the medial olfactory placode to the arcuate nucleus of the hypothalamus.35 The common origin of GnRH and olfactory neurons is demonstrated by Kallmann’s syndrome, where GnRH deficiency is associated with anosmia. Kallmann’s syndrome is believed to be caused by a variety of gene defects that affect neuronal cell migration.36
The common origin of GnRH and olfactory neurons is also suggestive of the relationship between pheromones and menstrual cyclicity. Pheromones are small airborne chemicals that when secreted externally by one individual may be perceived by other individuals of the same species, producing a change in sexual or social behavior. It is well recognized that women who work or live together often develop synchrony of their menstrual cycles.37 Moreover, it has been shown that odorless compounds from the axillae of cycling women can alter the cycle characteristics of recipient women.38 Presumably, these alterations occur through olfactory GnRH-mediated mechanisms.
The GnRH Receptor
The GnRH-I receptor is a G-protein receptor that utilizes inositol triphosphate and diacylglycerol as second messengers to stimulate protein kinase, release of calcium ions, and cyclic adenosine monophosphate (cAMP) activity. The GnRH-I receptor is encoded by a gene on chromosome 14q21.1 and is expressed in many parts of the body outside of the brain, including ovarian follicles and the placenta. In humans it appears that GnRH-II signaling occurs through the GnRH-I receptor.5 Although a GnRH-II receptor is present in many mammalian species, its functional capacity is limited due to a frame shift and a stop codon. GnRH receptors are regulated by many substances, including GnRH itself, inhibin, activin, estrogen, and progesterone.
Changes to the amino acid sequence of GnRH can extend its half-life to hours or days and can change its biologic activity from an agonist to an antagonist. All of the GnRH agonists currently available extend their half-life by substitutions at amino acid 6 and sometimes amino acid 10 of native GnRH (Table 1-5). Continuous activation of the GnRH receptor results in desensitization due to phosphorylation and conformational change of the receptor, uncoupling from G proteins, internalization of the receptor via endocytosis, and decreased receptor synthesis.39,40 On administration all GnRH agonists increase gonadotropin secretion (the flare effect). However, after 7 to 14 days GnRH receptor desensitization occurs and pituitary suppression is achieved.
In contrast, GnRH antagonists directly inhibit gonadotropin secretion. Structurally, GnRH antagonists are characterized by multiple amino acid substitutions to the natural GnRH decapeptide. The commercially available GnRH antagonists cetrorelix and ganirelix have large amino acid additions to position 1 of native GnRH. GnRH antagonists compete for and occupy pituitary GnRH receptors, thus competitively blocking endogenous GnRH–GnRH receptor binding. In contrast to GnRH agonists, there is no flare effect with GnRH antagonists. Because receptor loss does not occur, a constant supply of antagonist is necessary to ensure that all GnRH receptors are continuously occupied. Thus, the therapeutic dosage range for antagonists is typically higher than that for agonists (mg versus μg).
THE PITUITARY
The pituitary gland measures approximately 15 × 10 × 6 mm and weighs approximately 500 to 900 mg. The pituitary gland lies immediately beneath the third ventricle and just above the sphenoidal sinus in a bony cavity called the sella turcica (Turkish saddle). It consists of anterior and posterior lobes, each having different embryologic origins, functions, and control mechanisms (Fig. 1-3).
The Anterior Pituitary
The major cell types of the anterior pituitary are outlined in Table 1-6. The anterior pituitary is a classic endocrine gland in that it is composed of secretory cells of epithelial origin supported by connective tissue rich in blood and lymphatic capillaries. In accordance with their active synthetic function the endocrine cells are characterized by prominent nuclei and prolific mitochondria, endoplasmic reticulum, Golgi bodies, and secretory vesicles. Synthesis of gonadotropins takes place in the rough endoplasmic reticulum, after which the hormones are packaged within the Golgi apparatus and stored as secretory granules. In response to GnRH the secretory granules are extruded from the cell membrane. The endothelial lining of capillary sinusoids is fenestrated, facilitating the passage of pituitary hormones into the sinusoids.
Gonadotropins
FSH
The sialic acid content of FSH, LH, TSH, and hCG varies, and these differences are largely responsible for variations in half-life of the glycoprotein hormones. The liver is the major site of clearance for gonadotropins. Sialic acid prevents hepatic clearance; thus, the greater the sialic acid content, the longer the half-life.41 hCG, with 20 sialic acid residues, has the longest half-life (about 24 hours), whereas LH (1 to 2 sialic acid residues) has the shortest half-life (20 to 30 minutes). Addition of sialic acid residues in urinary-derived commercially available gonadotropins (e.g., hMG) is responsible for their longer half-life (30 hours).
In gonadotrophs of the anterior pituitary, GnRH signaling leads to transcription of the α and β subunits for both FSH and LH. The GnRH-dependent availability of the β subunits is the rate-limiting step in gonadotropin synthesis. Although both FSH and LH require GnRH stimulation, synthesis of the FSH β subunit also requires the presence of activin.42,43
FSH and LH Receptors
In contrast, FSH receptors are found exclusively on granulosa cells in the ovary. FSH binds to receptors on the cell surface of granulosa cells in antral follicles. Like LH, FSH acts via the cAMP-dependent protein kinase pathway.44 In response to FSH the androgens produced as a result of LH stimulation are then aromatized to estrogens in granulosa cells.
Opioid Modulation of Pituitary Hormones
Opioids (i.e., endogenous opiates) are natural occurring sedative narcotics produced in the brain whose structure and function are similar to opium. Opioids include enkephalins, endorphins, and dynorphins; they modulate every pituitary hormone by acting on the hypothalamus. An important action of opioids is to inhibit gonadotropin secretion by suppressing GnRH release.45
Opioid tone is an important regulator of menstrual cyclicity.46–49 Endorphins are at a nadir in the early follicular phase (menstruation) and gradually rise to peak levels in the luteal phase in response to the rise in estrogen and progesterone. It is believed that opioids mediate the negative feedback of ovarian steroids on gonadotropin release, particularly in the luteal phase.50
Endogenous opioids appear to play a central role in hypothalamic amenorrhea. Treatment of women suffering from this condition with an opioid receptor antagonist (e.g., naltrexone) results in the return to ovulatory menstrual patterns and even conception in some cases.51,52 Women with stress-related amenorrhea demonstrate increased hypothalamic corticotropin-releasing hormone and pituitary corticotropin, which manifests as hypercortisolism.53 The corticotropin precursor peptide, pro-opiomelanocortin, is also the precursor for endorphin synthesis. It is hypothesized that stress-related amenorrhea is the result of GnRH inhibition secondary to increased production of endogenous opioids. Opioids also rise during exercise (“runners’ high”), and this may contribute to hypothalamic amenorrhea in athletes.54,55
Ovarian Peptide Hormone Feedback on Gonadotropin Secretion
The ovary secrets two polypeptide hormones that inhibit or stimulate FSH secretion by the anterior pituitary. Inhibin and activin act as opposing nonsteroidal gonadal hormones that regulate FSH synthesis and secretion by the pituitary. They also have paracrine effects within the ovary, where they modulate follicle growth and steroidogenesis. Follistatin is a binding protein that modulates the effects of activin but not inhibin.
Inhibin and Activin
Inhibin and activin are members of the transforming growth factor-β (TGF-β) superfamily of ligands, which includes müllerian inhibiting substance (MIS; see Chapter 2). Like the gonadotropins, inhibin and activin are comprised of two subunits. Inhibin is comprised of an α and β subunit and has been isolated in two forms containing different β subunits, Inhibin-A and Inhibin-B. Activin is comprised of two beta subunits identical to those found in inhibin.
Inhibin is secreted by granulosa cells in response to FSH.56 However, mRNA for inhibin has also been found in pituitary gonadotrophs. Inhibin selectively inhibits FSH but not LH secretion.57 Thus, a negative feedback loop is created where FSH stimulates inhibin and in turn inhibin suppresses FSH.
Inhibin B is predominantly secreted in the follicular phase of the menstrual cycle, whereas inhibin A is predominantly secreted in the luteal phase.58 Peak levels of inhibin B in the follicular phase are in the range of 50 to 100 pg/mL. Peak levels of inhibin A in the luteal phase are between 40 and 60 pg/mL.
OVARIAN STEROIDOGENESIS DURING THE MENSTRUAL CYCLE
Ovarian steroidogenesis during the menstrual cycle occurs in granulosa and theca cells (Table 1-7 and Fig. 1-4). Before ovulation, theca cells are separated from granulosa cells in the same follicle by a basal membrane. Thus, granulosa cells of preovulatory follicles do not have a blood supply. However, at the time of the LH surge the preovulatory follicle undergoes luteinization with disappearance of the basal membrane and capillary invasion of the granulosa cells. Theca cells become theca-lutein cells, and granulosa cells become granulosa-lutein cells.
Table 1-7 Site of Synthesis of Major Steroidogenic Products of the Ovary
| Cell Type | Major Steroid Hormone Products |
|---|---|
| Theca cells | Androgens (androstenedione, DHEA, testosterone)* |
| Granulosa cells | Estrogens (estradiol, estrone) |
| Theca-lutein cells | Progestogens (progesterone, 17-hydroxyprogesterone)** |
| Granulosa-lutein cells | Estrogens (estradiol, estrone) |
