CHAPTER 1 The History and Impact of Genetics in Medicine
Presenting historical truth is at least as challenging as the pursuit of scientific truth and our view of human endeavors down the ages is heavily biased in favor of winners—those who have conquered on military, political, or, indeed, scientific battlefields. The history of genetics in relation to medicine is one of breathtaking discovery from which patients and families already benefit hugely, but in the future success will be measured by ongoing progress in translating discoveries into both treatment and prevention of disease. As this takes place, we should not neglect looking back with awe at what our forebears achieved with scarce resources and sheer determination, sometimes aided by serendipity, in order to lay the foundations of this dynamic science. A holistic approach to science can be compared with driving a car: without your eyes on the road ahead, you will crash and make no progress; however, the competent driver will glance in the rear and side mirrors regularly to maintain control.
Gregor Mendel and the Laws of Inheritance
Early Beginnings
Early Greek philosophers and physicians such as Aristotle and Hippocrates concluded, with typical masculine modesty, that important human characteristics were determined by semen, using menstrual blood as a culture medium and the uterus as an incubator. Semen was thought to be produced by the whole body; hence bald-headed fathers would beget bald-headed sons. These ideas prevailed until the seventeenth century, when Dutch scientists such as Leeuwenhoek and de Graaf recognized the existence of sperm and ova, thus explaining how the female could also transmit characteristics to her offspring.
Our present understanding of human genetics owes much to the work of the Austrian monk Gregor Mendel (1822–1884; Figure 1.1) who, in 1865, presented the results of his breeding experiments on garden peas to the Natural History Society of Brünn in Bohemia (now Brno in the Czech Republic). Shortly after, Mendel’s observations were published by that association in the Transactions of the Society, where they remained largely unnoticed until 1900, some 16 years after his death, when their importance was first recognized. In essence, Mendel’s work can be considered as the discovery of genes and how they are inherited. The term gene was first coined in 1909 by a Danish botanist, Johannsen, and was derived from the term ‘pangen’ introduced by De Vries. This term was itself a derivative of the word ‘pangenesis,’ coined by Darwin in 1868. In acknowledgement of Mendel’s enormous contribution, the term mendelian is now part of scientific vocabulary, applied both to the different patterns of inheritance shown by single-gene characteristics and to disorders found to be the result of defects in a single gene.

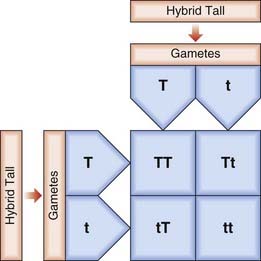
In his breeding experiments, Mendel studied contrasting characters in the garden pea, using for each experiment varieties that differed in only one characteristic. For example, he noted that when strains bred for a feature such as tallness were crossed with plants bred to be short all of the offspring in the first filial or F1 generation were tall. If plants in this F1 generation were interbred, this led to both tall and short plants in a ratio of 3 : 1 (Figure 1.2). Characteristics that were manifest in the F1 hybrids were referred to as dominant, whereas those that reappeared in the F2 generation were described as being recessive. On reanalysis it has been suggested that Mendel’s results were ‘too good to be true’ in that the segregation ratios he derived were suspiciously closer to the value of 3 : 1 than the laws of statistics would predict. One possible explanation is that he may have published only those results that best agreed with his preconceived single-gene hypothesis. Whatever the truth of the matter, events have shown that Mendel’s interpretation of his results was entirely correct.
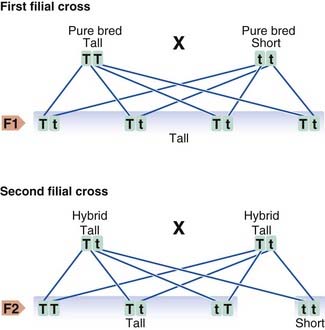
An alternative method for determining genotypes in offspring involves the construction of what is known as a Punnett square (Figure 1.3). This is used further in Chapter 8 when considering how genes segregate in large populations.
The Law of Segregation
The law of segregation refers to the observation that each person possesses two genes for a particular characteristic, only one of which can be transmitted at any one time. Rare exceptions to this rule can occur when two allelic genes fail to separate because of chromosome non-disjunction at the first meiotic division (p. 43).
The Law of Independent Assortment
The law of independent assortment refers to the fact that members of different gene pairs segregate to offspring independently of one another. In reality, this is not always true, as genes that are close together on the same chromosome tend to be inherited together, because they are ‘linked’ (p. 136). There are a number of other ways by which the laws of mendelian inheritance are breached but, overall, they remain foundational to our understanding of the science.
The Chromosomal Basis of Inheritance
As interest in mendelian inheritance grew, there was much speculation as to how it actually occurred. At that time it was also known that each cell contains a nucleus within which there are several threadlike structures known as chromosomes, so called because of their affinity for certain stains (chroma = color, soma = body). These chromosomes had been observed since the second half of the nineteenth century after development of cytologic staining techniques. Human mitotic figures were observed from the late 1880s, and it was in 1902 that Walter Sutton, an American medical student, and Theodour Boveri, a German biologist, independently proposed that chromosomes could be the bearers of heredity (Figure 1.4). Subsequently, Thomas Morgan transformed Sutton’s chromosome theory into the theory of the gene, and Alfons Janssens observed the formation of chiasmata between homologous chromosomes at meiosis. During the late 1920s and 1930s, Cyril Darlington helped to clarify chromosome mechanics by the use of tulips collected on expeditions to Persia. It was during the 1920s that the term genome entered the scientific vocabulary, being the fusion of genom (German for ‘gene’) and ome from ‘chromosome’.
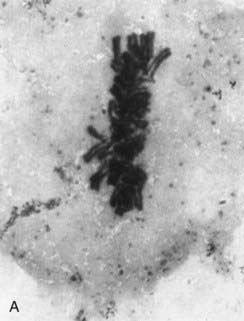
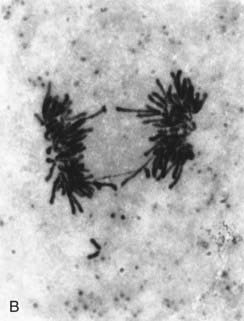
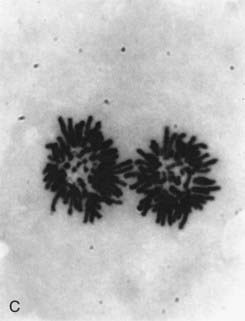
FIGURE 1.4 Chromosomes dividing into two daughter cells at different stages of cell division. A, Metaphase; B, anaphase; C, telophase. The behavior of chromosomes in cell division (mitosis) is described at length in Chapter 3.
(Photographs courtesy Dr. K. Ocraft, City Hospital, Nottingham.)
When the connection between mendelian inheritance and chromosomes was first made, it was thought that the normal chromosome number in humans might be 48, although various papers had come up with a range of figures. The number 48 was settled on largely as a result of a paper in 1921 from Theophilus Painter, an American cytologist who had been a student of Boveri. In fact, Painter himself had some preparations clearly showing 46 chromosomes, even though he finally settled on 48. These discrepancies were probably from the poor quality of the material at that time; even into the early 1950s, cytologists were counting 48 chromosomes. It was not until 1956 that the correct number of 46 was established by Tjio and Levan, 3 years after the correct structure of DNA had been proposed. Within a few years, it was shown that some disorders in humans could be caused by loss or gain of a whole chromosome as well as by an abnormality in a single gene. Chromosome disorders are discussed at length in Chapter 18. Some chromosome aberrations, such as translocations, can run in families (p. 44), and are sometimes said to be segregating in a mendelian fashion.
DNA as the Basis of Inheritance
Whilst James Watson and Francis Crick are justifiably credited with discovering the structure of DNA in 1953, they were attracted to working on it only because of its key role as the genetic material, as established in the 1940s. Formerly many believed that hereditary characteristics were transmitted by proteins, until it was appreciated that their molecular structure was far too cumbersome. Nucleic acids were actually discovered in 1849. In 1928 Fred Griffith, working on two strains of Streptococcus, realized that characteristics of one strain could be conferred on the other by something that he called the transforming principle. In 1944, at the Rockefeller Institute in New York, Oswald Avery, Maclyn McCarty, and Colin MacLeod identified DNA as the genetic material while working on the pneumococcus (Streptococcus pneumoniae). Even then, many in the scientific community were skeptical; DNA was only a simple molecule with lots of repetition of four nucleic acids—very boring! The genius of Watson and Crick, at Cambridge, was to hit on a structure for DNA that would explain the very essence of biological reproduction, and their elegant double helix has stood the test of time. Crucial to their discovery was the x-ray crystallography work of Maurice Wilkins and Rosalind Franklin at King’s College, London.
This was merely the beginning, for it was necessary to discover the process whereby DNA, in discrete units called genes, issues instructions for the precise assembly of proteins, the building blocks of tissues. The sequence of bases in DNA, and the sequence of amino acids in protein, the genetic code, was unravelled in some elegant biochemical experiments in the 1960s and it became possible to predict the base change in DNA that led to the amino-acid change in the protein. Further experiments, involving Francis Crick, Paul Zamecnik, and Mahlon Hoagland, identified the molecule transfer RNA (tRNA) (p. 20), which directs genetic instructions via amino acids to intracellular ribosomes, where protein chains are produced. Confirmation of these discoveries came with DNA sequencing methods and the advent of recombinant DNA techniques. Interestingly, however, the first genetic trait to be characterized at the molecular level had already been identified in 1957 by laborious sequencing of the purified proteins. This was sickle-cell anemia, in which the mutation affects the amino-acid sequence of the blood protein hemoglobin.
The Fruit Fly
In view of these unique properties, fruit flies were used extensively in early breeding experiments. Today their study is still proving of great value in fields such as developmental biology, where knowledge of gene homology throughout the animal kingdom has enabled scientists to identify families of genes that are important in human embryogenesis (see Chapter 6). When considering major scientific achievements in the history of genetics, it is notable that sequencing of the 180 million base pairs of the Drosophila melanogaster genome was completed toward the end of 1999.
The Origins of Medical Genetics
In 1900 Mendel’s work resurfaced. His papers were quoted almost simultaneously by three European botanists—De Vries (Holland), Correns (Germany), and Von Tschermak (Austria)—and this marked the real beginning of medical genetics, providing an enormous impetus for the study of inherited disease. Credit for the first recognition of a single-gene trait is shared by William Bateson and Archibald Garrod, who together proposed that alkaptonuria was a rare recessive disorder. In this relatively benign condition, urine turns dark on standing or on exposure to alkali because of the patient’s inability to metabolize homogentisic acid (p. 171). Young children show skin discoloration in the napkin (diaper) area and affected adults may develop arthritis in large joints. Realizing that this was an inherited disorder involving a chemical process, Garrod coined the term inborn error of metabolism in 1908. However, his work was largely ignored until the mid-twentieth century, when the advent of electrophoresis and chromatography revolutionized biochemistry. Several hundred such disorders have now been identified, giving rise to the field of study known as biochemical genetics (see Chapter 11). The history of alkaptonuria neatly straddles almost the entire twentieth century, starting with Garrod’s original observations of recessive inheritance in 1902 and culminating in cloning of the relevant gene on chromosome 3 in 1996.
Single-Gene Disorders
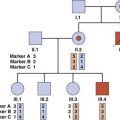
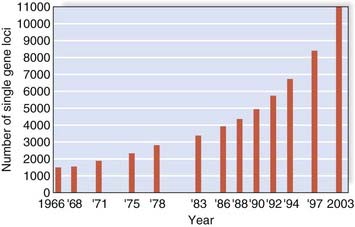
In addition to alkaptonuria, Garrod suggested that albinism and cystinuria could also show recessive inheritance. Soon other examples followed, leading to an explosion in knowledge and disease delineation. By 1966 almost 1500 single-gene disorders or traits had been identified, prompting the publication by an American physician, Victor McKusick (Figure 1.5), of a catalog of all known single-gene conditions. By 1998, when the 12th edition of this catalog was published, it contained more than 8500 entries (Figure 1.6). The growth of ‘McKusick’s Catalog’ has been exponential and is now available electronically as Online Mendelian Inheritance in Man (OMIM) (see Appendix). By 2010 OMIM contained a total of almost 20,000 entries.

Chromosome Abnormalities
Improved techniques for studying chromosomes led to the demonstration in 1959 that the presence of an additional number 21 chromosome (trisomy 21) results in Down syndrome. Other similar discoveries followed rapidly—Klinefelter and Turner syndromes—also in 1959. The identification of chromosome abnormalities was further aided by the development of banding techniques in 1970 (p. 33). These enabled reliable identification of individual chromosomes and helped confirm that loss or gain of even a very small segment of a chromosome can have devastating effects on human development (see Chapter 18).
Later it was shown that several rare conditions featuring learning difficulties and abnormal physical features are due to loss of such a tiny amount of chromosome material that no abnormality can be detected using even the most high-powered light microscope. These conditions are referred to as microdeletion syndromes (p. 280) and can be diagnosed using a technique known as FISH (fluorescent in-situ hybridization), which combines conventional chromosome analysis (cytogenetics) with newer DNA diagnostic technology (molecular genetics) (p. 34). Already, however, the latest technique of microarray CGH (comparative genomic hybridization) is revolutionizing clinical genetics through the detection of subtle genomic imbalances (p. 36).
Multifactorial Disorders
This model of quantitative inheritance is now widely accepted and has been adapted to explain the pattern of inheritance observed for many relatively common conditions (see Chapter 9). These include congenital malformations such as cleft lip and palate, and late-onset conditions such as hypertension, diabetes mellitus, and Alzheimer disease. The prevailing view is that genes at several loci interact to generate a susceptibility to the effects of adverse environmental trigger factors. Recent research has confirmed that many genes are involved in most of these adult-onset disorders, although progress in identifying specific susceptibility loci has been disappointingly slow. It has also emerged that in some conditions, such as type I diabetes mellitus, different genes can exert major or minor effects in determining susceptibility (p. 233). Overall, multifactorial or polygenic conditions are now known to make a major contribution to chronic illness in adult life (see Chapter 15).
Acquired Somatic Genetic Disease
Not all genetic errors are present from conception. Many billions of cell divisions (mitoses) occur in the course of an average human lifetime. During each mitosis, there is an opportunity for both single-gene mutations to occur, because of DNA copy errors, and for numerical chromosome abnormalities to arise as a result of errors in chromosome separation. Accumulating somatic mutations and chromosome abnormalities are now known to play a major role in causing cancer (see Chapter 14), and they probably also explain the rising incidence with age of many other serious illnesses, as well as the aging process itself. It is therefore necessary to appreciate that not all disease with a genetic basis is hereditary.
Before considering the impact of hereditary disease, it is helpful to introduce a few definitions.
Frequency
Frequency is a general term that lacks scientific specificity, although the word is often taken as being synonymous with incidence when calculating gene ‘frequencies’ (see Chapter 8).
Congenital
Congenital means that a condition is present at birth. Thus, cleft palate represents an example of a congenital malformation. Not all genetic disorders are congenital in terms of age of onset (e.g., Huntington disease), nor are all congenital abnormalities genetic in origin (e.g., fetal disruptions, as discussed in Chapter 16).
The Impact of Genetic Disease
During the twentieth century, improvements in all areas of medicine, most notably public health and therapeutics, resulted in changing patterns of disease, with increasing recognition of the role of genetic factors at all ages. For some parameters, such as perinatal mortality, the actual numbers of cases with exclusively genetic causes have probably remained constant but their relative contribution to overall figures has increased as other causes, such as infection, have declined. For other conditions, such as the chronic diseases of adult life, the overall contribution of genetics has almost certainly increased as greater life expectancy has provided more opportunity for adverse genetic and environmental interaction to manifest itself, for example in Alzheimer disease, macular degeneration, cardiomyopathy, and diabetes mellitus.
Consider the impact of genetic factors in disease at different ages from the following observations.
Spontaneous Miscarriages
A chromosome abnormality is present in 40% to 50% of all recognized first-trimester pregnancy loss. Approximately 1 in 6 of all pregnancies results in spontaneous miscarriage, thus around 5% to 7% of all recognized conceptions are chromosomally abnormal (p. 273). This value would be much higher if unrecognized pregnancies could also be included, and it is likely that a significant proportion of miscarriages with normal chromosomes do in fact have catastrophic submicroscopic genetic errors.
Newborn Infants
Of all neonates, 2% to 3% have at least one major congenital abnormality, of which at least 50% are caused exclusively or partially by genetic factors (see Chapter 16). The incidences of chromosome abnormalities and single-gene disorders in neonates are approximately 1 in 200 and 1 in 100, respectively.
Major New Developments
The study of genetics and its role in causing human disease is now widely acknowledged as being among the most exciting and influential areas of medical research. Since 1962 when Francis Crick, James Watson, and Maurice Wilkins gained acclaim for their elucidation of the structure of DNA, the Nobel Prize for Medicine and/or Physiology has been won on 22 occasions by scientists working in human and molecular genetics or related fields (Table 1.1), and for the first time in 2009 two such prizes were awarded in a single year. These pioneering studies have spawned a thriving molecular technology industry with applications as diverse as the development of genetically modified disease-resistant crops, the use of genetically engineered animals to produce therapeutic drugs, and the possible introduction of DNA-based vaccines for conditions such as malaria. Pharmaceutical companies are investing heavily in the DNA-based pharmacogenomics—drug therapy tailored to personal genetic makeup.
Table 1.1 Genetic Discoveries that Have led to the Award of the Nobel Prize for Medicine and/or Physiology and/or Chemistry, 1962–2009
| Year | Prize Winners | Discovery |
|---|---|---|
| 1962 | Francis Crick James Watson Maurice Wilkins |
The molecular structure of DNA |
| 1965 | François Jacob Jacques Monod André Lwoff |
Genetic regulation |
| 1966 | Peyton Rous | Oncogenic viruses |
| 1968 | Robert Holley Gobind Khorana Marshall Nireberg |
Deciphering of the genetic code |
| 1975 | David Baltimore Renato Dulbecco Howard Temin |
Interaction between tumor viruses and nuclear DNA |
| 1978 | Werner Arber Daniel Nathans Hamilton Smith |
Restriction endonucleases |
| 1980 | Baruj Benacerraf Jean Dausset George Snell |
Genetic control of immunologic responses |
| 1983 | Barbara McClintock | Mobile genes (transposons) |
| 1985 | Michael Brown Joseph Goldstein |
Cell receptors in familial hypercholesterolemia |
| 1987 | Susumu Tonegawa | Genetic aspects of antibodies |
| 1989 | Michael Bishop Harold Varmus |
Study of oncogenes |
| 1993 | Richard Roberts Phillip Sharp |
‘Split genes’ |
| 1995 | Edward Lewis Christiane Nüsslein-Volhard Eric Wieschaus |
Homeotic and other developmental genes |
| 1997 | Stanley Prusiner | Prions |
| 1999 | Günter Blobel | Protein transport signaling |
| 2000 | Arvid Carlsson Paul Greengard Eric Kandel |
Signal transduction in the nervous system |
| 2001 | Leland Hartwell Timothy Hunt Paul Nurse |
Regulators of the cell cycle |
| 2002 | Sydney Brenner Robert Horritz John Sulston |
Genetic regulation in development and programmed cell death (apoptosis) |
| 2006 | Andrew Fire Craig Mello |
RNA interference |
| 2007 | Mario Capecchi Martin Evans Oliver Smithies |
Gene modification by the use of embryonic stem cells |
| 2009 | Elizabeth Blackburn Carol Greider Jack Szostak |
The role of telomerase in protecting chromosome telomeres (Medicine prize) |
| Venkatraman Ramakrishnan Thomas A. Steitz Ada E. Yonath |
Structure and function of the ribosome (Chemistry prize) |
Gene Therapy
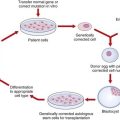
Most genetic disease is resistant to conventional treatment so that the prospect of successfully modifying the genetic code in a patient’s cells is extremely attractive. Despite major investment and extensive research, success in humans has so far been limited to a few very rare immunologic disorders. For more common conditions, such as cystic fibrosis, major problems have been encountered, such as targeting the correct cell populations, overcoming the body’s natural defense barriers, and identifying suitably non-immunogenic vectors. However, the availability of mouse models for genetic disorders, such as cystic fibrosis (p. 301), Huntington disease (p. 293), and Duchenne muscular dystrophy (p. 307), has greatly enhanced research opportunities, particularly in unraveling the cell biology of these conditions. In recent years there has been increasing optimism for novel drug therapies and stem cell treatment (p. 356), besides the prospects for gene therapy itself (p. 350).
The Internet
The availability of information in genetics has been enhanced greatly by the development of excellent online databases, and a selection of the well established is listed in the Appendix—GenBank, Ensembl, DDBJ. By 2010 there were more than a thousand molecular biology databases, so navigating this ever-growing maze can be daunting—and not just for the novice. This has developed into the exciting growth area of Bioinformatics, the science where biology, computer science, and information technology merge into a single discipline that encompasses gene maps, DNA sequences, comparative and functional genomics, and a lot more. Familiarity with interlinking databases is essential for the molecular geneticist, but is increasingly relevant for the keen clinician with an interest in genetics, who will find OMIM a good place to start for an account of all mendelian disorders, together with pertinent clinical details and extensive references. Although it is unlikely that more traditional sources of information, such as this textbook, will become completely obsolete, it is clear that only electronic technology can hope to match the explosive pace of developments in all areas of genetic research.
Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet. 1988;42:677-693.
A comprehensive study of the incidence of genetic disease in a large Western urban population.
Dunham I, Shimizu N, Roe BA, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489-495.
The first report of the complete sequencing of a human chromosome.
Emery AEH. Portraits in medical genetics—Joseph Adams 1756–1818. J Med Genet. 1989;26:116-118.
Garrod AE. The incidence of alkaptonuria: a study in chemical individuality. Lancet. 1902;ii:1916-1920.
Orel V. Gregor Mendel: the first geneticist. Oxford: Oxford University Press; 1995.
Ouellette F. Internet resources for the clinical geneticist. Clin Genet. 1999;56:179-185.
A guide to how to access some of the most useful online databases.
Shapiro R. The human blueprint: the race to unlock the secrets of our genetic script. New York: St Martin’s Press; 1991.
Watson J. The Double Helix. New York: Atheneum; 1968.
The story of the discovery of the structure of DNA, through the eyes of Watson himself.
Online Mendelian Inheritance in Man:
For literature For literature:
http://www.ncbi.nlm.nih.gov/PubMed/
www.ncbi.nlm.nih.gov/omim/GenBank
www.hgmd.cf.ac.uk (human, Cardiff)
www.ensembl.org (human, comparative, European, Cambridge)
http://genome.ucsc.edu (American browser)
Elements