[level-membership-for-basic-science-category]
CHAPTER 4 DNA Technology and Applications
In the history of medical genetics, the ‘chromosome breakthrough’ in the mid-1950s was revolutionary. In the past 4 decades, DNA technology has had a profound effect, not only in medical genetics (Figure 4.1), but also in many areas of biological science (Box 4.1).The seminal developments in the field are summarized in Table 4.1.
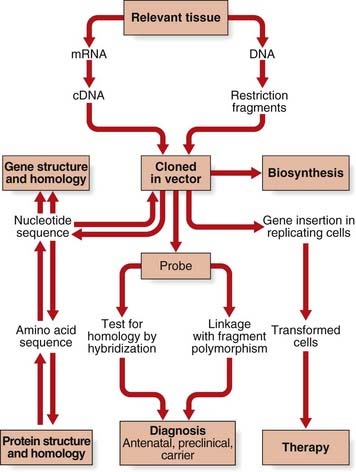
Table 4.1 Development of DNA Technology
| Decade | Development | Examples of Application |
|---|---|---|
| 1970s | Recombinant DNA technology, Southern blot, and Sanger sequencing | Recombinant erythropoietin (1987), DNA fingerprinting (1984), and DNA sequence of Epstein-Barr virus genome (1984) |
| 1980s | Polymerase chain reaction (PCR) | Diagnosis of genetic disorders |
| 1990s | Capillary sequencing and microarray technology | Draft human genome sequence (2001) |
| 2000s | Next-generation ‘clonal’ sequencing | First acute myeloid leukaemia (AML) cancer genome sequenced (2008) |
DNA technology can be split into two main areas: DNA cloning and methods of DNA analysis.
DNA Cloning
In-vivo Cell-Based DNA Cloning
There are six basic steps in in-vivo cell-based DNA cloning.
Generation of DNA Fragments
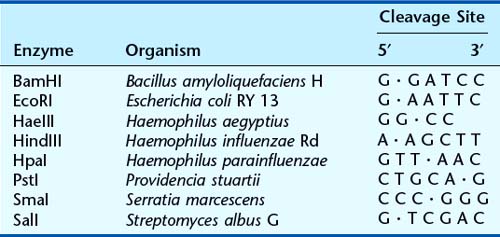
Although fragments of DNA can be produced by mechanical shearing techniques, this is a haphazard process producing fragments that vary in size. In the early 1970s, it was recognized that certain microbes contain enzymes that cleave double-stranded DNA in or near a particular sequence of nucleotides. These enzymes restrict the entry of foreign DNA into bacterial cells and were therefore called restriction enzymes. They recognize a palindromic nucleotide sequence of DNA of between four and eight nucleotides in length (i.e., the same sequence of nucleotides occurring on the two complementary DNA strands when read in one direction of polarity, e.g., 5′ to 3′) (Table 4.2). The longer the nucleotide recognition sequence of the restriction enzyme, the less frequently that particular nucleotide sequence will occur by chance and therefore the larger the average size of the DNA fragments generated.
Table 4.2 Some Examples of Restriction Endonucleases with Their Nucleotide Recognition Sequence and Cleavage Sites
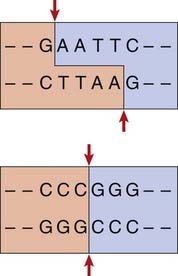
The complementary pairing of bases in the DNA molecule means that cleavage of double-stranded DNA by a restriction endonuclease always creates double-stranded breaks, which, depending on the cleavage points of the particular restriction enzyme used, results in either a staggered or a blunt end (Figure 4.2).
Vectors
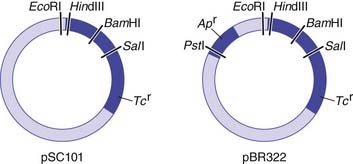
For naturally occurring vectors to be used for DNA cloning, they need to be modified to ensure that the target DNA is inserted at a specific location and that recombinant vectors containing target inserted DNA can be detected. Many of the early vectors were constructed so that insertion of the target DNA in a gene for antibiotic resistance resulted in loss of that function (Figure 4.3).
The five main types of vector commonly used include plasmids, bacteriophages, cosmids, and bacterial and yeast artificial chromosomes (BACs and YACs). The choice of vector used in cloning depends on a number of factors, such as the particular restriction enzyme being used and the size of the target DNA to be inserted. Some of the early vectors, such as plasmids and bacteriophages, were very limited in terms of the size of the target DNA fragment that could be inserted. Later generations of vectors, such as cosmids, can take inserts up to approximately 50 kb in size. A cosmid is essentially a plasmid that has had all but the minimum vector DNA necessary for propagation removed (i.e., the cos sequence), to enable insertion of the largest possible foreign DNA fragment and still allow replication.
The development of BACs and YACs allows the possibility of cloning DNA fragments of between 300 kb and 1000 kb in size. YACs consist of a plasmid that contains within it the minimum DNA sequences necessary for centromere and telomere formation plus DNA sequences known as autonomous replication sequences, all of which are necessary for accurate replication within yeast. YACs have the advantage that they can incorporate DNA fragments of up to 1000 kb in size as well as allow replication of eukaryotic DNA with repetitive DNA sequences, which often cannot take place in bacterial cells. Many eukaryotic genes are very large, being up to 2 to 3 million base pairs (bp) in length (p. 388). YACs allow detailed mapping of genes of this size and their flanking regions, whereas the use of conventional vectors would require an inordinate number of overlapping clones.
Transformation of the Host Organism
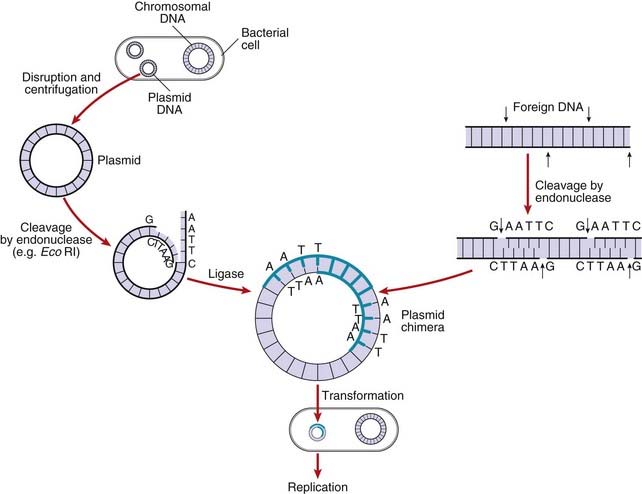
After introducing the target DNA fragment into the vector, the recombinant vector is introduced into specially modified bacterial or yeast host cells. The bacterial cell membrane is not normally permeable to large molecules such as DNA fragments but can be made permeable by a variety of different methods, including exposure to certain salts or high voltage; this is known as becoming competent. Usually only a single DNA molecule is taken up by a host cell undergoing the process known as transformation. If the transformed cells are allowed to multiply, large quantities of identical copies of the original single target DNA or clones will be produced (Figure 4.4).
Screening for Recombinant Vectors
After the transformed cells have multiplied in culture medium, they are plated out on a master plate of nutrient agar in a Petri dish. Recombinant vectors can be screened for by a detection system; for example, loss of antibiotic resistance can be screened for by replica plating on agar containing the appropriate antibiotic (see Figure 4.3). Thus, if the enzyme PstI were used to generate DNA fragments and to cut the plasmid pBR322, any recombinant plasmids produced would make the bacterial host cells they transform sensitive to ampicillin, as this gene would no longer be functional, but they would remain resistant to tetracycline. Replica plating of the master plates from the cultures allows identification of individual specific recombinant clones.
Selection of Specific Clones
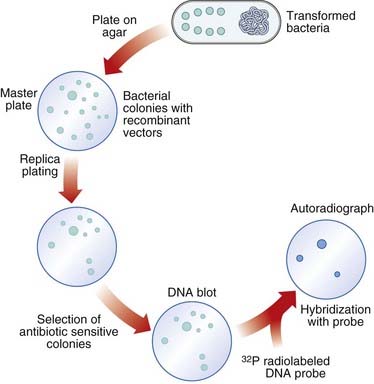
Several techniques have been developed to detect the presence of clones with specific DNA sequence inserts. The most widely used method is nucleic acid hybridization (p. 57). Colonies of transformed host bacteria with recombinant clones are used to make replica plates that are lyzed and then blotted on to a nitrocellulose filter to which nucleic acid binds. The DNA of the replica blot is then denatured to make the DNA single stranded, which will allow it to hybridize with single-stranded, radioactively labeled DNA or RNA probes (p. 58), which can then be detected by exposure to an x-ray film, or what is known as autoradiography. In this way, a transformed host bacterial colony containing a sequence complementary to the probe can be detected and, from its position on the replica plate, the colony containing that clone can be identified on the master plate, ‘picked’, and cultured separately (Figure 4.5).
DNA Libraries
Different sources of DNA can be used to make recombinant DNA molecules. DNA from nucleated cells is termed total or genomic DNA. DNA made by the action of the enzyme reverse transcriptase on messenger RNA (mRNA) is called complementary DNA or cDNA. It is possible to enrich for DNA sequences of particular interest by using a specific tissue or cell type as a source of mRNA; for instance, immature red blood cells (reticulocytes) containing predominantly globin mRNA resulted in cloning of the genes for the globin chains of hemoglobin (p. 156).
Cell-Free DNA Cloning
The PCR
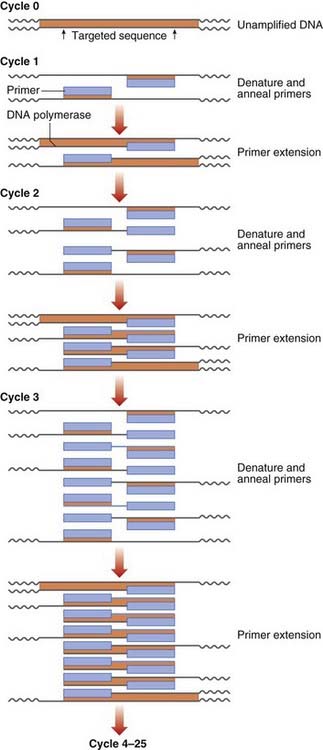
DNA sequence information is used to design two oligonucleotide primers (amplimers) of approximately 20 bp in length complementary to the DNA sequences flanking the target DNA fragment. The first step is to denature the double-stranded DNA by heating. The primers then bind to the complementary DNA sequences of the single-stranded DNA templates. DNA polymerase extends the primer DNA in the presence of the deoxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP) to synthesize the complementary DNA sequence. Subsequent heat denaturation of the double-stranded DNA, followed by annealing of the same primer sequences to the resulting single-stranded DNA, will result in the synthesis of further copies of the target DNA. Some 30 to 35 successive repeated cycles results in more than 1 million copies (amplicons) of the DNA target, sufficient for direct visualization by ultraviolet fluorescence after ethidium bromide staining, without the need to use indirect detection techniques (Figure 4.6).
PCR allows analysis of DNA from any cellular source containing nuclei; in addition to blood, this can include less invasive samples such as buccal scrapings or pathological archival material. It is also possible to start with quantities of DNA as small as that from a single cell, as is the case in preimplantation genetic diagnosis (p. 335). Great care has to be taken with PCR, however, because DNA from a contaminating extraneous source, such as desquamated skin from a laboratory worker, will also be amplified. This can lead to false-positive results unless the appropriate control studies are used to detect this possible source of error.
Techniques of DNA Analysis
Nucleic Acid Probes
Nucleic acid probes are usually single-stranded DNA sequences that have been radioactively or non-radioactively labeled and can be used to detect DNA or RNA fragments with sequence homology. DNA probes can come from a variety of sources, including random genomic DNA sequences, specific genes, cDNA sequences or oligonucleotide DNA sequences produced synthetically based on knowledge of the protein amino-acid sequence. A DNA probe can be labeled by a variety of processes, including isotopic labeling with 32P and non-isotopic methods using modified nucleotides containing fluorophores (e.g., fluorescein or rhodamine). Hybridization of a radioactively labeled DNA probe with cDNA sequences on a nitrocellulose filter can be detected by autoradiography, whereas DNA fragments that are fluorescently labeled can be detected by exposure to the appropriate wavelength of light, for example fluorescent in-situ hybridization (p. 34).
Nucleic Acid Hybridization
Southern Blotting
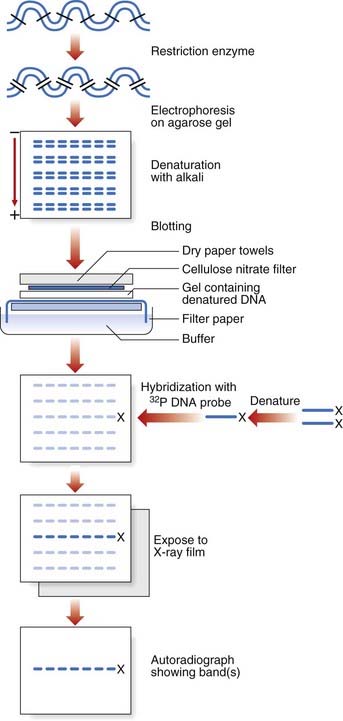
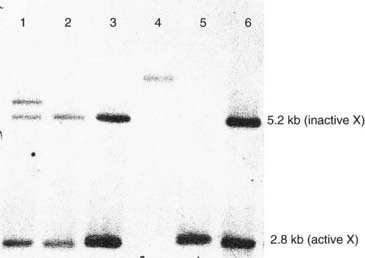
Southern blotting, named after Edwin Southern (who developed the technique), involves digesting DNA by a restriction enzyme that is then subjected to electrophoresis on an agarose gel. This separates the DNA or restriction fragments by size, the smaller fragments migrating faster than the larger ones. The DNA fragments in the gel are then denaturated with alkali, making them single stranded. A ‘permanent’ copy of these single-stranded fragments is made by transferring them on to a nitrocellulose filter that binds the single-stranded DNA, the so-called Southern blot. A particular target DNA fragment of interest from the collection on the filter can be visualized by adding a single-stranded 32P radioactively labeled DNA probe that will hybridize with homologous DNA fragments in the Southern blot, which can then be detected by autoradiography (Figure 4.7). Non-radioactive Southern blotting techniques have been developed with the DNA probe labeled with digoxigenin and detected by chemiluminescence. This approach is safer and generates results more rapidly. An example of the use of Southern blotting for diagnostic fragile X testing in patients is shown in Figure 4.8.
DNA Microarrays
DNA microarrays are based on the same principle of hybridization but on a miniaturized scale, which allows simultaneous analysis of several million targets. Short, fluorescently labeled oligonucleotides attached to a glass microscope slide can be used to detect hybridization of target DNA under appropriate conditions. The color pattern of the microarray is then analyzed automatically by computer. Four classes of application have been described: (1) expression studies to look at the differential expression of thousands of genes at the mRNA level; (2) analysis of DNA variation for mutation detection and single nucleotide polymorphism (SNP) typing (p. 67); (3) testing for genomic gains and losses by array comparative genomic hybridization (CGH) (p. 36); and (4) a combination of the latter two, SNP–CGH, which allows the detection of copy-neutral genetic anomalies such as uniparental disomy (p. 121).
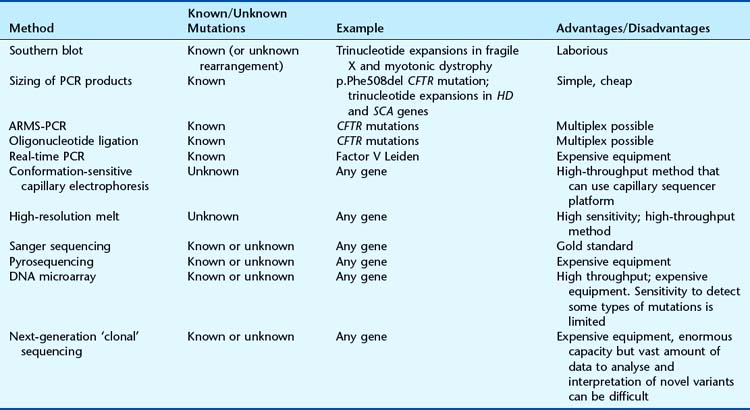
Mutation Detection
The choice of method depends primarily on whether the test is for a known sequence change or to identify the presence of any mutation within a particular gene. A number of techniques can be used to screen for mutations that differ in their ease of use and reliability. The choice of assay depends on many factors, including the sensitivity required, cost, equipment, and the size and structure (including number of polymorphisms) of the gene (Table 4.3). Identification of a possible sequence variant by one of the mutation screening methods requires confirmation by DNA sequencing. Some of the most common techniques in current use are described in the following section.
Size Analysis of PCR Products
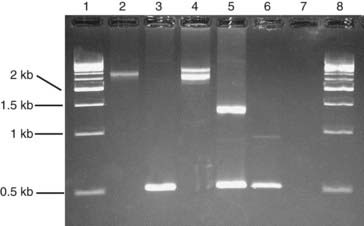
Deletion or insertion mutations can sometimes be detected simply by determining the size of a PCR product. For example, the most common mutation that causes cystic fibrosis, p.Phe508del, is a 3-bp deletion that can be detected on a polyacrylamide gel. Some trinucleotide repeat expansion mutations can be amplified by PCR (Figure 4.9).
Restriction Fragment Length Polymorphism
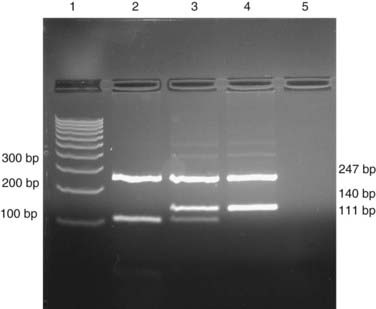
If a base substitution creates or abolishes the recognition site of a restriction enzyme, it is possible to test for the mutation by digesting a PCR product with the appropriate enzyme and separating the products by electrophoresis (Figure 4.10).
Amplification-Refractory Mutation System (ARMS) PCR
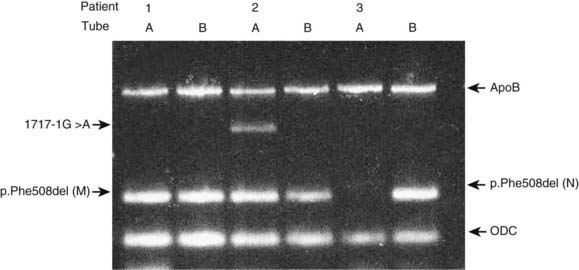
Allele-specific PCR uses primers specific for the normal and mutant sequences. The most common design is a two-tube assay with normal and mutant primers in separate reactions together with control primers to ensure that the PCR reaction has worked. An example of a multiplex ARMS assay to detect 12 different cystic fibrosis mutations is shown in Figure 4.11.
Oligonucleotide Ligation Assay
A pair of oligonucleotides is designed to anneal to adjacent sequences within a PCR product. If the pair is perfectly hybridized, they can be joined by DNA ligase. Oligonucleotides complementary to the normal and mutant sequences are differentially labeled and the products identified by computer software (Figure 4.12).
Real-Time PCR
There are multiple hardware platforms for real-time PCR and ‘fast’ versions that can complete a PCR reaction in less than 30 minutes. TaqManTM and LightCyclerTM use fluorescence technology to detect mutations by allelic discrimination of PCR products. Figure 4.13 illustrates the factor V Leiden mutation detected by TaqManTM methodology.
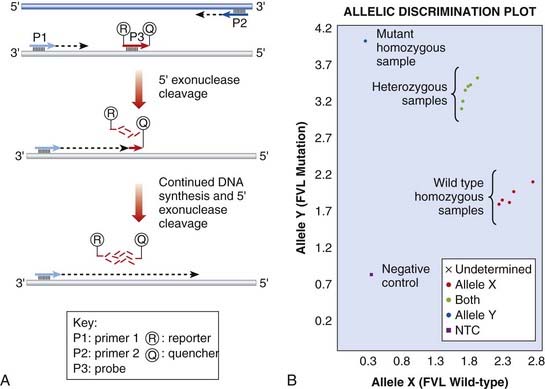
FIGURE 4.13 Real-time polymerase chain reaction (PCR) to detect the Factor V Leiden mutation. A, TaqMan technique. The sequence encompassing the mutation is amplified by PCR primers, P1 and P2. A probe, P3, specific to the mutation is labelled with two fluorophores. A reporter fluorophore, R, is attached to the 5′ end of the probe and a quencher fluorophore, Q, is attached to the 3′ end. During the PCR reaction, the 5′ exonuclease activity of the polymerase enzyme progressively degrades the probe, separating the reporter and quencher dyes, which results in fluorescent signal from the reporter fluorophore. B, TaqMan genotyping plot. Each sample is analysed with two probes, one specific for the wild-type and one for the mutation. The strength of fluorescence from each probe is plotted on a graph (wild-type on X-axis, mutant on Y-axis). Each sample is represented by a single point. The samples fall into 3 clusters representing the possible genotypes; homozygous wild-type, homozygous mutant or heterozygous.
(Courtesy Dr. E. Young, Department of Molecular Genetics, Royal Devon and Exeter Hospital, Exeter, UK.)
DNA Microarrays (DNA ‘Chips’)
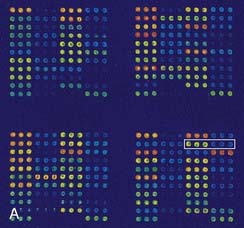
DNA microarrays hold the promise of rapid mutation testing. They involve synthesizing custom-designed 20 bp to 25 bp oligonucleotide sequences for both the normal DNA sequence and known and/or possible single nucleotide substitutions of a gene. These are attached to a ‘chip’ in a structured arrangement in what is known as a microarray. The sample DNA being screened for a mutation is amplified by PCR, fluorescently labeled, and hybridized with the oligonucleotides in the microarray (Figure 4.14). Computer analysis of the color pattern of the microarray generated after hybridization allows rapid automated mutation testing. The prospect of gene-specific DNA chip microarrays may lead to a revolution in the speed and reliability of mutation screening, provided the technology is affordable and the technique can be demonstrated to be robust. The detection of known base substitutions and SNPs has been very successful, but screening for insertion mutations is more limited.

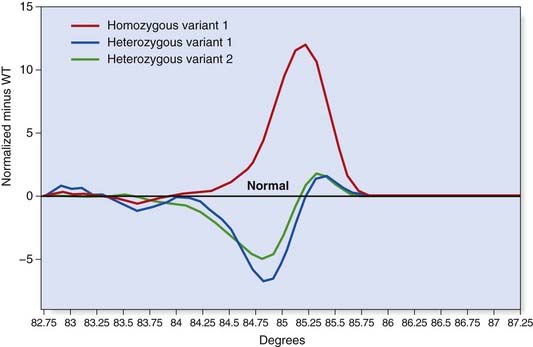
High-Resolution Melt Curve Analysis
This technique employs a class of fluorescent dyes that intercalate with double-stranded, but not single-stranded, DNA. The intercalating dye is incorporated in the PCR reaction and the products are then heated to separate the two strands. Fluorescence levels decrease as the DNA strands dissociate and this ‘melting’ profile depends on the PCR product size and sequence (Figure 4.15). High-resolution melt curve analysis appears to be very sensitive and can be used for high-throughput mutation screening.
Sanger Sequencing
The ‘gold standard’ method of mutation screening is DNA sequencing using the dideoxy chain termination method developed in the 1970s by Fred Sanger. This method originally employed radioactive labeling with manual interpretation of data. The use of fluorescent labels detected by computerized laser systems has improved ease of use and increased throughput and accuracy. Today’s capillary sequencers can sequence around 1 Mb (1 million bases) per day.
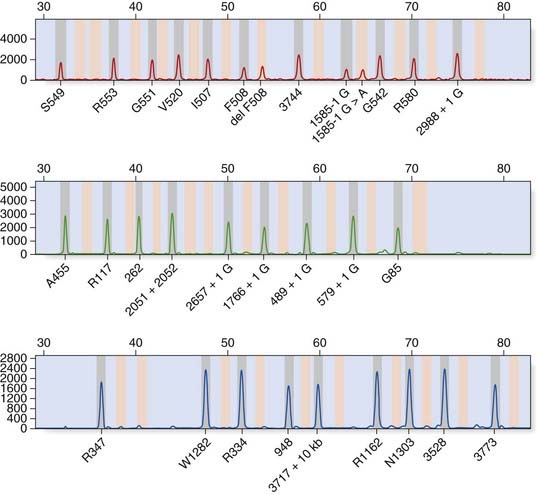
Dideoxy sequencing involves using a single-stranded DNA template (e.g., denatured PCR products) to synthesize new complementary strands using a DNA polymerase and an appropriate oligonucleotide primer. In addition to the four normal deoxynucleotides, a proportion of each of the four respective dideoxynucleotides is included, each labeled with a different fluorescent dye. The dideoxynucleotides lack a hydroxyl group at the 3′ carbon position; this prevents phosphodiester bonding, resulting in each reaction container consisting of a mixture of DNA fragments of different lengths that terminate in their respective dideoxynucleotide, owing to chain termination occurring at random in each reaction mixture at the respective nucleotide. When the reaction products are separated by capillary electrophoresis, a ladder of DNA sequences of differing lengths is produced. The DNA sequence complementary to the single-stranded DNA template is generated by the computer software and the position of a mutation may be highlighted with an appropriate software package (Figure 4.16).
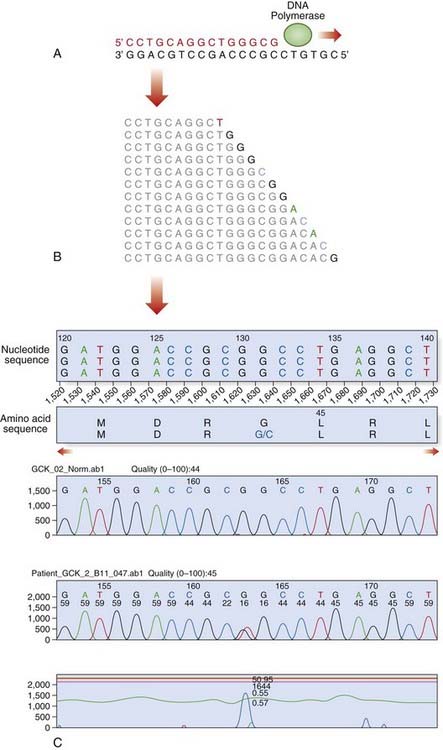
FIGURE 4.16 Fluorescent dideoxy DNA sequencing. The sequencing primer (shown in red) binds to the template and primes synthesis of a complementary DNA strand in the direction indicated (A). The sequencing reaction includes four dNTPs and four ddNTPs, each labeled with a different fluorescent dye. Competition between the dNTPs and ddNTPs results in the production of a collection of fragments (B), which are then separated by electrophoresis to generate an electropherogram (C). A heterozygous mutation, p.Gly44Cys (GGC > TGC; glycine > cysteine), is identified by the software.
Pyrosequencing
Pyrosequencing uses a sequencing by synthesis approach in which modified nucleotides are added and removed one at a time, with chemiluminescent signals produced after the addition of each nucleotide. This technology generates quantitative sequence data rapidly and an example of its application in the identification of KRAS mutations in patients with colorectal cancer is shown in Figure 4.17.
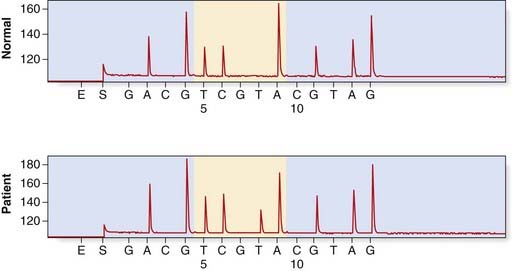
FIGURE 4.17 Detection of a KRAS mutation in a colorectal tumour by pyrosequencing. The upper panel shows a normal control, sequence A GGT CAA GAG G. In the lower panel is the tumour sample with the KRAS mutation p.Gln61Leu (c.182A > T).
(Courtesy Dr. L. Meredith, Institute of Medical Genetics, University Hospital of Wales, Cardiff.)
Next-Generation ‘Clonal’ Sequencing
The demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that produce millions of sequences at once. Next (or second) generation ‘clonal’ sequencers use an in vitro cloning step to amplify individual DNA molecules by emulsion or bridge PCR (Figure 4.18). The cloned DNA molecules are then sequenced in parallel, either by pyrosequencing, by using reversible terminators or with a sequencing by ligation approach. A comparison with Sanger sequencing is shown in Table 4.5 and an example of a mutation identified by next generation sequencing is shown in Figure 4.19. So-called ‘third generation’ sequencers have recently been developed. They can generate massively parallel sequence data from single molecules due to their extremely sensitive lasers.
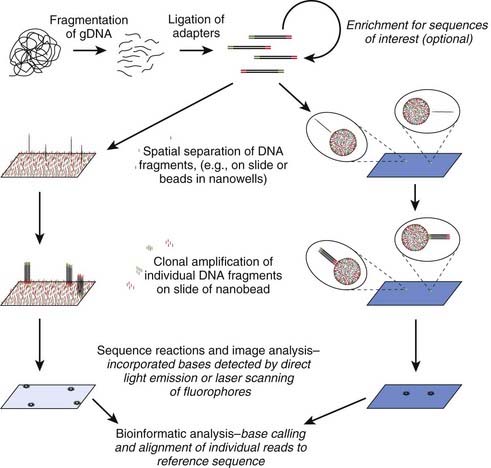

Table 4.5 Sanger Sequencing Compared to Next-Generation ‘Clonal’ Sequencing
| Sanger Sequencing | Next-Generation ‘Clonal’ Sequencing |
|---|---|
| One sequence read per sample | Massively parallel sequencing |
| 500–1000 bases per read | 100–400 bases per read |
| ∼1 million bases per day per machine | ∼2 billion bases per day per machine |
| ∼£1 per 1000 bases | ∼£0.02 per 1000 bases |

Dosage Analysis
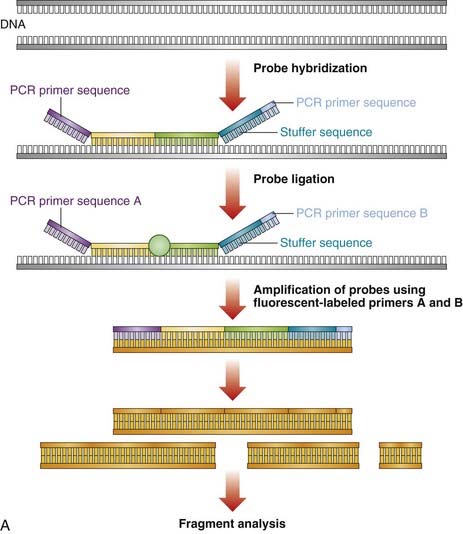
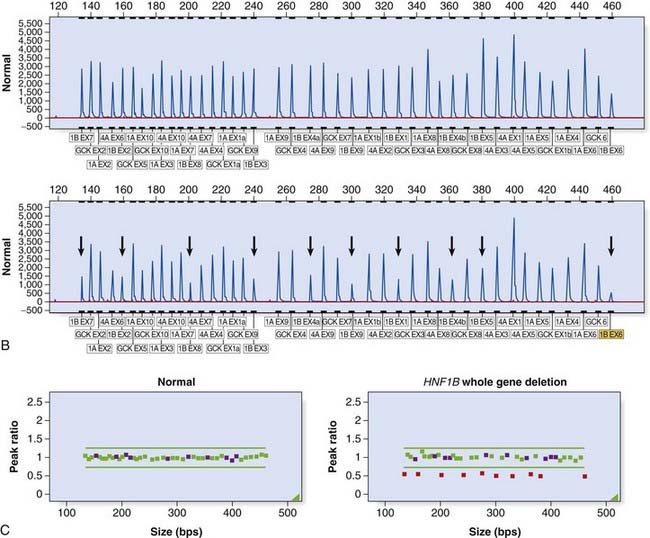
Large deletion and duplication mutations have been reported in a number of disorders and may encompass a single exon, several exons, or an entire gene (e.g., HNPP [p. 297]; HMSN type 1 [p. 296]). Several techniques have been developed to identify such mutations (see Table 4.4). Multiplex ligation-dependent probe amplification (MLPA) is a high-resolution method used to detect deletions and duplications (Figure 4.20). Each MLPA probe consists of two fluorescently labeled oligonucleotides that can hybridize, adjacent to each other, to a target gene sequence. When hybridized, the two oligonucleotides are joined by a ligase and the probe is then amplified by PCR (each oligonucleotide includes a universal primer sequence at its terminus). The probes include a variable-length stuffer sequence that enables separation of the PCR products by capillary electrophoresis. Up to 40 probes can be amplified in a single reaction.
Dosage analysis by quantitative fluorescent PCR (QF-PCR) is routinely used for rapid aneuploidy screening; for example, in prenatal diagnosis (p. 325). Microsatellites (see the following section) located on chromosomes 13, 18, and 21 may be amplified within a multiplex and trisomies detected, either by the presence of three alleles or by a dosage effect where one allele is overrepresented (Figure 4.21).
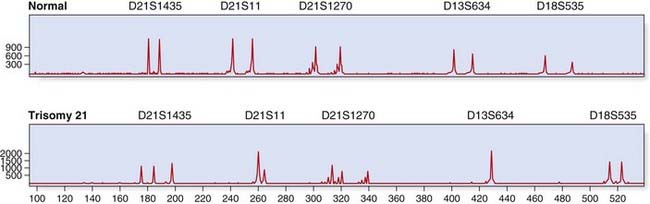
FIGURE 4.21 Quantitative fluorescent (QF)-polymerase chain reaction (PCR) for rapid prenatal aneuploidy testing. The upper panel shows a normal control, with two alleles for each microsatellite marker. The lower panel illustrates trisomy 21 with either three alleles (microsatellites D21S1435, D21S1270) or a dosage effect (D21S11). Microsatellite markers for chromosomes 13 and 18 show a normal profile.
(Courtesy Chris Anderson, Institute of Medical Genetics, University Hospital of Wales, Cardiff, UK.)
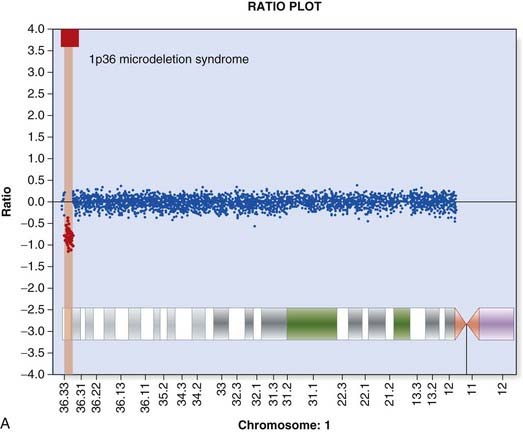
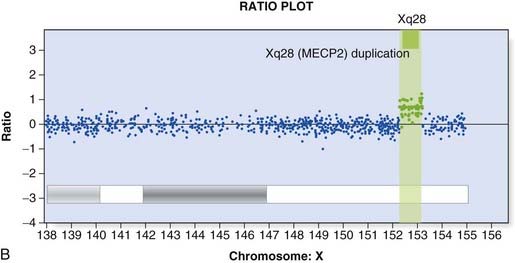
Array CGH was introduced in Chapter 3 (p. 36) and provides a way to detect deletions and duplications on a genome-wide scale (Figure 4.22). Arrays used in clinical diagnostic laboratories include both genome wide probes to detect novel mutations and probes targeted to known deletion/duplication syndromes. A comprehensive knowledge of normal copy number variation is essential for interpreting novel mutations.
Application of DNA Sequence Polymorphisms
There is an enormous amount of DNA sequence variation in the human genome (p. 13). Two main types, SNPs and hypervariable tandem repeat DNA length polymorphisms, are predominantly used in genetic analysis.
Single Nucleotide Polymorphisms
Around 1 in 1000 bases within the human genome shows variation. SNPs are most frequently biallelic and occur in coding and non-coding regions. If an SNP lies within the recognition sequence of a restriction enzyme, the DNA fragments produced by that restriction enzyme will be of different lengths in different people. This can be recognized by the altered mobility of the restriction fragments on gel electrophoresis, so-called restriction fragment length polymorphisms, or RFLPs. Early genetic mapping studies used Southern blotting to detect RFLPs, but current technology enables the detection of any SNP. DNA microarrays have led to the creation of a dense SNP map of the human genome and assist genome searches for linkage studies in mapping single-gene disorders (p. 293) and association studies in common diseases.
Variable Number Tandem Repeats
Variable number tandem repeats (VNTRs) are highly polymorphic and are due to the presence of variable numbers of tandem repeats of a short DNA sequence that have been shown to be inherited in a mendelian co-dominant fashion (p. 113). The advantage of using VNTRs over SNPs is the large number of alleles for each VNTR compared with SNPs, which are mostly biallelic.
Minisatellites
Alec Jeffreys identified a short 10-bp to 15-bp ‘core’ sequence with homology to many highly variable loci spread throughout the human genome (p. 17). Using a probe containing tandem repeats of this core sequence, a pattern of hypervariable DNA fragments could be identified. The multiple variable-size repeat sequences identified by the core sequence are known as minisatellites. These minisatellites are highly polymorphic, and a profile unique to an individual (unless they have an identical twin!) is described as a DNA fingerprint. The technique of DNA fingerprinting is used widely in paternity testing and for forensic purposes.
Microsatellites
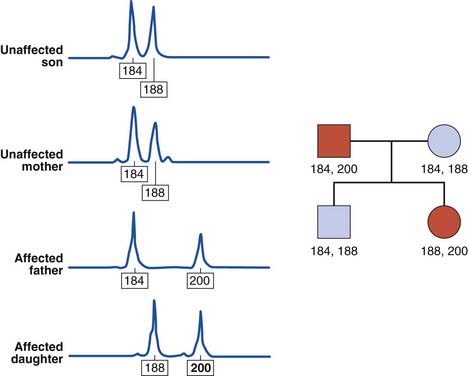
The human genome contains some 50,000 to 100,000 blocks of a variable number of tandem repeats of the dinucleotide CA:GT, so-called CA repeats or microsatellites (p. 18). The difference in the number of CA repeats at any one site between individuals is highly polymorphic and these repeats have been shown to be inherited in a mendelian co-dominant manner. In addition, highly polymorphic trinucleotide and tetranucleotide repeats have been identified, and can be used in a similar way (Figure 4.23). These microsatellites can be analyzed by PCR and the use of fluorescent detection systems allows relatively high-throughput analysis. Consequently, microsatellite analysis has replaced DNA fingerprinting for paternity testing and establishing zygosity.
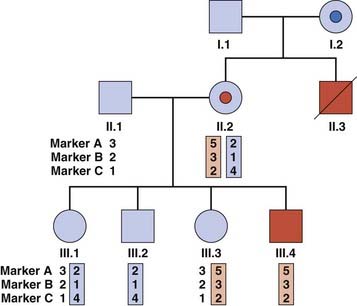
Clinical Applications of Gene Tracking
If a gene has been mapped by linkage studies but not identified, it is possible to use the linked markers to ‘track’ the mutant haplotype within a family. This approach may also be used for known genes where a familial mutation has not been found. Closely flanking or intragenic microsatellites are used most commonly, because of the lower likelihood of finding informative SNPs within families. Figure 4.24 illustrates a family in which gene tracking has been used to determine carrier risk in the absence of a known mutation. There are some pitfalls associated with this method: recombination between the microsatellite and the gene may give an incorrect risk estimate, and the possibility of genetic heterogeneity (where mutations in more than one gene cause a disease) should be borne in mind.
Elles R, Wallace A. Molecular diagnosis of genetic disease, 3rd ed. Clifton, NJ: Humana Press; 2010.
Key techniques used for genetic testing of common disorders in diagnostic laboratories.
Strachan T, Read AP. Human molecular genetics, 4th ed. London: Garland Science; 2011.
Weatherall DJ. The new genetics and clinical practice, 3rd ed. Oxford: Oxford Medical; 1991.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 4 DNA Technology and Applications
In the history of medical genetics, the ‘chromosome breakthrough’ in the mid-1950s was revolutionary. In the past 4 decades, DNA technology has had a profound effect, not only in medical genetics (Figure 4.1), but also in many areas of biological science (Box 4.1).The seminal developments in the field are summarized in Table 4.1.
Table 4.1 Development of DNA Technology
| Decade | Development | Examples of Application |
|---|---|---|
| 1970s | Recombinant DNA technology, Southern blot, and Sanger sequencing | Recombinant erythropoietin (1987), DNA fingerprinting (1984), and DNA sequence of Epstein-Barr virus genome (1984) |
| 1980s | Polymerase chain reaction (PCR) | Diagnosis of genetic disorders |
| 1990s | Capillary sequencing and microarray technology | Draft human genome sequence (2001) |
| 2000s | Next-generation ‘clonal’ sequencing | First acute myeloid leukaemia (AML) cancer genome sequenced (2008) |
DNA technology can be split into two main areas: DNA cloning and methods of DNA analysis.
DNA Cloning
In-vivo Cell-Based DNA Cloning
There are six basic steps in in-vivo cell-based DNA cloning.
Generation of DNA Fragments
Although fragments of DNA can be produced by mechanical shearing techniques, this is a haphazard process producing fragments that vary in size. In the early 1970s, it was recognized that certain microbes contain enzymes that cleave double-stranded DNA in or near a particular sequence of nucleotides. These enzymes restrict the entry of foreign DNA into bacterial cells and were therefore called restriction enzymes. They recognize a palindromic nucleotide sequence of DNA of between four and eight nucleotides in length (i.e., the same sequence of nucleotides occurring on the two complementary DNA strands when read in one direction of polarity, e.g., 5′ to 3′) (Table 4.2). The longer the nucleotide recognition sequence of the restriction enzyme, the less frequently that particular nucleotide sequence will occur by chance and therefore the larger the average size of the DNA fragments generated.
Table 4.2 Some Examples of Restriction Endonucleases with Their Nucleotide Recognition Sequence and Cleavage Sites
The complementary pairing of bases in the DNA molecule means that cleavage of double-stranded DNA by a restriction endonuclease always creates double-stranded breaks, which, depending on the cleavage points of the particular restriction enzyme used, results in either a staggered or a blunt end (Figure 4.2).
Vectors
For naturally occurring vectors to be used for DNA cloning, they need to be modified to ensure that the target DNA is inserted at a specific location and that recombinant vectors containing target inserted DNA can be detected. Many of the early vectors were constructed so that insertion of the target DNA in a gene for antibiotic resistance resulted in loss of that function (Figure 4.3).
The five main types of vector commonly used include plasmids, bacteriophages, cosmids, and bacterial and yeast artificial chromosomes (BACs and YACs). The choice of vector used in cloning depends on a number of factors, such as the particular restriction enzyme being used and the size of the target DNA to be inserted. Some of the early vectors, such as plasmids and bacteriophages, were very limited in terms of the size of the target DNA fragment that could be inserted. Later generations of vectors, such as cosmids, can take inserts up to approximately 50 kb in size. A cosmid is essentially a plasmid that has had all but the minimum vector DNA necessary for propagation removed (i.e., the cos sequence), to enable insertion of the largest possible foreign DNA fragment and still allow replication.
The development of BACs and YACs allows the possibility of cloning DNA fragments of between 300 kb and 1000 kb in size. YACs consist of a plasmid that contains within it the minimum DNA sequences necessary for centromere and telomere formation plus DNA sequences known as autonomous replication sequences, all of which are necessary for accurate replication within yeast. YACs have the advantage that they can incorporate DNA fragments of up to 1000 kb in size as well as allow replication of eukaryotic DNA with repetitive DNA sequences, which often cannot take place in bacterial cells. Many eukaryotic genes are very large, being up to 2 to 3 million base pairs (bp) in length (p. 388). YACs allow detailed mapping of genes of this size and their flanking regions, whereas the use of conventional vectors would require an inordinate number of overlapping clones.
Transformation of the Host Organism
After introducing the target DNA fragment into the vector, the recombinant vector is introduced into specially modified bacterial or yeast host cells. The bacterial cell membrane is not normally permeable to large molecules such as DNA fragments but can be made permeable by a variety of different methods, including exposure to certain salts or high voltage; this is known as becoming competent. Usually only a single DNA molecule is taken up by a host cell undergoing the process known as transformation. If the transformed cells are allowed to multiply, large quantities of identical copies of the original single target DNA or clones will be produced (Figure 4.4).
Screening for Recombinant Vectors
After the transformed cells have multiplied in culture medium, they are plated out on a master plate of nutrient agar in a Petri dish. Recombinant vectors can be screened for by a detection system; for example, loss of antibiotic resistance can be screened for by replica plating on agar containing the appropriate antibiotic (see Figure 4.3). Thus, if the enzyme PstI were used to generate DNA fragments and to cut the plasmid pBR322, any recombinant plasmids produced would make the bacterial host cells they transform sensitive to ampicillin, as this gene would no longer be functional, but they would remain resistant to tetracycline. Replica plating of the master plates from the cultures allows identification of individual specific recombinant clones.
Selection of Specific Clones
Several techniques have been developed to detect the presence of clones with specific DNA sequence inserts. The most widely used method is nucleic acid hybridization (p. 57). Colonies of transformed host bacteria with recombinant clones are used to make replica plates that are lyzed and then blotted on to a nitrocellulose filter to which nucleic acid binds. The DNA of the replica blot is then denatured to make the DNA single stranded, which will allow it to hybridize with single-stranded, radioactively labeled DNA or RNA probes (p. 58), which can then be detected by exposure to an x-ray film, or what is known as autoradiography. In this way, a transformed host bacterial colony containing a sequence complementary to the probe can be detected and, from its position on the replica plate, the colony containing that clone can be identified on the master plate, ‘picked’, and cultured separately (Figure 4.5).
DNA Libraries
Different sources of DNA can be used to make recombinant DNA molecules. DNA from nucleated cells is termed total or genomic DNA. DNA made by the action of the enzyme reverse transcriptase on messenger RNA (mRNA) is called complementary DNA or cDNA. It is possible to enrich for DNA sequences of particular interest by using a specific tissue or cell type as a source of mRNA; for instance, immature red blood cells (reticulocytes) containing predominantly globin mRNA resulted in cloning of the genes for the globin chains of hemoglobin (p. 156).
Cell-Free DNA Cloning
The PCR
DNA sequence information is used to design two oligonucleotide primers (amplimers) of approximately 20 bp in length complementary to the DNA sequences flanking the target DNA fragment. The first step is to denature the double-stranded DNA by heating. The primers then bind to the complementary DNA sequences of the single-stranded DNA templates. DNA polymerase extends the primer DNA in the presence of the deoxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP) to synthesize the complementary DNA sequence. Subsequent heat denaturation of the double-stranded DNA, followed by annealing of the same primer sequences to the resulting single-stranded DNA, will result in the synthesis of further copies of the target DNA. Some 30 to 35 successive repeated cycles results in more than 1 million copies (amplicons) of the DNA target, sufficient for direct visualization by ultraviolet fluorescence after ethidium bromide staining, without the need to use indirect detection techniques (Figure 4.6).
PCR allows analysis of DNA from any cellular source containing nuclei; in addition to blood, this can include less invasive samples such as buccal scrapings or pathological archival material. It is also possible to start with quantities of DNA as small as that from a single cell, as is the case in preimplantation genetic diagnosis (p. 335). Great care has to be taken with PCR, however, because DNA from a contaminating extraneous source, such as desquamated skin from a laboratory worker, will also be amplified. This can lead to false-positive results unless the appropriate control studies are used to detect this possible source of error.
