[level-membership-for-neurosurgery-category]
CHAPTER 102 The Genetic Origins of Brain Tumors
There are external causes that significantly increase the risk for development of a cancer, but for brain tumors the evidence for this is limited. A high dose of ionizing radiation to the brain has the strongest evidence for increasing the risk for certain brain tumors, as seen with radiation therapy involving the brain1 or increases in meningiomas in atomic bomb survivors.2,3 Environmental causes of brain tumor are not as evident. The main carcinogen in our society, tobacco, does not have evidence of producing risk for brain tumor as with many other cancers. Certain synthetic chemicals, such as fungicides and pesticides, show an association of exposure to increased incidence,4,5 but more research is needed to demonstrate a direct cause-and-effect relationship. Overall, it can be said that the vast majority of mutations that give rise to brain cancers are spontaneous, and except for family members with certain rare cancer syndromes or those who have previously undergone brain irradiation, it is not possible to accurately predict who in our society is at greater risk for a brain tumor.
There are more than 120 different pathologic classes of brain tumor.6 For some tumors, in particular glioblastoma multiforme (GBM), many of the mutations have now been identified, but little is known about the genetic basis of most brain tumors. It is clear, however, that the number and complexity of mutations arising during malignant tumor development, including brain cancers, are much greater than originally predicted. The field of cancer genomics, or oncogenomics, has advanced rapidly, and thousands of genes in a cancer genome are sequenced in a single project, as opposed to the time when only one candidate gene at a time could be evaluated. The current trend is to build comprehensive databases of cancer mutations for the common cancers, and as the cost of DNA sequencing decreases, such data will probably be generated for more types of brain tumors.
In this chapter the well-documented and commonly altered genes are reviewed for the few classes of brain tumor for which we have knowledge of their mutational basis. This does not imply by any means that we have a complete picture of the various gene mutation patterns for each cancer. In GBM, for example, an average of more than 60 acquired mutations can be observed per genome.7 Fortunately, these mutations cluster in a small number of molecular pathways that are altered to give rise to a cancer. Understanding the function and mechanisms of these pathways will produce better insight into how tumors proliferate and thus provide researchers with better means for molecular targeting.
Clonal Expansion of Malignant Tumors
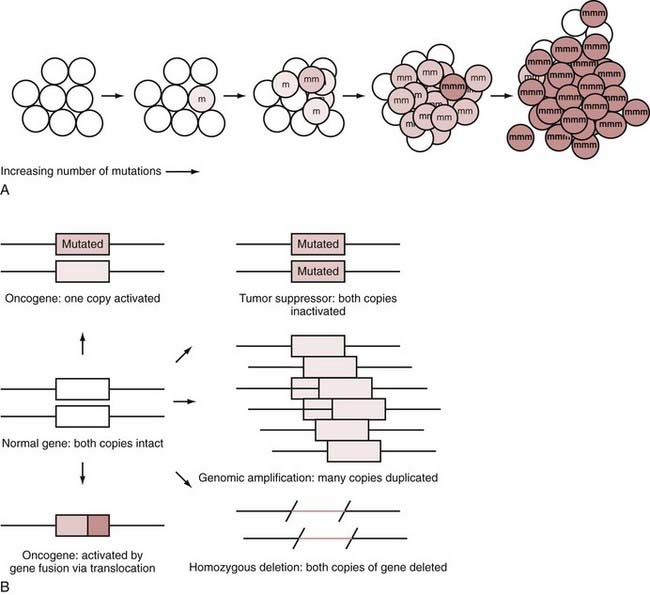
A fundamental concept in cancer is the clonal expansion of tumors, with each step in tumor formation based on adding another mutation to the tumor genome. Figure 102-1A illustrates this concept. The process starts with a single mutation in a single cell. As mutations in critical genes accumulate in a cell, a cancer develops in stepwise fashion. In brain cancers, it is not clear how long it takes for mutations to accumulate and in what order all the mutations might occur because the early stages of the tumors are not easily observed during tumorigenesis. It appears, however, that many mutations and changes in copy number, in any one of many different combinations, accumulate to form a GBM.7 Because this clonal expansion presumably arises from a single cell, all the cells in the tumor should have the same mutation. This is normally true, except in a few situations, such as the presence of unstable amplifications that give rise to heterogeneity for this change in the tumor or the acquisition of a late-occurring mutation in the latter stages of tumor cell clonal expansion. For the vast majority of mutations, however, the same mutations will appear in all parts of the tumor.
Inherited Mutations and Familial Syndromes
Because the first hit is inherited as a germline mutation, it can be transmitted to offspring. Although most malignant tumors observed in the clinic do not have an evident hereditary basis, it is important to recognize the possibility of familial clustering of brain tumors and consider genetic counseling and further evaluation if evident. Table 102-1 lists some of the more common cancer-associated mendelian disorders that have brain tumors as part of the phenotype. Mendelian disorders are genetic diseases that have a clear pattern of inheritance within families, such as dominant or recessive inheritance. Most of the syndromes that involve brain tumors have an autosomal dominant mendelian inheritance pattern. A complete catalogue and literature review of all the mendelian disorders identified is readily accessible at the Online Mendelian Inheritance in Man (OMIM) website.8

The mismatch repair (MMR) cancer syndrome is an important syndrome that involves several different types of cancers, including some GBMs, meningiomas, and medulloblastomas.8,11,12 It has an autosomal dominant pattern of inheritance, as do all the syndromes listed in Table 102-1. There are several DNA repair enzymes that are necessary for correct DNA MMR in a normal cell. In MMR cancer syndrome, an inactivating mutation or deletion of the MLH1, MSH2, MSH6, or PMS genes reduces the ability of the cell to identify and correctly repair DNA mismatches that occur during DNA replication. Therefore, a mutation in any of these genes can lead to an increased risk for brain and other cancers. The mutations in these “mutator genes” increase the cell’s mutation rate and accelerates the acquisition of mutations elsewhere in the genome, thus leading to a significantly increased risk for the development of cancer at an early age.
Different Types of DNA Mutations and Alterations
DNA alterations can range from single base pair changes all the way to entire chromosome gains or losses, as well as any size change in between. There does not seem to be any restriction to the DNA sequence changes that can occur during the development of cancer. In addition to DNA sequence changes, it also appears that epigenetic changes can contribute to tumor progression by altering gene expression without altering the nucleotide sequence.17
Some of the basic concepts of cancer genes, such as gene amplification, oncogenes, and tumor suppressors, are diagrammed in Figure 102-1B.
The Spectrum of Mutations that Underlie Brain Cancers
In a recent comprehensive genomic analysis of 22 GBM genomes, in excess of 20,000 genes were sequenced for acquired mutations and assayed for changes in copy number.7 More than 40 genes had acquired genetic changes that occurred at a statistically significant rate and very likely contributed to the development of GBM. The mutations occurred in many various combinations. In GBMs the mutations include not only the frequently mutated and well-known genes such as EGFR and p53 but also a complex mixture of lesser known genes that are mutated in only a small percentage of cancers. This mixture of a few highly mutated genes plus many more low-frequency mutations was also observed in colon and breast cancers,18,19 thus suggesting that it is a common theme in malignancies and will be observed in other brain cancers besides GBM.
Glioblastomas
About 5% to 10% of GBMs arise from grade II or III tumors.20,21 The secondary GBMs as a group, not surprisingly, have a different spectrum of mutations. Secondary GBMs have a higher p53 mutation frequency (65% versus 28%), lower frequency of PTEN mutations (4% versus 25%), and lower rate of EGFR amplification (8% versus 36%) than do primary GBMs.22 It also appears that the types of p53 mutations differ between primary and secondary GBMs,20 thus suggesting a different mode of mutagenesis.
Recent oncogenomic studies have resulted in the GBM genome being one of the best characterized for mutations.7,23 One study sequenced the coding region of 20,661 genes and the copy number changes in 22 glioblastoma genomes followed by sequencing of an additional 83 samples for mutated genes.7 The integrated analysis revealed that in GBMs an average of 60 acquired genomic changes could be observed per cancer genome. Not all these mutations are causative, and some fraction probably consists of passenger mutations that do not contribute to the development of cancer. However, an individual tumor still has many alterations that contribute to tumor development, in contrast to previous predictions.
In the screening of 20,661 genes, isocitrate dehydrogenase 1 (IDH1) was found to be mutated in 11% of tumors.7 These mutations were found in nearly all secondary GBMs (those that progress from lower grades) and not frequent in primary (de novo) GBMs.7 This suggests that IDH1 and related mutations may help define the secondary GBM class. It also appears that this mutation is very highly associated with lower grade astrocytomas and is maintained in relapsed higher grade tumors. Thus, it appears that some of the pathways that lead to GBM differ and that secondary GBM can be characterized by IDH1 mutations, in addition to having a lower EGFR and higher p53 mutation rate.6,20
As a group, there were 41 genes in the 22 GBM genomes sequenced that were statistically significant for their mutation frequency (CAN-genes)—strong evidence that at least 41 different genes can be mutated in GBMs that contribute in some way to progression of glioblastoma.7 Many of these genes are well-known oncogenes and tumor suppressors such as p53, but the function of the other genes with regard to cancer is still unknown. A list of the more commonly mutated genes in GBM, assembled from two major genomic studies,7,23 is presented in Table 102-2. It is clear that not only does each tumor have many mutations but that there is also a wide variety of genes that can be mutated, thus giving each tumor a spectrum of sequence alterations that is probably unique.
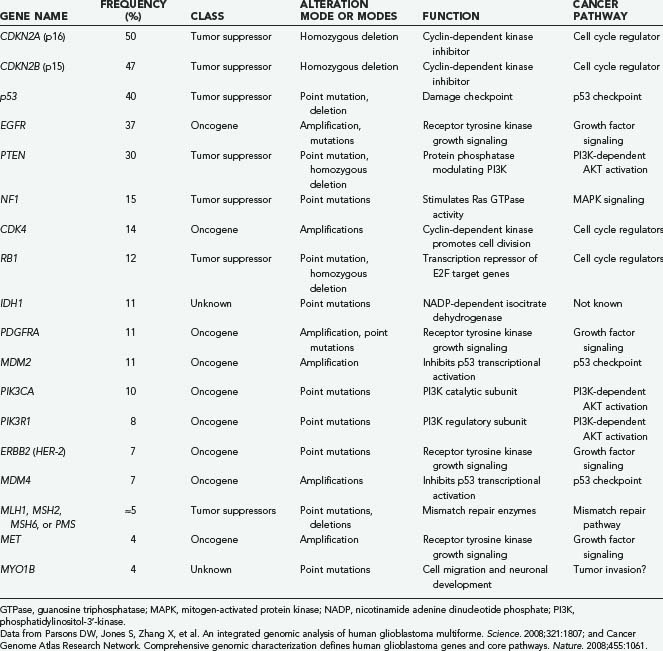
Despite the complexity caused by the numerous possible combinations of the many different genes that are mutated in cancer, the mutations can be clustered into functional groups. For example, genes that might serve the same function when mutated in cancer all activate a particular signal transduction pathway. One of the most commonly mutated pathways in cancer, the p53 checkpoint, can be disrupted either by inactivation via p53 mutation or by amplification of MDM2 or MDM4, which have a dominant-negative effect. In all, half of GBMs have a p53 pathway gene altered.7 A sketch of the three major pathways that contribute to GBM formation is shown in Figure 102-2.
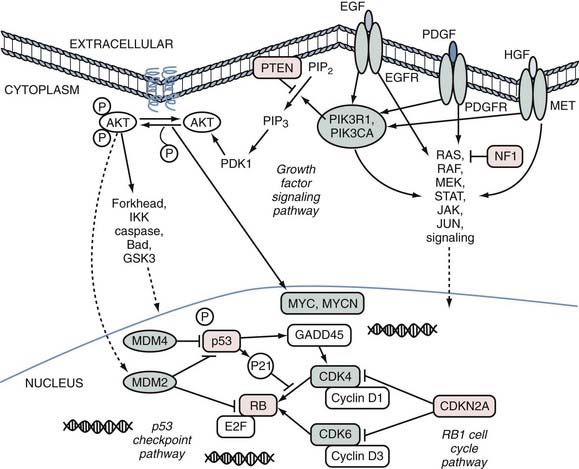
Another important group of genes in the development of GBM, as well as many other cancers, is the phosphatidylinositol-3′-kinases (PI3Ks).24 In half of GBMs there is either a PI3K-activating mutation, deletion/mutation of the negative regulator PTEN,25,26 or a mutated insulin-mediated activator of PI3K kinase, insulin substrate receptor 1 (IRS1). It is also possible that mutations in their receptor tyrosine kinases, in particular amplifications of EGFR, contribute to the signaling in this pathway. Together, these genes broadly form a growth factor signaling pathway that consists of the receptor tyrosine kinases and downstream signaling through AKT, mitogen-activated kinases, and other transducers.
Of the important genes shown in Figure 102-2, one deserves special attention as the most frequently mutated oncogene known in GBM. EGFR is genomically amplified27 in about 40% of tumors,28 and an additional number of GBMs have an activating point mutation.7,23 In all, nearly half of GBMs have an EGFR gene activated by a genomic change that occurs during development of the tumor.
After genomic amplification the EGFR gene may undergo further rearrangement.29,30 The most frequently observed genomic deletion in EGFR, present in two thirds of GBMs with EGFR amplification, is loss of the internal exons in the gene that correspond to the coding sequence for amino acids 6 to 273.31 This most common rearrangement is referred to as the EGFR type 3 rearrangement, or EGFRvIII.32 Removal of the deleted domain constitutively activates the tyrosine kinase activity of the mutant protein.33,34 Re-expression of the mutant protein in GBM cells increases the ability of the cells to invade through a simulated extracellular matrix.35
As a highly expressed oncogene on the cell surface of about half of GBMs, EGFR is in theory a good molecular target. Efforts to treat GBM patients with EGFR inhibitors in a single-agent regimen, however, have thus far not achieved reproducible increases in survival in clinical trials.36,37 Some of the reasons for this lack of success might include the inability to achieve sufficient intratumoral concentrations of the inhibitors or the ability of the cancer cell to signal growth through parallel pathways and thus overcome the effects of the EGFR inhibitors.
Although MMR gene mutations can be inherited, they can also occur sporadically to initiate tumor formation or promote resistance to chemotherapy.38–41 Because MMR mutations induce many nonfunctional as well as functional mutations, it is hard to precisely estimate the mutation rate of these genes in GBM, but about 5% of GBMs have microsatellite instability, a sign of a functional MMR mutation. It appears that MMR mutations are more common in relapsing patients, and this instability might be selected for as a means of more rapidly acquiring drug resistance.41,42
Low-Grade Astrocytomas and Oligodendrogliomas
The lower grade astrocytomas have not been evaluated for mutations as extensively as GBMs have, but there are some molecular similarities among GBM and grade II and grade III astrocytomas. For example, p53 mutations are found in grade II astrocytomas, similar to GBMs, although at a higher rate.7,23,43–45 It appears that of all the astrocytomas, gemistocytic astrocytomas (grade II) exhibit the highest rate of p53 mutation, with 88% having a mutation, followed next by fibrillary astrocytomas, with 53% mutated.22 Secondary GBMs have a higher p53 mutation rate than primary GBMs do.22 Yet progression of grade II astrocytomas to higher grade tumors does not seem to be dependent on p53 mutations inasmuch as grade II astrocytomas with and without p53 mutations progress at similar frequencies.43,44 Because p53 mutations are more prevalent in grade II astrocytomas, those that progress to GBMs result in secondary GBMs with a higher rate of p53 mutation. The mutations in p53 can be an initiating event for grade II astrocytomas, as evidenced by Li-Fraumeni syndrome, in which an inherited p53 mutation gives rise to higher risk for glioma along with many other cancers.
In contrast to high-grade astrocytomas, grade I pilocytic astrocytomas have a very low frequency of p53 mutations. The low frequency at which the mutations might occur has been reported differently in the literature and probably has yet to be accurately established.46–48 Overall, very little of the molecular basis of these mostly benign tumors is known. Despite sharing histologic features with other astrocytomas, there is little reported molecular similarity.
More is known about the molecular basis of oligodendrogliomas, and some of the chromosomal changes in this tumor help define this class. Oligodendrogliomas are now best defined at a molecular level by LOH on chromosomal arms 1p and 19q and a relative paucity of p53 mutations.22,49,50 The loss of 1p/19q in oligodendrogliomas very accurately predicts sensitivity to chemotherapy.50,51 It is one of the few examples of brain cancers in which a clinically useful prognostic marker has been identified.
Medulloblastomas
Table 102-3 summarizes the important known mutations for medulloblastomas. Because medulloblastoma is the most common malignant pediatric brain tumor, there have been numerous molecular studies on this cancer. Many important pathways and genes have been implicated in medulloblastoma, although systematic sequencing of the medulloblastoma coding genome has not been performed, as with GBM. Therefore, it is likely that there will be many new genes and pathways implicated in medulloblastoma in the future.
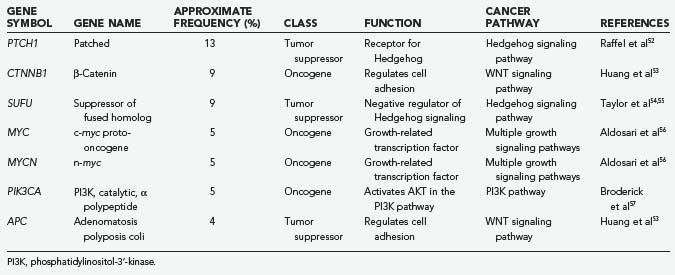
The pathway best studied thus far in medulloblastoma is the Hedgehog developmental signaling pathway. This pathway was first implicated in medulloblastoma by the discovery of Patched 1 (PTCH1) mutations.52,58–60 PTCH1 mutation in one allele was first identified as the hereditary basis of basal cell nevus syndrome (Gorlin’s syndrome) and, with both alleles mutated, as the basis of basal cell carcinoma.61 Patients with Gorlin’s syndrome also have a significant increase in the incidence of medulloblastoma, and such mutations were also found in these tumors. Sporadic medulloblastomas also have PTCH1 mutations, but this is limited mainly to the 15% or so medulloblastomas of the desmoplastic variant.52,58–60 By studying other genes in this developmental signaling pathway, additional mutations in desmoplastic medulloblastomas were discovered, for example, in the SUFU gene.54,55 It appears that Patched pathway mutations help define the desmoplastic variant of medulloblastomas.
Table 102-3 also shows a low frequency of medulloblastoma mutations in other well-studied cancer-related pathways. Mutations in APC and β-catenin implicate the important colon cancer–associated APC pathway in this brain tumor,53 although these mutations are found in only a small percentage of patients and are partially tied to inherited forms of this tumor. The PI3K pathway, a pathway activated in many cancers, appears to be activated in at least 5% of medulloblastomas.57 However, the molecular basis of the majority of medulloblastomas is still unknown. It is likely to be a mixture of the few genes listed in Table 102-3, plus a large group of genes yet to be implicated.
Perspectives in Brain Tumor Genomics and Genetics
The fields of cancer genetics and cancer genomics have for several decades identified many of the molecular changes that give rise to cancer. There has been a tremendous increase in the rate of discovery of mutated genes in brain and other cancers. This acceleration is due in part to advances in automated sequencing technology and completion of the human genome sequence.62–64 Starting with efforts such as linkage analysis, with which it took many years to identify a cancer-causing gene, technology has advanced to the point where tens of thousands of genes can be simultaneously evaluated in a cancer genome, thus enabling researchers to identify the more complete pattern of many acquired mutations in one project. This approach has already been applied to GBMs, but as the cost of these technologies decreases, one can expect to see it applied to other brain tumors. Now that we have or soon will have better knowledge of the mutations that give rise to brain cancers, attention can better be focused on understanding how these mutated pathways give rise to cancer.
There are several important reasons to understand the mutational basis of brain cancer. First, it helps us answer the question of why someone gets a brain cancer, a question frequently faced by patients and patient families. Second, the mutations and genomic alterations that occur in the more than 120 different types of brain tumor6 are starting to help us better classify these tumors for better diagnostic and prognostic purposes. Third, understanding of how the tumor differs at a molecular level from normal cells has helped us design successful new treatment strategies in other cancers. It is possible that this understanding will lead to better treatment options for brain tumors.
Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038.
Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061.
Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383.
Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839.
Kleihues P, Burger PC, Plate KH, et al. Pathology & Genetics: Tumours of the Nervous System. Lyon: International Agency for Research on Cancer; 2000.
Lal A, Glazer CA, Martinson HM, et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002;62:3335.
Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943.
McKusick V: Online Mendelian Inheritance in Man, OMIM (TM). in McKusick V, ed. Bethesda, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, Md) and National Center for Biotechnology Information, National Library of Medicine 2008, vol 2008.
Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892.
Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479.
Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther. 2007;6:1909.
Paraf F, Jothy S, Van Meir EG. Brain tumor–polyposis syndrome: two genetic diseases? J Clin Oncol. 1997;15:2744.
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807.
Pietsch T, Waha A, Koch A, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085.
Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636.
Voelzke WR, Petty WJ, Lesser GJ. Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr Treat Options Oncol. 2008;9:23.
Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108.
Yonehara S, Brenner AV, Kishikawa M, et al. Clinical and epidemiologic characteristics of first primary tumors of the central nervous system and related organs among atomic bomb survivors in Hiroshima and Nagasaki, 1958-1995. Cancer. 2004;101:1644.
1 Ron E, Modan B, Boice JDJr, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988;319:1033.
2 Yonehara S, Brenner AV, Kishikawa M, et al. Clinical and epidemiologic characteristics of first primary tumors of the central nervous system and related organs among atomic bomb survivors in Hiroshima and Nagasaki, 1958-1995. Cancer. 2004;101:1644.
3 Shintani T, Hayakawa N, Hoshi M, et al. High incidence of meningioma among Hiroshima atomic bomb survivors. J Radiat Res (Tokyo). 1999;40:49.
4 Musicco M, Sant M, Molinari S, et al. A case-control study of brain gliomas and occupational exposure to chemical carcinogens: the risk to farmers. Am J Epidemiol. 1988;128:778.
5 Pogoda JM, Preston-Martin S. Household pesticides and risk of pediatric brain tumors. Environ Health Perspect. 1997;105:1214.
6 Kleihues P, Burger PC, Plate KH, et al. Pathology & Genetics: Tumours of the Nervous System. Lyon: International Agency for Research on Cancer; 2000.
7 Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807.
8 McKusick V: Online Mendelian Inheritance in Man, OMIM (TM). in McKusick V, ed. Bethesda, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine 2008, vol 2008.
9 Thiagalingam S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology. 2004;111:568.
10 Upadhyaya M, Osborn MJ, Maynard J, et al. Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet. 1997;99:88.
11 Paraf F, Jothy S, Van Meir EG. Brain tumor–polyposis syndrome: two genetic diseases? J Clin Oncol. 1997;15:2744.
12 Turcot J, Despres JP, St Pierre F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959;2:465.
13 Li FP, Fraumeni JFJr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358.
14 Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839.
15 Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908.
16 Narod SA, Parry DM, Parboosingh J, et al. Neurofibromatosis type 2 appears to be a genetically homogeneous disease. Am J Hum Genet. 1992;51:486.
17 Issa JP, Ottaviano YL, Celano P, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536.
18 Leary RJ, Lin JC, Cummins J, et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:16224.
19 Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108.
20 Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892.
21 Dropcho EJ, Soong SJ. The prognostic impact of prior low grade histology in patients with anaplastic gliomas: a case-control study. Neurology. 1996;47:684.
22 Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479.
23 Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061.
24 Gallia GL, Rand V, Siu IM, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4:709.
25 Wang SI, Puc J, Li J, et al. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183.
26 Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943.
27 Wong AJ, Bigner SH, Bigner DD, et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84:6899.
28 Collins VP. Gene amplification in human gliomas. Glia. 1995;15:289.
29 Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965.
30 Bigner SH, Humphrey PA, Wong AJ, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017.
31 Frederick L, Wang XY, Eley G, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383.
32 Ekstrand AJ, Sugawa N, James CD, et al. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89:4309.
33 Moscatello DK, Holgado-Madruga M, Emlet DR, et al. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem. 1998;273:200.
34 Huang HS, Nagane M, Klingbeil CK, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927.
35 Lal A, Glazer CA, Martinson HM, et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002;62:3335.
36 Voelzke WR, Petty WJ, Lesser GJ. Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr Treat Options Oncol. 2008;9:23.
37 Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther. 2007;6:1909.
38 Leung SY, Chan TL, Chung LP, et al. Microsatellite instability and mutation of DNA mismatch repair genes in gliomas. Am J Pathol. 1998;153:1181.
39 Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038.
40 Alvino E, Fernandez E, Pallini R. Microsatellite instability in primary brain tumors. Neurol Res. 2000;22:571.
41 Rellecke P, Kuchelmeister K, Schachenmayr W, et al. Mismatch repair protein hMSH2 in primary drug resistance in in vitro human malignant gliomas. J Neurosurg. 2004;101:653.
42 Martinez R, Schackert HK, Plaschke J, et al. Molecular mechanisms associated with chromosomal and microsatellite instability in sporadic glioblastoma multiforme. Oncology. 2004;66:395.
43 Kraus JA, Bolln C, Wolf HK, et al. TP53 alterations and clinical outcome in low grade astrocytomas. Genes Chromosomes Cancer. 1994;10:143.
44 Watanabe K, Sato K, Biernat W, et al. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997;3:523.
45 del Arco A, Garcia J, Arribas C, et al. Timing of p53 mutations during astrocytoma tumorigenesis. Hum Mol Genet. 1993;2:1687.
46 Cheng Y, Pang JC, Ng HK, et al. Pilocytic astrocytomas do not show most of the genetic changes commonly seen in diffuse astrocytomas. Histopathology. 2000;37:437.
47 Hayes VM, Dirven CM, Dam A, et al. High frequency of TP53 mutations in juvenile pilocytic astrocytomas indicates role of TP53 in the development of these tumors. Brain Pathol. 1999;9:463.
48 Patt S, Gries H, Giraldo M, et al. p53 gene mutations in human astrocytic brain tumors including pilocytic astrocytomas. Hum Pathol. 1996;27:586.
49 Sonabend AM, Lesniak MS. Oligodendrogliomas: clinical significance of 1p and 19q chromosomal deletions. Expert Rev Neurother. 2005;5:S25.
50 Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636.
51 Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473.
52 Raffel C, Jenkins RB, Frederick L, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842.
53 Huang H, Mahler-Araujo BM, Sankila A, et al. APC mutations in sporadic medulloblastomas. Am J Pathol. 2000;156:433.
54 Taylor MD, Zhang X, Liu L, et al. Failure of a medulloblastoma-derived mutant of SUFU to suppress WNT signaling. Oncogene. 2004;23:4577.
55 Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306.
56 Aldosari N, Bigner SH, Burger PC, et al. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children’s Oncology Group. Arch Pathol Lab Med. 2002;126:540.
57 Broderick DK, Di C, Parrett TJ, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048.
58 Pietsch T, Waha A, Koch A, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085.
59 Vorechovsky I, Tingby O, Hartman M, et al. Somatic mutations in the human homologue of Drosophila patched in primitive neuroectodermal tumours. Oncogene. 1997;15:361.
60 Wolter M, Reifenberger J, Sommer C, et al. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57:2581.
61 Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299.
62 Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304.
63 Sanger F, Donelson JE, Coulson AR, et al. Use of DNA polymerase I primed by a synthetic oligonucleotide to determine a nucleotide sequence in phage f1 DNA. Proc Natl Acad Sci U S A. 1973;70:1209.
64 Smith LM, Sanders JZ, Kaiser RJ, et al. Fluorescence detection in automated DNA sequence analysis. Nature. 1986;321:674.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 102 The Genetic Origins of Brain Tumors
There are external causes that significantly increase the risk for development of a cancer, but for brain tumors the evidence for this is limited. A high dose of ionizing radiation to the brain has the strongest evidence for increasing the risk for certain brain tumors, as seen with radiation therapy involving the brain1 or increases in meningiomas in atomic bomb survivors.2,3 Environmental causes of brain tumor are not as evident. The main carcinogen in our society, tobacco, does not have evidence of producing risk for brain tumor as with many other cancers. Certain synthetic chemicals, such as fungicides and pesticides, show an association of exposure to increased incidence,4,5 but more research is needed to demonstrate a direct cause-and-effect relationship. Overall, it can be said that the vast majority of mutations that give rise to brain cancers are spontaneous, and except for family members with certain rare cancer syndromes or those who have previously undergone brain irradiation, it is not possible to accurately predict who in our society is at greater risk for a brain tumor.
There are more than 120 different pathologic classes of brain tumor.6 For some tumors, in particular glioblastoma multiforme (GBM), many of the mutations have now been identified, but little is known about the genetic basis of most brain tumors. It is clear, however, that the number and complexity of mutations arising during malignant tumor development, including brain cancers, are much greater than originally predicted. The field of cancer genomics, or oncogenomics, has advanced rapidly, and thousands of genes in a cancer genome are sequenced in a single project, as opposed to the time when only one candidate gene at a time could be evaluated. The current trend is to build comprehensive databases of cancer mutations for the common cancers, and as the cost of DNA sequencing decreases, such data will probably be generated for more types of brain tumors.
In this chapter the well-documented and commonly altered genes are reviewed for the few classes of brain tumor for which we have knowledge of their mutational basis. This does not imply by any means that we have a complete picture of the various gene mutation patterns for each cancer. In GBM, for example, an average of more than 60 acquired mutations can be observed per genome.7 Fortunately, these mutations cluster in a small number of molecular pathways that are altered to give rise to a cancer. Understanding the function and mechanisms of these pathways will produce better insight into how tumors proliferate and thus provide researchers with better means for molecular targeting.
Clonal Expansion of Malignant Tumors
A fundamental concept in cancer is the clonal expansion of tumors, with each step in tumor formation based on adding another mutation to the tumor genome. Figure 102-1A illustrates this concept. The process starts with a single mutation in a single cell. As mutations in critical genes accumulate in a cell, a cancer develops in stepwise fashion. In brain cancers, it is not clear how long it takes for mutations to accumulate and in what order all the mutations might occur because the early stages of the tumors are not easily observed during tumorigenesis. It appears, however, that many mutations and changes in copy number, in any one of many different combinations, accumulate to form a GBM.7 Because this clonal expansion presumably arises from a single cell, all the cells in the tumor should have the same mutation. This is normally true, except in a few situations, such as the presence of unstable amplifications that give rise to heterogeneity for this change in the tumor or the acquisition of a late-occurring mutation in the latter stages of tumor cell clonal expansion. For the vast majority of mutations, however, the same mutations will appear in all parts of the tumor.
Inherited Mutations and Familial Syndromes
Because the first hit is inherited as a germline mutation, it can be transmitted to offspring. Although most malignant tumors observed in the clinic do not have an evident hereditary basis, it is important to recognize the possibility of familial clustering of brain tumors and consider genetic counseling and further evaluation if evident. Table 102-1 lists some of the more common cancer-associated mendelian disorders that have brain tumors as part of the phenotype. Mendelian disorders are genetic diseases that have a clear pattern of inheritance within families, such as dominant or recessive inheritance. Most of the syndromes that involve brain tumors have an autosomal dominant mendelian inheritance pattern. A complete catalogue and literature review of all the mendelian disorders identified is readily accessible at the Online Mendelian Inheritance in Man (OMIM) website.8
The mismatch repair (MMR) cancer syndrome is an important syndrome that involves several different types of cancers, including some GBMs, meningiomas, and medulloblastomas.8,11,12 It has an autosomal dominant pattern of inheritance, as do all the syndromes listed in Table 102-1. There are several DNA repair enzymes that are necessary for correct DNA MMR in a normal cell. In MMR cancer syndrome, an inactivating mutation or deletion of the MLH1, MSH2, MSH6, or PMS genes reduces the ability of the cell to identify and correctly repair DNA mismatches that occur during DNA replication. Therefore, a mutation in any of these genes can lead to an increased risk for brain and other cancers. The mutations in these “mutator genes” increase the cell’s mutation rate and accelerates the acquisition of mutations elsewhere in the genome, thus leading to a significantly increased risk for the development of cancer at an early age.
Different Types of DNA Mutations and Alterations
DNA alterations can range from single base pair changes all the way to entire chromosome gains or losses, as well as any size change in between. There does not seem to be any restriction to the DNA sequence changes that can occur during the development of cancer. In addition to DNA sequence changes, it also appears that epigenetic changes can contribute to tumor progression by altering gene expression without altering the nucleotide sequence.17
[/not-level-membership-for-neurosurgery-category]
