Chapter 36 The Difficult Pediatric Airway
I Introduction
One of the most challenging aspects facing anesthesiologists is maintaining the technical skills that are necessary for the management of the difficult airway (DA). The American Society of Anesthesiologists (ASA) guidelines define a difficult airway as the clinical situation in which a conventionally trained anesthesiologist experiences difficulty with face mask ventilation of the upper airway, difficulty with endotracheal intubation, or both.1 Recent reports demonstrate how important skilled airway management is to the practice of pediatric anesthesia. Data from the ASA Pediatric Closed Claims Data Base demonstrate a greater frequency of adverse respiratory events in the pediatric population.2 In the pediatric closed claims analysis, respiratory events accounted for 43% of all adverse events, most frequently related to inadequate ventilation (20%). Esophageal intubation, airway obstruction, and difficult intubation (DI) combined accounted for 14% of the remaining adverse respiratory events. In the Pediatric Perioperative Cardiac Arrest (POCA) registry, 20% of all cardiac arrests were attributed to the respiratory system.3 Airway obstruction and DI were responsible for 27% and 13% of these events, respectively. Incidence of difficult mask ventilation in nonobese children is 2.1%. Most of the patients who experience arrests from airway obstruction or DI have an underlying disease or syndrome.
II Anatomy of the Pediatric Airway
The pediatric airway, particularly in infants, is different from the adult airway. Understanding these differences is important when managing the pediatric airway. Following is a brief review of the anatomy of the normal pediatric airway.4–7
A Larynx
The larynx is situated more cephalad at the third and fourth cervical vertebrae (C3-C4) level in the infant and migrates to the adult C5 level by 6 years.6 Because the infant’s larynx is more rostral (higher), the tongue is located closer to the palate and more easily apposes the palate. As a result, airway obstruction may occur during induction or emergence from anesthesia. A common misnomer is that the infant’s larynx is more “anterior” when it is really more “rostral” or “superior” in the neck, compared with the adult larynx. In syndromes associated with mandibular hypoplasia, such as Pierre Robin, the larynx is actually positioned more posteriorly than normal. This results in a greater acute angulation between the laryngeal inlet and the base of the tongue. In this circumstance, direct visualization of the glottis may be difficult or impossible. Because of the cephalad position of the larynx and the large occiput, the “sniffing” position does not assist in visualization of the pediatric larynx.4,7 Elevating the head only moves the larynx into a more anterior position. Infants should be positioned with the head and shoulders on a flat surface with the head in a neutral position and the neck neither flexed nor extended.5
B Epiglottis
The infant epiglottis is long, stiff, and often described as Ω or U shaped.6 It projects posteriorly above the glottis at a 45-degree angle. Because the epiglottis is more obliquely angled, visualization of the vocal cords may be difficult during direct laryngoscopy. It may be necessary to lift the tip of the epiglottis with a laryngoscope blade to visualize the vocal cords. Straight laryngoscope blades are often preferred for this reason. If the patient is not paralyzed, use of a Macintosh blade is less stimulating because it is not necessary to lift the epiglottis.
C Subglottis
The cricoid cartilage is the narrowest portion of the infant’s airway, about 5 mm in diameter, compared with the vocal cords of the adult airway.6 The infant’s larynx is funnel shaped with a narrow cricoid cartilage, whereas the adult airway is cylindrical. Tight-fitting endotracheal tubes that compress the mucosa at this level may cause edema and increase resistance to flow. Resistance to flow is inversely proportional to the radius of the lumen to the fourth power (r4). One millimeter (1 mm) of edema can reduce the cross-sectional area of the infant trachea by 75%, versus 44% in the adult trachea.
III Evaluation of the Pediatric Airway
The evaluation of the pediatric airway should begin with a history and physical examination of the head and neck. The examinations mostly involve subjective experience, and consistent evaluation criteria should improve the ability to predict the DA. Clues to a potentially difficult airway include snoring, noisy breathing, difficulty breathing with feeding or an upper respiratory tract infection, and recurrent croup. Review of previous anesthesia records should be performed if available. If a DA is encountered, documentation of events and the ability to mask-ventilate is helpful for future caregivers. A prior uneventful anesthesia does not guarantee success the next time.4,7
Knowledge of syndromes that may adversely affect the airway is crucial to the management of the difficult pediatric airway. The presence of one anomaly mandates a search for others. A common feature in patients with many of these syndromes is micrognathia. Micrognathia creates more difficulty with displacement of the tongue during direct laryngoscopy, thus increasing the chance that the glottis will be difficult to visualize.4,7 The ability to intubate often changes as the child grows. Intubation often becomes easier with syndromes associated with micrognathia (e.g., Pierre Robin) as the patient ages. In mucopolysaccharide disorders or abnormalities involving the cervical spine (e.g., Klippel-Feil syndrome), intubation may become more difficult as the child ages.5
Abnormalities of the ear or the presence of ear tags has been suggested as an indicator of DI.5 In one study, bilateral microtia was associated with an increased incidence of DI (42%, vs. 2% in unilateral microtia). Mandibular hypoplasia was associated with bilateral microtia 10 times more than with unilateral microtia (50% vs. 5%), thus allowing bilateral microtia to be used as an indirect predictor of DI.8
Physical examination must focus on the head, neck, and cervical spine. Many evaluations used to predict DA in adults have not been extrapolated to the pediatric population. Cooperation of the patient is necessary for precise evaluation. In the young or uncooperative child, appropriate evaluation is limited. Preliminary data indicate that the Mallampati classification may be an insensitive predictor of DI in the pediatric population.9 Pediatric anesthesiologists are at a disadvantage because they are anesthetizing patients with less objective airway information available. This underscores the need for a skilled approach to the difficult pediatric airway.
Evaluations should focus on the size and shape of the mandible, size of the mouth and tongue, absence or prominence of teeth, presence of loose teeth, and the neck length and range of motion. Berry10 suggests that the appropriate thyromental distance in infants is one finger breadth (1.5 cm). Lateral examinations of the head and neck may provide clues to the presence of micrognathia. Mandibular enlargement has also been identified as a risk factor for DI.
Cherubism is a childhood disease consisting of painless mandibular enlargement with or without maxillary involvement that progresses rapidly in early childhood and then regresses during puberty. In cherubism, the potential displacement space is encroached on by mandibular enlargement.11 Palpation of the soft tissue of the potential displacement area may reveal the problem.
A Diagnostic Evaluation
Magnetic resonance imaging (MRI) and computed tomography (CT) may be extremely helpful in the evaluation of airway pathology. Flexible fiberoptic endoscopy may be of benefit before intubation when visualization of vocal cords is thought to be difficult or when airway pathology is suspected. In patients with unilateral hemifacial microsomia, radiographic classification of the mandibular anatomy can help predict ease of intubation.12
Radiographic evaluation of patients with airway obstruction may be obtained in patients who present to the emergency room only if they are not in respiratory distress. Radiographs should be obtained in the upright position because obstruction may worsen in the supine position.13 In this situation, it is mandatory that a clinician skilled in airway management and capable of managing a difficult pediatric airway accompany the patient along with the appropriate equipment.
Radiographs have high sensitivity (>86%) for the diagnosis of airway foreign body, exudative tracheitis, and innominate artery compression. For laryngomalacia and tracheomalacia, radiography has much lower sensitivity (5% and 62%, respectively).13 Radiologic evaluation should not take precedence over airway control in patients with a compromised airway. Other physicians, especially the otolaryngologist, may be consulted and may support a DI.
IV Classification of the Difficult Pediatric Airway
Difficulty with ventilation, intubation, or both is the definition of a difficult airway according to the ASA DA management guidelines.1 Recognition of the DA along with the circumstances that predispose to airway problems is crucial to the safe management of the pediatric airway. Classification of the difficult pediatric airway may be made according to the anatomic location affected. Major anomalies of the head, face, mouth and tongue, nasopharynx, larynx, trachea, and neck are listed.
V Pediatric Airway Equipment
To manage a DA successfully, the appropriate equipment should be immediately available. We recommend the creation of a difficult pediatric airway cart stocked with equipment for patients ranging from premature infants to small adults. In addition, the American Academy of Pediatrics Section on Anesthesiology recommends the creation of a DA cart for all locations anesthetizing children.14 This cart should be dedicated only for use in a DA or a “cannot intubate, cannot ventilate” scenario (Box 36-1).
Box 36-1 Pediatric Difficult Airway Cart
A Face Mask
Face masks have been modified for fiberoptic intubation (FOI) in a variety of ways.15–18 Frei and colleagues16,17 described modifying a commercially available mask (Vital Signs) by drilling a hole into the lateral aspect of the mask and attaching a corrugated silicon tube. The center of the mask is fitted with a plastic ring covered by a silicon membrane. A hole 1 to 2 mm smaller than the outer diameter (OD) of the bronchoscope is punched into the membrane. This airway endoscopy mask has been used to facilitate fiberoptic bronchoscopy (FOB) in patients ranging in age from 3 days to 12 months with spontaneous ventilation and propofol sedation.15 A commercially available face mask with a ventilation side port (MERA, Senko Ika Kogyo, Tokyo) was modified and used successfully to intubate fiberoptically nine patients age 3 months to 11 years under inhalational anesthesia with continuous manual ventilation.18
B Oropharyngeal Airway
Upper airway obstruction may occur during induction of anesthesia because the infant’s tongue is large in relation to the oropharynx. Appropriately sized oropharyngeal airways are necessary for air exchange. Guedel and Berman airways are the most common airways available. By holding the airway next to the child’s face, the correct size can be estimated. If the airway is too short, obstruction may be worsened. If the airway is too long, the epiglottis or uvula may be damaged. Use of a tongue depressor to insert the oropharyngeal airway is recommended to avoid impaired lymphatic drainage of the tongue.4
C Nasopharyngeal Airway
Nasopharyngeal airways are available in sizes 12 to 36 French (F) and are used with caution in pediatric patients with hypertrophied adenoids. The modified nasal trumpet was first described by Beattie, followed by its use in pediatric airway management as described by Holm-Knudsen in 2005 (Fig. 36-1).
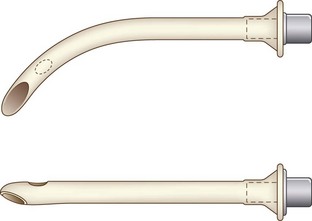
D Endotracheal Tube
Endotracheal tubes (ETTs) in a variety of sizes (2.5-7.0 mm) should be available for the pediatric patient. Laser-resistant, nasal/oral Ring-Adair-Elywn (RAE), and wire-reinforced ETTs are available for use depending on the surgical requirement. Determination of correct ETT size is based on the patient’s age and weight. ETTs one-half size larger and smaller than the calculated size should be available (Fig. 36-2). Traditional teaching advocates the use of uncuffed ETTs in patients younger than 8 years. Pediatric ETTs with low-pressure high-volume cuffs are available for use in patients with low lung compliance or those at risk for aspiration. For cuffed ETTs, a half-size smaller tube should be used because the OD of the tube is larger with the cuff.19
Maintenance of air leak pressure at less than 25 cm H2O with or without a cuff is recommended to minimize the occurrence of postintubation croup. Use of a manometer is recommended to avoid overinflation of the cuff. Koka and associates20 cite the incidence of postintubation croup as 1%. In a prospective study of more than 5000 children, however, Litman and Keon21 found that seven patients developed croup, defined as inspiratory stridor at least 30 minutes in duration, for an incidence of 0.1%. In that study, ETTs with air leak pressures greater than 40 cm H2O were replaced with the next smaller size.21 The presence or absence of a leak depends on the level of anesthesia and the use of muscle relaxants. Many clinicians use the degree of difficulty in passing the ETT below the vocal cords as the indicator of proper fit.
In general, there are many formulas to calculate the appropriate size of ETT. Formulas for selecting an uncuffed ETT in children older than 2 years include (age + 16)/4 or (age/4) + 4. The use of cuffed ETTs in newborns and children under 8 years has been studied. In a group of 488 patients, patients were randomly allocated to receive a cuffed or an uncuffed ETT.22 The formula for the cuffed tube was (age/4) + 3. This formula was appropriate for 99% of patients. In that study, three patients in each group were treated for croup symptoms. Formulas for length of insertion of an oral ETT include length (cm) + 3 times internal diameter (mm) or length (cm) = age (years)/2 + 12.19 In the premature or newborn infant, the rule is tip-to-lip distance in cm = 6 + weight (kg).23 Whatever method is chosen, correct ETT position should be confirmed by auscultation of bilateral breath sounds (Tables 36-1 and 36-2). Also, leak should be checked to a permissible pressure.
| Age | Size (mm ID) |
|---|---|
| Preterm (>1000 g) | 2.5 |
| Preterm (1000-2500 g) | 3.0 |
| Newborn to 6 months | 3.0-3.5 |
| 1-2 years | 4.0-4.5 |
| >2 years | (Age +16)/4 = ID |
ID, Internal diameter.
TABLE 36-2 Formula for Endotracheal Tube (ETT) Size and Depth of Insertion
| Type/Insertion | Formula |
|---|---|
| Uncuffed ETT | (Age + 16)/4 or ETT >2 years, Age/4 + 4 |
| Cuffed ETT | Age/4 + 3 |
| Length of insertion (oral) | Age (years)/2 + 12 or 3 × ID (mm) |
| Length of insertion (nasal) | 3 × ID (mm) + 2 |
ID, Internal diameter.
Double-lumen tubes are not available for use in pediatric patients younger than 6 to 8 years. The Arndt Endobronchial Blocker (Cook Critical Care, Bloomington, Ind) has been used to provide one-lung ventilation in infants.24 The 5.0-F blocker is available; the recommended ETT size is 4.5 mm. The Univent tube (Fuji Systems, Tokyo) is a single-lumen tube with an incorporated movable bronchial blocker inside.25 Pediatric sizes of the Univent tube are available: 3.5-mm internal diameter (ID) and 4.5-mm ID. The 3.5-mm Univent tube does not have a lumen for suctioning or administration of oxygen to the blocked lung. FOB is needed for placement. Further detail regarding one-lung ventilation is provided in Chapter 26.
E Endotracheal Tube Exchangers
Endotracheal tube (ETT) exchangers have multiple uses; they can be used to exchange damaged ETs and provide a conduit for reintubation, if necessary. Many different types of exchangers are available for use in adult patients. These tube exchangers are long, semirigid catheters that fit inside ETTs. The Frova Intubating Introducer (Cook Critical Care) is available in a pediatric size (8 F) that allows placement of a 3.0-mm ETT. It is 33 cm in length with a hollow lumen and a blunt curved tip that is shaped like the gum elastic bougie. The blunt curved tip can be passed “blindly” into the trachea when visualization of the glottis is inadequate. The Frova catheter has a hollow lumen and two side ports and is packaged with removable Rapi-Fit adapters that allow ventilation and a stiffening cannula26 (Fig. 36-2).
Cook also manufactures airway exchange catheters (AECs) in four sizes. These catheters are blunt tipped and hollow, with distal side ports and a Rapi-Fit adapter. The 8-F size is 45 cm in length and can be used in 3.0-mm ETTs.26
F Laryngoscopes
2 Oxyscope
The Oxyscope is a fiberoptic Miller no. 1 blade with a port for insufflation of oxygen during intubation. Oxygen insufflation during laryngoscopy in spontaneously breathing, anesthetized infants has been shown to minimize the decrease in transcutaneous oxygen tension, thus making airway instrumentation safer.27
3 Anterior Commissure Laryngoscope
The anterior commissure laryngoscope is frequently used by otolaryngologists for visualization of the glottis. It is a rigid, tubular, straight-blade laryngoscope with a distally located, recessed light source. This design permits enhanced visualization by preventing the tongue from obscuring the field of view.28
4 Bullard Laryngoscope
The Bullard laryngoscope (Circon ACMI, Stanford, Conn), developed by Dr. Roger Bullard at the Medical College of Georgia, is an indirect laryngoscope that utilizes fiberoptic and mirror technology to visualize the larynx.29 Use of fiberoptics and a curved blade enable visualization of the larynx “around the corner” of the blade, thus eliminating the need to align the oral, pharyngeal, and tracheal axes. A standard laryngoscope handle or a flexible fiberoptic cable connected to a light source powers the fiberoptic light source. This laryngoscope is manufactured in three sizes: adult, pediatric, and pediatric long.30 The adult size, with a blade that is 2.5 cm wide, is suitable for children older than 10 years. The pediatric version (newborn to age 2 years) has a blade 1.3 cm wide that extends 0.6 cm beyond the fiberoptics. This blade is recommended for use in neonates, infants, and smaller children. The pediatric long version is available for use in infants and small children up to age 10; it has a longer blade (1.4 cm) and a wider flange (1.6 cm). In the pediatric long version, a multifunctional stylet is attached to the fiberoptic bundle between the eyepiece and handle and aligns the tip of the ETT beneath the flange of the blade. The smallest ETT that passes over the stylet in the pediatric long version is 4.5 mm.30
The Bullard laryngoscope requires minimal mouth opening for its insertion (0.64 cm in cephalad-caudad axis). It has been used to intubate patients with unstable cervical spine or with Pierre Robin, Treacher Collins, Noonan’s, or Klippel-Feil syndrome, among others.29 The adult Bullard laryngoscope has been used successfully to intubate patients older than 12 months with normal airways.31 Contact with the right aryepiglottic fold and, in children, contact with the anterior vocal cord occurred.31,32 Compared with the Wis-Hipple 11/2, the adult Bullard laryngoscope provided a similar view and required a slightly longer time for intubation in children 1 to 5 years of age.32
5 Angulated Video-Intubation Laryngoscope
The angulated video-intubation laryngoscope (AVIL), invented by Dr. Marcus Weiss of Zurich, is an endoscopic intubation device. The AVIL consists of a cast-plastic Macintosh 4 laryngoscope, with the blade angulated distally, and an integrated fiberoptic endoscope (1.8 m long, OD 2.8 mm, VOLPI, Schlieren, Switzerland). The distal blade tip is angulated about 25 degrees to provide increased viewing for the fiberoptic lens. With the angulated tip, the AVIL resembles an activated McCoy blade. Flattening of the blade’s vertical flange enables the device to be used in children. The fiberoptic endoscope runs from the handle to the tip of the blade. The AVIL uses conventional laryngoscopy techniques coupled with video monitoring from the blade tip. Styletted ETTs, in a “hockey stick” configuration, are passed along the vertical flange of the blade under video control.33
The AVIL has been used in patients ranging in age from 3 months to 17 years with manual in-line neck stabilization. In infants and small children, care should be taken with insertion of the blade; initial insertion of the blade was too deep in these patients.34 Several reports document the use of this device in pediatric patients with a DA. The video laryngoscope has been used successfully to intubate children with Morquio’s syndrome as well as a 3-day-old neonate with Pierre Robin syndrome.33,35
6 Truview Laryngoscope
The Truview (Truphatek International, Netanya, Israel) is a recently introduced rigid laryngoscope that has an angulated tip and an optical assembly that provides an illuminated and magnified view of the larynx. The optical system consists of a lens and a prism, which extends the view beyond the tip of the blade. The tip of the device is narrow to accommodate small mouth openings and angulated 46 degrees anteriorly to provide a wider view of the larynx using light refraction. This laryngoscope also has a side port that allows for oxygen insufflation and can be attached to video monitoring to assist with training. The height of the laryngoscope blade is 8 mm. Compared with traditional laryngoscopes, the Truview EVO2 laryngoscope offers various advantages and improves laryngoscopic view. A study by Singh and colleagues35a of 60 neonates and infants comparing the Truview EVO2 with the Miller blade demonstrated an improved laryngoscopic view with the Truview blade, with an increased time to intubation that was statistically but not clinically significant.
7 GlideScope Video Laryngoscope
The GlideScope Video Laryngoscope (Cobalt, Verahon) has a reusable video baton and single-use laryngoscopy blades in two sizes. The laryngoscope comes with a monitor screen, and a video recording unit is also available. The GlideScope Cobalt model features a 10-mm laryngoscope blade. The blade is inserted in the midline without displacing the tongue.36 Two studies have been reported using the GlideScope in children with normal airways. Both studies found it suitable for intubation in pediatric patients.37,38 In one of the studies, the time required for intubation was longer.
8 Airtraq Optical Laryngoscope
The Airtraq Optical Laryngoscope (AOL; Prodol, Vizcaya, Spain) is a single-use indirect laryngoscope for tracheal intubation. The Airtraq comes in two pediatric sizes: infant (size 0) for ETT 2.5 to 3.5 and pediatric (size 1) for ETT sizes 3.5 to 5.5. Both sizes require a mouth opening of 12 to 13 mm. The rubber eyepiece may be used or a camera may be attached and used with a wireless monitor. Images from the distal tip of the blade are projected to the proximal eyepiece. The Airtraq is inserted midline, and the tip may be placed in the vallecula or used to lift the epiglottis. Once the glottis is visualized, the ETT is slowly advanced. For intubation, it is important to lubricate the ETT so that the tube advances easily. Problems with advancement of the ETT may be caused by too large diameter of the ETT, the guide channel, or incorrect angle of the ETT.36 Two case reports documented the use of the Airtraq in pediatric patients with difficult airways: a 9-year-old child with Treacher Collins syndrome who weighed 23 kg37 and a 4.8-kg infant with Pierre Robin syndrome.38 Other case reports have documented difficulty with advancement of the ETT into the trachea despite a good view of the larynx.39
G Stylets
There are various types of stylets available as adjuncts to endotracheal intubation, including the traditional malleable stylet, lighted stylets, and optical stylets. Stylets should be available for the DA. The stylet is inserted into the ETT until the distal end of the stylet is just short of the ETT tip. The ETT and stylet are bent into the desired shape, usually a hockey stick configuration. Complications associated with use of stylets include tracheal trauma, ETT obstruction, and shearing of the stylet. When removal of a stylet becomes difficult, the tip should be examined.40
1 Lighted Stylets
Several different types of lighted stylets, or lightwands, are currently commercially available, including the Vital Signs Light Wand Illuminating Stylet (Vital Signs, Totawa, NJ) and the Tube Stat Lighted Stylet (Xomed, Jacksonville, Fla). Pediatric versions are available for use with ETTs as small as 2.0 to 4.0 mm. The use of the lighted stylet to guide blind endotracheal intubation relies on the principle of transillumination. The presence of a well-defined glow in the neck indicates tracheal placement. Esophageal placement is indicated by the absence of a glow in the neck. Several different reports describe successful intubation of pediatric patients with the lightwand.41,42 Successful technique includes the following principles:
1. A small shoulder roll should be used to keep the head in a neutral to slightly extended position. This is extremely important in a small infant, whose neck naturally flexes when lying on a flat surface because of the large occiput.
2. The lightwand should be advanced in the midline; if the light deviates to one side, the lightwand should be withdrawn and repositioned.
3. The epiglottis is elevated by lifting the jaw with the nondominant hand.
4. Transillumination should be assessed before advancing the lightwand too far.
5. Blind nasal intubation in children is often easier with the rigid stylet left in place.
6. The wand is bent less sharply than for an oral intubation.
2 Optical Stylets
The first optical stylet, described in 1979, was a Hopkins telescope with a fiberoptic external light source (Karl Storz, Tuttlingen, Germany).43 The Seeing Optical Stylet (SOS) system (Clarus Medical, Minneapolis) is a new, reusable, high-resolution fiberoptic endoscope with a malleable stainless steel stylet.36 It combines the features of an FOB and a lightwand. The Shikani Seeing Stylet is portable, lightweight, and available in pediatric and adult versions. The pediatric version is compatible with ETTs 3.0 to 5.0 mm in size. The SOS can be inserted directly into an ETT, allowing intubation to be performed under direct vision. Illumination is provided by a standard green line fiberoptic laryngoscope handle or the included SITElite halogen handle. An adjustable tube stop with an oxygen port, which goes over the shaft of the stylet, allows supplemental oxygen to be delivered. Many factors do not affect the SOS, including cervical spine injury, small mouth, large tongue, and reduced jaw mobility.36
Pfitzner and colleagues44 described the use of the Shikani SOS on eight occasions in seven patients with DA. There were seven successful intubations; one patient, who had previous surgery and radiotherapy for a retropharyngeal rhabdomyosarcoma, could not be intubated by any method. Two patients with limited mouth opening and one patient with a C1-C2 subluxation were intubated on the first attempt. A patient with Hunter’s syndrome was intubated on the second attempt. A potential difficulty mentioned with the SOS is loss of the visual field, which occurs when the lens is next to a mucosal surface. Maneuvers to increase the operating space available are use of a laryngoscope to retract the base of the tongue, lifting the mandible, and pulling the tongue forward.45
The Shikani Stylet is inserted into the ETT after lubrication with silicon spray. The fiberoptic cable can be connected to a video monitor. The mandible is lifted with the left hand and displaced anteriorly until the lower teeth are anterior to the upper teeth.45 The stylet with the loaded ETT is advanced into the trachea under direct vision. Laryngoscopy may be useful in cases of DI (Fig. 36-3). The Shikani Optical Stylet (Clarus Medical) is a portable video stylet.

3 Video-Optical Intubation Stylet
Another video-optical intubating stylet (Acutronic Medical Systems, Hirzel, Switzerland) consists of a flexible fiberoptic endoscope (developed by Dr. Weiss of Zurich). A sliding connector locks the video stylet onto the ETT adapter; it does not require neck extension but does require mouth opening. One report documents successful use of the video-optical intubating stylet in patients age 6 to 16 years, with a simulated grade III laryngoscopic view; 46 of 50 patients were intubated on the first attempt; four attempts were considered failures because of prolonged intubation time (>60 seconds).46
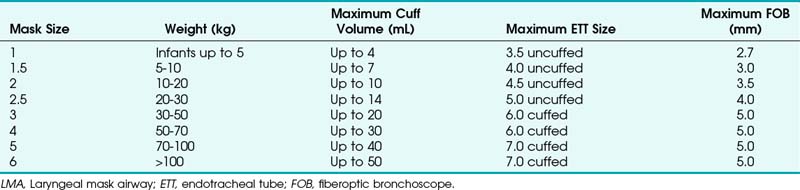
H Laryngeal Mask Airways
1 LMA Family
The laryngeal mask airway (LMA North America, San Diego), introduced in 1983 and approved for use in 1991 by the U.S. Food and Drug Administration (FDA), is a standard part of the ASA DA algorithm.1,47 Pediatric versions of the LMA Classic, as well as the disposable LMA, are available for use and are part of the pediatric DA algorithm, as described by Steward and Lerman.48 Application of the LMA requires minimum training and can be useful in neonatal resuscitation.49 The LMA Flexible is available in sizes 2 and 2.5, and the LMA ProSeal is available in a size 2. The size of the LMA in children is determined by the patient’s weight, although a new method has been suggested. With the hand extended and palm side facing up, the thumb and little finger are extended. The second, third, and fourth fingers are placed together. The fully inflated LMA is placed against the palmar side of the patient’s fingers, keeping the widest part of the LMA in line with the widest part of the three fingers. In a study of 163 children at birth to 14 years old, this method was correct in 78%. In the remaining patients, a difference of only one size was observed.50
The LMA has been described as a conduit for blind intubation as well as a conduit for fiberoptic intubation.51–55 Awake placement of the LMA has been described in an infant with Pierre Robin syndrome.56 Anterograde intubation through the LMA with a guidewire was also described in an infant with micrognathia who could not be intubated with conventional methods. A soft-tipped guidewire was advanced through the LMA and the position confirmed by fluoroscopy. An ETT was inserted over the guidewire, followed by removal of the LMA.57 A review of the literature demonstrates different insertion techniques.
The standard technique described with the cuff deflated for adults has also been advocated for children. In addition, a rotational or reverse technique has been described. The LMA is inserted with the cuff facing the hard palate and then rotated and advanced simultaneously. An alternative technique involves inserting the LMA with the cuff partially inflated. Reports on placement of the LMA with the different techniques are conflicting. In children, one study compared two insertion techniques. The partially inflated cuff insertion technique does not increase the incidence of downfolding of the epiglottis and is an acceptable alternative to the standard technique.58 In another study, insertion of the partially inflated LMA required less time and was associated with a higher success rate on first attempts compared with the standard (deflated) technique.59 Results from a study detailing the fiberoptic positioning of the LMA in children with a DA show that 29.5% of patients had a grade I (full) view of the glottis, 29.5% had a grade II (partial) view, and 41% a grade III (epiglottis only) view. Children with a mucopolysaccharide disorder had a grade III view 54% and a grade I view 14% of the time.60
The ProSeal LMA is now available in pediatric sizes. This LMA has a second mask to isolate the upper esophagus with a second dorsal cuff to increase the seal against the glottis. Lopez-Gill and coworkers61 found that it was easily inserted, and oropharyngeal leak pressure was greater than 40 cm H2O (Table 36-3).
I Rigid Ventilating Bronchoscope
The rigid ventilating bronchoscope is extremely useful for ventilating patients with a DA and is included in the most recent version of the ASA DA algorithm as an alternative device in the “cannot ventilate, cannot intubate” situation. In any situation of potential airway collapse, the otolaryngologist and the rigid ventilating bronchoscope should be immediately available (see Chapter 29).
VI Induction Technique
The principles outlined in the ASA guidelines for DA management apply to the pediatric patient. Evaluation, recognition, and preparation are key elements.1 Preoxygenation of pediatric patients, although difficult, should be attempted before any DA intervention, if possible. Studies have demonstrated that the optimal time for preoxygenation in pediatric patients is different from that in adults. Values ranging from 80 to 100 seconds have been reported for adequate preoxygenation in healthy children.62,63 Summoning help early, using awake intubation, and preserving spontaneous ventilation during intubation attempts are also important when managing the DA. The awake or awake sedated approach is preferred in most circumstances when managing the DA. However, in pediatric patients, the patient’s cooperation may limit the usefulness of awake intubation. One well-tolerated technique is placement of a lubricated LMA in awake infants, which provides an airway for inhalational induction.56
The traditional approach to the difficult pediatric airway has been maintenance of spontaneous ventilation under inhalational anesthesia. Premedication with oral or intravenous atropine (0.01-0.02 mg/kg) is indicated for vagolytic and antimuscarinic effects. Inhalation induction may be performed with sevoflurane in 100% oxygen. Sevoflurane has been used in the management of the DA with success.64,65 The low blood gas solubility of sevoflurane and consequent rapid induction and emergence are advantageous when managing the DA. When the ability to ventilate the patient by mask is demonstrated, a small dose of muscle relaxant or propofol may be given to facilitate intubation.
For patients who can tolerate an awake sedated intubation technique, a variety of drugs can be used. One must always keep in mind the risk/benefit ratio when sedating a patient with a DA. Sedatives may further compromise an airway. Sedatives should not be given to any patient in acute distress or with the potential for acute obstruction. Use of sedatives should be based on careful physical examination, anesthesiologist experience with agents involved, and overall patient condition. If no other options are available, slow titration of pharmacologic agents to effect, without loss of spontaneous ventilation, should be performed. Use of pharmacologic agents that are easily antagonized is recommended. For older children and adolescents, a combination of midazolam and fentanyl may be used. Remifentanil can also be used. Dexmedetomidine has been used successfully to perform an awake fiberoptic intubation in a morbidly obese patient with facial, cervical, and upper thoracic edema.66 In extreme circumstances, parental presence at induction may be allowed. Careful preparation of the parent must be performed prior to induction. As soon as the patient separates or begins to lose consciousness, a designated member of the operating room (OR) staff should immediately escort the parent out of the OR.
Another important aspect for successful airway management is topicalization of the airway with local anesthesia. In pediatric patients, this may be obtained by nebulizing, spraying, or swabbing local anesthetic solution or by applying viscous gel to a gloved finger. FOB with suction ports can be used to spray local anesthesia on the vocal cords under direct vision. The maximum dose of local anesthetic allowed should be calculated before topicalization. The drug of choice is lidocaine because it has the best safety profile. Maximum doses of lidocaine are 5 mg/kg. Agents containing benzocaine (e.g., Cetacaine spray; Americaine ointment; Hurricaine ointment, gel, or spray) should be avoided in infants and young children because of the risk of methemoglobinemia.7
VII Airway Management Techniques
A Techniques for Ventilation
Obstruction of the upper airway is a common occurrence in pediatric patients undergoing an inhalation induction. Techniques for overcoming this type of obstruction include insertion of an appropriate-size oropharyngeal airway or a nasopharyngeal airway, or both. Another common mistake is occlusion of the submandibular space with incorrect placement of the anesthesiologist’s hand. Care should be taken to position the hand on the tip of the mandible and not on the submandibular space. Chin lift or jaw thrust combined with continuous positive airway pressure (CPAP) at 10 cm H2O has been shown to improve upper airway patency.67
Additional techniques are available for mask ventilation. The two-person technique involves either one person holding the mask with both hands while an assistant compresses the reservoir bag or a second person assisting in jaw lift while the first person continues to compress the reservoir bag. Another option is using the anesthesia ventilator to provide ventilation so that one person can hold the face mask with both hands.68
B Techniques for Intubation
1 Direct Laryngoscopy
Tips for successful visualization of the larynx include proper use of external laryngeal pressure and positioning. Direct laryngoscopy involves alignment of the oral, pharyngeal, and laryngeal axes in order to visualize the glottis. Because the larynx is situated in a more cephalad position and the occiput is large, the sniffing position in infants does not assist in visualization of the larynx.4,7 The infant should be positioned with the head in a neutral position with the neck neither flexed nor extended.69 A small shoulder or neck roll may be beneficial. Optimum external laryngeal manipulation (OELM) should also be used with a poor laryngoscopic view to improve visualization. OELM may improve the laryngoscopic view by at least one whole grade in adults. This is not cricoid pressure but rather pressing posteriorly and cephalad over the thyroid, hyoid, and cricoid cartilages. Benumof and Cooper70 suggest that OELM should be an instinctive and reflex response to a poor laryngoscopic view. This maneuver has also proved effective in pediatric patients.71 The main mechanism seems to be shortening of the incisor-to-glottis distance.
The two-anesthesiologist technique involves manipulating the larynx under direct vision by the laryngoscopist and intubation by a second anesthesiologist. This technique has been used successfully to intubate a 6-month-old infant with Pierre Robin syndrome and concomitant tongue-tie (ankyloglossia).72
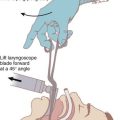
The retromolar or paraglossal technique has been advocated as useful in cases of DI related to a small mandible.73 A straight laryngoscope blade is introduced into the extreme right corner of the mouth overlying the molars, thus reducing the distance to the vocal cords. It is advanced in the space between the tongue and lateral pharyngeal wall until the epiglottis or glottis is visualized. The head is rotated to the left to improve visualization while applying external laryngeal pressure displacing the larynx to the right. Advancement of the ETT is facilitated by retracting the corner of the mouth to allow placement of the ETT. The styletted ETT should be shaped into the classic hockey stick configuration. An alternative approach involves placement of the ETT from the left side of the mouth.74 Lateral placement of the laryngoscope blade reduces the soft tissue compression because the tongue is essentially bypassed. The maxillary structures are also bypassed by the lateral blade placement, thus improving the view.4 Because there is a reduced space for displacement of the tongue in syndromes with micrognathia, this approach may be useful. The retromolar technique has been described as an alternative method for intubation of patients with Pierre Robin syndrome.75 A pediatric version of the Bonfils Retromolar Intubation Fiberscope is the Brambrink Intubation Scope (Karl Storz). It is an optical stylet that allows a retromolar approach to the DA.26
In adults the left molar approach with a Macintosh blade and OELM has been reported to improve the glottic view in cases of difficult laryngoscopy.76 Suspension laryngoscopy is often employed by otolaryngologists as an alternative technique for visualization of the difficult larynx. Intubation of an infant with Goldenhar’s syndrome was accomplished by suspension laryngoscopy.77 This method is similar to standard laryngoscopy by the retromolar technique.
2 Blind Intubation Technique
a Blind Nasotracheal Intubation
Blind nasotracheal intubation requires preservation of spontaneous ventilation either under general anesthesia or with adequate vasoconstriction and topicalization of the nasal mucosa. The tip of the ETT is directed toward the larynx by listening to the intensity of the breath sounds or by the capnograph tracing. This technique requires extensive practice before use. Tips for this technique include external pressure on the neck, which may direct the glottis toward the ETT; placement of a stylet through the ETT after passage through the nasopharynx to direct the tip to the glottis; inflating the ETT cuff with air to center it at the glottis and then deflating it for actual passage; and repositioning of the head (flexion/extension) if the initial intubation attempt fails.48 Pediatric patients with enlarged adenoids may be at risk for bleeding and trauma with this technique. Blind nasotracheal intubation of a neonate with Pierre Robin syndrome has been described.78
b Digital Endotracheal Intubation
Digital intubation is a blind technique that is relatively easy to learn. Intubation of an 8-day-old, 3.3-kg neonate with Pierre Robin syndrome has been reported.79 The left index finger is passed midline along the surface of the tongue until it passes the epiglottis. The left thumb may apply cricoid pressure to steady the larynx. The ETT, using the left index finger as a guide, is advanced into the trachea. This technique has been used as the primary method of intubating neonates in some neonatal centers.80 As with any new technique, practice is required.
c Lightwand Intubation
Intubation with a lightwand is a blind technique that has found success in management of the difficult pediatric airway.41,42 The success rate increases with experience; practice is thus required. As with any new technique, experience with patients who have normal anatomy should be gained first. Contraindications to the use of the lightwand include tumors, infections, trauma, and foreign bodies of the upper airway.37 Causes of failed intubation include entrapment in the vallecula or the aryepiglottic folds. A shoulder roll helps to extend the head and neck and increase the exposure of the anterior neck. After preparation of the lightwand, the jaw is lifted with the nondominant hand or a laryngoscope blade. The lightwand is inserted in the midline into the patient’s mouth, rotated around the patient’s tongue, then gently rocked back and forth. If the ETT wand is in the trachea, a well-defined bright light (size of a quarter) is visible at the level of the subglottis on the anterior surface of the neck.
3 Fiberoptic Laryngoscopy
Aids for fiberoptic intubation (FOI) include face masks, oropharyngeal airways, guidewires, and the LMA.81 The Frei mask previously described or variations of commercially available masks have been used with success.15,16,82 The Patil-Syracuse mask is available in a size 2, but it is difficult to achieve a good seal with this mask. An endoscopy mask can be made by attaching a swivel fiberoptic bronchoscope (FOB) adapter to a pediatric face mask in one of two ways81: a commercially available swivel adapter (Instrumentation Industries, Bethel Park, Pa) can be attached directly to the mask, or an adapter designed for attachment to the ETT (e.g., Portex bronchoscope adapter) can be connected to the face mask with a 15-mm to 22-mm adapter.
Oropharyngeal airways may also be modified for use in pediatric FOI. A strip may be cut from the convex surface of a Guedel-style airway to produce an aid for oral fiberoptic laryngoscopy, creating a channel. The fiberscope is placed in the channel, which helps maintain a midline position. The use of a smaller airway than predicted is suggested so that one may visualize the base of the tongue and epiglottis. Modified oropharyngeal airways are not effective as bite blocks, and one must be careful.81 Also, a nipple from a baby bottle has been modified to act as a conduit for FOI in an infant with an unstable cervical spine. In this case, a hole was cut obliquely into the end of the nipple. After topicalization of the airway with 2% lidocaine, FOI was performed with a 4.0-mm uncuffed ETT.83
Flexible fiberoptic laryngoscopy is one of the cornerstones of DA management. Preparation for fiberoptic laryngoscopy should include preparation of the patient (antisialogue) and checking of the FOB, light source, and suction as well as standard airway equipment. An assistant is necessary for monitoring of the patient and providing a jaw lift, which is useful because it elevates the tongue from the posterior pharynx.7 For older children and adolescents who will be sedated for the procedure, explanation and reassurance in a calm manner are helpful. A method of delivering oxygen is necessary as well. This can be accomplished in a variety of ways, either blowby from the anesthesia circuit or by nasal cannula. For patients who are anesthetized, an LMA or an endoscopy mask may be used to ventilate the patient while the intubation is being performed. Tips for successful oral intubation include midline placement of the FOB, advancement of the FOB only when recognizable structures are visualized, and retraction of the tongue with gauze or clamps if needed.7 If the view from the fiberscope is pink mucosa, the FOB is slowly pulled back until a recognizable structure is seen. If the nasal route is chosen, a topical vasoconstrictor may be used to reduce the chance of bleeding. In a series of 46 patients with DA, fiberoptic nasal intubation was successful on the first attempt in 37 patients (80.4%) and on the second or third attempt in 7 patients (15.2%). Two failures occurred: one related to bleeding and the other to inability to introduce the scope nasally.84
Flexible fiberoptic laryngoscopy may be performed in a variety of ways. The standard technique involves passage of the ETT over the FOB. The ultrathin fiberoptic laryngoscope with a directable tip allows FOI to be performed with ETTs as small as 2.5 mm. Intubation of a 3-month-old infant with the Pierre Robin syndrome has been successfully performed with an ultrathin fiberscope.85 A new 2.5-mm ultrathin flexible FOB with a 1.2-mm suction channel has been used to intubate a newborn with a DA.86 This FOB has a 2.5-mm OD, 1.2-mm working suction channel, angle of deflection of 160 degrees up and 130 degrees down, and a working length of 450 mm.
In scenarios where the available bronchoscope is too large for the required ETT, a staged technique may be employed.87 A FOB with a working channel, a cardiac catheter, and a guidewire are required. The guidewire is passed into the working channel of the fiberscope before intubation. The FOB guidewire assembly is then introduced into the mouth and positioned above the larynx. The guidewire is advanced into the trachea under direct visualization, followed by removal of the FOB. A cardiac catheter (used to stiffen the wire) is threaded over the guidewire. Finally, an ETT is advanced into the trachea over the guidewire-catheter assembly, which is then removed. A modification of this technique involves passage of the ETT over the guidewire without the reinforcing cardiac catheter. This has been used to intubate nasotracheally a 3-day-old infant with Pierre Robin syndrome.88
The fiberoptic bronchoscope may also be used as an aid for nasal intubation either under direct vision or with a guide. In these cases, FOB is introduced into one of the nares while the ETT is advanced into the trachea through the other naris.38 Alternatively, if the ETT cannot be manipulated into the glottis, a guide may be placed in the opposite naris and directed into the trachea. The ETT is then removed and threaded over the guide. A urethral catheter has been used in this manner to assist in the intubation of a 2-week-old neonate with Klippel-Feil syndrome, occipital meningocele, and microretrognathia.89 Another variation of the staged technique involves placement of a larger ETT into the larynx under fiberoptic visualization, followed by removal of the FOB, leaving the larger ETT in the larynx. A bougie is placed through the larger ETT into the trachea, and the ETT is removed. An appropriate-size ETT is then advanced over the bougie into the trachea.90
Flexible FOB intubation through the LMA has been successful.52,53,91 Staged intubation techniques involving the LMA, FOB, guidewires, and catheters (dilators) have been reported, including the use of LMA-assisted wire-guided fiberoptic endotracheal intubation. In a series of 15 cases, Heard and colleagues92 demonstrated that this technique was safe, successful, and easy to learn. After the FOB is placed through the LMA and the vocal cords are visualized, the guidewire is passed through the suction port of the bronchoscope and into the trachea. The LMA and FOB are carefully removed, and the ETT is advanced over the wire. A variation of this theme involves fiberoptic visualization of the glottis through the LMA followed by passage of a guidewire through the suction port of a FOB into the trachea as before. The fiberscope is then removed and an airway catheter or a ureteral dilator passed over the wire into the trachea through the LMA. The LMA is then removed and an ETT advanced over the catheter into the trachea.93 This technique has been used successfully to manage the airway in children with mucopolysaccharidoses. The use of an LMA, an airway exchange catheter (AEC), and a 2.2-mm–OD FOB has also been described.94 After placement of the LMA and visualization of the vocal cords, the fiberscope is removed. The fiberscope is placed into the lumen of a size 11 AEC, which had been cut to 25 cm. This combination was advanced through the LMA into the trachea by a connector. The LMA and FOB are removed, and an ETT is advanced over the Cook AEC.
Przybylo and coauthors95 reported the performance of a retrograde FOI through a tracheocutaneous fistula in a child with Nager’s syndrome. The ultrathin FOB was passed through the fistula in a cephalad direction past the vocal cords and exiting the nares. The ETT was then advanced over the FOB into the trachea.
4 Bullard Laryngoscope
The pediatric Bullard laryngoscope is placed into the oropharynx in the horizontal plane. After passing the tongue, the handle is rotated to a vertical position. One must be careful to stay in the midline as the blade slides around the tongue. The handle then is lifted to visualize the glottis.29 Once the glottis is visualized, a styletted ETT is advanced under direct vision into the trachea.
5 Dental Mirror–Assisted Laryngoscopy
The dental mirror can be used as an adjunct to direct laryngoscopy in order to view an anterior larynx. After direct laryngoscopy is performed, the dental mirror is inserted on the right side of the mouth and angled so that the vocal cords are seen. The handle of the mirror is moved to the left and held by the left hand. An appropriately shaped, styletted ETT then is advanced into the trachea while looking at the dental mirror. Practice is required to develop the necessary coordination. A Stortz no. 3 dental mirror and a Macintosh no. 1 laryngoscope have been used to intubate a 3.9-kg, 2.5-month-old infant.96
6 Retrograde Intubation
The classic retrograde technique involves percutaneous placement of an intravenous catheter through the cricothyroid membrane into the trachea followed by placement of a guidewire. The guidewire exits the mouth or nose and the ETT is then exchanged over the guidewire. If resistance to ETT passage occurs, counterclockwise rotation of the ETT may facilitate placement. This technique has been used for intubation of an infant with Goldenhar’s syndrome.97 A 14-F retrograde intubation set is commercially available from Cook for use with ETTs of ID 5.0 mm or greater.
A combined technique using the FOB and retrograde intubation has been used successfully in management of the difficult pediatric airway as well, as previously mentioned.98
A fiberoptic bronchoscope with a working channel is necessary for the combined technique. The guidewire is threaded into the suction port of an intubating FOB that has a preloaded softened ETT on it. The FOB is passed along the guidewire until it is past the vocal cords. When the scope is past the vocal cords, the wire is withdrawn and the ETT correctly positioned. This technique allows passage without obstruction from the arytenoid cartilage or epiglottis. Oxygen insufflation can be performed through the suction port as well, even with the wire in place. Care must be taken to limit flow to avoid tracheobronchial injury from excessive gas velocity. Audenaert and colleagues99 used this technique in 20 patients with DA age 1 day to 17 years and reported no major complications. Retrograde wire-guided direct laryngoscopy has also been reported for airway management in a 1-month-old infant.100 In that patient, attempts to pass a 2.5-mm ETT over the wire itself were unsuccessful, but endotracheal intubation was achieved over the wire with direct laryngoscopy.
7 Emergency Access
Emergency access is divided into the emergency surgical and the emergency nonsurgical airway.1 Emergency surgical airway access is often difficult and requires the presence of a skilled anesthesiologist. It is the last resort in the “cannot ventilate, cannot intubate” arm of the ASA DA algorithm.101 Three procedures are referred to in this category: emergency tracheostomy, emergency cricothyroidotomy, and percutaneous needle cricothyroidotomy. In children younger than 6 years, emergency tracheostomy is usually the procedure of choice because the cricothyroid membrane is too small for cannulation.99 In older children, percutaneous needle cricothyroidotomy is often preferred over a surgical approach because most anesthesiologists can perform this technique rapidly. Also, there is less risk of injury to surrounding structures.4 Emergency cricothyroidotomy kits are available from Cook with 3.5-, 4-, and 6-mm–ID airway catheters.
The emergency nonsurgical airway access includes use of the LMA, esophageal-tracheal Combitube, and transtracheal jet ventilation (TTJV).1 The Combitube is available in a small-adult size and is contraindicated in patients less than 4 feet tall.7 The LMA is useful in the management of the difficult pediatric airway, as stated previously, as an supraglottic airway device or as a conduit for intubation. However, in the presence of glottic or subglottic obstruction, the LMA is ineffective; TTJV is considered the technique of choice in this situation, as reported in two cases for laser endoscopic surgery.102 Caution with TTJV is urged because serious complications may result from its use.103 TTJV below a glottic or subglottic obstruction may result in barotrauma because the pathway for egress of air and oxygen is limited. Tension pneumothorax has been reported with jet ventilation through an AEC in an adult.104
VIII Complications of Airway Management
Complications that result from intubation in adults can occur in the pediatric population as well. Airway injury accounted for 6% of claims in the ASA closed-claims database.105 Four percent of the airway injury claims involved pediatric patients younger than 16 years. The most frequent sites of injury reported were the larynx (33%), pharynx (19%), and esophagus (18%). Injuries to the esophagus and trachea were more frequently associated with DI. Laryngeal injuries included vocal cord paralysis, granuloma, arytenoid dislocation, and hematoma. Pharyngeal injuries included lacerations, perforation, infection, sore throat, and miscellaneous injuries (foreign body, burn, hematoma, and diminished taste).
An oropharyngeal burn related to the laryngoscope lamp occurred in a term baby weighing 3.6 kg who was easily intubated at birth.106 The laryngoscope was switched on before intubation. Lightbulb laryngoscopes, in contrast to fiberoptic laryngoscopes, can reach temperatures that would result in burns to the oropharynx. Filaments may overlap with use, and it is common for two or more coils to touch.106 The resistance of the lamp decreases and the current increases, thus increasing the temperature. Koh and Coleman106 recommend that all lightbulb laryngoscopes be switched on for less than 1 minute; if left on, the temperature of the bulb should be manually checked before intubation.
Difficult intubation accounted for 62% of all esophageal injuries, with most involving esophageal perforation (90%). Esophageal perforation following DI has been reported in a neonate.107
Laryngotracheal stenosis may be classified as glottic, subglottic, or tracheal. Prolonged intubation seems to be the major etiology. The mechanism responsible seems to be ischemic necrosis caused by pressure from the ETT against the glottic and subglottic mucosa. This results in an inflammatory reaction with a secondary bacterial infection and scar formation. Risk factors include too large an ETT, prolonged intubation, repeated intubation, laryngeal trauma, sepsis, and chronic inflammatory disease.108
The incidence of postintubation croup varies from 0.1% to 1%.20,21 Risk factors include age under 4 years, tight-fitting ETT, repeated intubation attempts, duration of surgery exceeding 1 hour, patient’s position other than supine, and previous history of croup. Reports are conflicting concerning the risk from a concurrent upper respiratory tract infection. Classic treatment consists of humidified air, nebulized racemic epinephrine, and dexamethasone. In pediatric trauma patients, absence of an air leak at extubation was the strongest predictor of postextubation stridor requiring treatment.109
IX Airway Diseases and Implications
A Head Anomalies
2 Specific Anomalies
a Encephalocele
Patients with encephalocele may have other diseases that complicate airway management. The only two syndromes associated with encephalocele in which survival past infancy is likely are Roberts-SC phocomelia syndrome (includes pseudothalidomide syndrome, hypomelia-hypertrichosis–facial hemangioma syndrome) and facioauriculovertebral spectrum (includes first and second brachial arch syndrome, oculoauricular vertebral dysplasia, hemifacial microsomia, Goldenhar’s syndrome). Encephaloceles, or neural tube defects of the head, usually occur in the occipital area, although they may involve the frontal and nasal regions. When large, they affect airway management by interfering with mask fit or laryngoscopy.110
c Mucopolysaccharidoses
The anesthetic morbidity of the mucopolysaccharidoses is 20% to 30%.111 Morbidity is almost always related to respiratory difficulties. Intubation and maintenance of the airway might be difficult because of a variety of upper airway abnormalities, including micrognathia, macroglossia, patulous lips, restricted motion of the temporomandibular joints, friable tissues, and the presence of copious viscous secretions. Semenza and Pyeritz,112 in a retrospective study on 21 patients with the diagnosis of MPS, found that the anatomic factors affecting respiratory status included (1) upper airway narrowing by hypertrophied tongue, tonsils, adenoids, and mucous membranes; (2) lower airway narrowing by GAG deposition within the tracheobronchial mucosa; (3) decreased thoracic dimensions related to scoliosis and thoracic hyperkyphosis; and (4) decreased abdominal dimensions because of lumbar hyperlordosis, gibbus formation, and hepatosplenomegaly. In addition, a short neck and an anterior and narrowed larynx may lead to an increased incidence of difficult or failed intubations.113 In particular, patients with Hunter’s, Hurler’s, or Maroteaux-Lamy syndrome have significantly more airway difficulties as they grow older than MPS patients with other syndromes.114
The incidence of difficult intubation is high. In one review of 34 patients who underwent 89 anesthesias, the overall incidence of DI was 25% and failed intubation, 8%.113 In children with Hurler’s syndrome, incidence of DI was 54% and failed intubation, 23%. Herrick and Rhine115 administered 38 anesthetics to nine patients with MPS (Hunter’s, Hurler’s, Sanfilippo’s, and Morquio’s syndromes) and found an overall incidence of airway-related problems of 26%, with a 53% incidence in patients with the Hurler’s or Hunter’s syndrome.
Belani and associates116 reported their experience with 141 anesthetics in 30 patients with MPS. Visualization of the vocal cords during laryngoscopy was easier in children with Hurler’s syndrome when they were younger (23 vs. 41 months; P ≤ 2.01) and smaller (12 vs. 15 kg; P ≤ 2.05). Also, children with preoperative obstructive breathing had a significantly higher incidence of postextubation obstruction. A total of 28 children underwent bone marrow transplantation; this reversed upper airway obstruction and also reversed intracranial hypertension.
Failure to insert an LMA or nasopharyngeal airway and fatal outcomes have been reported.93,117,118 Consequently, nasotracheal intubation is not recommended, because of difficulties with the anatomy of nasal passages and potential hemorrhage from soft tissue trauma. Accumulation of mucopolysaccharides in the trachea may require a much smaller ETT than usual.117 Tracheostomy can also be difficult technically in these patients and in one case was impossible even postmortem.119
Cervical instability, potential spinal cord damage, and severe thoracic and lumbar skeletal abnormalities make positioning and intubation difficult. In their series, in children with Hurler’s syndrome, Belani and associates116 found a 94% incidence of odontoid dysplasia, whereas 38% demonstrated anterior C1-C2 subluxation. To avoid cervical cord damage in patients with cervical instability, Walker and colleagues113 described manual in-line stabilization during intubation and concluded that a pediatric FOB should be available for all known difficult intubations.113 Tzanova and coworkers120 reported successful anesthesia in a 23-month-old girl with Morquio’s syndrome and unstable neck. The Truview laryngoscope has been used successfully in patients with unstable neck and those with cervical collars. Fiberoptic nasal intubation with spontaneous ventilation has been suggested as the method of choice.
A number of other skeletal deformities should also be considered in MPS patients. Chest cage dysfunction related to kyphoscoliosis leads to reduced vital capacity and restrictive pulmonary disorder.69 Cardiac diseases are common in these patients. Both clinical and histologic studies of the cardiovascular system show progressive involvement of the coronary arteries, heart valves, and myocardium. The lumen of the coronary arteries is narrowed as a result of deposition of collagen and mucopolysaccharides in the intima. Coronary artery involvement and valvular involvement in patients with Hurler’s syndrome have been reported.114,121,122 Complications with 141 anesthetics in 30 patients with MPS included one child with intraoperative stroke and another with pulmonary edema; severe and extensive coronary obstruction was responsible for two intraoperative deaths, and coronary angiography underestimated coronary artery disease.116
Considering the high rate of DI in MPS patients, regional anesthesia seems a good alternative in older children. Failed epidural anesthesia was reported in one patient.117 The deposition of mucopolysaccharides in either the general epidural space or the sheath of the nerve fibers, preventing direct access of the local anesthetic to the nerve, was suspected.
B Facial Anomalies: Maxillary and Mandibular Disease
1 Tumors
a Cystic Hygroma
Cystic hygromas are multiloculated cystic structures that are benign in nature. They form as the result of budding lymphatics and thus may occur anywhere in the body, although most frequently in the neck (75%) and axilla (20%). As the tumor grows, it may cause symptoms from pressure on the trachea, pharynx, blood vessels, tongue, and nerves and eventually may severely compromise the airway. The tongue often protrudes outside the mouth and prevents its closure, making maintenance of the airway difficult if not impossible. Airway obstruction is the most critical complication of the cystic hygroma in the neck. The safest approach in these children seems to be nasal intubation,123 either blind or with fiberoptic assistance with the patient awake. In extreme cases, tracheostomy may be necessary.
b Neck Teratoma
Teratomas of the head and neck frequently arise with respiratory distress or even asphyxia at delivery, and a well-established plan for early airway management should be prepared. If they are untreated, the mortality of patients with these masses is 80% to 100%.124 Fetal ultrasonography has been used since the 1970s to aid in the prenatal diagnosis. Antenatal diagnosis is important for two reasons. First, elective cesarean section should be planned to avoid dystocia and fetal trauma. Second, because immediate establishment of a patent airway is essential for survival, a team of pediatric airway experts must be available.
The ex utero intrapartum technique (EXIT) allows the continuance of fetoplacental circulation during cesarean section. Initially, only the infant’s head and shoulders (but not the placenta) are delivered, thus maintaining uteroplacental blood flow. Intramuscular fentanyl and vecuronium are given, the infant’s airway is secured, and then the umbilical cord is clamped and delivery of the infant completed. The EXIT has proved useful in cases of anticipated DA instrumentation of the neonate (e.g., large fetal neck masses causing airway obstruction).125 Once the head of the neonate is delivered, a multitude of choices are available for airway management: direct laryngoscopy, FOI, pediatric Bullard laryngoscopy,124 or tracheostomy. The EXIT procedure has proved to be safe and efficacious, allowing establishment of an airway in a controlled manner because the placenta allows continued gas exchange during airway manipulation.126,127 Early identification of these masses allows controlled delivery of the neonate in a setting where pediatric anesthesiologists, surgeons, and neonatologists can develop strategies to minimize the risk of a postnatal respiratory death.
c Cherubism
Cherubism is a familial disease of childhood in which patients acquire mandibular and sometimes maxillary enlargement. The mandibular rami hypertrophy, limiting the submandibular space for displacement of the tongue and making visualization of the glottis during direct laryngoscopy difficult.11
2 Congenital Hypoplasia
a Acrocephalosyndactyly
Maxillary hypoplasia results from premature synostosis of facial and cranial sutures and usually manifests as one of multiple abnormal features in a group of rare but complex syndromes called acrocephalosyndactylies. Acrocephalosyndactyly encompasses a number of dysostoses, not all of which can be distinguished clearly. The midface retrusion gives the appearance of prognathia, although in reality the mandible is smaller than normal. In addition, there may be associated anomalies of the central nervous system (CNS; increased intracranial pressure, absent corpus callosum), the extremities, and in a small percentage of patients the heart.128 Both the upper and the lower airway may be compromised in these patients.129
Multiple pathologic conditions may be seen; maxillary regression may be associated with choanal stenosis or atresia, reduction in nasopharyngeal space,130 and palate deformity (narrow, high arched, or cleft). These features may cause respiratory compromise or obstructive apnea early in life, although as the child grows, obstruction can worsen because of continued restriction in growth of the maxillary region.131–134 In one series, upper airway obstruction arose more frequently in Crouzon’s disease and Pfeiffer’s syndrome than in Apert’s syndrome.
The incidence of airway obstruction has been addressed.135 Of a total 40 patients with severe “syndromic” craniosynostosis (13 had Apert’s syndrome and 27 had Crouzon’s disease), 40% presented with airway obstruction (12.5% severe and 27.5% mild obstruction). There was no significant difference in the distribution of airway status between patients with Apert’s syndrome and Crouzon’s disease. The severe obstruction in the five patients resulted from midface hypoplasia, lower airway obstruction, tonsillar and adenoid hypertrophy, and choanal atresia.
Lower airway disease in the acrocephalosyndactylies occurs in the form of tracheomalacia, bronchomalacia, solid cartilaginous trachea lacking tracheal rings, and tracheal stenosis. Patients with tubular cartilaginous trachea have displayed a propensity for easy tracheal injury, edema, and stenosis and a potential for lower airway infection (tracheitis and bronchitis) and mucous plugging, because tracheal ciliary activity may be deficient. Sleep apnea was described in association with tracheal cartilaginous sleeve in a patient with Pfeiffer’s syndrome.136
i Apert’s Syndrome
Apert’s syndrome is characterized by agenesis or premature closure of the cranial sutures, midface hypoplasia, and syndactyly of the hands and feet that is symmetrical and involves at least the second, third, and fourth digits. Prevalence is estimated at 1 in 65,000 live births (~15.5 per 1 million population). Apert’s syndrome accounts for 4.5% of all cases of craniostenosis. Concerning CNS abnormalities, intelligence varies from normal to mental deficiency, although a significant number of patients are mentally retarded. Malformations of the CNS may be responsible for most cases. Papilledema and optic atrophy with loss of vision may be present in cases of subtle increased intracranial pressure. Other abnormalities include cervical spine fusion, which is common and almost always involves C5-C6; osseous fusions may also be evident in other joints of the extremities and in the spine, tracheal cartilage anomalies, and diaphragmatic hernia.137
Airway Anomalies
These result from facial abnormalities, which include small nasopharynx and hypoplastic and retropositioned maxilla. DI in Apert’s syndrome has been reported. One of the suggested mechanisms is trismus related to temporalis muscle fibrosis.138 Both upper and lower airway can be compromised by complete or partial cartilage sleeve abnormalities of the trachea and obstructive sleep apnea.129
ii Crouzon’s Syndrome
Crouzon’s syndrome (Crouzon’s disease, craniofacial dysostosis) is closely related to Apert’s syndrome. In 1912, Crouzon described the triad of skull deformities, facial anomalies, and exophthalmos.139 Crouzon’s syndrome is an autosomal dominant disorder with complete penetrance and variable expressivity.140,141 About 50% of cases represent sporadic mutations, and 40% are familial. In the United States, prevalence is 1 per 60,000 live births (~16.5 per 1 million population). Crouzon’s syndrome makes up approximately 4.8% of all cases of craniosynostosis at birth.142 Crouzon’s disease is associated with acanthosis nigricans (5%) and CNS defects such as chronic tonsillar herniation (73%), progressive hydrocephalus (30%), and syringomyelia.143 Multiple sutural synostoses frequently involve premature fusion of the skull base sutures, causing midfacial hypoplasia, shallow orbits, a foreshortened nasal dorsum, maxillary hypoplasia, and occasional upper airway obstruction.144
iii Saethre-Chotzen Syndrome
Chotzen’s syndrome is an autosomal dominant acrocephalosyndactyly that affects between 1 and 2 of every 50,000 people. Craniosynostosis, facial asymmetry, low frontal hairline, ptosis, brachydactyly, and cutaneous syndactyly of the fingers and of the second and third toes are characteristic features.145
b Acrocephalopolysyndactyly
Acrocephalopolysyndactyly includes the following four types of syndromes:
• Noack’s syndrome: similar to acrocephalosyndactyly type V (Pfeiffer type)
• Carpenter’s syndrome: mental retardation, bradydactyly
• Sakati-Nyhan syndrome: hypoplastic tibias; deformed, displaced fibulas
• Goodman’s syndrome: congenital heart defect, clinodactyly, camptodactyly, ulnar deviation, intact intelligence
i Pfeiffer’s Syndrome (Type I)
Pfeiffer’s (Noack’s) syndrome (type I) is also a close relative of Apert’s syndrome, although it is less severe. Pfeiffer’s syndrome has three clinical subtypes and is manifested by craniosynostosis, broad thumbs and toes, variable maxillary retrusion, and partial soft tissue syndactyly. Type I is classic Pfeiffer’s syndrome; affected patients have normal intelligence and a good prognosis. Type II is associated with cloverleaf skull, severe proptosis, and ankylosis of the elbows (Fig. 36-4). Type III is manifested by the absence of cloverleaf skull but the presence of elbow ankylosis and high morbidity in infancy. Other abnormalities are severe exorbitism that puts patients at risk for corneal exposure and damage, high-arched palate, crowded teeth, hydrocephalus, and seizures.146
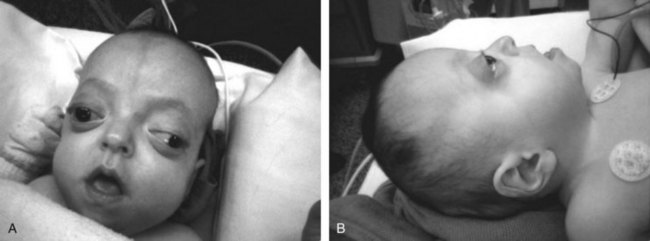
Airway Implications
As with Apert’s syndrome, Pfeiffer’s syndrome can arise with upper and lower airway obstruction. Congenital tracheal stenosis,147 tracheal obstruction related to congenital tracheomalacia,148 and obstructive sleep apnea have been reported.149 In addition to a high incidence of vertebral fusion (73%), other radiologic abnormalities include hypoplasia of the neural arches, hemivertebrae, and a “butterfly” vertebra.150 The C2-C3 level was most often involved, although fusion was noted at all levels of the cervical spine.
ii Carpenter’s Syndrome (Type II)
Carpenter’s syndrome (type II) is typically evident at or shortly after birth. Because of craniosynostosis, the top of the head may appear unusually conical (acrocephaly) or the head may seem short and broad (brachycephaly). In addition, the cranial sutures often fuse unevenly, causing the head and face to appear dissimilar from one side to the other (craniofacial asymmetry). Other malformations of the skull and facial (craniofacial) region may include downslanting eyelid folds (palpebral fissures), a flat nasal bridge, malformed (dysplastic), low-set ears, small dental malformations,151 and underdeveloped (hypoplastic) upper or lower jaw (maxilla or mandible), or both.
Additional abnormalities may include short stature, structural heart malformations (congenital heart defects), mild to moderate obesity, protrusion of portions of the intestine through an abnormal opening in the abdominal wall near the navel (umbilical hernia), or failure of the testes to descend into the scrotum (cryptorchidism) in affected males. Both normal intellect and mild mental retardation have been reported in patients with Carpenter’s syndrome.152,153
c Mandibular Hypoplasia
Mandibular hypoplasia is one of the main anomalies of the mandible, with a profound effect on airway management. Micrognathia results in posterior regression of the tongue and a small hyomental space. The mandible develops from the first branchial arch and is a feature in many rare syndromes (e.g., Pierre Robin, Treacher Collins, Goldenhar’s, Nager’s).154 Although micrognathia is a feature typically shared by these syndromes, they often present additional specific features with adverse effects on the airway.
Micrognathia may affect the airway in three ways: (1) the tongue may not be easily moved during laryngoscopy; (2) if the tongue is not pulled forward in the normal developmental manner, the laryngeal inlet appears more anterior and difficult to visualize; and (3) the oral aperture is not opened as easily or as widely.155 Glossoptosis may further complicate the airway in micrognathic children. Glossoptosis makes displacement of the tongue to the left difficult, so the airway is difficult to visualize.
i Pierre Robin Syndrome
Pierre Robin sequence, which affects 1 in 8500 newborns,156 was described in 1923 by Pierre Robin as airway obstruction associated with glossoptosis and hypoplasia of the mandible. At present, this syndrome is characterized by retrognathia or micrognathia, glossoptosis, and airway obstruction. An incomplete cleft of the palate is associated with the syndrome in approximately 50% of these patients (Fig. 36-5). Pierre Robin sequence results from failure of mandibular growth during the first several weeks of embryogenesis. This causes posterior displacement of the tongue, which prevents normal growth and closure of the palate.
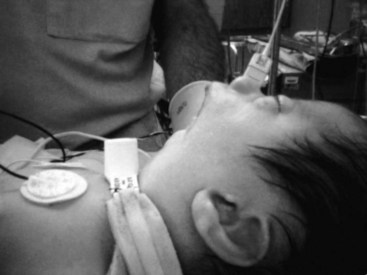
The Pierre Robin sequence represents a spectrum of anatomic anomalies whose common features include mandibular hypoplasia, glossoptosis, and cleft palate. Four types of airway obstruction have been described in patients with Pierre Robin sequence; in only 50% is the obstruction totally related to posterior positioning of the tongue.157 Therefore, glossopexy fails to relieve airway obstruction in approximately half of all symptomatic patients with the Pierre Robin sequence. This may explain why the use of an oral or nasopharyngeal airway alone may not improve an already-difficult mask airway. Patients who fail to improve after glossopexy or nasopharyngeal airway placement, or both, usually require tracheostomy.56
Airway Implications
A large body of literature details airway management of patients with Pierre Robin sequence. Preoperative or postoperative airway obstruction and mask ventilation difficulties have been a frequent problem in these patients. In a 10-year retrospective study of 26 infants with Pierre Robin syndrome, Benjamin and Walker158 found that awake intubation without general anesthesia proved to be safer and less difficult when a special-purpose slotted laryngoscope was used. Li and colleagues159 reviewed the airway management in 110 children with Robin sequence. Prone posturing was effective in the treatment of mild airway obstruction in 82 patients (90.2%) who had noisy breathing sounds. Only 30% of the patients required endotracheal intubation and 6.6% required tracheostomy (all were eventually decannulated).
Alternative intubation techniques used successfully in patients with Pierre Robin syndrome include LMA,56,160–162 FOB intubation,88,163,164 FOI through an LMA,52 rigid nasoendoscope with video camera or video intubation laryngoscope,35 Trachlight with a homemade lighted stylet,165–167 and retrograde intubation.168 Digitally assisted endotracheal intubation and elective endotracheal intubation in prone position have also been reported.78,79
ii Treacher Collins Syndrome
Most children with Treacher Collins syndrome have normal development and intelligence. However, additional physical findings have included a 40% hearing loss, dry eyes, cleft palate, and breathing problems. Both acute and obstructive sleep apneas have been described.169,170
Airway Implications
The airway of children with Treacher Collins syndrome had been successfully managed with an LMA,171–173 the Bullard intubating laryngoscope,30 Augustine stylet,174 and FOB. Rasch and coauthors175 recommend that children with obstructive symptoms have laryngoscopy before anesthetic induction. If the glottic opening is visualized, inhalational induction can proceed. If the glottic structures cannot be visualized, the anesthetist must choose between awake oral or nasal intubation, elective tracheostomy, or FOI.
iii Goldenhar’s Syndrome/Hemifacial Microsomia
Airway Implications
Difficulties in airway management result from mandibular hypoplasia, cleft or high-arched palate, cervical vertebral anomalies, and scoliosis.176 Suggested airway management approaches include using a lighted stylet,177 suspension laryngoscopy,77 or LMA under anesthesia or using awake FOI through a laryngeal mask.178,179
iv Nager’s Syndrome
Nager’s syndrome (mandibulofacial dysostosis) is a rare craniofacial disorder with fewer than 100 cases reported in the medical literature. The morphologic features of Nager’s syndrome include downslanted palpebral fissures, malar hypoplasia, a high nasal bridge, atretic external auditory canals, and micrognathia (severe underdevelopment of the lower jaw). Proximal limb malformations include absent or hypoplastic thumbs, hypoplasia of the radius, and shortened humeral bones.180 Many of the characteristic facial features may be similar to those of Treacher Collins syndrome. However, patients with Treacher Collins syndrome have more severe maxillary and zygomatic hypoplasia, downslanting palpebral fissures, and lower lid coloboma.
Among the additional problems of children with Nager’s syndrome are stomach and kidney reflux and hearing loss. Cardiac and spine defects have been also reported.181 Danziger and coworkers181 reported four patients with a cardiac defect (type unspecified), and tetralogy of Fallot was reported in another patient.154
Airway Implications
Difficulties with airway management and postoperative airway obstruction may occur secondary to mandibular hypoplasia with micrognathia, restricted jaw mobility, and microstomia. Associated cleft lip or cleft palate, or both, and maxillary hypoplasia with midface deformities may further complicate airway management and appropriate mask fit during mask ventilation. The airway has been successfully managed with LMA,182 retrograde intubation,95 and FOI.183
d Smith-Lemli-Opitz Syndrome
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive syndrome characterized by congenital anomalies affecting the airway; cardiorespiratory, gastrointestinal, and genitourinary systems; and CNS. SLOS has an incidence between 1 in 26,500 pregnancies in Canada and 1 in 50,000 pregnancies in the United States.184,185 The syndrome results from an inborn error of cholesterol biosynthesis involving a deficiency of 3β-hydroxysterol δ7-reductase, the enzyme that catalyzes the reduction of 7-dehydrocholesterol to cholesterol.186 Patients with SLOS can have severe growth failure, congenital anomalies affecting most organ systems, early death, developmental delay, and self-injurious and ritualistic behavior.187–189
Airway Implications
Patients with SLOS can be a challenge for airway management because of the typical dysmorphic facial features, including micrognathia, prominent incisors, cleft palate, and a small and abnormally hard tongue. There are several reports of DI and abnormal laryngoscopic views in patients with SLOS.190–192 An LMA was used successfully in managing the airway in a newborn infant with SLOS.193
Quezado and associates194 presented experience from a series of 20 anesthesias in 14 SLOS patients, prospectively deciding to use fiberoptic laryngoscopy as the initial technique of intubation in spite of the possible gastroesophageal reflux,184 muscle rigidity,192 and behavioral abnormalities in these patients.189 In all patients, adequate spontaneous ventilation was maintained throughout the airway management. One patient had laryngospasm during induction, and one was intubated by an otolaryngologist.
e Cornelia De Lange (Cryptophthalmos) Syndrome
Cornelia de Lange syndrome (CDLS) is a syndrome of multiple congenital anomalies transmitted in an autosomal dominant pattern, characterized by a distinctive facial appearance, prenatal and postnatal growth deficiency, feeding difficulties, psychomotor delay, behavioral problems, and associated malformations mainly involving the upper extremities. The incidence is 1 per 30,000 to 50,000 live births.195 A most important feature is a striking delay in the maturation of structure and function of most organ systems, including the CNS.196 CDLS patients are short in stature (the syndrome is also called Amsterdam dwarfism),197 have microcephaly (98%), and the facial features are perhaps the most diagnostic of all the physical signs. Cardiac defects occur in 15% of patients.198
Airway Implications
Intubation may be difficult because of a short (86%), often webbed neck; a high-arched (66%), sometimes cleft palate; and a small mouth with micrognathia (84%). There is also a high incidence of gastroesophageal reflux (58%) and hiatal hernia. There are a number of case reports of DI in CDLS; the airway was successfully managed by blind nasal intubation in one case.199 Lumb and Carli200 reported respiratory arrest in a 3-year-old child after caudal injection of bupivacaine and hypothesized that changes in intracranial pressure secondary to caudal injection might be the cause of the cardiac arrest.
f Hallermann-Streiff Syndrome
Hallermann-Streiff syndrome (oculomandibulodyscephaly with hypotrichosis or oculomandibulofacial syndrome) is rare, with approximately 150 cases reported.201 Cardinal features are dyscephaly with bird facies; frontal or parietal bossing; dehiscence of sutures with open fontanelles; hypotrichosis of scalp, eyebrows, and eyelashes; cutaneous atrophy of scalp and nose; mandibular hypoplasia; forward displacement of temporomandibular joints; high-arched palate; small mouth; multiple dental anomalies; and proportionate small stature.202,203 Children with Hallermann-Streiff syndrome can have a multitude of cardiorespiratory problems. The incidence of cardiac anomalies is 4.8% and includes septal defects, patent ductus arteriosus, and tetralogy of Fallot. Upper airway obstruction may result from small nares and glossoptosis secondary to micrognathia, which may lead to cor pulmonale.201
Airway Implications
The patients have natal teeth, which are brittle and may be easily broken or avulsed during laryngoscopy. The temporomandibular joint (TMJ) may be easily dislocated. At times, the TMJ is absent, making placement of the ETT by the oral route impossible. Small nostrils, deviated nasal septum, high-arched palate, and anterior larynx preclude blind nasotracheal intubation. The ascending ramus of the mandible is either underdeveloped or absent, resulting in a small mouth cavity. Intubation was achieved with difficulty in two cases with the patient under inhaled anesthesia. In both cases, mask ventilation was impossible.203,204 Most patients with Hallermann-Streiff syndrome may require elective tracheostomy because of respiratory difficulty.202
3 Inflammatory Disease
a Juvenile Rheumatoid Arthritis (Still’s Disease)
Difficulty in maintaining the airway patency and inability to intubate the trachea are the most serious anesthetic problems in these children. Severe respiratory distress requiring endotracheal intubation has been reported in children with JRA.207–210 Vetter210 reported an acute exacerbation of JRA, manifesting as acute arytenoiditis and resulting in marked upper airway obstruction. Symmetrical swelling of the arytenoids and moderate swelling of the epiglottis were noted at laryngoscopy. In another case, direct laryngoscopy demonstrated immobile vocal cords, which were approximated to each other in the midline secondary to arthritis of the cricoarytenoid joints.209 In both patients, intubation was achieved with some difficulty during direct laryngoscopy, and both recovered after large doses of steroids. Nevertheless, a fiberoptic bronchocope should always be available in case of failure.
C Mouth and Tongue Anomalies
1 Microstomia
Microstomia (a small mouth opening) is uncommon and may be congenital or acquired. Pediatric microstomia may be congenital (in Freeman-Sheldon [whistling face], Hallermann-Streiff, and otopalatodigital syndromes) but is more often acquired after accidental thermal injuries, such as biting an electrical extension cord or ingesting household lye.98
a Congenital Microstomia
i Freeman-Sheldon Syndrome
Airway Implications
Anesthetic challenges include DA management, intravenous cannulation, and regional technique. Patients may be at increased risk for malignant hyperthermia and postoperative pulmonary complications. Oral FOI is considered the preferred airway management technique; the nasal route cannot be used because of small nostrils.211 An LMA was used successfully in one patient after direct laryngoscopy proved to be impossible.212,213
ii Hallermann-Streiff Syndrome
Airway Implications
Again, these patients have brittle natal teeth that may be easily broken or avulsed during laryngoscopy. The TMJ may be easily dislocated214 and at times is absent, making oral intubation impossible. The small nostrils, deviated nasal septum, high-arched palate, and anterior larynx preclude blind nasotracheal intubation. The ascending ramus of the mandible may either be underdeveloped or absent, resulting in a small mouth cavity. The options available to circumvent these problems are awake intubation, intubation over a fiberoptic bronchoscope, retrograde intubation,214 and intubation under inhalational anesthesia. Even tracheostomy proved to be difficult in these cases; thus an experienced pediatric otolaryngologist should be available.215
b Acquired Microstomia
i Epidermolysis Bullosa Hereditaria Dystrophica
See “Pharyngeal Bullae or Scarring” under “Nasal and Palatal Anomalies.”
Postburn contractures of the neck following a burn injury may hamper cervical hyperextension and lifting of the mandible. Direct laryngoscopy may also be difficult because of rigid scar tissue, which obscures the mandibular and laryngeal anatomy, or microstomia after retraction of scar tissue in facial burns.216 Fiberoptic intubation is the method of choice for securing the airway,217 but LMA can also be used successfully. Kreulen and colleagues216 described a quick surgical neck release of contractures to facilitate endotracheal intubation in postburn patients. Bilateral commissurotomy to allow insertion of the laryngoscope into the mouth is also reported.98
ii Burns From Lye Ingestion
Microstomia from lye ingestion may be associated not only with limited mouth opening but also with such severe intraoral scarring that common landmarks guiding either rigid or flexible fiberoptic laryngoscopy are obscured, rendering oral and nasal intubation difficult or impossible.98,218
2 Diseases of the Tongue
Increase in tongue size is known as macroglossia, defined as a resting tongue that extends beyond the teeth or alveolar ridge.219
a Congenital Disease
i Hemangioma
Hemangiomas are the most common tumor seen during infancy and affect 10% to 12% of white children.220 Most hemangiomas (70%) are seen during the first weeks of life as an erythematous macula or a telangiectasia. All hemangiomas proliferate during the first year of life. Complications include ulceration, high-output cardiac failure, airway obstruction, and the Kasabach-Merritt syndrome, which results from platelet sequestration and destruction within the hemangioma as well as consumptive coagulopathy. It is fatal in 60% of children.
ii Lymphangioma
Lymphangioma is a rare congenital disease of unknown etiology.221 Cystic hygroma of the head and neck, with large lymphatic endothelium-lined cysts, is amenable to surgical excision. Cavernous or microcystic lymphangioma, however, is composed of small lymphatic spaces and poses a therapeutic dilemma by its propensity to cause airway and feeding difficulties and by its tendency to recur despite extensive surgery. All lymphangiomas are present at birth, even though they may not become apparent until the first or second year of life. Although the lymphatic malformation affects preferentially the submandibular space and the neck, it may extend cephalad and invade the tongue and surrounding structures.221
b Traumatic injurY
Burns of the face and mouth can affect the tongue and pharynx. Aspiration of hot liquid can occur in conjunction with upper-body scald burns, leading to acute compromise of the airway, “thermal epiglottitis.” Clinical features and radiologic findings are similar to those seen in patients with acute infectious epiglottitis.224 Thermal epiglottitis can be an extremely difficult problem if subtle signs of impending airway compromise are not appreciated. The treatment should be approached with the same caution and preparedness for emergency airway management as in acute infectious epiglottitis. Immediate endotracheal intubation should be performed in those with acute respiratory distress, and prompt investigation by direct laryngoscopy in the OR is appropriate in those who have not yet developed overt respiratory distress.225 Surprisingly, only 9.2% of 1092 burn patients admitted to the Shriners Burns Institute in Galveston, Texas, over a 5-year period needed endotracheal intubation or tracheostomy for more than 24 hours.226 A similar incidence of endotracheal intubation (10%) was found after accidental inhalation of caustic substances.227
i Lymphatic or Venous Obstruction
Tongue swelling may result from prolonged surgical traction and local mechanical pressure. This may be caused by transesophageal echocardiography probe or by dentures in adults.228,229 Angioneurotic edema or other reactions to drugs can cause marked swelling of the tongue, leading to life-threatening airway emergencies.230–232
c Metabolic Disorders
The Beckwith-Wiedemann syndrome (BWS) comprises a constellation of clinical features including the presence of omphalocele, macroglossia, hypoglycemia (related to hyperinsulinism), inguinal hernia with gigantism, organomegaly, renal medullary dysplasia, cardiac defects, and embryonic tumors occurring less frequently.233
Airway Implications
The anesthetic management of children with BWS may be complicated by a potentially difficult airway related to macroglossia.234–236 Because of the high rate of omphalocele in this syndrome, anesthetic care is frequently required during the neonatal period. The LMA was used successfully in children with BWS.233 Even though endotracheal intubation was possible in most case reports,234,236,237 airway obstruction presented a major concern, especially after extubation. Swelling, secretions, and blood may precipitate complete airway obstruction. Because of the size of the tongue, additional pathology (tongue hematoma and bleeding) can increase the difficulty of airway management and cause postoperative obstruction.235
i Glycogen Storage Diseases
Glycogen storage diseases (GSDs) are a heterogeneous group of inherited disorders involving one of the several steps of glycogen synthesis or degradation. They occur in approximately 1 in 20,000 live births. Isolated deficiencies of virtually all the enzymes involved in glycogen processing have been described. The glycogen present in patients with GSD is abnormal in structure, amount, or both. Of the 10 Cori-type GSDs, only Cori’s type II (Pompe’s disease, also known as generalized glycogenosis or [lysosomal] acid maltase deficiency) is associated with glycogen infiltration of the skeletal muscle of the tongue, which can lead to macroglossia and potential airway issues.238
Airway Implications
Severe macroglossia may lead to airway obstruction during anesthetic induction, emergence from anesthesia, or the postoperative period. Associated cardiomyopathy, myopathy, nervous system involvement (especially the motor neurons in the brainstem and spinal cord), and alterations in the regulation of serum glucose concentrations are part of the clinical presentation. Only a few reports address airway problems related to macroglossia in patients with GSD.239,240
ii Lipid Storage Diseases
Lipid storage diseases are characterized by abnormal sphingolecithin metabolism, which results in an abnormal amount of lipid products being stored in the cells of the reticuloendothelial system. The lipids include cholesterol (xanthomatosis), cerebroside (Gaucher’s disease), and sphingomyelin (Niemann-Pick disease). In Gaucher’s disease, accumulation of the substrate leads to multiorgan dysfunction involving the brain, spleen, liver, lymph node, and bone marrow. Airway difficulties may arise because of trismus, limited neck extension, and upper airway infiltration with glucocerebroside. Kita and colleagues241 found it was impossible to insert an LMA in a 9-year-old child with Gaucher’s disease because of trismus and a narrowed oral cavity. Subsequently, FOI was performed successfully.
iii Neurofibromatosis
Neurofibromatoses are a group of hereditary diseases transmitted in an autosomal dominant manner and characterized by a tendency to form tumors of ectodermal and mesodermal tissues. Two distinct forms recognized on clinical and genetic grounds are designated neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2).242 Von Recklinghausen’s neurofibromatosis (NF1) is one of the most common genetic disorders related to an autosomal dominant mutation and occurs at a frequency of 1 in 3000 to 4000 live births.243 The clinical features of NF1 include café au lait spots; neurofibromas involving the skin, deeper peripheral nerves, nerve roots, and blood vessels; intracranial and spinal cord tumors; kyphoscoliosis; short stature; and learning disability. One feature common to all patients is disease progression over time. NF1 also is associated with a higher incidence of malignant disease than NF2.
Airway Implications
Possible problems in airway management of the patient with NF1 include the presence of intraoral lesions, tumors compromising the airway, and the presence of thoracic deformities or neurologic lesions. Although their presence in the upper airway is rare, neurofibromas may pose a serious problem in airway management. An estimated 5% of NF1 patients have intraoral manifestations of the disease.244 Discrete neurofibromas may involve the tongue or the larynx.245,246 This may cause obstruction, as well as symptoms of dyspnea, stridor, loss or change of voice, or dysphagia, and should warn the anesthetist of potential airway problems.247 Airway obstruction after induction of anesthesia has been reported in patients with a tongue neurofibroma and a neurofibroma involving the laryngeal inlet.244,247 Both patients required emergency tracheostomy. Even if intraoral pathology is recognized preoperatively, elective awake fiberoptic endotracheal intubation may fail because of a grossly distorted anatomy. In addition, the presence of macroglossia, macrocephaly, mandibular abnormalities, and cervical spine involvement may contribute to difficulties of airway management.242
d Tongue Tumors
i Lingual Tonsil Hypertrophy
The lingual tonsil, a normal component of Waldeyer’s ring, consists of lymphoid tissue located at the base of the tongue. Acute inflammation and hypertrophy of lingual tonsils can occur and has been reported as one of the unusual causes of unexpected difficulty with both mask ventilation and endotracheal intubation.248,249 Lingual tonsil hypertrophy (LTH), or lingual tonsillar hyperplasia, has occasionally been reported in children but more often occurs in adults, particularly in atopic individuals.250,251 The etiology is unclear. However, LTH is thought to be a compensatory mechanism following removal of the palatine tonsils or secondary to a chronic, low-grade infection of the tonsils.251,252
Airway Implications
Clinically, LTH is not detectable on routine preoperative physical examination.253 Although many patients are asymptomatic, others may complain of a globus sensation, alteration of voice, chronic cough, choking, or dyspnea.254 Jones and Cohle253 were the first to report a death secondary to failed airway management in a patient with unrecognized LTH. Asai and colleagues248 reported a case of suboptimal ventilation and failed endotracheal intubation using various intubation strategies, including the intubating LMA and FOB.
Enlarged lingual tonsils can impinge against the epiglottis, displacing it posteriorly. This can make mobilization of the epiglottis difficult during direct laryngoscopy. Similarly, FOI is often equally difficult because the posterior displacement of the epiglottis causes interference with the insertion of the tip of the endoscope under it. These difficulties may be compounded by the presence of redundant pharyngeal tissue interfering with fiberoptic exposure and the use of muscle relaxants.253 With the onset of neuromuscular blockade, the pharyngeal musculature relaxes, causing further posterior movement of the tongue and epiglottis.255
In a retrospective study of unexpected DI in 33 patients, Ovassapian and coworkers256 reported that the only finding common to all patients was LTH observed on fiberoptic pharyngoscopy. Most of the patients had normal airway measurements (Mallampati class of I or II), and 36% of patients were difficult to ventilate.
The LMA has been used in “cannot intubate, cannot ventilate” situations caused by LTH with both success and partial success.257–260 Asai and colleagues248 highlighted that the LMA cannot always solve a truly glottic or subglottic problem; rather, the ventilatory mechanism must get below the lesion. If an airway cannot be established with an LMA, TTJV and cricothyrotomy are other options. Crosby and Skene258 recommended the Bullard laryngoscope (which can be fitted with a camera) as the airway device of choice for LTH patients because its robust construction permits gentle manipulation of airway tissues, allowing it to create the necessary endoscopic airspace.
D Nasal and Palatal Anomalies
Nasal obstruction in pediatric patients may result from choanal atresia or stenosis, nasal masses, foreign body, trauma, or adenoidal hypertrophy, as well as choanal stenosis combined with nasal mucosal edema.261,262 These lesions may become evident at birth or later in childhood. Nasopalatal anomalies can result in airway obstruction and feeding difficulty and can complicate airway management.
1 Choanal Atresia
Many patients have associated narrowed nasopharynx, widened vomer, medialized lateral nasal wall, or arched hard palate. Associated malformations occur in 47% of infants without chromosomal anomalies. Such malformations include cleft palate, cleft lip, and Treacher Collins syndrome. The upper airway abnormalities are present in 56% of patients with choanal atresia.263 Nonrandom association of malformations can be demonstrated using the CHARGE association, which appears to be overused in clinical practice. The components of the CHARGE association are coloboma, 80%; heart disease, 58%; atresia choanae, 100%; retarded growth, 87%; development, or CNS anomalies, 97%; genital hypoplasia, 75%; and ear anomalies or deafness, 88%. Other airway abnormalities, as part of the CHARGE association, may be present.264
Airway Implications
Roger and associates265 evaluated the need for a tracheostomy and its timing in 45 patients during the evolution of CHARGE association. They found a high percentage of associated airway abnormalities: pharyngolaryngeal anomalies leading to dyspnea (58%; discoordinate pharyngolaryngomalacia, glossoptosis, retrognathia, laryngeal paralysis, DI) and tracheobronchial anomalies (40%; tracheoesophageal fistula, esophageal atresia, tracheomalacia). Tracheostomy was necessary in 13 patients (29%) despite that the posterior nasal choanae were patent in 10 patients. The authors concluded that often a tracheostomy could not be avoided in these patients, regardless of choanal patency, and that tracheostomy needs to be performed early to avoid hypoxic events.
Asher and coworkers266 studied the association between catastrophic airway events and developmental delay in patients with CHARGE association. They found that children with CHARGE association have a propensity for airway instability, and that cerebral hypoxia contributed to the developmental delay in some of the patients. They recommended early tracheostomy rather than early choanal atresia repair in these patients to protect the CNS.
2 Nasal Masses
Nasal mass lesions are rare disorders in the pediatric population, with an incidence of 1 in 20,000 to 40,000 live births.267,268 Nasal mass lesions are a diverse group of lesions that include anomalies of embryogenesis, such as encephaloceles, dermal and nasolacrimal duct cysts, tumors, and inflammatory processes.267 Encephaloceles represent herniation of CNS tissue at the level of the cranium. Although most encephaloceles are located in the occipital area, some occur anteriorly and may contain various quantities of brain tissue. Encephaloceles may be associated with midline defects. Dermal cysts become evident as hard intranasal masses that result from herniation of dura and subsequent contact with the skin. These midline defects may manifest as a nasal obstruction without a facial mass. There is a risk of local abscess formation and intracranial infection.
Airway Implications
Nasal masses can affect the management of the airway by interfering with mask ventilation or with direct laryngoscopy and endotracheal intubation. Nasotracheal intubation in these patients should be avoided. Extension of a cephalocele through a palatal defect interfered with endotracheal intubation in one patient.269 All the airway implications previously discussed under choanal atresia with unilateral (or even bilateral) obstruction are valid in patients with nasal airway obstruction.
3 Palatal Anomalies
Anomalies of the palate include cleft and high-arched deformities and hypertrophy of the alveolar ridge area. In two studies of children undergoing palate repair, the incidence of difficult laryngoscopy (Cormack and Lehane grades III and IV) was 6.5% and 7.4%.263,270 Of the 59 patients with difficult laryngoscopy in Gunawardana’s study,263 2.95% had unilateral cleft lip, 45.76% had bilateral cleft lip, and 34.61% had retrognathia. Interestingly, endotracheal intubation was successful in 99% of patients in whom laryngoscopy was difficult (failed intubation was 1%). There was a significant association between age and laryngoscopic view: 66.1% of patients with difficult laryngoscopies were younger than 6 months, 20.3% were 6 to 12 months, and 13.6% were 1 to 5 years old.
The presence of other associated congenital anomalies, including cardiac and renal anomalies, should always be remembered, particularly in children with isolated cleft palate. More than 150 syndromes have been described in association with cleft lip or palate, but fortunately all are rare. Some, however, have considerable anesthetic implications, and many involve potential airway problems, including the well-known Pierre Robin, Treacher Collins, and Goldenhar syndromes. Other, such as Klippel-Feil syndrome, may include abnormalities of the cervical spine.271
Henriksson and Skoog272 reviewed the records of 154 patients who underwent closure of the palate and found that 84% had isolated cleft palate, 12% had Pierre Robin syndrome, and the rest had other identified syndromes. The risk of anesthetic complications was four times greater with surgery in children less than 1 year of age, with a sixfold increase when a more elaborate velopharyngoplasty technique was used.
The postoperative airway complications ranged between 5.6% and 8% in two surveys.273,274 As a rule, patients with cleft palate with the Pierre Robin sequence or other additional congenital anomalies had an increased risk for airway problems after palatoplasty.
Palatal edema or hematoma may also develop. Swelling limited to the soft palate or uvula can cause posture-dependent airway obstruction in children.275 Edema may result from instrumentation of the airway, burn injury, allergy, or infectious agents.
Many methods of management of DA in patients with cleft palate have been described. The use of firm pressure over the larynx (cricoid pressure) to aid laryngoscopy, with a bougie as a guide to endotracheal intubation, is relatively simple to perform by any competent anesthetist and is usually successful.263 Other techniques (e.g., LMA, FOB) have been described,271 especially when cleft palate is associated with different syndromes.
4 Adenotonsillar Disease
Together, the lingual tonsils anteriorly, the palatine tonsils laterally, and the pharyngeal tonsils (adenoids) posterosuperiorly form a ring of lymphoid or adenoid tissue at the upper end of the pharynx known as Waldeyer’s tonsillar ring. All the structures of Waldeyer’s ring have similar histology and function, and regarding airway management, they produce similar symptoms and require treatment. In response to recurrent infections, adenoids and tonsils can hypertrophy and lead to airway obstruction.276
Adenoidal hypertrophy peaks at 4 to 6 years of age and disappears by adolescence. Although a disease of the older child, hypertrophy can occur in the infant. One of the major complications of adenoidal hyperplasia is obstructive sleep apnea (OSA). Signs and symptoms of airway obstruction include snoring and restless sleep, somnolence during the day, noisy breathing, mouth breathing, hyponasal speech, persistent nasal secretions, apnea, choking during feeding, respiratory distress, and behavioral disturbances.163 If the condition is left untreated, failure to thrive; a characteristic, long adenoid facies with open mouth, palate, and dental malformations; and cardiovascular changes (cor pulmonale) reflective of chronic hypoxemia and hypercapnia may develop.277
Airway obstruction resulting from adenoid tissue is determined not by the absolute size of the adenoids but rather by their size relative to the volume of the pharynx.249 Patients with preexisting diseases that reduce nasopharyngeal size or alter its integrity may have airway obstruction with only mild degrees of adenoidal hyperplasia. Examples are children with craniofacial anomalies (in whom the nasopharynx may be reduced in size) and those with nasal polyps, septal or turbinate malformations, MPS, or deficient pharyngeal support (Down syndrome).
Tonsillar hyperplasia is a physiologic phenomenon of childhood that peaks at about 7 years of age. It can cause OSA with restless sleep and an irregular breathing pattern, snoring, and intermittent periods of apnea as well as daytime somnolence, irritability, and poor school performance.278 Long-standing partial obstruction of the airway can be associated with repeated hypoxic episodes and may result in pulmonary hypertension, cor pulmonale, and right-sided heart failure. Acute exacerbation of adenotonsillar hypertrophy may necessitate an emergency securing of the airway.279,280
The treatment of adenoidal and tonsillar hyperplasia is adenoidectomy and tonsillectomy. These are among the most common surgical procedures in children. There are multiple indications for excision of tonsils and adenoids.276 Upper airway obstruction is of most concern for the anesthesiologist because these patients may have airway obstruction both during induction of anesthesia and in the postoperative period.
Airway Implications
Upper airway obstruction may occur after premedication, during induction of anesthesia, or following tracheal extubation. Visualization of the glottis during direct laryngoscopy may be difficult with tonsillar hypertrophy. Resection of tonsils and adenoids may not result in immediate relief of airway obstruction. Bleeding and edema can make the child susceptible to postoperative airway obstruction. Although it usually causes chronic upper airway obstruction, adenotonsillar hypertrophy can result in acute airway obstruction.279–281 Airway assessment and management of patients with OSA caused by adenotonsillar hypertrophy are detailed in the next section.
Peritonsillar abscess in children manifests as a purulent mass surrounded by the tonsillar capsule. It occurs more frequently in untreated children with chronic tonsillitis or those who have been inadequately treated.276 Signs and symptoms include fever, sore throat, tonsillar mass, dysphagia, drooling (caused by odynophagia and dysphagia), muffled voice, trismus (caused by irritation of pterygoid muscle by pus and inflammation), and variable degrees of toxic state. Peritonsillar abscess requires intravenous antibiotic therapy. If symptoms of airway obstruction develop or the patient fails to respond to medical therapy, needle aspiration, incision, and drainage with tonsillectomy are recommended.276 In a prospective study of 50 adult patients with peritonsillar abscess, the Mallampati score did not correlate with the Cormack and Lehane glottic view during laryngoscopy because of palatopharyngeal arch distortion. There were no DIs in this study group.282
5 Obstructive Sleep Apnea
a Definition
Obstructive sleep apnea syndrome (OSAS) in children is a disorder of breathing during sleep characterized by prolonged partial upper airway obstruction or intermittent complete obstruction (obstructive apnea) that disrupts normal ventilation during sleep and normal sleep patterns.283
b Prevalence of Snoring
The prevalence of primary snoring ranges from 3.2% to 12.1%,39 whereas the prevalence of OSAS ranges from 0.7% to 10.3%.39,284,285 The ability to maintain upper airway patency during the normal respiratory circle is the result of a delicate equilibrium between various forces that promote airway closure and dilatation. This “balance of pressure” concept was first proposed independently by Remmers and colleagues286 in 1978 and Brouillette and Thach287 in 1979 and represents the current thought regarding the pathophysiologic mechanisms of OSAS.
The four major predisposing factors for upper airway obstruction are as follows:
• Anatomic narrowing. The upper airway behaves as predicted by the Sterling resistor model; the maximal inspiratory flow is determined by the pressure changes upstream (nasal) to a collapsible site of the upper airway, and flow is independent of downstream (tracheal) pressure generated by the diaphragm.
• Children with OSAS close their airways at the level of enlarged adenoids and tonsils at low positive pressures, whereas healthy children require subatmospheric pressures to induce upper airway closure.288
• Abnormal mechanical linkage between airway dilating muscles and airway walls. Control of the upper airway size and stiffness depends on the relative and rhythmic contraction of a host of paired muscles, which include palatal, pterygoid, tensor palatini, genioglossus, geniohyoid, and sternohyoid. With contraction, these muscles promote motion of the soft palate, mandible, tongue, and hyoid bone. The activity of these muscles is dependent in particular on the brainstem respiratory network. Wakefulness conveys a supervisory function that ensures airway patency, and sedative agents that compromise genioglossal muscle activity may result in significant upper airway compromise. Roberts and others289 demonstrated that mechanoreceptor- and chemoreceptor-mediated genioglossal activity is critical for maintenance of upper airway patency in both normal and micrognathic infants.
• Muscle weakness. There is little evidence to suggest that intrinsic muscle weakness is a major contributor to upper airway dysfunction. Nevertheless, in patients with neuromuscular disorders, airway obstruction is frequently observed during sleep.290
• Abnormal neural regulation. Subtle alterations in central chemoreceptor activity were found by different researchers. Gozal and others291 reported that arousal to hypercapnia was blunted, whereas Onal and coworkers292 found that upper airway musculature is more stimulated than the diaphragm.
c Pathophysiology and Clinical Picture
The etiology and pathophysiology of obstructive sleep apnea in children are multifactorial, with anatomic and neuromuscular abnormalities playing a major role in the disorder.293–297 Others, however, downplay the role of neuromuscular factors because the vast majority of children with OSAS can be cured by correcting anatomic obstructions. The narrowing of the airway lumen by hypertrophied lymphoid tissue, compliance, elasticity of the pharyngeal soft tissue, facial morphology, and the physiologic changes that occur in the pharyngeal dilators during sleep determine the severity of airway collapse.
One of the hallmarks of sleep-disordered breathing is fragmentation and disruption of normal sleep architecture. By definition, deeper levels of sleep, especially rapid eye movement (REM) sleep, are less susceptible to arousal from various stimuli, including adverse ventilatory events.298 Oxyhemoglobin desaturation therefore tends to be more frequent and more severe during REM sleep. The hypercapnia and hypoxemia and resulting arousals associated with OSAS, at least in part, often result in a reduction in REM sleep.298,299
Although OSAS and hypertension are often associated in adults, children with OSAS also tend to have higher diastolic blood pressure. The cardiovascular changes appear to be the result of an increase in sympathetic tone that results from the sleep arousals, which in turn are related to the obstructive respiratory events.300 The clinical presentation of OSAS in children has many similarities and important differences compared with the disorder in adults245,299,301 (Table 36-4).
TABLE 36-4 Adult vs. Childhood Obstructive Sleep Apnea Syndrome (OSAS)
| Features | Adult OSAS | Childhood OSAS |
|---|---|---|
| Snoring | Intermittent | Continuous |
| Mouth breathing | Uncommon | Continuous |
| Obesity | Common | Uncommon |
| Failure to thrive | — | Common |
| Daytime hypersomnolence | Common | Uncommon |
| Gender predilection | Male | None |
| Most common obstructive event | Apnea | Hypopnea |
| Arousal | Common | Uncommon treatment |
| Nonsurgical | CPAP in majority | CPAP in minority |
| Surgery | Selected cases in majority | T&A |
CPAP, Continuous positive airway pressure; T&A, tonsillectomy and adenoidectomy.
Unlike findings in adults, obesity is not a common factor in pediatric OSAS, although its role increases with the age of the child.302 Abnormal sleep positions with preference for an upright position and hyperextension of the neck have been noted in children with sleep-related breathing disorders.303
Prolonged exposure to hypoxia and hypercarbia results in compensatory changes in the pulmonary vasculature. Pulmonary vascular resistance increases, causing increased right ventricular strain.304 Severe cases may progress to pulmonary hypertension, dysrhythmias, and cor pulmonale.305
d Laboratory Evaluation
Polysomnography (PSG) remains the gold standard for the diagnosis of OSAS in adults and children. In 1995 the American Thoracic Society adopted guidelines for performing PSG in children.306 Use of PSG was recommended to differentiate primary snoring, which does not require any form of treatment, from OSAS, which can lead to cardiopulmonary dysfunction and functional impairment if left untreated.307 In general, studies show that history alone does not have sufficiently high diagnostic sensitivity or specificity to be the basis for recommending therapy.308
In a study of 50 healthy children, Marcus and colleagues309 reported normal PSG values for the various respiratory events. The apnea indices (number of apneas per hours of total sleep time, TST) were 0.1 ± 0.5, with the minimum oxygen saturation being 96%, maximal drop in saturation 4%, and CO2 over 55 mm Hg no more than 0.5% of TST. An abnormal study includes an apnea index greater than 1, oxygen desaturation greater than 4% more than three times an hour or associated with a greater than 25% change in heart rate, oxygen desaturation less than 92%, and elevation of end-tidal CO2 to more than 52 mm Hg for more than 8% of TST, or 45 mm Hg for more than 60% of TST (Table 36-5).
| Measurement | Normal Values |
|---|---|
| Sleep latency (minutes) | >10 |
| TST (hours) | >5.5 |
| REM sleep (%) | >15% TST |
| Stage 3 and 4 non-REM sleep (%) | >25% TST |
| Respiratory arousal index (no./hr TST) | >5 |
| Periodic leg movements (no./hr TST) | >1 |
| Apnea index (no./hr TST) | >1 |
| Hypopnea index (nasal/esophageal pressure catheter; no./hr TST) | <3 |
| Respiratory disturbance index (RDI) (Apnea/hypopnea index) |
<1 |
| Nadir oxygen saturation (%) | <92 |
| Mean oxygen saturation (%) | <95 |
| Desaturation index (<4% for 5 sec; no./hr TST) | <5 |
| Highest CO2 (mm Hg) | 52 |
| CO2 < 45 mm Hg | >20% TST |
TST, Total sleep time; REM, rapid eye movement.
e Airway Implications
Medical therapy of pediatric OSA is not considered to be consistently effective. Systemic or topical steroids may shrink lymphoid tissue, but the long-term effectiveness is not known, and a short course of systemic corticosteroids appears to be ineffective. Topical intranasal steroids appear to reduce the severity of OSAS.310
Adenotonsillectomy remains the mainstay of treatment for pediatric OSA.311 The optimal age for adenotonsillectomy is 4 to 7 years, although a young age, even under 1 year, is not a contraindication for surgery for airway obstruction or OSA. Children with Down syndrome deserve further comment because they frequently have severe OSA.312 Although data are conflicting on the usefulness of adenotonsillectomy in this group, it appears worthwhile if the tonsils or adenoids are obstructing the airway. If an adenotonsillectomy fails or is not considered appropriate therapy, uvulopalatopharyngoplasty may be effective.39
Minimal specific evidence exists for or against the use of opiates and sedatives in the perioperative period in children with OSAS. To date, there are only anecdotal reports of respiratory depression in children in response to sedatives such as chloral hydrate and in the postoperative period,313,314 including hypoxia.315–317 Children with OSAS appear to have increased sensitivity to opioids.
Waters and associates318 found that children with OSAS develop more pronounced respiratory depression than with aged-matched control subjects when breathing spontaneously under anesthetic with the upper airway secured.318 Addition of a small dose of opioids increased the respiratory depression in children with OSAS. The low dose of fentanyl used (0.5 µg/kg) precipitated central apnea in 46% of the OSAS group. In this study, the best predictor of opioid-induced central apnea was an increase in end-tidal CO2 to levels greater than 50 mm Hg during spontaneous breathing after anesthetic induction. In contrast to the previous studies,316,318–320 Wilson and coauthors321 found no correlation between the preoperative cardiorespiratory sleep study (PSG and home sleep studies) parameters and opioid administration and postoperative outcome.
Few studies provide data pertaining to complications of surgery in children undergoing adenotonsillectomy for upper airway obstruction. All specifically address the risk of postoperative respiratory obstruction315–317,319–324 (Table 36-6). The authors define “respiratory compromise” in various ways but generally consider the need for supplemental oxygen as a minimum criterion. The papers report a wide range for the incidence of postoperative respiratory complications (0-27%), primarily because their populations include different proportions of children with neuromuscular, chromosomal, and craniofacial disorders.
TABLE 36-6 Respiratory Compromise After Adenotonsillectomy in Children with Obstructive Sleep Apnea Syndrome (OSAS)
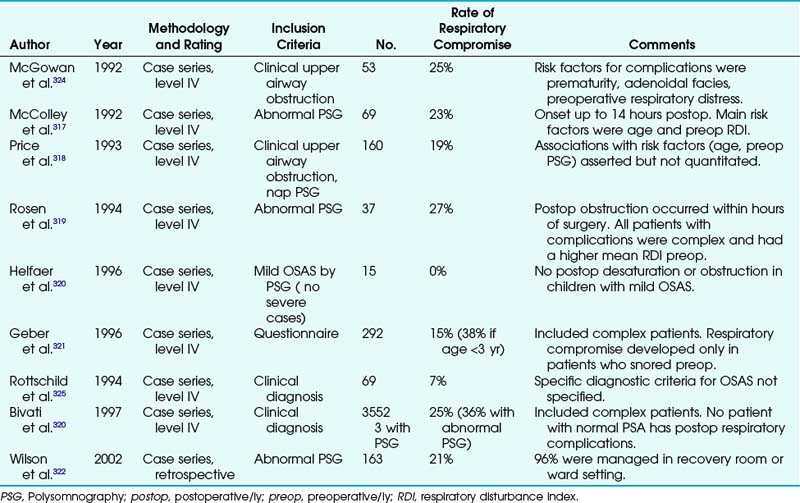
Young age (<3 years) and associated medical problems were found in most studies to define the highest-risk groups. A high preoperative respiratory disturbance index (apnea/hypopnea index) also seems to be a risk factor for postoperative complications.316,317 Time to onset of respiratory compromise after adenotonsillectomy appears to be brief, although McColley and others316 reported that one patient required 14 hours to manifest respiratory symptoms. Postobstructive pulmonary edema may develop in some children undergoing adenotonsillectomy for relief of upper airway obstruction. The incidence of this complication is unknown, and pulmonary edema often manifests immediately after endotracheal intubation.
The patient’s position, especially after extubation, seems to be important for the development of airway obstruction. Ishikawa and colleagues325 found that prone position increases upper airway collapsibility in anesthetized infants. Isono and associates,326 in a study of adult patients, reported that lateral position structurally improves maintenance of the passive pharyngeal airway in OSA patients. These findings are in concordance with the current practice of extubating and transporting children in the lateral position.
In a retrospective study of 163 OSAS children, Wilson and coworkers321 found a 21% incidence of respiratory compromise requiring medical interventions after adenotonsillectomy. Ninety-six percent of the children with OSAS were managed in a recovery room or ward setting. Six children required postoperative admission to the intensive care unit (ICU).
Most of the polysomnographic studies done weeks after adenotonsillectomy in children with OSAS reported a cure rate between 85% and 100%.327,328 A major concern in the immediate postoperative period is the effect of residual anesthesia, pain, sedative and analgesic medication, and edema of the pharyngeal tissues on the complication rate in this category of patients. Helfaer and coauthors315 tried to respond to this question by comparing preoperative and first-night postoperative polysomnograms in children with mild OSAS.315 Surprisingly, most of the children had improvements in their sleep studies on the night of surgery. These findings were not affected by the choice of intraoperative anesthetic. Specifically, intraoperative administration of narcotics was not associated with postoperative respiratory impairment. Even though this study was performed on a relatively small number of patients with mild disease, it was concluded that children with mild OSAS can be safely discharged home on the day of surgery (see Table 36-6).
6 Retropharyngeal and Parapharyngeal Abscesses
Retropharyngeal abscess is a rare but potentially fatal infection of the pharyngeal wall. It occurs primarily in pediatric patients; in one study more than half of the patients were younger than 12 months.329 In children, it usually results from suppurative involvement of lymph nodes located in the retropharyngeal space. These nodes drain lymph from the pharynx, nasopharynx, paranasal sinuses, and middle ear. The most common pathogens are Staphylococcus aureus (25%), Klebsiella species (13%), group A streptococci (8%), and a mixture of gram-negative and anaerobic organisms (38%).329,330 Other causes of retropharyngeal abscess include spread of infection from pharyngitis or peritonsillar abscess, penetrating trauma, and foreign body ingestion.
Clinical presentation of retropharyngeal abscess varies with the patient’s age. Most children have fever, some degree of toxic appearance, a hyperextended or stiff neck, dysphagia, drooling, trismus, muffled voice, and respiratory distress. Infants and young children may have stridor. Older children with mediastinal involvement may, in addition, complain of chest pain. Physical examination may reveal cervical lymphadenopathy and pharyngeal swelling. A lateral radiograph of the neck typically shows widening of the retropharyngeal prevertebral soft tissue. CT is helpful in the diagnosis of retropharyngeal abscess but has difficulty differentiating cellulites and abscess. Lateral neck radiography was found to be very specific when the air sign was present.331 Ultrasound imaging can also distinguish between suppurative and presuppurative stage.332 Chest radiographs may show mediastinal involvement and tracheal deviation.333
Airway Implications
The danger of retropharyngeal abscess is related to the potential for rapid progression to airway obstruction. In one report, 5 of 65 patients required tracheostomy.334 There is also an ever-present risk of abscess rupture and aspiration of pus into the airway. The clinical presentation of children with retropharyngeal abscess can mimic that of children with epiglottitis and croup. The mortality rate is high; exact incidence is not known. In a retrospective study, Ameh335 reported two deaths among 10 children surveyed; one child died before the abscess was drained, and the other died in the postoperative period because of laryngospasm. Coulthard and Isaacs329 reported two deaths in 31 children with retropharyngeal abscess.
7 Pharyngeal Bullae or Scarring
Epidermolysis bullosa (EB) describes a group of genetically determined mechanobullous disorders that vary in course and severity, ranging from relatively minor disability to death in early infancy.336,337 They are characterized by an excessive susceptibility of the skin and mucosa to separate from the underlying tissues and form bullae following minimal mechanical trauma. The affected areas can be considerable in size as the bullae enlarge by expanding and tracking along the natural tissue planes. As with all blisters, they can be extremely painful. More than 20 types of EB are described,338 with three major subtypes: dystrophic, simplex, and junctional, with each broad category of EB containing several subtypes.
a Dystrophic Epidermolysis Bullosa
Dystophic epidermolysis bullosa (DEB), which was first described by Fox in 1879, is probably the most frequent type of EB to have surgical treatment.339,340 The prevalence of DEB is approximately 2 in 100,000 children.341 The majority of DEB patients have wounds that are present at birth or shortly after, with a variety of blister sizes, some even exceeding 10 cm in diameter. The blisters of DEB are usually flaccid and filled with either a clear or a blood-stained fluid. New blisters tend to develop less frequently as the child ages. Scarring is unusual after a single episode of blistering, but blistering is much more easily provoked in previously blistered areas; it is this recurrence that causes atrophic scars to form. As a result of repeated skin infection, injury, and healing, patients with the dystrophic form develop contractures, which may involve the skin of the neck and mouth.
b Epidermolysis Bullosa Simplex
Almost all cases of epidermolysis bullosa simplex (EBS) are inherited in an autosomal dominant manner.339 Although the exact prevalence of EBS is not known, it is thought to be approximately 1 or 2 in 100,000 children.336 There are three major subtypes: Dowling-Meara, Weber-Cockayne, and Koebner.
d Airway Implications
Children with EB, especially DEB, are more likely to have airway management problems, with the risk of DI secondary to contracture formation. In addition to oral, pharyngeal, and laryngeal problems, head and neck skin involvement and contractures may make positioning for laryngoscopy difficult.342 A DI should always be suspected and contingency plans made before embarking on anesthesia. To avoid prolonged facial manipulation during the procedure, airway maintenance by intubation is often preferred.343 To reduce the risk of new laryngeal bullae formation, an ETT a half to one size smaller than predicted may be necessary. If a cuffed tube is required, the cuff should be slightly inflated. The risk of bullae formation after intubation is low because the larynx and trachea are lined with ciliated columnar epithelium rather than the squamous epithelium that lines the oropharynx and esophagus.344
Although securing the ETT by wiring it to a tooth has been advocated,345 a more conservative approach is to tie the tube in place with either ribbon gauze or Vaseline gauze and a collar of adhesive tape around the ETT to prevent the ties from slipping. Nasal intubation can be performed, preferably with a fiberoptic scope, but blind nasal intubation should be avoided. Blind techniques (e.g., blind oral intubation or lighted stylet) have been used successfully but may result in trauma to the laryngeal structures if multiple unsuccessful attempts are required and probably should be avoided.346
E Laryngeal Anomalies
1 Laryngomalacia
Laryngomalacia is the most common congenital abnormality of the larynx and is characterized by a long, narrow epiglottis and floppy aryepiglottic folds.341 It is the most common cause of noninfective stridor in children.341 Stridor, usually present at birth, may appear after weeks or months. It may appear only with crying or in the presence of an acute upper respiratory infection. The stridor is inspiratory, high pitched, and more obvious in the supine position.197 In the mild form, stridor peaks at 9 months and then levels off, declines, and disappears by 2 years of age.347 Severe laryngomalacia may cause upper airway obstruction, cyanosis, failure to thrive, and cor pulmonale. GER has been reported as well, and antireflux therapy is recommended.348
2 Epiglottitis
Epiglottitis, more appropriately called supraglottitis, is a life-threatening infection of the epiglottis, aryepiglottic folds, and arytenoids. It is a true airway emergency because supraglottitis may progress rapidly to complete airway obstruction. Supraglottitis is classically described as occurring between 2 and 8 years of age, although it can occur in infants, older children, and adults.351 Haemophilus influenzae type B (Hib) is the most common causative agent, although other organisms have been reported. Pseudomonas, group A β-hemolytic Streptococcus, and Candida have been reported in the literature as etiologies of epiglottitis as well.351–353 The introduction of the H. influenzae conjugate vaccine has dramatically reduced the incidence of supraglottitis, but vaccine failure does occur.353 A high index of suspicion for the diagnosis of supraglottitis should be maintained because the disease has not been completely eliminated.
Children with epiglottitis often present with the four Ds of supraglottitis: drooling, dyspnea, dysphagia, and dysphonia. These children are described as “toxic appearing” and anxious, preferring to rest in the tripod position (upright sitting position, leaning forward with the mouth open).352 High fevers and signs of respiratory distress evolve over a few hours. Stridor, if present, is usually inspiratory.354
Diagnosis is usually based on clinical findings. Radiographs are indicated only if the child has no respiratory distress and a physician capable of controlling the DA is in attendance. A lateral neck radiograph obtained with hyperextension during inspiration is the single best exposure. Classic findings include round, thick epiglottis (thumb sign), loss of the vallecular airspace, and thickening of the aryepiglottic folds.351 Definitive diagnosis is made at laryngoscopy in the OR. No one should attempt to visualize the posterior pharynx in the emergency room. Dynamic airway collapse may occur, and complete obstruction ensues.
Airway Implications
The mainstay of therapy for supraglottitis is to obtain an airway, usually with a multidisciplinary approach in an organized and controlled manner. An otolaryngologist capable of performing an emergency tracheostomy is present at the induction. The difficult pediatric airway cart, a rigid bronchoscope, and tracheostomy set must be in the OR. When dealing with the child with epiglottitis, it is vital that the child remain calm. If separation from the parents is too stressful, parental presence at induction, after proper preparation, should be considered. Sedation is not advised in this situation. After placement of a precordial stethoscope and pulse oximeter, a gradual inhalation induction with 100% oxygen is performed with the child in the sitting position. Maintenance of spontaneous ventilation is crucial; CPAP at 10 cm H2O may be beneficial in maintaining a patent airway. Once anesthesia is induced, an intravenous line is placed and a volume bolus of 10 to 30 mL/kg of lactated Ringer’s solution is given. The rest of the monitors are applied, and atropine or glycopyrrolate is given intravenously before laryngoscopy for its antimuscarinic effect. After an adequate depth of anesthesia is obtained, direct laryngoscopy is performed and an oral ETT is placed. Identification of a cherry-red edematous epiglottis is diagnostic. A tip for successful intubation is that gentle pressure applied to the chest may reveal expiratory gas bubbles. A styletted ETT, one or two sizes smaller than predicted, is placed into the trachea.354 If the patient cannot be intubated, the DA algorithm is followed. Rigid FOB may be attempted if the condition permits, or a surgical airway is obtained.
After the appropriate cultures are obtained, antibiotic therapy is initiated. Some advocate changing the oral ETT to a nasotracheal tube because of the greater stability of the nasotracheal tube. The mean duration of intubation ranges from 30 to 72 hours. Extubation is performed when the patient demonstrates clinical improvement and there is evidence of an air leak around the ETT. Some clinicians advocate the use of dexamethasone before extubation to reduce the incidence of postextubation stridor.351
3 Congenital Glottic Lesions
Congenital laryngeal anomalies include laryngomalacia, vocal cord paralysis, laryngeal web, and atresia. Vocal cord paralysis is the second most common cause of congenital laryngeal malformations.355 Bilateral vocal cord paralysis is often associated with CNS abnormalities such as Arnold-Chiari malformation. Birth trauma may also induce vocal cord paralysis. The presentation of bilateral vocal cord paralysis is high-pitched inspiratory stridor and a normal or mildly hoarse cry. Severe airway obstruction may develop that requires emergency intubation or tracheostomy.355 Occasionally, vocal cord paralysis resolves spontaneously or after a ventriculoperitoneal shunt is placed.356 In unilateral paralysis, the left side is more frequently affected. Cardiovascular and mediastinal problems are often associated with unilateral paralysis.355 Unilateral paralysis arises with a weak cry.276 It seldom requires surgery.356
Laryngeal atresia is a rare and often fatal anomaly. Survival depends on the presence of an associated tracheoesophageal fistula or immediate tracheostomy at birth.355
4 Recurrent Respiratory Papillomatosis
Laryngeal papillomatosis, or recurrent respiratory papillomatosis (RRP), is the most frequent benign tumor of the larynx, with an incidence in the United States of 4.3 per 100,000 children. It is caused by the human papillomavirus (HPV) types 6 and 11. It is also the second most common cause of hoarseness in children.357 Laryngeal papillomas are located primarily in the larynx on the vocal cord margins and epiglottis; however, any part of the respiratory tract may be affected.358 RRP may affect children and adults. The juvenile form is often more aggressive than the adult form of the disease. Pediatric patients with RRP often wheeze and the diagnosis may be delayed. The primary symptom of RRP is hoarseness or a weak cry. Stridor is often the second symptom to develop, usually starting as inspiratory and progressing to biphasic with advancing disease.357 Other symptoms may include chronic cough, paroxysms of choking, failure to thrive, and respiratory fatigue.358 Diagnosis is made with a flexible fiberoptic nasopharyngoscope. If patient’s cooperation limits the examination, general anesthesia may be needed. Treatment consists of CO2 laser microlaryngoscopy, which vaporizes the lesions and causes minimal bleeding. Frequent surgical procedures may be required to control the disease. Medical management includes the use of acyclovir, interferon-α, cidofovir, and indole 3-carbinol.358
Airway Implications
Airway obstruction has been reported with induction of anesthesia in patients with RRP.359 Anesthetic evaluation should include careful preoperative assessment of the airway and the emotional status of the child.360 Sedation is necessary because these patients require frequent surgeries, but it should be avoided in patients with respiratory compromise. In appropriate cases, parental presence in the OR may be beneficial. Anesthesia should be induced with an inhalational induction in 100% oxygen while maintaining spontaneous respirations. Patients may be apprehensive about the mask, and an alternative technique, such as cupping the hands around the circuit to increase the concentration of the inhalational agent, may be useful.360 This is a recognized DA, and appropriate equipment and personnel should be in the OR before induction.
5 Laryngeal Granulomas
Laryngeal granulomas are frequently the result of prolonged endotracheal intubation.108 However, granuloma formation has been reported after short-term intubation as well. Other factors contributing to granuloma formation include female gender, size of the ETT, position of the ETT, traumatic intubation, and excessive cuff pressure.82 The incidence in adults has been described as 1 in 800 to 1 in 20,000.361 Typically, granulomas form in the posterior glottis on the medial aspect of the arytenoids.106 Hoarseness is a common feature. Treatment consists of inhaled steroids, antireflux measures, antibiotics, and surgical removal under direct visualization.82
6 Congenital and Acquired Subglottic Disease
a Subglottic Stenosis
Subglottic stenosis may be classified as congenital or acquired. It is defined as the presence of an abnormally small subglottic lumen (<3.5 mm in diameter in newborn).355 Congenital subglottic stenosis is the third most common congenital anomaly.356 Patients may present with mild or severe airway obstruction. Another common presentation is recurrent croup.355 Patients who develop recurrent croup with upper respiratory tract infections during the first years of life should be evaluated for congenital subglottic stenosis. Acquired subglottic stenosis is usually the result of endotracheal intubation. Definitive diagnosis is made with rigid endoscopy. Treatment consists of anterior or multiple cricoid splitting with cartilage graft interpositioning (mitomycin). The success rate for these procedures is approximately 90%.356
b Croup
Croup, or laryngotracheobronchitis, is the most common cause of infectious airway obstruction in children. The incidence of croup in the United States is 18 per 1000 children annually. The peak incidence is 60 per 1000 among children 1 to 2 years of age.351 Croup affects children between the ages of 6 months and 4 years, with peak incidence in early fall and winter. Parainfluenza type I is the most common etiologic agent responsible for croup. This is a viral infection that affects the subglottic region of the larynx, causing edema. The disease has a gradual onset, usually arising after an upper respiratory tract infection. Symptoms include inspiratory stridor; suprasternal, intercostal, and subcostal retractions; and a croupy or barking cough. Anteroposterior films of the neck show the classic church steeple sign (symmetrical narrowing of the subglottic air).351
For mild cases, treatment consists of breathing humidified air or oxygen.351 In severe cases, treatment with nebulized racemic epinephrine (0.25-0.5 mL in 2 mL of saline) is indicated. Repeated treatments, every 1 to 2 hours, may be necessary. Because the duration of action is brief (<2 hours), rebound respiratory distress may develop after treatment, and observation is necessary. Studies suggest that patients may be discharged from the emergency room after a 3-hour observation period provided that the parents are reliable and easy access to the ER is available. Racemic epinephrine should be used with caution in patients with tachycardia or underlying cardiac abnormalities, such as tetralogy of Fallot or idiopathic hypertrophic subaortic stenosis.362
After years of debate, the use of steroids in the treatment of mild to moderate viral croup has gained acceptance.363–365 Treatment with steroids has been associated with a reduction in admissions and length of stay.365 Dexamethasone, 0.6 mg/kg (maximum dose, 10 mg) intravenously, is the standard dose. Dexamethasone (0.6 mg/kg) given orally was associated with more rapid resolution of symptoms than nebulized dexamethasone.363 Heliox, a mixture of helium and oxygen, has also been used in the treatment of viral croup. Helium is an inert, nontoxic gas that has low specific gravity, low viscosity, and low density. Because of these properties, helium reduces airway resistance by decreasing turbulent flow in the airway.366 If the preceding measures fail, intubation is necessary.
F Tracheobronchial Anomalies
1 Tracheomalacia
Tracheomalacia is characterized by weakness of the tracheal wall related to softness of the cartilaginous support. This allows the affected portion to collapse under conditions where the extraluminal pressure exceeds the intraluminal pressure.367 Tracheomalacia may be classified into either congenital (primary) or acquired (secondary). Congenital tracheomalacia may be further subdivided into idiopathic or syndromic conditions. Tracheoesophageal fistula, CHARGE syndrome, and DiGeorge’s syndrome are associated with congenital tracheomalacia. Acquired tracheomalacia is typically caused by extrinsic compression of great vessels or is secondary to bronchopulmonary dysplasia. Symptoms include episodic respiratory distress, persistent dry cough, wheezing, dysphagia, and recurrent respiratory infections. Failure to wean from the ventilator or failure of extubation may also be indicative of tracheomalacia.367
Airway Implications
Airway obstruction has been reported in patients with tracheomalacia during general anesthesia, even in asymptomatic patients.368,369 Collapse of the affected segment occurs during expiration and with particularly forceful expiration or coughing. CPAP with or without intermittent positive-pressure ventilation (PPV) can alleviate the obstruction.370 Noninvasive PPV through a face mask has been used successfully to prevent reintubation in an infant with tracheomalacia postoperatively.371
3 Bacterial Tracheitis
Bacterial tracheitis, formerly called pseudomembranous tracheitis or membranous laryngotracheobronchitis, is a potentially life-threatening disease. It is an infection of the subglottic region, and progression to full airway obstruction is possible. Bacterial tracheitis is believed to result from a bacterial superinfection preceded by a viral upper respiratory tract infection.372 The peak incidence is in the fall and winter, affecting children from age 6 months to 8 years. S. aureus, H. influenzae, α-hemolytic Streptococcus, and group A Streptococcus are the usual causative agents. Patients usually present with a several day history of viral upper respiratory symptoms followed by rapid deterioration. The patient develops high fever, respiratory distress, and a toxic appearance. In contrast to those with supraglottitis, these patients have a substantial cough, appear comfortable when supine, and tend not to drool.351
In contrast to those with laryngotracheobronchitis, patients with bacterial tracheitis do not respond to racemic epinephrine or corticosteroids. Radiographs of the airway often show irregular tracheal densities and subglottic narrowing.372 Patients with severe respiratory distress should be taken to the OR for rigid endoscopy and intubation.
Airway Implications
Patients with bacterial tracheitis have the potential for airway obstruction. Preparations for management of the difficult pediatric airway must be made, including a rigid bronchoscope. Inhalation induction with maintenance of spontaneous respirations is preferred. Endoscopy is performed with removal of the sloughed mucosa. Intubation is performed, and specimens for culture and Gram stain are taken. Broad-spectrum antibiotics are started and continued for 10 to 14 days. Intubation is usually required for 3 to 7 days.351
4 Mediastinal Masses
Anesthesia for patients with mediastinal masses, usually anterior mediastinal masses, is associated with a high risk of airway obstruction, hemodynamic instability, or even death from extrinsic compression of three structures: the heart, great vessels (primarily superior vena cava), and the trachea and bronchi.373 Induction of anesthesia and PPV may exacerbate the airway compression in a variety of ways. Loss of intrinsic muscle tone, reduced lung volumes, and a reduced transpleural pressure gradient combine to increase the effects of extrinsic compression. Cardiac arrest, superior vena cava syndrome, and airway occlusion are problems that can occur during induction of anesthesia.374–376 Airway compression during induction of anesthesia can occur even in asymptomatic patients.375 These complications may be unresponsive to position changes or open cardiac massage.
Mediastinal masses may be divided into anterosuperior, visceral, and posterior. The anatomic location of the mediastinal mass varies with age. In children, mediastinal masses are predominantly found in the posterior mediastinum. Neurogenic tumors, especially neuroblastomas, are the most common mediastinal tumor in young children. Germ cell tumors are the second most common anterior mediastinal mass in children. In adolescents, lymphomas are the most common anterior mediastinal mass.373
Symptoms such as orthopnea, stridor, and wheezing are ominous signs of airway obstruction.377 Positional dyspnea, tachyarrhythmia, and syncope suggest right-sided heart and pulmonary vascular compression. Syncope during a Valsalva maneuver suggests significant vascular encroachment.373 Children usually display symptoms earlier than adults. Small decreases in airway diameter result in increased resistance. Preoperative evaluation should focus on symptoms of respiratory compromise in the supine and standing positions. Intolerance of the supine position indicates compression by the mass on the trachea, heart, pulmonary artery, or superior vena cava. Preoperative CT scan should be obtained. Minimum criteria for safe administration of general anesthesia should be a tracheal cross-sectional area at least 50% of predicted and a peak expiratory flow rate at least 50% of predicted value.378
Airway Implications
Patients with a mediastinal mass are considered difficult to ventilate. Avoidance of general anesthesia, muscle relaxants, and PPV are the mainstay of anesthetic management for patients presenting for biopsy before irradiation or chemotherapy. Biopsies should be performed, if at all possible, under local anesthesia.379 Ketamine, local anesthesia, and a 50 : 50 mixture of O2 and nitrous oxide (N2O) while maintaining spontaneous ventilation have been used successfully for a diagnostic biopsy in a 13-year-old patient.380 Placing the patient in reverse Trendelenburg position may help.
With pediatric patients, general anesthesia may be needed for biopsy. Recommendations have been made for a rigid pediatric bronchoscopy and femoral-to-femoral bypass standby.375,381 If possible, irradiation of the mass before general anesthesia may reduce the risk associated with anesthesia. Peripheral shielding of the mediastinum may allow subsequent tissue biopsy.382 For older children, an awake fiberoptic intubation or FOB should be performed to assess the degree of obstruction after topicalization of the airway. In small infants and children, an awake intubation is not practical. In these patients, an inhalation induction with maintenance of spontaneous ventilation is recommended. Intravenous access must be obtained in a lower extremity before induction.381 Induction of anesthesia in the lateral semi-Fowler position has been recommended.376 Maintenance of spontaneous ventilation is vital; however, this is not foolproof.383 Heliox (80% helium, 20% O2) has been used for induction with sevoflurane and an LMA for successful airway management in a 3-year-old patient with severe respiratory distress related to a massive mediastinal mass.384
If airway obstruction or hemodynamic collapse occurs with induction, the following steps are suggested. First, one attempts to pass the ETT down the least obstructed bronchus. If passage of the ETT is not possible, rigid bronchoscopy to bypass the obstruction is attempted. Position changes to the lateral or prone position may alleviate the obstruction by changing the weight distribution of the tumor. Finally, cardiopulmonary bypass has been recommended.375 Airway obstruction may occur during emergence as well. Extubation should be performed with the patient awake. These patients should be monitored postoperatively in the ICU.
5 Vascular Malformations
Vascular malformations result from abnormal development of the arterial component of the branchial arch system, resulting in complete or incomplete encirclement of the trachea or esophagus, or both.385 In 1945, Gross386 introduced the term vascular ring to describe this anomaly. Patients with vascular rings may present with symptoms of respiratory distress or dysphagia because of tracheoesophageal compression. Patients may present with respiratory distress after birth or may be asymptomatic for life. Most children with vascular rings present with nonspecific symptoms such as stridor, dyspnea, cough, or recurrent respiratory tract infection.387 Dysphagia is often the primary symptom in adults with vascular ring.385 In a retrospective review of vascular rings, 74% of the malformations were symptomatic, with inspiratory stridor and wheezing as the main complaints.387
Various types of vascular rings have been described, including double aortic arch and right aortic arch with aberrant left subclavian artery. The double aortic arch usually arises earlier than other varieties requiring surgical correction.385 Associated cardiac anomalies are often present with the vascular ring. Diagnosis is confirmed by radiologic studies. A chest radiograph may indicate the site of the ascending and descending aorta. A barium esophagogram may disclose extrinsic compression of the esophagus. Angiography has been considered the gold standard for identifying vascular rings. CT and MRI scans are able to assist in the diagnosis of vascular ring and determine the anatomy. The diagnosis of vascular ring may be delayed because of the nonspecific symptoms.387 Patients who are symptomatic should undergo surgery. Surgical correction is by a left thoracotomy, right thoracotomy, or median sternotomy.385
6 Foreign Body Aspiration
Foreign body aspiration is a cause of significant morbidity and mortality in the pediatric population. Young children are at increased risk for foreign body aspiration, with children less than 2 years old most often affected.388 A second peak of aspiration occurs between ages 10 and 11 years.389 Most of the deaths occur in children younger than 1 year. The objects most frequently aspirated are food products. There is only a slight propensity for the object to lodge on the right side because of symmetrical bronchial angles in children under 15 years old. The left main stem bronchus is displaced by the aortic knob by age 15, creating a more obtuse angle at the carina.390
Witnessed events are easier to diagnose. A history of choking, gagging, or coughing is usually given. Patients may be asymptomatic at the time or may develop symptoms of acute distress. A persistent cough, wheezing, or recurrent pneumonia may be the initial sign if the aspiration occurred in the past. The American Academy of Pediatrics has developed guidelines for the management of choking episodes. For children under 1 year, back blows and abdominal thrusts with the child in a head-down position are recommended. The Heimlich maneuver is reserved for older children and adults.391
Classically, peanuts should be removed promptly because of the inflammatory reaction to the peanut oil. Emergency removal is indicated if the patient is in distress or if the foreign body is in a precarious location. If the patient is stable, radiographs may be taken to assist in localizing and identifying the foreign body. If the foreign body is radiopaque, it is easily identified. Radiolucent foreign bodies may demonstrate soft tissue density in or narrowing of the airway.13 Indirect signs of air trapping, mediastinal shift, or atelectasis may be present. Lateral decubitus films are helpful in infants and younger children because they cannot cooperate with expiratory films.390 The downside lung should be deflated unless it is obstructed with a foreign body.389
Airway Implications
In general, inhalation induction without cricoid pressure is the favored technique for removal of foreign bodies in the airway, regardless of the type of object, according to a postal survey of members of the Society for Pediatric Anesthesia.388 (For foreign bodies in the upper esophagus, a rapid-sequence induction without cricoid pressure was the preferred technique, whereas for objects in the lower esophagus and stomach, a rapid-sequence induction with cricoid pressure was chosen.) Cricoid pressure may cause harm if the foreign body is sharp or positioned in the larynx. If the case is not an emergency, one can wait until the appropriate nothing-by-mouth time has passed. In a retrospective review of anesthetic management for tracheobronchial foreign body removal, neither spontaneous nor controlled ventilation was associated with an increased incidence of adverse events.392
With an inhalation induction, a prolonged induction may occur because of airway obstruction. CPAP at 5 to 10 cm H2O and assisted ventilation may be needed at times to maintain a patent airway. After an adequate level of anesthesia is obtained, topicalization of the airway may decrease the incidence of coughing or laryngospasm. Use of a ventilating rigid bronchoscope allows ventilation during the procedure. High oxygen flow rates may be needed to overcome the presence of an air leak around the FOB. Communication between the anesthesiologist and the endoscopist is crucial because this is a shared airway. The patient may require intermittent ventilation if desaturation occurs during the FOB. When the foreign body is grasped, the glottis should be relaxed for removal. Short-acting muscle relaxants, propofol, or deeper inhalational anesthesia may be used. The forceps and the bronchoscope are removed from the trachea as a single unit.354 Dislodgement of foreign bodies at the glottic or subglottic area has been reported.393 If a foreign body is dislodged and obstructs the trachea, the bronchoscope must be used to push the foreign body into a main stem bronchus to enable ventilation of one lung. FOB with tracheotomy removal of a bronchial foreign body has been used successfully to remove an object that was too large to pass through the subglottis.394
7 Other Tracheal Disease
Tracheal stenosis is congenital or acquired. Congenital stenosis may be associated with congenital airway malformations such as tracheoesophageal fistula, hypoplastic lungs, and tracheomalacia. Congenital complete tracheal rings are also a cause of tracheal stenosis. In this condition, the rings are fused posteriorly and there is no posterior membranous wall. Acquired stenosis is usually attributed to prolonged intubation, inhalational injuries, trauma, or tumors. Symptoms include stridor, wheezing, croup, tachypnea, and cough. For mild lesions, conservative therapy is warranted. Surgical treatment involves tracheal resection with primary reanastomosis for short-segment lesions. Anterotracheal split procedures may be used for longer segmental lesions. Laser excision of granulation tissue at the repair site may be needed.276
Airway Implications
Tracheal stenosis may cause difficulty with ventilation or ETT advancement. Minimal trauma to the airway can lead to acute airway obstruction.276 Use of an LMA has been described in two patients with subglottic stenosis in whom the stenotic areas were 2 mm or less.395,396 Passage of the rigid bronchoscope should be performed only at definitive repair.
G Cervical Spine Anomalies
1 Limited Cervical Spine Mobility
a Klippel-Feil Syndrome
Klippel-Feil syndrome is characterized by fusion of two or more cervical vertebrae. Other features include short neck, a low posterior hairline, scoliosis, and congenital heart disease.397 Difficulty with airway management usually arises in the latter half of the first decade of life. The degree of difficulty with airway management depends on the severity of neck fixation.
c Juvenile Rheumatoid Arthritis
Juvenile rheumatoid arthritis is a chronic arthritis with variable manifestations. Several different subgroups of disease have been identified: systemic onset (Still’s disease), polyarticular, and oligoarticular (see under “Facial Anomalies: Maxillary and Mandibular Disease”).
d Airway Implications
Careful preoperative evaluation of patients with limited cervical mobility must be done before anesthetic induction. Previous anesthetic records, if available, should be reviewed for any relevant information. Because a DA is presumed to exist, preparation for management of the difficult pediatric airway must be made. In cases of limited cervical mobility, the ability to align the oral, pharyngeal, and laryngeal axes for visualization of the glottis is impaired. The presence of TMJ involvement may limit mouth opening as well. Awake endotracheal intubation is recommended in this scenario. Many techniques are available for use, including FOB, Bullard laryngoscope, retrograde wire technique, and lightwand. This may not be suitable in younger patients. For patients who will not cooperate with an awake technique, a mask induction with 100% O2 and spontaneous ventilation is indicated. Retrograde intubation, suspension laryngoscopy, and fiberoptic intubation through the LMA have all been reported in pediatric patients with Goldenhar’s syndrome.77, 97, 398 The lightwand was used to intubate an 18-day-old infant with right hemifacial microsomia.42
2 Congenital Cervical Spine Instability
Cervical spine instability, if unrecognized, is a potential cause of serious morbidity and even mortality during airway management. Cervical spine instability or subluxation most often involves the atlanto-occipital joint. Congenital syndromes such as trisomy 21, Hurler’s syndrome, Hunter’s syndrome, and Morquio’s syndrome are associated with cervical spine instability.399 Of these, trisomy 21 is the syndrome most often encountered by anesthesiologists.
a Down Syndrome
Trisomy 21 (Down syndrome) occurs in approximately 1 of every 660 live births. Mental retardation, congenital heart disease, OSA, and congenital subglottic stenosis may be present. Approximately 20% of patients have ligamentous laxity of the atlantoaxial joint, which may allow atlantoaxial instability. This may predispose them to cervical spinal cord compression. Children are at risk for injury during hyperextension, hyperflexion, or increased rotation of the neck.275,400 Signs of cervical spinal cord compression include loss of ambulatory function, spasticity, hyperreflexia of the lower extremities, extensor plantar reflexes, and loss of bowel and bladder control. Other signs may include increased fatigue with walking and torticollis.400 Preoperative evaluation of the patient with Down syndrome must attempt to discover any preexisting signs or symptoms of spinal cord compression. The issue of screening for atlantoaxial instability in patients with Down syndrome is controversial. The American Academy of Pediatrics Committee on Sports Medicine and Fitness decided that the value of cervical spine radiographs is uncertain in screening for possible catastrophic neck injury in athletes with Down syndrome.400 However, Pueschel401 argued that patients should be screened for atlantoaxial instability. A survey of the Society of Pediatric Anesthesia found that members obtain preoperative radiographs (18%) or subspecialty consultation (8%), or both, for asymptomatic patients. For symptomatic patients, radiographs and preoperative consultations are obtained 64% and 74% of the time, respectively. The majority of respondents attempt to maintain the head in a neutral position for both symptomatic and asymptomatic patients.402
Airway Implications
Airway management for patients with Down syndrome should consider the possibility of cervical spine instability with cord compression. In addition, the large tongue and potential for OSA can lead to upper airway obstruction. Patients who have symptoms of cord compression should have radiographic evaluation before any elective surgical procedure. Lateral extension and flexion radiographs of the upper cervical spine can reveal atlantoaxial subluxation. An odontoid process (axis) to anterior arch (atlas) distance greater than 4.5 mm indicates abnormal instability.400
3 Acquired Cervical Spine Instability
Acquired cervical spine instability in pediatric patients can result from multiple trauma or head and neck trauma. Any pediatric patient with a severe head injury should be treated as though a cervical spine injury is present.403 An estimated 1% to 2% of pediatric patients with multiple trauma have a cervical injury.391 Pediatric patients with underlying medical conditions such as Down syndrome may be more susceptible to cervical cord injury.391 Pediatric patients less than 8 years old are at increased risk for injury to the upper cervical spine and craniovertebral junction. Only 30% of cervical injuries occur below C3 in children younger than 8. They also have a higher incidence of spinal cord injury without radiographic abnormality (SCIWORA).404 Immobilization of a patient with suspected cervical injury is crucial so that further damage to the cord is prevented. A hard collar, spine board, and soft spacing devices between the head and securing straps are needed. The occiput is large, and a blanket under the torso allows the neck to rest in a neutral position.
Airway Implications
For a nonurgent intubation, further evaluation of the cervical spine is warranted. When the cervical spine has been “cleared” by the neurosurgeon or trauma surgeon, a rapid-sequence induction with cricoid pressure may be performed after adequate preoxygenation if the airway appears reasonable. If the cervical spine is unstable, a rapid-sequence induction with cricoid pressure may be performed with in-line stabilization. Fluoroscopy was used to assist the intubation of an 11-year-old patient with an unstable subluxation of C1-C2 after a motor vehicle crash.405 Awake techniques such as flexible fiberoptic laryngoscopy, Bullard laryngoscope, lightwand, SOS, or retrograde intubation may be indicated if the patient has an unstable cervical spine and a DA.