[level-membership-for-neurology-category]
Chapter 67 The Cerebellum and the Hereditary Ataxias
Basic Cerebellar Structure, Function, and Dysfunction
Cerebellar Structure
The cerebellum can be divided anatomically in the anteroposterior and mediolateral dimensions (Figure 67-1A and B). Fissures divide the rostrocaudal length of the cerebellum into ten lobules, which are analogous to the gyri of the cerebral cortex. These lobules can be grouped into three lobes: the anterior lobe (lobules I–V), the posterior lobe (lobules VI–IX), and the flocculonodular lobe (lobule X) (Figure 67–1C). The mediolateral divisions are the vermis, which lies medially, and the hemispheres, which lie laterally and are separated from the vermis by shallow grooves.
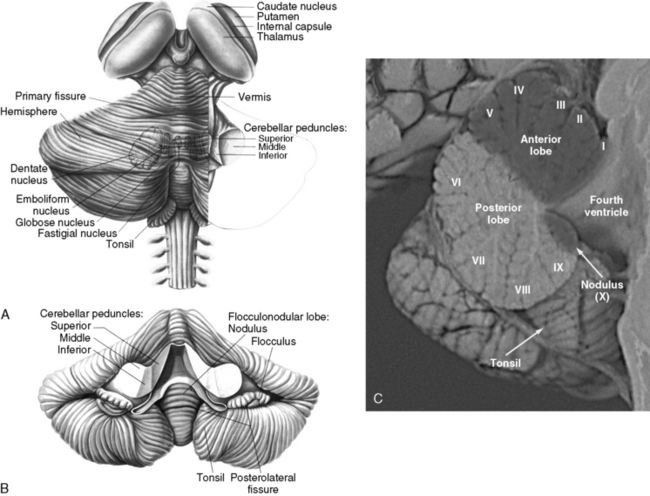
Fig. 67-1 The human cerebellum.
(A and B, From Kandel ER and Schwartz, JH, eds. Principles of neural science, 2nd edn. New York: Elsevier, 1985, pp 504 and 505. C, Courtesy of Dr. Dawna Armstrong.)
The cerebellar cortex is formed by five cell types, arranged in the same relative orientation, regardless of anatomical or functional location (Figure 67-2A). Purkinje cells are among the largest neurons in the central nervous system (CNS), and represent the sole output of the cortex. Cerebellar granule cells, which reside in the internal granular layer (IGL), are the most numerous neuronal population in the CNS. (Their numbers are nearly equal to all other CNS neurons combined.) Granule cells extend long, bifurcated axons (parallel fibers) into the molecular layer, where they travel for several millimeters and synapse with 1000–2000 Purkinje cells on their dendritic arbors; in turn, each Purkinje cell forms synapses with as many as 1,000,000 parallel fibers. Granule cells are the only excitatory cell type in the cerebellar cortex. Three local inhibitory interneurons also reside in the cortex. Golgi type II cells are large neurons found in the IGL that receive mossy fiber inputs and inhibit granule cells. Basket and stellate cells are found in the molecular layer, where they receive excitatory inputs from parallel fibers and are inhibitory to Purkinje cells.
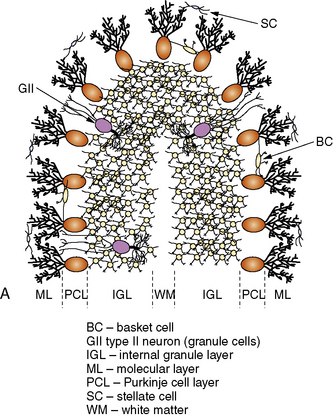
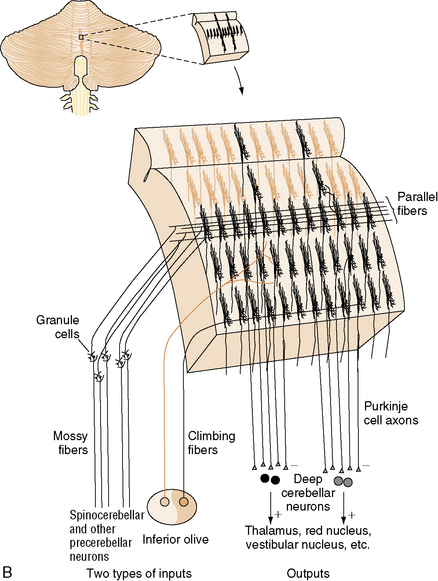
Fig. 67-2 Major cell types of the cerebellar cortex.
(B, From Kandel ER et al., eds. Principles of neural science, 4th edn. New York: McGraw-Hill, 2003, p 838.)
The neurons of the deep cerebellar nuclei (DCN) form four separate clusters in the cerebellar white matter (see Figure 67-1A). These cells receive inhibitory cortical input from the Purkinje cells, and excitatory extracerebellar afferents from climbing and mossy fibers. The fastigial nuclei lie most medially, and receive input from vermal Purkinje cells. The interposed nuclei (nucleus globose and nucleus emboliform) receive input from paravermal Purkinje cells. The dentate nuclei, which are the largest DCN in primates, receive input from the cerebellar hemispheres. Along with some Purkinje cells found in the flocculonodular lobe, the DCN represent the sole efferent output of the cerebellum, projecting to multiple brainstem nuclei in the medulla and pons, as well as the red nucleus and thalamus.
Cerebellar input consists of three classes of afferents. Climbing fibers originate in the inferior olives, which project to the contralateral cerebellar hemisphere. They provide strong excitatory input to Purkinje cells and neurons of the DCN. Each climbing fiber synapses on no more than ten Purkinje cells, while each Purkinje cell receives input from only one climbing fiber. Mossy fibers originate in multiple locations in the thalamus, brainstem, and spinal cord, and form excitatory synapses on granule cells and Golgi type II neurons in the IGL, as well as on neurons of the DCN. Climbing fibers, and to a lesser extent mossy fibers, are arranged in a series of parasagittal bands that “stack” next to one another in the mediolateral dimension (Figure 67-2B); the functional significance of this arrangement will be discussed in the next section. Finally, multilayer fibers originate in the hypothalamus, locus ceruleus, and raphe nuclei, and form a diffuse fiber network in the cortex and DCN.
Like the cerebral cortex, cerebellar cortical afferents and efferents are arranged in a somatotopic manner (reviewed in Manni and Petrosini [2004]). This organization is more complex than that of the cerebral cortex in two ways. First, at least three separate homunculi (one in the anterior lobe and one in each of the paravermian lobules) can be identified (Figure 67-3A). These are believed to converge on separate, continuous somatotopic maps in the four cerebellar nuclei. This arrangement, which was initially described in other species, has recently been directly demonstrated in humans using functional neuroimaging [Bushara et al., 2001; Manni and Petrosini, 2004]. Second, the sensory map within a cerebellar folium is not continuous. Instead, a fractured somatotopy exists, where each region of the face or limb has multiple representations within the folium that may not be adjacent to one another (Figure 67-3B). The reason for this arrangement is unknown, but it may be necessary to bring climbing fiber and mossy fiber inputs into register with one another.
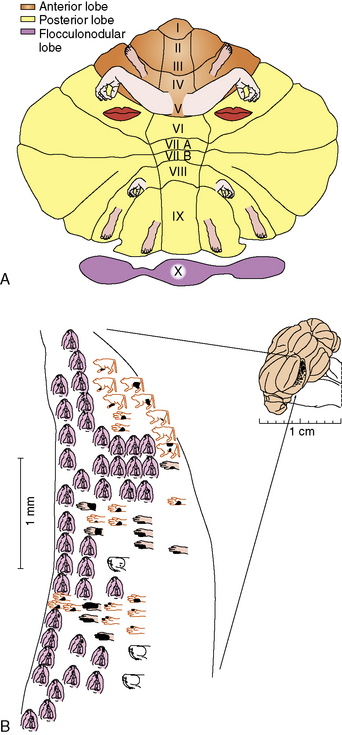
Cerebellar Function
As noted above, cerebellar afferent input is segregated into a series of parasagittal bands. There are two main hypotheses as to how this parasagittal arrangement might be functionally important. First, comparison of climbing fiber to mossy fiber excitatory inputs within these bands could contribute to refining movements and motor learning [Ito, 2002]. It has also been proposed that mossy fiber input, which is translated through granule cells and their parallel fibers, could transform spatial relationships into temporal ones as the signal passes from Purkinje cells closest to the granule cells to those farther down the parallel fiber path. The receptive Purkinje cells could then detect timing differences between the incoming granule cell and climbing fiber signals. Thus, the cerebellum could act as an alarm clock, stopwatch, or coincidence/time shift detector [Braitenberg, 1967].
Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) suggest that the cerebellum is involved in several nonmotor functions. These include sensory discrimination, attention, working memory, semantic association, verbal learning and memory, and complex problem solving [Allen et al., 1997]. A number of other studies have also linked the cerebellum to various higher-order cognitive processes [Leiner et al., 1991; Middleton and Strick, 1994]. Most of these functions are subserved by the cerebellar hemispheres and the dentate nuclei.
Cerebellar Dysfunction
Cerebellar (ataxic) dysarthria produces abnormalities in articulation (slurring and inaccuracy in range, force, and timing; patients sound as if inebriated), phonation (vocal quality can be harsh or uneven), resonation (a nasal quality is common), and prosody (patients tend to place equal and excessive emphasis on each syllable). Other kinds of dysarthria exist; there is, for example, the hypokinetic dysarthria seen in Parkinson’s disease, in which bradykinesia due to loss of dopaminergic neurons in the substantia nigra tends to produce low volume of speech or hoarseness, reduced pitch variability, palilalia (repetition of syllables), and very slow speech. There is evidence that left cerebellar lesions interfere with speech prosody because of disruptions of interconnections with the right cerebral hemisphere, which mediates this process [Brazis et al., 2001]. Cerebellar mutism is sometimes seen after removal of vermian tumors, and may result from bilateral involvement of the dentatorubrothalamic tracts.
Nonmotor manifestations of cerebellar disease can also occur. A cerebellar cognitive affective syndrome has been described in patients with isolated cerebellar lesions of a number of different etiologies [Schmahmann and Sherman, 1998]. In this syndrome, executive functions, spatial cognition, personality changes, and language deficits are all present. The postulated neural substrate involves circuits that link prefrontal, posterior parietal, superior temporal, and limbic cortices to the cerebellum.
Differential Diagnosis of Ataxia
The underlying cause of ataxia is virtually always primary or secondary dysfunction of the cerebellum. Cerebellar disease is a result of a number of underlying conditions, many of which are listed in Box 67-1. The most prevalent causes of acute cerebellar ataxia are viruses (e.g., coxsackievirus, rubeola, varicella), toxins (e.g., alcohol, barbiturates, antiepileptic drugs), and traumatic insults. Tumors of the cerebellum (vermal and hemispheric) or the adjacent brainstem, particularly pontine gliomas, are also relatively common. Numerous metabolic conditions affecting the central and peripheral nervous systems lead to ataxia. Congenital abnormalities of the nervous system, such as Chiari malformation, Dandy–Walker malformation, and basilar impression, are associated with ataxia. Endocrinologic abnormalities, particularly hypothyroidism, may also present with ataxia as the predominant manifestation. Hemorrhage, infarction, and embolism are unusual causes of cerebellar damage and often occur in conjunction with damage to neighboring brain regions. Angioblastomas of the cerebellum are rare; von Hippel–Lindau disease, a neurocutaneous condition, is associated with vascular lesions of the cerebellum, as is the PHACE (posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of aorta and cardiac defects, and eye abnormalities) syndrome [Frieden et al., 1996].
Box 67-1 Selected Causes of Ataxia in Childhood










Degenerative and/or Genetic

 Ataxia, retinitis pigmentosa, deafness, vestibular abnormality, and intellectual deterioration [Francois and Descampas*]
Ataxia, retinitis pigmentosa, deafness, vestibular abnormality, and intellectual deterioration [Francois and Descampas*]

 Cerebellar ataxia with deafness, anosmia, absent caloric responses, nonreactive pupils, and hyporeflexia [Brown, 1959]
Cerebellar ataxia with deafness, anosmia, absent caloric responses, nonreactive pupils, and hyporeflexia [Brown, 1959]



 Hereditary cerebellar ataxia, intellectual retardation, choreoathetosis, and eunuchoidism [Altchule and Kotowski*]
Hereditary cerebellar ataxia, intellectual retardation, choreoathetosis, and eunuchoidism [Altchule and Kotowski*]



















































































Management of Cerebellar Dysfunction and Ataxia
When ataxia is the result of certain metabolic disorders or toxins, trauma, or neoplasia, specific therapies for the underlying condition can be curative (Table 67-1). Direct neurological causes of ataxia, however, such as inherited diseases, are not yet amenable to treatment. Patients can benefit from various aids for ambulation, as well as physical therapy and occupational therapy; though these approaches will not restore mobility in advanced disease, they none the less alleviate other symptoms such as spasticity and can greatly improve quality of life.
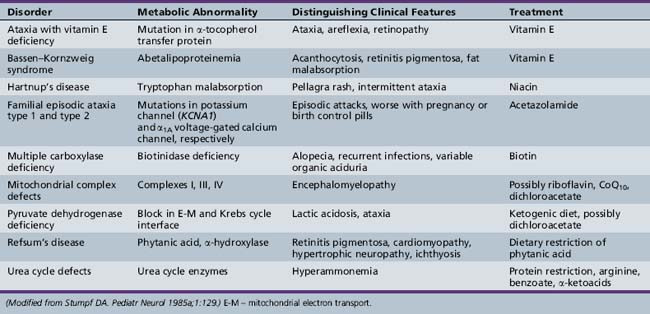
The Hereditary Ataxias
The classification of the hereditary ataxias has long posed a clinical challenge. Prior to the advent of gene cloning technology, these conditions were classified based on their mode of inheritance, clinical features, and pathological findings. These approaches ultimately proved to be unsatisfactory, and often led to more confusion than clarity. Patients with known hereditary ataxia have a range of cerebellar dysfunction that may be exclusively cerebellar in origin or combined with other features, including brainstem dysfunction, spinal cord abnormalities, extrapyramidal findings, neuropathy, retinopathy, deafness, cataracts, seizures, and/or dementia [Greenfield, 1954; Koeppen and Barron, 1984; Zoghbi et al., 1993]. The age of onset and clinical manifestations of any given ataxia may differ significantly from family to family and even in the same kindred, making specific assignment beyond mode of inheritance difficult [Currier et al., 1972]. Likewise, attempts to classify ataxia according to the anatomic site of the most dominant pathology (e.g., spinal cord, cerebellum) are also flawed. Descriptors such as “olivopontocerebellar atrophy” (OPCA), a pathological finding seen most predominantly in the dominantly inherited ataxias, do not adequately serve to distinguish between them.
Autosomal-Recessive Inherited Syndromes
The autosomal recessive inherited ataxias (summarized in Table 67-2) are a varied group of disorders that tend to have an early age of onset in infancy or childhood. They can involve multiple regions of the central and peripheral nervous system. In general, earlier age of onset heralds a more aggressive disease course. Several causative mutations have been identified, leading to a clear genetic classification scheme.
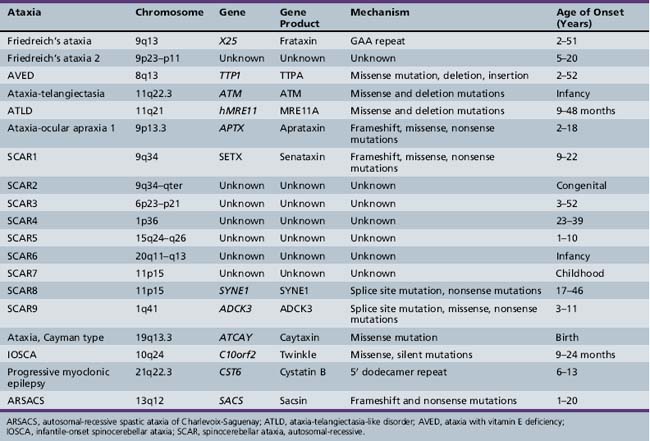
Friedreich’s Ataxia (Freidreich’s Ataxia; Spinocerebellar Ataxia – OMIM 229300)
Friedreich ataxia is one of the most common hereditary ataxias, with a prevalence of approximately 1–2 per 100,000 in the general population [Harding, 1984]. Caucasians are affected more often than other racial groups, with a prevalence closer to 1 per 50,000 [Cossee et al., 1997]. Clinically, the disease is signaled by the presence of ataxia and corticospinal tract dysfunction. Other associated features include nystagmus, kyphoscoliosis, and an unusual cardiomyopathy. The disease was originally reported by Friedreich [Friedreich, 1863], who believed it was associated only with spinal cord abnormalities, subsequent findings demonstrated involvement of the medulla, peripheral nerves, and cerebral cortex in some cases.
Clinical manifestations
Loss of cells in cranial nerves VII, X, and XII causes facial weakness, with cerebellar dysarthria and dysphagia. Lateral or horizontal nystagmus may be prominent, and visual pursuit is punctuated with small jerks. Impairment of pupillary responsivity and limited extraocular movements are also common. Additional ophthalmologic abnormalities include cataracts, retinitis pigmentosa, and optic atrophy. Auditory dysfunction is common [Cassandro et al., 1986], and is documented by abnormal brainstem auditory-evoked potentials that are likely the result of impairment of both central and peripheral acoustic pathways [Knezevic and Stewart-Wynne, 1985]. The degree of hearing compromise is variable; in addition, some patients experience vertigo. Although higher cortical function appears intact by clinical measures, some studies suggest a decrease in information processing speed [Hart et al., 1985].
The lower limbs typically show loss of deep tendon reflexes, but reflexes may be preserved for a longer period in young children [Salih et al., 1990], and patients with late-onset disease can be spastic with hyperreflexia. Bilateral extensor toe signs are easily elicited. Cremasteric and abdominal reflexes may be lost. The masseter reflex remains intact [Auger, 1992]. Occasionally, weakness, muscle atrophy, and profound hypotonia may be evident, usually associated with spinal nerve disintegration. Virtually all facets of cerebellar function are involved, and both gross and fine coordination are affected. Position sense, vibration sense, and other skills requiring normal posterior column function are grossly impaired. Autonomic innervation of superficial layers of skin may be involved. Pain, touch, and temperature sensation are usually intact, but diminish over the course of the illness. Evidence of axonal sensory neuropathy, indicated by electrodiagnostic studies, is invariably present. Somatosensory-evoked potentials are abnormal, as is central motor function, as determined by magnetic stimulation [Claus et al., 1988].
Patients with Friedreich’s ataxia often develop talipes equinovarus or pes cavus. Deformities of the hand can also occur. Flexor spasms can become a great source of pain. Many patients suffer from kyphoscoliosis [Labelle et al., 1986], and may be unable to walk secondary to poor balance and an abnormally shifted center of gravity (Figure 67-4). Surgical intervention can correct the abnormal spinal curvature, and this is likely to help with ventilation and autonomic functions, even if it does not restore ambulation.

Fig. 67-4 Friedreich’s ataxia and moderate scoliosis in a 13-year-old boy who is unable to walk independently.
Cardiomyopathies are the most common accompanying cardiac abnormalities, although congenital heart disease has been described on occasion. The cardiomyopathy is often progressive, and is documented even before clinical manifestations by the presence of electrocardiogram (EKG) abnormalities (e.g., deep Q waves, low-wave QRS complexes, S-T segment changes, T-wave inversion) [Harding and Hewer, 1983; Child et al., 1986]. Arrhythmias may be particularly worrisome and life-threatening. EKG and vector cardiographic changes are demonstrable in more than 90 percent of patients [Child et al., 1986]. Left ventricular hypertrophy or symmetric ventricular hypertrophy is frequently evident on echocardiography [Pentland and Fox, 1983]. Hypertrophic cardiomyopathy is the leading cause of death in Friedreich’s ataxia patients.
Diabetes mellitus is associated with Friedreich’s ataxia; even in its absence, the glucose tolerance curve may be abnormal. Insulin resistance appears to be the explanation [Khan et al., 1986]. Unaffected relatives may also manifest insulin resistance. Episodic hypothermia, intermittent vomiting, and ventilatory dysfunction have also been reported [Thoren, 1962].
Imaging studies show both structural and functional changes that correlate with pathologic progression. MRI studies document atrophy of the upper cervical spinal cord and cerebellum [Wessel et al., 1989]. PET imaging demonstrates a widespread increase in cerebral metabolic rate for glucose in the brain of ambulatory patients with Friedreich’s ataxia. As the disease progresses, glucose metabolism decreases in a regionally specific manner; only the caudate and lenticular nuclei have increased metabolic rate in nonambulatory patients [Gilman et al., 1990].
Diagnosis is suggested by the presence of the expected clinical manifestations and sometimes by family history. Distinct diagnostic criteria for Friedreich’s ataxia were advanced by Geoffroy and colleagues [Geoffroy et al., 1976]: autosomal-recessive inheritance, onset before 10 years of age, gait ataxia, dysarthria, absent deep tendon reflexes, dorsal column signs, and weakness. The presence of an abnormal 5-hour glucose tolerance curve is corroborative. Spinal fluid findings may include elevated protein content and mild pleocytosis.
Pathology
The primary site of involvement on pathologic examination is the spinal cord. Gross examination reveals a shrunken cord, with the posterior columns, spinocerebellar tracts, corticospinal tracts, and posterior rootlets strikingly affected by varying degrees of fiber loss, demyelination, and gliosis. Conditional Freidreich’s ataxia gene inactivation in experimental animals showed that anomalies observed in the DRG (dorsal root ganglion) neurons are primary events, whereas neuronal loss in the posterior thoracic nucleus (Clarke’s column) and degeneration in the posterior columns may be secondary events [Simon et al., 2004]. Axons and myelin alike are involved, although the medullary nuclei (Burdach’s nuclei) are usually spared [Greenfield, 1954]. Structures in the medial root zone, including ganglion cells and the posterior roots that terminate within them, manifest varying degrees of involvement but are typically decreased in number. The lumbar and sacral roots are most severely affected, and the large, myelinated lumbar spinal nerves lose many axons. In contrast, anterior horn cells and their rootlets are usually uninvolved; however, minor pathologic alterations may be evident. Myopathic changes are rare.
The density of large myelinated fibers in sural nerve biopsies is reduced, even in young children, and often worsens with age [Said et al., 1986].
Pathologic changes in the heart include evidence of cardiomyopathy and involvement of blood vessels, nerves, and ganglia [James et al., 1987].
Genetics
Friedreich’s ataxia is transmitted as an autosomal-recessive disorder; the finding of consanguinity among several families clearly supported the autosomal-recessive inheritance pattern. The disease affects males and females equally. The establishment of the criteria proposed by Geoffroy and co-workers [Geoffroy et al., 1976] provided a basis for research studies and allowed clinical identification of families for linkage studies. Genetic linkage studies mapped the Friedreich’s ataxia gene to chromosome 9 and confirmed that the “Acadian” form of ataxia, characterized by a slower course of degeneration [Barbeau et al., 1984; Keats et al., 1989], also maps to chromosome 9 [Chamberlain et al., 1988; Keats et al., 1989]. Friedreich’s ataxia is caused by decreased expression of the mitochondrial protein, frataxin, encoded by the X25 gene located at 9q13 [Bidichandani et al., 1998; Koeppen, 1998].
Campuzano et al. [1996] were the first to identify an unstable GAA trinucleotide repeat in the first intron of X25. They also identified three different point mutations. In the group of 74 patients without a point mutation, 71 were homozygous for expanded alleles, and 3 were heterozygous for the expanded repeat. Five patients with point mutations were heterozygous for the expansion. The size of the repeat ranged from 7 to 22 on normal alleles and from 200 to 900 on Friedreich’s ataxia chromosomes. Further studies revealed that the triplet repeat could get as large as 1700 units, and that the lengths of both the larger and the smaller expanded alleles correlated inversely with the age of onset and severity of disease [Durr et al., 1996; Filla et al., 1996]. The mean allele length is significantly higher in Friedreich’s ataxia patients with diabetes, cardiomyopathy, and loss of reflexes in the upper extremities. Of 187 patients with autosomal-recessive ataxia, 140 were homozygous for a GAA expansion [Durr et al., 1996]. Furthermore, age-dependent accumulation of large GAA expansions contributes to the progressive pathology involving in the DRGs. Therefore, somatic instability of the expanded GAA triple repeat sequence may contribute directly to disease pathogenesis and progression in specific tissues [De Biase et al., 2007].
Frataxin is localized to the inner mitochondrial membrane and is thought to play a role in mitochondrial respiration and iron accumulation. Deletion of the frataxin homolog in yeast leads to mitochondrial iron accumulation and increased susceptibility to oxidative stress, with an increased production of free radicals (reviewed in Voncken et al. [2004]). These observations, coupled with the fact that expanded trinucleotide repeats interfere with transcription elongation of the X25 gene and cause a frataxin deficiency state, might explain the human phenotype [Bidichandani et al., 1998]. The high evolutionary conservation of frataxin has enabled the development of disease models in various organisms, from the unicellular eukaryote Saccharomyces cerevisiae to the mouse. Yeast was an important model system for identifying the crucial role of frataxin in the regulation of intracellular iron trafficking, iron-sulfur (Fe-S) cluster and heme biogenesis, and oxidative metabolism. Several Caenorhabditis elegans models with knockdown of the C. elegans FXN homolog frh-1 show conflicting results in response to oxidative stress, but all of them show reduced life span and may be useful for drug screening in Freidreich’s ataxia. Moderate reduction of the Drosophila frataxin homolog (dfh) causes a hypersensitive response to hypoxia, which upholds a causative role for oxidative stress in Freidreich’s ataxia. Mouse models of Freidreich’s ataxia have provided the most insight, however (reviewed in Puccio [2009]). The knockout model of frataxin suffers early embryonic lethality without iron accumulation, while heterozygosity for the frataxin null allele shows no pathological phenotype. These results suggest that residual frataxin expression associated with expansion mutation is critical for survival [Cossee et al., 2000]. Conditional animal models are viable and reproduce certain morphological and biochemical features observed in patients with Freidreich’s ataxia, including cardiac hypertrophy without skeletal muscle involvement, large sensory neuron dysfunction without alteration of small sensory and motor neurons, and deficient activities of complexes I through III of the respiratory chain and of the aconitases, enzymes essential in the citric acid cycle (reviewed in Puccio [2009]). Humanized GAA repeat expansion mouse models were obtained by combining the constitutive knockout model with the transgenic expression of a yeast artificial chromosome (YAC) carrying the human locus. These animals exhibit oxidative stress, leading to progressive neuronal and cardiac pathology [Al-Mahdawi et al., 2006]. Although none of these mouse models is authentic, a multimodel approach facilitates the study of different pathophysiological mechanisms and the examination of different therapeutic strategies. Why only selective neuronal systems are vulnerable to the loss of frataxin function remains unknown.
Treatment
Given the proposed function of frataxin protein in iron-sulfur cluster biosynthesis and the increased oxidative stress found with decreased protein levels, several groups have initiated treatment with idebenone, a short-chain analog of coenzyme Q10 (reviewed in Schulz et al. [2009]). All but one of these trials (the shortest trial) with idebenone at 5 mg/kg and 10 mg/kg/day have demonstrated improved cardiac function and decreased ventricular mass: important findings, since cardiac dysfunction is the leading cause of death in Friedreich’s ataxia patients. In these low-dose trials, idebenone treatment has no effect on the progression of ataxia [Schulz et al., 2009], but Di Prospero et al. showed that higher doses of idebenone (up to 60 mg/kg/day) are well tolerated, provide neurological benefit, and improve activities of daily life (ADL) in Freidreich’s ataxia patients who are still ambulatory [Di Prospero et al., 2007].
A high proportion of Freidreich’s ataxia patients have decreased serum CoQ10 levels, which was the best predictor of a positive clinical response to CoQ10/vitamin E therapy. Low- and high-dose CoQ10/vitamin E therapies were equally effective in improving ICARS scores (International Cooperative Ataxia Rating Scale) [Cooper et al., 2008]. Therapy with other antioxidants, iron chelators, and/or glutathione peroxidase mimetics has been suggested (and in some cases tried), but no clear beneficial effect has been established.
Expansion of intronic GAA repeat likely induces chromatin changes and in turn FXN silencing in Freidreich’s ataxia [Herman et al., 2006]. Treatment with a histone deacetylase (HDAC) inhibitor restored wild-type frataxin levels in the nervous system and heart, and increased histone H3 and H4 acetylation in chromatin near the GAA repeat [Rai et al., 2008]. These findings provide a very promising prospect for Freidreich’s ataxia treatment using HDAC inhibitors.
Friedreich’s Ataxia 2 (OMIM 601992)
Other families with a Friedreich’s ataxia disease phenotype, but without point mutations or GAA expansions in the X25 gene, have been described [Kostrzewa et al., 1997; Christodoulou et al., 2001]. In one family, linkage to a locus on chromosome 9p23–p11 has been found [Christodoulou et al., 2001]. The biological basis for the genocopy is not known, but it has been proposed that mutations in genes that lie in the iron metabolism pathway may be responsible.
Vitamin E Deficiency and Related Syndromes
Numerous mutations have been identified in the tocopherol transfer protein gene (TTP1), which encodes the α-tocopherol transfer protein (TTPA) [Ouahchi et al., 1995; Cavalier et al., 1998]. TTPA is expressed in the liver and is responsible for the incorporation of tocopherol (vitamin E) into very-low-density lipoprotein (VLDL) [Traber et al., 1990]. Mutated TTPA proteins fail to facilitate the secretion of vitamin E from cells, and the severity of the AVED pathology corresponds to the degree of impairment associated with each mutation [Qian et al., 2006].
Ataxia-Telangiectasia (AT; Louis-Bar Syndrome – OMIM 208900)*
The initial clinical description of this disease was reported by Syllaba and Henner [1926], who called it congenital double athetosis-aconjunctival vascular plexus syndrome. Louis-Bar [1941] reported a 9-year-old male with associated ataxia and oculocutaneous telangiectasias, classifying the case as a previously unrecognized phakomatosis. Two additional independent reports subsequently appeared in the literature [Biemond, 1957; Boder and Sedgwick, 1957], and Boder and Sedgewick named the condition ataxia-telangiectasia.
Clinical manifestations
Early motor development appears to be normal until around the time that the child starts walking, when ataxia is noted. The ataxia is progressive and ultimately leads to an inability to ambulate by the beginning of the second decade. Choreoathetosis and dystonia occur in up to 90 percent of patients, and these motor findings become more prominent with increasing age. Facial weakness leads to the characteristic impassive faces, as well as drooling and dysarthria. While strength is initially normal, many patients in their 20s and 30s develop progressive spinal muscular atrophy, predominantly affecting the hands and feet [Gatti et al., 1991]. Peripheral neuropathy in the form of diminished deep tendon reflexes and loss of large fiber sensation is also seen. Mental function is well preserved, although deficits in short-term memory can occur in the third and fourth decades [Gatti et al., 1991].
Oculomotor apraxia is a distinguishing feature of the disease. The apraxia commonly presents before the appearance of conjunctival telangiectasias and is characterized by defects of initiating voluntary saccades, hypometric voluntary saccades accompanied by compensatory eye-blinking and/or head-thrusting movements, and disrupted smooth pursuit movements [Smith and Cogan, 1959; Baloh et al., 1978]. Involuntary saccade initiation and optokinetic nystagmus may be impaired as well, and oculovestibular reflexes may be increased.
Telangiectasias are usually first observed in patients between the ages of 2 and 4 years, although they can occur as early as birth and as late as 14 years of age [Centerwall and Miller, 1958; McFarlin et al., 1972] (Figure 67-5). In addition to the conjunctivae, they appear on exposed areas of the skin, particularly areas of friction and trauma, such as the auricle, nasal bridge, and antecubital and popliteal spaces. Exposure to sun enhances their appearance. Premature aging of hair and skin is frequent, as are skin infections, including chronic blepharitis. Other skin changes, consisting of vitiligo and café-au-lait spots, can be seen. Rarely, scleroderma-like lesions occur.
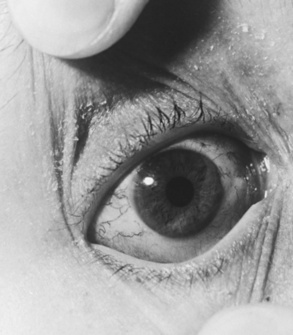
Fig. 67-5 Characteristic conjunctival telangiectasias observed in a 12-year-old with ataxia-telangiectasia.
Recurrent sinopulmonary infections are common, affecting 90 percent of patients, and usually result in chronic bronchitis, bronchiectasis, or both. The impairment of cellular immunity is also manifest by abnormally developed or absent adenoids, tonsils, lymphoid tissue, and thymus gland. Patients have an impaired delayed hypersensitivity response to skin-sensitizing antigens and a delayed homograft-rejection response [McFarlin and Oppenheim, 1969].
Patients with AT are prone to develop tumors and are more likely than the general population to have Hodgkin’s disease, leukemia, lymphoma, and lymphosarcoma. Other associated neoplasms include brain tumors, gastric adenocarcinomas, ovarian dysgerminomas, gonadoblastomas, cystic adenofibromas, uterine leiomyomas, and thyroid adenomas [Miller and Chatten, 1967; Gatti and Good, 1971].
Patients with less severe variant forms of AT have been described [Gilad et al., 1998; Saviozzi et al., 2002]. Immunodeficiency, telangiectasias, cancer, and sinopulmonary infections may be absent or reduced, but the neurological manifestations still occur. These individuals have a later onset and slower progression of neurological signs, longer life spans, and decreased chromosomal instability and cellular radiosensitivity.
Laboratory findings
Fibroblast and lymphoid cell lines from AT patients show increased radiosensitivity, leading to spontaneous and radiation-induced chromosomal breakage and rearrangement, which can be assessed in culture [Shiloh et al., 1985]. In lymphoid cell lines, the chromosomal breaks occur at the loci of T-cell antigen receptors and immunoglobulin genes, where DNA rearrangements and deletions naturally occur [Hecht and Hecht, 1985].
Pathology
Cerebellar atrophy primarily affects the Purkinje and granular cells, although basket cells can also be involved. Neuronal degeneration of the dentate and olivary nuclei and the substantia nigra has been demonstrated, and nuclear changes occur in the cells of the oculomotor complex, pretectal nuclei, and hypothalamus. Pituitary cells often have enlarged or dysplastic nuclei. Older patients have denervation of the posterior columns of the spinal cord. Degeneration of the anterior horn cells, decreased numbers of satellite cells in the dorsal root ganglia, and nucleomegaly of Schwann cells have been described [De Leon et al., 1976].
Genetics
The worldwide incidence of AT is approximately 1 in 40,000–100,000 live births; the carrier frequency ranges from 0.5 to 1.0 percent. The gene locus maps to chromosome 11q22–23 [Gatti et al., 1988]. The ATM (ataxia telengiectasia mutated) gene is very large, with 66 exons spanning 150 kb of genomic DNA [Uziel et al., 1996]; this fact makes mutation detection a challenge. In addition, because of the large number of different mutations, the majority of patients are compound heterozygotes. Still, using a combination of techniques, the estimated mutation detection rate has been detected to be over 95 percent in affected individuals [Buzin et al., 2003]. It is estimated that truncating mutations account for about 85 percent of mutations, while point mutations are responsible for the rest [Buzin et al., 2003].
ATM is a nuclear phosphoprotein homologous to a family of phosphatidylinositol kinase-related proteins, and it functions in the DNA damage response and in cell cycle regulation [Savitsky et al., 1995a, b]. Individuals with classic AT have little or no active ATM protein, while most individuals with variant AT have one severe mutation coupled with a mild mutation. In this latter condition, some ATM activity is preserved, which probably explains the milder phenotype [Saviozzi et al., 2002].
Loss of ATM activity could lead to neuronal degeneration in at least two ways. As ATM is a monitor of DNA strand breaks important for initiating repair, it has been suggested that ATM is necessary for the elimination of neural cells with genomic damage, and that the preservation of those cells may contribute to neuronal dysfunction [Herzog et al., 1998]. Several other diseases that affect DNA repair (ataxia-telangiectasia-like disorder, ataxia-ocular apraxia 1, xeroderma pigmentosum, Cockayne’s syndrome, Nijmegen breakage syndrome, and de Sanctis–Caccione syndrome) have a cerebellar phenotype, suggesting that cerebellar neurons may be very sensitive to DNA damage. This hypothesis highlights a nuclear role for the ATM protein. In neurons, however, a large percentage of the protein is found in the cytoplasm and axons. Increased numbers of lysosomes are found in Purkinje cell bodies and axons in ATM-deficient mice [Barlow et al., 2000]. This, coupled with an observed interaction with β-adaptin, a protein important for clathrin-mediated endocytosis, suggests that vesicle/protein transport may also be disrupted in ATM-defective cells [Lim et al., 1998].
Treatment
AT is a multisystem disease and its treatment requires attention to multiple types of interventions. Currently, we are not able to halt progressive neurodegeneration, although some features of the disease might respond to treatment. l-DOPA derivatives and anticholinergics may improve basal ganglia dysfunction. Amantadine, fluoxetine, or buspirone may help with the loss of balance and impaired speech; furthermore, tremors can often be controlled by gabapentin, clonazepam, or propranolol (reviewed in Lavin et al. [2007]).
Patients should receive vigorous supportive therapy, with particular attention directed to recurrent sinopulmonary infection. Attempts to improve immunologic status by plasma transfusion of thymosin and provision of fetal thymus transplants have not altered the course of the neurologic signs and symptoms [Wara and Ammann, 1978]. Treatment of neoplasms is a delicate proposition because patients are extremely sensitive to radiation and chemotherapy, and demonstrate resultant ulcerative dermatitis, severe esophagitis, dysphagia, and deep-tissue necrosis [Gotoff et al., 1967].
Ataxia-Telangiectasia-Like Disorder (ATLD – OMIM 604391)
ATLD patients have been described with clinical presentations including cerebellar ataxia and oculomotor apraxia, which are essentially identical to that of ataxia-telangiectasia except that telangiectasias are absent [Klein et al., 1996; Fernet et al., 2005]. Two of the patients, who were brothers, presented first with chorea that later progressed to ataxia [Klein et al., 1996]. In 2005, Fernet et al. described ten ATLD patients from three unrelated Saudi Arabian families. These patients had slowly progressive ataxia and ocular apraxia, but they did not develop tumors. Furthermore, they did not have telangiectasia, raised AFP, or reduced immunoglobulin levels [Fernet et al., 2005].
Increased susceptibility to DNA damage was noted in all four patients. Stewart et al. [1999] showed that the disorder maps to chromosome 11q21 in all four patients. Loss-of-function mutations in the hMRE11A gene are causative. Like ATM, MRE11A is important for DNA repair, and may act as a sensor of DNA damage. Stewart et al. estimate that approximately 6 percent of AT patients may in fact have ATLD [Stewart et al., 1999].
Ataxia-Oculomotor Apraxia 1 (AOA1; Early-Onset Ataxia with Ocular Motor Apraxia and Hypoalbuminemia – OMIM 208920)
This autosomal-recessive ataxia was initially described in Japanese families but has subsequently been described in other areas of the world as well [Tranchant et al., 2003]. It is the most frequent cause of autosomal-recessive cerebellar ataxia in Japan, and the second most common cause in Portugal. The presentation is nearly identical to that of AT without the non-neurological features [Aicardi et al., 1988], and accounts for up to 10 percent of autosomal-recessive cerebellar ataxias. Gait imbalance and dysarthria are the typical presenting features, although chorea can sometimes be the presenting symptom. Ocular apraxia typically occurs a few years after symptom onset; with time, progressive ophthalmoplegia occurs as well. Areflexia, dystonia, choreoathetosis, sensory and motor neuropathy, cognitive impairment, and retinal/macular lesions seen on funduscopy can also be part of the presentation. Life span is not affected.
Laboratory studies reveal hypoalbuminemia and hypercholesterolemia in the majority of cases as a later manifestation of the disease. Neuroimaging demonstrates cerebellar and sometimes brainstem atrophy. Pathology studies show loss of Purkinje cells in the cerebellum, and degeneration of the posterior columns, spinocerebellar tracts, and anterior horn cells of the spinal cord [Sekijima et al., 1998].
The gene, APTX, maps to chromosome 9p13.3 and is expressed in all body tissues [Date et al., 2001; Moreira et al., 2001]. Several mutations have been described, as have compound heterozygotes; frameshift and nonsense mutations result in more severe phenotypes than missense mutations. The APTX gene product, aprataxin, has domains that suggest a potential role in single-strand DNA repair, and has been shown to interact with another protein implicated in this process [Sano et al., 2004]. CoQ10 is often low in patients with AOA1 [Le Ber et al., 2007], and this deficiency responds to CoQ10 supplementation [Quinzii et al., 2005].
Spinocerebellar Ataxia, Autosomal-Recessive 1 (SCAR1 – OMIM 606002); Ataxia-Oculomotor Apraxia 2 (AOA2)
A second AT-like spinocerebellar ataxia with slightly different features than AOA1 has been described in families of several different ethnic backgrounds. The clinical features of AOA2 include gait ataxia, sensorimotor neuropathy, and ocular apraxia, the latter occurring about 50 percent of the time. While overall cognitive function is normal, subtle changes in executive function can be seen on neuropsychologic testing [Le Ber et al., 2004]. Neuroimaging demonstrates cerebellar atrophy. AOA2 is distinguished from AOA1 by the later age of onset, less common ocular apraxia, high levels of serum AFP, and normal serum albumin. The functional prognosis is also better in AOA2. The gene (SETX) maps to chromosome 9q34 [Nemeth et al., 2000]. Mutations cause premature termination of the senataxin protein, which is a member of the helicase family and is involved in RNA maturation and termination [Moreira et al., 2004]. Recently, Suraweera et al. [2009] have proposed a role for senataxin in the regulation/modulation of transcription.
Interestingly, other mutations in senataxin are transmitted in an autosomal-dominant manner and cause juvenile amyotrophic lateral sclerosis (ALS4) [Chen et al., 2004a]; the basis for the dramatic phenotypic difference between the two diseases is unknown.
Spinocerebellar Ataxia, Autosomal-Recessive 2 (SCAR2 – OMIM 213200); Cerebellar Hypoplasia, Nonprogressive Norman Type
Norman [1940] described three siblings in one family and two siblings in another who had had cerebellar ataxia and mental deficiency since early life. Postmortem examinations showed severe cerebellar granule cell loss. One child showed delayed motor development and mental deficiency in infancy. Other features included small head, cataracts, increased knee jerks, and intention tremor [Norman, 1940]. The disease locus was assigned on chromosome 9q34–qter [Delague et al., 2001]. It is not certain if SCAR2 is distinct from the disorder called cerebellar hypoplasia (OMIM 213000).
Spinocerebellar Ataxia, Autosomal-Recessive 3 (SCAR3 – OMIM 271250); Spinocerebellar Ataxia with Blindness and Deafness (SCABD)
This recessively inherited spinocerbellar ataxia accompanies optic and cochlear degeneration, leading to blindness and deafness [Spoendlin, 1974; van Bogaert and Martin, 1974]. Bomont et al. [2000] described an Israeli man and his niece, who were affected by an early-onset recessive ataxia and subsequently developed hearing impairment and optic atrophy. They demonstrated its linkage to the chromosome 6p23–p21.
Spinocerebellar Ataxia, Autosomal-Recessive 4 (SCAR4 – OMIM 607317); Spinocerebellar Ataxia with Saccadic Intrusions (SCASI)
Five of 14 siblings of a family of Slovenian descent have been described with progressive ataxia, gait unsteadiness, and difficulty reading starting in the third decade. All patients developed gait, trunk, and limb ataxia, as well as pyramidal tract signs. They eventually required walking aids by the sixth decade (ages 49–56 years). Moreover, patients had myoclonic jerks, fasciculations, impaired joint position sense, cerebellar dysarthria, and mild pes cavus. There was also striking disturbance of eye movements, with horizontal macrosaccadic oscillations of a high velocity that were induced with each gaze shift. By genome-wide linkage analysis a candidate locus on chromosome 1p36 was identified [Swartz et al., 2003].
Spinocerebellar Ataxia, Autosomal-Recessive 5 (SCAR5 – OMIM 606937); Nonprogressive Congenital Cerebellar Ataxia with Mental Retardation, Optic Atrophy, and Skin Abnormalities (CAMOS)
Megarbane et al. [2001] described five children from a large inbred Lebanese Druze family, with a new form of nonprogressive autosomal-recessive congenital ataxia associated with optic atrophy, severe mental retardation, short stature, speech defect, abnormal osmiophilic pattern of skin vessels, and cerebellar atrophy. There was no evidence of metabolic disease, and analysis of respiratory chain complex abnormalities was unremarkable [Megarbane et al., 2001]. The disease locus has been mapped to a 3.6-cM interval on chromosome 15q24–q26 using identity by descent and DNA pooling [Delague et al., 2002].
Spinocerebellar Ataxia, Autosomal-Recessive 6 (SCAR6 – OMIM 608029); Norwegian Nonprogressive Infantile Cerebellar Ataxia (CLA3)
SCAR6 was first described in seven consanguineous patients, four adults and three children from a large inbred Norwegian family, ranging in age from 4 years to 59 years. All patients showed a nonprogressive cerebellar ataxia manifesting in early childhood. The phenotype of this disorder includes defects in motor coordination of limbs, slow speech, poor eye movement coordination, slight spastic signs, short stature, but normal intelligence [Kvistad et al., 1985]. The disease locus has been mapped to a 19.5-cM interval on chromosome 20q11–q13 [Tranebjaerg et al., 2003].
Spinocerebellar Ataxia, Autosomal-Recessive 7 (SCAR7 – OMIM 609270)
Breedveld et al. [2004] identified a nonconsanguineous Dutch family with slowly progressive spinocerebellar ataxia, nystagmus, saccadic pursuit eye movements, a postural tremor, hyperreflexia, extensor plantar responses, and absence of other (non)neurological features. All patients with this childhood-onset disease were studied as adults, from 46 to 64 years of age, but the severity of symptoms and progression of the disease within this family were remarkably variable. Using a systematic genome-wide scan, the responsible gene for SCAR7 was mapped to a 5.9 cM interval on chromosome 11p15 [Breedveld et al., 2004].
Spinocerebellar Ataxia, Autosomal-Recessive 8 (SCAR8 – OMIM 610743); Cerebellar Ataxia, Autosomal-Recessive, Type 1 (ARCA1); Recessive Ataxia of Beauce
SCAR8 is identified in 26 French Canadian families, including 53 affected members, mostly from the Beauce and Bas-St-Laurent regions of the province of Quebec in Canada and mapped on the chromosome 11p15. SCAR8 is a fairly pure cerebellar ataxia that is caused by various mutations in SYNE1 [Gros-Louis et al., 2007]. This autosomal-recessive disease starts at 17–46 years of age. Various mutations produce a fairly homogeneous phenotype among patients. A splice-site mutation was the most common mutation, occurring at a frequency of 50.8 percent among 124 patients [Dupre et al., 2007]. SCAR8 shows slow progression and causes moderate disability. Affected individuals have significant dysarthria, mild oculomotor abnormalities, occasional brisk reflexes in the lower extremities, normal nerve conduction studies, and diffuse cerebellar atrophy on imaging [Dupre et al., 2007].
SYNE1 encodes a very large protein with 8797 amino acid residues. The protein contains two N-terminal actin-binding regions that comprise tandem-paired calponin-homology domains, a transmembrane domain, multiple spectrin repeats, and a C-terminal klarsicht domain (KASH). It is highly expressed in Purkinje cells and neuromuscular junctions. SYNE1 is a part of the spectrin family, a group of structural proteins that share a common function of linking the plasma membrane to the actin cytoskeleton [Gros-Louis et al., 2007].
Spinocerebellar Ataxia, Autosomal-Recessive 9 (SCAR9 – OMIM 612016); Cerebellar Ataxia, Autosomal-Recessive, Type 2 (ARCA2)
SCAR9 has been described in four patients from a consanguineous Algerian family and in three other sporadic patients with slowly progressive childhood-onset gait ataxia and cerebellar atrophy. Five patients had pes cavus, 4 had brisk reflexes, 2 had Huffmann’s sign, 3 had mild psychomotor retardation, 3 had exercise intolerance and elevated serum lactate, and 1 patient had mild axonal degeneration of the sural nerve. Single nucleotide polymorphism (SNP)-based genome-wide scanning revealed a locus for autosomal-recessive ataxia at chromosome 1q41, and further sequencing led to the identification of the causative mutation as a homozygous splice-site mutation in the aarF-domain-containing kinase 3 gene (ADCK3). ADCK3 is a mitochondrial protein homologous to the yeast COQ8 and the bacterial UbiB proteins, which are required for muscle coenzyme Q10 (CoQ10 or ubiquinone) biosynthesis [Lagier-Tourenne et al., 2008].
CoQ10 deficiency has been reported in some patients with childhood-onset cerebellar ataxia [Lamperti et al., 2003]. CoQ10 deficiency causes mitochondrial dysfunction because CoQ10 carries electrons from complex I and complex II to complex III in the mitochondrial respiratory chain. Most of the SCAR9 patients showed a decreased level of CoQ10 [Lagier-Tourenne et al., 2008]. CoQ10 supplementation might be beneficial for SCAR9 patients, as well as for others with mitochondrial respiratory chain dysfunction-related ataxias.
Cerebellar Ataxia, Cayman Type (ATCAY – OMIM 601238)
Cerebellar ataxia, Cayman type, is a recessive disorder described in an isolated population from Grand Cayman Islands, mapping to chromosome 19q13.3 [Johnson et al., 1978; Brown et al., 1984; Nystuen et al., 1996]. The disorder causes hypotonia from birth, followed by psychomotor retardation, nystagmus, intention tremor, dysarthria, and ataxic gait. The cerebellar findings are typically nonprogressive. Brain imaging shows isolated cerebellar atrophy. The ATCAY gene is expressed only in neurons in the brain and in components of the spinal cord and peripheral nervous system. Two nucleotide substitutions, one in exon 9 that results in a serine to arginine substitution, and one in intron 9 that interferes with splicing, have been identified as causative [Bomar et al., 2003]. ATCAY codes for the caytaxin protein, a protein that shares a conserved domain with TTPA (see Vitamin E deficiency syndromes, above). However, it does not appear that caytaxin binds vitamin E, and its function is currently unknown.
Infantile-Onset Spinocerebellar Ataxia (IOSCA – OMIM 271245)
IOSCA, the second most common inherited ataxia in Finland with a population carrier frequency of more than 1:230, maps to chromosome 10q24 [Nikali et al., 2005]. Dysfunction starts at 9–18 months of age, with muscle hypotonia, athetosis, ataxia, ophthalmoplegia, sensorineural hearing deficit, sensory axonal neuropathy, epileptic encephalopathy, and female hypogonadism [Koskinen et al., 1994; Nikali et al., 2005]. Sensory axonal neuropathy and progressive atrophy of the cerebellum, brainstem, and spinal cord are neuropathological characteristics of IOSCA [Koskinen et al., 1994]. The disease results from a recessive mutation in C10Orf2, which encodes Twinkle (Y508C), a mitochondrial (mt)DNA-specific helicase [Nikali et al., 2005]. Different mutations in this same gene cause autosomal-dominant progressive external ophthalmoplegia (adPEO) with multiple mtDNA deletions (OMIM 606075), a neuromuscular disorder sharing a spectrum of symptoms with IOSCA [Nikali et al., 2005]. The depletion of mtDNA has been shown in IOSCA patients’ brains. This finding categorizes IOSCA as a new member of the mtDNA depletion syndrome family [Hakonen et al., 2008].
Ramsay Hunt Syndrome: Progressive Myoclonic Ataxia and Progressive Myoclonic Epilepsy (Baltic Myoclonus, Mediterranean Myoclonus, Unverricht–Lundborg Syndrome)
Ramsay Hunt syndrome is a heterogeneous group of disorders whose clinical hallmarks are cerebellar ataxia and myoclonus [Hunt, 1922]. The presentation and severity are variable, and the differential diagnosis includes myoclonic epilepsy with ragged red fibers (MERRF), sialidosis, and progressive myoclonic epilepsy. These disorders are described in other chapters in this text.
Progressive myoclonic epilepsy (OMIM 254800) was first described in families from Estonia and Eastern Sweden, but is most common in Finland. The disease typically presents with generalized tonic-clonic seizures and myoclonus, with ataxia seen later in the course. Dodecamer repeats in the promoter region of the cystatin B gene are the most frequent cause [Lafreniere et al., 1997; Virtaneva et al., 1997].
Other Childhood-Onset Ataxias
Other early-onset ataxias with associated deafness and varying degrees of mental retardation have been reported. In Behr’s syndrome, mental retardation, ataxia, sensory neuropathy, spasticity, and optic atrophy are the predominant findings [Thomas et al., 1984]. Marinesco–Sjögren syndrome is characterized by mental retardation, ataxia, cataracts, short stature, and limited sexual maturation.
Other conditions have been associated with atypical findings of dementia, muscular atrophy, ichthyosis, and familial spastic paralysis. Both autosomal-dominant and X-linked transmission have been described in these patients [Spira et al., 1979; Farlow et al., 1987].
Autosomal-Dominant Inherited Ataxias (Spinocerebellar Ataxias)
The dominantly inherited spinocerebellar ataxias (SCAs; summarized in Table 67-3) are a group of conditions characterized by premature cerebellar neuronal loss, with some types involving additional structures such as the optic nerve, basal ganglia, brainstem, and spinal cord. These heterogeneous disorders have been designated the olivopontocerebellar atrophies (OPCAs) because of the frequent presence of atrophy involving the cerebellum and pontine and olivary nuclei [Koeppen and Barron, 1984]. In parallel with the pathologic abnormalities, patients with SCA display various clinical features of progressive ataxia, motor impairment, extrapyramidal symptoms, retinal degeneration, deafness, ophthalmoplegia, dorsal column dysfunction, and peripheral neuropathy. The overall incidence of SCA is 1–5 per 100,000, with an average age of onset in the third decade of life.
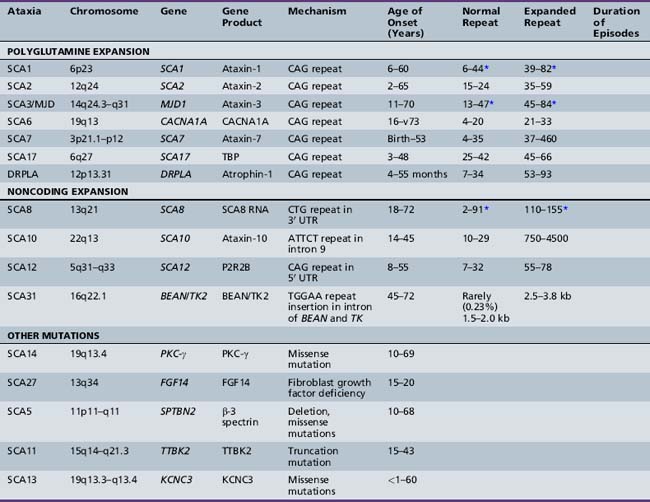
Harding proposed a classification system for the autosomal-dominant cerebellar ataxias (ADCAs) that uses the constellation of clinical symptoms for categorization [Harding, 1993]. Type I ADCAs have a cerebellar syndrome plus pyramidal signs, supranuclear ophthalmoplegia, extrapyramidal signs, and dementia. Type II ADCAs show a cerebellar syndrome plus pigmentary maculopathy. Type III ADCAs are “pure” cerebellar syndromes that have mild, if any, pyramidal symptoms. Some authors have attempted to keep these clinical categories and assign genetically defined SCAs to them. The clinical findings are so variable that they are unreliable for precise diagnosis [Schut, 1954; Currier et al., 1972; Nino et al., 1980; Zoghbi et al., 1988].
Spinocerebellar Ataxia Type 1 (SCA1 – OMIM 164400)
SCA1 is characterized by progressive loss of balance and coordination, mild cognitive impairments, oculomotor deficits (including gaze palsy and slowing of saccades), dysarthria, dysphagia, and eventual respiratory failure. The primary sites of neurodegeneration in SCA1 include cerebellar Purkinje cells, brainstem cranial nerve nuclei, the inferior olive, and spinocerebellar tracts [Zoghbi and Orr, 2000]. Onset is typically in the third or fourth decade of life, but patients presenting as young as 4 years of age and as late as 60 years of age have been reported [Zoghbi and Orr, 2000]. The disorder has been described in many ethnic groups, including whites, blacks, Asians and Iakuts [Ranum et al., 1994a].
The SCA1 gene was mapped to chromosome 6p22–23 and the mutation identified using a positional cloning approach [Orr et al., 1993; Banfi et al., 1994]. A highly polymorphic CAG repeat encoding a polyglutamine tract was found to be unstable and expanded in all individuals with SCA1 [Orr et al., 1993]. The number of CAG repeats ranges from 6 to 44 on normal chromosomes, and from 39 to 82 on SCA1 chromosomes [Servadio et al., 1995]. The distinction between disease-causing and “normal” alleles in the range of overlap is made on the basis of the fact that the repeat tract on normal chromosomes is interrupted by 1–4 CAT trinucleotides in alleles with 20 or more repeats, whereas SCA1 chromosomes contain a “pure,” uninterrupted CAG tract [Chung et al., 1993]. This observation led to the hypothesis that loss of the CAT interruption rendered the SCA1 CAG repeat unstable and more likely to expand [Chung et al., 1993].
There is a strong inverse correlation between repeat size and age of onset; individuals with 70 or more CAG repeats have childhood onset, a rapidly deteriorating course, and death within 4–5 years, whereas adult-onset patients have fewer than 70 repeats and typically live 10–20 years after symptoms appear [Zoghbi and Orr, 2000]. Genetic anticipation, or the tendency of affected individuals in each successive generation of a kindred to show earlier and earlier disease onset, is a prominent feature of all polyglutamine repeat diseases. The mechanism involves progressive expansion of the polyglutamine repeats in disease alleles, particularly those that are paternally transmitted [Orr et al., 1993].
The SCA1 CAG repeat falls within the open reading frame of a transcript that is widely expressed and whose size is approximately 11.5 kb [Orr et al., 1993]. The CAG repeat codes for polyglutamine within ataxin-1, a protein predicted to contain 792–830 amino acids, depending on the size of the repeat [Banfi et al., 1994]. Ataxin-1 is predominantly nuclear in neurons and cytoplasmic in peripheral cells; neurons might shuttle the protein between the two compartments [Servadio et al., 1995]. A 120-residue region of the protein (563–694 in alleles containing 30 glutamines) shares significant sequence similarity with the HMG-box transcription factor HBP1 and has been implicated in protein–protein interactions and possibly in protein–RNA interactions [Chen et al., 2004b].
The exact cellular functions of ataxin-1 are currently unknown. Mice lacking ataxin-1 have spatial and motor learning deficiencies, as well as impairment of short-term plasticity [Matilla et al., 1998], but do not display ataxia or neuronal degeneration. These findings show that loss of function of ataxin-1 cannot explain SCA1 pathogenesis. Support for a gain-of-function pathogenic mechanism first came from mice overexpressing a mutant SCA1 cDNA with 82 glutamines (82Q), specifically in Purkinje cells. These mice developed progressive ataxia and Purkinje cell degeneration [Burright et al., 1995]. The mutant ataxin-1 accumulates in nuclear inclusions (NIs), along with components of the proteasome, suggesting that mutant ataxin-1 might misfold and resist degradation [Cummings et al., 1998]. Similarly, fruitflies that overexpress human ataxin-1 (82Q) also developed progressive neuronal degeneration and ataxin-1-containing NIs that co-localized with chaperones and ubiquitin. To characterize the effects of the SCA1 mutation on all ataxin-1-expressing neurons, an expanded CAG tract of 154 repeats was targeted in place of the typical two CAGs present in the endogenous mouse locus. Sca1154Q/+ mice reproduced all features of human SCA1, including ataxia, brainstem dysfunction, Purkinje cell degeneration, and premature death [Watase et al., 2002]. Consistent with the in vivo data, cellular and in vitro studies demonstrated that ataxin-1 is ubiquitinated, that the expanded polyglutamine tract retards its degradation, and that inhibition of the proteasome enhances its accumulation [Cummings et al., 1998, 1999]. Chaperone overexpression, which presumably aids in the folding and/or elimination of the expanded protein, improved motor coordination and suppressed Purkinje cell degeneration in SCA1 (82Q) transgenic mice [Cummings et al., 2001]. The important roles of protein misfolding and degradation have also been demonstrated in several other polyglutamine diseases [Opal and Zoghbi, 2002].
Furthermore, neuropathogenesis of ataxin-1 depends on native, not novel, protein interactions [Lam et al., 2006]. Lim et al. [2008] proposed a two-pronged model of SCA1 neurodegeneration, in which increased function of a particular stable endogenous protein complex is combined with a simultaneous loss of function of other stable endogenous protein complexes.
Continuous expression of mutant ataxin-1 is necessary for progression of SCA1 pathogenesis. Given the reversibility of the symptoms and cell pathology in an SCA1 mouse model, there is a window of opportunity when therapies can ameliorate this disease [Zu et al., 2004]. No treatment is currently available for SCA1. Patients experience progressive limitations in their activities, lose the ability to walk, and eventually become bedridden. Based on the studies in SCA1 animal models, it appears that lithium and neural stem cell transplantation in the cerebellum could be viable therapeutic approaches for SCA1 patients [Watase et al., 2007; Chintawar et al., 2009].
Spinocerebellar Ataxia Type 2 (SCA2 – OMIM 183090)
In 1989, Orozco et al. described a form of dominantly inherited spinocerebellar ataxia that occurs at an estimated frequency of 41 per 100,000 in the province of Holguín, Cuba [Orozco et al., 1989]. Age of onset varies from 2 to 65 years, with 40 percent of the patients presenting with symptoms before 25 years of age. Onset before the age of 20 years correlates with a more aggressive disease course. The clinical features include ataxia, dysarthria, tremor, and extremely slow saccades. Hyporeflexia of the upper limbs and ophthalmoparesis are seen in over half the patients. Dementia occurs in a significant minority of patients. The locus was termed SCA2 and mapped to chromosome 12q24 [Gispert et al., 1993]. Families with this disorder from other regions of the world have also been described [Lopes-Cendes et al., 1994].
Pathology specimens show severe loss of Purkinje cells and of neurons in the substantia nigra and basis pontis. Mild to moderate neuronal loss is seen in the inferior and accessory olives; dentate, arcuate, gracile, and accessory cuneate nuclei; internal granule cell layer of the cerebellum; and the anterior horns of the spinal cord. Axonal loss from the dorsal roots and posterior columns, the dorsal spinocerebellar tract, and often the anterior spinal roots is also found. Reactive gliosis can be seen in the globus pallidus, thalamus, subthalamus, and periaqueductal regions [Koeppen, 1998; Huynh et al., 1999].
Using three different approaches, the SCA2 gene was identified, and the mutation was determined to involve an expansion of a CAG trinucleotide repeat that lies within the coding region of a novel protein termed ataxin-2 [Imbert et al., 1996; Pulst et al., 1996; Sanpei et al., 1996]. Normal ataxin-2 alleles contain 15–24 CAG repeats (with 22 repeats found 94 percent of the time), while disease alleles have 35–59 repeats. The range of pathogenic alleles in SCA2 is shifted toward shorter repeats compared with other SCAs, suggesting that the expanded repeat within ataxin-2 is more deleterious to neurons. The CAG repeat in the SCA2 gene is interrupted by 1–3 CAA units only on normal alleles, suggesting that, as in SCA1, interruption of CAG repeats confers stability. Finally, there is a strong inverse correlation between the size of the repeat and the age of symptom onset.
The SCA2 gene, ataxin-2, contains 25 exons and spans approximately 130 kb of genomic DNA. Two alternately spliced forms of messenger (m)RNAs are generated, one of which results in a protein that is truncated by 70 amino acids [Sahba et al., 1998]. Ataxin-2 mRNA is found in multiple tissues and in all regions of the CNS [Imbert et al., 1996; Pulst et al., 1996; Sanpei et al., 1996], where it is present predominantly in neurons and is highly expressed in Purkinje cells [Huynh et al., 1999]. Ataxin-2 regulates the intracellular concentration of its interaction partner, the poly(A)-binding protein, a stress granule component and a key factor for translational control [Nonhoff et al., 2007]. Ataxin-2 is associated with rough endoplasmic reticulum in a complex that is RNA, salt, and phosphorylation-dependent [van de Loo et al., 2009]. Ataxin-2 associates with polyribosomes under normal conditions and is recruited to stress granules upon environmental stress. Mutant ataxin-2 sensitizes Purkinje cells to glutamate-induced apoptosis. Glutamate-induced cell death of 58Q PC (Purkinje cell) cultures was attenuated by dantrolene, a clinically relevant ryanodine receptor (an intracellular calcium channel) inhibitor and Ca2 stabilizer. Thus, neuronal Ca2 signaling may play an important role in SCA2 pathology and the ryanodine receptor could be a potential therapeutic target for treatment of SCA2 patients [Liu et al., 2009].
Machado–Joseph Disease (SCA3 – OMIM 109150)
SCA3, also known as Machado–Joseph disease (MJD), was first described in families with Azorean-Portuguese origin and is the predominate form of autosomal-dominant SCA in Europe, Japan, and the United States (reviewed in Riess et al. [2008]). The clinical features of MJD include progressive ataxia, areflexia, peripheral amyotrophy, external ophthalmoplegia, bulging eyes, facial and lingual fasciculations, muscle atrophy, parkinsonian features, dystonia, and spasticity. Age of onset varies from 5 to 75 years of age, and correlates inversely with the CAG repeat length [Riess et al., 2008]. Early attempts to categorize this disorder into different clinical subtypes were thwarted by the variability in presentation and the tendency of individuals to “evolve” from one subtype to another [Rosenberg et al., 1976; Coutinho and Andrade, 1978; Barbeau et al., 1984].
The MJD gene, which encodes ataxin-3, was mapped to the long arm of chromosome 14 (14q24.3–q32) [Takiyama et al., 1993]. Subsequently, Stevanin et al. [1994] mapped the gene in a family with a dominantly inherited spinocerebellar ataxia to chromosome 14q24.3–qter. Normal MJD1 alleles contain from 13 to 47 CAG repeats, while expanded alleles contain 45–84 repeats [Riess et al., 2008].
Several ataxin-3 isoforms have been described. It is the smallest of the polyglutamine proteins with a ubiquitin interaction motif (UIM) situated near the polyglutamine domain. Ataxin-3 interacts with Rad23 and valosin-containing protein (VCP) [Doss-Pepe et al., 2003]. Rad23, a member of the proteasomal protein degradation machinery, in turn interacts with the proteasome subunit S5a, suggesting a role of the protein complex in “protein shuttling” for translocating proteins to the proteasome for degradation. VCP, also called CDC48, is an ATPase associated with various activities and has been implicated in multiple cellular functions ranging from organelle biogenesis to ubiquitin-dependent protein degradation (reviewed in Riess et al. [2008]. Mutation of VCP causes inclusion body myopathy with Paget’s disease and frontotemporal dementia (IBMPFD – OMIM 167320). Most interestingly, Parkin, the E3 ubiquitin ligase that is frequently mutated in early-onset autosomal-recessive Parkinson’s disease, promotes ubiquitination and degradation of ataxin-3 [Tsai et al., 2003].
The pathogenesis of MJD/SCA3 is still poorly understood. The introduction of ataxin-3 with an expanded polyglutamine tract into cultured cells induces apoptosis, suggesting that the mutant protein is either directly or indirectly involved with a cellular suicide pathway [Ikeda et al., 1996]. Other studies suggest that protein misfolding, presumably initiated by the expanded polyglutamine tract, leads to ubiquitination and subsequent formation of intranuclear inclusions [Chai et al., 1999]. This is a common pathway in polyglutamine expansion diseases. How this leads to cell dysfunction and death, and why the clinical time course takes so long to evolve, remain unknown.
Spinocerebellar Ataxia Type 4 (OMIM 600223)
Seven kindreds (one from Utah and six from Japan) have been described thus far [Gardner et al., 1994; Nagaoka et al., 2000]. Clinically, affected individuals in the Japanese families have a pure cerebellar ataxia, while those in the Utah kindred also have dysarthria and sensory axonal neuropathy [Flanigan et al., 1996]. Detailed genetic mapping studies show that the gene involved in SCA4 maps to the long arm of human chromosome 16. Anticipation does not occur.
Spinocerebellar Ataxia Type 5 (OMIM 600224)
The locus for SCA5 was localized to the centromeric region of human chromosome 11, based on detailed mapping studies of two major branches of a family descended from the paternal grandparents of President Abraham Lincoln [Ranum et al., 1994b]. Ikeda et al. [2006] found that SCA5 is caused by mutations in SPTBN2, which encodes beta-3 spectrin. Beta-3 spectrin is highly expressed in Purkinje cells and stabilizes the glutamate transporter EAAT4. The phenotype is that of pure cerebellar ataxia and dysarthria. The disease is milder than that observed in other SCAs, without substantial effects on life span.
Spinocerebellar Ataxia Type 6 (SCA6 – OMIM 183086)
SCA6 accounts for 1–13 percent of ADCA families from European centers, 6–31 percent from Japanese centers, and up to 11 percent in Chinese series [Schols et al., 1998] (see review in Mantuano et al. [2003]). The mutation is occasionally found in sporadic cases as well. The clinical features consist predominantly of mild but slowly progressive cerebellar ataxia of the limbs, gait ataxia, dysarthria, nystagmus, and mild vibratory and proprioceptive sensory loss. Cognitive function is not affected. Patients often have acute episodes of ataxia or vertigo that are responsive to acetazolamide. Onset is later than the other dominantly inherited SCAs, typically occurring in the third decade at the earliest. Brain imaging reveals atrophy of the cerebellum.
Pathology specimens demonstrate loss of Purkinje and granule cells, with proliferation of Bergmann glia, that is more prominent in the vermis than in the hemispheres [Gomez et al., 1997; Tsuchiya et al., 1998; Takahashi et al., 2004]. Loss of inferior olive neurons also occurs, but may be secondary to the cerebellar changes.
The genetic locus was mapped to chromosome 19q13, where a CAG repeat expansion in exon 47 at the 3′ region of the CACNA1A gene was identified as the causative mutation [Zhuchenko et al., 1997]. CACNA1A codes for the brain-specific, voltage-sensitive α1A (Cav2.1) subunit of the P/Q-type calcium channel, which is highly expressed in Purkinje cells [Ishikawa et al., 1999]. Alternative splicing leads to six isoforms, three of which contain the polyglutamine sequence. Repeat expansions are the smallest of the triplet repeat diseases, with 21 or more repeats being pathogenic. Individuals who are homozygous for expanded repeats show no phenotypic or age-of-onset differences [Takiyama et al., 1998]. Phenotypic severity is inversely correlated with the number of CAG repeats, as is the age of onset [Takahashi et al., 2004]. Clinical anticipation has been observed, but does not always appear to stem from CAG repeat instability. No CAA interruptions are present in the repeats.
Proteins with the expanded repeat aggregate specifically in the cytoplasm of Purkinje cells [Ishikawa et al., 1999]. These inclusions are not ubiquitinated and lack several other components found in the inclusions of other CAG repeat disorders. It is not clear whether SCA6 is caused by a “gain-of-function” mechanism similar to that in other polyglutamine diseases, or by a channelopathy that alters calcium homeostasis. As will be discussed later (see “Episodic Ataxias” later in this chapter), point mutations and deletions in the CACNA1A coding sequence cause episodic ataxia type 2 and familial hemiplegic migraine type 1. Families with significant phenotypic overlap have been identified, leading some to postulate that the three disorders represent a spectrum of disease, rather than truly separate entities. The identification of small triplet expansions (20 and 23 repeats) in two kindreds whose members presented with episodic ataxia type 2, and the identification of a point mutation leading to early hemiplegic migraine and later progressive ataxia, are supportive of this interpretation [Jodice et al., 1997; Alonso et al., 2003]. Gabapentin, which interacts with the α2δ subunit of the P/Q-type voltage-dependent calcium channel, demonstrated clinical benefit in a small trial on SCA6 patients [Nakamura et al., 2006]; establishing the efficacy of gabapantin will require further controlled trials.
Spinocerebellar Ataxia Type 7 (SCA7 – OMIM 164500)
SCA7 is distinguished clinically from the other SCAs by the presence of macular dystrophy leading to pigmentary retinal degeneration (reviewed in Michalik et al. [2004]). The retinal changes in SCA7 start in a central location and move peripherally as the disease progresses, thus distinguishing the eye findings from those in retinitis pigmentosa. Progressive ataxia/dysmetria, pyramidal tract signs, supranuclear ophthalmoplegia, and dysarthria/dysphagia are also present, and occasionally dementia and deafness are seen as well [Benton et al., 1998]. Either ataxia or visual changes can be the presenting symptom, depending upon the number of CAG repeats in the expanded allele (see below). Brain imaging in all cases typically demonstrates cerebellar atrophy; ventricular dilatation and delayed myelination can be found in infantile cases. Brain pathological specimens show atrophy of the cerebellum, with loss of dentate neurons and Purkinje cells, and loss of inferior olivary neurons in the brainstem; posterior column, and dorsal and ventral spinocerebellar tract loss in the spinal cord; and macular degeneration with loss of photoreceptive cells in the retina.
An extremely aggressive infantile-onset form of SCA7 has been described [Enevoldson et al., 1994; Benton et al., 1998]. It presents with hypotonia, dysphagia, myoclonic seizures, and visual disturbances that typically lead to rapid mental deterioration and severe physical disability, culminating in death by age 3 or earlier. The infantile-onset form is also associated with cardiac abnormalities, particularly patent ductus arteriosus [Benton et al., 1998; Johansson et al., 1998; van de Warrenburg et al., 2001]. Childhood-onset SCA7 is less aggressive than the infantile-onset form but more progressive than the adult-onset form.
SCA7 maps to the short arm of chromosome 3 [Benomar et al., 1995; Gouw et al., 1995; Holmberg et al., 1998]. By using an antibody that selectively detects proteins containing an expanded polyglutamine tract, it was demonstrated that the mutational mechanism in SCA7 is expansion of a CAG repeat [Trottier et al., 1995]. SCA7 encodes a novel protein, ataxin-7, with a polyglutamine tract at the amino-terminus [David et al., 1997]. The SCA7 CAG repeat ranges in size from 4 to 35 repeats in normal alleles, and from 37 to 306 repeats in expanded alleles. Typically, larger expansions are paternally inherited and result from significant intergenerational repeat instability. An inverse correlation exists between repeat length and age of onset, and repeat expansions of over 59 repeats tend to develop visual impairment prior to ataxia [Johansson et al., 1998]. Alleles ranging in size from 34 to 36 repeats have been observed in individuals who are at risk but asymptomatic at the time of evaluation [David et al., 1997; Benton et al., 1998; Holmberg et al., 1998; Johansson et al., 1998].
Mouse models of SCA7 have provided some insight into the pathogenesis of the disease. Expression of the expanded ataxin-7 protein in transgenic mice leads to the development of intranuclear inclusions and the degeneration of rod photoreceptors and Purkinje cells, findings that are consistent with the human phenotype [Yvert et al., 2000]. In retinal photoreceptor cells, the expression of expanded ataxin-7 is associated with alterations in gene expression that predate photoreceptor degeneration [La Spada et al., 2001; Yoo et al., 2003]. Another interesting finding is that the speed of ataxin-7 accumulation varies in different neuronal populations, suggesting that intrinsic differences in the ability to clear mutant protein may explain the variable susceptibility of different cell types in this and other polyglutamine diseases [Yoo et al., 2003]. Cellular dysfunction predates the appearance of neuronal inclusions, suggesting that, while they are part of the pathogenesis, inclusions in and of themselves are not the primary cause of dysfunction [Yoo et al., 2003].
Recently, it has been shown that ataxin-7 is a core component of a transcriptional co-activator complex called STAGA (reviewed in Garden and La Spada [2008]). STAGA is the mammalian equivalent of a yeast transcriptional co-activator named SAGA, which contains histone acetyltransferase (HAT) activity, mediated by the Gcn5 enzyme. Sgf73, the yeast homolog of ataxin-7, is a component of the SAGA complex with essential function in yeast [Sanders et al., 2002]. Two independent groups have shown that ataxin-7 is indeed a core component of STAGA complex in mammals [Helmlinger et al., 2004; Palhan et al., 2005], but why, in spite of ubiquitous expression of the STAGA complex, neurodegeneration happens only in specific group of neurons remains to be understood. The answer might underlie identification of STAGA-dependent transcription factors whose functional impairment underlies the production of the SCA7 phenotype.
Spinocerebellar Ataxia Type 8 (SCA8 – OMIM 608768)
Described predominantly in Caucasian families from the United States and Canada, the clinical presentation of SCA8 is similar to that of the other slowly progressive SCAs. Ataxic dysarthria, nystagmus, limb and gait ataxia, limb spasticity, and diminished vibratory sense are present [Koob et al., 1999]. MRI of the brain demonstrates cerebellar atrophy. SCA8 was mapped to chromosome 13q21, and the vast majority of disease alleles in individuals of European descent share a common haplotype [Ikeda et al., 2004].
Unlike other repeat disorders caused by polyglutamine expansions, SCA8 is caused by a CTG repeat in a noncoding gene; that is, no open reading frame is present in the gene, suggesting that it is transcribed into mRNA but not translated into protein [Koob et al., 1999]. This is only one of several unusual characteristics of SCA8. There is high variability in the repeat length of wild-type alleles; more than 99 percent of unaffected individuals have repeat lengths less than 74, but a few unaffected individuals have 800 repeats, by far the largest number of normal repeats of any SCA. This remarkable overlap of repeat size in pathogenic and nonpathogenic alleles is unique among the SCAs. Furthermore, homozygosity for an expanded allele does not appear to exacerbate the disease phenotype, as it does in SCA1 and dentatorubral-pallidoluysian atrophy. Repeat lengths contract with paternal transmission and expand with maternal transmission, another unusual finding of this disease. Further confusing the picture is the coexistence of SCA8 expansions with those of SCA1 or SCA6 in some kindreds [Izumi et al., 2003; Sulek et al., 2003]. SCA8 expansions have also been found in patients with Alzheimer’s disease, Parkinson’s disease, and an individual with vitamin E deficiency heterozygous for the TTPA mutation [Sobrido et al., 2001; Cellini et al., 2002; Izumi et al., 2003]. How these factors influence the pathogenesis of the disease is unknown, and has led some authors to question the pathogenesis of the expansion itself [Stevanin et al., 2000; Worth et al., 2000; Schols et al., 2003].
The genomic arrangement of the SCA8 locus suggests a potential function of the gene. The SCA8 gene lies in a reverse orientation to and partially overlaps the locus of another gene (KLHL1). It has been suggested that the SCA8 mRNA functions as an endogenous inhibitory RNA for KLHL1, or as a regulator of RNA-binding proteins [Mutsuddi et al., 2004].
Although a relationship between repeat size and clinical signs was demonstrated in the initial report, other families with this disorder have shown greater variability in penetrance [Ikeda et al., 2004]. Sequence interruptions of the CTG expansion and the size of the CTA tract that precedes the CTG expansion are among the multiple factors that may influence SCA8 disease penetrance. A similar RNA gain of function to that seen in myotonic dystrophy types 1 and 2 (DM1, OMIM 160900; DM2, OMIM 602668) has been suggested for SCA8 pathogenesis. Recent evidence suggests that SCA8 CUGexp transcripts form hallmark ribonuclear inclusions that co-localize with MBNL1 in humans and mice, and that genetic loss of Mbnl1 enhances motor coordination deficits in SCA8 BAC-EXP mice [Daughters et al., 2009]. Furthermore, ATXN8OS CUGexp transcripts dysregulate MBNL1-CUGBP1 pathways in the CNS and trigger downstream molecular changes in gamma-aminobutyric acid (GABA)A transporter 4 (Gabt4) regulation through an RNA gain-of-function mechanism. Unlike DMPK and ZNF9 (the genes responsible for DM1 and DM2, respectively), which have broad expression patterns, SCA8 is almost exclusively expressed in the CNS; the spatial and temporal expression differences between SCA8, DMPK, and ZNF9 could explain the phenotypic differences between these three diseases (reviewed in Dick et al. [2006]).
Spinocerebellar Ataxia Type 10 (SCA10 – OMIM 603516)
Grewal et al. [1998] described a dominantly inherited SCA in a single four-generation Mexican kindred. The locus was independently mapped to chromosome 22q13 by two different groups [Matsuura et al., 1999; Zu et al., 1999]. SCA10 is the second most common form of dominantly inherited ataxia after SCA2 in people of Mexican descent and MJD in southern Brazil; this fact, coupled with the rarity of expanded alleles in other populations, has led to the hypothesis that the mutation arose in the New World [Matsuura et al., 2002]. The phenotype consists of ataxia, dysarthria, nystagmus, extrapyramidal signs, and epilepsy. Mood disorders and polyneuropathy diagnosed by nerve conduction studies can also be seen. A pentanucleotide (ATTCT) repeat in intron 9 of the ataxin-10 gene is responsible for the phenotype; instability of the repeat region can lead to as many as 4500 repeats [Matsuura et al., 2000]. There is an inverse correlation between expansion size and the age of onset, and genetic anticipation is present. The repeats are highly unstable when transmitted paternally, while maternal transmission results in more stable repeats. Brain imaging reveals cerebellar atrophy.
Ataxin-10 is a protein that is widely expressed throughout the brain and in several other tissues [Marz et al., 2004]. It is localized to the cytosol and perinuclear region in neurons. Whether the disease phenotype stems from genomic disruption, RNA gain of function, or ataxin-10 loss of function is unknown, although decreased ataxin-10 RNA levels in primary cerebellar and cortical neuronal culture result in increased apoptosis, suggesting that loss of function may be the pathogenetic mechanism [Marz et al., 2004].
Spinocerebellar Ataxia Type 11 (SCA11 – OMIM 604432)
Worth et al. [1999] described a single eight-generation family with 34 affected individuals, suffering from a benign, slowly progressive, pure cerebellar syndrome linked to chromosome 15. Mean age of onset was about 25 years; genetic anticipation did not appear to be present. The most prevalent symptoms were gait, truncal, and limb ataxia, dysarthria, nystagmus, and hyperreflexia in the lower limbs [Edener et al., 2009]. In 2007, truncation mutations in the tau tubulin kinase 2 gene (TTBK2) were described as being associated with spinocerebellar ataxia type 11: a one-base insertion in a British pedigree and a deletion of a dinucleotide in a family of Pakistani ancestry [Houlden et al., 2007]. Neuropathological examination did not reveal neuronal inclusions using hematoxylin and eosin, p62, ubiquitin or 1C2 immunohistochemistry, but neurofibrillary tangles, neuropil threads, and tau-positive neuritis were visible in the medullary tegmentum, nigra, midbrain tegmentum and tectum, and putamen. The cerebellum showed cell loss but no other visible pathology, perhaps due to the high resistance of the cerebellum to forming tangles [Houlden et al., 2007].
Spinocerebellar Ataxia Type 12 (SCA12 – OMIM 604326)
Identified in a family of German descent [Holmes et al., 1999], SCA12 is unique, in that action tremor is the most distinguishing clinical feature and is typically the initial symptom. Slow progression over several decades leads to head tremor, gait ataxia, dysmetria, dysarthria, hyperreflexia, parkinsonian signs, abnormal eye movements, and occasionally dementia. Two patients in the initial kindred were reported with childhood-onset symptoms; one had nystagmus from birth, while the other had lower-extremity dystonia that developed in childhood [O’Hearn et al., 2001]. Brain imaging reveals both cortical and cerebellar atrophy. Pathology is available only on a single brain, and revealed diffuse atrophy of cerebral and cerebellar cortices, and specifically loss of Purkinje cells. SCA12 is rare, except in India, where it is the third most common SCA [Srivastava et al., 2001; Holmes et al., 2003].
Genetic analysis revealed a triplet CAG expansion on chromosome 5q31–q33. No apparent relationship exists between repeat size and age of onset. Unlike other SCAs with CAG expansions, the expanded allele does not lead to a polyglutamine tract. Rather, the CAG expansion is found 133 nucleotides upstream of the transcription start site for PPP2R2B. The SCA12 gene product is a brain-specific regulatory subunit of protein phosphatase 2A that regulates neuronal survival through mitochondrial fission/fusion balance [Dagda et al., 2008].
Spinocerebellar Ataxia Type 13 (SCA13 – OMIM 605259)
SCA13, initially described in a four-generation French family [Herman-Bert et al., 2000], has also been characterized in a large Filipino pedigree [Waters et al., 2005]. The SCA13 gene maps to chromosome 19q13, and mutations in the voltage-gated potassium channel KCNC3 are causative for the disease [Waters et al., 2005]. Sequence analysis of KCNC3 revealed mutations 1554 G→A (R420H) in the Filipino family and 1639C→A (F448L) in the French pedigrees. Both mutations alter KCNC3 function in a Xenopus laevis oocyte expression system. The F448L mutation would be expected to be more severe because it alters key gating properties of KCNC3 channels. In contrast, R420H would be expected to reduce channel activity without changing the functional properties of the residual current [Waters et al., 2006]. This is consistent with the childhood onset with concurrent mental retardation (IQ = 62–76) and seizures in members of the French pedigree and their absence in the Filipino patients. These mutations are expected to change the output characteristics of fast-spiking cerebellar neurons, where KCNC channels confer capacity for high-frequency repetitive firing.
Spinocerebellar Ataxia Type 14 (SCA14 – OMIM 605361)
Three families (one Japanese, one Dutch, and one English/Dutch) have been described, all with a locus mapping to 19q13.4 telomeric to the locus for SCA13 [Yamashita et al., 2000; Brkanac et al., 2002a; van de Warrenburg et al., 2003]. The phenotype varies from family to family, and within families as well. In the Japanese kindred, subjects with onset of symptoms before 27 years of age presented with axial myoclonus prior to the development of ataxia, while those with onset after age 39 presented with a pure cerebellar ataxia. Gait ataxia, dysarthria, horizontal gaze nystagmus, abnormal smooth pursuit movements, hyper- and hyporeflexia, and peripheral neuropathy were described in all of the families [Yamashita et al., 2000; Brkanac et al., 2002a; van de Warrenburg et al., 2003]. Imaging reveals atrophy of the cerebellar vermis and/or hemispheres. Life span does not appear to be affected in any of the kindreds, and ataxia displays a slowly progressive course. Genetic anticipation occurs. Limited pathologic specimens suggest a primary Purkinje cell defect [Brkanac et al., 2002b].
Molecular studies reveal that missense point mutations in exon 4 of the protein kinase C gamma (PRKCG) gene are responsible. PRKCG is a member of a serine/threonine kinase family that is involved in cell proliferation and signal transduction processes. The mechanism of pathogenesis remains unknown [Chen et al., 2003; van de Warrenburg et al., 2003; Yabe et al., 2003].
Spinocerebellar Ataxia Types 15 and 16 (SCA15 – OMIM 606658 and 606364)
Spinocerebellar ataxia type 15 (SCA15) is characterized by pure cerebellar ataxia, very slow progression, and distinct cerebellar atrophy. Additional clinical features include mild titubation and postural or action tremor. The locus for SCA15 was first mapped to 3p24.2–3pter in an Australian family [Storey et al., 2001]. Hara et al. mapped two Japanese families presenting with ataxia and postural tremor of the head, arm, or trunk. The first family had partial deletions involving both the type 1 inositol 1,4,5-triphosphate receptor (ITPR1) and sulfatase-modifying factor 1 (SUMF1) genes. and the second family had only a point mutation in ITPR1 (P1059L). The proline was highly conserved and the mutation was absent in 234 chromosomes in Japanese controls [Hara et al., 2008]. These results suggest that ITPR1 but not SUMF1 is the causative gene for SCA15. On the other hand, Iwaki et al. showed that haploinsufficiency in ITPR1 alone is the cause of what had been thought to be a different disease, SCA16 (OMIM 606364) [Iwaki et al., 2008]. Since these discoveries, SCA16 has been considered a “vacant SCA” and families in Iwaki et al. are regarded as actually having SCA15.
Spinocerebellar Ataxia Type 17 (SCA17 – OMIM 607136)
This SCA variably presents with predominant symptoms of cerebellar ataxia associated with dysarthria and extrapyramidal symptoms (e.g., parkinsonism and dystonia), with mental deterioration/dementia and psychiatric symptoms such as depression and hallucinations, or as a Huntington’s disease phenocopy with chorea as the major manifestation (reviewed in Stevanin and Brice [2008]). Different individuals in the same family can have dramatically different presenting phenotypes and, as the disease progresses, symptoms from each of the subtypes can occur in the same individual. Neuroimaging demonstrates diffuse cortical and cerebellar atrophy that is most pronounced in the vermis. Pathologic specimens reveal neuronal loss in many brain areas, including loss of Purkinje cells [Nakamura et al., 2001; Rolfs et al., 2003].
SCA17 is caused by CAG repeat expansions in the TATA-binding protein (TBP) gene on chromosome 6q27 [Nakamura et al., 2001]. Like other SCAs, short CAA tracts interrupt the CAG repeats; however, both the CAG and CAA tracts are replicated in the expansions, distinguishing this SCA. Marked anticipation was observed in an Italian kindred, with the youngest family member in the fourth generation presenting with dysarthria and ataxia at age 3 [Maltecca et al., 2003]. Repeat sizes of 42–48 repeats show variable penetrance. Like other expansion diseases, an inverse correlation exists between repeat size and age of onset.
TBP is the DNA binding subunit of the RNA polymerase II transcription factor D complex, which is important in general gene transcription [Tsuji, 2004]. Immunostaining for ubiquitin, TBP, and polyglutamine tracts demonstrates their presence in intranuclear inclusions in several neuronal cell types, including Purkinje cells [Nakamura et al., 2001; Rolfs et al., 2003].
Spinocerebellar Ataxia Type 18 (SCA18; Sensory/Motor Neuropathy with Ataxia – OMIM 607458)
SCA18, or sensorimotor neuropathy with ataxia (SMNA), was described in a five-generation American family of Irish descent [Brkanac et al., 2002b]. Clinically, affected individuals present first with gait instability in the dark, followed by limb ataxia, nystagmus, decreased vibratory and position sense, decreased deep tendon reflexes, and proximal or distal muscle weakness and atrophy. Upgoing plantar responses and pes cavus are occasionally seen. Neuroimaging is either normal or demonstrates mild cerebellar atrophy. Electromyography/nerve conduction velocity studies show evidence of denervation and axonal sensory neuropathy. Thus, the overall picture is one of a mixed cerebellar degeneration and peripheral neuropathy. The disorder was mapped to chromosome 7q31–32. Interferon-related developmental regulator gene 1 (IFRD1) has been proposed as a candidate gene [Brkanac et al., 2009].
Spinocerebellar Ataxia Type 19 (SCA19 – OMIM 607346)
A single Dutch family with ataxia, cognitive impairment, irregular postural tremor, myoclonus and poor performance on the Wisconsin Card Sorting Test has been described. Imaging shows atrophy of the cerebellar hemispheres or vermis [Schelhaas and van de Warrenburg, 2005]. The genetic locus maps to chromosome 1p21–q21, where it overlaps with the locus for SCA22 (see below).
Spinocerebellar Ataxia Type 20 (SCA20 – OMIM 608687)
A dominantly inherited cerebellar ataxia associated with palatal tremor and a characteristic dysphonia has been described in a single Anglo-Celtic kindred [Knight et al., 2004]. Imaging shows mild to moderate cerebellar atrophy and the unique finding of dentate calcification in the absence of basal ganglia calcification. The genetic locus overlaps with that of SCA5, so the separate identity of this SCA remains to be established.
Spinocerebellar Ataxia Type 21 (SCA21 – OMIM 607454)
This SCA was described in a single French kindred with gait ataxia, extrapyramidal signs such as tremor, akinesia and cogwheeling, and mental impairment mapping to chromosome 7p21.3–p15.1 [Devos et al., 2001; Vuillaume et al., 2002]. Eye movements are normal, and disease progression is slow. Anticipation is suggested. Brain imaging shows marked atrophy of the cerebellum. The responsible gene has not yet been identified.
Spinocerebellar Ataxia Type 22 (SCA22)
Chung et al. [2003] reported a pure cerebellar ataxia syndrome consisting of gait and limb ataxia, dysarthria, and nystagmus in a single Chinese kindred, mapping to chromosome 1p21–q23. Neuroimaging showed diffuse involvement of the cerebellum. Anticipation is suggested. Based on their chromosomal loci, it has been suggested that SCA19 and SCA22 might be the same disorder [Schelhaas and van de Warrenburg, 2005]; identification of the responsible gene(s) should answer this question.
Spinocerebellar Ataxia Type 23 (SCA23)
A single, three-generation Dutch family with a late onset (>40 years), slowly progressive SCA was identified with locus linkage at 20p13–p12.2 [Verbeek et al., 2004]. The clinical presentation includes gait and limb ataxia, disturbance of oculomotor control, dysarthria, and hyperreflexia. Neuroimaging demonstrates severe cerebellar atrophy. Pathology in a single individual who died at 80 years of age showed loss of Purkinje cells, neurons in the dentate nuclei and inferior olives; thinning of the cerebellopontine tracts; demyelination of the posterior and lateral columns in the spinal cord; and ubiquitin-positive, polyglutamine-negative intranuclear inclusions in nigral neurons that resembled Marinesco bodies. The pathogenic significance of this last finding is uncertain. A candidate gene approach has thus far failed to identify the responsible gene.
Spinocerebellar Ataxia Type 25 (SCA25 – OMIM 608703)
Stevanin et al. [2004] reported a single French kindred with cerebellar ataxia and peripheral sensory neuropathy manifest as loss of vibratory, light touch, and pain sensation. Initial symptoms often include vomiting and gastrointestinal features. No evidence of anticipation was found. Brain imaging shows global cerebellar atrophy. A candidate gene search failed to identify the affected gene.
Spinocerebellar Ataxia Type 26 (SCA26 – OMIM 609306)
Yu et al. [2005] studied a six-generation kindred of Norwegian ancestry with pure cerebellar ataxia signs, including slowly progressive gait ataxia, trunk and limbs ataxia, dysarthria, and irregular visual pursuit movements. Age at onset ranged from 26 to 60 years. Genome-wide linkage analysis identified the disease locus as chromosome 19p13.3, close to SCA6 (OMIM 183086). Genetic analysis, however, showed that these patients have wild-type CACNA1A alleles [Yu et al., 2005]. The region consists of approximately 100 known and predicted genes, and the responsible gene for SCA26 has not been identified.
Spinocerebellar Ataxia Type 27 (SCA27 – OMIM 609307); Fibroblast Growth Factor 14-SCA
Van Swieten et al. described a large Dutch family with early-onset hand tremor, dyskinesia, and slowly progressive cerebellar ataxia. Limb ataxia develops between the ages of 15 and 20 years. Ataxia, dysmetric saccades and smooth pursuit movements, nystagmus, small-amplitude hand tremor, psychiatric symptoms, and decreased cognitive performance are part of SCA27. Brain imaging is normal or shows cerebellar atrophy. The disease is associated with an F145S mutation in the fibroblast growth factor 14 (FGF14), a gene on chromosome 13q34 [van Swieten et al., 2003]. FGF14 knockout mice show ataxia and paroxysmal dyskinesia [Wang et al., 2002]. FGF14 regulates the intrinsic excitability of Purkinje cells [Shakkottai et al., 2009]. Other patients with a truncating mutation or a chromosome translocation have been reported to have similar phenotypes to SCA27.
Spinocerebellar Ataxia Type 28 (SCA28 – OMIM 610246)
SCA28 has been described in 13 patients in two Italian families with a juvenile-onset, slowly progressive ataxia. The mean age at onset for balance and gait abnormalities was 19.6 years (range 12–36). Other features included limb ataxia, dysarthria, and lower limb hyperreflexia [Cagnoli et al., 2006; Mariotti et al., 2008]. The disease locus was found on chromosome 18p11.22–q11.2. Two different missense mutations have been found in the AFG3L2 (ATPase family gene 3-like 2) protein [Mariotti et al., 2008]. AFG3L2 and paraplegin are nuclear-encoded mitochondrial proteins that form a hetero-oligomer. These ubiquitous proteins are m-AAA proteases and their complex ensures protein quality control in the inner mitochondrial membrane, jointly with a chaperone-like activity on the respiratory chain complexes. Mutations in paraplegin are responsible for a recessive form of hereditary spastic paraplegia (SPG7 – OMIM 607259), whereas mutant forms of AFG3L2 cause dominantly inherited SCA28. Mice haploinsufficient for AFG3L2 recapitulate important pathophysiological features of the human disease. Respiratory chain dysfunction and increased production of reactive oxygen species might explain the pathophysiology of SCA28 [Maltecca et al., 2009].
Spinocerebellar Ataxia Type 30 (SCA30)
Storey et al. [2009] have described an Australian family with slowly progressive ataxia in middle to late decades of life, with only minor pyramidal signs. MRI studies revealed cerebellar atrophy with preservation of the nodulus/uvula and brainstem. The patients did not have nystagmus or neuropathy. Linkage analysis identified chromosome 4q34.3–q35.1 as the locus, which is distinct from previously reported loci [Storey et al., 2009].
Spinocerebellar Ataxia Type 31 (SCA31 – OMIM 117210)
Nagaoka et al. [2000] reported 28 affected individuals from six Japanese families with cerebellar dysarthria, limb ataxia, decreased muscle tone, and horizontal gaze nystagmus. The disease starts at 45–72 years of age. The disease locus mapped to chromosome 16q22.1, the same locus as in SCA4. But these patients, unlike SCA4 patients, did not have any sign of pyramidal tract involvement, such as exaggerated tendon reflexes, Babinski signs, or spasticity [Nagaoka et al., 2000]. Recently, Sato et al. [2009] studied 160 affected individuals from 98 SCA31 families and 430 control subjects, including 5 SCA4 patients. They found an insertion, from 2.5–3.8 kb long, consisting of complex pentanucleotide repeats, including a long (TGGAA)n stretch. In controls, shorter (1.5–2.0 kb) insertions lacking (TGGAA)n were found only rarely. One family showed anticipation, and the length of the repeat inversely correlated with age of onset; an expansion was documented in a single family showing anticipation. The repeat insertion was located in introns of TK2 and BEAN (brain expressed, associated with Nedd4) expressed in the brain and formed RNA foci in Purkinje cell nuclei. They also found a single-nucleotide change in an intron of the thymidine kinase 2 gene (TK2). However, this did not appear to affect splicing or expression patterns [Sato et al., 2009].
Episodic Ataxias
Episodic ataxia type 1 (EA1 – OMIM 160120), also known as episodic ataxia with myokymia, is characterized by intermittent episodes of ataxia that can occur spontaneously but are often precipitated by exercise, fever, stress, or even sudden movement. The attacks can last seconds to minutes and may occur several times per day. Myokymia, a rippling movement of muscle, is usually observed in the periorbital and small hand muscles and persists between ataxic attacks. EA1 maps to human chromosome 12p13, and the disease is caused by point mutations in the potassium channel gene, KCNA1 (Kv1.1) [Browne et al., 1994; Comu et al., 1996]. All affected individuals are heterozygous.
Episodic ataxia type 2 (EA2 – OMIM 108500), also known as hereditary paroxysmal cerebellar ataxia, is characterized by intermittent attacks of ataxia and dysarthria lasting from a few minutes to a few days. Nausea, migraine, weakness, vertigo, diplopia, oscillopsia, and/or dystonia may accompany an attack. Attacks are often followed by a period of fatigue. Interictal neurologic findings include nystagmus (either gaze-evoked or downbeat), mild cerebellar ataxia, and occasionally epilepsy. A progressive cerebellar ataxia is seen in some kindreds. The ataxia and number of attacks typically decrease with acetazolamide treatment; 4-aminopyridine may be a useful alternative if acetazolamide fails or loses effectiveness [Strupp et al., 2004].
The gene locus for this disease was mapped to the short arm of chromosome 19 in a position that overlaps the locus for familial hemiplegic migraine (FHM) [Vahedi et al., 1995; von Brederlow et al., 1995]. Ophoff et al. subsequently demonstrated that both EA2 and FHM are caused by mutations in the CACNA1A gene [Ophoff et al., 1996]. Thus, EA2, FHM, and SCA6 are allelic disorders. CACNA1A codes for the brain-specific, voltage-sensitive α1A (Cav2.1) subunit of the P/Q-type calcium channel, which is highly expressed in Purkinje cells [Ishikawa et al., 1999]. Several different missense and nonsense mutations in the gene are associated with EA2 [Jen et al., 2004]. Although there initially appeared to be a clear genotype–phenotype relationship, further studies have demonstrated that the nature of the mutation in the gene is not predictive of whether an individual will clinically present with EA2 or FHM.
EA3 (OMIM 606554), also called periodic vestibulocerebellar ataxia, was described in a large Canadian kindred [Steckley et al., 2001]. Clinical attacks consist of vertigo, incoordination, imbalance, tinnitus, diplopia, and/or visual blurring lasting for 1 minute to 6 hours. Interictal myokymia and/or ataxia are also present. Attacks often occur 1–2 times per day, followed by a refractory period. Generalized seizures occur in some members of the kindred, suggesting that a channelopathy might be responsible. Acetazolamide was useful for reducing the frequency of attacks in one patient. EA1 and EA2 loci were excluded by mapping studies. By a genome-wide screen Cader et al. [2005] found linkage to a 4-cM region on chromosome 1q42 in the family reported by Steckley et al. [2001]. Although Steckley et al. [2001] referred to periodic vestibulocerebellar ataxia (PATX) as episodic ataxia-3 (EA3) and the disorder in OMIM 606554 as EA4, the same group [Cader et al., 2005] later referred to the disorder described by Steckley et al. [2001] in OMIM 606554 as EA3. Thus episodic ataxia 4 (EA4) is designated for PATX [Steckley et al., 2001; Cader et al., 2005].
EA4, or periodic vestibulocerebellar ataxia (PATX – OMIM 606552), has been described in two white families from North Carolina [Farmer and Mustian, 1963; Vance et al., 1984]. Clinically, the disorder consists of episodic attacks of vertigo, nausea, tinnitus, horizontal nystagmus, oscillopsia, and ataxia that begin in the third decade and may evolve into a constant condition. The visual sensation of objects moving past the patient, such as occurs while riding in a car, can exacerbate attacks, while lying quietly for 15–30 minutes can alleviate an attack. Acetazolamide is not therapeutic, although antihistamines have been anecdotally reported to decrease the frequency and severity of attacks in at least some individuals [Farmer and Mustian, 1963]. Neuroimaging and pathology studies have not been done. Several SCA loci, as well as EA1 and EA2, have been eliminated as causative [Damji et al., 1996]. The causative gene locus is not known.
EA5 (OMIM 601949) was identified when a series of families with episodic ataxia were screened for mutations in the calcium channel b4 subunit CACNB4 on chromosome 2q [Escayg et al., 2000]. This family had clinical features similar to EA2 but no mutation was found in CACNA1A. The episodes last for hours. Complicating matters, the same mutation was found in a German family with generalized epilepsy (but no ataxia) and functional studies showed only subtle changes in calcium channel function (reviewed in Jen et al. [2007]).
EA6 (OMIM 612656) was identified in a single child with episodic and progressive ataxia, episodes of seizures, and hemiplegia that lasted from hours to days. Through a candidate gene approach, a de novo mutation was identified from a screen of the candidate gene SLC1A3 on chromosome 5p13, a glutamate transporter localized to astrocytes [Jen et al., 2005]. De Vries et al. [2009] reported a Dutch family in which three individuals had a milder version of this type of episodic ataxia. The mutation altered a strictly conserved amino acid residue, and functional studies of the mutated protein showed an almost complete loss of function with a dominant negative effect on the wild-type allele.
In the case of EA7 (OMIM 611907), Kerber et al. [2007] described seven patients with episodic ataxia from a four-generation family. Symptoms started before 20 years of age; attacks lasted hours to days and were associated with weakness and dysarthria. Exercise and excitement were triggers. Two affected family members reported vertigo during attacks. The frequency ranged from monthly to yearly and tended to decrease with age. By genome-wide linkage and haplotype analysis, chromosome 19q13 has been identified as the EA7 gene locus [Kerber et al., 2007].
Dentatorubral-Pallidoluysian Atrophy (DRPLA – OMIM 125370)
DRPLA is a rare autosomal-dominant, neurodegenerative disorder characterized by progressive ataxia, myoclonus, epilepsy, chorea, athetosis, and dementia [Smith et al., 1958]. The hereditary nature of the disease was confirmed through detailed clinical, genetic, and neuropathologic studies of five Japanese families with familial myoclonic epilepsy and choreoathetosis [Naito and Oyanagi, 1982]. Clinically, age of onset is linked to disease phenotype. Early onset cases (<20 years of age) tend to present with a progressive myoclonic epilepsy syndrome consisting of epilepsy, myoclonus, cerebellar ataxia, and mental retardation, while adult-onset cases (>20 years of age) tend to present with cerebellar ataxia, choreoatheotosis, and/or dementia. The phenotypic diversity within and among families is striking, as is the overlap with the clinical presentation of Huntington’s chorea.
Imaging studies demonstrate cerebellar, tegmental, and cerebral atrophy, as well as signal changes in the white matter. In late-onset cases, signal change can also be found in the pons, midbrain, thalamus, and globus pallidus (reviewed in Kanazawa [1998]). Pathologic findings in DRPLA include neuronal loss in the dentate nucleus, red nucleus, globus pallidus, and subthalamic nucleus. There is also diffuse loss of myelin staining in the cerebral white matter [Kanazawa, 1998; Munoz et al., 2004]. The disorder is very rare, except in Japan, where it accounts for a significant proportion of autosomal-dominant spinocerebellar ataxias [Watanabe et al., 1998].
By using a candidate gene approach, the mutation causing DRPLA was identified by two research groups [Koide et al., 1994; Nagafuchi et al., 1994]. They both found linkage to chromosome 12 and identified a CAG repeat as responsible for the disorder. An inverse correlation exists between the number of repeat units and the age of onset. Anticipation has been demonstrated in both paternal and maternal transmission of the disease allele, and extreme expansions (>90 repeats) can lead to infantile disease [Shimojo et al., 2001]. Gene dosage also plays a role in severity of phenotype, with homozygosity for a pathogenic allele causing more severe clinical manifestations than would otherwise be predicted based on repeat length [Sato et al., 1995].
The clinical heterogeneity of DRPLA belies its genetic cause. Haw River syndrome shares many clinical and neuropathologic features with DRPLA, Huntington’s disease, and the SCAs. However, this clinical entity differs from DRPLA in the absence of myoclonic epilepsy, as well as the presence of basal ganglia calcifications and neuroaxonal dystrophy. Despite these phenotypic differences, the DRPLA expansion was detected in an African American family with Haw River syndrome [Burke et al., 1994]. Similarly, Warner et al. [1994] reported the CAG repeat expansion in a family of Maltese origin with variable clinical features that included dementia and seizures in younger patients, and dementia, psychosis, ataxia, and chorea in older patients. The neuropathologic findings in this family were not consistent with DRPLA. These examples illustrate the wide phenotypic diversity encountered in DRPLA. Genetic testing for the disorder is therefore indicated in patients with an autosomal-dominant disorder causing dementia, ataxia, seizures, psychiatric symptoms, dystonia, chorea, or myoclonus. Also, patients with features of Huntington’s disease but no evidence of expansion of the Huntington’s disease CAG repeat should be evaluated for the CAG repeat at the DRPLA locus.
The pathogenesis of DRPLA remains a mystery. The number of CAG repeats varies in different tissues and tends to be larger in the brain, suggesting somatic instability of the repeat [Ueno et al., 1995]. However, the degree of expansion does not seem to parallel neuropathologic involvement. Intranuclear inclusions are found in both neurons and glia; their presence in oligodendrocytes may cause the white-matter lesions that are sometimes seen [Hayashi et al., 1998]. Atrophin-1, the DRPLA gene product, is expressed ubiquitously in human tissue. Several proteins proposed to interact with atrophin-1 have been identified using a yeast two-hybrid screen [Wood et al., 1998]. Recent evidence suggests that the wild-type atrophin-1 protein is involved in regulation of transcription, both positively and negatively [Shimohata et al., 2000; Zhang et al., 2002]. Therapeutically, hypoactivation of histone H3 in brain tissue of Atro-118Q mice, a transgenic DRPLA mouse model, as well as administration of sodium butyrate, a histone deacetylase inhibitor, ameliorated histone acetylation defects, significantly improved motor performance, and extended the average life span of Atro-118Q mice [Ying et al., 2006].
Hereditary Spastic Ataxia (HSA)
Autosomal-dominant hereditary spastic ataxia (SAX1 – OMIM 108600)
This disorder was described in three kindreds from Newfoundland. These patients typically present first with progressive leg spasticity, followed by dysarthria and ocular movement abnormalities [Meijer et al., 2002]. Fine mapping of the SAX1 locus on chromosome 12p13 reduced the critical interval to 1.9 Mb [Grewal et al., 2004]. The DRPLA locus has not been excluded, although genetic anticipation is not observed.
Autosomal-recessive spastic ataxia of charlevoix-saguenay (ARSACS – OMIM 270550)
Originally described in French Canadians from the Charlevoix-Saguenay region of Quebec, this autosomal-recessive disorder has also been found in a large Tunisian family [Bouchard et al., 1978; Mrissa et al., 2000]. Early onset is a rule, with the condition first being noticed when a child begins to walk; however, age of onset can be as late as 20 years. In addition to spasticity and ataxia, affected individuals also have sensory disturbances in the lower limbs (electromyography shows absent sensory nerve conduction and reduced motor nerve velocity), as well as hypermyelination of retinal nerve fibers. Physical exam often reveals absent or reduced tendon reflexes in the lower limbs with present Babinski; claw-hand deformity and pes cavus can develop. Eye movements are not affected. Nonverbal IQ scores are often at the low end of normal.
The SACS gene was mapped to chromosome 13q12. Frameshift and nonsense mutations lead to truncations in the gene product, sacsin, which is expressed throughout the CNS and may be involved in chaperone-mediated protein folding [Engert et al., 2000].
Spastic Paraplegia, Ataxia, and Mental Retardation (SPAR – OMIM 607565)
Hedera et al. [2002] identified a single kindred whose older members presented with uncomplicated spastic paraplegia, while later generations had either spastic paraplegia and ataxia, or spastic paraplegia, ataxia, and mental retardation [Hedera et al., 2002]. Multiple SCAs and hereditary spastic paraplegias were excluded by gene tests and linkage analysis, demonstrating that SPAR is a distinct disorder. Neuroimaging demonstrates dorsal column atrophy in all three types, while only the latter two had cerebellar atrophy. Pathologic studies are not available. Genetic anticipation, both in age of onset and disease severity, was suggested. The causative gene is unknown.
X-Linked Spinocerebellar Ataxia
A group of hereditary ataxias can be classified as X-linked based on the pattern of inheritance in specific kindreds. Shokeir [1970] described three families with an X-linked recessive pattern of inheritance and onset of ataxia in the late teens or early 20s. No extrapyramidal or posterior column deficits were noted. Another syndrome of X-linked cerebellar ataxia, with hypotonia, optic atrophy, and sensorineural deafness, has been described [Schmidley et al., 1987]. The disease occurs in infancy, is progressive, and causes death in childhood. Remarkable degeneration occurs in the red nucleus, dorsal motor nucleus of the vagus nerve, and auditory pathway, with some neuronal loss and gliosis in the dentate nucleus and inferior olive. Apak et al. [1989] described a progressive ataxia combined with paraplegia in a family in which seven males were affected; death typically occurred in the third or fourth decade. A pure X-linked cerebellar ataxia with slow progression has also been seen [Lutz et al., 1989], as has an X-linked recessive disease characterized by ataxia, early-onset floppiness, liability to infections (especially of the upper respiratory tract), deafness, and eventually flaccid quadriplegia and areflexia [Arts et al., 1993]. This latter disease is rapidly progressive and results in death in early childhood. No precise genetic localization of the X-linked ataxias has been established.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Aicardi J., Barbosa C., et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol. 1988;24(4):497-502.
Al-Mahdawi S., Pinto R.M., et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88(5):580-590.
Allen G., Buxton R.B., et al. Attentional activation of the cerebellum independent of motor involvement. Science. 1997;275(5308):1940-1943.
Alonso I., Barros J., et al. Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family. Arch Neurol. 2003;60(4):610-614.
Apak S., Yuksel M., et al. Heterogeneity of X-linked recessive (spino)cerebellar ataxia with or without spastic diplegia. Am J Med Genet. 1989;34(2):155-158.
Arts W.F., Loonen M.C., et al. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann Neurol. 1993;33(5):535-539.
Auger R.G. Preservation of the masseter reflex in Friedreich’s ataxia. Neurology. 1992;42(4):875-878.
Baloh R.W., Yee R.D., et al. Eye movements in ataxia-telangiectasia. Neurology. 1978;28(11):1099-1104.
Banfi S., Servadio A., et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet. 1994;7(4):513-520.
Barbeau A., Roy M., et al. Recessive ataxia in Acadians and “Cajuns”. Can J Neurol Sci. 1984;11(4 Suppl):526-533.
Barlow C., Ribaut-Barassin C., et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA. 2000;97(2):871-876.
Benomar A., Krols L., et al. The gene for autosomal dominant cerebellar ataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nat Genet. 1995;10(1):84-88.
Benton C.S., de Silva R., et al. Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology. 1998;51(4):1081-1086.
Bidichandani S.I., Ashizawa T., et al. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62(1):111-121.
Biemond A. [Acute ataxia in children]. Ned Tijdschr Geneeskd. 1957;101(12):529-532.
Boder E., Sedgwick R.P. Ataxia-telangiectasia. A familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. A preliminary report on 7 children, an autopsy, and a case history. USC Med Bull. 1957;9:15-28.
Bomar J.M., Benke P.J., et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35(3):264-269.
Bomont P., Watanabe M., et al. Homozygosity mapping of spinocerebellar ataxia with cerebellar atrophy and peripheral neuropathy to 9q33-34, and with hearing impairment and optic atrophy to 6p21-23. Eur J Hum Genet. 2000;8(12):986-990.
Bouchard J.P., Barbeau A., et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. 1978;5(1):61-69.
Braitenberg V. Is the cerebellar cortex a biological clock in the millisecond range? Prog Brain Res. 1967;25:334-346.
Brazis P., Masdeu J., et al. Localization in Clinical Neurology. Philadelphia: Lippincott Williams & Wilkins; 2001.
Breedveld G.J., van Wetten B., et al. A new locus for a childhood onset, slowly progressive autosomal recessive spinocerebellar ataxia maps to chromosome 11p15. J Med Genet. 2004;41(11):858-866.
Brkanac Z., Bylenok L., et al. A new dominant spinocerebellar ataxia linked to chromosome 19q13.4-qter. Arch Neurol. 2002;59(8):1291-1295.
Brkanac Z., Fernandez M., et al. Autosomal dominant sensory/motor neuropathy with Ataxia (SMNA): Linkage to chromosome 7q22-q32. Am J Med Genet. 2002;114(4):450-457.
Brkanac Z., Spencer D., et al. IFRD1 is a candidate gene for SMNA on chromosome 7q22-q23. Am J Hum Genet. 2009;84(5):692-697.
Brown J.R. Degenerative cerebellar ataxias. Neurology. 1959;9:799-805.
Brown J.R. Disorders of the cerebellum. In: Baker A.B., Baker L.H., editors. Clinical neurology. New York: Harper & Row, 1962.
Brown L., Mueller M., et al. A non-progressive cerebellar ataxia on Grand Cayman Island. Neurology. 1984;34:273.
Browne D.L., Gancher S.T., et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8(2):136-140.
Burke J.R., Wingfield M.S., et al. The Haw River syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African-American family. Nat Genet. 1994;7(4):521-524.
Burright E.N., Clark H.B., et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82(6):937-948.
Bushara K.O., Wheat J.M., et al. Multiple tactile maps in the human cerebellum. Neuroreport. 2001;12(11):2483-2486.
Buzin C.H., Gatti R.A., et al. Comprehensive scanning of the ATM gene with DOVAM-S. Hum Mutat. 2003;21(2):123-131.
Cader M.Z., Steckley J.L., et al. A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia. Neurology. 2005;65(1):156-158.
Cagnoli C., Mariotti C., et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain. 2006;129(Pt 1):235-242.
Campuzano V., Montermini L., et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423-1427.
Cassandro E., Mosca F., et al. Otoneurological findings in Friedreich’s ataxia and other inherited neuropathies. Audiology. 1986;25(2):84-91.
Cavalier L., Ouahchi K., et al. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet. 1998;62(2):301-310.
Cellini E., Piacentini S., et al. A family with spinocerebellar ataxia type 8 expansion and vitamin E deficiency ataxia. Arch Neurol. 2002;59(12):1952-1953.
Centerwall W.R., Miller M.M. Ataxia, telangiectasia, and sinopulmonary infections; a syndrome of slowly progressive deterioration in childhood. AMA J Dis Child. 1958;95(4):385-396.
Chai Y., Koppenhafer S.L., et al. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet. 1999;8(4):673-682.
Chamberlain S., Shaw J., et al. Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature. 1988;334(6179):248-250.
Chen D.H., Brkanac Z., et al. Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet. 2003;72(4):839-849.
Chen Y.W., Allen M.D., et al. The structure of the AXH domain of spinocerebellar ataxin-1. J Biol Chem. 2004;279(5):3758-3765.
Chen Y.Z., Bennett C.L., et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74(6):1128-1135.
Child J.S., Perloff J.K., et al. Cardiac involvement in Friedreich’s ataxia: a clinical study of 75 patients. J Am Coll Cardiol. 1986;7(6):1370-1378.
Chintawar S., Hourez R., et al. Grafting neural precursor cells promotes functional recovery in an SCA1 mouse model. J Neurosci. 2009;29(42):13126-13135.
Christodoulou K., Deymeer F., et al. Mapping of the second Friedreich’s ataxia (FRDA2) locus to chromosome 9p23-p11: evidence for further locus heterogeneity. Neurogenetics. 2001;3(3):127-132.
Chung M.Y., Lu Y.C., et al. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain. 2003;126(Pt 6):1293-1299.
Chung M.Y., Ranum L.P., et al. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet. 1993;5(3):254-258.
Claus D., Harding A.E., et al. Central motor conduction in degenerative ataxic disorders: a magnetic stimulation study. J Neurol Neurosurg Psychiatry. 1988;51(6):790-795.
Comu S., Giuliani M., et al. Episodic ataxia and myokymia syndrome: a new mutation of potassium channel gene Kv1.1. Ann Neurol. 1996;40(4):684-687.
Cooper J.M., Korlipara L.V., et al. Coenzyme Q10 and vitamin E deficiency in Friedreich’s ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol. 2008;15(12):1371-1379.
Cossee M., Puccio H., et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9(8):1219-1226.
Cossee M., Schmitt M., et al. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA. 1997;94(14):7452-7457.
Coutinho P., Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology. 1978;28(7):703-709.
Cummings C.J., Mancini M.A., et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19(2):148-154.
Cummings C.J., Reinstein E., et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24(4):879-892.
Cummings C.J., Sun Y., et al. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10(14):1511-1518.
Currier R.D., Glover G., et al. Spinocerebellar ataxia: study of a large kindred. I. General information and genetics. Neurology. 1972;22(10):1040-1043.
Dagda R.K., Merrill R.A., et al. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bbeta2 antagonizes neuronal survival by promoting mitochondrial fission. J Biol Chem. 2008;283(52):36241-36248.
Damji K.F., Allingham R.R., et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol. 1996;53(4):338-344.
Date H., Onodera O., et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184-188.
Daughters R.S., Tuttle D.L., et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5(8):e1000600.
David G., Abbas N., et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17(1):65-70.
De Biase I., Rasmussen A., et al. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61(1):55-60.
De Leon G.A., Grover W.D., et al. Neuropathologic changes in ataxia-telangiectasia. Neurology. 1976;26(10):947-951.
de Vries B., Mamsa H., et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66(1):97-101.
Delague V., Bareil C., et al. Nonprogressive autosomal recessive ataxia maps to chromosome 9q34-9qter in a large consanguineous Lebanese family. Ann Neurol. 2001;50(2):250-253.
Delague V., Bareil C., et al. A new autosomal recessive non-progressive congenital cerebellar ataxia associated with mental retardation, optic atrophy, and skin abnormalities (CAMOS) maps to chromosome 15q24-q26 in a large consanguineous Lebanese Druze Family. Neurogenetics. 2002;4(1):23-27.
Devos D., Schraen-Maschke S., et al. Clinical features and genetic analysis of a new form of spinocerebellar ataxia. Neurology. 2001;56(2):234-238.
Di Prospero N.A., Baker A., et al. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol. 2007;6(10):878-886.
Dick K.A., Margolis J.M., et al. Dominant non-coding repeat expansions in human disease. Genome Dyn. 2006;1:67-83.
Doss-Pepe E.W., Stenroos E.S., et al. Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol Cell Biol. 2003;23(18):6469-6483.
Dupre N., Gros-Louis F., et al. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol. 2007;62(1):93-98.
Durr A., Cossee M., et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335(16):1169-1175.
Edener U., Kurth I., et al. Missense exchanges in the TTBK2 gene mutated in SCA11. J Neurol. 2009;256(11):1856-1859.
Emamian E.S., Kaytor M.D., et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38(3):375-387.
Enevoldson T.P., Sanders M.D., et al. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy. A clinical and genetic study of eight families. Brain. 1994;117(Pt 3):445-460.
Engert J.C., Berube P., et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000;24(2):120-125.
Escayg A., De Waard M., et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66(5):1531-1539.
Farlow M.R., DeMyer W., et al. X-linked recessive inheritance of ataxia and adult-onset dementia: clinical features and preliminary linkage analysis. Neurology. 1987;37(4):602-607.
Farmer T.W., Mustian V.M. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol. 1963;8:471-480.
Fernet M., Gribaa M., et al. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet. 2005;14(2):307-318.
Filla A., De Michele G., et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet. 1996;59(3):554-560.
Flanigan K., Gardner K., et al. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet. 1996;59(2):392-399.
Frieden I.J., Reese V., et al. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132(3):307-311.
Friedreich N. Über degenerative Atrophie der spinalen Hinterstrange. Virchows Arch Pathol Anat. 1863;26:433.
Garden G.A., La Spada A.R. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7(2):138-149.
Gardner K., Alderson K., et al. Autosomal dominant spinocerebellar ataxia: clinical description of a distinct hereditary ataxia and genetic localization to chromosome 16 (SCA4) in a Utah kindred. Neurology. A361, 1994.
Gatti R.A., Berkel I., et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. 1988;336(6199):577-580.
Gatti R.A., Boder E., et al. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore). 1991;70(2):99-117.
Gatti R.A., Good R.A. Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer. 1971;28(1):89-98.
Geoffroy G., Barbeau A., et al. Clinical description and roentgenologic evaluation of patients with Friedreich’s ataxia. Can J Neurol Sci. 1976;3(4):279-286.
Gilad S., Chessa L., et al. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet. 1998;62(3):551-561.
Gilman S., Junck L., et al. Cerebral glucose hypermetabolism in Friedreich’s ataxia detected with positron emission tomography. Ann Neurol. 1990;28(6):750-757.
Gispert S., Twells R., et al. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1. Nat Genet. 1993;4(3):295-299.
Gomez C.M., Thompson R.M., et al. Spinocerebellar ataxia type 6: gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann Neurol. 1997;42(6):933-950.
Gotoff S.P., Amirmokri E., et al. Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am J Dis Child. 1967;114(6):617-625.
Gouw L.G., Kaplan C.D., et al. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat Genet. 1995;10(1):89-93.
Greenfield J. The spino-cerebellar degenerations. Springfield, IL: Charles C. Thomas; 1954.
Grewal K.K., Stefanelli M.G., et al. A founder effect in three large Newfoundland families with a novel clinically variable spastic ataxia and supranuclear gaze palsy. Am J Med Genet A. 2004;131(3):249-254.
Grewal R.P., Tayag E., et al. Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar ataxia. Neurology. 1998;51(5):1423-1426.
Gros-Louis F., Dupre N., et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39(1):80-85.
Hakonen A.H., Goffart S., et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet. 2008;17(23):3822-3835.
Hara K., Shiga A., et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology. 2008;71(8):547-551.
Harding A. The Hereditary Ataxias and Related Disorders. Edinburgh: Churchill Livingstone; 1984.
Harding A.E. Clinical features and classification of inherited ataxias. Adv Neurol. 1993;61:1-14.
Harding A.E., Hewer R.L. The heart disease of Friedreich’s ataxia: a clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q J Med. 1983;52(208):489-502.
Hart R.P., Kwentus J.A., et al. Information processing speed in Friedreich’s ataxia. Ann Neurol. 1985;17(6):612-614.
Hayashi Y., Kakita A., et al. Hereditary dentatorubral-pallidoluysian atrophy: detection of widespread ubiquitinated neuronal and glial intranuclear inclusions in the brain. Acta Neuropathol. 1998;96(6):547-552.
Hecht F., Hecht B.K. Ataxia-telangiectasia breakpoints in chromosome rearrangements reflect genes important to T and B lymphocytes. Kroc Found Ser. 1985;19:189-195.
Hedera P., Rainier S., et al. Spastic paraplegia, ataxia, mental retardation (SPAR): a novel genetic disorder. Neurology. 2002;58(3):411-416.
Helmlinger D., Hardy S., et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13(12):1257-1265.
Herman-Bert A., Stevanin G., et al. Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am J Hum Genet. 2000;67(1):229-235.
Herman D., Jenssen K., et al. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2(10):551-558.
Herzog K.H., Chong M.J., et al. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280(5366):1089-1091.
Holmberg M., Duyckaerts C., et al. Spinocerebellar ataxia type 7 (SCA7): a neurodegenerative disorder with neuronal intranuclear inclusions. Hum Mol Genet. 1998;7(5):913-918.
Holmes S.E., O’Hearn E., et al. Why is SCA12 different from other SCAs? Cytogenet Genome Res. 2003;100(1–4):189-197.
Holmes S.E., O’Hearn E.E., et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet. 1999;23(4):391-392.
Houlden H., Johnson J., et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39(12):1434-1436.
Hunt J. Dyssynergia cerebellaris myoclonica – primary atrophy of the dentate system: contribution to the pathology and symptomatology of the cerebellum. Brain. 1922;44:490.
Huynh D.P., Del Bigio M.R., et al. Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer’s disease and spinocerebellar ataxia 2. Ann Neurol. 1999;45(2):232-241.
Ikeda H., Yamaguchi M., et al. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet. 1996;13(2):196-202.
Ikeda Y., Dalton J.C., et al. Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am J Hum Genet. 2004;75(1):3-16.
Ikeda Y., Dick K.A., et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38(2):184-190.
Imbert G., Saudou F., et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14(3):285-291.
Ishikawa K., Fujigasaki H., et al. Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum Mol Genet. 1999;8(7):1185-1193.
Ito M. Historical review of the significance of the cerebellum and the role of Purkinje cells in motor learning. Ann N Y Acad Sci. 2002;978:273-288.
Iwaki A., Kawano Y., et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45(1):32-35.
Izumi Y., Maruyama H., et al. SCA8 repeat expansion: large CTA/CTG repeat alleles are more common in ataxic patients, including those with SCA6. Am J Hum Genet. 2003;72(3):704-709.
James T.N., Cobbs B.W., et al. Coronary disease, cardioneuropathy, and conduction system abnormalities in the cardiomyopathy of Friedreich’s ataxia. Br Heart J. 1987;57(5):446-457.
Jen J., Kim G.W., et al. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62(1):17-22.
Jen J.C., Graves T.D., et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130(Pt 10):2484-2493.
Jen J.C., Wan J., et al. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65(4):529-534.
Jodice C., Mantuano E., et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6(11):1973-1978.
Johansson J., Forsgren L., et al. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998;7(2):171-176.
Johnson W., Murphy M., et al. Recessive congenital cerebellar disorder in a genetic isolate: CPD type VII? Neurology. 1978;28:352.
Kanazawa I. Dentatorubral-pallidoluysian atrophy or Naito-Oyanagi disease. Neurogenetics. 1998;2(1):1-17.
Keats B.J., Ward L.J., et al. “Acadian” and “classical” forms of Friedreich ataxia are most probably caused by mutations at the same locus. Am J Med Genet. 1989;33(2):266-268.
Kerber K.A., Jen J.C., et al. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. 2007;64(5):749-752.
Khan R.J., Andermann E., et al. Glucose intolerance in Friedreich’s ataxia: association with insulin resistance and decreased insulin binding. Metabolism. 1986;35(11):1017-1023.
Klein C., Wenning G.K., et al. Ataxia without telangiectasia masquerading as benign hereditary chorea. Mov Disord. 1996;11(2):217-220.
Knezevic W., Stewart-Wynne E.G. Brainstem auditory evoked responses in hereditary spinocerebellar ataxias. Clin Exp Neurol. 1985;21:149-155.
Knight M.A., Gardner R.J., et al. Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain. 2004;127(Pt 5):1172-1181.
Koeppen A.H. The hereditary ataxias. J Neuropathol Exp Neurol. 1998;57(6):531-543.
Koeppen A.H., Barron K.D. The neuropathology of olivopontocerebellar atrophy. Adv Neurol. 1984;41:13-38.
Koide R., Ikeuchi T., et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet. 1994;6(1):9-13.
Koob M.D., Moseley M.L., et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet. 1999;21(4):379-384.
Koskinen T., Sainio K., et al. Sensory neuropathy in infantile onset spinocerebellar ataxia (IOSCA). Muscle Nerve. 1994;17(5):509-515.
Kostrzewa M., Klockgether T., et al. Locus heterogeneity in Friedreich ataxia. Neurogenetics. 1997;1(1):43-47.
Kvistad P.H., Dahl A., et al. Autosomal recessive non-progressive ataxia with an early childhood debut. Acta Neurol Scand. 1985;71(4):295-302.
La Spada A.R., Fu Y.H., et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31(6):913-927.
Labelle H., Tohme S., et al. Natural history of scoliosis in Friedreich’s ataxia. J Bone Joint Surg Am. 1986;68(4):564-572.
Lafreniere R.G., Rochefort D.L., et al. Unstable insertion in the 5′ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat Genet. 1997;15(3):298-302.
Lagier-Tourenne C., Tazir M., et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82(3):661-672.
Lam Y.C., Bowman A.B., et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127(7):1335-1347.
Lamperti C., Naini A., et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60(7):1206-1208.
Lavin M.F., Gueven N., et al. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br Med Bull. 2007;81–82:129-147.
Le Ber I., Bouslam N., et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127(Pt 4):759-767.
Le Ber I., Dubourg O., et al. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology. 2007;68(4):295-297.
Leiner H.C., Leiner A.L., et al. The human cerebro-cerebellar system: its computing, cognitive, and language skills. Behav Brain Res. 1991;44(2):113-128.
Lim D.S., Kirsch D.G., et al. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci USA. 1998;95(17):10146-10151.
Lim J., Crespo-Barreto J., et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452(7188):713-718.
Liu J., Tang T.S., et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29(29):9148-9162.
Lopes-Cendes I., Andermann E., et al. Confirmation of the SCA-2 locus as an alternative locus for dominantly inherited spinocerebellar ataxias and refinement of the candidate region. Am J Hum Genet. 1994;54(5):774-781.
Louis-Bar D. Sur un syndrome professif comprenant des telangiectasis capillaries cutanees et conjunctivales symetriques a disposition naevoide et des troubles cerebelleux. Confin Neurol. 1941;4:32.
Lutz R., Bodensteiner J., et al. X-linked olivopontocerebellar atrophy. Clin Genet. 1989;35(6):417-422.
Maltecca F., Filla A., et al. Intergenerational instability and marked anticipation in SCA-17. Neurology. 2003;61(10):1441-1443.
Maltecca F., Magnoni R., et al. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29(29):9244-9254.
Manni E., Petrosini L. A century of cerebellar somatotopy: a debated representation. Nat Rev Neurosci. 2004;5(3):241-249.
Mantuano E., Veneziano L., et al. Spinocerebellar ataxia type 6 and episodic ataxia type 2: differences and similarities between two allelic disorders. Cytogenet Genome Res. 2003;100(1–4):147-153.
Mariotti C., Brusco A., et al. Spinocerebellar ataxia type 28: a novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum. 2008;7(2):184-188.
Marz P., Probst A., et al. Ataxin-10, the spinocerebellar ataxia type 10 neurodegenerative disorder protein, is essential for survival of cerebellar neurons. J Biol Chem. 2004;279(34):35542-35550.
Matilla A., Roberson E.D., et al. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998;18(14):5508-5516.
Matsuura T., Achari M., et al. Mapping of the gene for a novel spinocerebellar ataxia with pure cerebellar signs and epilepsy. Ann Neurol. 1999;45(3):407-411.
Matsuura T., Ranum L.P., et al. Spinocerebellar ataxia type 10 is rare in populations other than Mexicans. Neurology. 2002;58(6):983-984.
Matsuura T., Yamagata T., et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26(2):191-194.
McFarlin D.E., Oppenheim J.J. Impaired lymphocyte transformation in ataxia-telangiectasia in part due to a plasma inhibitory factor. J Immunol. 1969;103(6):1212-1222.
McFarlin D.E., Strober W., et al. Ataxia-telangiectasia. Medicine (Baltimore). 1972;51(4):281-314.
Megarbane A., Delague V., et al. New autosomal recessive cerebellar ataxia disorder in a large inbred Lebanese family. Am J Med Genet. 2001;101(2):135-141.
Meijer I.A., Hand C.K., et al. A locus for autosomal dominant hereditary spastic ataxia, SAX1, maps to chromosome 12p13. Am J Hum Genet. 2002;70(3):763-769.
Michalik A., Martin J.J., et al. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet. 2004;12(1):2-15.
Middleton F.A., Strick P.L. Anatomical evidence for cerebellar and basal ganglia involvement in higher cognitive function. Science. 1994;266(5184):458-461.
Miller M.E., Chatten J. Ovarian changes in ataxia telangiectasia. Acta Paediatr Scand. 1967;56(5):559-561.
Moreira M.C., Barbot C., et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189-193.
Moreira M.C., Klur S., et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36(3):225-227.
Mrissa N., Belal S., et al. Linkage to chromosome 13q11-12 of an autosomal recessive cerebellar ataxia in a Tunisian family. Neurology. 2000;54(7):1408-1414.
Munoz E., Campdelacreu J., et al. Severe cerebral white matter involvement in a case of dentatorubropallidoluysian atrophy studied at autopsy. Arch Neurol. 2004;61(6):946-949.
Mutsuddi M., Marshall C.M., et al. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol. 2004;14(4):302-308.
Nagafuchi S., Yanagisawa H., et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet. 1994;6(1):14-18.
Nagaoka U., Takashima M., et al. A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia. Neurology. 2000;54(10):1971-1975.
Naito H., Oyanagi S. Familial myoclonus epilepsy and choreoathetosis: hereditary dentatorubral-pallidoluysian atrophy. Neurology. 1982;32(8):798-807.
Nakamura H., Yasui Y., et al. DNA repair defect in AT cells and their hypersensitivity to low-dose-rate radiation. Radiat Res. 2006;165(3):277-282.
Nakamura K., Jeong S.Y., et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10(14):1441-1448.
Nemeth A.H., Bochukova E., et al. Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxia-telangiectasia-like syndrome) is linked to chromosome 9q34. Am J Hum Genet. 2000;67(5):1320-1326.
Nikali K., Suomalainen A., et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. 2005;14(20):2981-2990.
Nino H.E., Noreen H.J., et al. A family with hereditary ataxia: HLA typing. Neurology. 1980;30(1):12-20.
Nonhoff U., Ralser M., et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18(4):1385-1396.
Norman R.M.. Primary degeneration of the granular layer of the cerebellum: an unusual form of familial cerebellar atrophy occurring in early life. Brain. 1940(63):365-379.
Nystuen A., Benke P.J., et al. A cerebellar ataxia locus identified by DNA pooling to search for linkage disequilibrium in an isolated population from the Cayman Islands. Hum Mol Genet. 1996;5(4):525-531.
O’Hearn E., Holmes S.E., et al. SCA-12: Tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology. 2001;56(3):299-303.
Opal P., Zoghbi H.Y. The role of chaperones in polyglutamine disease. Trends Mol Med. 2002;8(5):232-236.
Ophoff R.A., Terwindt G.M., et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543-552.
Orozco G., Estrada R., et al. Dominantly inherited olivopontocerebellar atrophy from eastern Cuba. Clinical, neuropathological, and biochemical findings. J Neurol Sci. 1989;93(1):37-50.
Orr H.T., Chung M.Y., et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4(3):221-226.
Ouahchi K., Arita M., et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet. 1995;9(2):141-145.
Palhan V.B., Chen S., et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA. 2005;102(24):8472-8477.
Pentland B., Fox K.A. The heart in Friedreich’s ataxia. J Neurol Neurosurg Psychiatry. 1983;46(12):1138-1142.
Puccio H. Multicellular models of Friedreich ataxia. J Neurol. 2009;256(Suppl 1):18-24.
Pulst S.M., Nechiporuk A., et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14(3):269-276.
Qian J., Atkinson J., et al. Biochemical consequences of heritable mutations in the alpha-tocopherol transfer protein. Biochemistry. 2006;45(27):8236-8242.
Quinzii C.M., Kattah A.G., et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64(3):539-541.
Rai M., Soragni E., et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3(4):e1958.
Ranum L.P., Chung M.Y., et al. Molecular and clinical correlations in spinocerebellar ataxia type I: evidence for familial effects on the age at onset. Am J Hum Genet. 1994;55(2):244-252.
Ranum L.P., Schut L.J., et al. Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat Genet. 1994;8(3):280-284.
Riess O., Rub U., et al. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7(2):125-137.
Rolfs A., Koeppen A.H., et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol. 2003;54(3):367-375.
Rosenberg R.N., Nyhan W.L., et al. Autosomal dominant striatonigral degeneration. A clinical, pathologic, and biochemical study of a new genetic disorder. Neurology. 1976;26(8):703-714.
Sahba S., Nechiporuk A., et al. Genomic structure of the human gene for spinocerebellar ataxia type 2 (SCA2) on chromosome 12q24.1. Genomics. 1998;47(3):359-364.
Said G., Marion M.H., et al. Hypotrophic and dying-back nerve fibers in Friedreich’s ataxia. Neurology. 1986;36(10):1292-1299.
Salih M.A., Ahlsten G., et al. Friedreich’s ataxia in 13 children: presentation and evolution with neurophysiologic, electrocardiographic, and echocardiographic features. J Child Neurol. 1990;5(4):321-326.
Sanders S.L., Jennings J., et al. Proteomics of the eukaryotic transcription machinery: identification of proteins associated with components of yeast TFIID by multidimensional mass spectrometry. Mol Cell Biol. 2002;22(13):4723-4738.
Sano Y., Date H., et al. Aprataxin, the causative protein for EAOH is a nuclear protein with a potential role as a DNA repair protein. Ann Neurol. 2004;55(2):241-249.
Sanpei K., Takano H., et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14(3):277-284.
Sato K., Kashihara K., et al. Does homozygosity advance the onset of dentatorubral-pallidoluysian atrophy? Neurology. 1995;45(10):1934-1936.
Sato N., Amino T., et al. Spinocerebellar Ataxia Type 31 Is Associated with “Inserted” Penta-Nucleotide Repeats Containing (TGGAA)(n). Am J Hum Genet. 2009;85(5):544-557.
Saviozzi S., Saluto A., et al. A late onset variant of ataxia-telangiectasia with a compound heterozygous genotype, A8030G/7481insa. J Med Genet. 2002;39(1):57-61.
Savitsky K., Bar-Shira A., et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749-1753.
Savitsky K., Sfez S., et al. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet. 1995;4(11):2025-2032.
Schelhaas H.J., van de Warrenburg B.P. Clinical, psychological, and genetic characteristics of spinocerebellar ataxia type 19 (SCA19). Cerebellum. 2005;4(1):51-54.
Schmahmann J.D., Sherman J.C. The cerebellar cognitive affective syndrome. Brain. 1998;121(Pt 4):561-579.
Schmidley J.W., Levinsohn M.W., et al. Infantile X-linked ataxia and deafness: a new clinicopathologic entity? Neurology. 1987;37(8):1344-1349.
Schols L., Bauer I., et al. Do CTG expansions at the SCA8 locus cause ataxia? Ann Neurol. 2003;54(1):110-115.
Schols L., Kruger R., et al. Spinocerebellar ataxia type 6: genotype and phenotype in German kindreds. J Neurol Neurosurg Psychiatry. 1998;64(1):67-73.
Schulz J.B., Di Prospero N.A., et al. Clinical experience with high-dose idebenone in Friedreich ataxia. J Neurol. 2009;256(Suppl 1):42-45.
Schut J. Hereditary ataxia: clinical study through six generations. Arch Neurol Psychiatr. 1954;63:535.
Sekijima Y., Ohara S., et al. Hereditary motor and sensory neuropathy associated with cerebellar atrophy (HMSNCA): clinical and neuropathological features of a Japanese family. J Neurol Sci. 1998;158(1):30-37.
Servadio A., Koshy B., et al. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genet. 1995;10(1):94-98.
Shakkottai V.G., Xiao M., et al. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33(1):81-88.
Shiloh Y., Tabor E., et al. In vitro phenotype of ataxia-telangiectasia (AT) fibroblast strains: clues to the nature of the “AT DNA lesion” and the molecular defect in AT. Kroc Found Ser. 1985;19:111-121.
Shimohata T., Nakajima T., et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26(1):29-36.
Shimojo Y., Osawa Y., et al. Severe infantile dentatorubral pallidoluysian atrophy with extreme expansion of CAG repeats. Neurology. 2001;56(2):277-278.
Shokeir M. X-linked cerebellar ataxia. Clin Genet. 1970;1:225.
Simon D., Seznec H., et al. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J Neurosci. 2004;24(8):1987-1995.
Smith J.K., Gonda V.E., et al. Unusual form of cerebellar ataxia; combined dentato-rubral and pallido-Luysian degeneration. Neurology. 1958;8(3):205-209.
Smith J.L., Cogan D.G. Ataxia-telangiectasia. Arch Ophthalmol. 1959;62:364-369.
Sobrido M.J., Cholfin J.A., et al. SCA8 repeat expansions in ataxia: a controversial association. Neurology. 2001;57(7):1310-1312.
Spira P.J., McLeod J.G., et al. A spinocerebellar degeneration with X-linked inheritance. Brain. 1979;102(1):27-41.
Spoendlin H. Optic cochleovestibular degenerations in hereditary ataxias. II. Temporal bone pathology in two cases of Friedreich’s ataxia with vestibulo-cochlear disorders. Brain. 1974;97(1):41-48.
Srivastava A.K., Choudhry S., et al. Molecular and clinical correlation in five Indian families with spinocerebellar ataxia 12. Ann Neurol. 2001;50(6):796-800.
Steckley J.L., Ebers G.C., et al. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology. 2001;57(8):1499-1502.
Stevanin G., Bouslam N., et al. Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann Neurol. 2004;55(1):97-104.
Stevanin G., Brice A. Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4). Cerebellum. 2008;7(2):170-178.
Stevanin G., Herman A., et al. Are (CTG)n expansions at the SCA8 locus rare polymorphisms? Nat Genet. 2000;24(3):213-215.
Stevanin G., Le Guern E., et al. A third locus for autosomal dominant cerebellar ataxia type I maps to chromosome 14q24.3-qter: evidence for the existence of a fourth locus. Am J Hum Genet. 1994;54(1):11-20.
Stewart G.S., Maser R.S., et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577-587.
Storey E., Bahlo M., et al. A new dominantly inherited pure cerebellar ataxia, SCA 30. J Neurol Neurosurg Psychiatry. 2009;80(4):408-411.
Storey E., Gardner R.J., et al. A new autosomal dominant pure cerebellar ataxia. Neurology. 2001;57(10):1913-1915.
Strupp M., Kalla R., et al. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62(9):1623-1625.
Sulek A., Hoffman-Zacharska D., et al. SCA8 repeat expansion coexists with SCA1–not only with SCA6. Am J Hum Genet. 2003;73(4):972-974.
Suraweera A., Lim Y., et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet. 2009;18(18):3384-3396.
Swartz B.E., Li S., et al. Pathogenesis of clinical signs in recessive ataxia with saccadic intrusions. Ann Neurol. 2003;54(6):824-828.
Syllaba L., Henner K. Contribution a l’independance de l’athetose double idiopathique et congenitale: atteinte familiale, syndrome dystrophique, signe du reseau vasculaire conjunctival, integrite psychique. Rev Neurol. 1926;1:541.
Takahashi H., Ishikawa K., et al. A clinical and genetic study in a large cohort of patients with spinocerebellar ataxia type 6. J Hum Genet. 2004;49(5):256-264.
Takiyama Y., Nishizawa M., et al. The gene for Machado-Joseph disease maps to human chromosome 14q. Nat Genet. 1993;4(3):300-304.
Takiyama Y., Sakoe K., et al. A Japanese family with spinocerebellar ataxia type 6 which includes three individuals homozygous for an expanded CAG repeat in the SCA6/CACNL1A4 gene. J Neurol Sci. 1998;158(2):141-147.
Thomas P.K., Workman J.M., et al. Behr’s syndrome. A family exhibiting pseudodominant inheritance. J Neurol Sci. 1984;64(2):137-148.
Thoren C. Diabetes mellitus in Friedreich’s ataxia. Acta Paediatr Suppl. 1962;135:239-247.
Traber M.G., Sokol R.J., et al. Impaired ability of patients with familial isolated vitamin E deficiency to incorporate alpha-tocopherol into lipoproteins secreted by the liver. J Clin Invest. 1990;85(2):397-407.
Tranchant C., Fleury M., et al. Phenotypic variability of aprataxin gene mutations. Neurology. 2003;60(5):868-870.
Tranebjaerg L., Teslovich T.M., et al. Genome-wide homozygosity mapping localizes a gene for autosomal recessive non-progressive infantile ataxia to 20q11-q13. Hum Genet. 2003;113(3):293-295.
Trottier Y., Lutz Y., et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature. 1995;378(6555):403-406.
Tsai Y.C., Fishman P.S., et al. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003;278(24):22044-22055.
Tsuchiya K., Ishikawa K., et al. A clinical, genetic, neuropathological study in a Japanese family with SCA 6 and a review of Japanese autopsy cases of autosomal dominant cortical cerebellar atrophy. J Neurol Sci. 1998;160(1):54-59.
Tsuji S. Spinocerebellar ataxia type 17: latest member of polyglutamine disease group highlights unanswered questions. Arch Neurol. 2004;61(2):183-184.
Ueno S., Kondoh K., et al. Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (DRPLA). Hum Mol Genet. 1995;4(4):663-666.
Uziel T., Savitsky K., et al. Genomic Organization of the ATM gene. Genomics. 1996;33(2):317-320.
Vahedi K., Joutel A., et al. A gene for hereditary paroxysmal cerebellar ataxia maps to chromosome 19p. Ann Neurol. 1995;37(3):289-293.
van Bogaert L., Martin L. Optic and cochleovestibular degenerations in the hereditary ataxias. I. Clinico-pathological and genetic aspects. Brain. 1974;97(1):15-40.
van de Loo S., Eich F., et al. Ataxin-2 associates with rough endoplasmic reticulum. Exp Neurol. 2009;215(1):110-118.
van de Warrenburg B.P., Frenken C.W., et al. Striking anticipation in spinocerebellar ataxia type 7: the infantile phenotype. J Neurol. 2001;248(10):911-914.
van de Warrenburg B.P., Verbeek D.S., et al. Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology. 2003;61(12):1760-1765.
van Swieten J.C., Brusse E., et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet. 2003;72(1):191-199.
Vance J., Pericak-Vance M., et al. Linkage and genetic analysis in adult onset periodic vestibulo-cerebellar ataxia: report of a new family. Am J Hum Genet. 1984;36:78S.
Verbeek D.S., van de Warrenburg B.P., et al. Mapping of the SCA23 locus involved in autosomal dominant cerebellar ataxia to chromosome region 20p13-12.3. Brain. 2004;127(Pt 11):2551-2557.
Virtaneva K., D’Amato E., et al. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat Genet. 1997;15(4):393-396.
von Brederlow B., Hahn A.F., et al. Mapping the gene for acetazolamide responsive hereditary paryoxysmal cerebellar ataxia to chromosome 19p. Hum Mol Genet. 1995;4(2):279-284.
Voncken M., Ioannou P., et al. Friedreich ataxia-update on pathogenesis and possible therapies. Neurogenetics. 2004;5(1):1-8.
Vuillaume I., Devos D., et al. A new locus for spinocerebellar ataxia (SCA21) maps to chromosome 7p21.3-p15.1. Ann Neurol. 2002;52(5):666-670.
Wang Q., Bardgett M.E., et al. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35(1):25-38.
Wara D.W., Ammann A.J. Thymosin treatment of children with primary immunodeficiency disease. Transplant Proc. 1978;10(1):203-209.
Warner T.T., Williams L., et al. DRPLA in Europe. Nat Genet. 1994;6(3):225.
Watanabe H., Tanaka F., et al. Frequency analysis of autosomal dominant cerebellar ataxias in Japanese patients and clinical characterization of spinocerebellar ataxia type 6. Clin Genet. 1998;53(1):13-19.
Watase K., Gatchel J.R., et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007;4(5):e182.
Watase K., Weeber E.J., et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron. 2002;34(6):905-919.
Waters M.F., Fee D., et al. An autosomal dominant ataxia maps to 19q13: Allelic heterogeneity of SCA13 or novel locus? Neurology. 2005;65(7):1111-1113.
Waters M.F., Minassian N.A., et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38(4):447-451.
Wessel K., Schroth G., et al. Significance of MRI-confirmed atrophy of the cranial spinal cord in Friedreich’s ataxia. Eur Arch Psychiatry Neurol Sci. 1989;238(4):225-230.
Wood J.D., Yuan J., et al. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci. 1998;11(3):149-160.
Worth P.F., Giunti P., et al. Autosomal dominant cerebellar ataxia type III: linkage in a large British family to a 7.6-cm region on chromosome 15q14-21.3. Am J Hum Genet. 1999;65(2):420-426.
Worth P.F., Houlden H., et al. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. 2000;24(3):214-215.
Yabe I., Sasaki H., et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol. 2003;60(12):1749-1751.
Yamashita I., Sasaki H., et al. A novel locus for dominant cerebellar ataxia (SCA14) maps to a 10.2-cm interval flanked by D19S206 and D19S605 on chromosome 19q13.4-qter. Ann Neurol. 2000;48(2):156-163.
Ying M., Xu R., et al. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. 2006;281(18):12580-12586.
Yoo S.Y., Pennesi M.E., et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37(3):383-401.
Yu G.Y., Howell M.J., et al. Spinocerebellar ataxia type 26 maps to chromosome 19p13.3 adjacent to SCA6. Ann Neurol. 2005;57(3):349-354.
Yvert G., Lindenberg K.S., et al. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet. 2000;9(17):2491-2506.
Zhang S., Xu L., et al. Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell. 2002;108(1):45-56.
Zhuchenko O., Bailey J., et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62-69.
Zoghbi H.Y., Frontali M., et al. Linkage studies in dominantly inherited ataxias. Adv Neurol. 1993;61:133-137.
Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217-247.
Zoghbi H.Y., Pollack M.S., et al. Spinocerebellar ataxia: variable age of onset and linkage to human leukocyte antigen in a large kindred. Ann Neurol. 1988;23(6):580-584.
Zu L., Figueroa K.P., et al. Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am J Hum Genet. 1999;64(2):594-599.
Zu T., Duvick L.A., et al. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24(40):8853-8861.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 67 The Cerebellum and the Hereditary Ataxias
Basic Cerebellar Structure, Function, and Dysfunction
Cerebellar Structure
The cerebellum can be divided anatomically in the anteroposterior and mediolateral dimensions (Figure 67-1A and B). Fissures divide the rostrocaudal length of the cerebellum into ten lobules, which are analogous to the gyri of the cerebral cortex. These lobules can be grouped into three lobes: the anterior lobe (lobules I–V), the posterior lobe (lobules VI–IX), and the flocculonodular lobe (lobule X) (Figure 67–1C). The mediolateral divisions are the vermis, which lies medially, and the hemispheres, which lie laterally and are separated from the vermis by shallow grooves.
Fig. 67-1 The human cerebellum.
(A and B, From Kandel ER and Schwartz, JH, eds. Principles of neural science, 2nd edn. New York: Elsevier, 1985, pp 504 and 505. C, Courtesy of Dr. Dawna Armstrong.)
The cerebellar cortex is formed by five cell types, arranged in the same relative orientation, regardless of anatomical or functional location (Figure 67-2A). Purkinje cells are among the largest neurons in the central nervous system (CNS), and represent the sole output of the cortex. Cerebellar granule cells, which reside in the internal granular layer (IGL), are the most numerous neuronal population in the CNS. (Their numbers are nearly equal to all other CNS neurons combined.) Granule cells extend long, bifurcated axons (parallel fibers) into the molecular layer, where they travel for several millimeters and synapse with 1000–2000 Purkinje cells on their dendritic arbors; in turn, each Purkinje cell forms synapses with as many as 1,000,000 parallel fibers. Granule cells are the only excitatory cell type in the cerebellar cortex. Three local inhibitory interneurons also reside in the cortex. Golgi type II cells are large neurons found in the IGL that receive mossy fiber inputs and inhibit granule cells. Basket and stellate cells are found in the molecular layer, where they receive excitatory inputs from parallel fibers and are inhibitory to Purkinje cells.
Fig. 67-2 Major cell types of the cerebellar cortex.
(B, From Kandel ER et al., eds. Principles of neural science, 4th edn. New York: McGraw-Hill, 2003, p 838.)
The neurons of the deep cerebellar nuclei (DCN) form four separate clusters in the cerebellar white matter (see Figure 67-1A). These cells receive inhibitory cortical input from the Purkinje cells, and excitatory extracerebellar afferents from climbing and mossy fibers. The fastigial nuclei lie most medially, and receive input from vermal Purkinje cells. The interposed nuclei (nucleus globose and nucleus emboliform) receive input from paravermal Purkinje cells. The dentate nuclei, which are the largest DCN in primates, receive input from the cerebellar hemispheres. Along with some Purkinje cells found in the flocculonodular lobe, the DCN represent the sole efferent output of the cerebellum, projecting to multiple brainstem nuclei in the medulla and pons, as well as the red nucleus and thalamus.
Cerebellar input consists of three classes of afferents. Climbing fibers originate in the inferior olives, which project to the contralateral cerebellar hemisphere. They provide strong excitatory input to Purkinje cells and neurons of the DCN. Each climbing fiber synapses on no more than ten Purkinje cells, while each Purkinje cell receives input from only one climbing fiber. Mossy fibers originate in multiple locations in the thalamus, brainstem, and spinal cord, and form excitatory synapses on granule cells and Golgi type II neurons in the IGL, as well as on neurons of the DCN. Climbing fibers, and to a lesser extent mossy fibers, are arranged in a series of parasagittal bands that “stack” next to one another in the mediolateral dimension (Figure 67-2B); the functional significance of this arrangement will be discussed in the next section. Finally, multilayer fibers originate in the hypothalamus, locus ceruleus, and raphe nuclei, and form a diffuse fiber network in the cortex and DCN.
Like the cerebral cortex, cerebellar cortical afferents and efferents are arranged in a somatotopic manner (reviewed in Manni and Petrosini [2004]). This organization is more complex than that of the cerebral cortex in two ways. First, at least three separate homunculi (one in the anterior lobe and one in each of the paravermian lobules) can be identified (Figure 67-3A). These are believed to converge on separate, continuous somatotopic maps in the four cerebellar nuclei. This arrangement, which was initially described in other species, has recently been directly demonstrated in humans using functional neuroimaging [Bushara et al., 2001; Manni and Petrosini, 2004]. Second, the sensory map within a cerebellar folium is not continuous. Instead, a fractured somatotopy exists, where each region of the face or limb has multiple representations within the folium that may not be adjacent to one another (Figure 67-3B). The reason for this arrangement is unknown, but it may be necessary to bring climbing fiber and mossy fiber inputs into register with one another.
Cerebellar Function
As noted above, cerebellar afferent input is segregated into a series of parasagittal bands. There are two main hypotheses as to how this parasagittal arrangement might be functionally important. First, comparison of climbing fiber to mossy fiber excitatory inputs within these bands could contribute to refining movements and motor learning [Ito, 2002]. It has also been proposed that mossy fiber input, which is translated through granule cells and their parallel fibers, could transform spatial relationships into temporal ones as the signal passes from Purkinje cells closest to the granule cells to those farther down the parallel fiber path. The receptive Purkinje cells could then detect timing differences between the incoming granule cell and climbing fiber signals. Thus, the cerebellum could act as an alarm clock, stopwatch, or coincidence/time shift detector [Braitenberg, 1967].
Positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) suggest that the cerebellum is involved in several nonmotor functions. These include sensory discrimination, attention, working memory, semantic association, verbal learning and memory, and complex problem solving [Allen et al., 1997]. A number of other studies have also linked the cerebellum to various higher-order cognitive processes [Leiner et al., 1991; Middleton and Strick, 1994]. Most of these functions are subserved by the cerebellar hemispheres and the dentate nuclei.
Cerebellar Dysfunction
Cerebellar (ataxic) dysarthria produces abnormalities in articulation (slurring and inaccuracy in range, force, and timing; patients sound as if inebriated), phonation (vocal quality can be harsh or uneven), resonation (a nasal quality is common), and prosody (patients tend to place equal and excessive emphasis on each syllable). Other kinds of dysarthria exist; there is, for example, the hypokinetic dysarthria seen in Parkinson’s disease, in which bradykinesia due to loss of dopaminergic neurons in the substantia nigra tends to produce low volume of speech or hoarseness, reduced pitch variability, palilalia (repetition of syllables), and very slow speech. There is evidence that left cerebellar lesions interfere with speech prosody because of disruptions of interconnections with the right cerebral hemisphere, which mediates this process [Brazis et al., 2001]. Cerebellar mutism is sometimes seen after removal of vermian tumors, and may result from bilateral involvement of the dentatorubrothalamic tracts.
Nonmotor manifestations of cerebellar disease can also occur. A cerebellar cognitive affective syndrome has been described in patients with isolated cerebellar lesions of a number of different etiologies [Schmahmann and Sherman, 1998]. In this syndrome, executive functions, spatial cognition, personality changes, and language deficits are all present. The postulated neural substrate involves circuits that link prefrontal, posterior parietal, superior temporal, and limbic cortices to the cerebellum.
Differential Diagnosis of Ataxia
The underlying cause of ataxia is virtually always primary or secondary dysfunction of the cerebellum. Cerebellar disease is a result of a number of underlying conditions, many of which are listed in Box 67-1. The most prevalent causes of acute cerebellar ataxia are viruses (e.g., coxsackievirus, rubeola, varicella), toxins (e.g., alcohol, barbiturates, antiepileptic drugs), and traumatic insults. Tumors of the cerebellum (vermal and hemispheric) or the adjacent brainstem, particularly pontine gliomas, are also relatively common. Numerous metabolic conditions affecting the central and peripheral nervous systems lead to ataxia. Congenital abnormalities of the nervous system, such as Chiari malformation, Dandy–Walker malformation, and basilar impression, are associated with ataxia. Endocrinologic abnormalities, particularly hypothyroidism, may also present with ataxia as the predominant manifestation. Hemorrhage, infarction, and embolism are unusual causes of cerebellar damage and often occur in conjunction with damage to neighboring brain regions. Angioblastomas of the cerebellum are rare; von Hippel–Lindau disease, a neurocutaneous condition, is associated with vascular lesions of the cerebellum, as is the PHACE (posterior fossa malformations, hemangiomas, arterial anomalies, coarctation of aorta and cardiac defects, and eye abnormalities) syndrome [Frieden et al., 1996].
Box 67-1 Selected Causes of Ataxia in Childhood
Degenerative and/or Genetic
Ataxia, retinitis pigmentosa, deafness, vestibular abnormality, and intellectual deterioration [Francois and Descampas*] Cerebellar ataxia with deafness, anosmia, absent caloric responses, nonreactive pupils, and hyporeflexia [Brown, 1959] Hereditary cerebellar ataxia, intellectual retardation, choreoathetosis, and eunuchoidism [Altchule and Kotowski*]Management of Cerebellar Dysfunction and Ataxia
When ataxia is the result of certain metabolic disorders or toxins, trauma, or neoplasia, specific therapies for the underlying condition can be curative (Table 67-1). Direct neurological causes of ataxia, however, such as inherited diseases, are not yet amenable to treatment. Patients can benefit from various aids for ambulation, as well as physical therapy and occupational therapy; though these approaches will not restore mobility in advanced disease, they none the less alleviate other symptoms such as spasticity and can greatly improve quality of life.
The Hereditary Ataxias
The classification of the hereditary ataxias has long posed a clinical challenge. Prior to the advent of gene cloning technology, these conditions were classified based on their mode of inheritance, clinical features, and pathological findings. These approaches ultimately proved to be unsatisfactory, and often led to more confusion than clarity. Patients with known hereditary ataxia have a range of cerebellar dysfunction that may be exclusively cerebellar in origin or combined with other features, including brainstem dysfunction, spinal cord abnormalities, extrapyramidal findings, neuropathy, retinopathy, deafness, cataracts, seizures, and/or dementia [Greenfield, 1954; Koeppen and Barron, 1984; Zoghbi et al., 1993]. The age of onset and clinical manifestations of any given ataxia may differ significantly from family to family and even in the same kindred, making specific assignment beyond mode of inheritance difficult [Currier et al., 1972]. Likewise, attempts to classify ataxia according to the anatomic site of the most dominant pathology (e.g., spinal cord, cerebellum) are also flawed. Descriptors such as “olivopontocerebellar atrophy” (OPCA), a pathological finding seen most predominantly in the dominantly inherited ataxias, do not adequately serve to distinguish between them.
Autosomal-Recessive Inherited Syndromes
The autosomal recessive inherited ataxias (summarized in Table 67-2) are a varied group of disorders that tend to have an early age of onset in infancy or childhood. They can involve multiple regions of the central and peripheral nervous system. In general, earlier age of onset heralds a more aggressive disease course. Several causative mutations have been identified, leading to a clear genetic classification scheme.
Friedreich’s Ataxia (Freidreich’s Ataxia; Spinocerebellar Ataxia – OMIM 229300)
Friedreich ataxia is one of the most common hereditary ataxias, with a prevalence of approximately 1–2 per 100,000 in the general population [Harding, 1984]. Caucasians are affected more often than other racial groups, with a prevalence closer to 1 per 50,000 [Cossee et al., 1997]. Clinically, the disease is signaled by the presence of ataxia and corticospinal tract dysfunction. Other associated features include nystagmus, kyphoscoliosis, and an unusual cardiomyopathy. The disease was originally reported by Friedreich [Friedreich, 1863], who believed it was associated only with spinal cord abnormalities, subsequent findings demonstrated involvement of the medulla, peripheral nerves, and cerebral cortex in some cases.
Clinical manifestations
Loss of cells in cranial nerves VII, X, and XII causes facial weakness, with cerebellar dysarthria and dysphagia. Lateral or horizontal nystagmus may be prominent, and visual pursuit is punctuated with small jerks. Impairment of pupillary responsivity and limited extraocular movements are also common. Additional ophthalmologic abnormalities include cataracts, retinitis pigmentosa, and optic atrophy. Auditory dysfunction is common [Cassandro et al., 1986], and is documented by abnormal brainstem auditory-evoked potentials that are likely the result of impairment of both central and peripheral acoustic pathways [Knezevic and Stewart-Wynne, 1985]. The degree of hearing compromise is variable; in addition, some patients experience vertigo. Although higher cortical function appears intact by clinical measures, some studies suggest a decrease in information processing speed [Hart et al., 1985].
The lower limbs typically show loss of deep tendon reflexes, but reflexes may be preserved for a longer period in young children [Salih et al., 1990], and patients with late-onset disease can be spastic with hyperreflexia. Bilateral extensor toe signs are easily elicited. Cremasteric and abdominal reflexes may be lost. The masseter reflex remains intact [Auger, 1992]. Occasionally, weakness, muscle atrophy, and profound hypotonia may be evident, usually associated with spinal nerve disintegration. Virtually all facets of cerebellar function are involved, and both gross and fine coordination are affected. Position sense, vibration sense, and other skills requiring normal posterior column function are grossly impaired. Autonomic innervation of superficial layers of skin may be involved. Pain, touch, and temperature sensation are usually intact, but diminish over the course of the illness. Evidence of axonal sensory neuropathy, indicated by electrodiagnostic studies, is invariably present. Somatosensory-evoked potentials are abnormal, as is central motor function, as determined by magnetic stimulation [Claus et al., 1988].
Patients with Friedreich’s ataxia often develop talipes equinovarus or pes cavus. Deformities of the hand can also occur. Flexor spasms can become a great source of pain. Many patients suffer from kyphoscoliosis [Labelle et al., 1986], and may be unable to walk secondary to poor balance and an abnormally shifted center of gravity (Figure 67-4). Surgical intervention can correct the abnormal spinal curvature, and this is likely to help with ventilation and autonomic functions, even if it does not restore ambulation.
Fig. 67-4 Friedreich’s ataxia and moderate scoliosis in a 13-year-old boy who is unable to walk independently.
Cardiomyopathies are the most common accompanying cardiac abnormalities, although congenital heart disease has been described on occasion. The cardiomyopathy is often progressive, and is documented even before clinical manifestations by the presence of electrocardiogram (EKG) abnormalities (e.g., deep Q waves, low-wave QRS complexes, S-T segment changes, T-wave inversion) [Harding and Hewer, 1983; Child et al., 1986]. Arrhythmias may be particularly worrisome and life-threatening. EKG and vector cardiographic changes are demonstrable in more than 90 percent of patients [Child et al., 1986]. Left ventricular hypertrophy or symmetric ventricular hypertrophy is frequently evident on echocardiography [Pentland and Fox, 1983]. Hypertrophic cardiomyopathy is the leading cause of death in Friedreich’s ataxia patients.
Diabetes mellitus is associated with Friedreich’s ataxia; even in its absence, the glucose tolerance curve may be abnormal. Insulin resistance appears to be the explanation [Khan et al., 1986]. Unaffected relatives may also manifest insulin resistance. Episodic hypothermia, intermittent vomiting, and ventilatory dysfunction have also been reported [Thoren, 1962].
Imaging studies show both structural and functional changes that correlate with pathologic progression. MRI studies document atrophy of the upper cervical spinal cord and cerebellum [Wessel et al., 1989]. PET imaging demonstrates a widespread increase in cerebral metabolic rate for glucose in the brain of ambulatory patients with Friedreich’s ataxia. As the disease progresses, glucose metabolism decreases in a regionally specific manner; only the caudate and lenticular nuclei have increased metabolic rate in nonambulatory patients [Gilman et al., 1990].
Diagnosis is suggested by the presence of the expected clinical manifestations and sometimes by family history. Distinct diagnostic criteria for Friedreich’s ataxia were advanced by Geoffroy and colleagues [Geoffroy et al., 1976]: autosomal-recessive inheritance, onset before 10 years of age, gait ataxia, dysarthria, absent deep tendon reflexes, dorsal column signs, and weakness. The presence of an abnormal 5-hour glucose tolerance curve is corroborative. Spinal fluid findings may include elevated protein content and mild pleocytosis.
Pathology
The primary site of involvement on pathologic examination is the spinal cord. Gross examination reveals a shrunken cord, with the posterior columns, spinocerebellar tracts, corticospinal tracts, and posterior rootlets strikingly affected by varying degrees of fiber loss, demyelination, and gliosis. Conditional Freidreich’s ataxia gene inactivation in experimental animals showed that anomalies observed in the DRG (dorsal root ganglion) neurons are primary events, whereas neuronal loss in the posterior thoracic nucleus (Clarke’s column) and degeneration in the posterior columns may be secondary events [Simon et al., 2004]. Axons and myelin alike are involved, although the medullary nuclei (Burdach’s nuclei) are usually spared [Greenfield, 1954]. Structures in the medial root zone, including ganglion cells and the posterior roots that terminate within them, manifest varying degrees of involvement but are typically decreased in number. The lumbar and sacral roots are most severely affected, and the large, myelinated lumbar spinal nerves lose many axons. In contrast, anterior horn cells and their rootlets are usually uninvolved; however, minor pathologic alterations may be evident. Myopathic changes are rare.
The density of large myelinated fibers in sural nerve biopsies is reduced, even in young children, and often worsens with age [Said et al., 1986].
Pathologic changes in the heart include evidence of cardiomyopathy and involvement of blood vessels, nerves, and ganglia [James et al., 1987].
Genetics
Friedreich’s ataxia is transmitted as an autosomal-recessive disorder; the finding of consanguinity among several families clearly supported the autosomal-recessive inheritance pattern. The disease affects males and females equally. The establishment of the criteria proposed by Geoffroy and co-workers [Geoffroy et al., 1976] provided a basis for research studies and allowed clinical identification of families for linkage studies. Genetic linkage studies mapped the Friedreich’s ataxia gene to chromosome 9 and confirmed that the “Acadian” form of ataxia, characterized by a slower course of degeneration [Barbeau et al., 1984; Keats et al., 1989], also maps to chromosome 9 [Chamberlain et al., 1988; Keats et al., 1989]. Friedreich’s ataxia is caused by decreased expression of the mitochondrial protein, frataxin, encoded by the X25 gene located at 9q13 [Bidichandani et al., 1998; Koeppen, 1998].
Campuzano et al. [1996] were the first to identify an unstable GAA trinucleotide repeat in the first intron of X25. They also identified three different point mutations. In the group of 74 patients without a point mutation, 71 were homozygous for expanded alleles, and 3 were heterozygous for the expanded repeat. Five patients with point mutations were heterozygous for the expansion. The size of the repeat ranged from 7 to 22 on normal alleles and from 200 to 900 on Friedreich’s ataxia chromosomes. Further studies revealed that the triplet repeat could get as large as 1700 units, and that the lengths of both the larger and the smaller expanded alleles correlated inversely with the age of onset and severity of disease [Durr et al., 1996; Filla et al., 1996]. The mean allele length is significantly higher in Friedreich’s ataxia patients with diabetes, cardiomyopathy, and loss of reflexes in the upper extremities. Of 187 patients with autosomal-recessive ataxia, 140 were homozygous for a GAA expansion [Durr et al., 1996]. Furthermore, age-dependent accumulation of large GAA expansions contributes to the progressive pathology involving in the DRGs. Therefore, somatic instability of the expanded GAA triple repeat sequence may contribute directly to disease pathogenesis and progression in specific tissues [De Biase et al., 2007].
Frataxin is localized to the inner mitochondrial membrane and is thought to play a role in mitochondrial respiration and iron accumulation. Deletion of the frataxin homolog in yeast leads to mitochondrial iron accumulation and increased susceptibility to oxidative stress, with an increased production of free radicals (reviewed in Voncken et al. [2004]). These observations, coupled with the fact that expanded trinucleotide repeats interfere with transcription elongation of the X25 gene and cause a frataxin deficiency state, might explain the human phenotype [Bidichandani et al., 1998]. The high evolutionary conservation of frataxin has enabled the development of disease models in various organisms, from the unicellular eukaryote Saccharomyces cerevisiae to the mouse. Yeast was an important model system for identifying the crucial role of frataxin in the regulation of intracellular iron trafficking, iron-sulfur (Fe-S) cluster and heme biogenesis, and oxidative metabolism. Several Caenorhabditis elegans models with knockdown of the C. elegans FXN homolog frh-1 show conflicting results in response to oxidative stress, but all of them show reduced life span and may be useful for drug screening in Freidreich’s ataxia. Moderate reduction of the Drosophila frataxin homolog (dfh) causes a hypersensitive response to hypoxia, which upholds a causative role for oxidative stress in Freidreich’s ataxia. Mouse models of Freidreich’s ataxia have provided the most insight, however (reviewed in Puccio [2009]). The knockout model of frataxin suffers early embryonic lethality without iron accumulation, while heterozygosity for the frataxin null allele shows no pathological phenotype. These results suggest that residual frataxin expression associated with expansion mutation is critical for survival [Cossee et al., 2000]. Conditional animal models are viable and reproduce certain morphological and biochemical features observed in patients with Freidreich’s ataxia, including cardiac hypertrophy without skeletal muscle involvement, large sensory neuron dysfunction without alteration of small sensory and motor neurons, and deficient activities of complexes I through III of the respiratory chain and of the aconitases, enzymes essential in the citric acid cycle (reviewed in Puccio [2009]). Humanized GAA repeat expansion mouse models were obtained by combining the constitutive knockout model with the transgenic expression of a yeast artificial chromosome (YAC) carrying the human locus. These animals exhibit oxidative stress, leading to progressive neuronal and cardiac pathology [Al-Mahdawi et al., 2006]. Although none of these mouse models is authentic, a multimodel approach facilitates the study of different pathophysiological mechanisms and the examination of different therapeutic strategies. Why only selective neuronal systems are vulnerable to the loss of frataxin function remains unknown.
Treatment
Given the proposed function of frataxin protein in iron-sulfur cluster biosynthesis and the increased oxidative stress found with decreased protein levels, several groups have initiated treatment with idebenone, a short-chain analog of coenzyme Q10 (reviewed in Schulz et al. [2009]). All but one of these trials (the shortest trial) with idebenone at 5 mg/kg and 10 mg/kg/day have demonstrated improved cardiac function and decreased ventricular mass: important findings, since cardiac dysfunction is the leading cause of death in Friedreich’s ataxia patients. In these low-dose trials, idebenone treatment has no effect on the progression of ataxia [Schulz et al., 2009], but Di Prospero et al. showed that higher doses of idebenone (up to 60 mg/kg/day) are well tolerated, provide neurological benefit, and improve activities of daily life (ADL) in Freidreich’s ataxia patients who are still ambulatory [Di Prospero et al., 2007].
A high proportion of Freidreich’s ataxia patients have decreased serum CoQ10 levels, which was the best predictor of a positive clinical response to CoQ10/vitamin E therapy. Low- and high-dose CoQ10/vitamin E therapies were equally effective in improving ICARS scores (International Cooperative Ataxia Rating Scale) [Cooper et al., 2008]. Therapy with other antioxidants, iron chelators, and/or glutathione peroxidase mimetics has been suggested (and in some cases tried), but no clear beneficial effect has been established.
Expansion of intronic GAA repeat likely induces chromatin changes and in turn FXN silencing in Freidreich’s ataxia [Herman et al., 2006]. Treatment with a histone deacetylase (HDAC) inhibitor restored wild-type frataxin levels in the nervous system and heart, and increased histone H3 and H4 acetylation in chromatin near the GAA repeat [Rai et al., 2008]. These findings provide a very promising prospect for Freidreich’s ataxia treatment using HDAC inhibitors.
Friedreich’s Ataxia 2 (OMIM 601992)
Other families with a Friedreich’s ataxia disease phenotype, but without point mutations or GAA expansions in the X25 gene, have been described [Kostrzewa et al., 1997; Christodoulou et al., 2001]. In one family, linkage to a locus on chromosome 9p23–p11 has been found [Christodoulou et al., 2001]. The biological basis for the genocopy is not known, but it has been proposed that mutations in genes that lie in the iron metabolism pathway may be responsible.
Vitamin E Deficiency and Related Syndromes
Numerous mutations have been identified in the tocopherol transfer protein gene (TTP1), which encodes the α-tocopherol transfer protein (TTPA) [Ouahchi et al., 1995; Cavalier et al., 1998]. TTPA is expressed in the liver and is responsible for the incorporation of tocopherol (vitamin E) into very-low-density lipoprotein (VLDL) [Traber et al., 1990]. Mutated TTPA proteins fail to facilitate the secretion of vitamin E from cells, and the severity of the AVED pathology corresponds to the degree of impairment associated with each mutation [Qian et al., 2006].
Ataxia-Telangiectasia (AT; Louis-Bar Syndrome – OMIM 208900)*
The initial clinical description of this disease was reported by Syllaba and Henner [1926], who called it congenital double athetosis-aconjunctival vascular plexus syndrome. Louis-Bar [1941] reported a 9-year-old male with associated ataxia and oculocutaneous telangiectasias, classifying the case as a previously unrecognized phakomatosis. Two additional independent reports subsequently appeared in the literature [Biemond, 1957; Boder and Sedgwick, 1957], and Boder and Sedgewick named the condition ataxia-telangiectasia.
Clinical manifestations
Early motor development appears to be normal until around the time that the child starts walking, when ataxia is noted. The ataxia is progressive and ultimately leads to an inability to ambulate by the beginning of the second decade. Choreoathetosis and dystonia occur in up to 90 percent of patients, and these motor findings become more prominent with increasing age. Facial weakness leads to the characteristic impassive faces, as well as drooling and dysarthria. While strength is initially normal, many patients in their 20s and 30s develop progressive spinal muscular atrophy, predominantly affecting the hands and feet [Gatti et al., 1991]. Peripheral neuropathy in the form of diminished deep tendon reflexes and loss of large fiber sensation is also seen. Mental function is well preserved, although deficits in short-term memory can occur in the third and fourth decades [Gatti et al., 1991].
Oculomotor apraxia is a distinguishing feature of the disease. The apraxia commonly presents before the appearance of conjunctival telangiectasias and is characterized by defects of initiating voluntary saccades, hypometric voluntary saccades accompanied by compensatory eye-blinking and/or head-thrusting movements, and disrupted smooth pursuit movements [Smith and Cogan, 1959; Baloh et al., 1978]. Involuntary saccade initiation and optokinetic nystagmus may be impaired as well, and oculovestibular reflexes may be increased.
Telangiectasias are usually first observed in patients between the ages of 2 and 4 years, although they can occur as early as birth and as late as 14 years of age [Centerwall and Miller, 1958; McFarlin et al., 1972] (Figure 67-5). In addition to the conjunctivae, they appear on exposed areas of the skin, particularly areas of friction and trauma, such as the auricle, nasal bridge, and antecubital and popliteal spaces. Exposure to sun enhances their appearance. Premature aging of hair and skin is frequent, as are skin infections, including chronic blepharitis. Other skin changes, consisting of vitiligo and café-au-lait spots, can be seen. Rarely, scleroderma-like lesions occur.
Fig. 67-5 Characteristic conjunctival telangiectasias observed in a 12-year-old with ataxia-telangiectasia.
Recurrent sinopulmonary infections are common, affecting 90 percent of patients, and usually result in chronic bronchitis, bronchiectasis, or both. The impairment of cellular immunity is also manifest by abnormally developed or absent adenoids, tonsils, lymphoid tissue, and thymus gland. Patients have an impaired delayed hypersensitivity response to skin-sensitizing antigens and a delayed homograft-rejection response [McFarlin and Oppenheim, 1969].
Patients with AT are prone to develop tumors and are more likely than the general population to have Hodgkin’s disease, leukemia, lymphoma, and lymphosarcoma. Other associated neoplasms include brain tumors, gastric adenocarcinomas, ovarian dysgerminomas, gonadoblastomas, cystic adenofibromas, uterine leiomyomas, and thyroid adenomas [Miller and Chatten, 1967; Gatti and Good, 1971].
Patients with less severe variant forms of AT have been described [Gilad et al., 1998; Saviozzi et al., 2002]. Immunodeficiency, telangiectasias, cancer, and sinopulmonary infections may be absent or reduced, but the neurological manifestations still occur. These individuals have a later onset and slower progression of neurological signs, longer life spans, and decreased chromosomal instability and cellular radiosensitivity.
Laboratory findings
Fibroblast and lymphoid cell lines from AT patients show increased radiosensitivity, leading to spontaneous and radiation-induced chromosomal breakage and rearrangement, which can be assessed in culture [Shiloh et al., 1985]. In lymphoid cell lines, the chromosomal breaks occur at the loci of T-cell antigen receptors and immunoglobulin genes, where DNA rearrangements and deletions naturally occur [Hecht and Hecht, 1985].
Pathology
Cerebellar atrophy primarily affects the Purkinje and granular cells, although basket cells can also be involved. Neuronal degeneration of the dentate and olivary nuclei and the substantia nigra has been demonstrated, and nuclear changes occur in the cells of the oculomotor complex, pretectal nuclei, and hypothalamus. Pituitary cells often have enlarged or dysplastic nuclei. Older patients have denervation of the posterior columns of the spinal cord. Degeneration of the anterior horn cells, decreased numbers of satellite cells in the dorsal root ganglia, and nucleomegaly of Schwann cells have been described [De Leon et al., 1976].
Genetics
The worldwide incidence of AT is approximately 1 in 40,000–100,000 live births; the carrier frequency ranges from 0.5 to 1.0 percent. The gene locus maps to chromosome 11q22–23 [Gatti et al., 1988]. The ATM (ataxia telengiectasia mutated) gene is very large, with 66 exons spanning 150 kb of genomic DNA [Uziel et al., 1996]; this fact makes mutation detection a challenge. In addition, because of the large number of different mutations, the majority of patients are compound heterozygotes. Still, using a combination of techniques, the estimated mutation detection rate has been detected to be over 95 percent in affected individuals [Buzin et al., 2003]. It is estimated that truncating mutations account for about 85 percent of mutations, while point mutations are responsible for the rest [Buzin et al., 2003].
ATM is a nuclear phosphoprotein homologous to a family of phosphatidylinositol kinase-related proteins, and it functions in the DNA damage response and in cell cycle regulation [Savitsky et al., 1995a, b]. Individuals with classic AT have little or no active ATM protein, while most individuals with variant AT have one severe mutation coupled with a mild mutation. In this latter condition, some ATM activity is preserved, which probably explains the milder phenotype [Saviozzi et al., 2002].
Loss of ATM activity could lead to neuronal degeneration in at least two ways. As ATM is a monitor of DNA strand breaks important for initiating repair, it has been suggested that ATM is necessary for the elimination of neural cells with genomic damage, and that the preservation of those cells may contribute to neuronal dysfunction [Herzog et al., 1998]. Several other diseases that affect DNA repair (ataxia-telangiectasia-like disorder, ataxia-ocular apraxia 1, xeroderma pigmentosum, Cockayne’s syndrome, Nijmegen breakage syndrome, and de Sanctis–Caccione syndrome) have a cerebellar phenotype, suggesting that cerebellar neurons may be very sensitive to DNA damage. This hypothesis highlights a nuclear role for the ATM protein. In neurons, however, a large percentage of the protein is found in the cytoplasm and axons. Increased numbers of lysosomes are found in Purkinje cell bodies and axons in ATM-deficient mice [Barlow et al., 2000]. This, coupled with an observed interaction with β-adaptin, a protein important for clathrin-mediated endocytosis, suggests that vesicle/protein transport may also be disrupted in ATM-defective cells [Lim et al., 1998].
Treatment
AT is a multisystem disease and its treatment requires attention to multiple types of interventions. Currently, we are not able to halt progressive neurodegeneration, although some features of the disease might respond to treatment. l-DOPA derivatives and anticholinergics may improve basal ganglia dysfunction. Amantadine, fluoxetine, or buspirone may help with the loss of balance and impaired speech; furthermore, tremors can often be controlled by gabapentin, clonazepam, or propranolol (reviewed in Lavin et al. [2007]).
Patients should receive vigorous supportive therapy, with particular attention directed to recurrent sinopulmonary infection. Attempts to improve immunologic status by plasma transfusion of thymosin and provision of fetal thymus transplants have not altered the course of the neurologic signs and symptoms [Wara and Ammann, 1978]. Treatment of neoplasms is a delicate proposition because patients are extremely sensitive to radiation and chemotherapy, and demonstrate resultant ulcerative dermatitis, severe esophagitis, dysphagia, and deep-tissue necrosis [Gotoff et al., 1967].
Ataxia-Telangiectasia-Like Disorder (ATLD – OMIM 604391)
ATLD patients have been described with clinical presentations including cerebellar ataxia and oculomotor apraxia, which are essentially identical to that of ataxia-telangiectasia except that telangiectasias are absent [Klein et al., 1996; Fernet et al., 2005]. Two of the patients, who were brothers, presented first with chorea that later progressed to ataxia [Klein et al., 1996]. In 2005, Fernet et al. described ten ATLD patients from three unrelated Saudi Arabian families. These patients had slowly progressive ataxia and ocular apraxia, but they did not develop tumors. Furthermore, they did not have telangiectasia, raised AFP, or reduced immunoglobulin levels [Fernet et al., 2005].
Increased susceptibility to DNA damage was noted in all four patients. Stewart et al. [1999] showed that the disorder maps to chromosome 11q21 in all four patients. Loss-of-function mutations in the hMRE11A gene are causative. Like ATM, MRE11A is important for DNA repair, and may act as a sensor of DNA damage. Stewart et al. estimate that approximately 6 percent of AT patients may in fact have ATLD [Stewart et al., 1999].
Ataxia-Oculomotor Apraxia 1 (AOA1; Early-Onset Ataxia with Ocular Motor Apraxia and Hypoalbuminemia – OMIM 208920)
This autosomal-recessive ataxia was initially described in Japanese families but has subsequently been described in other areas of the world as well [Tranchant et al., 2003]. It is the most frequent cause of autosomal-recessive cerebellar ataxia in Japan, and the second most common cause in Portugal. The presentation is nearly identical to that of AT without the non-neurological features [Aicardi et al., 1988], and accounts for up to 10 percent of autosomal-recessive cerebellar ataxias. Gait imbalance and dysarthria are the typical presenting features, although chorea can sometimes be the presenting symptom. Ocular apraxia typically occurs a few years after symptom onset; with time, progressive ophthalmoplegia occurs as well. Areflexia, dystonia, choreoathetosis, sensory and motor neuropathy, cognitive impairment, and retinal/macular lesions seen on funduscopy can also be part of the presentation. Life span is not affected.
Laboratory studies reveal hypoalbuminemia and hypercholesterolemia in the majority of cases as a later manifestation of the disease. Neuroimaging demonstrates cerebellar and sometimes brainstem atrophy. Pathology studies show loss of Purkinje cells in the cerebellum, and degeneration of the posterior columns, spinocerebellar tracts, and anterior horn cells of the spinal cord [Sekijima et al., 1998].
The gene, APTX, maps to chromosome 9p13.3 and is expressed in all body tissues [Date et al., 2001; Moreira et al., 2001]. Several mutations have been described, as have compound heterozygotes; frameshift and nonsense mutations result in more severe phenotypes than missense mutations. The APTX gene product, aprataxin, has domains that suggest a potential role in single-strand DNA repair, and has been shown to interact with another protein implicated in this process [Sano et al., 2004]. CoQ10 is often low in patients with AOA1 [Le Ber et al., 2007], and this deficiency responds to CoQ10 supplementation [Quinzii et al., 2005].
Spinocerebellar Ataxia, Autosomal-Recessive 1 (SCAR1 – OMIM 606002); Ataxia-Oculomotor Apraxia 2 (AOA2)
A second AT-like spinocerebellar ataxia with slightly different features than AOA1 has been described in families of several different ethnic backgrounds. The clinical features of AOA2 include gait ataxia, sensorimotor neuropathy, and ocular apraxia, the latter occurring about 50 percent of the time. While overall cognitive function is normal, subtle changes in executive function can be seen on neuropsychologic testing [Le Ber et al., 2004]. Neuroimaging demonstrates cerebellar atrophy. AOA2 is distinguished from AOA1 by the later age of onset, less common ocular apraxia, high levels of serum AFP, and normal serum albumin. The functional prognosis is also better in AOA2. The gene (SETX) maps to chromosome 9q34 [Nemeth et al., 2000]. Mutations cause premature termination of the senataxin protein, which is a member of the helicase family and is involved in RNA maturation and termination [Moreira et al., 2004]. Recently, Suraweera et al. [2009] have proposed a role for senataxin in the regulation/modulation of transcription.
Interestingly, other mutations in senataxin are transmitted in an autosomal-dominant manner and cause juvenile amyotrophic lateral sclerosis (ALS4) [Chen et al., 2004a]; the basis for the dramatic phenotypic difference between the two diseases is unknown.
Spinocerebellar Ataxia, Autosomal-Recessive 2 (SCAR2 – OMIM 213200); Cerebellar Hypoplasia, Nonprogressive Norman Type
Norman [1940] described three siblings in one family and two siblings in another who had had cerebellar ataxia and mental deficiency since early life. Postmortem examinations showed severe cerebellar granule cell loss. One child showed delayed motor development and mental deficiency in infancy. Other features included small head, cataracts, increased knee jerks, and intention tremor [Norman, 1940
[/not-level-membership-for-neurology-category]
